 Ilaria Roato
Ilaria Roato Massimo Vitale
Massimo Vitale- 1Center for Research and Medical Studies (CeRMS), A.O.U. Città della Salute e della Scienza di Torino, Turin, Italy
- 2UOC Immunologia, IRCCS Ospedale Policlinico San Martino Genova, Genoa, Italy
Bone is one of the main metastatic sites of solid tumors like breast, lung, and prostate cancer. Disseminated tumor cells (DTCs) and cancer stem cells (CSCs) represent the main target to counteract bone metastatization. These cells often localize in bone marrow (BM) at level of pre-metastatic niche: they can remain dormant for years or directly grow and create bone lesion, according to the different stimulations received in BM. The immune system in bone marrow is dampened and represents an appealing site for DTCs/CSCs. NK cells have an important role in controlling tumor progression, but their involvement in bone metastasis formation is an interesting and not fully investigated issue. Indeed, whether NK cells can interfere with CSC formation, kill them at the site of primary tumor, during circulation or in the pre-metastic niche needs to be elucidated. This review focuses on different aspects that regulate DTC/CSC life in bone and how NK cells potentially control bone metastasis formation.
Introduction
Although many cancer patients benefit from more efficient treatments of primary tumors and become long survivors, the overall probability to develop metastases is increasing, making this aspect of the disease a key target for researchers and clinicians (1). Bone is one of the main metastatic sites for different solid tumors including breast, lung, and prostate cancer (2). Bone metastasis formation and evolution is strongly influenced by a complex cross talk occurring among tumor, immune, and bone cells (3, 4). BM, besides cell precursors, contains different types of resident or recirculating mature immune cells, including Dendritic cells (DC), macrophages, granulocytes, myeloid derived suppressor cells (MDSCs), NK cells, and different T and B lymphocyte subsets. Although some of these cells (i.e., macrophages, granulocytes, lymphocytes, and NK cells) are endowed with effector functions and directly involved in pathogen elimination, virtually all BM immune cells can produce a variety of cytokines, chemokines, or other factors possibly influencing the local tissue homeostasis. Moreover, subtypes of bone cells, such as osteoclasts (OCs), originate from immune progenitors and use “immune” receptor/ligand pairs to rule their maturation and also to govern their bone degradation activity, a process significantly involved in metastasis formation (5).
Immune cells are poorly effective in the control of metastasis formation and growth, and this is true also for bone metastases, in spite of the consistency of the immune system in the bone niches. The definition of immunotherapeutic approaches in the metastatic disease is nevertheless attractive, especially considering NK cells, a subset of powerful effectors of the innate immunity endowed with anti-tumor activity. These cells have been shown to kill pro-metastatic tumor initiating cells and, recently, also to control metastases in animal models. On the other hand, NK cell heterogeneity and the complexity of their functional interactions with the local tumor microenvironment indicate that specific studies need to be addressed to define their role in bone metastases.
DTCs Colonize Bone Marrow and Activate the Bone Metastatic Vicious Cycle
In primary tumors, genetic, and epigenetic changes favor the switch of malignant cells to less differentiated forms through a process called epithelial-to-mesenchymal transition (EMT) (6). Cells rising from this switch can leave the primary tumor site becoming disseminated tumor cells (DTCs). DTCs can express cancer stem cell (CSC) profiles and properties such as resistance to chemotherapy and ability to home in BM for long time. Indeed, DTCs can migrate to distant organs and establish in BM at level of the premetastatic niches, which are induced by soluble factors or extra-cellular vescicles released in circulation by the primary tumor (7). The presence of DTCs in BM has clinical relevance, since it is associated to an increased risk to develop bone metastases (8–11).
In the BM, DTCs can also compete with hematopoietic stem cells (HSC) (12, 13) and establish in the niche by interacting with different elements including osteoblasts (OBs), endothelial cells, and Extracellular Matrix (ECM). OBs constitutively express CXCL12 and attract CXCR4-expressing tumor cells (14). Using mouse models, it has been shown that breast, lung, and prostate cancer cells overexpressing CXCR4 and CXCR7 increased their ability to extravasate and colonize bone (15, 16), and CXCR4 inhibition decreased bone and lung metastases (17, 18). Integrins and cadherins are other crucial factors for the interactions between DTCs and niches (16, 19). In breast cancer, the vascular-endothelial molecule-1 (VCAM-1) binds with high affinity α4β7 and α4β1 on OC precursors, leading to osteoclastogenesis, and α4 or VCAM-1 blocking antibodies effectively inhibit bone metastasis (20). Integrins can also interact with osteopontin (OPN), an ECM protein overexpressed in tumors and associated to tumor cell migration, metastases, and poor prognosis (21, 22).
Breast and prostate cancer DTCs can live in BM in a dormant state in pre-metastatic niche for years before starting to grow and to form metastases (5, 23). Indeed, the outgrowth of DTCs from dormant state, depends both on factors released by bone microenvironment, such as fibronectin, collagen I, and periostin (24), and by physical factors such as acid pH, hypoxia, high extracellular calcium concentration (25), which also cause disruption of the balanced physiological bone remodeling due to OC and OB activity (26). Remarkably, an increased OC activity generates the physical space for tumor expansion and induces the release from the bone matrix of molecules that further stimulate tumor cell proliferation, creating the vicious cycle (27, 28). Tumor cells in turn secrete PTHrP, activated vitamin D, tumor necrosis factor (TNF), matrix metalloproteinases (MMPs), interleukin-6 (IL-6), and other factors, which stimulate the expression of the receptor activator of nuclear factor NF-kB ligand (RANKL) on OBs, leading to the final stimulation of osteoclastogenesis from local OC precursors (3, 20).
