
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Endocrinol. , 30 April 2019
Sec. Pediatric Endocrinology
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00263
 Eloïse Giabicani1*
Eloïse Giabicani1* Sandra Chantot-Bastaraud1,2Adeline Bonnard2Myriam Rachid2
Sandra Chantot-Bastaraud1,2Adeline Bonnard2Myriam Rachid2 Sandra Whalen3Irène Netchine1
Sandra Whalen3Irène Netchine1 Frédéric Brioude1
Frédéric Brioude1We report an original association of complex genetic defects in a patient carrying both an 11p paternal duplication, resulting in the double expression of insulin-like growth factor 2 (IGF2), as reported in Beckwith-Wiedemann syndrome, and a 15q terminal deletion, including the type 1 IGF receptor gene (IGF1R), resulting in haploinsufficiency for this gene. The patient was born with measurements appropriate for her gestational age but experienced growth retardation in early childhood, allowing a better comprehension of the IGF system in the pathophysiology of growth. It is possible that IGF-II plays a key role in fetal growth, independently of IGF1R signaling, and that its role is less important in post-natal growth, leaving IGF-I and growth hormone as the main actors.
Fetal and postnatal growth is a complex process, involving genetic, endocrine, and environmental factors. The insulin-like growth factor (IGF) system includes two ligands (IGF-I and IGF-II), two receptors [the IGF receptor type I (IGF1R) and the mannose-6-phosphate cation independent (M6PCI) receptor or IGF2R] (1). Circulating IGF-I is mainly produced by the liver and its production is stimulated by growth hormone (GH) after birth. IGF2 is located in the 11p15 region in humans and is an imprinted gene expressed only from the paternal allele (Figure 1) (2). During fetal life, IGF2 exhibits monoallelic expression and IGF-II acts as an auto/paracrine factor. After birth, circulating IGF-II is produced from both alleles by the liver, whereas IGF2 is expressed from one allele in most other tissues and acts as a paracrine factor (3, 4). The role of circulating IGF-II after birth in humans is unclear. IGF-I and IGF-II both act through the IGF1R, which is a ubiquitously expressed tyrosine kinase receptor.
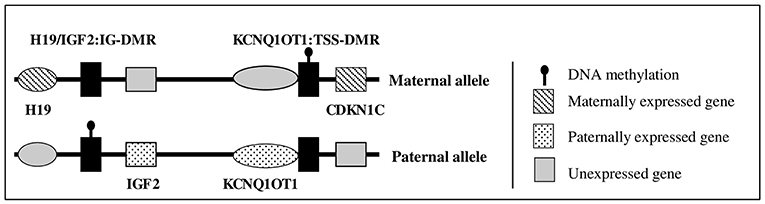
Figure 1. Schematic representation of 11p15 region gene expression.
In humans, molecular anomalies of the 11p15 region have been observed in two rare diseases characterized by abnormal fetal and postnatal growth: Beckwith-Wiedemann syndrome (BWS, OMIM #130650) and Silver Russell syndrome (SRS, OMIM #180860). BWS is characterized by fetal and postnatal overgrowth, macroglossia, exomphalos, organomegaly, lateralized overgrowth, and an increased risk of embryonic tumors during early life (5). In contrast, SRS is characterized by fetal and postnatal growth retardation, with a relatively conserved head circumference at birth, hemihypotrophy, feeding difficulties, and a protruding forehead (6, 7). The 11p15 region contains two domains: the telomeric domain, containing IGF2, only expressed from the paternal allele, and the maternally expressed H19 gene (a long non-coding RNA); and the centromeric domain, which includes the maternally expressed cyclin-dependent kinase inhibitor 1C (CDKN1C) gene (a negative regulator of the cell cycle, which reduces fetal growth) (Figure 1) (2, 8, 9). The expression of H19 and IGF2 is controlled by an imprinting center, called the H19/IGF2 intergenic differentially methylated region (IG-DMR) (previously called IC1), which is methylated on the paternal allele. CDKN1C expression is controlled by a second imprinting center called KCNQ1OT1:TSS-DMR (or IC2), which is methylated on the maternal allele. Abnormal methylation of IC1 or IC2 or uniparental disomy can lead to abnormal expression of IGF2 and/or CDKN1C, resulting in abnormal fetal/postnatal growth (2, 6). Duplications of 11p15 have been rarely reported, with either overgrowth or growth retardation, depending on the gene content and the parental origin of the duplication (9–11).
