 Valentina Salvestrini
Valentina Salvestrini Christian Sell
Christian Sell Antonello Lorenzini
Antonello Lorenzini
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 03 May 2019
Sec. Endocrinology of Aging
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00266
This article is part of the Research Topic Adipose Tissue: Which Role in Aging and Longevity? View all 8 articles
Lines of evidence from several studies have shown that increases in life expectancy are now accompanied by increased disability rate. The expanded lifespan of the aging population imposes a challenge on the continuous increase of chronic disease. The prevalence of overweight and obesity is increasing at an alarming rate in many parts of the world. Further to increasing the onset of metabolic imbalances, obesity leads to reduced life span and affects cellular and molecular processes in a fashion resembling aging. Nine key hallmarks of the aging process have been proposed. In this review, we will review these hallmarks and discuss pathophysiological changes that occur with obesity, that are similar to or contribute to those that occur during aging. We present and discuss the idea that obesity, in addition to having disease-specific effects, may accelerate the rate of aging affecting all aspects of physiology and thus shortening life span and health span.
The world population is aging at a rapid pace (1), we face a future where the number of elderly people will exceed that of children and there will be more people at extreme old age than ever before. The significant rise in average life expectancy during the 20th century ranks as one of society's greatest achievements. In addition, the increase in life expectancy is accompanied by a change in principal causes of disease and death, creating an “epidemiologic transition”. This transition is based on a decline in infectious and acute disease and an increase in chronic and degenerative disease (2).
The question whether living longer represents more years of healthier life or an increase in years of disability is an important question (3, 4). Evidence from several studies indicate that the recent increase in life expectancy is accompanied by an increased disability rate. The net result is no net difference in the length of a healthy life span in some estimations (5–7) and potentially a decrease in other estimates (8, 9). In a large-scale study of 187 countries examining the global burden of disease, Salomone and colleagues indicated that as life expectancy rose between 1990 and 2010, the number of healthy years lost to disability has also increased (10). This trend, which is typical of industrialized countries, has a significant impact on public health, due to the cost of care imposed by the increase in years lost to disability and underscores the need to make healthy aging a priority.
A major factor contributing to the increase in disability is an increase in obesity, creating a new and pressing challenge for public health (11–17). Obesity is expanding at a worrisome rate, the frequency of overweight and obesity combined increased by 27.5% for adults and 47.1% for children between 1980 and 2013. In developing countries, the proportion of obese adults rose from about 15% in 1980 to more than 20% in 2013 (18). Obesity is associated with an increased risk of cardiovascular disease, type 2 diabetes mellitus, cancer, osteoarthritis, work disability and sleep apnea (19).
It has been suggested that obesity not only increases the onset of metabolic imbalances, but also decreases life span and impacts cellular processes in a manner similar to aging (20).
A defining characteristic of aging is the gradual loss of physiological integrity, which results in increased vulnerability to disease and death. This loss of physiological integrity underlies multiple pathologies, including cancer, diabetes, cardiovascular disorders and neurodegenerative disease (21). Recently, nine hallmarks which define the aging process have been described (21). We will briefly discuss each of the hallmarks of aging, the potential interactions between each hallmark and obesity and, where available, the effect of caloric restriction (CR).
Telomeres are repetitive, non-coding, chromosomal regions located at the end of each chromosome. These telomeric regions are assembled into higher order structures, which prevent both chromosomal fusions and activation of the DNA damage response. In human somatic cells, telomere erosion occurs with each cell division creating a trigger for senescence when a critical length is reached and the telomere structure is destabilized (22).
Telomere length is inversely correlated with lifespan (23), and telomere dysfunction accelerates the aging process (21). Telomere shortening, moreover, can be accelerated by factors that induce aging and attenuated by factors that improve health (24). On these grounds, telomere length has been proposed as marker of biological aging (21). Inflammation and oxidative stress have been associated with aging in general (25), and shortening of telomeres in particular.
Obesity causes oxidative stress and inflammation, which may increase the rate of telomere shortening (24). Although the association is weak or moderate, the results of a systematic review by Mundstock and colleagues show a trend toward a negative association between obesity, in particular central obesity, and telomere length (24). Human studies indicate that telomere shortening is directly correlated to adiposity (26), and telomere length is inversely associated with BMI (19). However, this association is not linear across the age and it is stronger in younger compared to older individuals (26). Interestingly, telomere length measured from subcutaneous adipocytes was significantly lower in obese patients compared to never-obese ones (27), and it appears that physical activity may protect patients from telomere shortening due to obesity, although extended periods of overweight/obesity seem to mitigate this protection (28). It should be noted that these results are not consistent in all studies (29).
Telomere length in leukocytes is linked to both obesity and smoking. For example, telomere length was inversely correlated with serum concentration of leptin, an adipokine which may contribute to an inflammatory state and elevated oxidative stress (30). Importantly for the current discussion, leukocyte telomere length, a common aging biomarker, has been shown to negatively correlate with BMI, although the association was relatively weak and gender specific (females only) (31).
A dedicated review on the possible links between obesity, telomeres and aging concludes: “obesity may affect telomere dynamics and accelerate the aging process” (32). We feel that although the results cumulatively show a tendency toward an inverse correlation between obesity and telomere length; it is more prudent to conclude that the available studies are heterogeneous and show a weak statistical significance (24, 26).
Epigenetic modifications such as DNA methylation, histone modification and chromatin remodeling refer to alterations in gene expression that are inherited in descendant cells or organisms (33).
Epigenetic changes occur with age and there appears to be a relationship between epigenetic changes and age-related health problems (21). One of the strongest correlations between epigenetics and aging involves changes in a subset of methylation sites throughout the genome. These sites were identified as having an altered methylation pattern during aging and these changes have been proposed to represent an “epigenetic clock,” that may be tied to the aging process (34).
There is evidence that lifestyle changes, including weight loss/gain, affect gene expression by altering the DNA methylation pattern (35) and increasing the risk of developing diseases in later life. In this context, nutrition, among other environmental factors, plays a key role in inducing epigenetic changes and these changes can influence the phenotype of subsequent generations (36). Among nutrients, methyl donors play a central role. For example, folate supplementation during gestation increased DNA methylation at imprinted loci within the IGF2 gene and was associated with lower birth weight, while loss of imprinting at the IGF1 gene correlates with somatic overgrowth (36).
High fat diets (HFD) have been shown to alter the epigenome. For example, in utero, HFD feeding and maternal obesity alters DNA methylation patterns and histone modifications while increasing susceptibility to obesity in offspring (37). The offspring of mice exposed to a HFD exhibit modifications such as histone acetylation, in genes involved in metabolic pathways, such as glycemic homeostatic regulation. These modifications can affect the gluconeogenic capacity and potentially lead to excessive glucose production and altered insulin sensitivity in adulthood (38).
Another example of epigenetic alteration due to HFD is the increased expression of the histone deacetylase HDAC5. Increased expression of this deacetylase reduces BDNF chromatin accessibility and consequently its transcription. BDNF is a key regulator of synaptogenesis, essential for learning and memory. It has been demonstrated that HDAC5 is significantly increased in the brains of diabetic patients and in the brains of mice chronically fed a HFD (39). These epigenetic changes in the brain may persist, even after a return to normal diet, leading to pathological alterations in the cognitive machinery (40). In this study, Wang and coll. demonstrated that limited, early presence of obesity and insulin resistance may have long-term deleterious consequences in the brain, leading to a more susceptible, less resilient cognitive machinery, and contributing to the onset/progression of cognitive dysfunction, such as impairment in learning and memory formation, during aging (40).
Besides the impact of nutrition, the direct role on aging and life span of BMI and obesity associated epigenetic changes, have also been studied. Several studies demonstrated that obesity is associated with extensive changes in gene expression in multiple tissues (41) and that increased BMI is associated with an altered methylation of specific genes (42–44). For instance, Nevalainen et al. showed that obesity is associated with methylation changes in blood leukocyte DNA that could lead to immune dysfunction. They also investigated the association between BMI and epigenetic age in blood cells and demonstrated that BMI is positively associated with epigenetic aging in middle-aged individuals (44). The impact of obesity on epigenetic aging is also described by Horvath et al. They showed that obesity accelerates epigenetic changes associated with aging in the human liver resulting in an apparent age acceleration of 2.7 years for a 10-point increase in BMI (45), supporting the idea that obesity may accelerate the aging process.
CR as well as overnutrition can induce epigenetic alterations, which potentially impact aging. CR clearly retarded the methylation drift, thus resulting in a significantly younger “methylation age” (46). Another example is SIRT6, a stress responsive deacetylase that represents a potentially significant enzyme for aging (47, 48). Functionally, SIRT6 plays an important role in DNA repair, telomerase function, genomic stability, cellular senescence and in regulation of the transcription factor nuclear factor-κB (NF-κB), which is involved in inflammation and aging (21, 49). SIRT6 activity is significantly modulated by CR (49). Nutrient depletion or long-term CR increase SIRT6 activity in the brain, white adipose tissue, muscle, liver and kidney in mice (49, 50).
Several reports demonstrate that nutrition and obesity are able to modulate the epigenetic signature of an individual, even during prenatal development. The observed alterations do not always overlap those seen in aging, however some studies show a close correlation between epigenetic alteration induced by obesity and an acceleration of tissue aging. This suggests that obesity could accelerate age-related dysfunction by inducing epigenetic alterations that are not necessarily the same as those observed during aging in non-obese individuals.
Mitochondria play a central role in bioenergetic metabolism and ATP production, and maintenance of their function across lifespan is essential for general homeostasis.
Because of their key role in multiple cellular functions, these organelles are involved in multiple distinct processes with relation to aging, including: inflammation, mitophagy and proteolysis, the mitochondrial unfolded protein response, cellular senescence, stem cell function, accumulation of DNA mutations, and bioenergetics alterations [reviewed in (51)]. During the aging process, a reduction in the efficiency of mitochondrial bioenergetics has been observed and several involved mechanisms have been described, such as a reduced biogenesis, mutation in mtDNA, alteration in mitochondrial dynamics (imbalance fission/fusion) or defective mitophagy (52).
Both excessive nutrient consumption and obesity have been linked with mitochondrial dysfunctions.
During excessive nutrient consumption, the metabolism shifts toward increased lipid storage and glycolytic ATP synthesis while concurrently decreasing mitochondrial biogenesis (53). Mitochondria play an essential role in nutrient adaptation; excessive consumption of nutrients affects their functions in those tissues that participate to nutrient metabolism: adipose tissue, liver, and skeletal muscle. Excessive nutrient intake also increases the concentration of free fatty acids and mitochondrial ROS production, leading to hyperglycemia and adipocyte mitochondrial dysfunction. In this tissue, mitochondrial biogenesis, mtDNA content and the rate of β-oxidation are reduced while adipogenesis, fatty acids esterification and lipolysis are altered. These modifications contribute to alteration of insulin sensitivity (54).
Obesity has also been associated with mitochondrial dysfunctions (54). CR, conversely, which increases longevity, maintains mitochondrial function (55). Several studies showed that obesity induces a reduction in mitochondrial biogenesis and a decreased mitochondrial oxidative capacity in adipocytes of both rodents (56) and humans (54). In obese individuals, reduced mitochondrial biogenesis is associated with metabolic alterations, low-grade inflammation, and insulin resistance (57). Several lines of evidence (54) suggest that obesity induces a shift toward a fission process linked to mitochondrial dysfunction in liver and skeletal muscle. In skeletal muscle of obese mice, an increased mitochondrial fission was observed and the activity of protein involved in mitochondrial dynamic was altered (58).
Some of the adaptations observed under excessive nutrient consumption are also observed in obesity; mitochondria of obese individuals show a reduced oxidation of fatty acids, have less defined internal membranes, a lower energy generation capacity and an increased glucose dependence for ATP synthesis (59).
