
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 09 April 2019
Sec. Reproduction
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00215
This article is part of the Research Topic Causes and Consequences of Intrauterine Growth Restriction View all 22 articles
 Immaculate M. Langmia1,2,3
Immaculate M. Langmia1,2,3 Kristin Kräker1,2,4,5,6
Kristin Kräker1,2,4,5,6 Sara E. Weiss1,2,4
Sara E. Weiss1,2,4 Nadine Haase1,2,4,5,6
Nadine Haase1,2,4,5,6 Till Schütte2,6,7
Till Schütte2,6,7 Florian Herse1,2,5
Florian Herse1,2,5 Ralf Dechend1,2,4,6,8*
Ralf Dechend1,2,4,6,8*Intrauterine growth restriction (IUGR) is a condition whereby a fetus is unable to achieve its genetically determined potential size. IUGR is a global health challenge due to high mortality and morbidity amongst affected neonates. It is a multifactorial condition caused by maternal, fetal, placental, and genetic confounders. Babies born of diabetic pregnancies are usually large for gestational age but under certain conditions whereby prolonged uncontrolled hyperglycemia leads to placental dysfunction, the outcome of the pregnancy is an intrauterine growth restricted fetus with clinical features of malnutrition. Placental dysfunction leads to undernutrition and hypoxia, which triggers gene modification in the developing fetus due to fetal adaptation to adverse utero environmental conditions. Thus, in utero gene modification results in future cardiovascular programming in postnatal and adult life. Ongoing research aims to broaden our understanding of the molecular mechanisms and pathological pathways involved in fetal programming due to IUGR. There is a need for the development of effective preventive and therapeutic strategies for the management of growth-restricted infants. Information on the mechanisms involved with in utero epigenetic modification leading to development of cardiovascular disease in adult life will increase our understanding and allow the identification of susceptible individuals as well as the design of targeted prevention strategies. This article aims to systematically review the latest molecular mechanisms involved in the pathogenesis of IUGR in cardiovascular programming. Animal models of IUGR that used nutrient restriction and hypoxia to mimic the clinical conditions in humans of reduced flow of nutrients and oxygen to the fetus will be discussed in terms of cardiac remodeling and epigenetic programming of cardiovascular disease. Experimental evidence of long-term fetal programming due to IUGR will also be included.
Cardiovascular disease (CVD) is the main cause of mortality and morbidity in the twenty-first century (1–3). According to the World Health Organization (WHO), coronary heart disease and stroke accounted for ~15.2 million deaths in 2016 globally. Previously, CVD was assumed to be caused by a series of events that occur after birth, such as a person's lifestyle, age, and other disease conditions. In other words, postnatal gene-environment interactions are important factors promoting CVD, but recently we began to understand that in utero environmental changes that occur in the first few months of prenatal development epigenetically program the fetus for development of CVD in adulthood (4–6). Therefore, apart from postnatal gene-environment interaction, information on in utero gene-environment interactions is needed to improve our understanding of CVD.
The placenta performs many functions ranging from growth of the fetus, prevention of fetal rejection by maternal immune system, as well as the transport/exchange of gases, nutrients, and waste products between mother and fetus (7). In addition, the placenta is involved in metabolism and production of many hormones that play vital roles in the maintenance of pregnancy (7). In a normal pregnancy the placental weight and birth weight are highly correlated (8). In adverse situations where the maternal-fetal circulation is altered due to disease conditions such as diabetes, the development of the placenta also changes (9). The determining factor in placental dysfunction is the extent of exposure to hyperglycemia during fetal and placental development (10). Depending on the degree of hyperglycemia, the growth of the placenta and the fetus can be severely affected (10). Long-term uncontrolled hyperglycemia can lead to placental vascular dysfunction in women suffering from diabetes (10). Endothelial dysfunction was observed in hyperglycemic human umbilical vein cells (HUVEC) compared to control cells (11). According to studies by Starikov et al. pre-gestational diabetes led to placentas that were small for gestational age in 20% of pregnancies and placentas that were large for gestational age in 30% of cases (9). In conditions where maternal hyperglycemia is not controlled, it results in histological changes of the placental tissues (10, 12). Placentas from mothers with diabetes display different histological features such as immature villous, increased number of fetal capillaries, and fibrinoid necrosis of the placental villi (7). Prolonged maternal insults such as hyperglycemia, dyslipidemia, and hyperinsulinemia may exceed the placental capacity to adapt and respond leading to placental dysfunction and adverse fetal outcome such as in IUGR (13, 14). Placental dysfunction is the most common cause of asymmetrical intrauterine growth restriction (15–17). Uteroplacental malperfusion, decidual vasculopathy, placental infarct, and maternal vasculopathy were observed in women with diabetes (12) and are associated with reduced fetal growth and adverse outcome (17, 18). Abnormal placental vascular development decreases normal placental blood supply, leading to reduced oxygen (hypoxia) and nutrient delivery (undernutrition) to the fetus and a subsequent IUGR fetus (16, 17). As a result of reduced oxygen and nutrition, the fetus redistributes its cardiac output to increase oxygen and nutrient supply to the brain, which is referred to as brain sparing (19). This prenatal cerebral circulatory adaptation has been associated with long-term behavioral problems in schoolchildren who were growth-restricted at birth. These difficulties include problems in memory, cognition, motor skills, and neuropsychological malfunction (20). IUGR offspring experience the highest rate of coronary heart disease, myocardial dysfunction, type 2 diabetes, hypertension, and stroke as adults (21, 22). The underlying molecular mechanisms leading to fetal susceptibility to adult disease are still under investigation. Interestingly, we now know that clinical symptoms of cardiovascular disease may not appear until adult life (4). In 1989, David Barker demonstrated this aspect amongst individuals with low birth weight, showing a direct association between low birth weight and CVD in adulthood. This study demonstrated that the incidence of ischemic heart disease and death were three times higher among men with low birth weight compared to men with high birth weight (5). Epidemiological investigations of adults born at the time of the Dutch famine between 1944 and 1945 revealed an association between maternal starvation and a low infant birth weight with a high incidence of hypertension and coronary heart disease in these adults (23). Furthermore, Painter et al. reported the incidence of early onset coronary heart disease among persons conceived during the Dutch famine (24). In that regard, Barker's findings led to the concept of fetal adaptation to prenatal environmental changes and vulnerability to chronic diseases in adult life, a concept known as “fetal programming.” In 2003, the concept of “Developmental origins of health and disease (DOHaD)” was published (25). DOHaD describes the scenario whereby in utero maternal insults result in structural and functional changes in fetal organs extending in postnatal life and increasing susceptibility of chronic disease in adulthood. Fetal programming of other organs such as the brain (19), lungs (26), and hypothalamic-pituitary-adrenal (HPA) axis have been greatly studied, however the heart is also very important and few studies have investigated fetal programming of the heart as summarized below (27).