IGF1R is located on chromosome 15q26 and spans 315kb. IGF1R disruption (OMIM#270450) is usually responsible for fetal and postnatal growth retardation, with paradoxically high levels of plasma IGF-I (defining IGF-I resistance) and can be associated with microcephaly, variable levels of cognitive impairment, micrognathia, and feeding difficulties (12, 13). The phenotype is highly heterogeneous. In most cases, the anomaly is present in a heterozygous state, but rare homozygous or compound heterozygous mutation carriers have been reported (13–15).
We report here a patient with postnatal growth retardation and a complex chromosomal rearrangement, including a distal 15q26.3-qter deletion, encompassing the telomeric part of IGF1R, and a mosaic paternal duplication of the entire 11p15 region. Although the 11p duplication should have led to BWS, the patient presented with growth retardation, microcephaly, and intellectual disability, which is in accordance with the IGF1R disruption phenotype. We discuss the impact of these two rare genetic defects on the growth phenotype, which highlights the major role of IGF1R in IGF-II signaling.
The patient was sent to a reference tertiary center because of intellectual disability. She was born after 36 weeks of amenorrhea (WA), with birth parameters appropriate for gestational age (AGA). Her birth weight was 2380 g [−0.6 standard deviation score (SDS)]. Her birth length was not recorded, but at 1 month of age (equivalent to 40 WA), it was 50 cm (in the normal range). Her head circumference at birth was 30 cm (−2.4 SDS). She was born from unrelated healthy parents of Romanian origin. The mother is 162 cm (−0.2 SDS) and the father 170 cm (−0.8 SDS) tall; both had birth parameters AGA. The target height was 159.5 cm (−0.7 SDS) and there was no familial history of short stature. The growth curve is shown in Figure 2. At the age of 8 years and 9 months, her height was 117.8 cm (−2.1 SDS), weight 24.5 kg [body mass index (BMI) of 17.7 kg/m2 (1.2 SDS)], and head circumference 47.5 cm (−3.2 SDS). She had no clinical signs of BWS according to the consensus clinical scoring system proposed in 2018 (5). Only two out of four items for the clinical scoring system for IGF1R defects were present (16). She presented with strabismus and interventricular communication, with no cardiac failure. She acquired motor skills with normal timing but experienced an early delay in language and cognitive development and required specialized education.
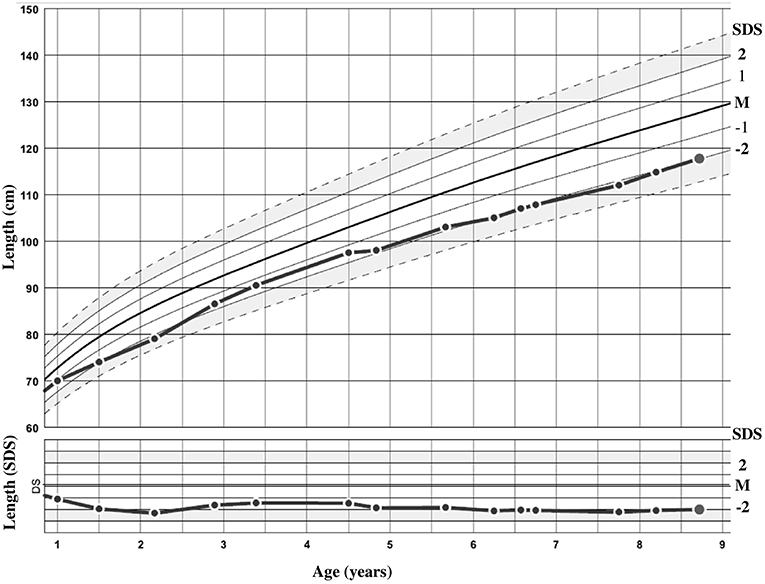
Figure 2. Growth curve of the patient in centimeters and standard deviation score (SDS).