Mitochondria are also a central players in apoptosis (60), and the availability or ingestion of nutrients is related to the regulation of cell death. Excessive food intake impairs mitochondrial respiratory capacity and sensitizes mitochondria to apoptotic stimuli (59). Obesity upregulates apoptotic pathways proteins in rodent and humans, and increased apoptosis in adipocytes, as demonstrated by an association between body fat and a pro-apoptotic state in adipose tissue of obese patients (54).
Mitochondrial dysfunction occurs in aged tissues, in response to excessive nutrient intake, and in obesity, contributing to inflammation and insulin resistance. Aging and obesity appear superimposable in their impact on mitochondria and it is reasonable to hypothesize that they could exert additive effects.
Cellular senescence is an irreversible block of the cell cycle that limits the proliferative potential of cells (61). Cellular senescence, along with apoptosis, is a physiological process that plays a crucial role in the removal of damaged cells and tissue remodeling; it is a crucial mechanism for development but becomes deleterious when it affects stem and immune cell function, impacting tissue homeostasis. Senescence can be triggered by several stress stimuli, such as telomere uncapping, DNA damage and oncogene activation (62). Senescent cells have a large flattened morphology, stop DNA replication, show increased levels of proteins involved in cell cycle arrest and tumor suppression (such as the tumor suppressor p53 and cyclin-dependent kinase inhibitors [CDKi] p16INK4A, p21CIP1/WAF1, and p15INK4B), and are positive for the senescence-associated β-galactosidase (SA β-gal). They also display altered histone modification profiles and an altered secretome consisting of pro-inflammatory factors, growth factors and proteases, the so called senescence associate secretory phenotype: SASP (63). SASP factors influence the behavior of neighboring cells, resulting in the paracrine induction of senescence, tissue remodeling, and recruitment of immune cells (e.g., T lymphocyte and macrophage) (63). Although senescent cells are resistant to apoptosis, their activation of immune system causes removal of nearby cells as well as the senescent cells themselves (64).
During the initial description of replicative senescence, Hayflick proposed that the process may contribute to organismal aging (65). Although two key axioms of this idea, i.e., the relationship of replicative senescence with donor age or with species longevity are not supported by subsequent experiments [reviewed in (66)], an increase in senescent cells has been observed in vivo in different tissues (67, 68). Perhaps more importantly, the clearance of accumulated senescent cells in tissue during aging has been demonstrated to extend median lifespan and to attenuate age-related deterioration of organs in mice (69).
It has been demonstrated that SA β-gal+ cells are more abundant in pre-adipocyte and endothelial cells isolated from obese compared to lean rats and human, moreover there is a positive correlation between BMI and adipose tissue SA β-gal activity and p53 [reviewed in (64)]. There is an accumulation of senescent T cells and an increased number of macrophages in the inflammatory foci of the visceral adipose tissue of HFD-fed obese mice (70), and obese mice accumulate senescent glial cells in the brain (71).
Senescent pre-adipocytes are defective in their differentiation capacity; it has been shown that senescent adipose-derived stromal/progenitor cells express reduced levels of adipogenic regulators and altered expression of adipogenic differentiation gene patterns in response to adipogenic hormone stimuli (72). In the heterochronic parabiosis model, blood from 3 months old mice is able to reduce the levels of pro-inflammatory cytokines in the visceral adipose tissue of 18 months old mice (73).
There appears to be a strong relationship between obesity and senescence. Reports like the ones described above suggest that obesity may promote the aging process by inducing senescence. Conversely, senescence and the resulting pro-inflammatory secretory phenotype could contribute to the morbidity associated with obesity and plays a role in the development of insulin resistance and diabetes. There is vast literature in support of this view, and we refer the interested readers to gather valuable in depth reviews (74–76). Finally, Fontana et al. have proposed that CR might exert its anti-aging capacities by limiting senescent cell accumulation (77).
There is increasing evidence that the aging process can have adverse effects on stem cells. As stem cells age, their renewal ability deteriorates and their ability to differentiate into the various cell types is altered (78). The life-long persistence of stem cells in the body makes them particularly susceptible to the accumulation of cellular damage, which ultimately can lead to cell death, senescence or loss of regenerative function (79). These changes translate into reduced effectiveness of cell replacement and tissue regeneration in aged organisms.
Obesity is associated with a pro-inflammatory response in a wide variety of tissues. Inflammation can activate the stem cell compartment with negative consequences. For example, a reduction in functionally active stem cells has been observed in subcutaneous adipose tissue from obese patients (80). Adipose-derived stem cells (ASC) isolated from obese patients demonstrated a reduced proliferative ability and a loss of viability together with changes in telomerase activity and telomere length (81). Moreover, their mitochondrial content and function are altered. Specifically, ASC contain a greater number of mitochondria and produce more ROS, however their mitochondria show a reduced respiration capacity, concomitant with a shift toward β-oxidation instead of glycolysis for energy production.
ASC from obese patients have reduced differentiation potential and are less proangiogenic (80), which is reflected in differences in their gene expression profile (82). Onate and colleague, demonstrated that obesity impairs the expression of genes involved in regulation of cell proliferation, differentiation and angiogenic potential of ASC, rendering them less multipotent. Moreover, obesity seems to affect ASC trafficking and homing (82), changes which may reduce the capacity of these cells for tissue repair (80).
Obesity also influences bone marrow (BM) homeostasis, increasing adipocyte formation. Bone marrow adipose tissue (BMAT) originates from bone marrow stromal stem cells (BM-MSC), which give rise to adipocyte, osteoblast and hematopoietic-supporting stroma (83). BMAT is an endocrine-active fat depot capable of influencing BM stem cells. Chronic low-grade inflammation associated with obesity is a stressor for BM stem cells due to the continuous response to inflammatory cytokines. In turn, inflammation causes alterations in the microenvironment with implications for cell production. In mice, HFD-induced obesity leads to a progenitor cell exhaustion and impairs osteoblast recruitment and bone formation, decreasing proliferative potential of progenitor cells and enhancing adipocytic differentiation of BM-MSC (83).
Obesity has a direct effect on the hematopoietic stem cell (HSC) compartment; however, obesity and aging seem to have different effects. HSC aging leads to a paradoxical increase in the stem cell pool and decline in stem cell function. One of the prominent modifications of HSC properties with age is their biased differentiation toward myeloid lineage at the expense of their lymphoid potential (84). None of these characteristics are observed in obesity. An elegant study by Lee et al. demonstrated that obesity leads to changes in the cellular architecture of the stem cell compartment. HSCs acquire an immature phenotype, remain quiescent, and are refractory to the low-grade inflammation signals, while differentiated progenitors are more greatly affected. Through the use of a genetic mouse model, the authors demonstrated that obesity affects the long-term reconstitution ability of HSC while also leading to an exacerbated proliferative response of multipotent progenitors. These effects are linked to the upregulation of Gfi1, a key regulator of HSC quiescence and self-renewal, in response to the oxidative stress associated with obesity (85). The aberrant HSCs activity is progressively acquired during weight gain but it is long lasting after weight loss, demonstrating that obesity induces lasting changes in the HSC compartment (85).
It has been demonstrated that postnatal overnutrition reduces myogenic stem cell frequency and function (86) and that HFD fed mice show a reduced number of neural stem cells in the hypothalamus with a reduced differentiation capacity (87).
Stem cells are adapting their metabolism in response to environmental changes, they skew toward a quiescent state in case of stress, or begin to proliferate-differentiate in response to injury (88). The effects of diet on stem cell metabolism and function have been assessed in response to CR. CR slows down age-related decline and enhances stem cell activity by altering their metabolic activity, promoting oxidative phosphorylation over glycolysis (88). CR potentially shifts the balance toward self-renewal while reducing the numbers of differentiated cells, thus preserving the stem cell pool and preventing stem cell exhaustion (89). In both young and old mice, CR increases the frequency and function of skeletal muscle stem cells by increasing mitochondrial content and promoting oxidative metabolism (90).
With the exception represented by the effects on the HSC compartment, both obesity and aging, negatively impact ASC, neural stem cells and BM homeostasis. In contrast, CR promotes self-renewal and prevents stem cell exhaustion. Overall, obesity does not mimic aging in terms of stem cells compartments but, similar to aging, has a disrupting influence on their tissue maintenance functions.
The major signal pathways that participate in nutrient sensing are: the insulin/ insulin-like growth factor (IGF-1) signaling (IIS) pathway which informs the cell of the presence of glucose (and IGF-1); mTOR, for sensing amino acid concentrations (and integrating this information with growth factor signals from the IIS); AMPK which senses low-energy state by detecting low level of ATP; and sirtuins which sense nutrient scarcity by detecting high NAD+ levels.
Trophic signals that activate IIS or the mTOR pathways are now considered major accelerators of aging. Multiple studies in mutant mice show that a reduction in the growth hormone (GH)/ IGF-1 signaling extends life span [reviewed in (91)] and at least one study points to an IGF-1 independent role of GH (92). mTOR activity is now regarded by many as a central player in dictating the pace of aging (93). On the opposite side, upregulation of the AMPK and sirtuins pathways may mediate lifespan extension (21).
Mediators of inflammatory signals such as c-Jun NH2-terminal kinase (JNK) and kB kinase-B (IKKβ) impair insulin signal pathways, in turn interfering with the phosphorylation of receptor substrate 1 (IRS1), reducing the interaction with PI3K and consequently reducing glucose uptake (94). In obesity, altered insulin action and the consequental PI3K/Akt signaling pathway alteration in skeletal muscle, liver and adipose tissue may cause systemic insulin resistance (95). In skeletal muscle, insulin resistance leads to a decreased glucose transport, and a reduction in glycogen synthesis. In liver, insulin resistance results in a failure of gluconeogenesis suppression, however it stimulates fatty acid synthesis. Adipose tissue shows altered insulin-stimulated glucose transport and lipolysis. However, not all insulin signaling is diminished. For example, in liver tissue, the gluconeogenic pathway becomes insulin resistant, although insulin dependent lipogenesis stays sensitive (96).
When caloric restriction is present, the liver produces less IGF-1 and it is refractory to GH stimulation (97). While it has been clearly demonstrated that hyperinsulinaemia in obesity leads to significantly reduced GH secretion, which affects insulin's ability to maintain normal glucose homeostasis (98). The effects of obesity on IGF-1 levels are more controversial. Chronic hyperinsulinemia is associated with increased circulating IGF-1 levels. Insulin suppresses IGF binding protein (IGFBP)-1 and -2, which reduce the bioavailability of IGF-1 in the peripheral tissues (98). Increasing BMI is associated with a reduction of IGFBP-1 and IGFBP-2 expression and consequently with high circulating free IGF-1 levels (98). However, while it has been reported that IGF-1 levels are high in obesity, other studies show that it is not increased or may even be decreased (98). These differences may be due to methodological challenges associated with IGF-1 measurements.
Diet and surgical induced weight loss can revert the defects in the GH/IGF-I axis in obesity (99).
Obesity promotes mTOR activity in adipose tissue, leading to exacerbated hyperlipidemia and insulin resistance (100). For example, mTORC1 is hyperactivated in tissue of obese and HFD fed rodents (101) and genetic variation in Raptor, an mTOR-interacting partner, is associated with overweight/obesity in American men of Japanese ancestry (102). High adiposity is closely associated with development of insulin resistance, and it has been demonstrated that in the state of overnutrition, one of the molecular factors involved in insulin resistance is the ribosomal protein S6 kinase 1 (S6K1), a downstream target of mTOR signaling (103).
