 Blake M. Hauser
Blake M. Hauser Ashley Lau1
Ashley Lau1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 14 May 2019
Sec. Pituitary Endocrinology
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00290
This article is part of the Research TopicMolecular network study of pituitary adenomasView all 16 articles
Background: The vast majority of pituitary tumors are benign and behave accordingly; however, a fraction are invasive and are more aggressive, with a very small fraction being frankly malignant. The cellular pathways that drive transformation in pituitary neoplasms are poorly characterized, and current classification methods are not reliable correlates of clinical behavior. Novel techniques in epigenetics, the study of alterations in gene expression without changes to the genetic code, provide a new dimension to characterize tumors, and may hold implications for prognostication and management.
Methods: We conducted a review of primary epigenetic studies of pituitary tumors with a focus on histone modification, DNA methylation, and transcript modification.
Results: High levels of methylation have been identified in invasive and large pituitary tumors. DNA methyltransferase overexpression has been detected in pituitary tumors, especially in macroadenomas. Methylation differences at CpG sites in promoter regions may distinguish several types of tumors from normal pituitary tissue. Histone modifications have been linked to increased p53 expression and longer progression-free survival in pituitary tumors; sirtuins are expressed at higher values in GH-expressing compared to nonfunctional adenomas and correlate inversely with size in somatotrophs. Upregulation in citrullinating enzymes may be an early pathogenic marker of prolactinomas. Numerous genes involved with cell growth and signaling show altered methylation status for pituitary tumors, including cell cycle regulators, components of signal transduction pathways, apoptotic regulators, and pituitary developmental signals.
Conclusions: The limited clinical predictive capacity of the current pituitary tumor classification system suggests that tumor subclasses likely remain to be discovered. Ongoing epigenetic studies could provide a basis for adding methylation and/or acetylation screening to standard pituitary tumor workups. Identifying robust correlations between tumor epigenetics and corresponding histological, radiographic, and clinical course information could ultimately inform clinical decision-making.
Pituitary tumors constitute at least 15% of intracranial neoplasms (1–4). The anterior pituitary is composed of several hormone-producing cell types, including corticotrophs, somatotrophs, lactotrophs, mammosomatotrophs, thyrotrophs, and gonadotrophs, all of which can give rise to tumors, leading to the heterogeneous group of neoplasms encompassed by the diagnosis of pituitary adenomas (5, 6). Recent work suggests that the term “pituitary tumor” may be more appropriate than “pituitary adenoma,” but “adenoma” has been used in this review in some instances to accurately reflect findings reported in the literature (7). These tumors can be functional—producing hormones that reflect their lineage with concordant systemic effects—or nonfunctional, producing systemic sequelae through compromised pituitary function. Each general group can produce symptoms by offending any of a number of adjacent anatomical structures. These groups and individual tumors can have a wide range of clinical behaviors, from benign to highly invasive. Their long-term behavior and response to therapy are not reliably predicted by current classification methods.
The biological underpinnings of pituitary tumors have been investigated to predict and manage them with more precision. The accumulation of genetic mutations confers downstream oncogenic changes such as sustained proliferation, invasion, angiogenesis, growth suppression evasion, and cell death resistance (8). These mutations can consist of changes to the DNA sequence, as well as chromosomal alterations and copy number changes (Figure 1; Table 1). Large scale genomic sequencing has revealed several mutations in subtypes of pituitary adenomas (9–18). However, on the whole, mutations that drive oncogenesis are sparse across pituitary tumors. Consequently, non-mutational sources of gene expression alteration in pituitary tumors are undergoing investigation.
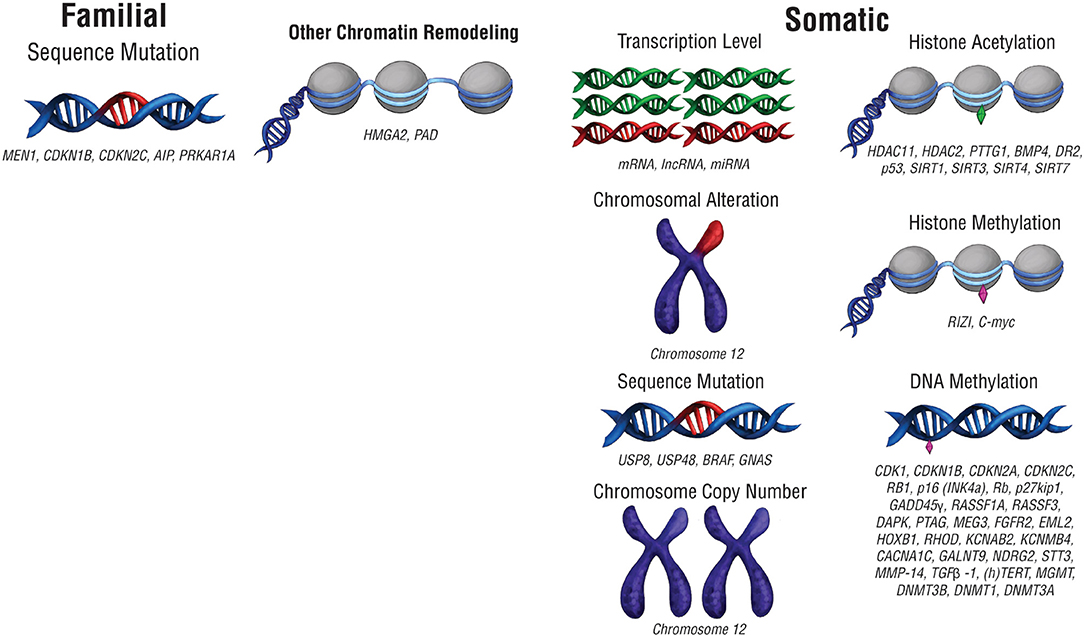
Figure 1. Pre-translational modifications that affect gene expression. Modifications that affect gene expression can occur at any point along the path from packaged DNA to protein expression. Alterations to chromosome structure or changes in chromosome copy number can affect expression at a chromosomal level. Epigenetic chromatin modifications, including histone acetylation and histone methylation, can also alter gene expression levels. At the DNA level, mutations in DNA sequence or DNA methylation can change the nature of the genes expressed as well as their level of expression. RNA transcript copy number also affects protein expression.

Table 1. Genetic, regulatory, and epigenetic mutations.
Epigenetics—the study of alterations in gene expression without changes to DNA sequence—provides an alternative avenue of tumorigenesis and disease characterization (Figure 1). DNA methylation by DNA methyltransferases (DMNTs), amongst other enzymes, typically silences gene expression by reducing the access of transcriptional machinery to methylated segments of DNA (Table 1). Changes to histone placement can also affect DNA transcription. Histone deacetylases (HDACs) and sirtuins modulate histone acetylation, which generally improves transcriptional access to surrounding DNA, while histone methyltransferases like RIZI alter methylation, which either improves or restricts transcriptional access depending on the methylation site (Table 1). Histone citrullination can also affect chromatin expression, and it can be mediated by peptidylarginine deaminase (PAD) enzymes (Table 1).