The heart is the first organ to develop and function during embryogenesis (28). Fetal organs that develop early are more vulnerable to changes of the in utero environment than organs that develop later (29). There is a direct correlation between the amount of oxygen and nutrient supply to the fetus and cardiomyocytes development and function (29). Chronic hyperglycemia causes placental vascular dysfunction, which leads to reduced nutrient and oxygen supply to the fetus resulting in IUGR (10). Neonates exposed to IUGR present significant changes in cardiac morphology and subclinical myocardial dysfunction at birth (29). As such, in placental dysfunction due to maternal diabetes involving undernutrition and hypoxia, the developing myocardium undergoes structural and functional changes referred to as cardiac remodeling (30). In 1982, Hochman and Bulkley were the first to use the term “remodeling” to describe the substitution of infarcted tissue with scar tissue in a model of myocardial infarction (31). Later, in the year 2000, an international forum defined cardiac remodeling as molecular, cellular, and interstitial changes that manifest clinically as changes in size, shape, and function of the heart resulting from cardiac load and injury (30). Therefore, the clinical phenotype is a consequence of genomic changes that occur due to in utero gene environment interaction. Few clinical studies and experimental animal models have investigated the impact of IUGR on cardiac remodeling as discussed below.
Epidemiological studies have shown changes in the structure and function of the heart in IUGR infants and neonates (29, 32). Clinical investigations have reported that postnatal catch-up growth in IUGR infants can result in abnormal cardiac function (33–35). Cardiovascular evaluation using echocardiographic assessment of IUGR fetuses revealed cardiac hypertrophy characterized by globular cardiac shape, higher intraventricular septum thickness, and increased pressure overload leading to both systolic and diastolic dysfunction (36–38). Primary cardiac and vascular changes observed at birth in IUGR fetus can persist from a few months up to a few years of age (35, 37).
Recently, clinical investigations have shown evidence of cardiac remodeling in IUGR offspring (32). For example, high systolic blood pressure and smaller aortic diameter (ascending aorta; IUGR:24.4 ± 1.5 mm, Control: 26.3 ± 2.2 mm, P < 0.05) were observed 20 years later in young adults (age 22–25 years) who were born growth restricted due to placental dysfunction (32). Compared to healthy controls, the diameter of the ascending aorta was smaller in IUGR subjects and a high aortic pressure augmentation index was observed (32). Electrocardiography (ECG) measurement revealed electrical remodeling in preadolescence who were born with IUGR (39). These studies evaluated signs of ventricular electrical remodeling in subjects with IUGR compared to controls by assessing changes in ventricular depolarization and repolarization using the QRS complex and T-wave, respectively (39). The classical frontal QRS-T angle was significantly narrower in IUGR preadolescents compared to control subjects (39). They also reported wider angles between the depolarization dominant vector and the frontal XY body plane in preterm-IUGR subjects, resulting in significantly wider angles between depolarization and repolarization vectors (39). The studies concluded that ventricular electrical remodeling observed in the pre-adolescent subjects might predispose to cardiovascular disease in later life (39). Electrical remodeling is induced by functional (altered electrical activation) and structural stressors (such as in hypertrophy and heart failure). These electrophysiological changes generate a substrate that is vulnerable to malignant ventricular arrhythmias. Cardiac remodeling and dysfunction increases with greater severity of IUGR (33, 40). The above-mentioned studies show evidence of cardiac remodeling in neonates, pre-adolescents, and young adults who were born with IUGR (41–44). These findings are supported by animal models; IUGR programming resulted in myocardial remodeling, reduced systolic and diastolic function and premature aging of the heart in growth restricted baboons (45). These structural and functional changes of the heart can be associated to changes in fetal cardiac gene expression that have occurred because of fetal adaptations to intrauterine environmental changes (46). These in utero changes, which result in reduced nutrition and oxygen, are due to placental dysfunction caused by maternal insults such as maternal vasculopathy in a diabetic pregnancy (46).
Pre-gestational and gestational diabetes have been associated with maternal vasculopathy which can be caused by long-term poor glycemic control (12). Fetal growth in diabetic pregnancy might be affected in two different ways; maternal hyperglycemia stimulates fetal overgrowth and maternal vasculopathy may be associated with placental dysfunction leading to a reduced nutrient and oxygen supply and subsequent IUGR (12). The majority of animal models use either maternal undernutrition and/or hypoxia to mimic IUGR (6, 47, 48). Hypoxia, undernutrition, or both interventions induce cardiac remodeling in adult rats (47). IUGR models have linked cardiac remodeling with changes in fetal gene expression. Several genes that play key roles in cardiac development have been studied in detail. One prominent example is the hypoxia-inducible factor 1 (HIF-1), which is required for normal growth of the myocardium and coronary blood vessels in conditions of low oxygen (49, 50). High levels of HIF-1 were reported in fetal rodent hearts exposed to hypoxia (49, 50). Abnormal cardiogenesis was seen in HIF-1 alpha-deficient mice (51). Therefore, elevated expression of HIF1 and its downstream genes is essential for fetal adaptation and proper cardiac development in conditions of placental dysfunction in which oxygen supply to the fetus is relatively low (51–55). In a guinea pig model of maternal nutrient restriction, hearts of growth restricted offspring revealed increased expression of Poly [ADP-ribose] polymerase 1 (PARP1) gene and a reduction in cardiomyocyte number as well as hypertrophy (56). Another interesting class of genes associated with cardiac remodeling are the cardioprotective genes such as heat shock protein 70 (HSP70) and protein kinase C epsilon (PKCε) (57). Male rats exposed to hypoxia had increased expression of HSP70 and ischemic injury (48, 58–60). In addition, hypoxia in rat hearts resulted in a significant increase in PKCε expression and increased susceptibility to ischemia and perfusion injury in a sex-dependent manner (61). Furthermore, the mammalian target of the rapamycin complex 1 (mTORC1) pathway has also been associated with cardiac remodeling in IUGR mice, and prenatal mTORC1 inhibition caused a reduction in cardiomyocyte number in adult hearts compared to controls (62). In a rabbit model, IUGR was associated with cardiac mitochondrial Complex II dysfunction and an increase in Sirtuin 3 expression (63). In a rat model, maternal diabetes was associated with increased expression of forkhead box protein O1 (FOXO1) and its target genes in the heart of the offspring (64). Under conditions of undernutrition and hypoxia, changes that occur to enable the growing fetus to adapt and survive the inadequate substrate supply results in programming of fetal cardiac genes (4, 6, 65–67).