At the age of 3 years, her serum IGF-I level was in the upper range of the norm (145 ng/ml, 0.9 SDS), with elevated basal GH (45 mUI/L). At 8 years and 9 months, her hormonal status was as follows: IGF-I 345.3 ng/mL (2.1 SDS), IGF-binding protein (IGF-BP3) 4,638 ng/mL (normal range from 2,146 to 5,801 ng/mL), acid-labile subunit 2,145 mU/mL (normal range from 813 to 1,729 mU/mL) and IGF-II 710 ng/mL (normal range from 433 to 997 ng/mL). These data are in favor of IGF-I resistance with high levels of IGF-I and ALS.
Karyotype analysis revealed a mosaic karyotype 45,XX,dic(15;21)(q26.3;q10)[4]/46,XX[20] with two cell lines: (1) a 45,XX cell line with a dicentric chromosome dic(15;21) due to an apparently balanced mosaic structural rearrangement involving one chromosome 15q and one chromosome 21p (Figure 3A left) and (2) a 46,XX cell line. The karyotypes of the parents were normal.
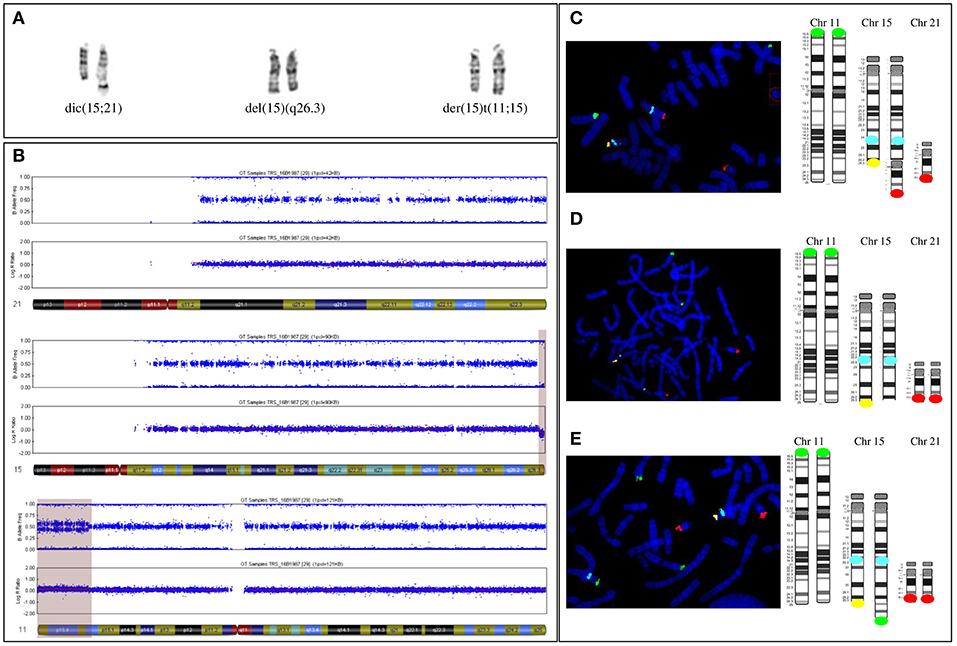
Figure 3. (A) Paired homologous chromosomes15 with Derivative chromosome 15 (left couple, on the right) and apparently normal 15 G-banded chromosomes 15 (middle couple) and (right couple). The two apparently normal chromosomes were then shown to carry a 15q terminal deletion (middle couple, on the right) and be a derivative chromosome 11 (right couple, on the right). (B) The SNP array profile shows a normal chromosome 21 (top), a heterozygous homogeneous 15q terminal deletion (middle, light purple area), and a mosaic gain on chromosome 11p (bottom, light purple area). (C) FISH [with a specific 11p subtelomeric probe (green), specific 15q22 probe (aqua blue), specific 15q subtelomeric probe (yellow), and specific subtelomeric 21q probe (red)] and schematic representation showing chromosomes 11, 15, and 21 in the three cell lines. First cell line with an unbalanced 15q;21p translocation. (D) Second cell line with a deleted 15q chromosome. The derivative chromosome 15 is missing a yellow signal. (E) Third cell line with an unbalanced 11p;15q translocation. The derivative chromosome 15 has a supernumerary green signal and no yellow signal.