Decreased activation of mTOR/S6K1 has been associated with increased insulin sensitivity (104). S6K1 is hyperactivated in the adipose tissue, liver and muscle of different genetic mouse model of obesity. It has been described that HFD fed s6k1 deficient mice are protected from developing obesity and insulin resistance (103). Chronic activation of the mTOR/S6K1 pathway by insulin, TNF-α and amino acids promote insulin resistance in obese mice and primary cultures of skeletal muscle cells from patients with type 2 diabetes through increased IRS1 serine phosphorylation and degradation (105). The HFD fed s6k1 deficient mice show a strong reduction of phosphorylation of these sites; suggesting that S6K1 inhibits insulin signaling by mediating IRS1 phosphorylation (104). Moreover, obese patients express increased levels of RPS6KB1, the human gene encoding S6K1, in visceral fat compared to lean volunteers (103). Fat mass reduction after CR is associated with adipose tissue mTOR inhibition. Accordingly, pharmacological inhibition of mTORC1 pathway is associated with a reduction of both adipocyte size and number (106). Deletion of the mTORC1 target p70S6K protects against age- and diet-induced obesity (104).
Obesity induces a broad, non-tissue, or isoform specific decrease in AMPK activity (107). HFD substantially inhibits AMPK activity in white adipose tissue, heart and liver, and this reduced activity is associated with systemic insulin resistance and hyperlipidemia (107). It has been reported that AMPK activity is lower in morbidly obese humans who are insulin resistant than in comparably obese individuals who are insulin sensitive (108). AMPK activity is also reduced in the paraventricular nucleus of mice with diet-induced obesity (109). In addition, some studies demonstrated the impairment of AMPK activation in skeletal muscle of individuals with obesity and diabetes (110, 111), and in visceral adipose tissue of centrally obese humans with Cushings syndrome, a disorder associated with insulin resistance (112).
There is a correlation between obesity and reduced Sirt1 levels. Adipose tissue from HFD fed mice (113) and db/db leptin resistant obese mice (114) show a significant reduction of Sirt1. Lower levels of Sirt1 have been reported in obese pigs compared to lean ones (115), and Choi et al. demonstrated that microRNA mir34a, which is elevated in obesity, reduces NAD+ levels and Sirt1 activity (116). Observational studies demonstrated the association between changes in sirtuins and obesity in human. Reduced mRNA levels of Sirt1 were observed in adipose tissue from obese women compared to lean women (117) and in peripheral blood mononuclear cells of diabetic subjects with insulin resistance (118). Furthermore, an increased expression of Sirt1 and Sirt3 was observed in adipose tissue of severely obese patients who experienced weight loss after gastric banding surgery (119).
In biogerontology, the IIS and mTOR pathway are considered “accelerators” of the aging process. There is accumulating literature suggesting that in obesity, these pathways are over-activated. In contrast, there is also accumulating literature showing that pro longevity pathways, such as the AMPK and sirtuins pathways are dampened by obesity. In conclusion, there is solid evidence that obesity deregulates cellular mechanisms related to nutrient sensing.
It is accepted that aging impacts the organism at the cellular level, but also decreases the capacity of cells of an organism to interact.
During aging, there is a decreased communication at the neuronal, neuroendocrine and endocrine levels. Two of the most compelling examples of impaired communication are inflammaging and immunosenescence (120). Inflammaging refers to the concept that aging is accompanied by a proinflammatory state, which is the consequence of multiple conditions, SASP, defects in autophagy and mitophagy, an enhanced activation of the inflammatory mediator, NF-κB. This phenotype results in elevated cytokines such as: IL-1b, tumor necrosis factor, and interferons. These cytokines can accelerate and propagate the aging process. Immunosenescence refers to the decreasing efficiency of the adaptive immune system with aging.
With obesity, the adipocyte secretome changes toward greater secretion of pro-inflammatory mediators and reduced production of anti-inflammatory or insulin sensitizing factors (121). More precisely, hypertrophic conditions induce adipocyte stress, activating Jun N-terminal kinase (JNK), NF-κB, Ask1, and MKK4. Activation of these pathways induce adipocytes, endothelial cells and immune cells to produce pro-inflammatory cytokines, endothelial adhesion molecules, proatherogenic and chemotactic mediators [IL-6, tumor necrosis factor- α (TNF-α), IL-1β, MCP-1, PAI-1, Csf-1, progranulin, chemerin, and others] in adipose tissue (122). These changes impact both number and function of immune cells, increasing the number and the activity of a subset (macrophages, neutrophils, mast cells, B, and T lymphocytes) and other subtypes [eosinophils, T helper 2, Treg and natural killer T cells (NKT)] (123, 124).
It has been demonstrated that macrophage number increases with adiposity, and the accumulation is greatest in visceral fat in humans (124). Chemoattractant molecules, such as MCP-1, secreted by adipocytes, recruit monocytes from peripheral blood to adipose tissue, where they differentiate into macrophages (125). Moreover, MCP-1 promotes the local proliferation of adipose-resident macrophages. Monocytes migrate in adipose tissue in response to adipocyte-derived cell stress markers, including CCL5, IL-6, IFN-γ and TNF-α which enhance macrophage accumulation and their polarization toward a pro-inflammatory M2 phenotype (124). The increased number of macrophages has a positive correlation with the degree of insulin resistance in both mice and humans (124). Elevated chemokine ligand (CXCL)-2 release by adipose tissue promotes neutrophil infiltration, which are 20-fold more abundant in adipose tissue from HFD fed mice compared to chow-fed ones (124). On the contrary, obesity decreases AT eosinophil numbers leading to reduced insulin sensitivity while an increase in eosinophils in response to IL-15 overexpression improves obesity-induced insulin resistance (123).
CD4+ Th1 cells increase in human subcutaneous adipose tissue with obesity and exhibit an activated CD25+ phenotype. HFD fed mice show an increased IFN- γ secretion, that impaired insulin signaling and promoted macrophage infiltration, as a consequence of Th1 cell predominance (124). As with CD4+, obesity also increases CD8+ T cell levels along with their products, granzyme B and IFN-γ. In obesity, Treg cells, suppressors of inflammatory reactions, are decreased both in their proliferative capacity and in number. Dendritic cells accumulate in AT of HFD fed mice, and induce a pro-inflammatory microenvironment by secreting IL-6 and promoting macrophage recruitment/proliferation, following enhanced INF signaling and MCP-1 production (124).
The resulting imbalance in immunological phenotypes leads to development of local inflammation that further spreads into systemic circulation affecting other organs. For example, the adipokine Haptoglobin (Hp), a clinical marker of inflammation increased in the cerebral spinal fluid of patients with neurodegenerative disorders, has an abundance positively related to body fat in adipose tissue and plasma (126, 127).
Because inflammaging contributes to immunosenescence, the obesity-derived inflammatory status reduces the efficiency of the immune system, consistent with the observation that obese people are more susceptible to infection from bacteria, fungi or viruses [see an article part of this Research Topic, (128)]. For a more in depth discussion of the interconnections between adipokines and aging we refer the reader to two additional articles which are part of this Research Topic (129, 130).
Extracellular vesicles (EV, micro-vesicle and exosomes) are nanoparticles that contain protein and nucleic acids, which interact with target tissues. Exosomes are increased in many inflammatory conditions (131), and increased numbers of microvescicles have been associated with obesity (132), while a significant reduction occurs in CR or following bariatric surgery in obese patients (133).
Several studies have demonstrated that EVs collected from adipose tissue of ob/ob obese mice induce, in a target cell population, changes consistent with the obese phenotype (134). EV treated monocytes were more activated, secreted more IL-6 and TNF-α compared to those treated with EV from wild type mice, and macrophages were more activated and had an increased homing capacity to adipose tissue and liver (134). In humans, EVs isolated from adipose tissue induced monocytes to adopt properties characteristic of adipose tissue macrophages (135).
Finally, it has been demonstrated that obesity reduces the pro-angiogenic potential of adipose tissue stem cell-derived EV by reducing VEGF, MMP-2 and miR-126 content (136).
The literature persuasively suggests that the accumulation of pro-inflammatory cells, in the adipose tissue of obese patients, through cytokines and extracellular vesicles, accelerates the rate of aging both in the adipose tissue itself and the entire organism.
The hypothesis that aging may result from the accumulation of DNA damage is one of the classical theories of aging (137), and is supported by considerable evidence. For example, accumulation of DNA damage (138) and mutations (139) with increasing chronological age. Longevity differs by several orders of magnitude among animals and long life spans seem to associate with a greater capacity to detect the presence of DNA damage at the cellular level [reviewed in (140)]. Enhanced recognition of damage should allow enhanced DNA repair. In mammals, there is an exponential relationship between longevity and the capacity to perform the first step of non-homologous end-joining, i.e., the recognition of linear DNA ends (141), resulting in improved genomic stability (140, 142).
The impact of obesity on genomic instability has been analyzed in a recent review by Setayesh et al. (143). Results from animal studies and from 39 studies in humans, monitoring DNA damage in lymphocytes and sperm, were analyzed. However, heterogeneity in the study design, methodology, and confounding factors, preclude the conclusion that an association exists between obesity and DNA damage. Nevertheless, the causal relation between excess of body weight and genomic instability is supported by mechanistic studies.
Several molecular mechanism may cause genetic instability in overweight/obese individuals; one of them is oxidative stress. Oxygen derived free radicals may act as potential cytotoxic intermediates inducing inflammatory and degenerative processes, or as signal messengers for the regulation of gene expression. Many articles show evidence for the induction of oxidative DNA damage and a decreased antioxidant capacity in obesity. High glucose levels directly, and high insulin levels, through the over activation of its signaling pathways, could be responsible for increased ROS formation. Othman et al. demonstrated that insulin causes DNA damage in kidney cells (144); it has been demonstrated that insulin and glucose blood level correlate with DNA damage in Korean men (145), as well as DNA damage in sperm in a mouse model (146). Other studies provide evidence for a reduction of antioxidant enzymes and a subsequent oxidative stress as a consequence of obesity, in human and mice (143).
Oxidative stress leads to oxidation of fatty acids, and some metabolites of lipid peroxidation are molecules that attack DNA and are involved in the etiology of cancer. Elevated levels of lipid peroxidation markers are observed in blood, muscles and adipose tissue of obese individuals. Excess body weight causes hormonal imbalance and it has been demonstrated that alterations of hormonal status plays a role in some cancers, including breast and endometrial cancer (143). Hormones increase the mitotic activity of breast cells, leading to accumulation of errors in DNA replication that are frequently converted in persistent mutations (147). Some products of estrogen metabolism cause direct DNA damage. Metabolites of estradiol are mutagenic in rat and human cells, and the association between genotoxic estrogen metabolites and breast cancer is well documented (143). Another mechanism by which ROS are generated is glycation. Glycation end products cause DNA damage directly and via interaction with signaling pathways. They bind specific receptors and activate NADH-oxidase to induce ROS formation (148). Elevated concentrations of glycation end products have been observed in adipose tissue and the livers of HFD fed mice (149), as well as human adipose tissue (150). In patients with metabolic syndrome, elevated serum levels of glycation end products correlate with markers of insulin resistance and inflammation (151).
The persistent production of ROS by inflammatory cells present in adipose tissue damages macromolecules (DNA, RNA, lipids, carbohydrates and proteins), induces genomic instability and tips the balance from an antitumor activity of ROS to a tumor promoting one (152).
Obesity is strongly associated with an increased incidence of cancer both in humans (153) and in rodents (143). An impact of obesity has been described also on infertility, and some studies (154, 155) demonstrated an increase in DNA damage in the sperm of obese men.
Obesity, moreover, impacts the DNA repair process. For example, the offspring of mice fed a low folate diet showed a reduced base excision repair capacity in several brain regions when exposed to HFD (156). There is an inverse association between adiposity and nucleotide excision repair (157), while some studies demonstrate an altered methylation pattern of genes involved in DNA repair in overweight individuals (143). Significantly, HFD reduces the expression of MLH1, a protein involved in DNA mismatch repair, and elevate CpG methylation in mice (158).