Interaction Between Immune System and Bone FAVOR Tumor Cell Survival and Proliferation
A fundamental molecular link between immune system and bone is represented by the axis comprising RANKL, its receptor RANK, and the natural decoy receptor osteoprotegerin (OPG) (29, 30). RANK/RANKL interaction activates osteoclastogenesis, while OPG counteracts this effect by competing with RANK to bind RANKL (31). OBs and BM stromal cells are the main producers of both RANKL and OPG in physiological conditions (32), however, B or activated T cells can influence the RANKL/OPG ratio, end eventually osteoclastogenesis by producing OPG or RANKL, respectively (33). Literature data report that T cells could directly carry on a modulatory action on OCs through production of different factors such as IL-7, RANKL, TNFα (34–38). Circulating OC precursors from bone metastatic patients have been shown to differentiate into mature OCs in a T cell dependent way, in the absence of the classical OC inducers M-CSF, and RANKL (39). On the other hand, in mouse models, it has been shown that T cells exert a fundamental anti-tumor effect, regardless of OC status. Indeed, PLCγ2-KO mice, with dysfunctional OCs and impaired T-cell activation, showed increased bone tumor growth despite protection from bone loss, whereas Lyn-KO mice with numerous OCs and increased T-cell responses, showed impaired tumor growth in bone despite enhanced OC activity and osteolysis. The injection of antigen-specific wild-type cytotoxic CD8(+) T cells in both these mouse models normalized tumor growth in bone, suggesting their important role in the regulation of tumor bone metastases (40). T cells can limit tumor cell diffusion by releasing IFNγ, which also affects osteoclastogenesis, indeed lack of IFNγ has been related to the increase of bone metastases (41).
Tumor cells modify the surrounding microenvironment, indeed it has been shown that BM from breast cancer patients differed from that of healthy subjects in its cellular composition as well as the activation status of cells from the innate immune system (macrophages, NK cells) and from the adaptive immune system (T cell subsets) (42). Many immature and suppressor immune cell types are present in bone, such as T regulatory cells, which must maintain a balanced immune-reactivity (43), and MDSCs, which stimulate osteoclastogenesis (44). In breast cancer, infiltrating T regulatory cells produce RANKL, promoting OC differentiation, activity, and subsequent bone lesions (45).
MDSCs are increased in cancer patients from 2 up to 25% (46) suppressing innate and adaptive immune response, thus sustaining tumor growth and metastatization (47). In breast cancer, MDSCs, derived from bone metastatic microenvironment, can differentiate into mature and functional OCs in vitro (48).
NK Cells are Endowed With Powerful Anti-Tumor Functions
NK cells can kill a variety of tumor cells of different origin and types (49–52). This wide range of reactivity is ensured by the expression at the cell surface of several receptors capable of activating or inhibiting the main functions of NK cells, including the release of cytolytic granules (49, 53). Thus, thanks to their HLA-I-specific inhibitory receptors and a complex and heterogeneous group of activating receptors, NK cells can sense the HLA-I expression decrease that often characterizes tumor cells and recognize different ligands that can be variably induced on cells undergoing tumor transformation (Table 1). Different patterns of NK receptors are engaged during contact with pathological or non-pathological cells, regulating the activation, and the intensity of the cytolytic response (49, 50, 53, 54). Most NK cells express the FcγIII-receptor (CD16), which is a strong activator of cytotoxicity and enables NK cells to mediate the Antibody-Dependent Cellular Cytotoxicity (ADCC).
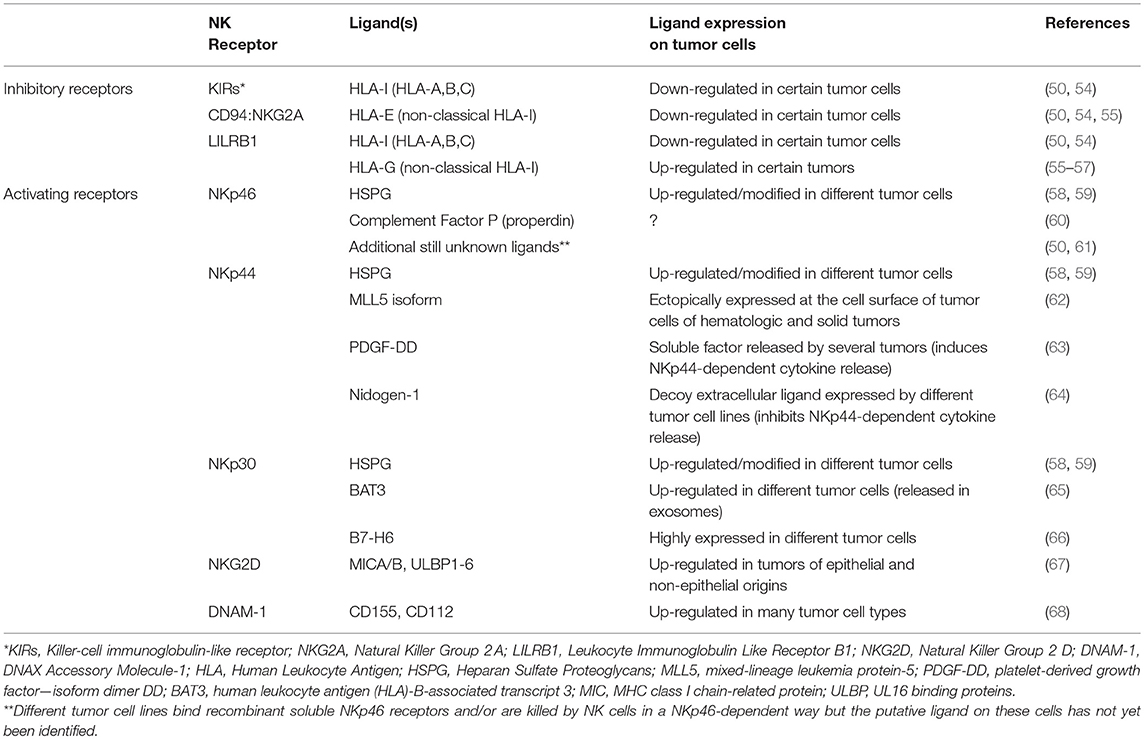
Table 1. Overview of the major NK cell receptors and Ligands involved in tumor cell recognition.
NK cells can attack tumor cells by releasing pro-apoptotic factors, including TNF-α and Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) (69, 70), or cytokines capable of inhibiting tumor cell proliferation and promoting the inflammatory response, such as IFN-γ. In addition, NK cells can release chemokines (CCL3, CCL4, CCL5, and XCL1) capable of attracting T cells, DC, and monocytes (71, 72) and give rise to specific cross-talks promoting and regulating the adaptive anti-tumor response (73–75). Finally, NK cells can also amplify their recruitment at the tumor site by releasing a chemotactic form of HMGB1 molecule upon interaction with tumor cells (76).