Epigenetic changes can also alter mRNA transcript levels, resulting in either upregulation or downregulation of gene expression. Changes in messenger RNA (mRNA) transcript levels can occur as part of oncogenic transformation (Figure 1; Table 1). Alterations in mRNA expression can modulate downstream changes in protein expression levels, which in turn drive cellular function. Additionally, differential expression of long non-coding RNA (lncRNA) and microRNA (miRNA) can result in or accompany oncogenesis (Table 1).
Incorporating epigenetics into tumor classification schemes for other types of cancer has improved clinical reliability. In breast cancer, DNA hypermethylation of promoter CpG islands corresponds to the presence of certain hormonal receptors as well as clinical tumor progression (19). Similarly, promoter methylation in glioblastoma correlates with response to therapy (20), and DNA methylation-based classification schemes have shown utility in tumor subclassification and prognosis in meningioma (21). For pituitary tumors, subclassification is a more complicated problem given that the multiple cell types present in the pituitary can give rise to tumors with varied secretory properties. Additionally, with the exception of metastasis, criteria for pituitary tumor malignancy remain unclear. Recent studies profiling epigenetic changes in pituitary tumors have shed new insights into the classification of pituitary tumors and may possibly augment prediction of clinical behavior.
The World Health Organization (WHO) classification draws upon pituitary adenohypophyseal cell lineage to categorize tumors into acidophilic, corticotroph, and gonadotroph subtypes based on transcription factor and hormone expression (22, 23). The absence of hormones and transcription factors defines a null cell adenoma. Subtypes with a propensity to exhibit invasiveness, rapid growth, recurrence, and resistance are categorized as clinically aggressive. Terms such as “aggressive,” “invasive,” and “large” are sometimes used interchangeably in the literature, even though these terms refer to distinct features of pituitary tumors. This review maintains the same terminology accompanying the discussed finding from the cited literature. Clinically aggressive tumors often have high mitotic activity and Ki-67 expression but are not defined by a single biomarker (24). Invasive tumors are loosely defined by a combination of clinical, radiological, and histopathological findings, and do not necessarily imply clinical aggressiveness in terms of disease control or recurrence risk (24). Aggressive tumors are defined by clinical characteristics, and primarily reflect a tumor's rate of recurrence (25). Invasion of the dura can be seen in up to 45.5% of pituitary adenomas (26). Combining multiple modes of information to classify tumors likely provides more accurate prognostic information (27). Novel biomarkers may facilitate the division of pituitary tumors into more clinically useful categories.
Large-scale genomic studies to identify molecular alterations have been thorough, but pituitary tumors display relatively few genetic aberrations compared to other tumor types and cancers. As a result, pituitary tumor genetic information has limited potential to inform the course of treatment for the numerous pituitary tumors without these identified genetic aberrations. Genome-wide association studies (GWAS) have been used to identify genetic markers associated with pituitary tumor development. GWAS has revealed common variants (10p12.31, 10q21.1, and 13q12.13) that are associated with sporadic pituitary tumors (17).
Recurrent genetic mutations have been identified in small subsets of pituitary tumors. The first category of tumors with recurrent genetic mutations are those that arise due to familial syndromes (Table 2) which include McCune-Albright syndrome, multiple endocrine neoplasia types 1 and 4 (MEN1 and MEN4), familial isolated pituitary adenomas (FIPA), and Carney complex (28). Interestingly, only a small percentage of sporadic pituitary tumors harbor mutations in the genes implicated in familial pituitary tumor disorders [MEN1, Cyclin Dependent Kinase Inhibitor 1B (CDKN1B), Cyclin Dependent Kinase Inhibitor 2C (CDKN2C), Aryl-Hydrocarbon Receptor Interacting Protein (AIP), and Protein Kinase cAMP-Dependent Type 1 Regulatory Subunit Alpha (PRKAR1A) (9–11)]. Select somatic genetic alterations have been identified in several subtypes of adenomas, including high mobility group A 2 (HMGA2) amplification via focal amplification or abnormalities of chromosome 12 in prolactinomas (12), Ubiquitin Specific Peptidase 8 (USP8), Ubiquitin Specific Peptidase 48 (USP48), and BRAF in corticotroph adenomas (13, 15, 29), and activating mutations in GNAS in GH-secreting pituitary adenomas (14, 16). Chromosome arm-level copy-number alterations also recur within a subset of pituitary tumors, the majority of which are functional macroadenomas (18). In some cases, familial mutations and chromosome abnormalities have been associated with larger tumor size. Genetic associations offer limited utility beyond distinguishing tumor subtype, which may indicate that epigenetic regulation plays a role in the clinical course of pituitary tumors.
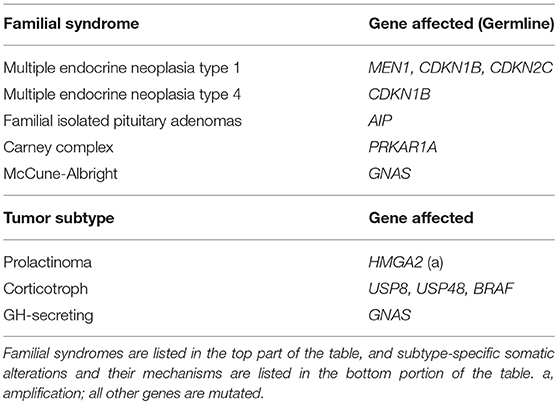
Table 2. Familial and somatic mutations associated with pituitary tumors.
The distinct gene expression profiles of pituitary tumors correlate to some extent with hormone expression status. Additionally, gene expression profiles may have some predictive value with respect to clinical aggressiveness (18). Tumor classification systems with a molecular basis often yield more insight into tumor origin, tumor behavior, and probable clinical outcomes than purely histological approaches (21, 30, 31). The idea that gene expression signatures may provide insights about tumor behavior and outcome has motivated transcriptomic studies in pituitary tumors to gain a better understanding of how signatures correlate with tumor properties and patient outcomes. These studies also permit the evaluation of the effect of genetic mutations on a protein level, which can improve the clinical utility of tumor genetic information.
