
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 19 March 2019
Sec. Reproduction
Volume 10 - 2019 | https://doi.org/10.3389/fendo.2019.00136
This article is part of the Research TopicFollicle-Stimulating Hormone: Fertility and BeyondView all 22 articles
 Daria Lizneva1
Daria Lizneva1 Alina Rahimova1
Alina Rahimova1 Se-Min Kim1
Se-Min Kim1 Ihor Atabiekov1
Ihor Atabiekov1 Seher Javaid1
Seher Javaid1 Bateel Alamoush1
Bateel Alamoush1 Charit Taneja1Ayesha Khan1Li Sun1
Charit Taneja1Ayesha Khan1Li Sun1 Ricardo Azziz2
Ricardo Azziz2 Tony Yuen1Mone Zaidi1*
Tony Yuen1Mone Zaidi1*The traditional view of follicle-stimulating hormone (FSH) as a reproductive hormone is changing. It has been shown that FSH receptors (FSHRs) are expressed in various extra-gonadal tissues and mediate the biological effects of FSH at those sites. Molecular, animal, epidemiologic, and clinical data suggest that elevated serum FSH may play a significant role in the evolution of bone loss and obesity, as well as contributing to cardiovascular and cancer risk. This review summarizes recent data on FSH action beyond reproduction.
Follicle-stimulating hormone (FSH) is long thought to exert its effects in gonadal tissues, mainly limited to Sertoli cells in testes and granulosa cells in ovaries (1). Recently, using methods such as RT-PCR, Sanger sequencing, immunohistochemistry, and competitive binding assays, FSH receptors (FSHRs) have been shown to be expressed in extragonadal tissues, including endothelium, monocytes, developing placenta, endometrium, malignant tissues, bone and fat (2–10).
Our group first demonstrated that by increasing bone resorption by osteoclasts, FSH regulates bone mass in mouse models (11). Moreover, we found that FSH exerts action on adipocytes. In particular, a novel FSH antibody blocks the action of FSH on FSHRs (10, 11), causing an increase in bone mass, a reduction of body fat and induction of beiging of white adipocytes (9). These findings are consistent with large epidemiologic data. Indeed, the Study of Women's Health Across the Nation (SWAN) has shown significant reductions in bone mineral density (BMD) and high resorption rates ~2–3 years prior to menopause, which was also associated with increased body weight and visceral adiposity (12, 13). It is important to note that these changes take place when serum FSH level is increasing and estrogen level remains normal (14). Emerging epidemiologic evidence also suggests a relationship between FSH and several cardiovascular risk factors such as coronary artery calcium deposition, carotid intima-media thickness, and the number of aortic plaques (15–17). In particular, FSH interacts with its receptor on monocytes, up-regulates RANK expression and promotes monocytic infiltration of atherosclerotic plaque (18).
With this new evidence, the view that FSH acts solely as a gonadal hormone has changed rapidly over the past decade. It also provides perspectives on new roles that FSH might have in the pathophysiology of certain diseases and how treatment approaches targeting FSH may open up new possibilities for prevention and treatment. For example, we showed that a FSH blocking antibody could prevent bone loss and visceral adiposity in various mouse models (10). These data provide the foundation for future human studies. Similarly, detection of vascular endothelial FSHR in various types of solid tumors and sarcomas has prompted a debate as to whether an anti-FSH antibody could serve as a treatment modality in future anti-cancer drugs (7, 19).
Here, we review the epidemiologic, molecular and animal data on FSH action in normal physiology and the pathophysiology of osteoporosis, obesity, cardiovascular disorders, and cancer.
FSH, luteinizing hormone (LH), thyroid-stimulating hormone (TSH) and human chorionic gonadotropin (hCG) are all glycoprotein hormones, which share the same alpha subunit and differ in their beta polypeptide units, specific for each of aforementioned molecules (20). Pulsatile release of gonadotropin-releasing hormone (GnRH) from the hypothalamus stimulates the release of FSH and LH. Inhibin B and estradiol are the primary inhibitors of FSH secretion (21–23). Several other pituitary-regulatory proteins, such as activin and follistatin, have been implicated in FSH secretion and action (22). The activity of FSH is regulated in part by glycosylation.