Several studies demonstrated a beneficial impact of CR on genomic stability. CR slows down the rate of DNA damage by decreasing the levels of oxidative stress and by increasing the expression of stress response genes to enhance DNA repair (159). Although the vast majority of available studies are on rodents, there is also evidence in humans; for example, after 12 months of bariatric surgery, using the comet assay, a significant reduction in DNA damage is observed in peripheral blood cells (160).
Although much research has been performed, the assumption of a relation between obesity and genomic instability is not supported unequivocally. Oxidative damage seems as the one mechanism regarded as the most relevant (161).
Proteins represent a key components of cells and tissues, and protein quality control and homeostasis are critical to the organism. Proteostasis is maintained through multiple mechanisms ensuring stabilization of correctly folded proteins and degradation of misfolded proteins. The first task is performed by chaperones, most prominently the heat shock protein (HSP) family, and the second task is performed by proteasome and lysosome mediated degradation (162). Proteins are synthetized in the endoplasmic reticulum (ER) where unfolded proteins are bound by the ER stress sensor Binding immunoglobulin protein (BiP/GRP78) to trigger the unfolded protein response (UPR). The existence of this evolutionarily conserved mechanism provides evidence of the critical role of proteostasis.
With age, the ability of many cells and organs to preserve proteostasis under resting and stressful conditions is gradually compromised (162). Key pathways affected by the aging process alter components of the proteostasis machinery, e.g., by inducing reduction of chaperones or proteasomal degradation (163, 164). The consequent increase in misfolded or degraded proteins can lead do the development of age-related pathologies such as Alzheimer and Parkinson diseases (165).
Obesity can induce prolonged or chronic UPR response (166) possibly mediated by proteasome dysfunctions (167). In the livers of mouse models of obesity and in HFD fed mice, proteasome activity is reduced and polyubiquinated proteins accumulate. In these mice, impaired proteasome function leads to hepatic steatosis, hepatic insulin resistance, and UPR activation. Treatment with chemical chaperones partially reverted this phenotype (168). Elevated free fatty acids in obesity activate the UPR in both adipose tissue and liver (167). It has been demonstrated that ER protein folding is impaired in the liver and within adipose tissue of obese mice. Overexpression of the chaperone BiP/GRP78 in the liver of ob/ob mice reduces UPR activation markers, hepatic steatosis, and improved insulin action (169). Obesity-induced changes in ER calcium store can induce a reduction in chaperone-mediated protein folding activity, ER stress and UPR activation (167). Cholesterol and free fatty acids induce ER stress via increased reactive oxygen species, and ER Ca2+depletion from sarco/endoplasmic reticulum calcium ATPase (SERCA) dysfunction. Diminished SERCA expression and activity were observed in livers and macrophages of obese and insulin resistant mice, which also have higher level of ER stress (170). Overexpression of SERCA in obese mice reduced UPR activation and improved glucose homeostasis (167).
Other key regulators of ER homeostasis are the three luminal sensor inositole, requiring protein 1 (IRE1α), protein kinase RNA-like ER kinase (PERK) and activating transcription factor 6 (ATF6). Obesity leads to a disproportionate production of these key molecules. A reduction of ATF6 in the presence of a sustained PERK activation was observed (171). Hyperactivation of the IRE1α-XBP1 pathway has been documented in the adipose tissue of obese human (172). Shan et al. found that IRE1α was activated in adipose tissue macrophages and adipocytes of HFD fed mice (172). Their observations are consistent with other studies showing that in genetic and diet-induced models of obesity, IRE1α undergoes prominent activation (173).
A link between HSPs and obesity was suggested by observations that HSP70 was reduced in muscles of insulin-resistance obese patients with type 2 diabetes (174). Subsequently, several studies confirmed an altered expression of HSPs in obese humans and animals (94). Binding of extracellular HSP60 to TLR4 triggers a proinflammatory response and promotes insulin sensitivity (175). Obese subjects with or without type 2 diabetes have high HSP60 plasma levels. Interestingly, a reduced expression was observed in morbidly obese individuals after weight loss due to bariatric surgery (94).
Upregulation of HSP72 and HSP25 was previously shown to inhibit JNK and IKK-β activation, improve glucose tolerance, restore insulin-stimulated glucose transport, and increase insulin signaling in skeletal muscles from rats fed at high-fat diet (176).
Misfolded proteins can also be removed through autophagy, which is enhanced by CR (177). Autophagy is regulated by the integrated action of insulin and mTOR, both altered in obesity (178). Bugliani et al. have recently reported that promotion of autophagy increases survival of human pancreatic beta cells under ER stress and in type 2 diabetes (179). Yang et al. demonstrated that defective autophagy is causal to impaired hepatic insulin sensitivity and glucose homeostasis (178). Persistent IIS signaling in cell culture decreases autophagy and cell viability (164). Significant autophagy defects are observed related to ER stress in the liver tissue of HFD fed mice, and restoration of autophagy reduced ER stress (178).
Here we have briefly reviewed the abundant literature indicating that obesity significantly decreases mechanisms associated with proteome maintenance.
We have reviewed and organized the literature with the intent of showing the existing parallels between excessive fat accumulation and the aging process. We have categorized these reports following what have been proposed to be the nine hallmarks of aging (21) (Figure 1). Based on the evidence, two distinct hypotheses can be proposed. One is that the cellular responses provoked by an excess of nutrients cause obesity, and that obesity is responsible for accelerating the pace of aging. Supporting this hypothesis are the observations that knocking out the fat-specific insulin receptor, to produce extremely lean mice (180), and removal of visceral fat in rats (181) increased life span; additionally, CR on lean strains of rats, had only a minor effects on lifespan (182, 183). The alternative possibility is that the cellular responses provoked by an excess of nutrients are responsible for increasing the pace of aging. This common soil shared by both aging and obesity has been named “adipaging” (184), and there is some evidence of commonalities: hyperglycaemia, for example, induces senescence and the SASP in endothelial cells and macrophages (185) while glucose reduction prevents replicative senescence in human mesenchymal stem cells (186). The more abundant macronutrients (by weight and by calories) in the diet are usually carbohydrates and lipids, and specific reviews are available that focus on the possible toxic effects of their respective excess: carbotoxicity (187) and lipotoxicity (188). In this second scenario, obesity represents only a side effect of the excess nutrient status and the fulcrum are the cellular nutrient sensing pathways; see for example the possible central role of mTOR (189, 190). Whether adipose tissue hyper-function/dysfunction is causative of aging functional decline or whether it represents simply a marker of the advancing aging process will become clearer with future studies. In addition, not all fat depots are equal in their impact to health (191) and it could also turn out that both hypotheses are concurrently true (192).
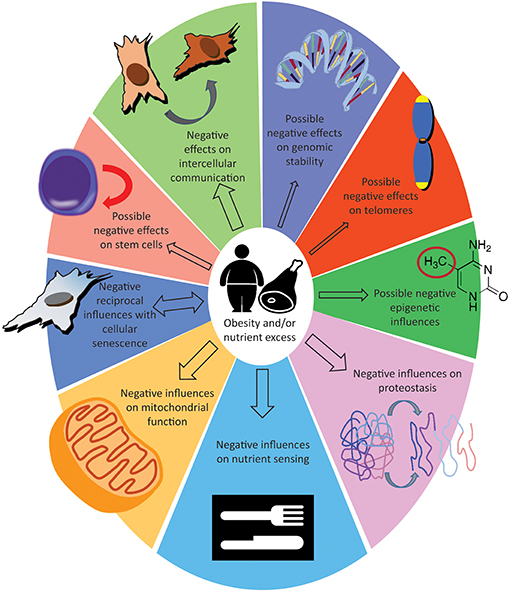
Figure 1. Effect of obesity or nutrient excess on the hallmarks of aging. The size of the arrows indicate how solid are the evidences, see “take home summaries” in the text. The double-headed arrow for cellular senescence indicate that detrimental influences can feedback from senescence to obesity.
While large epidemiological analyses indicate obesity will threaten any future gains in life expectancy (193), caloric restriction is considered a powerful tool to slow down the pace of aging. The effect of CR on increasing life span were first observed in rodents in 1935 (194). Although there are important differences between mouse, rat and human fat depots (195), the CR regiment has a clear impact in reducing fat mass in all these species (196). Caloric restriction studies are undoubtedly highly valuable for understanding the aging process; however, how to translate the data on animal models to human is a highly debated issue (197–204).
One caveat is that the response of our species could not be as strong as the one observed in experimental species. In fact, although the extension of life span with CR seems a universal biological response, this response is particularly evident in model species (Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster and Rodents) suggesting involuntary selection caused by human husbandry (205). Additionally, while wild-Drosophila respond to CR (206), wild-mice do not (207).
Moreover, it is not clear if all humans will benefit from CR. Data on diets mimicking fasting (208), a more recent approach to caloric restriction, suggest that this approach is effective, particularly on subjects at risk for metabolic disease (209). Additionally, while extreme obesity consistently increases all-cause mortality, overweight and even mild obesity appears protective toward cardiovascular disease. A phenomenon well described and referred to as the obesity paradox (210). This stresses the importance of carefully considering the starting BMI level before suggesting CR to people. Lorenzini has proposed that initial BMI levels could be the main reason why the two larger CR studies on rhesus monkeys gave discordant result on longevity (201).
If the possibilities raised in this review are correct, we will have to conclude that it is not CR that is slowing down aging but it is the ad libitum feeding, coupled with the lack of physical activity (the typical condition of laboratory animals) that are actually accelerating aging (181). If this is true, we should call ad libitum feeding “overfeeding,” an insight that has profound implications well beyond geroscience (211), and translating to humans the evidence from CR on animals will be as simple as to say: “avoid obesity”.
VS and AL wrote and revised the article. AL wrote Introduction and Conclusions. VS wrote all the other paragraphs. CS implemented and revised each section. All authors gave final approval of the submitted version.
This work was supported by RFO funding of University of Bologna to AL.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to all three reviewers for their valuable inputs.
BMI, body mass index; CR, caloric restriction; ASC, Adipose-derived stem cells; UPR, unfolded protein response; HSC, hematopoietic stem cells; BM, bone marrow; BMAT, bone marrow adipose tissue; BM-MSC, bone marrow stromal stem cell; HFD, high fat diet; GH, growth hormone; IGF-1, insulin-like growth factor; IGFBP, IGF binding protein; HSP, heat shock protein; ER, endoplasmic reticulum; UPR, unfolded protein response.