Single-nucleotide polymorphism (SNP) array revealed a more complex unbalanced autosomal structural rearrangement than expected from the karyotype analysis: (1) no copy number variation for chromosome 21, (2) a 3 Mb homogeneous heterozygous 15qter deletion, and (3) a 13.4 Mb mosaic 11pter duplication in approximately 30% of the cells analyzed (Figure 3B from top to bottom) (10).
Subsequent fluorescent in situ hybridization (FISH) analysis with specific subtelomeric 11p, 15q, and 21q probes showed a supernumerary signal for 11p at the end of a deleted 15qter chromosome in ~30% of cells, accounting for an unbalanced translocation t(11p;15q), with a normal signal on the two telomeres of 21q (Figures 3A right, E). In approximately 20% of cells, one signal for the chromosome 21q telomere was located at the end of the deleted 15qter chromosome with a normal signal for the two 11p telomeric probes, accounting for an unbalanced translocation t(15q;21q) (Figures 3A left, C). In the remaining cells, with a normal signal on telomeres 11p and 21q, only one signal from the telomeric 15q probe was detected, corresponding to a 15q deletion (Figures 3A middle, D). Thus, the karyotype of the patient was finally amended to ISCN 2016 as de novo mos 45,XX,dic(15;21)(q26.3;q10)/46,XX.ish.der(15)(11;15)(p15.2;q26.3)(RP11-889I17+,D15S936-)/46,XX.ish.del(15)(q26.3)(D15S936-).arr[hg19]11p15.5p15.2(204,062-13,618,804)x3[0.3],15q26.3(99,434,357-102,461,162)x1dn.
Thus, the patient's mosaic included three abnormal cell lines: the first (30%) with 46 chromosomes and a chromosome 15 derived from an unbalanced translocation between the chromosome 11p region and the terminal 15q26 region of chromosome 15; the second (20%) with 45 chromosomes and an unbalanced dicentric chromosome derived from both chromosomes 21 and 15, with a 15q26 terminal deletion, and the third (50%) with 46 chromosomes that only carried the 15q26 terminal deletion of one chromosome.
Methylation studies (using methylation-specific multiplex ligation dependent probe amplification after bisulfite treatment of DNA) of the 11p15 locus revealed a partial gain of methylation of the H19/IGF2:IG-DMR (methylation index of 55; normal range 46 à 51) and a partial loss of methylation of the KCNQ1OT1:TSS-DMR (methylation index of 43; normal range 48- 53), in favor of a paternal origin of the 11p duplication (17).
We report a patient with a complex chromosomal rearrangement which includes both IGF2 and the IGF1R. In this case, two rare conditions coexist: a mosaic 11p15 paternal duplication, which usually leads to overgrowth (BWS), and a 15qter deletion including IGF1R, which usually leads to fetal and post-natal growth restriction. The major role of IGF-I and IGF–II as major actors in the control of fetal growth, through their binding to the IGF1R, has been known for years. Indeed, in humans, genetic defects of IGF1 and the IGF1R or alterations in IGF2 expression, through genetic or epigenetic mechanisms (SRS) leads to fetal growth restriction (16–18). Conversely, overexpression of IGF2 (BWS) or the IGF1R lead to overgrowth (19, 20).
Moreover, murine models of invalidation of these genes confirmed these observations, as knockout models for either Igf1, Igf2, or Igf1r present with growth restriction at birth (21, 22). Interestingly, the double knockout for Igf1 and Igf2 or Igf2 and Igf1r are smaller at birth than the knockout for Igf1r, whereas double knockouts for Igf1 and Igf1r are the same size as the Igf1r knockout mice. These data suggest that, conversely to IGF-I, which only interacts with the IGF1R, IGF-II may also act through other signaling pathways (22).
In humans, many findings have highlighted the role of IGF-I in stimulating postnatal growth: patients with GH deficiency or GH resistance [Laron (OMIM#262500) or Noonan (OMIM#163950) syndromes] have extremely low levels of IGF-I and present with growth failure after birth (12). Conversely, patients with acromegalogigantism (because of GH pituitary adenoma) have extremely high levels of IGF-I and tall stature.