FSH exerts its biological action via a G protein-coupled receptor, FSH receptor (FSHR). A stimulatory Gαs protein initiates signal transduction via the cAMP/protein kinase A (PKA) pathway (1, 24). This cascade of events leads to the activation of cAMP regulatory element-binding protein (CREB) (24). In addition to CREB, cAMP-activated PKA activates several other factors such as p38 MAP kinases, p70-S6 kinase and phosphoinositide-3 kinase (PI3K), PKB/Akt and FOXO1 and regulates gene expression in target tissues (25, 26). According to recent data, the effect of FSHR activation is not limited to the classical pathway, but also produces its action through Gαi (27), Gαq (28), and via other molecules, including β-arrestins (29) and an adapter protein having pleckstrin homology and phosphotyrosine binding domains together with a leucine zipper motif (30). In this case, the signal transduction is accomplished though inositol trisphosphate (IP3), Akt and ERK1/2.
FSH plays a pivotal role in the development and regulation of both the male and female reproductive systems by acting on the FSHR which is predominantly expressed in granulosa and Sertoli cells (24). In females, FSH induces follicular growth and maturation, and contributes to LH-triggered ovulation and luteinization (31–33). In males, FSH regulates the mitotic proliferation of Sertoli cells, supports their growth and maturation and prompts the release of androgen-binding protein, which regulates the overall process of spermatogenesis (34). Moreover, in testis, endothelial FSHR mediates FSH transport across gonadal endothelial barrier (35). Below, we will discuss the role of FSH on bone, fat, cardiovascular system and cancer cells.
Traditionally, bone loss in peri- and postmenopausal women has been attributed primarily to reduced estrogen production due to ovarian senescence. Estrogen replacement therapy has been considered a logical therapeutic choice in an attempt to slow postmenopausal bone loss and reduce fracture risk (36). However, FSH has been implicated in bone loss in reproductive and non-reproductive age women, as well as in women undergoing menopausal transition (37, 38).
While data from placebo-controlled randomized clinical trials is not available, the multi-center multi-ethnic cohort SWAN showed a compelling correlation between FSH action and bone loss during the menopausal transition. SWAN demonstrated that changes in bone turnover markers and bone mass density (BMD) in perimenopausal women undergoing menopausal transition were independent of serum estradiol, but were inversely related to changes in the FSH level. The levels of serum FSH over a 4-year time period predicted BMD reduction in these women (14, 39). Moreover, lower levels of bone loss in the lumbar spine during perimenopause were noted in women with higher estrogen-to-FSH ratio (40). All of these observations may suggest that bone loss during perimenopause is not solely dependent on estrogen, and may be due in part to FSH action on bone.
Epidemiological data from across the US, Europe, and China further substantiate findings from SWAN (41–46). The US NHANES III cohort study documented the relationship between serum FSH and femoral neck BMD among woman between the ages of 42 and 60 (41). Likewise, using univariate regression analyses, another US cross-sectional study confirmed the inverse relation of FSH to BMD in perimenopausal women (42). The Italian Bone Turnover Range of Normality (BONTURNO) study compared women undergoing menopausal transition, and showed significantly increased bone loss in the group with FSH>30 IU/L vs. age-matched controls, although both had regular menses (43). Yet another cross-sectional study conducted in Spain included 92 postmenopausal female participants and showed a positive correlation between serum FSH and C-terminal telopeptide of type I collagen (CTX) and serum osteocalcin, but no relation to estradiol. Several Chinese studies have reported a negative relationship between bone loss, bone turnover markers and serum FSH levels in perimenopausal women (45–47), with those in the highest quartile of serum FSH showing bone loss at a rate that was 1.3–2.3-fold higher than those in the lowest quartile (48).
The detrimental and deleterious effect of FSH on bone during a woman's reproductive years can be observed in instances of hypergonadotropic conditions. For example, lower lumbar spine bone density was reported in a hypergonadotropic amenorrheic group as compared to hypogonadotropic European patients under 40 years of age (49). Groups did not differ in estradiol or progesterone levels; however, in hypergonadotropic women, FSH levels had a negative relationship with lumbar spine BMD. Interestingly, females diagnosed with functional hypothalamic amenorrhea tend to develop less severe bone loss (50, 51).