2. Omran AR. The epidemiologic transition: a theory of the epidemiology of population change. Milbank Q. (2005) 83:731–57. doi: 10.1111/j.1468-0009.2005.00398.x
3. Beltrán-Sánchez H, Soneji S, Crimmins EM. Past, present, and future of healthy life expectancy. Cold Spring Harb Perspect Med. (2015) 5:a025957. doi: 10.1101/cshperspect.a025957
4. Lorenzini A. Quantifying life style impact on lifespan. Annal Kinesiol. (2012) 2:139–48. Available online at: http://ojs.zrs.upr.si/index.php/AK/article/view/86
5. Crimmins EM, Beltrán-Sánchez H. Mortality and morbidity trends: is there compression of morbidity? J Gerontol B Psychol Sci Soc Sci. (2011) 66:75–86. doi: 10.1093/geronb/gbq088
6. Lakdawalla DN, Bhattacharya J, Goldman DP. Are the young becoming more disabled? Health Aff. (2004) 23:168–76. doi: 10.1377/hlthaff.23.1.168
7. Bhattacharya J, Cutler DM, Goldman DP, Hurd MD, Joyce GF, Lakdawalla DN, et al. Disability forecasts and future Medicare costs. Front Health Policy Res. (2004) 7:75–94. doi: 10.2202/1558-9544.1052
8. Murray CJ, Barber RM, Foreman KJ, Abbasoglu Ozgoren A, Abd-Allah F, Abera SF, et al. Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990–2013: quantifying the epidemiological transition. Lancet. (2015) 386:2145–91. doi: 10.1016/s0140-6736(15)61340-x
9. Hulsegge G, Picavet HS, Blokstra A, Nooyens AC, Spijkerman AM, van der Schouw YT, et al. Today's adult generations are less healthy than their predecessors: generation shifts in metabolic risk factors: the doetinchem cohort study. Eur J Prev Cardiol. (2014) 21:1134–44. doi: 10.1177/2047487313485512
10. Salomon JA, Wang H, Freeman MK, Vos T, Flaxman AD, Lopez AD, et al. Healthy life expectancy for 187 countries, 1990–2010: a systematic analysis for the Global Burden Disease Study (2010) Lancet. (2012) 380:2144–62. doi: 10.1016/S0140-6736(12)61690-0
11. Koster A, Leitzmann MF, Schatzkin A, Mouw T, Adams KF, van Eijk JT, et al. Waist circumference and mortality. Am J Epidemiol. (2008) 167:1465–75. doi: 10.1093/aje/kwn079
12. Manson JE, Willett WC, Stampfer MJ, Colditz GA, Hunter DJ, Hankinson SE, et al. Body weight and mortality among women. N Engl J Med. (1995) 333:677–85. doi: 10.1056/NEJM199509143331101
13. Olshansky SJ, Passaro DJ, Hershow RC, Layden J, Carnes BA, Brody J, et al. A potential decline in life expectancy in the United States in the 21st century. N Engl J Med. (2005) 352:1138–45. doi: 10.1056/NEJMsr043743
14. Willett WC, Dietz WH, Colditz GA, Guidelines for healthy weight. N Engl J Med. (1999) 341:427–34. doi: 10.1056/NEJM199908053410607
15. Seidell JC, Halberstadt J. The global burden of obesity and the challenges of prevention. Ann Nutr Metab. (2015) 66 (Suppl. 2):7–12. doi: 10.1159/000375143
16. Tiso D, Baldini M, Piaggesi N, Ferrari P, Biagi P, Malaguti M, et al. “7 days for my health” A new tool to evaluate kids' lifestyle. Agro Food Industry Hi-Tech. (2010) 21:47–50.
17. Pasqui F, Baldini M, Lorenzini A, Bordoni A, Malaguti M, Di Carlo M, et al. Evaluation of some nutritional and sport aspects in sedentary and active adolescents: analysis in Italian and Romanian school. Prog Nutr. (2009) 11:47–56.
18. Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. (2014) 384:766–81. doi: 10.1016/S0140-6736(14)60460-8
19. Kim S, Parks CG, DeRoo LA, Chen H, Taylor JA, Cawthon RM, et al. Obesity and weight gain in adulthood and telomere length. Cancer Epi Biomark Prev. (2009) 18:816–20. doi: 10.1158/1055-9965.EPI-08-0935
20. Ahima RS. Connecting obesity, aging and diabetes. Nat Med. (2009) 15:996–7. doi: 10.1038/nm0909-996
21. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
22. DiLoreto R, Murphy CT. The cell biology of aging. Mol Biol Cell. (2015) 26:4524–31. doi: 10.1091/mbc.E14-06-1084
23. Gomes NM, Ryder OA, Houck ML, Charter SJ, Walker W, Forsyth NR, et al. Comparative biology of mammalian telomeres: hypotheses on ancestral states and the roles of telomeres in longevity determination. Aging Cell. (2011) 10:761–8. doi: 10.1111/j.1474-9726.2011.00718.x
24. Mundstock E, Sarria EE, Zatti H, Mattos Louzada F, Kich Grun L, Herbert Jones M, et al. Effect of obesity on telomere length: systematic review and meta-analysis. Obesity (Silver Spring). (2015) 23:2165–74. doi: 10.1002/oby.21183
25. Chung HY, Kim HJ, Kim JW, Yu BP. The inflammation hypothesis of aging: molecular modulation by calorie restriction. Ann N Y Acad Sci. (2001) 928:327–35. doi: 10.1111/j.1749-6632.2001.tb05662.x
26. Lee M, Martin H, Firpo MA, Demerath EW. Inverse association between adiposity and telomere length: the Fels longitudinal study. Am J Hum Biol. (2011) 23:100–6. doi: 10.1002/ajhb.21109
27. Moreno-Navarrete JM, Ortega F, Sabater M, Ricart W, Fernández-Real JM. Telomere length of subcutaneous adipose tissue cells is shorter in obese and formerly obese subjects. Int J Obes (Lond). (2010) 34:1345–8. doi: 10.1038/ijo.2010.49
28. Zou Z, Mao L, Wang L, Cai W. The impact of overweight/obesity duration and physical activity on telomere length: an application of the WATCH paradigm. Obes Res Clin Pract. (2017) 11:247–252. doi: 10.1016/j.orcp.2016.11.002
29. Pérez LM, Amaral MA, Mundstock E, Barbé-Tuana FM, Guma FTCR, Jones MH, et al. Effects of diet on telomere length: systematic review and meta-analysis. Public Health Genomics. (2017) 20:286–292. doi: 10.1159/000486586
30. Valdes AM, Andrew T, Gardner JP, Kimura M, Oelsner E, Cherkas LF, et al. Obesity, cigarette smoking, and telomere length in women. Lancet. (2005) 366:662–4. doi: 10.1016/S0140-6736(05)66630-5
31. Nordfjäll K, Eliasson M, Stegmayr B, Melander O, Nilsson P, Roos G. Telomere length is associated with obesity parameters but with a gender difference. Obesity (Silver Spring). (2008) 16:2682–9. doi: 10.1038/oby.2008.413
32. Tzanetakou IP, Katsilambros NL, Benetos A, Mikhailidis DP, Perrea DN. “Is obesity linked to aging?”: adipose tissue and the role of telomeres. Ageing Res Rev. (2012) 11:220–9. doi: 10.1016/j.arr.2011.12.003
33. Drummond EM, Gibney ER. Epigenetic regulation in obesity. Curr Opin Clin Nutr Metab Care. (2013) 16:392–7. doi: 10.1097/MCO.0b013e3283620f45
34. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. (2013) 14:R115. doi: 10.1186/gb-2013-14-10-r115
35. de Mello VD, Pulkkinen L, Lalli M, Kolehmainen M, Pihlajamäki J, Uusitupa M, et al. DNA methylation in obesity and type 2 diabetes. Ann Med. (2014) 46:103–13. doi: 10.3109/07853890.2013.857259
36. Niculescu MD, Lupu DS. Nutritional influence on epigenetics and effects on longevity. Curr Opin Clin Nutr Metab Care. (2011) 14:35–40. doi: 10.1097/MCO.0b013e328340ff7c
37. Taylor EM, Jones AD, Henagan TM. A review of mitochondrial-derived fatty acids in epigenetic regulation of obesity and Type 2 diabetes. J Nutrit Health Food Sci. (2014) 2:1–4. doi: 10.15226/jnhfs.2014.00127
38. Khalyfa A, Carreras A, Hakim F, Cunningham JM, Wang Y, Gozal D. Effects of late gestational high-fat diet on body weight, metabolic regulation and adipokine expression in offspring. Int J Obes (Lond). (2013) 37:1481–9. doi: 10.1038/ijo.2013.12
39. Wang J, Gong B, Zhao W, Tang C, Varghese M, Nguyen T, et al. Epigenetic mechanisms linking diabetes and synaptic impairments. Diabetes. (2014) 63:645–54. doi: 10.2337/db13-1063
40. Wang J, Freire D, Knable L, Zhao W, Gong B, Mazzola P, et al. Childhood and adolescent obesity and long-term cognitive consequences during aging. J Comp Neurol. (2015) 523:757–68. doi: 10.1002/cne.23708
41. Kaushik P, Anderson JT. Obesity: epigenetic aspects. Biomol Concepts. (2016) 7:145–55. doi: 10.1515/bmc-2016-0010
42. Richmond RC, Sharp GC, Ward ME, Fraser A, Lyttleton O, McArdle WL, et al. DNA Methylation and BMI: investigating identified methylation sites at HIF3A in a causal framework. Diabetes. (2016) 65:1231–44. doi: 10.2337/db15-0996
43. Wang X, Zhu H, Snieder H, Su S, Munn D, Harshfield G, et al. Obesity related methylation changes in DNA of peripheral blood leukocytes. BMC Med. (2010) 8:87. doi: 10.1186/1741-7015-8-87
44. Nevalainen T, Kananen L, Marttila S, Jylhävä J, Mononen N, Kähönen M, et al. Obesity accelerates epigenetic aging in middle-aged but not in elderly individuals. Clin Epigenetics. (2017) 9:20. doi: 10.1186/s13148-016-0301-7
45. Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schönfels W, Ahrens M, et al. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci USA. (2014) 111:15538–43. doi: 10.1073/pnas.1412759111
46. Maegawa S, Lu Y, Tahara T, Lee JT, Madzo J, Liang S, et al. Caloric restriction delays age-related methylation drift. Nat Commun. (2017) 8:539. doi: 10.1038/s41467-017-00607-3
47. Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. (2006) 124:315–29. doi: 10.1016/j.cell.2005.11.044
48. Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. (2012) 483:218–21. doi: 10.1038/nature10815
49. Zhang N, Li Z, Mu W, Li L, Liang Y, Lu M, et al. Calorie restriction-induced SIRT6 activation delays aging by suppressing NF-κB signaling. Cell Cycle. (2016) 15:1009–18. doi: 10.1080/15384101.2016.1152427
50. Kuang J, Chen L, Tang Q, Zhang J, Li Y, He J. The role of Sirt6 in obesity and diabetes. Front Physiol. (2018) 9:135. doi: 10.3389/fphys.2018.00135
51. Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell. (2016) 61:654–666. doi: 10.1016/j.molcel.2016.01.028
52. Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy. (2011) 7:297–300. doi: 10.4161/auto.7.3.14502
53. Kusminski CM, Scherer PE. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol Metab. (2012) 23:435–43. doi: 10.1016/j.tem.2012.06.004
54. de Mello AH, Costa AB, Engel JDG, Rezin GT. Mitochondrial dysfunction in obesity. Life Sci. (2018) 192:26–32. doi: 10.1016/j.lfs.2017.11.019
55. Lee HC, Wei YH, Mitochondria and aging. Adv Exp Med Biol. (2012) 942:311–27. doi: 10.1007/978-94-007-2869-1_;14
56. Rong JX, Qiu Y, Hansen MK, Zhu L, Zhang V, Xie M, et al. Adipose mitochondrial biogenesis is suppressed in db/db and high-fat diet-fed mice and improved by rosiglitazone. Diabetes. (2007) 56:1751–60. doi: 10.2337/db06-1135
57. Heinonen S, Buzkova J, Muniandy M, Kaksonen R, Ollikainen M, Ismail K, et al. Impaired mitochondrial biogenesis in adipose tissue in acquired obesity. Diabetes. (2015) 64:3135–45. doi: 10.2337/db14-1937
58. Zorzano A, Liesa M, Palacín M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol. (2009) 41:1846–54. doi: 10.1016/j.biocel.2009.02.004
59. Hernández-Aguilera A, Rull A, Rodríguez-Gallego E, Riera-Borrull M, Luciano-Mateo F, Camps J, et al. Mitochondrial dysfunction: a basic mechanism in inflammation-related non-communicable diseases and therapeutic opportunities. Mediators Inflamm. (2013) 2013:13. doi: 10.1155/2013/135698
60. Pintus F, Floris G, Rufini A. Nutrient availability links mitochondria, apoptosis, and obesity. Aging (Albany NY). (2012) 4:734–41. doi: 10.18632/aging.100505
61. Cristofalo VJ, Lorenzini A, Allen RG, Torres C, Tresini M. Replicative senescence: a critical review. Mech Ageing Dev. (2004) 125:827–48. doi: 10.1016/j.mad.2004.07.010
62. Bitto A, Crowe EP, Lerner C, Torres C, Sell C. The senescence arrest program and the cell cycle. Methods Mol Biol. (2014) 1170:145–54. doi: 10.1007/978-1-4939-0888-2_;8
63. Aravinthan A. Cellular senescence: a hitchhiker's guide. Hum Cell. (2015) 28:51–64. doi: 10.1007/s13577-015-0110-x
64. Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, et al. Fat tissue, aging, and cellular senescence. Aging Cell. (2010) 9:667–84. doi: 10.1111/j.1474-9726.2010.00608.x
65. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. (1965) 37, 614–36. doi: 10.1016/0014-4827(65)90211-9
66. Lorenzini A, Maier AB. Influence of donor age and species longevity on replicative cellular senescence. In: Rattan S, Hayflick L, editors. Cellular Ageing and Replicative Senescence. Healthy Ageing and Longevity. Cham; Heidelberg; New York, NY; Dordrecht; London: Springer (2016). p. 49–70.