Researchers have been investigating the mechanisms by which IUGR caused changes in fetal cardiac genes. Epigenetic regulation is one principle underlying molecular mechanism that causes differential gene expression in IUGR fetuses compared to normally grown fetuses. Epigenetic activities can alter gene expression throughout an individual's lifetime; thus, the function of the corresponding protein products as well as the organs involved are affected. Gene modifications occur in response to environmental changes, and genes are switched on and off via epigenetic mechanisms. Epigenetic alterations could be altered by lifestyle (for example overnutrition or undernutrition) or possibly by using treatments strategies postnatally (68). Hence, it is proposed that epigenetic mechanisms are important for understanding pathophysiology as well as potential targets for diagnosis and treatment of cardiovascular diseases (69, 70). Epigenetic regulation occurs by DNA methylation, histone modification, or regulation of genes by non-coding RNAs (ncRNAs) such as small microRNA and long non-coding RNA (71). So far, DNA methylation and histone acetylation are the most studied epigenetic mechanisms and clinical evidence of epigenetic programming is reported in adult patients with various cardiovascular diseases as reviewed by Muka et al. (72). Other studies have investigated the posttranscriptional regulatory role of non-coding RNA in relation to cardiovascular disease susceptibility in adult cardiac patients (73–75). We will focus on DNA methylation in IUGR leading to cardiovascular disease.
DNA methylation is the covalent addition of methyl groups to the C5 position of cytosine in dinucleotide CpG islands (76). CpG islands are regions with a high frequency of CpG sites where a cytosine nucleotide is followed by a guanine nucleotide. Methylation of CpG sites is catalyzed by DNA methyl transferases (DNMTs), such as DNMT1, DNMT3a, and DNMT3b. Very few studies have examined DNA methylation mechanisms in IUGR leading to cardiovascular remodeling or cardiac dysfunction. In a UK cohort of 144 children, investigators used umbilical cord blood to examine the relationship between prenatal antisense non-coding RNA in the INK4 locus (ANRIL) promoter DNA methylation and risk markers of coronary heart disease (76). Hypermethylation at CpG5 of the ANRIL promoter was associated with increased childhood pulse wave velocity, indicating increased arterial stiffness and a risk of coronary heart disease in these children at 9 years old (76). The ANRIL promoter is present on chromosome 2p21 which is considered a strong candidate for coronary heart disease in adult patients (77). Methylation of ANRIL and other genes on this chromosome might contribute to the prolonged cardiovascular programming in coronary disease patients. However, it is difficult to conclude the epigenetic contribution unless studies involving long-term follow-up of IUGR individuals are performed. Several studies have investigated epigenetic disturbances of nitric oxide synthase (NOS), which may predispose individuals to cardiovascular disease by modulating endothelial dysfunction (78–80). Human endothelial cells isolated from umbilical arteries and veins of IUGR fetuses were analyzed for a DNA methylation pattern in the promoter region of the endothelial nitric oxide synthase 3 (eNOS3) and arginine-2 (ARG2) genes. Compared to control cells, differential DNA methylation at the CpG-352 site of NOS3 gene promoter of IUGR-endothelial cells was observed (80). Differential eNOS expression could be normalized by simply silencing the DNA methylation machinery (80). Hypoxia downregulates eNOS gene expression and activity, resulting in a reduced production of nitric oxide and endothelial dysfunction (78). Endothelial dysfunction is a very early stage in the development of atherosclerosis, which appears prior to the existence of atherosclerotic plaques or cardiovascular outcomes (81–84). This favors the concept that an epigenetic marker detectable as early as during prenatal and early postnatal developmental periods might be capable of identifying persons at risk of endothelial-related cardiovascular end organ damage. Furthermore, it has been shown that hypoxia-related IUGR changes the methylation pattern and expression of the PKCε gene in rat hearts, causing ischemic injuries in offspring (85). Hypoxia causes increased production of reactive oxygen species (ROS) which induces epigenetic repression of the PKCε gene leading to susceptibility of the heart to ischemic injury in the offspring (85). Hypermethylation patterns were observed within the promoter region of the PKCε gene in IUGR male rat hearts compared to controls (85). PKCε promoter hypermethylation was associated with a corresponding down regulation of PKCε gene expression in the heart of male rats but not in females. Activation of the PKCε gene in the heart restored hypoxia-induced cardiac vulnerability to ischemic injury in male rats (85). The transcription factor, early growth response factor (Egr-1), is involved in the regulation of PKCe promoter activity (86) since it binds to the PKCε promoter region, increasing its activity. Further studies showed that the absence of methylation in females was due to high levels of estrogen receptors in the female rat heart. Estrogen receptors bind to the regulatory gene Egr-1 and inhibit PKCε promoter methylation activity (86). Therefore, compared to males, the female hearts are protected against hypoxia-induced ischemic injury due to high levels of estrogen. These data help to explain the differences in the prevalence of CVD between men and women and gives a molecular mechanism for the role of sex hormone (87). Postmenopausal women seem to lose this hormonally-induced cardioprotective ability and as such, hormone therapy is suggested as one of the remedies for prevention of cardiovascular disease in this group of women (88). However, large studies do not favor hormone replacement therapy for prevention of cardiovascular disease since it also has side effects such as increased risk for breast cancer and pulmonary embolism (88). For IUGR epigenetically programmed hearts, the best preventive strategy would be the use of epigenetic therapies (drugs) that aim to reverse fetal heart programming in early postnatal life (89). Postnatal drugs are capable of altering the epigenetic modifications that occurred during prenatal life as a result of fetal adaptations to changes in the intrauterine environment (89). Animal models and human studies of IUGR revealed epigenetic changes in 10 genes which result in a predisposition to type 2 diabetes as reviewed in detail by Liguori et al. (90). These epigenetic mechanisms may further contribute to the high prevalence of cardiovascular disease in type 2 diabetes patients (90). Another gene involved in growth and development of the fetal heart is insulin-like growth factor (IGF). Upregulation of cardiac IGF receptor is associated with an increase in ventricular mass in a sheep model of nutrient restriction (91). Moreover, epigenetic mechanisms in the renin angiotensin system are linked to cardiovascular programming. Animal models revealed epigenetic modification of the renin-angiotensin system in fetal programming of hypertension (92, 93). Significantly lower renal expression of angiotensin II type 2 receptor, impairment of renal development and elevation of blood pressure were observed (93). Maternal insults during pregnancy induce placental dysfunction, leading to IUGR (Figure 1) (10). So far, it is well established that IUGR leads to cardiac remodeling via epigenetic modulation of cardiac genes which play important roles in cardiac development and function. However, many epigenetic pathways are still to be elucidated.