Evidence from genetic studies further explores the function of FSHR in humans. In particular, women with an activating FSHRN680S polymorphism have an increased risk of developing postmenopausal osteoporosis, independent of circulating levels of FSH and estrogens (52). Likewise, in a multicenter study of postmenopausal Spanish women two-gene combinations of wild type IVS4 or 3′UTR markers of CYP19A1 with FSHR and BMP15 genes yielded skeletal protection (53). Therefore, epidemiologic data derived from several cross-sectional and cohort studies, together with genetic association studies, suggest a detrimental effect of FSH on bone.
In contrast, a couple of clinical studies in humans using GnRH agonists failed to demonstrate any effect of FSH suppression on bone. For instance, FSH suppression with leuprolide acetate in a group of postmenopausal women has not being associated with any significant changes in bone resorption markers (54). In another study, eugonadal men receiving goserelin acetate combined with daily topical testosterone gel did not demonstrate any changes in serum N-terminal telopeptide, C-terminal telopeptide, and osteocalcin compared to control (55). However, both studies were relatively small and the duration of the intervention was short (approximately 4 months).
In 2006, we were the first to observe the direct regulation of bone mass by FSH, which resulted mainly from osteoclastic bone resorption in rodents (11). Accumulating evidence now shows that FSH acts directly on bone via a specific shorter isoform of the FSHR (identified in humans), which then increases osteoclastogenesis and stimulates bone resorption (4, 11, 56–58). Studies failed to identify the expression of FSHRs on osteoclast lineage cells most likely used PCR primers designed to target the full-length gonadal FSHR (59, 60). FSH binding to the bone FSHR has subsequently been proven in vivo through the binding of fluorophore-tagged FSH to gonads and bone. A molar excess of unlabeled FSH displaced tagged FSH underscoring the specificity of FSH binding to bone (10, 61). The level of FSH glycosylation is important, as fully glycosylated (i.e., 24 kD) recombinant FSH isoform has a higher affinity to the bone FSHR, as compared to the partially glycosylated FSH molecule (i.e., 21 kD isoform), which is more active in gonads (62, 63).
FSH acts on FSHRs on osteoclasts, stimulating NFκB, MEK/Erk, and AKT pathways and, thus, promoting osteoclast formation, function and survival. The osteoclastic FSHR is coupled to Gαi2, so that its activation causes intracellular cAMP reductions, in contrast to the ovaries where the FSHR couples with a Gαs-protein and triggers an increase in cAMP. Blocking the aforementioned pathway or absence of Gαi2 leads to bone unresponsiveness to FSH (11). Stimulation of osteoclasts by FSH also occurs via an indirect pathway—the upregulation of receptor activator NFκB (RANK) increases the synthesis of interleukin-1β (IL-1β), interleukin-6 (IL-6) and tumor necrosis factor alpha (TNFα) proportionately to FSHR expression (64, 65). Moreover, FSH can interact with an immunoreceptor tyrosine-based activation motif (ITAM) adapter to enhance osteoclastogenesis (57).
In vivo FSH injection caused enhanced bone loss, whereas FSH inhibitor administration decreased bone resorption in ovariectomized rats (66, 67). Mice with an absent or deficient allele of FSHR or FSHβ had higher bone mass and diminished bone loss, which may be partially explained by high serum androgens (68). However, mice lacking aromatase, despite elevated androgen levels, still showed dramatic bone loss (69). Moreover, when FSH inhibitor was injected into male mice they also developed increased bone mass (9). To prevent confounding, generated by the opposite effects of FSH and estrogens on bone resorption, we developed a specific antibody to FSHβ (70, 71), which was shown to decrease osteoclastogenesis in vitro (10, 71), and decrease bone loss and stimulate bone formation in vivo (11, 70, 71). It is also known that FSH acts via the FSHR on mesenchymal stem cells to suppress their differentiation into osteoblasts (70).