In order to appropriately evaluate the role of NK cells in the control of tumors it should be also considered that the NK cell population is rather heterogeneous as it includes different cell subsets, each characterized by peculiar functional capabilities (77). In humans, the CD56brightCD16dim/neg (CD56bright) and the CD56dim/CD16bright (CD56dim) cells represent the two most studied NK cell types. The CD56bright NK cells largely produce IFN-γ in response to monokines but are poorly cytotoxic. These cells constitute 5–10% of circulating NK cells, and, in line with their pattern of chemokine and homing receptors (i.e., CD62L, CCR7, CXCR3, and CXCR4), represent most LN-NK cells and an important fraction of tissue NK cells in different organs. The CD56dim cells release IFN-γ upon triggering of major activating receptors (NKp46, NKp30, NKp44, and CD16) and are highly cytotoxic. They represent 90–95% of PB NK cells and predominate in spleen, lungs, and kidney although in different percentages. Moreover, CD56dim NK cells express chemokine receptors (CXCR1, CX3CR1, and CXCR4) that allow their possible recruitment to inflamed peripheral tissues (77, 78). The assessment of NK cells in tissues and the definition of their anti-tumor potential are rather complicated. Indeed, tissues comprise both potentially cytotoxic NK cells that recirculate from PB, but also stably resident cells expressing specific markers of tissue retention (CD69, CD49a, and CD103) and chemokine receptors (CCR5, CXCR6) (79–82). These latter cells may display unique functions, possibly organ-specific, not necessarily oriented to tumor cell killing.
Role of NK Cells in the Control of Solid Tumors and Metastatic Spread
Several studies using different mice models have documented that NK cells can control tumor insurgence, growth, and metastasis dissemination (83–86). Remarkably, the role of NK cells in the control of tumors has also been suggested in different human studies. In a 11-year follow-up study on more than 8,000 healthy individuals, Imai et al. initially showed that insurgence of tumors of different types inversely correlated with the levels of natural cytotoxic activity of peripheral blood lymphocytes (87). More recently, different groups have found correlations between the quantity and the quality of tumor infiltrating or PB-NK cells and a more favorable prognosis or the lower number of metastases at diagnosis (88). In this last decade, it has also become evident that a plethora of mechanisms of tumor escape can strongly reduce the efficacy of NK cells. Within the tumor microenvironment, different immune suppressor cells (including Tregs and MDSCs), tumor-associated fibroblasts (TAF), and tumor cells can produce soluble factors (TGF-β, PGE2, IDO-derived kynurenine) which inhibit expression and function of the major activating receptors (89, 90). Similar effects on activating receptors are induced also by soluble decoy ligands shed by tumor cells or released as extracellular molecules (64, 89, 91, 92). Finally, exposure to hypoxia, which often characterizes tumor tissues, can also cause activating receptor down-regulation (93). Remarkably, some of these suppressive mechanisms, such as those induced by hypoxia and TAFs, appear to minimally affect the ADCC function (94). On the other hand, hypoxia and tumor cells can modulate the repertoire of chemokine receptors on NK cells and favor the preferential recruitment of CD56bright cells (poorly cytotoxic and unable to mediate ADCC) (95). The NK-cell recruitment into neoplastic tissues may also be influenced by the chemokine profile induced in the tumor microenvironment. Human lung and breast tumors have been shown to express higher CCL19 (a CD56bright cell attracting chemokine) and lower CXCL12 compared to their normal tissue counterpart (96), while in mice, BM with MM showed increased CXCL9 and CXCL10, decreased CXCL12, down-modulation of CXCR3 on NK cells, and selective reduction of KLRG1− cytotoxic NK cells (97). Collectively, the above-described mechanisms can account for the observation that in different tumor tissues the NK cell infiltrate is often limited or constituted by CD56bright or altered (poorly functional) CD56dim cells (50, 89).
Another important issue regards the so-called immune-checkpoints. Different pairs of receptor-ligands are available to the immune system to regulate or terminate excessive (dangerous) responses. Some of these receptors, such as PD-1, TIM-3, TIGIT, and SIGIRR, can be also expressed by NK cells, especially by those associated to tumors, and control different NK cell functions including cytotoxicity. Blocking or overcoming these checkpoints, by specific monoclonal antibodies or activating cytokines can improve the NK-mediated control of carcinogenesis or metastasis formation (98–101).
The Ambiguous Role of the NK:CSC Cross-Talk in the Control of Metastasis Formation
Whether NK cells can interfere with CSC formation, or kill CSC at the site of primary tumor, during circulation, or in the pre-metastatic niches represents an interesting and still incompletely investigated issue. Several reports have indicated that NK cells can kill tumor cells with features of CSC derived from different tumors (glioma, melanoma, colon, prostate, and breast) (102). Consistent with these findings, CSCs of different origins have been shown to express or even up-regulate the ligands for NKG2D, DNAM1, and NKp30 NK-activating receptors and cells undergoing EMT showed up-regulated NKG2D-Ls (88, 102). In addition, EMT induction in lung cancer cells could promote increased NK cell-mediated metastasis-specific immunosurveillance in RAG1−/− mice (103). On the other hand, it has also been shown that NK cells could induce melanoma cells to undergo EMT, upregulate the expression of stemness markers, reduce proliferative capability, thus acquiring characteristics reminiscent of the CSC phenotype. Moreover, EMT increased ability of melanoma cells to suppress NK cell cytotoxicity against tumor cells (104).
Is there any Role for NK Cells in Bone Metastases?
Although the role of NK cells in contrasting bone metastases has been recently suggested in breast cancer preclinical models (105), an established knowledge on this issue is still lacking. BM is where NK cells mature and differentiate from CD34+ progenitors; therefore, it contains precursors at different stages. Once NK cells have matured, changes in the expression of the key receptors CXCR4 and S1P5 (down- and up-regulated, respectively) drive their egress from the CXCL12-containing BM and their recruitment to blood where the S1P5-ligand S1P is abundant (106). Besides the immature NK cell precursors, BM also contains a reservoir of mature NK cells, recirculating from the blood, which can be mobilized upon inflammatory stimuli (107). BM also includes a substantial population of resident CXCR6+CD69+ NK (BMrNK) cells, which may be poorly effective against tumor cells. Indeed, compared to classical NK cells, BMrNK cells display lower proliferative capacity, cytolytic granule content, DNAM1, and higher TIGIT expression (108).