67. Erusalimsky JD, Kurz DJ. Cellular senescence in vivo: its relevance in ageing and cardiovascular disease. Exp Gerontol. (2005) 40:634–42. doi: 10.1016/j.exger.2005.04.010
68. Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev. (2007) 128:36–44. doi: 10.1016/j.mad.2006.11.008
69. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. (2016) 530:184–9. doi: 10.1038/nature16932
70. Shirakawa K, Yan X, Shinmura K, Endo J, Kataoka M, Katsumata Y, et al. Obesity accelerates T cell senescence in murine visceral adipose tissue. J Clin Invest. (2016) 126:4626–39. doi: 10.1172/JCI88606
71. Ogrodnik M, Zhu Y, Langhi LGP, Tchkonia T, Krüger P, Fielder E, et al. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab. (2019) doi: 10.1016/j.cmet.2018.12.008. [Epub ahead of print].
72. Mitterberger MC, Lechner S, Mattesich M, Zwerschke W. Adipogenic differentiation is impaired in replicative senescent human subcutaneous adipose-derived stromal/progenitor cells. J Gerontol A Biol Sci Med Sci. (2014) 69:13–24. doi: 10.1093/gerona/glt043
73. Ghosh AK, O'Brien M, Mau T, Qi N, Yung R. Adipose tissue senescence and inflammation in aging is reversed by the young milieu. J Gerontol A Biol Sci Med Sci. (2018) doi: 10.1093/gerona/gly290. [Epub ahead of print].
74. Prattichizzo F, De Nigris V, La Sala L, Procopio AD, Olivieri F, Ceriello A. “Inflammaging” as a druggable target: a senescence-associated secretory phenotype-centered view of Type 2 diabetes. Oxid Med Cell Longev. (2016) 2016:1810327. doi: 10.1155/2016/1810327
75. Burton DGA, Faragher RGA. Obesity and type-2 diabetes as inducers of premature cellular senescence and ageing. Biogerontology. (2018) 19:447–59. doi: 10.1007/s10522-018-9763-7
76. Franceschi C. Healthy ageing in 2016: obesity in geroscience - is cellular senescence the culprit? Nat Rev Endocrinol. (2017) 13:76–78. doi: 10.1038/nrendo.2016.213
77. Fontana L, Nehme J, Demaria M. Caloric restriction and cellular senescence. Mech Ageing Dev. (2018) 176:19–23. doi: 10.1016/j.mad.2018.10.005
78. Ahmed AS, Sheng MH, Wasnik S, Baylink DJ, Lau KW. Effect of aging on stem cells. World J Exp Med. (2017) 7:1–10. doi: 10.5493/wjem.v7.i1.1
79. Oh J, Lee YD, Wagers AJ. Stem cell aging: mechanisms, regulators and therapeutic opportunities. Nat Med. (2014) 20:870–80. doi: 10.1038/nm.3651
80. Oñate B, Vilahur G, Ferrer-Lorente R, Ybarra J, Díez-Caballero A, Ballesta-López C, et al. The subcutaneous adipose tissue reservoir of functionally active stem cells is reduced in obese patients. FASEB J. (2012) 26:4327–36. doi: 10.1096/fj.12-207217
81. Pérez LM, Bernal A, de Lucas B, San Martin N, Mastrangelo A, García A, et al. Altered metabolic and stemness capacity of adipose tissue-derived stem cells from obese mouse and human. PLoS ONE. (2015) 10:e0123397. doi: 10.1371/journal.pone.0123397
82. Oñate B, Vilahur G, Camino-López S, Díez-Caballero A, Ballesta-López C, Ybarra J, et al. Stem cells isolated from adipose tissue of obese patients show changes in their transcriptomic profile that indicate loss in stemcellness and increased commitment to an adipocyte-like phenotype. BMC Genomics. (2013) 14:625. doi: 10.1186/1471-2164-14-625
83. Tencerova M, Figeac F, Ditzel N, Taipaleenmäki H, Nielsen TK, Kassem M. High-fat diet-induced obesity promotes expansion of bone marrow adipose tissue and impairs skeletal stem cell functions in mice. J Bone Miner Res. (2018) 33:1154–65. doi: 10.1002/jbmr.3408
84. Rimmelé P, Bigarella CL, Liang R, Izac B, Dieguez-Gonzalez R, Barbet G, et al. Aging-like phenotype and defective lineage specification in SIRT1-deleted hematopoietic stem and progenitor cells. Stem Cell Rep. (2014) 3:44–59. doi: 10.1016/j.stemcr.2014.04.015
85. Lee JM, Govindarajah V, Goddard B, Hinge A, Muench DE, Filippi MD, et al. Obesity alters the long-term fitness of the hematopoietic stem cell compartment through modulation of. J Exp Med. (2018) 215:627–644. doi: 10.1084/jem.20170690
86. Woo M, Isganaitis E, Cerletti M, Fitzpatrick C, Wagers AJ, Jimenez-Chillaron J, et al. Early life nutrition modulates muscle stem cell number: implications for muscle mass and repair. Stem Cells Dev. (2011) 20:1763–9. doi: 10.1089/scd.2010.0349
87. Livesey FJ. A potential link between obesity and neural stem cell dysfunction. Nat Cell Biol. (2012) 14:987–9. doi: 10.1038/ncb2599
88. Murphy T, Thuret S. The systemic milieu as a mediator of dietary influence on stem cell function during ageing. Ageing Res Rev. (2015) 19:53–64. doi: 10.1016/j.arr.2014.11.004
89. Yilmaz ÖH, Katajisto P, Lamming DW, Gültekin Y, Bauer-Rowe KE, Sengupta S, et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature. (2012) 486:490–5. doi: 10.1038/nature11163
90. Cerletti M, Jang YC, Finley LW, Haigis MC, Wagers AJ. Short-term calorie restriction enhances skeletal muscle stem cell function. Cell Stem Cell. (2012) 10:515–9. doi: 10.1016/j.stem.2012.04.002
91. Brown-Borg HM, Sharma S, Borg KE, Rakoczy SG. Growth hormone and aging in mice. In: Sell C, Lorenzini A, Brown-Borg H, editors. Life-Span Extension. Aging Medicine. Humana Press (2009). doi: 10.1007/978-1-60327-507-1_;7
92. Lorenzini A, Salmon AB, Lerner C, Torres C, Ikeno Y, Motch S, et al. Mice producing reduced levels of insulin-like growth factor type 1 display an increase in maximum, but not mean, life span. J Gerontol A Biol Sci Med Sci. (2014) 69:410–9. doi: 10.1093/gerona/glt108
93. Blagosklonny MV. TOR-driven aging: speeding car without brakes. Cell Cycle. (2009) 8:4055–9. doi: 10.4161/cc.8.24.10310
94. Moura CS, Lollo PCB, Morato PN, Amaya-Farfan J. Dietary nutrients and bioactive substances modulate heat shock protein (HSP) expression: a review. Nutrients. (2018) 10:E683. doi: 10.3390/nu10060683
95. McArdle MA, Finucane OM, Connaughton RM, McMorrow AM, Roche HM. Mechanisms of obesity-induced inflammation and insulin resistance: insights into the emerging role of nutritional strategies. Front Endocrinol. (2013) 4:52. doi: 10.3389/fendo.2013.00052
96. Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. (2012) 148:852–71. doi: 10.1016/j.cell.2012.02.017
97. Franco C, Bengtsson BA, Johannsson G. The GH/IGF-1 Axis in Obesity: Physiological and Pathological Aspects. Metab Syndr Relat Disord. (2006) 4:51–6. doi: 10.1089/met.2006.4.51
98. Lewitt MS, Dent MS, Hall K. The insulin-like growth factor system in obesity, insulin resistance and Type 2 diabetes mellitus. J Clin Med. (2014) 3:1561–74. doi: 10.3390/jcm3041561
99. Rasmussen MH. Obesity, growth hormone and weight loss. Mol Cell Endocrinol. (2010) 316:147–53. doi: 10.1016/j.mce.2009.08.017
100. Cai H, Dong LQ, Liu F. Recent advances in adipose mTOR signaling and function: therapeutic prospects. Trends Pharmacol Sci. (2016) 37:303–317. doi: 10.1016/j.tips.2015.11.011
101. Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology. (2005) 146:1473–81. doi: 10.1210/en.2004-0921
102. Morris BJ, Carnes BA, Chen R, Donlon TA, He Q, Grove JS, et al. Genetic variation in the raptor gene is associated with overweight but not hypertension in American men of Japanese ancestry. Am J Hypertens. (2015) 28:508–17. doi: 10.1093/ajh/hpu188
103. Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Andrada P, Rotellar F, et al. Expression of S6K1 in human visceral adipose tissue is upregulated in obesity and related to insulin resistance and inflammation. Acta Diabetol. (2015) 52:257–66. doi: 10.1007/s00592-014-0632-9
104. Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. (2004) 431:200–5. doi: 10.1038/nature02866
105. Bouzakri K, Roques M, Gual P, Espinosa S, Guebre-Egziabher F, Riou JP, et al. Reduced activation of phosphatidylinositol-3 kinase and increased serine 636 phosphorylation of insulin receptor substrate-1 in primary culture of skeletal muscle cells from patients with type 2 diabetes. Diabetes. (2003) 52:1319–25. doi: 10.2337/diabetes.52.6.1319
106. Fernández-Veledo S, Vázquez-Carballo A, Vila-Bedmar R, Ceperuelo-Mallafré V, Vendrell J. Role of energy- and nutrient-sensing kinases AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) in adipocyte differentiation. IUBMB Life. (2013) 65:572–83. doi: 10.1002/iub.1170
107. Lindholm CR, Ertel RL, Bauwens JD, Schmuck EG, Mulligan JD, Saupe KW, et al. A high-fat diet decreases AMPK activity in multiple tissues in the absence of hyperglycemia or systemic inflammation in rats. J Physiol Biochem. (2013) 69:165–75. doi: 10.1007/s13105-012-0199-2
108. Gauthier MS, O'Brien EL, Bigornia S, Mott M, Cacicedo JM, Xu XJ, et al. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem Biophys Res Commun. (2011) 404:382–7. doi: 10.1016/j.bbrc.2010.11.127
109. Martin TL, Alquier T, Asakura K, Furukawa N, Preitner F, Kahn BB. Diet-induced obesity alters AMP kinase activity in hypothalamus and skeletal muscle. J Biol Chem. (2006) 281:18933–41. doi: 10.1074/jbc.M512831200
110. Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM. Increased malonyl-CoA levels in muscle from obese and type 2 diabetic subjects lead to decreased fatty acid oxidation and increased lipogenesis; thiazolidinedione treatment reverses these defects. Diabetes. (2006) 55:2277–85. doi: 10.2337/db06-0062
111. Sriwijitkamol A, Coletta DK, Wajcberg E, Balbontin GB, Reyna SM, Barrientes J, et al. Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes. (2007) 56:836–48. doi: 10.2337/db06-1119
112. Kola B, Christ-Crain M, Lolli F, Arnaldi G, Giacchetti G, Boscaro M, et al. Changes in adenosine 5′-monophosphate-activated protein kinase as a mechanism of visceral obesity in Cushing's syndrome. J Clin Endocrinol Metab. (2008) 93:4969–73. doi: 10.1210/jc.2008-1297
113. Chalkiadaki A, Guarente L. High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metab. (2012) 16:180–8. doi: 10.1016/j.cmet.2012.07.003
114. Qiao L, Shao J. SIRT1 regulates adiponectin gene expression through Foxo1-C/enhancer-binding protein alpha transcriptional complex. J Biol Chem. (2006) 281:39915–24. doi: 10.1074/jbc.M607215200