There is strong correlative evidence between high FSH and body fat in postmenopausal women. A Michigan sub-study of the SWAN, which included women undergoing menopausal transition, showed a positive relationship between fat mass and serum FSH. Participants with higher FSH had increased fat mass and waist circumference, even after adjusting for baseline measurements, and lower lean and skeletal muscle mass (72). In addition, the Oklahoma Postmenopausal Health Disparities Study, which included a large group of postmenopausal women, showed that the best predictors of waist-to-hip ratio were serum FSH, estradiol and body mass index (BMI) (73). A similar positive relation between FSH to central obesity in infertile females of reproductive age has also been reported (74).
FSH has also been independently associated with lean mass in 94 postmenopausal participants after adjustment for estrogen, testosterone, LH, parathyroid hormone, sex hormone binding globulin (SHBG) and urine N-telopeptide (75). The Study of Women Entering and in Endocrine Transition (SWEET) found significantly higher lean mass in premenopausal Sub-Saharan African females, as compared to postmenopausal females, with a negative correlation between FSH and lean mass (76).
However, several groups reported an inverse relationship between FSH levels and BMI in women, particularly those in the reproductive age (77–82). This phenomenon can be explained by feedback FSH inhibition by estrogens arising from adipose tissue. For example, a study from France reported that non-obese reproductive-age females undergoing infertility workups had higher levels of gonadotropins and estradiol compared to obese women (78). Another study found an inverse relationship between FSH and BMI in reproductive age females over 326 IVF cycles (77). Overweight/obese fertile women from Italy had lower FSH, LH, estradiol and inhibin B in the early follicular phase (79). The same scenario was reported in post- and perimenopausal females. For instance, Penn Ovarian Aging Study compared abdominal MR images and hormonal levels in women at different time points and demonstrated a positive relationship between estradiol and visceral fat, but a negative one was found between FSH and visceral fat (13). Furthermore, data from the 11-year follow-up SWAN study demonstrated that obesity is associated with low FSH trajectory in women of all ethnicities (80). According to The Pan-Asia Menopause (PAM) Study, gonadotropins and estradiol had a strong positive correlation with BMI. Interestingly, estrogen and LH levels were dependent on age, whereas FSH was not (81). Another study, conducted among 73 postmenopausal Serbian women, found higher FSH in normal weight individuals than in obese females (82).
These observations are consistent with those in girls, particularly among pubertal girls who underwent bioelectric impedance measures of body fat >29%. Sorensen and Juul demonstrated that girls within this cohort had significantly lower LH and FSH levels vs. normal weight comparators (83). Likewise, Bouvattier et al. observed a negative correlation between LH, FSH and GnRH responses regarding body mass index among perimenarchial and young adult girls (84).
No significant correlation between BMI and FSH was identified in observational studies of males regardless of age (85–88), except that one cross-sectional study reported that body mass index was negatively related to FSH, inhibin B, and testosterone levels in adult men (89). However, very recent data from a randomized clinical trial suggest that high serum FSH levels cause an increase in body fat in the absence of changes in other hormones. A two-arm open-label randomized clinical study included 58 men with prostate cancer, who were randomly assigned to orchiectomy or GnRH agonist treatment for 24 weeks (90). Notably, serum FSH levels increased after orchiectomy, while GnRH agonist injections inhibited FSH secretion (91). Men treated with orchiectomy experienced greater increases in total fat mass, subcutaneous adipose tissue mass, and weight at 48 weeks as compared to men treated with GnRH agonist (90). This is the first intervention study to demonstrate that FSH regulates body fat in human.
Limited data also suggests that serum FSH may be related to metabolic syndrome. For example, one cross-sectional study, of 320 Polish women reported FSH to be a better indicator of increased risk for metabolic syndrome than SHBG levels (92). Serum FSH also appeared to be more accurate in metabolic syndrome prediction compared with leptin or C-reactive peptide in menopausal females (93).
The role of FSH in non-alcoholic fatty liver disease (NAFLD) has not been well-established. However, a few studies have reported an association between serum FSH levels and fat deposition in the liver, detected by ultrasonography (94, 95). For example, the 2014 Survey on Prevalence in East China for Metabolic Diseases conducted among women over 55 years of age have revealed that serum FSH levels were negatively associated with NAFLD (94). In an adjusted model for waist circumference and HOMA-IR, FSH levels were not associated with mild hepatic steatosis, however the association of FSH with moderate-severe hepatic steatosis remained evident (P for trend <0.01) (94). Similarly, another cross-sectional study conducted among 71 elderly (i.e., 60 years of age or older) patients from China showed that the “normal” diurnal rhythm of FSH was independently associated with NAFLD (95).