The heterogeneity of the NK cells in the BM and their still poorly defined interaction with the metastatic niche, together with the possible cross-talk between PB-NK and CSC/EMT cells add a layer of complexity to the issue of how NK cells can influence bone metastasis formation and progression (Figure 1). In the bone, the RANK-RANK-L axis, whose deregulation is important in metastasis formation, also influences NK cells. The signaling of RANK-L in leukemia cells can induce the release of NK-suppressing factors (109), whereas, under inflammatory conditions (knee arthritis) NK cells can stimulate OCs through activation of RANKL pathway (110). NK cells can either favor or inhibit generation of OCs depending on the release of TNF-α or IFN-γ, respectively (111, 112). On the other hand, OCs have been shown to contribute to the induction of efficient NK cells, capable of inhibiting growth of poorly differentiated tumors in humanized BLT mice (113). This effect is in line with the ability of OCs to produce NK-stimulating cytokines such as IL-12, IL-15, and IL-18. Finally, OCs are targets of NK cells, as they express MHC class I molecules at low levels and are killed by IL-2 treated NK cells (114).
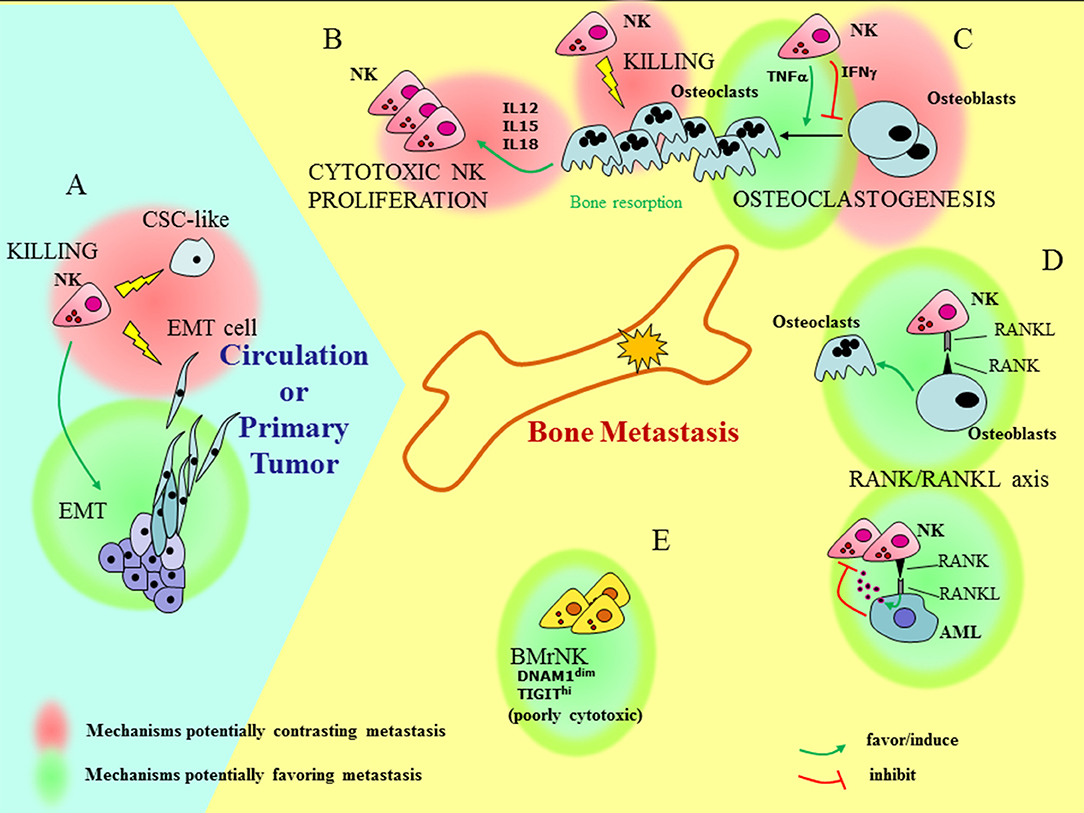
Figure 1. Understanding how NK cells can influence bone metastasis. Outside and inside the bone (light blue and yellow fields, respectively), different functional interactions involving BM resident, recirculating, or PB NK cells can have contrasting effects on bone metastasis formation and tumor progression. (A) At the site of primary tumors NK cells may favor EMT. On the other hand, NK cells can kill pro-metastatic tumor cells, such as cells that have undergone EMT (EMT cells), and cells with features of CSC (CSC-like). (B) OCs can induce proliferation of highly cytotoxic NK cells via the release of IL12, IL15, and IL18. (C) The interaction of NK cells with OCs gives rise to contrasting effects: NK cells can either favor or inhibit generation of OCs depending on the release of TNF-α or IFN-γ, respectively and kill OCs. (D) The RANK/RANKL axis may play a role in the cross-talk between NK cells and bone microenvironment: the signaling of RANK-L in leukemia cells (AML) can induce the release of NK-suppressing factors; on the other hand, RANKL-expressing NK cells provide signals for OC generation. (E) Besides recirculating PB-NK cells, BM also contains resident poorly cytotoxic NK cells (BMrNK) which may hardly eliminate tumor cells.
Concluding Remarks
Understanding the reason why and how in many patients' metastases can overcome the surveillance of NK cells is still poorly understood. Studies are rapidly progressing to define how to properly activate NK cells by cytokine combinations and unleash their potential by blocking their checkpoint receptors. The crucial mechanisms that govern entrance and egress of NK cells in the bone metastatic niche and modulate the NK cell killing capability within the bone lesions are lacking. Addressing these questions will significantly increase the therapeutic options for NK cells in the treatment of bone metastatic disease.
Author Contributions
IR revised the literature and wrote the paragraphs concerning bone metastasis. MV revised the literature and wrote the paragraphs concerning NK cells.
Funding
This work was supported by the CRT Foundations, Fondazione Ricerca Molinette ONLUS (IR) and Associazione Italiana Ricerca sul Cancro AIRC under grants: IG 2014 project n. 15428 (MV), 5 × 1000 Min Sal. 2013 (MV) and Roche Foundation: Bando per la Ricerca Roche 2017.