Subtypes of pituitary tumors express distinct transcriptional profiles from each other and from normal pituitary gland tissue as assessed by gene microarrays and RNA-Seq. Given that transcription profile differences correlate with tumor presence and subtype, it is possible that they also offer a molecular approach to improving classification schemes. Relative to normal pituitary tissue, pituitary tumors have differentially expressed mRNA transcripts (32–37), lncRNA transcripts (36, 38), and miRNA transcripts (39–42). Notably, investigations have found that two miRNAs (miR196a-2 and miR-212) which target HMGA transcripts can be deregulated in all tumor types (43–47). Changes in expression profile also manifest in different subclasses of tumors (48–50). In particular, deregulation of miR-183 in prolactinomas has been associated with clinical aggressiveness (51).
However, large-scale transcriptome analyses often produce gene expression results that conflict with the findings in other studies. Heterogeneity within tumor samples, a small patient sample that fails to capture a representative selection of tumor samples, and different experimental conditions may contribute to divergent study results. Furthermore, one potential shortcoming of transcriptome studies is the use of predetermined histological or radiographic categories to partition gene expression results. This approach can identify gene expression differences representative of each tumor type or subclass but precludes identification of novel classes within the tumor population that are independent of histological characteristics. The disparate clinical trajectories of pituitary tumor subtypes suggest that there are likely to be subclasses with varying degrees of invasiveness and aggressiveness that remain to be discovered. However, no system for classifying pituitary tumors accurately correlates genetic markers with clinical outcomes, so tumor genetic information still has limited clinical utility.
Given the centrality of gene expression changes and the relative dearth of genetic abnormalities in pituitary tumors, epigenetic modifications have received considerable attention.
DNA methylation was examined (Figure 2), particularly at CpG islands in gene promoters, where methylation often correlates with gene silencing (52). Approximately two-thirds of reference epigenomes have been found to contain quiescent signatures, whereas only 5% of genomes contain promoter and enhancer signatures (53). The “histone code” hypothesis, which states that certain patterns of post-translational modifications on histone tails can function as signals in gene regulatory processes (54) (Figure 2), has also led to a small number of studies that assay histone modifications in pituitary tumors.
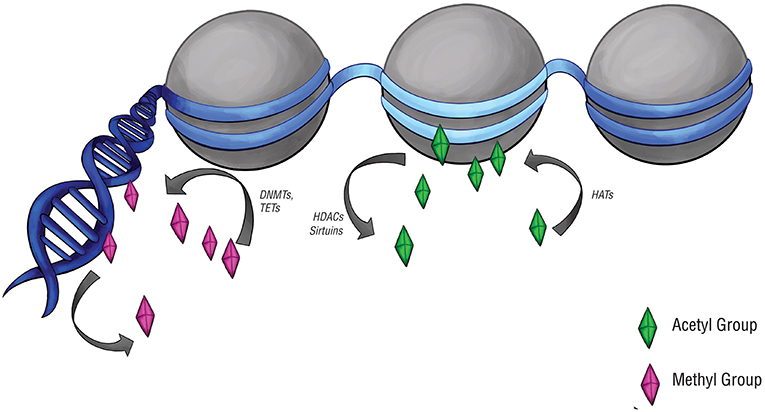
Figure 2. Epigenetic modifications and regulators. Epigenetic modifications are carried out by a set of specialized enzymes that act directly on DNA or on the histones around which chromatin is wrapped. Histone acetyltransferases (HATs) place acetyl groups on chromatin, while histone deacetylases (HDACs) and sirtuins do the opposite. Acetylation of histones typically upregulates gene expression, denoted here by the light blue region of DNA. DNA methyltransferases (DNMTs) and ten-eleven translocation enzymes (TETs) can alter the level of DNA or histone methylation, which typically downregulates gene expression. This is denoted by the navy-blue regions of DNA.
Methylation of gene promoters is frequently deregulated in tumors (55), and appears to be a common mechanism for gene inactivation in pituitary tumors. Given the simplicity of detecting DNA methylation at targeted locations within the genome, clinically meaningful epigenetic findings could be implemented rapidly to guide treatment plan development.
DNA methyltransferase (DNMT) enzymes catalyze methylation at CpG dinucleotides, with DNMT3A and DNMT3B serving as the de novo methyltransferases, and DNMT1 as the maintenance methyltransferase. Ten-eleven translocation (TET) enzymes may also participate in regulating methylation as removers of methylation modifications (56). Early observations that classic oncogene and tumor suppressor mutations were absent in pituitary tumors led to the realization that promoter methylation changes constituted an alternative mechanism by which causative genes could be deregulated. Numerous genes involved with cell growth and signaling show altered methylation status, including cell cycle regulators [Cyclin Dependent Kinase 1 (CDK1) (57), CDKN1B (58), Cyclin Dependent Kinase Inhibitor 2A (CDKN2A) (59), CDKN2C (59, 60), Retinoblastoma Transcriptional Corepressor 1 (RB1) (58, 61), CDKN2A protein (p16INK4a) (58), Retinoblastoma (Rb) (62), CDKN1B protein (p27kip1) (63), Growth Arrest and DNA Damage 45γ (GADD45γ) (64, 65)]; components of signal transduction pathways [Ras Associated Domain Family Member 1A (RASSF1A) (66) and Ras Associated Domain Family Member 3 (RASSF3)]; apoptotic regulators [Death-Associated Protein Kinase (DAPK) (67) and Pituitary Tumor Apoptosis Gene (PTAG) (68)]; developmental gene Maternally Expressed 3 (MEG3) (69); and the growth factor signaling component Fibroblast Growth Factor Receptor 2 (FGFR2) (70).
High levels of methylation may be associated with clinically aggressive behavior in pituitary tumors (Table 3). DNMT1 and DNMT3A overexpression has been detected in pituitary tumors (77). Both were significantly associated with more aggressive tumors, with DNMT1 levels also significantly higher in macroadenomas. Relatively higher levels of expression of DNMT3B has also been found in pituitary tumors in comparison to normal tissue with no difference in DNMT1 and DNMT3A expression (71). It is possible that the transfer of methyl groups will also result in regions of DNA being hypomethylated and therefore expressed at a higher level. As DNA hypomethylation has also shown some association with cancerous behavior, high levels of DNMT expression could theoretically increase the risk of malignancy through hypomethylation mechanism as well (79).
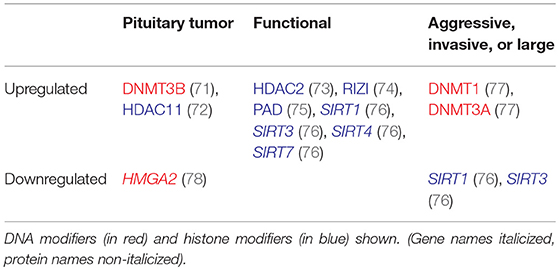
Table 3. Altered regulation of epigenetic modifiers in aggressive, invasive, or large, and functional tumors.