There is compelling evidence for FSHR expression in chicken, murine and human adipocytes (9, 96, 97). FSH directly stimulates primary murine adipocytes and 3T3-L1 cells through Gαi-coupled FSHR (Figure 1), resulting in the up-regulation of core fat genes, such as Fas, Lpl, and Pparg, and the induction of lipid biosynthesis (9). Moreover, FSHR activation leads to cAMP reduction and subsequently UCP1 inactivation in ThermoMouse-derived differentiated brown fat cells (9).
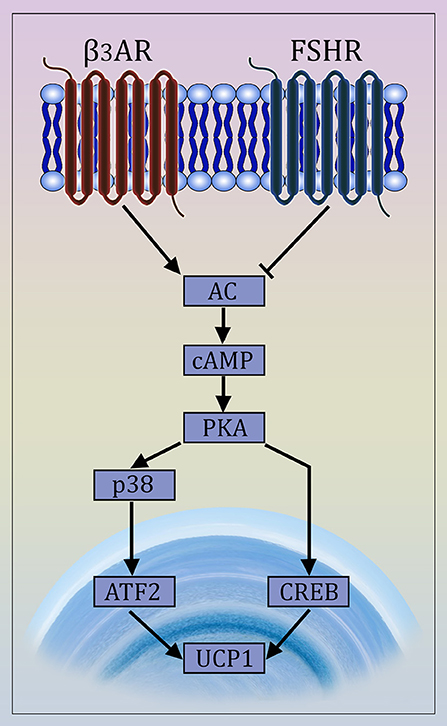
Figure 1. Mechanism of Action of FSH on Adipocytes. The newly described FSH signaling pathway opposes β3 adrenergic signaling. The latter is known to cause the transdifferentiation of white to beige adipocytes via interaction of the β3 receptor with a Gαs protein that stimulates cAMP production and activates the MAP kinase p38 and the transcription factor ATF2, which then translocates to the nucleus causing the transcriptional activation of the Ucp1 gene (98, 99). FSH opposes this action by interacting with a Gαi-coupled FSH receptor, also involving CREB-mediated pathway (9, 97).
We have fine-mapped the receptor-binding epitope of FSHβ and developed a blocking antibody capable of binding to this motif to prevent FSHβ/FSHR interaction (9, 10). Injection of this anti-FSHβ antibody in various murine models, including ovariectomized mice and mice either pair-fed on a high-fat diet or allowed ad libitum access to normal chow, caused a significant reduction in visceral, bone marrow and subcutaneous fat (9). The FSH antibody also significantly increased bone mass in ovariectomized mice (9, 70). These phenotypes were recapitulated in haploinsufficient Fshr+/− mice, indicating a dominant role of FSH in bone and body fat regulation. Interestingly, anti-FSHβ antibody failed to decrease adiposity in Fshr+/− male mice fed on a high-fat diet, proving FSH specificity (9). These observations were reproduced contemporaneously in other centers, using various laboratory methods (9, 100).
FSH blockade in mice also led to the up-regulation of brown adipocyte genes, such as Cox7, Cox8a, Ucp1, and Cidea, in visceral fat indicating “beiging,” which, we found, was occurring independently of sympathetic tone (9). However, it is not clear as to whether the beige adipocytes were the product of transdifferentiation of white adipocytes or if they were formed from a specific precursor (101, 102). Using in vivo fluorescence imaging on the IVIS platform, FSH blockade in ThermoMice triggered UCP1 transcription in brown-fat-rich areas initially with increases in white fat-predominant regions. The production of mitochondria-rich, thermogenic “beige” adipose tissue with the anti-FSHβ antibody was further substantiated using PhAM mice, and by documenting elevated basal energy expenditure in metabolic cage studies (9). Interestingly, FSH blockade did not affect glucose or insulin metabolism (92, 93).