The role of IGF-II in postnatal growth is unclear. Indeed, SRS patients with a loss of methylation at the H19/IGF2:IG-DMR present with persistent post-natal growth failure, despite normal circulating IGF-II levels (23, 24). Such normal circulating IGF-II levels may be due to the biallelic expression of IGF2 from a non-imprinted promotor in the liver. However, IGF2 is still imprinted in other tissues, and loss of methylation at 11p15 leads to the loss of IGF2 expression in these tissues (4, 17). Thus, these plasmatic levels of IGF-II do not reflect the local levels and activity of IGF-II. 11p15.5 paternal duplications have been recently reviewed (10). In such duplications, either growth retardation (SRS), overgrowth (BWS), or a normal phenotype can be observed, depending on the extent of the duplication and the parental origin of the duplicated allele. Usually, duplications of the paternal telomeric domain of 11p15 leads to IGF2 overexpression and thus BWS, whereas a gain of CDKN1C expression, due to maternal duplication of the centromeric domain of 11p15, leads to SRS (10). In our patient, we demonstrate that the duplicated 11p15 allele was of paternal origin and encompassed both IC1 and IC2. Thus, IGF2 is likely overexpressed in the contingent of cells with the 11p15 duplication. However, in this patient plasmatic IGF-II levels were within the normal range which could be secondary to a different mosaicism in the liver with a lower rate of cells carrying the 11p15 duplication.
The growth retardation, microcephaly, developmental delay, and high plasma levels of IGF-I observed in our patient are more concordant with the IGF1R deletion phenotype. Here, fetal growth was not affected, despite IGF2 overexpression and the coexisting IGF1R defect. These two opposite mechanisms may compensate each other and finally lead to normal birth parameters. Nevertheless, we should be cautious when speculating on the respective role of genetic anomalies impact here since we do not know the exact proportion of mosaicism in the different tissues. Information on placenta, for example, would have been of major interest since IGF-II and IGF1R play a major role on placental development and function (25). It reinforces the hypothesis that IGF-II can signal through a pathway that is independent of the IGF1R, at least during fetal life (22). Finally, in vitro studies have also shown a stronger affinity of IGF-I than IGF-II for IGF1R and IGF1R is activated with lower concentrations of IGF-I compared to IGF-II (26, 27).
The growth retardation our patient experienced suggests that the IGF1R defect prevails over IGF-II overexpression after birth. Indeed, post-natal growth retardation with a biological IGF-I resistance profile is concordant with the predominant dysfunction of the IGF1R. In patients with low IGF2 expression (SRS), the fetal growth restriction is obvious, whereas their growth velocity is usually unaffected after birth (despite no catch-up) (23). Conversely a comparison of patients with BWS shows birth length to generally be greater in patients with IGF2 overexpression (gain of methylation in IC1), whereas height during childhood is usually greater in patients with an isolated IC2 loss of methylation (no IGF2 overexpression) (19). This favors a predominant role of IGF-II in fetal rather than in post-natal growth. For the patient reported here, another explanation for the normal measurements at birth may lie in the mosaicism presented by the patient, since all her circulating cells carried the IGF1R deletion, but only approximately 30% had the 11p duplication. This mosaicism may vary across various tissues, and the growth plate may have a different proportion of cells with the 11p duplication.
Patients with IGF1 or IGF1R defects usually present with microcephaly, which distinguishes them from SRS patients, for whom head circumference is relatively conserved at birth. This would favor a major role of IGF-I in cerebral development over that of IGF-II (28). Another possibility is the absence of maternal imprinting of IGF2 in brain, as several studies found either biallelic expression in different cerebral regions or, more recently a maternal expression (4, 21, 29). In the latter case, IGF2 would not be overexpressed in the brain of the patient (because of the paternal origin of the duplication) conversely to the other tissues. Thus, IGF1R haploinsufficiency would have led to the microcephaly, in accordance with previous observations of IGF1R disruption.
We report here an original association of multiple chromosomal rearrangements involving IGF2 and IGF1R, two critical genes involved in the regulation of fetal and post-natal growth regulation. It favors the predominance of IGF-II during fetal growth and IGF-I during post-natal growth.
A written informed consent was obtained from the parents of the participant for the publication of this case report.
EG, SC-B, and FB shared writing of the manuscript. AB, MR, and SC-B performed the molecular tests and analysis. SW and IN participated in the discussion and revisions during the writing process.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer TE declared a past co-authorship with the authors to the handling Editor.
We would like to thank Dr. Blandine Esteva and Dr. Diane Doummar who helped us in the management of this patient and Laurence Perin and Marilyne Le Jules for their technical help.