115. Pang W, Wang Y, Wei N, Xu R, Xiong Y, Wang P, et al. Sirt1 inhibits akt2-mediated porcine adipogenesis potentially by direct protein-protein interaction. PLoS ONE. (2013) 8:e71576. doi: 10.1371/journal.pone.0071576
116. Choi SE, Fu T, Seok S, Kim DH, Yu E, Lee KW, et al. Elevated microRNA-34a in obesity reduces NAD+ levels and SIRT1 activity by directly targeting NAMP. Aging Cell. (2013) 12:1062–72. doi: 10.1111/acel.12135
117. Pedersen SB, Ølholm J, Paulsen SK, Bennetzen MF, Richelsen B. Low Sirt1 expression, which is upregulated by fasting, in human adipose tissue from obese women. Int J Obes (Lond). (2008) 32:1250–5. doi: 10.1038/ijo.2008.78
118. de Kreutzenberg SV, Ceolotto G, Papparella I, Bortoluzzi A, Semplicini A, Dalla Man C, et al. Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: potential biochemical mechanisms. Diabetes. (2010) 59:1006–15. doi: 10.2337/db09-1187
119. Moschen AR, Wieser V, Gerner RR, Bichler A, Enrich B, Moser P, et al. Adipose tissue and liver expression of SIRT1, 3, and 6 increase after extensive weight loss in morbid obesity. J Hepatol. (2013) 59:1315–22. doi: 10.1016/j.jhep.2013.07.027
120. Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol. (2017) 8:1960. doi: 10.3389/fimmu.2017.01960
121. Skurk T, Alberti-Huber C, Herder C, Hauner H. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab. (2007) 92:1023–33. doi: 10.1210/jc.2006-1055
122. Blüher M. Adipose tissue inflammation: a cause or consequence of obesity-related insulin resistance? Clin Sci (Lond). (2016) 130:1603–14. doi: 10.1042/CS20160005
123. Mraz M, Haluzik M. The role of adipose tissue immune cells in obesity and low-grade inflammation. J Endocrinol. (2014) 222:R113–27. doi: 10.1530/JOE-14-0283
124. Trim W, Turner JE, Thompson D. Parallels in immunometabolic adipose tissue dysfunction with ageing and obesity. Front Immunol. (2018) 9:169. doi: 10.3389/fimmu.2018.00169
125. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. (2006) 116:1793–801. doi: 10.1172/JCI29069
126. Maffei M, Barone I, Scabia G, Santini F. The multifaceted haptoglobin in the context of adipose tissue and metabolism. Endocr Rev. (2016) 37:403–16. doi: 10.1210/er.2016-1009
127. Spagnuolo MS, Maresca B, La Marca V, Carrizzo A, Veronesi C, Cupidi C, et al. Haptoglobin interacts with apolipoprotein E and beta-amyloid and influences their crosstalk. ACS Chem Neurosci. (2014) 5:837–47. doi: 10.1021/cn500099f
128. Frasca D, McElhaney J. Influence of obesity on pneumococcus infection risk in the elderly. Front Endocrinol. (2019) 10:71. doi: 10.3389/fendo.2019.00071
129. Arai Y, Kamide K, Hirose N. Adipokine and aging: findings from centenarians and the very old. Front. Endocrinol. (2019) 10:142. doi: 10.3389/fendo.2019.00142
130. Mancuso P, Bouchard B. The impact of aging on adipose function and adipokine synthesis. Front Endocrinol. (2019) 10:137. doi: 10.3389/fendo.2019.00137
131. Suades R, Padró T, Badimon L. The role of blood-borne microparticles in inflammation and hemostasis. Semin Thromb Hemost. (2015) 41:590–606. doi: 10.1055/s-0035-1556591
132. Stepanian A, Bourguignat L, Hennou S, Coupaye M, Hajage D, Salomon L, et al. Microparticle increase in severe obesity: not related to metabolic syndrome and unchanged after massive weight loss. Obesity (Silver Spring). (2013) 21:2236–43. doi: 10.1002/oby.20365
133. Cheng V, Kashyap SR, Schauer PR, Kirwan JP, McCrae KR. Restoration of glycemic control in patients with type 2 diabetes mellitus after bariatric surgery is associated with reduction in microparticles. Surg Obes Relat Dis. (2013) 9:207–12. doi: 10.1016/j.soard.2011.09.026
134. Huang-Doran I, Zhang CY, Vidal-Puig A. Extracellular vesicles: novel mediators of cell communication in metabolic disease. Trends Endocrinol Metab. (2017) 28:3–18. doi: 10.1016/j.tem.2016.10.003
135. Kranendonk ME, Visseren FL, van Balkom BW, Nolte-'t Hoen EN, van Herwaarden JA, de Jager W, et al. Human adipocyte extracellular vesicles in reciprocal signaling between adipocytes and macrophages. Obesity (Silver Spring). (2014) 22:1296–308. doi: 10.1002/oby.20679
136. Togliatto G, Dentelli P, Gili M, Gallo S, Deregibus C, Biglieri E, et al. Obesity reduces the pro-angiogenic potential of adipose tissue stem cell-derived extracellular vesicles (EVs) by impairing miR-126 content: impact on clinical applications. Int J Obes (Lond). (2016) 40:102–11. doi: 10.1038/ijo.2015.123
137. Szilard L. On the nature of the aging process. Proc Natl Acad Sci USA. (1959) 45:30–45. doi: 10.1073/pnas.45.1.30
138. Waaijer ME, Croco E, Westendorp RG, Slagboom PE, Sedivy JM, Lorenzini A, et al. DNA damage markers in dermal fibroblasts in vitro reflect chronological donor age. Aging (Albany NY). (2016) 8:147–57. doi: 10.18632/aging.100890
139. Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, et al. Somatic mutant clones colonize the human esophagus with age. Science. (2018) 362:911–7. doi: 10.1126/science.aau3879
140. Croco E, Marchionni S, Storci G, Bonafè M, Franceschi C, Stamato TD, et al. Convergent adaptation of cellular machineries in the evolution of large body masses and long life spans. Biogerontology. (2017) 18:485–497. doi: 10.1007/s10522-017-9713-9
141. Lorenzini A, Johnson FB, Oliver A, Tresini M, Smith JS, Hdeib M, et al. Significant correlation of species longevity with DNA double strand break recognition but not with telomere length. Mech Ageing Dev. (2009) 130:784–92. doi: 10.1016/j.mad.2009.10.004
142. Croco E, Marchionni S, Bocchini M, Angeloni C, Stamato T, Stefanelli C, et al. DNA Damage detection by 53BP1: relationship to species longevity. J Gerontol A Biol Sci Med Sci. (2017) 72:763–770. doi: 10.1093/gerona/glw170
143. Setayesh T, Nersesyan A, Mišík M, Ferk F, Langie S, Andrade VM, et al. Impact of obesity and overweight on DNA stability: Few facts and many hypotheses. Mutat Res. (2018) 777:64–91. doi: 10.1016/j.mrrev.2018.07.001
144. Othman EM, Kreissl MC, Kaiser FR, Arias-Loza PA, Stopper H. Insulin-mediated oxidative stress and DNA damage in LLC-PK1 pig kidney cell line, female rat primary kidney cells, and male ZDF rat kidneys in vivo. Endocrinology. (2013) 154:1434–43. doi: 10.1210/en.2012-1768
145. Jang Y, Kim OY, Ryu HJ, Kim JY, Song SH, Ordovas JM, et al. Visceral fat accumulation determines postprandial lipemic response, lipid peroxidation, DNA damage, and endothelial dysfunction in nonobese Korean men. J Lipid Res. (2003) 44:2356–64. doi: 10.1194/jlr.M300233-JLR200
146. Palmer NO, Bakos HW, Owens JA, Setchell BP, Lane M. Diet and exercise in an obese mouse fed a high-fat diet improve metabolic health and reverse perturbed sperm function. Am J Physiol Endocrinol Metab. (2012) 302:E768–80. doi: 10.1152/ajpendo.00401.2011
147. Martino S, Cauley JA, Barrett-Connor E, Powles TJ, Mershon J, Disch D, et al. Continuing outcomes relevant to Evista: breast cancer incidence in postmenopausal osteoporotic women in a randomized trial of raloxifene. J Natl Cancer Inst. (2004) 96:1751–61. doi: 10.1093/jnci/djh319
148. Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol. (2014) 2:411–29. doi: 10.1016/j.redox.2013.12.016
149. Song F, Hurtado del Pozo C, Rosario R, Zou YS, Ananthakrishnan R, Xu X, et al. RAGE regulates the metabolic and inflammatory response to high-fat feeding in mice. Diabetes. (2014) 63:1948–65. doi: 10.2337/db13-1636
150. Gaens KH, Goossens GH, Niessen PM, van Greevenbroek MM, van der Kallen CJ, Niessen HW, et al. Nε-(carboxymethyl)lysine-receptor for advanced glycation end product axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arterioscler Thromb Vasc Biol. (2014) 34:1199–208. doi: 10.1161/ATVBAHA.113.302281
151. Uribarri J, Cai W, Woodward M, Tripp E, Goldberg L, Pyzik R, et al. Elevated serum advanced glycation endproducts in obese indicate risk for the metabolic syndrome: a link between healthy and unhealthy obesity? J Clin Endocrinol Metab. (2015) 100:1957–66. doi: 10.1210/jc.2014-3925
152. Pálmai-Pallag T, Bachrati CZ. Inflammation-induced DNA damage and damage-induced inflammation: a vicious cycle. Microbes Infect. (2014) 16:822–32. doi: 10.1016/j.micinf.2014.10.001
153. Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. (2008) 371:569–78. doi: 10.1016/S0140-6736(08)60269-X
154. La Vignera S, Condorelli RA, Vicari E, Calogero AE. Negative effect of increased body weight on sperm conventional and nonconventional flow cytometric sperm parameters. J Androl. (2012) 33:53–8. doi: 10.2164/jandrol.110.012120
155. Fariello RM, Pariz JR, Spaine DM, Cedenho AP, Bertolla RP, Fraietta R. Association between obesity and alteration of sperm DNA integrity and mitochondrial activity. BJU Int. (2012) 110:863–7. doi: 10.1111/j.1464-410X.2011.10813.x
156. Langie SA, Achterfeldt S, Gorniak JP, Halley-Hogg KJ, Oxley D, van Schooten FJ, et al. Maternal folate depletion and high-fat feeding from weaning affects DNA methylation and DNA repair in brain of adult offspring. FASEB J. (2013) 27:3323–34. doi: 10.1096/fj.12-224121
157. Tyson J, Caple F, Spiers A, Burtle B, Daly AK, Williams EA, et al. Inter-individual variation in nucleotide excision repair in young adults: effects of age, adiposity, micronutrient supplementation and genotype. Br J Nutr. (2009) 101:1316–23. doi: 10.1017/S0007114508076265
158. Remely M, Ferk F, Sterneder S, Setayesh T, Kepcija T, Roth S, et al. Vitamin E modifies high-fat diet-induced increase of DNA strand breaks, and changes in expression and DNA methylation of Dnmt1 and MLH1 in C57BL/6J Male Mice. Nutrients. (2017) 9:E607. doi: 10.3390/nu9060607
159. Heydari AR, Unnikrishnan A, Lucente LV, Richardson A. Caloric restriction and genomic stability. Nucleic Acids Res. (2007) 35:7485–96. doi: 10.1093/nar/gkm860
160. Bankoglu EE, Seyfried F, Arnold C, Soliman A, Jurowich C, Germer CT, et al. Reduction of DNA damage in peripheral lymphocytes of obese patients after bariatric surgery-mediated weight loss. Mutagenesis. (2018) 33:61–67. doi: 10.1093/mutage/gex040