Conflict of Interest Statement
IR was supported by Roche Foundation. The funder played no role in the study design, the collection, analysis or interpretation of data, the writing of this paper or the decision to submit it for publication.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Macedo F, Ladeira K, Pinho F, Saraiva N, Bonito N, Pinto L, et al. Bone metastases: an overview. Oncol Rev. (2017) 11:321. doi: 10.4081/oncol.2017.321
2. Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. (2006) 12 (20 Pt 2):6243s−9s. doi: 10.1158/1078-0432.CCR-06-0931
3. Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. (2002) 2:584–93. doi: 10.1038/nrc867
4. D'Amico L, Roato I. The impact of immune system in regulating bone metastasis formation by osteotropic tumors. J Immunol Res. (2015) 2015:143526. doi: 10.1155/2015/143526
5. Li L, Bhatia R. Stem cell quiescence. Clin Cancer Res. (2011) 17:4936–41. doi: 10.1158/1078-0432.CCR-10-1499
6. Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. (2013) 19:1438–49. doi: 10.1038/nm.3336
7. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. (2017) 17:302–17. doi: 10.1038/nrc.2017.6
8. Braun S, Vogl FD, Naume B, Janni W, Osborne MP, Coombes RC, et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N Engl J Med. (2005) 353:793–802. doi: 10.1056/NEJMoa050434
9. Pantel K, Brakenhoff RH, Brandt B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat Rev Cancer. (2008) 8:329–40. doi: 10.1038/nrc2375
10. Bartkowiak K, Kwiatkowski M, Buck F, Gorges TM, Nilse L, Assmann V, et al. Disseminated tumor cells persist in the bone marrow of breast cancer patients through sustained activation of the unfolded protein response. Cancer Res. (2015) 75:5367–77. doi: 10.1158/0008-5472.CAN-14-3728
11. Janni W, Vogl FD, Wiedswang G, Synnestvedt M, Fehm T, Juckstock J, et al. Persistence of disseminated tumor cells in the bone marrow of breast cancer patients predicts increased risk for relapse–a European pooled analysis. Clin Cancer Res. (2011) 17:2967–76. doi: 10.1158/1078-0432.CCR-10-2515
12. Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. (2003) 425:841–6. doi: 10.1038/nature02040
13. Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. (2003) 425:836–41. doi: 10.1038/nature02041
14. Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. (2005) 201:1307–18. doi: 10.1084/jem.20041385
15. Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. (2003) 3:537–49. doi: 10.1016/S1535-6108(03)00132-6
16. Wang H, Yu C, Gao X, Welte T, Muscarella AM, Tian L, et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell. (2015) 27:193–210. doi: 10.1016/j.ccell.2014.11.017
17. Richert MM, Vaidya KS, Mills CN, Wong D, Korz W, Hurst DR, et al. Inhibition of CXCR4 by CTCE-9908 inhibits breast cancer metastasis to lung and bone. Oncol Rep. (2009) 21:761–7. doi: 10.3892/or_00000282
18. Bertolini G, D'Amico L, Moro M, Landoni E, Perego P, Miceli R, et al. Microenvironment-modulated metastatic CD133+/CXCR4+/EpCAM- lung cancer-initiating cells sustain tumor dissemination and correlate with poor prognosis. Cancer Res. (2015) 75:3636–49. doi: 10.1158/0008-5472.CAN-14-3781
19. Chu K, Cheng CJ, Ye X, Lee YC, Zurita AJ, Chen DT, et al. Cadherin-11 promotes the metastasis of prostate cancer cells to bone. Mol Cancer Res. (2008) 6:1259–67. doi: 10.1158/1541-7786.MCR-08-0077
20. Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell. (2011) 20:701–14. doi: 10.1016/j.ccr.2011.11.002
21. Rudland PS, Platt-Higgins A, El-Tanani M, De Silva Rudland S, Barraclough R, Winstanley JH, et al. Prognostic significance of the metastasis-associated protein osteopontin in human breast cancer. Cancer Res. (2002) 62:3417–27.
22. Anborgh PH, Mutrie JC, Tuck AB, Chambers AF. Role of the metastasis-promoting protein osteopontin in the tumour microenvironment. J Cell Mol Med. (2010) 14:2037–44. doi: 10.1111/j.1582-4934.2010.01115.x
23. Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. (2011) 121:1298–312. doi: 10.1172/JCI43414
24. Oskarsson T, Batlle E, Massague J. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell. (2014) 14:306–21. doi: 10.1016/j.stem.2014.02.002
25. Lou Y, McDonald PC, Oloumi A, Chia S, Ostlund C, Ahmadi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. (2011) 71:3364–76. doi: 10.1158/0008-5472.CAN-10-4261
26. Chiang AC, Massague J. Molecular basis of metastasis. N Engl J Med. (2008) 359:2814–23. doi: 10.1056/NEJMra0805239
27. Roodman GD. Mechanisms of bone metastasis. N Engl J Med. (2004) 350:1655–64. doi: 10.1056/NEJMra030831
28. Bussard KM, Gay CV, Mastro AM. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. (2008) 27:41–55. doi: 10.1007/s10555-007-9109-4
29. Hofbauer LC, Kuhne CA, Viereck V. The OPG/RANKL/RANK system in metabolic bone diseases. J Musculoskelet Neuronal Interact. (2004) 4:268–75.