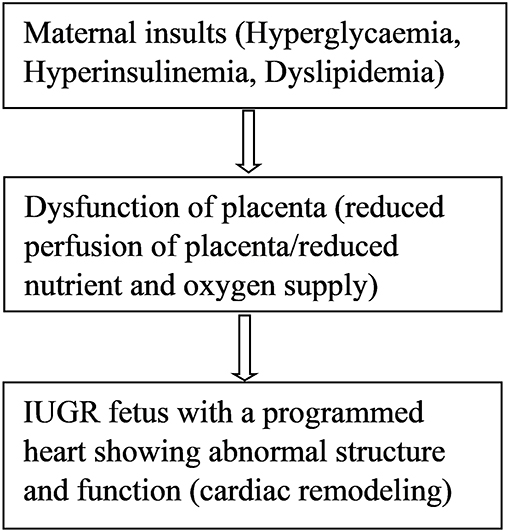
Figure 1. Maternal insults result in placental dysfunction, IUGR, and cardiac remodeling.
Placental dysfunction due to maternal insults can result in IUGR, which result in an altered fetal epigenome leading to cardiac remodeling and subclinical symptoms of cardiovascular phenotypes in offspring. It is well-proven that IUGR leads to cardiac remodeling, likewise evidence linking altered genome expression and cardiovascular disease in IUGR has been published (85, 86, 91, 94). However, research involving epigenetic mechanisms of cardiovascular programming in growth restricted fetuses has just commenced. To date, most research has focused on fetal programming leading to CVD-related risk factors such as a predisposition to metabolic syndrome (95–97). Although such studies improve our understanding of how to prevent cardiovascular disease in carriers of cardiovascular risk factors, it is important to fully understand the epigenetic mechanisms acting directly on cardiovascular structure and function in IUGR offspring. It seems IUGR results in DNA methylation in a tissue-specific manner; thus, understanding the epigenetic mechanisms in various tissues that have direct impact on the cardiac system will be beneficial (98). The greatest task is to uncover the specific pathways of prenatal methylation of gene expression, as it is vital to the development and function of heart tissues, and to be able to link these pathways to specific cardiac diseases. Even though one can find DNA methylation in interesting CpG island in clinical samples obtained from affected IUGR offspring, it is also difficult to conclude a causal link between these epigenetic modifications and the various heart diseases. However, animal models may help to improve our understanding. Another challenge is linking affected pathways based on the severity of the IUGR since the extent of cardiac remodeling is based on the severity of IUGR (40). Epidemiological time point experiments have been performed to widen our understanding on the role of IUGR in fetal vulnerability to cardiovascular disease (33–35); however, there is need for longitudinal studies involving long-term follow-up of affected IUGR offspring. The use of epigenetic therapies in treatment of CVD is promising but it is still at its early experimental stage. In other diseases such as cancer, systemic lupus erythematosus, acute myeloid leukemia, and Alzheimer's disease, many epigenetic-related therapeutic agents are being tested for their possible use in clinical practice, but many are still awaiting approval to begin clinical trials or to receive approval by the FDA (99–102). In terms of DNA methylation mechanisms, therapeutic agents such as inhibitors of DNA methyl transferase (DNMT), hormonal therapy, and certain dietary compounds have been suggested for treatment of cardiovascular disease (89). Early prediction of cardiovascular disease might reduce risk as well as improve follow-up and treatment of susceptible patients. Effective biomarkers of risk are needed for development of CVD prevention strategies (103). Epigenetic biomarkers could be useful for early prediction of high-risk individuals (103).
Growth restricted newborns are at high risk of respiratory distress syndrome, retinopathy of prematurity, and long-term complications such as metabolic syndrome and cardiovascular disease (104, 105). Therefore, it is vital to identify and follow those who are likely to be affected. Studies also show that an inheritable altered fetal cardiac genome can be transferred from one generation to another, which is referred to as transgenerational programing of the cardiac system (106). “Healthy aging starts with a healthy pregnancy,” summarizes very beautifully this important concept and was first introduced in an editorial of Scioscia (107). A timely diagnosis of IUGR pregnancies and application of various therapeutic measures to treat and monitor affected neonates will help to reduce cardiac problems in future generations.
RD and IL conceived the presented idea. IL wrote the first draft of the manuscript. KK, SW, NH, TS, FH, and RD contributed to the manuscript revision, read and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Ofori SN, Odia OJ. Risk assessment in the prevention of cardiovascular disease in low-resource settings. Ind Heart J. (2016) 68:391–8. doi: 10.1016/j.ihj.2015.07.004
2. Hariram V, Arramraju SK. Assessment of cardiovascular risk in low resource settings “So much to do - So little done”. Ind Heart J. (2016) 68:260–2. doi: 10.1016/j.ihj.2015.07.046
3. Sanchis-Gomar F, Perez-Quilis C, Leischik R, Lucia A. Epidemiology of coronary heart disease and acute coronary syndrome. Ann Transl Med. (2016) 4:256. doi: 10.21037/atm.2016.06.33
4. Barker DJ. In utero programming of cardiovascular disease. Theriogenology. (2000) 53:555–74. doi: 10.1016/S0093-691X(99)00258-7
5. Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. (1989) 2:577–80. doi: 10.1016/S0140-6736(89)90710-1
6. Beauchamp B, Thrush AB, Quizi J, Antoun G, McIntosh N, Al-Dirbashi OY, et al. Undernutrition during pregnancy in mice leads to dysfunctional cardiac muscle respiration in adult offspring. Biosci Rep. (2015) 35:e00200. doi: 10.1042/BSR20150007
7. Gude NM, Roberts CT, Kalionis B, King RG. Growth and function of the normal human placenta. Thromb Res. (2004) 114:397–407. doi: 10.1016/j.thromres.2004.06.038
8. Roland MC, Friis CM, Voldner N, Godang K, Bollerslev J, Haugen G, et al. Fetal growth versus birthweight: the role of placenta versus other determinants. PLos ONE. (2012) 7:e39324. doi: 10.1371/journal.pone.0039324
9. Starikov R, Inman K, Chen K, Lopes V, Coviello E, Pinar H, et al. Comparison of placental findings in type 1 and type 2 diabetic pregnancies. Placenta. (2014) 35:1001–6. doi: 10.1016/j.placenta.2014.10.008
10. Leach L, Taylor A, Sciota F. Vascular dysfunction in the diabetic placenta: causes and consequences. J Anatomy. (2009) 215:69–76. doi: 10.1111/j.1469-7580.2009.01098.x
11. Patel H, Chen J, Das KC, Kavdia M. Hyperglycemia induces differential change in oxidative stress at gene expression and functional levels in HUVEC and HMVEC. Cardiovasc Diabetol. (2013) 12:142. doi: 10.1186/1475-2840-12-142
12. Huynh J, Yamada J, Beauharnais C, Wenger JB, Thadhani RI, Wexler D, et al. Type 1, type 2 and gestational diabetes mellitus differentially impact placental pathologic characteristics of uteroplacental malperfusion. Placenta. (2015) 36:1161–6. doi: 10.1016/j.placenta.2015.08.004