Genome-wide methylome studies have found methylation differences at promoter region CpG sites to distinguish several types of adenomas from normal pituitary tissue (80). A subset of genes are hypermethylated in nonfunctional adenomas as well as growth hormone (GH) and prolactin (PRL) secreting adenomas. However, only Echinoderm Microtubule Associated Protein Like 2 (EML2), Homeobox B1 (HOXB1), and Rho-Related GTP-Binding Protein (RHOD), also demonstrate a corresponding decrease in expression, suggesting that the degree of promoter methylation may not always translate to actual changes in gene expression. HOXB1 has been identified as a tumor suppressor gene in glioma (81), and RHOD may affect cytoskeletal reorganization and transportation (82).
Variations in methylation may also exist at CpG sites across the genome, including intergenic sites and gene body regions (83, 84). Nonfunctional tumors have displayed global hypermethylation relative to hormonally active tumors (84), particularly GH (83). Genes involved in ion channel signaling, including Voltage-Gated Potassium Channel Subunit Beta-2 (KCNAB2), Calcium-Activated Potassium Channel Subunit Beta-4 (KCNMB4), and Calcium Voltage-Gated Channel Subunit Alpha1 C (CACNA1C), can be hypermethylated in nonfunctional tumors (83). However, expression does not consistently correlate with methylation at promoter CpG sites, so it is unclear to what extent such epigenetic changes affect phenotype. Hypomethylated CpGs are significantly more common in invasive NF pituitary adenomas than hypermethylated CpGs (84). One differentially methylated site was associated with Polypeptide N-acetylgalactosaminyltransferase 9 (GALNT9), and its expression was downregulated significantly in the invasive tumors. A number of other differentially methylated sites correspond to genes involved in cell adhesion, indicating a possible mechanism by which methylation changes influence tumor phenotype. Characteristic methylation patterns can also be associated with GH-secreting, ACTH-secreting and NF pituitary tumor subtypes, but further investigation is required to better elucidate the extent to which differential methylation exists across subtypes of pituitary tumors (85).
The lack of correlation between hypermethylation and gene expression implies that additional regulatory mechanisms beyond methylation remain to be discovered. Methylation and expression levels of the N-myc Downstream-Regulated Gene 2 (NDRG2) and Signal Transducer and Activator of Transcription 3 (STAT3) promoters are also uncorrelated, along with a lack of correlation with clinical factors (86, 87). Methylation of the Matrix Metallopeptidase 14 (MMP-14) and Transforming Growth Factor Beta 1 (TGFβ-1) promoters was also not associated with tumor functionality or recurrence (88). Hypermethylation of the Human Telomerase Reverse Transcriptase (hTERT) promoter, which can contribute to cellular immortalization and tumorigenesis, has also been noted across different pituitary tumor subtypes but has not been associated with significant differences in tumor parameters, tumor subtype, or prognosis (89). However, differential methylation of TERT promoters has not been consistently observed (90). Larger patient sample sizes are required to better understand the clinical impact of specific epigenetic changes in pituitary tumors.
Methylation at the O6-Methylguanine-DNA Methyltransferase (MGMT) promoter is of particular interest given its utility epigenetic modification in determining response to temozolomide (TMZ) in glioblastoma (20). Temozolomide has been used for aggressive pituitary adenomas, with mixed results (91, 92). Thus far, MGMT expression, rather than promoter methylation, appears to better correlate with TMZ response in pituitary adenomas; however, a limited number of studies have examined this relationship, and TMZ is still administered, particularly in the context of aggressive pituitary tumors, regardless of MGMT status (24, 93). MGMT promoter methylation has also been associated with tumor regrowth in pituitary adenoma (94). Even though MGMT methylation does not offer as much predictive value for pituitary tumors as glioblastoma, finding clinically informative methylation markers remains the goal.
Acetylation of histone tails, particularly H3 and H4, is generally seen as a mark of active regions of the genome, whereas methylation of histone tails, particularly lysine 9 on H3 (H3K9), is associated with inactive heterochromatin (95). Epigenetic markers can be dynamically modified by chromatin regulators including the histone acetyltransferases (HATs) and histone deacetylases (HDACs).
Multiple chromatin regulators are differentially regulated in pituitary tumors, including HMGA2 and HDAC2 (73, 78), suggesting that pituitary tumors likely have altered patterns of histone modifications (Table 3). HDAC11 has been shown to interfere with p53 expression in pituitary tumor cells (72). Global acetylation resulted in an increase in Pituitary Tumor-Transforming Gene 1 (PTTG1), Bone Morphogenic Protein 4 (BMP4), and Dopamine Receptor 2 (DR2) expression in pituitary tumor cells, suggesting that global alterations in epigenetic modifications may result in gene expression changes (96, 97). Given the nonspecific effects of global acetylation modification, it is unclear how relevant global acetylation findings are to pituitary tumor pathogenesis and therapeutic applications.
The differential expression of some members of the sirtuin family (SIRT) of HDACs has been observed in somatotropinomas as compared to NF pituitary adenomas (76). SIRT1 was overexpressed in somatotropinomas, while SIRT3, SIRT4, and SIRT7 were under-expressed in NF pituitary adenomas. SIRT1 overexpression correlated with smaller tumor size, while SIRT3 under-expression correlated with larger tumor size. There was no association between sirtuin levels and invasiveness or Ki-67 proliferative index.
Non-invasive pituitary adenomas express significantly higher levels of RIZI, which acts as a tumor-suppressor as well as a possible histone methyltransferase, and lower levels of C-myc, as compared to invasive pituitary adenomas (74). Increased RIZI expression also correlates with significant differences in methylation at four CpG sites, reduced H3K4/H3K9 methylation, and enhanced H3K27 methylation, as well as significantly longer progression-free survival (74). Additionally, p53 mis-expression correlates with H3K9 methylation (98). Specific examples of correlations between epigenetic modifications and gene expression further affirm the possibility that histone modifications may alter gene expression in pituitary tumors.
Peptidylarginine deaminase (PAD) enzymes facilitate histone citrullination, which can modulate chromatin expression (Table 3). Increased PAD prevalence in prolactinomas and somatoprolactinomas has been associated with increased mRNA targeting of oncogenes HMGA, Insulin-like Growth Factor 1 (IGF-1), and Neuroblastoma MYC Oncogene (N-MYC) by miRNAs, which may yield insight into the etiology of the affected tumor subtypes (75).
The limited ability of existing pituitary tumor classification systems to predict clinical behavior motivates investigation into a molecular taxonomy with clinical implications. Though epigenetic signatures are not yet incorporated into clinical decision making for pituitary tumors, the importance of methylation and epigenetic signatures is increasingly appreciated across brain tumors with clinical implications (20, 21). Given the absence of recurrent oncogenic mutations and copy number alterations in many pituitary tumors, epigenetic mechanisms present an intriguing biological avenue for further exploration.