Other studies have found a direct correlation between abdominal fat mass and FSHR mRNA expression in female chickens (96). FSH was found to alter lipid metabolism by affecting the expression of Dci, Lpl, RarB, Rdh10, Dgat2, and Acsl3 genes, shifting fatty acid and retinol metabolism, and altering PPARγ signaling (96). Interestingly, FSH has also been shown to inhibit hepatic cholesterol metabolism. FSH was found to interact with FSHRs in HepG2 cells, reducing LDLR levels (103). Moreover, FSHR knockdown with specific siRNA in mice demonstrated lower LDLR (103), suggesting that FSH may be indirectly involved in the pathogenesis of NAFLD. Finally, in a Chinese cohort, rising FSH levels correlated positively with serum cholesterol and LDL levels in postmenopausal women (104).
Males receiving androgen deprivation therapy (ADT) for prostate cancer have an increased risk of cardiovascular dysfunction, atherosclerosis and thrombosis (105–107). For example, it has been shown recently that FSH promotes the development of cardiovascular risk in ADT-treated males (18). Moreover, several studies in females have demonstrated effects of FSH on cardiovascular risk measures, such as coronary artery calcium deposition and carotid intima-media thickness. For example, the SWAN study showed that in 856 women who never reported a stroke or a heart attack, FSH trajectory was correlated positively with intima-media thickness(17). Furthermore, a sub-study of the Prospective Army Coronary Calcium project, called the Assessment of the Transition of Hormonal Evaluation with Non-invasive Imaging of Atherosclerosis, showed that serum FSH levels were associated with the number of aortic plaques in 126 women undergoing menopausal transition using contrast-enhanced CT angiography and carotid ultrasound (16). However, a 22-site population-based Survey on Prevalence in East China for Metabolic Diseases and Risk Factors, showed a negative association between FSH levels and cardiovascular risk (108). The study had a cross-sectional design and any causal relationship between FSH levels and cardiovascular risk factor factors could not be established.
Mice receiving ADT have been used to study the relation of serum FSH and cardiovascular disease (CVD) development. The interaction of FSH with FSHR on monocytes has been shown to up-regulate RANK expression and promote monocytic infiltration of atherosclerotic plaques (18). Th1 helper cells then release RANKL, which activates RANK on monocytes, leading to osteoclast formation. Osteoclasts resorb calcified areas and provoke atherosclerotic plaque instability, increasing the risk of rupture and thrombosis (18). In a second study, serum FSH levels were also found to be significantly lower in mice treated with GnRH antagonists, as compared to animals getting GnRH agonist or orchiectomized (109). The first group displayed less fat mass, at least a two-fold lower atherosclerotic plaque burden, high levels of high-density lipoproteins (HDL), and reduced serum low-density lipoproteins (LDL) compared to the latter two groups. Although all animals developed fatty changes in the aortic wall, the necrotic regions were dramatically smaller in the first group (109). This suggests that increased CVD risk in ADT cannot be explained solely by hypoandrogenemia, and may relate to changes in serum FSH. Furthermore, it has also been surmised that, as atherosclerotic plaque development is dependent on neovascularization (110), FSH may act by stimulating new vessel formation [as effectively as vascular endothelial growth factor (111, 112)] via FSHR present on vascular endothelial cells (7). The mechanism includes the stimulation of VCAM-1 synthesis by FSHR expressed on endothelial cells. VCAM-1 then recruits monocytes to affect their migration and differentiation into macrophages that accumulate lipid droplets and eventually become foam cells (17, 113). Finally, FSH may elevate production of cytokines, namely IL-6 and TNFα, from macrophages to cause low-grade inflammation, atherosclerosis development and insulin resistance (114). We have documented this direct action in osteoclasts (115).
FSH levels are elevated in ovarian cancer (116, 117). Furthermore, epithelial and endothelial FSHRs have been detected in various cancer types, including prostate (118, 119) and ovarian cancers (120–122), as well as in established cancer cell lines, namely prostate cancer cell lines DU145 (118) and PC-3 (118, 119), ovarian cancer cell lines including OVTOKO, CaOV-3, RNG1, OVCAR-3, and TOV-21G (121–126). Endothelial FSHR was detected by immunohistochemical and immunoblotting analysis in samples obtained from >1,000 patients with breast, prostate, colon, pancreas, urinary bladder, kidney, lung, liver, stomach, testis, and ovarian cancer (7). Recent data indicates that these FSHRs are signaling-efficient. In particular, endothelial FSHR expression is associated with vascular remodeling and tumor angiogenesis (6, 7), whereas epithelial FSHR induces cell proliferation (118–120), migration, and cancer cell invasion (127).