30. Santini D, Perrone G, Roato I, Godio L, Pantano F, Grasso D, et al. Expression pattern of receptor activator of NFkappaB (RANK) in a series of primary solid tumors and related bone metastases. J Cell Physiol. (2011) 226:780–4. doi: 10.1002/jcp.22402
31. Kostenuik PJ, Shalhoub V. Osteoprotegerin: a physiological and pharmacological inhibitor of bone resorption. Curr Pharm Des. (2001) 7:613–35. doi: 10.2174/1381612013397807
32. Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. (2000) 21:115–37. doi: 10.1210/er.21.2.115
33. Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood. (2007) 109:3839–48. doi: 10.1182/blood-2006-07-037994
34. Weitzmann MN, Roggia C, Toraldo G, Weitzmann L, Pacifici R. Increased production of IL-7 uncouples bone formation from bone resorption during estrogen deficiency. J Clin Invest. (2002) 110:1643–50. doi: 10.1172/JCI0215687
35. Lee SK, Kalinowski JF, Jastrzebski SL, Puddington L, Lorenzo JA. Interleukin-7 is a direct inhibitor of in vitro osteoclastogenesis. Endocrinology. (2003) 144:3524–31. doi: 10.1210/en.2002-221057
36. Colucci S, Brunetti G, Rizzi R, Zonno A, Mori G, Colaianni G, et al. T cells support osteoclastogenesis in an in vitro model derived from human multiple myeloma bone disease: the role of the OPG/TRAIL interaction. Blood. (2004) 104:3722–30. doi: 10.1182/blood-2004-02-0474
37. Roato I, Brunetti G, Gorassini E, Grano M, Colucci S, Bonello L, et al. IL-7 up-regulates TNF-alpha-dependent osteoclastogenesis in patients affected by solid tumor. PLoS ONE. (2006) 1:e124. doi: 10.1371/journal.pone.0000124
38. Giuliani N, Colla S, Sala R, Moroni M, Lazzaretti M, La Monica S, et al. Human myeloma cells stimulate the receptor activator of nuclear factor-kappa B ligand (RANKL) in T lymphocytes: a potential role in multiple myeloma bone disease. Blood. (2002) 100:4615–21. doi: 10.1182/blood-2002-04-1121
39. Roato I, Grano M, Brunetti G, Colucci S, Mussa A, Bertetto O, et al. Mechanisms of spontaneous osteoclastogenesis in cancer with bone involvement. FASEB J. (2005) 19:228–30. doi: 10.1096/fj.04-1823fje
40. Zhang K, Kim S, Cremasco V, Hirbe AC, Collins L, Piwnica-Worms D, et al. CD8+ T cells regulate bone tumor burden independent of osteoclast resorption. Cancer Res. (2011) 71:4799–808. doi: 10.1158/0008-5472.CAN-10-3922
41. Xu Z, Hurchla MA, Deng H, Uluckan O, Bu F, Berdy A, et al. Interferon-gamma targets cancer cells and osteoclasts to prevent tumor-associated bone loss and bone metastases. J Biol Chem. (2009) 284:4658–66. doi: 10.1074/jbc.M804812200
42. Feuerer M, Rocha M, Bai L, Umansky V, Solomayer EF, Bastert G, et al. Enrichment of memory T cells and other profound immunological changes in the bone marrow from untreated breast cancer patients. Int J Cancer. (2001) 92:96–105. doi: 10.1002/1097-0215(200102)9999:9999<::AID-IJC1152>3.0.CO;2-Q
43. Zou L, Barnett B, Safah H, Larussa VF, Evdemon-Hogan M, Mottram P, et al. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res. (2004) 64:8451–5. doi: 10.1158/0008-5472.CAN-04-1987
44. Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. (2001) 166:678–89. doi: 10.4049/jimmunol.166.1.678
45. Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, et al. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature. (2011) 470:548–53. doi: 10.1038/nature09707
46. Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. (2005) 65:3044–8. doi: 10.1158/0008-5472.CAN-04-4505
47. Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother. (2006) 55:237–45. doi: 10.1007/s00262-005-0048-z
48. Sawant A, Ponnazhagan S. Myeloid-derived suppressor cells as osteoclast progenitors: a novel target for controlling osteolytic bone metastasis. Cancer Res. (2013) 73:4606–10. doi: 10.1158/0008-5472.CAN-13-0305
49. Moretta L, Pietra G, Montaldo E, Vacca P, Pende D, Falco M, et al. Human NK cells: from surface receptors to the therapy of leukemias and solid tumors. Front Immunol. (2014) 5:87. doi: 10.3389/fimmu.2014.00087
50. Cantoni C, Grauwet K, Pietra G, Parodi M, Mingari MC, Maria AD, et al. Role of NK cells in immunotherapy and virotherapy of solid tumors. Immunotherapy. (2015) 7:861–82. doi: 10.2217/imt.15.53
51. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. (2016) 17:1025–36. doi: 10.1038/ni.3518
52. Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. (2018) 18:671–88. doi: 10.1038/s41577-018-0061-z
53. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science. (2011) 331:44–9. doi: 10.1126/science.1198687
54. Moretta L, Bottino C, Pende D, Vitale M, Mingari MC, Moretta A. Different checkpoints in human NK-cell activation. Trends Immunol. (2004) 25:670–6. doi: 10.1016/j.it.2004.09.008
55. Lopez-Botet M, Llano M, Navarro F, Bellon T. NK cell recognition of non-classical HLA class I molecules. Semin Immunol. (2000) 12:109–19. doi: 10.1006/smim.2000.0213
56. Carosella ED, Moreau P, Lemaoult J, Rouas-Freiss N. HLA-G: from biology to clinical benefits. Trends Immunol. (2008) 29:125–32. doi: 10.1016/j.it.2007.11.005
57. Rebmann V, Nuckel H, Duhrsen U, Grosse-Wilde H. HLA-G in B-chronic lymphocytic leukaemia: clinical relevance and functional implications. Semin Cancer Biol. (2007) 17:430–5. doi: 10.1016/j.semcancer.2007.06.011
58. Hecht ML, Rosental B, Horlacher T, Hershkovitz O, De Paz JL, Noti C, et al. Natural cytotoxicity receptors NKp30, NKp44 and NKp46 bind to different heparan sulfate/heparin sequences. J Proteome Res. (2009) 8:712–20. doi: 10.1021/pr800747c
59. Blackhall FH, Merry CL, Davies EJ, Jayson GC. Heparan sulfate proteoglycans and cancer. Br J Cancer. (2001) 85:1094–8. doi: 10.1054/bjoc.2001.2054
60. Narni-Mancinelli E, Campisi L, Bassand D, Cazareth J, Gounon P, Glaichenhaus N, et al. Memory CD8+ T cells mediate antibacterial immunity via CCL3 activation of TNF/ROI+ phagocytes. J Exp Med. (2007) 204:2075–87. doi: 10.1084/jem.20070204