Further elucidation of the mechanisms underlying gene deregulation is needed before viable therapeutic strategies can be developed. Several compounds that inhibit epigenetic modifications are FDA approved, though it remains to be determined whether compounds that globally affect DNA methylation and histone modifications can provide specificity and efficacy in targeting the genetic pathways deregulated in pituitary tumors. More targeted strategies for modulating epigenetic modifications, though currently still in early development, may hold promise for treatment of pituitary tumors (99). Ultimately, an integrated classification of epigenetic, genetic, and histopathologic features may augment the collective predictive power of molecular taxonomy in translating to clinical practice.
BH, AL, and SG contributed to the review of the literature and the drafting of the manuscript. WB and ID conceived of the project and provided guidance throughout the writing process.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors acknowledge Natalie Charewicz for contributing the original illustrations shown in Figures 1, 2. BH was supported by award Number T32GM007753 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
1. Gittleman H, Ostrom QT, Farah PD, Ondracek A, Chen Y, Wolinsky Y, et al. Descriptive epidemiology of pituitary tumors in the United States, 2004-2009. J Neurosurg. (2014) 121:527–35. doi: 10.3171/2014.5.JNS131819
2. Fernandez A, Karavitaki N, Wass JA. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol. (2010) 72:377–82. doi: 10.1111/j.1365-2265.2009.03667.x
3. Daly AF, Rixhon M, Adam C, Dempegioti A, Tichomirowa MA, Beckers A. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab. (2006) 91:4769–75. doi: 10.1210/jc.2006-1668
4. Raappana A, Koivukangas J, Ebeling T, Pirila T. Incidence of pituitary adenomas in Northern Finland in 1992-2007. J Clin Endocrinol Metab. (2010) 95:4268–75. doi: 10.1210/jc.2010-0537
5. Melmed S. Pathogenesis of pituitary tumors. Nat Rev Endocrinol. (2011) 7:257–66. doi: 10.1038/nrendo.2011.40
6. Asa SL, Ezzat S. The pathogenesis of pituitary tumours. Nat Rev Cancer. (2002) 2:836–49. doi: 10.1038/nrc926
7. Asa SL, Casar-Borota O, Chanson P, Delgrange E, Earls P, Ezzat S, et al. From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): an International Pituitary Pathology Club proposal. Endocr Relat Cancer. (2017) 24:C5–8. doi: 10.1530/ERC-17-0004
8. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
9. Lecoq AL, Kamenicky P, Guiochon-Mantel A, Chanson P. Genetic mutations in sporadic pituitary adenomas–what to screen for? Nat Rev Endocrinol. (2015) 11:43–54. doi: 10.1038/nrendo.2014.181
10. Wenbin C, Asai A, Teramoto A, Sanno N, Kirino T. Mutations of the MEN1 tumor suppressor gene in sporadic pituitary tumors. Cancer Lett. (1999) 142:43–7. doi: 10.1016/S0304-3835(99)00111-1
11. Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. (2006) 312:1228–30. doi: 10.1126/science.1126100
12. Finelli P, Pierantoni GM, Giardino D, Losa M, Rodeschini O, Fedele M, et al. The High Mobility Group A2 gene is amplified and overexpressed in human prolactinomas. Cancer Res. (2002) 62:2398–405.
13. Ma ZY, Song ZJ, Chen JH, Wang YF, Li SQ, Zhou LF, et al. Recurrent gain-of-function USP8 mutations in Cushing's disease. Cell Res. (2015) 25:306–17. doi: 10.1038/cr.2015.20
14. Ronchi CL, Peverelli E, Herterich S, Weigand I, Mantovani G, Schwarzmayr T, et al. Landscape of somatic mutations in sporadic GH-secreting pituitary adenomas. Eur J Endocrinol. (2016) 174:363–72. doi: 10.1530/EJE-15-1064
15. Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F, et al. Mutations in the deubiquitinase gene USP8 cause Cushing's disease. Nat Genet. (2015) 47:31–8. doi: 10.1038/ng.3166
16. Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. (1989) 340:692–6. doi: 10.1038/340692a0
17. Ye Z, Li Z, Wang Y, Mao Y, Shen M, Zhang Q, et al. Common variants at 10p12.31, 10q21.1 and 13q12.13 are associated with sporadic pituitary adenoma. Nat Genet. (2015) 47:793–7. doi: 10.1038/ng.3322
18. Bi WL, Horowitz P, Greenwald NF, Abedalthagafi M, Agarwalla PK, Gibson WJ, et al. Landscape of genomic alterations in pituitary adenomas. Clin Cancer Res. (2017) 23:1841–51. doi: 10.1158/1078-0432.CCR-16-0790
19. Wu Y, Sarkissyan M, Vadgama JV. Epigenetics in breast and prostate cancer. Methods Mol Biol. (2015) 1238:425–66. doi: 10.1007/978-1-4939-1804-1_23
20. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. (2005) 352:997–1003. doi: 10.1056/NEJMoa043331
21. Sahm F, Schrimpf D, Stichel D, Jones DTW, Hielscher T, Schefzyk S, et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. (2017) 18:682–94. doi: 10.1016/S1470-2045(17)30155-9
22. Lopes MBS. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathol. (2017) 134:521–35. doi: 10.1007/s00401-017-1769-8
23. Mete O, Lopes MB. Overview of the 2017 WHO classification of pituitary tumors. Endocr Pathol. (2017) 28:228–43. doi: 10.1007/s12022-017-9498-z
24. Di Ieva A, Rotondo F, Syro LV, Cusimano MD, Kovacs K. Aggressive pituitary adenomas–diagnosis and emerging treatments. Nat Rev Endocrinol. (2014) 10:423–35. doi: 10.1038/nrendo.2014.64
25. Raverot G, Burman P, McCormack A, Heaney A, Petersenn S, Popovic V, et al. European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumours and carcinomas. Eur J Endocrinol. (2018) 178:G1–24. doi: 10.1530/EJE-17-0796
26. Meij BP, Lopes MB, Ellegala DB, Alden TD, Laws ER Jr. The long-term significance of microscopic dural invasion in 354 patients with pituitary adenomas treated with transsphenoidal surgery. J Neurosurg. (2002) 96:195–208. doi: 10.3171/jns.2002.96.2.0195
27. Trouillas J, Roy P, Sturm N, Dantony E, Cortet-Rudelli C, Viennet G, et al. A new prognostic clinicopathological classification of pituitary adenomas: a multicentric case-control study of 410 patients with 8 years post-operative follow-up. Acta Neuropathol. (2013) 126:123–35. doi: 10.1007/s00401-013-1084-y
28. Bi WL, Larsen AG, Dunn IF. Genomic alterations in sporadic pituitary tumors. Curr Neurol Neurosci Rep. (2018) 18:4. doi: 10.1007/s11910-018-0811-0
29. Chen J, Jian X, Deng S, Ma Z, Shou X, Shen Y, et al. Identification of recurrent USP48 and BRAF mutations in Cushing's disease. Nat Commun. (2018) 9:3171. doi: 10.1038/s41467-018-05275-5
30. Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell. (2015) 27:728–43. doi: 10.1016/j.ccell.2015.04.002
31. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation-based classification of central nervous system tumours. Nature. (2018) 555:469–74. doi: 10.1038/nature26000