13. Gabbay-Benziv R, Baschat AA. Gestational diabetes as one of the “great obstetrical syndromes”–the maternal, placental, and fetal dialog. Best Pract Res Clin Obstet Gynaecol. (2015) 29:150–5. doi: 10.1016/j.bpobgyn.2014.04.025
14. Golic M, Stojanovska V, Bendix I, Wehner A, Herse F, Haase N, et al. Diabetes mellitus in pregnancy leads to growth restriction and epigenetic modification of the Srebf2 gene in rat fetuses. Hypertension. (2018) 71:911–20. doi: 10.1161/HYPERTENSIONAHA.117.10782
15. Gagnon R. Placental insufficiency and its consequences. Eur J Obstetr Gynecol Reprod Biol. (2003) 110(Suppl. 1):S99–107. doi: 10.1016/S0301-2115(03)00179-9
16. Dicke JM. Placenta: chronicle of intrauterine growth restriction. F1000 Med Rep. (2010) 2:69. doi: 10.3410/M2-69
17. Silver RM. Examining the link between placental pathology, growth restriction, and stillbirth. Best Pract Res Clin Obstet Gynaecol. (2018) 49:89–102. doi: 10.1016/j.bpobgyn.2018.03.004
18. Mifsud W, Sebire NJ. Placental pathology in early-onset and late-onset fetal growth restriction. Fetal Diagn Ther. (2014) 36:117–28. doi: 10.1159/000359969
19. Cohen E, Baerts W, van Bel F. Brain-sparing in intrauterine growth restriction: considerations for the neonatologist. Neonatology. (2015) 108:269–76. doi: 10.1159/000438451
20. Miller SL, Huppi PS, Mallard C. The consequences of fetal growth restriction on brain structure and neurodevelopmental outcome. J Physiol. (2016) 594:807–23. doi: 10.1113/JP271402
21. Lal MK, Manktelow BN, Draper ES, Field DJ. Chronic lung disease of prematurity and intrauterine growth retardation: a population-based study. Pediatrics. (2003) 111:483–7. doi: 10.1542/peds.111.3.483
22. Danhaive O, Margossian R, Geva T, Kourembanas S. Pulmonary hypertension and right ventricular dysfunction in growth-restricted, extremely low birth weight neonates. J Perinatol. (2005) 25:495–99. doi: 10.1038/sj.jp.7211299
23. Stein AD, Zybert PA, van der Pal-de Bruin K, Lumey LH. Exposure to famine during gestation, size at birth, and blood pressure at age 59 y: evidence from the Dutch Famine. Eur J Epidemiol. (2006) 21:759–65. doi: 10.1007/s10654-006-9065-2
24. Painter RC, de Rooij SR, Bossuyt PM, Simmers TA, Osmond C, Barker DJ, et al. Early onset of coronary artery disease after prenatal exposure to the Dutch famine. Am J Clin Nutr. (2006) 84:322–7; quiz 466–327. doi: 10.1093/ajcn/84.1.322
25. Gillman MW, Barker D, Bier D, Cagampang F, Challis J, Fall C, et al. Meeting report on the 3rd International Congress on Developmental Origins of Health and Disease (DOHaD). Pediatr Res. (2007) 61(5 Pt 1):625–9. doi: 10.1203/pdr.0b013e3180459fcd
26. Mestan KK, Steinhorn RH. Fetal origins of neonatal lung disease: understanding the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. (2011) 301:L858–9. doi: 10.1152/ajplung.00314.2011
27. Kapoor A, Dunn E, Kostaki A, Andrews MH, Matthews SG. Fetal programming of hypothalamo-pituitary-adrenal function: prenatal stress and glucocorticoids. J Physiol. (2006) 572(Pt 1):31–44. doi: 10.1113/jphysiol.2006.105254
28. Burton GJ, Jauniaux E. Development of the human placenta and fetal heart: synergic or independent? Front Physiol. (2018) 9:373. doi: 10.3389/fphys.2018.00373
29. Fouzas S, Karatza AA, Davlouros PA, Chrysis D, Alexopoulos D, Mantagos S, et al. Neonatal cardiac dysfunction in intrauterine growth restriction. Pediatr Res. (2014) 75:651–7. doi: 10.1038/pr.2014.22
30. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. (2000) 35:569–82. doi: 10.1016/S0735-1097(99)00630-0
31. Hochman JS, Bulkley BH. Expansion of acute myocardial infarction: an experimental study. Circulation. (1982) 65:1446–50. doi: 10.1161/01.CIR.65.7.1446
32. Bjarnegard N, Morsing E, Cinthio M, Lanne T, Brodszki J. Cardiovascular function in adulthood following intrauterine growth restriction with abnormal fetal blood flow. Ultrasound Obstet Gynecol. (2013) 41:177–84. doi: 10.1002/uog.12314
33. Crispi F, Hernandez-Andrade E, Pelsers MM, Plasencia W, Benavides-Serralde JA, Eixarch E, et al. Cardiac dysfunction and cell damage across clinical stages of severity in growth-restricted fetuses. Am J Obstetr Gynecol. (2008) 199:254.e1–8. doi: 10.1016/j.ajog.2008.06.056
34. Kaijser M, Bonamy AK, Akre O, Cnattingius S, Granath F, Norman M, et al. Perinatal risk factors for ischemic heart disease: disentangling the roles of birth weight and preterm birth. Circulation. (2008) 117:405–10. doi: 10.1161/CIRCULATIONAHA.107.710715
35. Crispi F, Bijnens B, Figueras F, Bartrons J, Eixarch E, Le Noble F, et al. Fetal growth restriction results in remodeled and less efficient hearts in children. Circulation. (2010) 121:2427–36. doi: 10.1161/CIRCULATIONAHA.110.937995
36. Leipälä JA, Boldt T, Turpeinen U, Vuolteenaho O, Fellman V. Cardiac hypertrophy and altered hemodynamic adaptation in growth-restricted preterm infants. Pediatr Res. (2003) 53:989–93. doi: 10.1203/01.PDR.0000061564.86797.78
37. Cruz-Lemini M, Crispi F, Valenzuela-Alcaraz B, Figueras F, Sitges M, Bijnens B, et al. Fetal cardiovascular remodeling persists at 6 months in infants with intrauterine growth restriction. Ultrasound Obstet Gynecol. (2016) 48:349–56. doi: 10.1002/uog.15767
38. Azevedo PS, Polegato BF, Minicucci MF, Paiva SAR, Zornoff LAM. Cardiac remodeling: concepts, clinical impact, pathophysiological mechanisms and pharmacologic treatment. Arq Bras Cardiol. (2016) 106:62–9. doi: 10.5935/abc.20160005
39. Ortigosa N, Rodriguez-Lopez M, Bailon R, Sarvari SI, Sitges M, Gratacos E, et al. Heart morphology differences induced by intrauterine growth restriction and preterm birth measured on the ECG at preadolescent age. J Electrocardiol. (2016) 49:401–9. doi: 10.1016/j.jelectrocard.2016.03.011
40. Akazawa Y, Hachiya A, Yamazaki S, Kawasaki Y, Nakamura C, Takeuchi Y, et al. Cardiovascular remodeling and dysfunction across a range of growth restriction severity in small for gestational age infants- implications for fetal programming. Circ J. (2016) 80:2212–20. doi: 10.1253/circj.CJ-16-0352
41. Menendez-Castro C, Rascher W, Hartner A. Intrauterine growth restriction - impact on cardiovascular diseases later in life. Mol Cell Pediatr. (2018) 5:4. doi: 10.1186/s40348-018-0082-5
42. Jendryczko A, Poreba R. [Effect of fetal and neonatal growth on the occurrence of some diseases in adults]. Ginekol Polska. (1996) 67:34–6.