Interestingly, murine T-cells directed against FSHR- positive ovarian cancer cells showed increased survival without causing toxicity (122). FSHR stimulation upregulated Oct4 expression via the Erk1/2 pathway in epithelial ovarian cancer (128). Epithelial-to-mesenchymal transition in ovarian cancer was also stimulated through PI3K/Akt-Snail signaling (129). It has been suggested that FSH stimulates ovarian cancer cell proliferation via FSHR isoform 3, which is not coupled with G-proteins and not associated with cAMP production, but activates the Erk pathway in a Ca2+-dependent manner (130, 131).
Cancer cells express abundant receptors to various growth factors, suggesting the potential possibility of restricting cancer growth through antibody-mediated blockade of these receptors (132, 133). Unfortunately, the delivery of antibodies through the endothelium is poor and high doses are prone to cause toxicity (134, 135). To avoid this problem, a different approach, notably targeting the tumor vasculature, has been proposed. However, two major groups of extensively studied agents targeting tumor vessels have proven limitations and lack efficacy. Antiangiogenic agents reduce the action of various growth factors inside the tumor, preventing new blood vessel formation (136–139). Their maximum effect is tumor shrinkage and these agents have failed to improve survival (140, 141). The second group of agents, namely vascular disrupting agents, affect mature vessels, rearranging the endothelial cytoskeleton and increasing vascular wall permeability (142), thus disrupting blood supply and leading to extensive central necrosis of a tumor (143); this nonetheless leaves viable peripheral neoplastic tissue that subsequently repopulates the necrotic area (144–146). A new promising direction for anticancer target therapy is to cause peritumoral infarction using truncated tissue factor (tTF) coupled to ligands that are highly specific for FSHR (147). Antihuman FSHR antibody, conjugated with tTF, binds the FSHR, which is abundant in peritumoral endothelium, initiating blood clotting with subsequent blood supply disruption and tumor necrosis (148). Interestingly, the vasculature of bone and fat has not been shown to express FSHRs: thus, such therapy will most likely cause no issues (8, 149). However, their presence in the female reproductive system may limit anti-FSHR-tTF treatment. This approach still needs extensive investigation in the future and provokes extensive discussion on the development of a cancer therapies based on agents tethered to anti-FSHR antibodies (19).
In the transitional phase of a women's reproductive life to menopause, the risk for osteoporosis, obesity and CVDs increase concurrently. Along with declining estrogen levels, sharply rising FSH levels have now been implicated in the pathogenesis of these diseases. It is now well-known that bone loss begins even before estrogen levels are altered in the perimenopause (150).
Several key findings have emerged relating serum FSH to bone loss, obesity, and perhaps even cardiovascular risk and cancer. First, it is clear that FSH directly impacts bone cells—osteoclasts and osteoblast precursors. The underlying mechanisms include a direct action on osteoclasts through the enhancement of RANKL signaling, and indirect actions to increase the expression of RANK in osteoclasts (151) and stimulate the synthesis of pro-resorptive cytokines, including TNFα, IL1β, and IL-6. Studies also conclusively demonstrate the expression of functional FSHRs on adipose tissue (97), which, when blocked by an FSH antibody, result in a profound reduction of body fat and generation of thermogenic “beige” adipose tissue (152). Together, the studies form the framework for using a humanized FSH antibody for the simultaneous treatment of two public health hazards—obesity and osteoporosis—with a single agent. Admittedly speculative at this stage, an increased risk of cardiovascular event(s) among postmenopausal women may also be in part attributable to subclinical atherosclerosis promoted by sharply rising FSH levels (153). Finally, certain cancers, prominently ovarian tumors in which oncogenic signaling through the FSHR can be proven may be amenable to novel FSH-based therapeutic agents.