32. Evans CO, Moreno CS, Zhan X, McCabe MT, Vertino PM, Desiderio DM, et al. Molecular pathogenesis of human prolactinomas identified by gene expression profiling, RT-qPCR, and proteomic analyses. Pituitary. (2008) 11:231–45. doi: 10.1007/s11102-007-0082-2
33. Evans CO, Young AN, Brown MR, Brat DJ, Parks JS, Neish AS, et al. Novel patterns of gene expression in pituitary adenomas identified by complementary deoxyribonucleic acid microarrays and quantitative reverse transcription-polymerase chain reaction. J Clin Endocrinol Metab. (2001) 86:3097–107. doi: 10.1210/jcem.86.7.7616
34. Moreno CS, Evans CO, Zhan X, Okor M, Desiderio DM, Oyesiku NM. Novel molecular signaling and classification of human clinically nonfunctional pituitary adenomas identified by gene expression profiling and proteomic analyses. Cancer Res. (2005) 65:10214–22. doi: 10.1158/0008-5472.CAN-05-0884
35. Morris DG, Musat M, Czirjak S, Hanzely Z, Lillington DM, Korbonits M, et al. Differential gene expression in pituitary adenomas by oligonucleotide array analysis. Eur J Endocrinol. (2005) 153:143–51. doi: 10.1530/eje.1.01937
36. Li J, Li C, Wang J, Song G, Zhao Z, Wang H, et al. Genome-wide analysis of differentially expressed lncRNAs and mRNAs in primary gonadotrophin adenomas by RNA-seq. Oncotarget. (2017) 8:4585–606. doi: 10.18632/oncotarget.13948
37. Ibanez-Costa A, Gahete MD, Rivero-Cortes E, Rincon-Fernandez D, Nelson R, Beltran M, et al. In1-ghrelin splicing variant is overexpressed in pituitary adenomas and increases their aggressive features. Sci Rep. (2015) 5:8714. doi: 10.1038/srep08714
38. Yu G, Li C, Xie W, Wang Z, Gao H, Cao L, et al. Long non-coding RNA C5orf66-AS1 is downregulated in pituitary null cell adenomas and is associated with their invasiveness. Oncol Rep. (2017) 38:1140–8. doi: 10.3892/or.2017.5739
39. Stilling G, Sun Z, Zhang S, Jin L, Righi A, Kovacs G, et al. MicroRNA expression in ACTH-producing pituitary tumors: up-regulation of microRNA-122 and−493 in pituitary carcinomas. Endocrine. (2010) 38:67–75. doi: 10.1007/s12020-010-9346-0
40. He W, Huang L, Li M, Yang Y, Chen Z, Shen X. MiR-148b, MiR-152/ALCAM axis regulates the proliferation and invasion of pituitary adenomas cells. Cell Physiol Biochem. (2017) 44:792–803. doi: 10.1159/000485342
41. Zhen W, Qiu D, Zhiyong C, Xin W, Mengyao J, Dimin Z, et al. MicroRNA-524-5p functions as a tumor suppressor in a human pituitary tumor-derived cell line. Horm Metab Res. (2017) 49:550–7. doi: 10.1055/s-0043-106437
42. Seltzer J, Ashton CE, Scotton TC, Pangal D, Carmichael JD, Zada G. Gene and protein expression in pituitary corticotroph adenomas: a systematic review of the literature. Neurosurg Focus. (2015) 38:E17. doi: 10.3171/2014.10.FOCUS14683
43. Bottoni A, Zatelli MC, Ferracin M, Tagliati F, Piccin D, Vignali C, et al. Identification of differentially expressed microRNAs by microarray: a possible role for microRNA genes in pituitary adenomas. J Cell Physiol. (2007) 210:370–7. doi: 10.1002/jcp.20832
44. Palmieri D, D'Angelo D, Valentino T, De Martino I, Ferraro A, Wierinckx A, et al. Downregulation of HMGA-targeting microRNAs has a critical role in human pituitary tumorigenesis. Oncogene. (2012) 31:3857–65. doi: 10.1038/onc.2011.557
45. Kitchen MO, Yacqub-Usman K, Emes RD, Richardson A, Clayton RN, Farrell WE. Epidrug mediated re-expression of miRNA targeting the HMGA transcripts in pituitary cells. Pituitary. (2015) 18:674–84. doi: 10.1007/s11102-014-0630-5
46. Esposito F, De Martino M, D'Angelo D, Mussnich P, Raverot G, Jaffrain-Rea ML, et al. HMGA1-pseudogene expression is induced in human pituitary tumors. Cell Cycle. (2015) 14:1471–5. doi: 10.1080/15384101.2015.1021520
47. Wierinckx A, Roche M, Legras-Lachuer C, Trouillas J, Raverot G, Lachuer J. MicroRNAs in pituitary tumors. Mol Cell Endocrinol. (2017) 456:51–61. doi: 10.1016/j.mce.2017.01.021
48. Ruebel KH, Leontovich AA, Jin L, Stilling GA, Zhang H, Qian X, et al. Patterns of gene expression in pituitary carcinomas and adenomas analyzed by high-density oligonucleotide arrays, reverse transcriptase-quantitative PCR, and protein expression. Endocrine. (2006) 29:435–44. doi: 10.1385/ENDO:29:3:435
49. Hussaini IM, Trotter C, Zhao Y, Abdel-Fattah R, Amos S, Xiao A, et al. Matrix metalloproteinase-9 is differentially expressed in nonfunctioning invasive and noninvasive pituitary adenomas and increases invasion in human pituitary adenoma cell line. Am J Pathol. (2007) 170:356–65. doi: 10.2353/ajpath.2007.060736
50. Galland F, Lacroix L, Saulnier P, Dessen P, Meduri G, Bernier M, et al. Differential gene expression profiles of invasive and non-invasive non-functioning pituitary adenomas based on microarray analysis. Endocr Relat Cancer. (2010) 17:361–71. doi: 10.1677/ERC-10-0018
51. Roche M, Wierinckx A, Croze S, Rey C, Legras-Lachuer C, Morel AP, et al. Deregulation of miR-183 and KIAA0101 in aggressive and malignant pituitary tumors. Front Med. (2015) 2:54. doi: 10.3389/fmed.2015.00054
52. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. (2002) 16:6–21. doi: 10.1101/gad.947102
53. Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. (2015) 518:317–30. doi: 10.1038/nature14248
54. Strahl BD, Allis CD. The language of covalent histone modifications. Nature. (2000) 403:41–5. doi: 10.1038/47412
55. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. (2011) 11:726–34. doi: 10.1038/nrc3130
56. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. (2012) 13:484–92. doi: 10.1038/nrg3230
57. Lidhar K, Korbonits M, Jordan S, Khalimova Z, Kaltsas G, Lu X, et al. Low expression of the cell cycle inhibitor p27Kip1 in normal corticotroph cells, corticotroph tumors, and malignant pituitary tumors. J Clin Endocrinol Metab. (1999) 84:3823–30. doi: 10.1210/jcem.84.10.6066
58. Yoshino A, Katayama Y, Ogino A, Watanabe T, Yachi K, Ohta T, et al. Promoter hypermethylation profile of cell cycle regulator genes in pituitary adenomas. J Neurooncol. (2007) 83:153–62. doi: 10.1007/s11060-006-9316-9
59. Kirsch M, Morz M, Pinzer T, Schackert HK, Schackert G. Frequent loss of the CDKN2C (p18INK4c) gene product in pituitary adenomas. Genes Chromosomes Cancer. (2009) 48:143–54. doi: 10.1002/gcc.20621
60. Hossain MG, Iwata T, Mizusawa N, Qian ZR, Shima SW, Okutsu T, et al. Expression of p18(INK4C) is down-regulated in human pituitary adenomas. Endocr Pathol. (2009) 20:114–21. doi: 10.1007/s12022-009-9076-0
61. Simpson DJ, Hibberts NA, McNicol AM, Clayton RN, Farrell WE. Loss of pRb expression in pituitary adenomas is associated with methylation of the RB1 CpG island. Cancer Res. (2000) 60:1211–6.