161. Usman M, Volpi EV. DNA damage in obesity: Initiator, promoter and predictor of cancer. Mutat Res. (2018) 778:23–37. doi: 10.1016/j.mrrev.2018.08.002
163. Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem. (2015) 84:435–64. doi: 10.1146/annurev-biochem-060614-033955
164. Bitto A, Lerner C, Torres C, Roell M, Malaguti M, Perez V, et al. Long-term IGF-I exposure decreases autophagy and cell viability. PLoS One. (2010) 5:e12592. doi: 10.1371/journal.pone.0012592
165. Bitto A, Lerner CA, Nacarelli T, Crowe E, Torres C, Sell C, et al. P62/SQSTM1 at the interface of aging, autophagy, and disease. Age (Dordr). (2014) 36:9626. doi: 10.1007/s11357-014-9626-3
166. Takamura T. Remodeling of nutrient homeostasis by unfolded protein response. Diabetes. (2014) 63:841–3. doi: 10.2337/db13-1721
167. Pagliassotti MJ, Kim PY, Estrada AL, Stewart CM, Gentile CL. Endoplasmic reticulum stress in obesity and obesity-related disorders: an expanded view. Metabolism. (2016) 65:1238–46. doi: 10.1016/j.metabol.2016.05.002
168. Otoda T, Takamura T, Misu H, Ota T, Murata S, Hayashi H, et al. Proteasome dysfunction mediates obesity-induced endoplasmic reticulum stress and insulin resistance in the liver. Diabetes. (2013) 62:811–24. doi: 10.2337/db11-1652
169. Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. (2009) 119:1201–15. doi: 10.1172/JCI37007
170. Lee J, Ozcan U. Unfolded protein response signaling and metabolic diseases. J Biol Chem. (2014) 289:1203–11. doi: 10.1074/jbc.R113.534743
171. Yang L, Calay ES, Fan J, Arduini A, Kunz RC, Gygi SP, et al. Metabolism. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science. (2015) 349:500–6. doi: 10.1126/science.aaa0079
172. Shan B, Wang X, Wu Y, Xu C, Xia Z, Dai J, et al. The metabolic ER stress sensor IRE1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat Immunol. (2017) 18:519–529. doi: 10.1038/ni.3709
173. Bujisic B, Martinon F. IRE1 gives weight to obesity-associated inflammation. Nat Immunol. (2017) 18:479–480. doi: 10.1038/ni.3725
174. Henstridge DC, Whitham M, Febbraio MA. Chaperoning to the metabolic party: The emerging therapeutic role of heat-shock proteins in obesity and type 2 diabetes. Mol Metab. (2014) 3:781–93. doi: 10.1016/j.molmet.2014.08.003
175. Ohashi K, Burkart V, Flohé S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. (2000) 164:558–61. doi: 10.4049/jimmunol.164.2.558
176. Gupte AA, Bomhoff GL, Swerdlow RH, Geiger PC. Heat treatment improves glucose tolerance and prevents skeletal muscle insulin resistance in rats fed a high-fat diet. Diabetes. (2009) 58:567–78. doi: 10.2337/db08-1070
177. Wohlgemuth SE, Julian D, Akin DE, Fried J, Toscano K, Leeuwenburgh C, et al. Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res. (2007) 10:281–92. doi: 10.1089/rej.2006.0535
178. Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. (2010) 11:467–78. doi: 10.1016/j.cmet.2010.04.005
179. Bugliani M, Mossuto S, Grano F, Suleiman M, Marselli L, Boggi U, et al. Autophagy regulates the function and survival of human pancreatic beta cells under endoplasmic reticulum stress and in type 2 diabetes. Front Endocr. (2019) 10:52. doi: 10.3389/fendo.2019.00052
180. Blüher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. (2003) 299:572–4. doi: 10.1126/science.1078223
181. Muzumdar R, Allison DB, Huffman DM, Ma X, Atzmon G, Einstein FH, et al. Visceral adipose tissue modulates mammalian longevity. Aging Cell. (2008) 7:438–40. doi: 10.1111/j.1474-9726.2008.00391.x
182. Teillet L, Gouraud S, Corman B. Does food restriction increase life span in lean rats? J Nutr Health Aging. (2004) 8:213−8.
183. Aydin C, Jarema KA, Phillips PM, Gordon CJ. Caloric restriction in lean and obese strains of laboratory rat: effects on body composition, metabolism, growth and overall health. Exp Physiol. (2015) 100:1280–97. doi: 10.1113/EP085469
184. Pérez LM, Pareja-Galeano H, Sanchis-Gomar F, Emanuele E, Lucia A, Gálvez BG. ‘Adipaging’: ageing and obesity share biological hallmarks related to a dysfunctional adipose tissue. J Physiol. (2016) 594:3187–207. doi: 10.1113/JP271691
185. Prattichizzo F, De Nigris V, Mancuso E, Spiga R, Giuliani A, Matacchione G, et al. Short-term sustained hyperglycaemia fosters an archetypal senescence-associated secretory phenotype in endothelial cells and macrophages. Redox Biol. (2018) 15:170–181. doi: 10.1016/j.redox.2017.12.001
186. Lo T, Ho JH, Yang MH, Lee OK. Glucose reduction prevents replicative senescence and increases mitochondrial respiration in human mesenchymal stem cells. Cell Transplant. (2011) 20:813–25. doi: 10.3727/096368910X539100
187. Kroemer G, López-Otín C, Madeo F, de Cabo R. Carbotoxicity-noxious effects of carbohydrates. Cell. (2018) 175:605–614. doi: 10.1016/j.cell.2018.07.044
188. Engin AB. What is lipotoxicity? Adv Exp Med Biol. (2017) 960:197–220. doi: 10.1007/978-3-319-48382-5_;8
189. Blagosklonny MV. TOR-centric view on insulin resistance and diabetic complications: perspective for endocrinologists and gerontologists. Cell Death Dis. (2013) 4:e964. doi: 10.1038/cddis.2013.506
190. Guillén C, Benito M. mTORC1 overactivation as a key aging factor in the progression to type 2 diabetes mellitus. Front Endocrinol (Lausanne). (2018) 9:621. doi: 10.3389/fendo.2018.00621
191. Huffman DM, Barzilai N. Contribution of adipose tissue to health span and longevity. Interdiscip Top Gerontol. (2010) 37:1–19. doi: 10.1159/000319991
192. Stout MB, Justice JN, Nicklas BJ, Kirkland JL. Physiological aging: links among adipose tissue dysfunction, diabetes, and frailty. Physiology (Bethesda). (2017) 32:9–19. doi: 10.1152/physiol.00012.2016
193. Preston SH, Vierboom YC, Stokes A. The role of obesity in exceptionally slow US mortality improvement. Proc Natl Acad Sci USA. (2018) 115:957–961. doi: 10.1073/pnas.1716802115
194. McCay CM, Crowell MF, Maynard LA. The effect of retarded growth upon the length of life span and upon the ultimate body size 1935. Nutrition. (1989) 5:155–71; discussion 172. doi: 10.1093/jn/10.1.63
195. Chusyd DE, Wang D, Huffman DM, Nagy TR. Relationships between rodent white adipose fat pads and human white adipose fat depots. Front Nutr. (2016) 3:10. doi: 10.3389/fnut.2016.00010
196. Barzilai N, Gupta G. Revisiting the role of fat mass in the life extension induced by caloric restriction. J Gerontol A Biol Sci Med Sci. (1999) 54:B89–96; discussion B97–8. doi: 10.1093/gerona/54.3.B89
197. Phelan JP, Rose MR. Why dietary restriction substantially increases longevity in animal models but won't in humans. Ageing Res Rev. (2005) 4:339–50. doi: 10.1016/j.arr.2005.06.001
198. Speakman JR, Hambly C. Starving for life: what animal studies can and cannot tell us about the use of caloric restriction to prolong human lifespan. J Nutr. (2007) 137:1078–86. doi: 10.1093/jn/137.4.1078
199. Roth LW, Polotsky AJ. Can we live longer by eating less? a review of caloric restriction and longevity. Maturitas. (2012) 71:315–9. doi: 10.1016/j.maturitas.2011.12.017
200. Cava E, Fontana L. Will calorie restriction work in humans? Aging (Albany NY). (2013) 5:507–14. doi: 10.18632/aging.100581
201. Lorenzini A. How much should we weigh for a long and healthy life span? the need to reconcile caloric restriction versus longevity with body mass index versus mortality data. Front Endocrinol (Lausanne). (2014) 5:121. doi: 10.3389/fendo.2014.00121
202. Sohal RS, Forster MJ. Caloric restriction and the aging process: a critique. Free Radic Biol Med. (2014) 73:366–82. doi: 10.1016/j.freeradbiomed.2014.05.015
203. Le Bourg E, Redman LM. Do-it-yourself calorie restriction: the risks of simplistically translating findings in animal models to humans. Bioessays. (2018) 40:e1800087. doi: 10.1002/bies.201800087
204. Le Bourg E. Does calorie restriction in primates increase lifespan? revisiting studies on Macaques (Macaca mulatta) and Mouse Lemurs (Microcebus murinus). Bioessays. (2018) 40:e1800111. doi: 10.1002/bies.201800111
205. Nakagawa S, Lagisz M, Hector KL, Spencer HG. Comparative and meta-analytic insights into life extension via dietary restriction. Aging Cell. (2012) 11:401–9. doi: 10.1111/j.1474-9726.2012.00798.x
206. Metaxakis A, Partridge L. Dietary restriction extends lifespan in wild-derived populations of Drosophila melanogaster. PLoS One. (2013) 8:e74681. doi: 10.1371/journal.pone.0074681
207. Harper JM, Leathers CW, Austad SN. Does caloric restriction extend life in wild mice? Aging Cell. (2006) 5:441–9. doi: 10.1111/j.1474-9726.2006.00236.x
208. Brandhorst S, Choi IY, Wei M, Cheng CW, Sedrakyan S, Navarrete G, et al. A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab. (2015) 22:86–99. doi: 10.1016/j.cmet.2015.05.012
209. Wei M, Brandhorst S, Shelehchi M, Mirzaei H, Cheng CW, Budniak J, et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci Transl Med. (2017) 9:eaai8700. doi: 10.1126/scitranslmed.aai8700
210. Lavie CJ, De Schutter A, Parto P, Jahangir E, Kokkinos P, Ortega FB, et al. Obesity and prevalence of cardiovascular diseases and prognosis-the obesity paradox updated. Prog Cardiovasc Dis. (2016) 58:537–47. doi: 10.1016/j.pcad.2016.01.008
Keywords: obesity, overweight, aging, aging hallmarks, caloric restriction
Citation: Salvestrini V, Sell C and Lorenzini A (2019) Obesity May Accelerate the Aging Process. Front. Endocrinol. 10:266. doi: 10.3389/fendo.2019.00266
Received: 09 November 2018; Accepted: 10 April 2019;
Published: 03 May 2019.
Edited by:
Alessandro Cellerino, Scuola Normale Superiore di Pisa, ItalyReviewed by:
Alessandro Bitto, University of Washington, United StatesCopyright © 2019 Salvestrini, Sell and Lorenzini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonello Lorenzini, YW50b25lbGxvLmxvcmVuemluaUB1bmliby5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.