61. Sivori S, Vitale M, Morelli L, Sanseverino L, Augugliaro R, Bottino C, et al. p46, a novel natural killer cell-specific surface molecule that mediates cell activation. J Exp Med. (1997) 186:1129–36. doi: 10.1084/jem.186.7.1129
62. Baychelier F, Sennepin A, Ermonval M, Dorgham K, Debre P, Vieillard V. Identification of a cellular ligand for the natural cytotoxicity receptor NKp44. Blood. (2013) 122:2935–42. doi: 10.1182/blood-2013-03-489054
63. Barrow AD, Edeling MA, Trifonov V, Luo J, Goyal P, Bohl B, et al. Natural killer cells control tumor growth by sensing a growth factor. Cell. (2018) 172:534–48.e19. doi: 10.1016/j.cell.2017.11.037
64. Gaggero S, Bruschi M, Petretto A, Parodi M, Zotto GD, Lavarello C, et al. Nidogen-1 is a novel extracellular ligand for the NKp44 activating receptor. Oncoimmunology. (2018) 7:e1470730. doi: 10.1080/2162402X.2018.1470730
65. Pogge von Strandmann E, Simhadri VR, von Tresckow B, Sasse S, Reiners KS, Hansen HP, et al. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity. (2007) 27:965–74. doi: 10.1016/j.immuni.2007.10.010
66. Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J Exp Med. (2009) 206:1495–503. doi: 10.1084/jem.20090681
67. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. (2013) 31:413–41. doi: 10.1146/annurev-immunol-032712-095951
68. Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surfaceligands for the human DNAM-1 (CD226) activating molecule. J Exp Med. (2003) 198:557–67. doi: 10.1084/jem.20030788
69. Takeda K, Hayakawa Y, Smyth MJ, Kayagaki N, Yamaguchi N, Kakuta S, et al. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med. (2001) 7:94–100. doi: 10.1038/83416
70. Plenker D, Bertrand M, de Langen AJ, Riedel R, Lorenz C, Scheel AH, et al. Structural alterations of MET trigger response to MET kinase inhibition in lung adenocarcinoma patients. Clin Cancer Res. (2018) 24:1337–43. doi: 10.1158/1078-0432.CCR-17-3001
71. Fauriat C, Long EO, Ljunggren HG, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood. (2010) 115:2167–76. doi: 10.1182/blood-2009-08-238469
72. Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. (2018) 172:1022–37.e14. doi: 10.1016/j.cell.2018.01.004
73. Moretta L, Ferlazzo G, Bottino C, Vitale M, Pende D, Mingari MC, et al. Effector and regulatory events during natural killer-dendritic cell interactions. Immunol Rev. (2006) 214:219–28. doi: 10.1111/j.1600-065X.2006.00450.x
74. Bellora F, Castriconi R, Dondero A, Reggiardo G, Moretta L, Mantovani A, et al. The interaction of human natural killer cells with either unpolarized or polarized macrophages results in different functional outcomes. Proc Natl Acad Sci USA. (2010) 107:21659–64. doi: 10.1073/pnas.1007654108
75. Ardolino M, Zingoni A, Cerboni C, Cecere F, Soriani A, Iannitto ML, et al. DNAM-1 ligand expression on Ag-stimulated T lymphocytes is mediated by ROS-dependent activation of DNA-damage response: relevance for NK-T cell interaction. Blood. (2011) 117:4778–86. doi: 10.1182/blood-2010-08-300954
76. Parodi M, Pedrazzi M, Cantoni C, Averna M, Patrone M, Cavaletto M, et al. Natural Killer (NK)/melanoma cell interaction induces NK-mediated release of chemotactic High Mobility Group Box-1 (HMGB1) capable of amplifying NK cell recruitment. Oncoimmunology. (2015) 4:e1052353. doi: 10.1080/2162402X.2015.1052353
77. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. (2017) 47:820–33. doi: 10.1016/j.immuni.2017.10.008
78. Bjorkstrom NK, Ljunggren HG, Michaelsson J. Emerging insights into natural killer cells in human peripheral tissues. Nat Rev Immunol. (2016) 16:310–20. doi: 10.1038/nri.2016.34
79. Marquardt N, Beziat V, Nystrom S, Hengst J, Ivarsson MA, Kekalainen E, et al. Cutting edge: identification and characterization of human intrahepatic CD49a+ NK cells. J Immunol. (2015) 194:2467–71. doi: 10.4049/jimmunol.1402756
80. Cuff AO, Robertson FP, Stegmann KA, Pallett LJ, Maini MK, Davidson BR, et al. Eomeshi NK cells in human liver are long-lived and do not recirculate but can be replenished from the circulation. J Immunol. (2016) 197:4283–91. doi: 10.4049/jimmunol.1601424
81. Hudspeth K, Donadon M, Cimino M, Pontarini E, Tentorio P, Preti M, et al. Human liver-resident CD56(bright)/CD16(neg) NK cells are retained within hepatic sinusoids via the engagement of CCR5 and CXCR6 pathways. J Autoimmun. (2016) 66:40–50. doi: 10.1016/j.jaut.2015.08.011
82. Aw Yeang HX, Piersma SJ, Lin Y, Yang L, Malkova ON, Miner C, et al. Cutting Edge: human CD49e- NK cells are tissue resident in the liver. J Immunol. (2017) 198:1417–22. doi: 10.4049/jimmunol.1601818
83. Smyth MJ, Thia KY, Street SE, Cretney E, Trapani JA, Taniguchi M, et al. Differential tumor surveillance by natural killer (NK) and NKT cells. J Exp Med. (2000) 191:661–8. doi: 10.1084/jem.191.4.661
84. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity. (2008) 28:571–80. doi: 10.1016/j.immuni.2008.02.016
85. Lakshmikanth T, Burke S, Ali TH, Kimpfler S, Ursini F, Ruggeri L, et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J Clin Invest. (2009) 119:1251–63. doi: 10.1172/JCI36022
86. Iguchi-Manaka A, Kai H, Yamashita Y, Shibata K, Tahara-Hanaoka S, Honda S, et al. Accelerated tumor growth in mice deficient in DNAM-1 receptor. J Exp Med. (2008) 205:2959–64. doi: 10.1084/jem.20081611
87. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet. (2000) 356:1795–9. doi: 10.1016/S0140-6736(00)03231-1
88. Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell. (2017) 32:135–54. doi: 10.1016/j.ccell.2017.06.009
89. Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur J Immunol. (2014) 44:1582–92. doi: 10.1002/eji.201344272
90. Vacca P, Munari E, Tumino N, Moretta F, Pietra G, Vitale M, et al. Human natural killer cells and other innate lymphoid cells in cancer: friends or foes? Immunol Lett. (2018) 201:14–9. doi: 10.1016/j.imlet.2018.11.004