62. Tateno T, Nakano-Tateno T, Ezzat S, Asa SL. NG2 targets tumorigenic Rb inactivation in Pit1-lineage pituitary cells. Endocr Relat Cancer. (2016) 23:445–56. doi: 10.1530/ERC-16-0013
63. Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, et al. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. (1996) 85:707–20. doi: 10.1016/S0092-8674(00)81237-4
64. Zhang X, Sun H, Danila DC, Johnson SR, Zhou Y, Swearingen B, et al. Loss of expression of GADD45 gamma, a growth inhibitory gene, in human pituitary adenomas: implications for tumorigenesis. J Clin Endocrinol Metab. (2002) 87:1262–7. doi: 10.1210/jcem.87.3.8315
65. Bahar A, Bicknell JE, Simpson DJ, Clayton RN, Farrell WE. Loss of expression of the growth inhibitory gene GADD45gamma, in human pituitary adenomas, is associated with CpG island methylation. Oncogene. (2004) 23:936–44. doi: 10.1038/sj.onc.1207193
66. Qian ZR, Sano T, Yoshimoto K, Yamada S, Ishizuka A, Mizusawa N, et al. Inactivation of RASSF1A tumor suppressor gene by aberrant promoter hypermethylation in human pituitary adenomas. Lab Invest. (2005) 85:464–73. doi: 10.1038/labinvest.3700248
67. Simpson DJ, Clayton RN, Farrell WE. Preferential loss of Death Associated Protein kinase expression in invasive pituitary tumours is associated with either CpG island methylation or homozygous deletion. Oncogene. (2002) 21:1217–24. doi: 10.1038/sj.onc.1205195
68. Bahar A, Simpson DJ, Cutty SJ, Bicknell JE, Hoban PR, Holley S, et al. Isolation and characterization of a novel pituitary tumor apoptosis gene. Mol Endocrinol. (2004) 18:1827–39. doi: 10.1210/me.2004-0087
69. Zhao J, Dahle D, Zhou Y, Zhang X, Klibanski A. Hypermethylation of the promoter region is associated with the loss of MEG3 gene expression in human pituitary tumors. J Clin Endocrinol Metab. (2005) 90:2179–86. doi: 10.1210/jc.2004-1848
70. Zhu X, Lee K, Asa SL, Ezzat S. Epigenetic silencing through DNA and histone methylation of fibroblast growth factor receptor 2 in neoplastic pituitary cells. Am J Pathol. (2007) 170:1618–28. doi: 10.2353/ajpath.2007.061111
71. Zhu X, Mao X, Hurren R, Schimmer AD, Ezzat S, Asa SL. Deoxyribonucleic acid methyltransferase 3B promotes epigenetic silencing through histone 3 chromatin modifications in pituitary cells. J Clin Endocrinol Metab. (2008) 93:3610–7. doi: 10.1210/jc.2008-0578
72. Wang W, Fu L, Li S, Xu Z, Li X. Histone deacetylase 11 suppresses p53 expression in pituitary tumor cells. Cell Biol Int. (2017) 41:1290–5. doi: 10.1002/cbin.10834
73. Bilodeau S, Vallette-Kasic S, Gauthier Y, Figarella-Branger D, Brue T, Berthelet F, et al. Role of Brg1 and HDAC2 in GR trans-repression of the pituitary POMC gene and misexpression in Cushing disease. Genes Dev. (2006) 20:2871–86. doi: 10.1101/gad.1444606
74. Xue Y, Chen R, Du W, Yang F, Wei X. RIZ1 and histone methylation status in pituitary adenomas. Tumour Biol. (2017) 39:1010428317711794. doi: 10.1177/1010428317711794
75. DeVore SB, Young CH, Li G, Sundararajan A, Ramaraj T, Mudge J, et al. Histone citrullination represses miRNA expression resulting in increased oncogene mRNAs in somatolactotrope cells. Mol Cell Biol. (2018) 38:e00084-18. doi: 10.1128/MCB.00084-18
76. Grande IPP, Amorim P, Freire A, Jallad RS, Musolino NR, Cescato VA, et al. Differential gene expression of sirtuins between somatotropinomas and nonfunctioning pituitary adenomas. Pituitary. (2018) 21:355–61. doi: 10.1007/s11102-018-0881-7
77. Ma HS, Wang EL, Xu WF, Yamada S, Yoshimoto K, Qian ZR, et al. Overexpression of DNA (Cytosine-5)-methyltransferase 1 (DNMT1) and DNA (Cytosine-5)-methyltransferase 3A (DNMT3A) is associated with aggressive behavior and hypermethylation of tumor suppressor genes in human pituitary adenomas. Med Sci Monit. (2018) 24:4841–50. doi: 10.12659/MSM.910608
78. D'Angelo D, Esposito F, Fusco A. Epigenetic mechanisms leading to overexpression of HMGA proteins in human pituitary adenomas. Front Med. (2015) 2:39. doi: 10.3389/fmed.2015.00039
79. Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. (2009) 1:239–59. doi: 10.2217/epi.09.33
80. Duong CV, Emes RD, Wessely F, Yacqub-Usman K, Clayton RN, Farrell WE. Quantitative, genome-wide analysis of the DNA methylome in sporadic pituitary adenomas. Endocr Relat Cancer. (2012) 19:805–16. doi: 10.1530/ERC-12-0251
81. Han L, Liu D, Li Z, Tian N, Han Z, Wang G, et al. HOXB1 is a tumor suppressor gene regulated by miR-3175 in glioma. PLoS ONE. (2015) 10:e0142387. doi: 10.1371/journal.pone.0142387