1. LeRoith D, Werner H, Beitner-Johnson D, Roberts CT. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. (1995) 16:143–63.
2. Eggermann T, Perez de Nanclares G, Maher ER, Temple IK, Tümer Z, Monk D, et al. Imprinting disorders: a group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin Epigenetics. (2015) 7:123. doi: 10.1186/s13148-015-0143-8
3. Rotwein P. Large-scale analysis of variation in the insulin-like growth factor family in humans reveals rare disease links and common polymorphisms. J Biol Chem. (2017) 292:9252–61. doi: 10.1074/jbc.M117.783639
4. Baran Y, Subramaniam M, Biton A, Tukiainen T, Tsang EK, Rivas MA, et al. The landscape of genomic imprinting across diverse adult human tissues. Genome Res. (2015) 25:927–36. doi: 10.1101/gr.192278.115
5. Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, et al. Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. (2018) 14:229–49. doi: 10.1038/nrendo.2017.166
6. Wakeling EL, Brioude F, Lokulo-Sodipe O, O'Connell SM, Salem J, Bliek J, et al. Diagnosis and management of Silver–Russell syndrome: first international consensus statement. Nat Rev Endocrinol. (2017) 13:105–24. doi: 10.1038/nrendo.2016.138
7. Azzi S, Salem J, Thibaud N, Chantot-Bastaraud S, Lieber E, Netchine I, et al. A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J Med Genet. (2015) 52:446–53. doi: 10.1136/jmedgenet-2014-102979
8. Brioude F, Oliver-Petit I, Blaise A, Praz F, Rossignol S, Le Jule M, et al. CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J Med Genet. (2013) 50:823–30. doi: 10.1136/jmedgenet-2013-101691
9. Brioude F, Netchine I, Praz F, Le Jule M, Calmel C, Lacombe D, et al. Mutations of the imprinted CDKN1C gene as a cause of the overgrowth Beckwith-Wiedemann syndrome: clinical spectrum and functional characterization. Hum Mutat. (2015) 36:894–902. doi: 10.1002/humu.22824
10. Heide S, Chantot-Bastaraud S, Keren B, Harbison MD, Azzi S, Rossignol S, et al. Chromosomal rearrangements in the 11p15 imprinted region: 17 new 11p15.5 duplications with associated phenotypes and putative functional consequences. J Med Genet. (2017) 55:205–13. doi: 10.1136/jmedgenet-2017-104919
11. Abi Habib W, Brioude F, Azzi S, Salem J, Das Neves C, Personnier C, et al. 11p15 ICR1 Partial deletions associated with IGF2/H19 DMR Hypomethylation and Silver-Russell Syndrome. Hum Mutat. (2017) 38:105–11. doi: 10.1002/humu.23131
12. David A, Hwa V, Metherell LA, Netchine I, Camacho-Hübner C, Clark AJL, et al. Evidence for a continuum of genetic, phenotypic, and biochemical abnormalities in children with growth hormone insensitivity. Endocr Rev. (2011) 32:472–97. doi: 10.1210/er.2010-0023
13. Abuzzahab MJ, Schneider A, Goddard A, Grigorescu F, Lautier C, Keller E, et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N Engl J Med. (2003) 349:2211–22. doi: 10.1056/NEJMoa010107
14. Gannage-Yared M-H, Klammt J, Chouery E, Corbani S, Megarbane H, Abou Ghoch J, et al. Homozygous mutation of the IGF1 receptor gene in a patient with severe pre- and postnatal growth failure and congenital malformations. Eur J Endocrinol. (2012) 168:K1–7. doi: 10.1530/EJE-12-0701
15. Yang L, Xu D, Sun C, Wu J, Wei H, Liu Y, et al. IGF1R variants in patients with growth impairment: four novel variants and genotype-phenotype correlations. J Clin Endocrinol Metab. (2018) 103:3939–44. doi: 10.1210/jc.2017-02782
16. Walenkamp MJE, Robers JML, Wit JM, Zandwijken GRJ, van Duyvenvoorde HA, Oostdijk W, et al. Phenotypic features and response to growth hormone treatment of patients with a molecular defect of the IGF-1 receptor. J Clin Endocrinol Metab. (2019). doi: 10.1210/jc.2018-02065. [Epub ahead of print].