43. Crispi F, Bijnens B, Sepulveda-Swatson E, Cruz-Lemini M, Rojas-Benavente J, Gonzalez-Tendero A, et al. Postsystolic shortening by myocardial deformation imaging as a sign of cardiac adaptation to pressure overload in fetal growth restriction. Circu Cardiovasc Imaging. (2014) 7:781–7. doi: 10.1161/CIRCIMAGING.113.001490
44. Cruz-Lemini M, Crispi F, Valenzuela-Alcaraz B, Figueras F, Gomez O, Sitges M, et al. A fetal cardiovascular score to predict infant hypertension and arterial remodeling in intrauterine growth restriction. Am J Obstetr Gynecol. (2014) 210: 552.e1–552.e22. doi: 10.1016/j.ajog.2013.12.031
45. Kuo AH, Li C, Huber HF, Clarke GD, Nathanielsz PW. Intrauterine growth restriction results in persistent vascular mismatch in adulthood. J Physiol. (2017) 596:5777–90. doi: 10.1113/JP275139
46. Scifres CM, Parks WT, Feghali M, Caritis SN, Catov JM. Placental maternal vascular malperfusion and adverse pregnancy outcomes in gestational diabetes mellitus. Placenta. (2017) 49:10–5. doi: 10.1016/j.placenta.2016.11.004
47. Xu Y, Williams SJ, O'Brien D, Davidge ST. Hypoxia or nutrient restriction during pregnancy in rats leads to progressive cardiac remodeling and impairs postischemic recovery in adult male offspring. FASEB J. (2006) 20:1251–3. doi: 10.1096/fj.05-4917fje
48. Li G, Xiao Y, Estrella JL, Ducsay CA, Gilbert RD, Zhang L. Effect of fetal hypoxia on heart susceptibility to ischemia and reperfusion injury in the adult rat. J Soc Gynecol Invest. (2003) 10:265–74. doi: 10.1016/S1071-55760300074-1
49. Bae S, Xiao Y, Li G, Casiano CA, Zhang L. Effect of maternal chronic hypoxic exposure during gestation on apoptosis in fetal rat heart. Am J Physiol Heart Circul Physiol. (2003) 285:H983–90. doi: 10.1152/ajpheart.00005.2003
50. Sugishita Y, Leifer DW, Agani F, Watanabe M, Fisher SA. Hypoxia-responsive signaling regulates the apoptosis-dependent remodeling of the embryonic avian cardiac outflow tract. Dev Biol. (2004) 273:285–96. doi: 10.1016/j.ydbio.2004.05.036
51. Compernolle V, Brusselmans K, Franco D, Moorman A, Dewerchin M, Collen D, et al. Cardia bifida, defective heart development and abnormal neural crest migration in embryos lacking hypoxia-inducible factor-1alpha. Cardiovasc Res. (2003) 60:569–79. doi: 10.1016/j.cardiores.2003.07.003
52. Wenger RH, Gassmann M. Oxygen(es) and the hypoxia-inducible factor-1. Biol Chem. (1997) 378:609–16.
53. Gordan JD, Simon MC. Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev. (2007) 17:71–7. doi: 10.1016/j.gde.2006.12.006
54. Maloyan A, Eli-Berchoer L, Semenza GL, Gerstenblith G, Stern MD, Horowitz M. HIF-1alpha-targeted pathways are activated by heat acclimation and contribute to acclimation-ischemic cross-tolerance in the heart. Physiol Genom. (2005) 23:79–88. doi: 10.1152/physiolgenomics.00279.2004
55. Ladoux A, Frelin C. Cardiac expressions of HIF-1 alpha and HLF/EPAS, two basic loop helix/PAS domain transcription factors involved in adaptative responses to hypoxic stresses. Biochem Biophys Res Commun. (1997) 240:552–6. doi: 10.1006/bbrc.1997.7708
56. Masoumy EP, Sawyer AA, Sharma S, Patel JA, Gordon PMK, Regnault TRH, et al. The lifelong impact of fetal growth restriction on cardiac development. Pediatr Res. (2018) 84:537–44. doi: 10.1038/s41390-018-0069-x
57. Snoeckx LH, Cornelussen RN, Van Nieuwenhoven FA, Reneman RS, Van Der Vusse GJ. Heat shock proteins and cardiovascular pathophysiology. Physiol Rev. (2001) 81:1461–97. doi: 10.1152/physrev.2001.81.4.1461
58. Saurin AT, Pennington DJ, Raat NJ, Latchman DS, Owen MJ, Marber MS. Targeted disruption of the protein kinase C epsilon gene abolishes the infarct size reduction that follows ischaemic preconditioning of isolated buffer-perfused mouse hearts. Cardiovasc Res. (2002) 55:672–80. doi: 10.1016/S0008-6363(02)00325-5
59. Inagaki K, Begley R, Ikeno F, Mochly-Rosen D. Cardioprotection by epsilon-protein kinase C activation from ischemia: continuous delivery and antiarrhythmic effect of an epsilon-protein kinase C-activating peptide. Circulation. (2005) 111:44–50. doi: 10.1161/01.CIR.0000151614.22282.F1
60. Inagaki K, Churchill E, Mochly-Rosen D. Epsilon protein kinase C as a potential therapeutic target for the ischemic heart. Cardiovasc Res. (2006) 70:222–30. doi: 10.1016/j.cardiores.2006.02.015
61. Xue Q, Zhang L. Prenatal hypoxia causes a sex-dependent increase in heart susceptibility to ischemia and reperfusion injury in adult male offspring: role of protein kinase C epsilon. J Pharmacol Exp Ther. (2009) 330:624–32. doi: 10.1124/jpet.109.153239
62. Hennig M, Fiedler S, Jux C, Thierfelder L, Drenckhahn JD. Prenatal mechanistic target of rapamycin complex 1 (m TORC1) inhibition by rapamycin treatment of pregnant mice causes intrauterine growth restriction and alters postnatal cardiac growth, morphology, and function. J Am Heart Assoc. (2017) 6:e005506. doi: 10.1161/JAHA.117.005506