82. Gad AK, Nehru V, Ruusala A, Aspenstrom P. RhoD regulates cytoskeletal dynamics via the actin nucleation-promoting factor WASp homologue associated with actin Golgi membranes and microtubules. Mol Biol Cell. (2012) 23:4807–19. doi: 10.1091/mbc.e12-07-0555
83. Ling C, Pease M, Shi L, Punj V, Shiroishi MS, Commins D, et al. A pilot genome-scale profiling of DNA methylation in sporadic pituitary macroadenomas: association with tumor invasion and histopathological subtype. PLoS ONE. (2014) 9:e96178. doi: 10.1371/journal.pone.0096178
84. Gu Y, Zhou X, Hu F, Yu Y, Xie T, Huang Y, et al. Differential DNA methylome profiling of nonfunctioning pituitary adenomas suggesting tumour invasion is correlated with cell adhesion. J Neurooncol. (2016) 129:23–31. doi: 10.1007/s11060-016-2139-4
85. Salomon MP, Wang X, Marzese D, Hsu SC, Nelson N, Zhang X, et al. The epigenomic landscape of pituitary adenomas reveals specific alterations and differentiates among acromegaly, Cushing's disease and endocrine-inactive subtypes. Clin Cancer Res. (2018) 24:4126–36. doi: 10.1158/1078-0432.CCR-17-2206
86. Vaitkiene P, Valiulyte I, Glebauskiene B, Liutkeviciene R. N-myc downstream-regulated gene 2 (NDRG2) promoter methylation and expression in pituitary adenoma. Diagn Pathol. (2017) 12:33. doi: 10.1186/s13000-017-0622-7
87. Valiulyte I, Steponaitis G, Skiriute D, Tamasauskas A, Vaitkiene P. Signal transducer and activator of transcription 3 (STAT3) promoter methylation and expression in pituitary adenoma. BMC Med Genet. (2017) 18:72. doi: 10.1186/s12881-017-0434-3
88. Ruskyte K, Liutkeviciene R, Vilkeviciute A, Vaitkiene P, Valiulyte I, Glebauskiene B, et al. MMP-14 and TGFbeta-1 methylation in pituitary adenomas. Oncol Lett. (2016) 12:3013–7. doi: 10.3892/ol.2016.4919
89. Kochling M, Ewelt C, Furtjes G, Peetz-Dienhart S, Koos B, Hasselblatt M, et al. hTERT promoter methylation in pituitary adenomas. Brain Tumor Pathol. (2016) 33:27–34. doi: 10.1007/s10014-015-0230-8
90. Boresowicz J, Kober P, Rusetska N, Maksymowicz M, Goryca K, Kunicki J, et al. Telomere length and TERT abnormalities in pituitary adenomas. Neuro Endocrinol Lett. (2018) 39:49–55.
91. McCormack A, Dekkers OM, Petersenn S, Popovic V, Trouillas J, Raverot G, et al. Treatment of aggressive pituitary tumours and carcinomas: results of a European Society of Endocrinology (ESE) survey 2016. Eur J Endocrinol. (2018) 178:265–76. doi: 10.1530/endoabs.49.OC12.2
92. Syro LV, Rotondo F, Camargo M, Ortiz LD, Serna CA, Kovacs K. Temozolomide and pituitary tumors: current understanding, unresolved issues, and future directions. Front Endocrinol. (2018) 9:318. doi: 10.3389/fendo.2018.00318
93. Salehi F, Scheithauer BW, Kros JM, Lau Q, Fealey M, Erickson D, et al. MGMT promoter methylation and immunoexpression in aggressive pituitary adenomas and carcinomas. J Neurooncol. (2011) 104:647–57. doi: 10.1007/s11060-011-0532-6
94. Arya S, Majaid MA, Shwetha SD, Sravani K, Arivazhagan A, Sampath S, et al. Implications of MGMT methylation status in pituitary adenoma. Pathol Res Pract. (2014) 210:407–11. doi: 10.1016/j.prp.2014.02.010
95. Jenuwein T, Allis CD. Translating the histone code. Science. (2001) 293:1074–80. doi: 10.1126/science.1063127
96. Yacqub-Usman K, Duong CV, Clayton RN, Farrell WE. Preincubation of pituitary tumor cells with the epidrugs zebularine and trichostatin A are permissive for retinoic acid-augmented expression of the BMP-4 and D2R genes. Endocrinology. (2013) 154:1711–21. doi: 10.1210/en.2013-1061
97. Li T, Huang H, Huang B, Huang B, Lu J. Histone acetyltransferase p300 regulates the expression of human pituitary tumor transforming gene (hPTTG). J Genet Genomics. (2009) 36:335–42. doi: 10.1016/S1673-8527(08)60122-8
98. Ebrahimi A, Schittenhelm J, Honegger J, Schluesener HJ. Histone acetylation patterns of typical and atypical pituitary adenomas indicate epigenetic shift of these tumours. J Neuroendocrinol. (2011) 23:525–30. doi: 10.1111/j.1365-2826.2011.02129.x
Keywords: pituitary tumor, pituitary adenoma, epigenetics, precision medicine, endocrine surgery, epigenome
Citation: Hauser BM, Lau A, Gupta S, Bi WL and Dunn IF (2019) The Epigenomics of Pituitary Adenoma. Front. Endocrinol. 10:290. doi: 10.3389/fendo.2019.00290
Received: 15 December 2018; Accepted: 23 April 2019;
Published: 14 May 2019.
Edited by:
Xianquan Zhan, Xiangya Hospital, Central South University, ChinaReviewed by:
Yona Greenman, Tel Aviv Sourasky Medical Center, IsraelCopyright © 2019 Hauser, Lau, Gupta, Bi and Dunn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenya Linda Bi, d2JpQGJ3aC5oYXJ2YXJkLmVkdQ==
Ian F. Dunn, aWFuLWR1bm5Ab3Voc2MuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.