17. Azzi S, Blaise A, Steunou V, Harbison MD, Salem J, Brioude F, et al. Complex tissue-specific epigenotypes in Russell-Silver Syndrome associated with 11p15 ICR1 hypomethylation. Hum Mutat. (2014) 35:1211–20. doi: 10.1002/humu.22623
18. Gicquel C, Rossignol S, Cabrol S, Houang M, Steunou V, Barbu V, et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat Genet. (2005) 37:1003–7. doi: 10.1038/ng1629
19. Mussa A, Russo S, Larizza L, Riccio A, Ferrero GB. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome: a paradigm for genomic medicine: phenotypes in Beckwith-Wiedemann syndrome. Clin Genet. (2016) 89:403–15. doi: 10.1111/cge.12635
20. Okubo Y, Siddle K, Firth H, O'Rahilly S, Wilson LC, Willatt L, et al. Cell proliferation activities on skin fibroblasts from a short child with absence of one copy of the type 1 Insulin-Like Growth Factor Receptor (IGF1R) gene and a tall child with three copies of the IGF1R gene. J Clin Endocrinol Metab. (2003) 88:5981–8. doi: 10.1210/jc.2002-021080
21. DeChiara TM, Efstratiadis A, Robertsen EJ. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature. (1990) 345:78–80.
22. Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell. (1993) 75:59–72.
23. Binder G, Liebl M, Woelfle J, Eggermann T, Blumenstock G, Schweizer R. Adult height and epigenotype in children with Silver-Russell syndrome treated with GH. Horm Res Pædiatrics. (2013) 80:193–200. doi: 10.1159/000354658
24. Binder G, Seidel A-K, Weber K, Haase M, Wollmann HA, Ranke MB, et al. IGF-II serum levels are normal in children with silver-russell syndrome who frequently carry epimutations at the IGF2 locus. J Clin Endocrinol Metab. (2006) 91:4709–12. doi: 10.1210/jc.2006-1127
25. Sferruzzi-Perri AN, Sandovici I, Constancia M, Fowden AL. Placental phenotype and the insulin-like growth factors: resource allocation to fetal growth. J Physiol. (2017) 595:5057–93. doi: 10.1113/JP273330
26. Tian D, Mitchell I, Kreeger PK. Quantitative analysis of insulin-like growth factor 2 receptor and insulin-like growth factor binding proteins to identify control mechanisms for insulin-like growth factor 1 receptor phosphorylation. BMC Syst Biol. (2016) 10:15. doi: 10.1186/s12918-016-0263-6
27. Henderson ST, Brierley GV, Surinya KH, Priebe IK, Catcheside DEA, Wallace JC, et al. Delineation of the IGF-II C domain elements involved in binding and activation of the IR-A, IR-B and IGF-IR. Growth Horm IGF Res. (2015) 25:20–7. doi: 10.1016/j.ghir.2014.09.004
28. Werner H, LeRoith D. Insulin and insulin-like growth factor receptors in the brain: physiological and pathological aspects. Eur Neuropsychopharmacol. (2014) 24:1947–53. doi: 10.1016/j.euroneuro.2014.01.020
Keywords: Beckwith-Wiedemann syndrome, IGF1 receptor, IGF-II, fetal growth restriction, imprinting disease, 11p duplication
Citation: Giabicani E, Chantot-Bastaraud S, Bonnard A, Rachid M, Whalen S, Netchine I and Brioude F (2019) Roles of Type 1 Insulin-Like Growth Factor (IGF) Receptor and IGF-II in Growth Regulation: Evidence From a Patient Carrying Both an 11p Paternal Duplication and 15q Deletion. Front. Endocrinol. 10:263. doi: 10.3389/fendo.2019.00263
Received: 04 March 2019; Accepted: 09 April 2019;
Published: 30 April 2019.
Edited by:
Tsutomu Ogata, Hamamatsu University School of Medicine, JapanReviewed by:
Masayo Kagami, National Center for Child Health and Development (NCCHD), JapanCopyright © 2019 Giabicani, Chantot-Bastaraud, Bonnard, Rachid, Whalen, Netchine and Brioude. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eloïse Giabicani, ZWxvaXNlLmdpYWJpY2FuaUBhcGhwLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.