63. Guitart-Mampel M, Gonzalez-Tendero A, Niñerola S, Morén C, Catalán-Garcia M, González-Casacuberta I, et al. Cardiac and placental mitochondrial characterization in a rabbit model of intrauterine growth restriction. Biochim Biophys Acta (BBA) Gen Subj. (2018) 1862:1157–67. doi: 10.1016/j.bbagen.2018.02.006
64. Musikant D, Sato H, Capobianco E, White V, Jawerbaum A, Higa R. Altered FOXO1 activation in the programming of cardiovascular alterations by maternal diabetes. Mol Cell Endocrinol. (2018) 479:78–86. doi: 10.1016/j.mce.2018.09.003
65. Thompson JA, Piorkowska K, Gagnon R, Richardson BS, Regnault TR. Increased collagen deposition in the heart of chronically hypoxic ovine fetuses. J Dev Origins Health Dis. (2013) 4:470–8. doi: 10.1017/S2040174413000299
66. Barker DJ, Osmond C, Law CM. The intrauterine and early postnatal origins of cardiovascular disease and chronic bronchitis. J Epidemiol Commun Health. (1989) 43:237–40. doi: 10.1136/jech.43.3.237
67. Bassareo PP, Marras AR, Cugusi L, Zedda AM, Mercuro G. The reasons why cardiologists should consider prematurity at birth and intrauterine growth retardation among risk factors. J Cardiovasc Med. (2016) 17:323–9. doi: 10.2459/JCM.0000000000000338
68. Li CC, Maloney CA, Cropley JE, Suter CM. Epigenetic programming by maternal nutrition: shaping future generations. Epigenomics. (2010) 2:539–49. doi: 10.2217/epi.10.33
69. Schleithoff C, Voelter-Mahlknecht S, Dahmke IN, Mahlknecht U. On the epigenetics of vascular regulation and disease. Clin Epigenetics. (2012) 4:7. doi: 10.1186/1868-7083-4-7
70. Kim M, Long TI, Arakawa K, Wang R, Yu MC, Laird PW. DNA methylation as a biomarker for cardiovascular disease risk. PLoS ONE. (2010) 5:e9692. doi: 10.1371/journal.pone.0009692
71. Handy DE, Castro R, Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation. (2011) 123:2145–56. doi: 10.1161/CIRCULATIONAHA.110.956839
72. Muka T, Koromani F, Portilla E, O'Connor A, Bramer WM, Troup J, et al. The role of epigenetic modifications in cardiovascular disease: a systematic review. Int J Cardiol. (2016) 212:174–83. doi: 10.1016/j.ijcard.2016.03.062
73. Gangwar RS, Rajagopalan S, Natarajan R, Deiuliis JA. Noncoding RNAs in cardiovascular disease: pathological relevance and emerging role as biomarkers and therapeutics. Am J Hypertension. (2018) 31:150–65. doi: 10.1093/ajh/hpx197
74. Gurha P. MicroRNAs in cardiovascular disease. Curr Opin Cardiol. (2016) 31:249–54. doi: 10.1097/HCO.0000000000000280
75. Joladarashi D, Thandavarayan RA, Babu SS, Krishnamurthy P. Small engine, big power: microRNAs as regulators of cardiac diseases and regeneration. Int J Mol Sci. (2014) 15:15891–911. doi: 10.3390/ijms150915891
76. Murray R, Bryant J, Titcombe P, Barton SJ, Inskip H, Harvey NC, et al. DNA methylation at birth within the promoter of ANRIL predicts markers of cardiovascular risk at 9 years. Clin Epigenet. (2016) 8:90. doi: 10.1186/s13148-016-0259-5
77. Zhuang J, Peng W, Li H, Wang W, Wei Y, Li W, et al. Methylation of p15(INK4b) and Expression of ANRIL on Chromosome 9p21 Are Associated with Coronary Artery Disease. PLoS ONE. (2012) 7:e47193. doi: 10.1371/journal.pone.0047193
78. Widmer RJ, Lerman A. Endothelial dysfunction and cardiovascular disease. Global Cardiol Sci Pract. (2014) 2014:291–308. doi: 10.5339/gcsp.2014.43
79. Barthelmes J, Nägele MP, Ludovici V, Ruschitzka F, Sudano I, Flammer AJ. Endothelial dysfunction in cardiovascular disease and Flammer syndrome—similarities and differences. EPMA J. (2017) 8:99–109. doi: 10.1007/s13167-017-0099-1
80. Krause BJ, Costello PM, Munoz-Urrutia E, Lillycrop KA, Hanson MA, et al. Role of DNA methyltransferase 1 on the altered eNOS expression in human umbilical endothelium from intrauterine growth restricted fetuses. Epigenetics. (2013) 8:944–52. doi: 10.4161/epi.25579
81. Mudau M, Genis A, Lochner A, Strijdom H. Endothelial dysfunction: the early predictor of atherosclerosis. Cardiovasc J Afr. (2012) 23:222–31. doi: 10.5830/CVJA-2011-068
82. Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circul Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
83. Park K-H, Park WJ. Endothelial dysfunction: clinical implications in cardiovascular disease and therapeutic approaches. J Korean Med Sci. (2015) 30:1213–25. doi: 10.3346/jkms.2015.30.9.1213
84. Celermajer DS, Sorensen KE, Gooch VM, Spiegelhalter DJ, Miller OI, Sullivan ID, et al. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. (1992) 340:1111–5. doi: 10.1016/0140-6736(92)93147-F
85. Patterson AJ, Xiao D, Xiong F, Dixon B, Zhang L. Hypoxia-derived oxidative stress mediates epigenetic repression of PKCepsilon gene in foetal rat hearts. Cardiovasc Res. (2012) 93:302–10. doi: 10.1093/cvr/cvr322
86. Chen M, Xiong F, Zhang L. Promoter methylation of Egr-1 site contributes to fetal hypoxia-mediated PKCε gene repression in the developing heart. Am J Physiol Regul Integrat Compar Physiol. (2013) 304:R683–9. doi: 10.1152/ajpregu.00461.2012
87. Pilote L, Dasgupta K, Guru V, Humphries KH, McGrath J, Norris C, et al. A comprehensive view of sex-specific issues related to cardiovascular disease. CMAJ Can Med Assoc J. (2007) 176:S1–44. doi: 10.1503/cmaj.051455
88. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women's Health Initiative randomized controlled trial. JAMA. (2002) 288:321–33. doi: 10.1001/jama.288.3.321
89. Schiano C, Vietri MT, Grimaldi V, Picascia A, De Pascale MR, Napoli C. Epigenetic-related therapeutic challenges in cardiovascular disease. Trends Pharmacol Sci. (2015) 36:226–35. doi: 10.1016/j.tips.2015.02.005
90. Liguori A, Puglianiello A, Germani D, Deodati A, Peschiaroli E, Cianfarani S. Epigenetic changes predisposing to type 2 diabetes in intrauterine growth retardation. Front Endocrinol. (2010) 1:5. doi: 10.3389/fendo.2010.00005
91. Dong F, Ford SP, Fang CX, Nijland MJ, Nathanielsz PW, Ren J. Maternal nutrient restriction during early to mid gestation up-regulates cardiac insulin-like growth factor (IGF) receptors associated with enlarged ventricular size in fetal sheep. Growth Horm IGF Res. (2005) 15:291–9. doi: 10.1016/j.ghir.2005.05.003
92. Bogdarina I, Welham S, King PJ, Burns SP, Clark AJL. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circul Res. (2007) 100:520–6. doi: 10.1161/01.RES.0000258855.60637.58
93. McMullen S, Gardner DS, Langley-Evans SC. Prenatal programming of angiotensin II type 2 receptor expression in the rat. Br J Nutr. (2004) 91:133–40. doi: 10.1079/BJN20031029
94. Patterson AJ, Zhang L. Hypoxia and fetal heart development. Curr Mol Med. (2010) 10:653–66. doi: 10.2174/156652410792630643
95. Smith CJ, Ryckman KK. Epigenetic and developmental influences on the risk of obesity, diabetes, and metabolic syndrome. Diabetes Metab Syndr Obes. (2015) 8:295–302. doi: 10.2147/DMSO.S61296
96. Kampmann U, Madsen LR, Skajaa GO, Iversen DS, Moeller N, Ovesen P. Gestational diabetes: a clinical update. World J Diabetes. (2015) 6:1065–72. doi: 10.4239/wjd.v6.i8.1065
97. Kawasaki M, Arata N, Miyazaki C, Mori R, Kikuchi T, Ogawa Y, et al. Obesity and abnormal glucose tolerance in offspring of diabetic mothers: a systematic review and meta-analysis. PLoS ONE. (2018) 13:e0190676. doi: 10.1371/journal.pone.0190676
98. Ma B, Wilker EH, Willis-Owen SAG, Byun H-M, Wong KCC, Motta V, et al. Predicting DNA methylation level across human tissues. Nucleic Acids Res. (2014) 42:3515–28. doi: 10.1093/nar/gkt1380
99. Wang Z, Chang C, Peng M, Lu Q. Translating epigenetics into clinic: focus on lupus. Clin Epigenet. (2017) 9:78. doi: 10.1186/s13148-017-0378-7
100. Mazzone R, Zwergel C, Mai A, Valente S. Epi-drugs in combination with immunotherapy: a new avenue to improve anticancer efficacy. Clin Epigenet. (2017) 9:59. doi: 10.1186/s13148-017-0358-y
101. Yun S, Vincelette ND, Abraham I, Robertson KD, Fernandez-Zapico ME, Patnaik MM. Targeting epigenetic pathways in acute myeloid leukemia and myelodysplastic syndrome: a systematic review of hypomethylating agents trials. Clin Epigenet. (2016) 8:68. doi: 10.1186/s13148-016-0233-2
102. Cuadrado-Tejedor M, Garcia-Barroso C, Sanzhez-Arias J, Mederos S, Rabal O, Ugarte A, et al. Concomitant histone deacetylase and phosphodiesterase 5 inhibition synergistically prevents the disruption in synaptic plasticity and it reverses cognitive impairment in a mouse model of Alzheimer's disease. Clin Epigenet. (2015) 7:108. doi: 10.1186/s13148-015-0142-9
103. Cardona-Monzonís A, Beltrán-García J, Ibañez-Cabellos JS, Pérez-Machado G, Malkani K, Sanchis-Gomar F, et al. Epigenetic biomarkers in cardiovascular disease. J Lab Precis Med. (2018) 3:24. doi: 10.21037/jlpm.2018.02.04
104. Salam RA, Das JK, Bhutta ZA. Impact of intrauterine growth restriction on long-term health. Curr Opin Clin Nutr Metab Care. (2014) 17:249–54. doi: 10.1097/MCO.0000000000000051
105. Longo S, Bollani L, Decembrino L, Di Comite A, Angelini M, Stronati M. Short-term and long-term sequelae in intrauterine growth retardation (IUGR). J Matern Fetal Neonatal Med. (2013) 26:222–5. doi: 10.3109/14767058.2012.715006
106. Master JS, Thouas GA, Harvey AJ, Sheedy JR, Hannan NJ, Gardner DK, et al. Fathers that are born small program alterations in the next-generation preimplantation rat embryos. J Nutr. (2015) 145:876–83. doi: 10.3945/jn.114.205724
Keywords: diabetes, placental dysfunction, gene modification, intra uterine growth restriction, cardiovascular programming
Citation: Langmia IM, Kräker K, Weiss SE, Haase N, Schütte T, Herse F and Dechend R (2019) Cardiovascular Programming During and After Diabetic Pregnancy: Role of Placental Dysfunction and IUGR. Front. Endocrinol. 10:215. doi: 10.3389/fendo.2019.00215
Received: 07 November 2018; Accepted: 18 March 2019;
Published: 09 April 2019.
Edited by:
Suzanne Lee Miller, Monash University, AustraliaReviewed by:
Charles Evans Wood, University of Florida, United StatesCopyright © 2019 Langmia, Kräker, Weiss, Haase, Schütte, Herse and Dechend. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralf Dechend, cmFsZi5kZWNoZW5kQGNoYXJpdGUuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.