91. Reinart N, Nguyen PH, Boucas J, Rosen N, Kvasnicka HM, Heukamp L, et al. Delayed development of chronic lymphocytic leukemia in the absence of macrophage migration inhibitory factor. Blood. (2013) 121:812–21. doi: 10.1182/blood-2012-05-431452
92. Schlecker E, Fiegler N, Arnold A, Altevogt P, Rose-John S, Moldenhauer G, et al. Metalloprotease-mediated tumor cell shedding of B7-H6, the ligand of the natural killer cell-activating receptor NKp30. Cancer Res. (2014) 74:3429–40. doi: 10.1158/0008-5472.CAN-13-3017
93. Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, et al. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur J Immunol. (2013) 43:2756–64. doi: 10.1002/eji.201343448
94. Costa D, Vene R, Benelli R, Romairone E, Scabini S, Catellani S, et al. Targeting the epidermal growth factor receptor can counteract the inhibition of natural killer cell function exerted by colorectal tumor-associated fibroblasts. Front Immunol. (2018) 9:1150. doi: 10.3389/fimmu.2018.01150
95. Parodi M, Raggi F, Cangelosi D, Manzini C, Balsamo M, Blengio F, et al. Hypoxia modifies the transcriptome of human NK cells, modulates their immunoregulatory profile, and influences NK cell subset migration. Front Immunol. (2018) 9:2358. doi: 10.3389/fimmu.2018.02358
96. Carrega P, Bonaccorsi I, Di Carlo E, Morandi B, Paul P, Rizzello V, et al. CD56(bright)perforin(low) noncytotoxic human NK cells are abundant in both healthy and neoplastic solid tissues and recirculate to secondary lymphoid organs via afferent lymph. J Immunol. (2014) 192:3805–15. doi: 10.4049/jimmunol.1301889
97. Ponzetta A, Benigni G, Antonangeli F, Sciume G, Sanseviero E, Zingoni A, et al. Multiple myeloma impairs bone marrow localization of effector natural killer cells by altering the chemokine microenvironment. Cancer Res. (2015) 75:4766–77. doi: 10.1158/0008-5472.CAN-15-1320
98. Molgora M, Bonavita E, Ponzetta A, Riva F, Barbagallo M, Jaillon S, et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature. (2017) 551:110–4. doi: 10.1038/nature24293
99. Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: a phenotypic and functional characterization. J Allergy Clin Immunol. (2017) 139:335–46 e3. doi: 10.1016/j.jaci.2016.04.025
100. Seo H, Jeon I, Kim BS, Park M, Bae EA, Song B, et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nat Commun. (2017) 8:15776. doi: 10.1038/ncomms15776
101. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
102. Voutsadakis IA. Expression and function of immune ligand-receptor pairs in NK cells and cancer stem cells: therapeutic implications. Cell Oncol. (2018) 41:107–21. doi: 10.1007/s13402-018-0373-9
103. Chockley PJ, Chen J, Chen G, Beer DG, Standiford TJ, Keshamouni VG. Epithelial-mesenchymal transition leads to NK cell-mediated metastasis-specific immunosurveillance in lung cancer. J Clin Invest. (2018) 128:1384–96. doi: 10.1172/JCI97611
104. Huergo-Zapico L, Parodi M, Cantoni C, Lavarello C, Fernandez-Martinez JL, Petretto A, et al. NK-cell editing mediates epithelial-to-mesenchymal transition via phenotypic and proteomic changes in melanoma cell lines. Cancer Res. (2018) 78:3913–25. doi: 10.1158/0008-5472.CAN-17-1891
105. Bottos A, Gotthardt D, Gill JW, Gattelli A, Frei A, Tzankov A, et al. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat Commun. (2016) 7:12258. doi: 10.1038/ncomms12258
106. Mayol K, Biajoux V, Marvel J, Balabanian K, Walzer T. Sequential desensitization of CXCR4 and S1P5 controls natural killer cell trafficking. Blood. (2011) 118:4863–71. doi: 10.1182/blood-2011-06-362574
107. Bernardini G, Sciume G, Santoni A. Differential chemotactic receptor requirements for NK cell subset trafficking into bone marrow. Front Immunol. (2013) 4:12. doi: 10.3389/fimmu.2013.00012
108. Melsen JE, Lugthart G, Vervat C, Kielbasa SM, van der Zeeuw SAJ, Buermans HPJ, et al. Human bone marrow-resident natural killer cells have a unique transcriptional profile and resemble resident memory CD8(+) T cells. Front Immunol. (2018) 9:1829. doi: 10.3389/fimmu.2018.01829
109. Schmiedel BJ, Werner A, Steinbacher J, Nuebling T, Buechele C, Grosse-Hovest L, et al. Generation and preclinical characterization of a Fc-optimized GITR-Ig fusion protein for induction of NK cell reactivity against leukemia. Mol Ther. (2013) 21:877–86. doi: 10.1038/mt.2013.11
110. Soderstrom K, Stein E, Colmenero P, Purath U, Muller-Ladner U, de Matos CT, et al. Natural killer cells trigger osteoclastogenesis and bone destruction in arthritis. Proc Natl Acad Sci USA. (2010) 107:13028–33. doi: 10.1073/pnas.1000546107
111. Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. (2000) 106:1481–8. doi: 10.1172/JCI11176
112. Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. (2000) 408:600–5. doi: 10.1038/35046102
113. Kaur K, Topchyan P, Kozlowska AK, Ohanian N, Chiang J, Maung PO, et al. Super-charged NK cells inhibit growth and progression of stem-like/poorly differentiated oral tumors in vivo in humanized BLT mice; effect on tumor differentiation and response to chemotherapeutic drugs. Oncoimmunology. (2018) 7:e1426518. doi: 10.1080/2162402X.2018.1426518
Keywords: bone metastases, NK cells, cancer stem cells, dormancy, immune response
Citation: Roato I and Vitale M (2019) The Uncovered Role of Immune Cells and NK Cells in the Regulation of Bone Metastasis. Front. Endocrinol. 10:145. doi: 10.3389/fendo.2019.00145
Received: 17 December 2018; Accepted: 18 February 2019;
Published: 07 March 2019.
Edited by:
Giacomina Brunetti, Università degli Studi di Bari, ItalyReviewed by:
Jacques Zimmer, Luxembourg Institute of Health (LIH), LuxembourgHanna Taipaleenmäki, University Medical Center Hamburg-Eppendorf, Germany
Copyright © 2019 Roato and Vitale. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ilaria Roato, cm9hdG83OEBsaWJlcm8uaXQ=