Thus, positive correlations between rising FSH levels and a plethora of illnesses like obesity, osteoporosis, cardiovascular pathology, and cancer changes our view of FSH from monogamously associated with fertility to a much broader view of the role of this “gonadotropin” in other medical conditions and in human physiology. It is therefore now conceivable that we question whether FSH is a true aging hormone. By developing new treatment approaches that target this gonadotropin, we may in the future be able to treat multiple age-related diseases perhaps even with a single drug.
DL wrote the manuscript with support from S-MK, AR, IA, SJ, BA, CT, AK, and LS. TY, MZ, and RA helped supervise the project. All authors provided critical feedback and helped shape the manuscript.
Work at Icahn School of Medicine at Mount Sinai was supported by the National Institutes of Health (NIH) by R01 Grants DK113627 to (MZ and LS), AG40132 (to MZ), AR65932 (to MZ), AR67066 (to MZ).
MZ is a named inventor on a patent related to FSH and bone, owned by Icahn School of Medicine at Mount Sinai. MZ will receive royalties and/or licensing fees per Mount Sinai policies, in the event the patent is commercialized. MZ also consults for Merck, Roche, and a number of financial consulting platforms.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors acknowledge Mount Sinai Innovation Partner for their collaboration on the actions of FSH on bone.
1. Ulloa-Aguirre A, Reiter E, Crepieux P. FSH receptor signaling: complexity of interactions and signal diversity. Endocrinology. (2018) 159:3020–35. doi: 10.1210/en.2018-00452
2. Stilley JA, Christensen DE, Dahlem KB, Guan R, Santillan DA, England SK, et al. FSH receptor (FSHR) expression in human extragonadal reproductive tissues and the developing placenta, and the impact of its deletion on pregnancy in mice. Biol Reprod. (2014) 91:74. doi: 10.1095/biolreprod.114.118562
3. Ponikwicka-Tyszko D, Chrusciel M, Stelmaszewska J, Bernaczyk P, Sztachelska M, Sidorkiewicz I, et al. Functional expression of FSH receptor in endometriotic lesions. J Clin Endocrinol Metab. (2016) 101:2905–14. doi: 10.1210/jc.2016-1014
4. Robinson LJ, Tourkova I, Wang Y, Sharrow AC, Landau MS, Yaroslavskiy BB, et al. FSH-receptor isoforms and FSH-dependent gene transcription in human monocytes and osteoclasts. Biochem Biophys Res Commun. (2010) 394:12–7. doi: 10.1016/j.bbrc.2010.02.112
5. Cannon JG, Kraj B, Sloan G. Follicle-stimulating hormone promotes RANK expression on human monocytes. Cytokine. (2011) 53:141–4. doi: 10.1016/j.cyto.2010.11.011
6. Planeix F, Siraj MA, Bidard FC, Robin B, Pichon C, Sastre-Garau X, et al. Endothelial follicle-stimulating hormone receptor expression in invasive breast cancer and vascular remodeling at tumor periphery. J Exp Clin Cancer Res. (2015) 34:12. doi: 10.1186/s13046-015-0128-7
7. Radu A, Pichon C, Camparo P, Antoine M, Allory Y, Couvelard A, et al. Expression of follicle-stimulating hormone receptor in tumor blood vessels. N Engl J Med. (2010) 363:1621–30. doi: 10.1056/NEJMoa1001283
8. Siraj A, Desestret V, Antoine M, Fromont G, Huerre M, Sanson M, et al. Expression of follicle-stimulating hormone receptor by the vascular endothelium in tumor metastases. BMC Cancer. (2013) 13:246. doi: 10.1186/1471-2407-13-246
9. Liu P, Ji Y, Yuen T, Rendina-Ruedy E, DeMambro VE, Dhawan S, et al. Blocking FSH induces thermogenic adipose tissue and reduces body fat. Nature. (2017) 546:107–12. doi: 10.1038/nature22342
10. Ji Y, Liu P, Yuen T, Haider S, He J, Romero R, et al. Epitope-specific monoclonal antibodies to FSHbeta increase bone mass. Proc Natl Acad Sci USA. (2018) 115:2192–7. doi: 10.1073/pnas.1718144115
11. Sun L, Peng Y, Sharrow AC, Iqbal J, Zhang Z, Papachristou DJ, et al. FSH directly regulates bone mass. Cell. (2006) 125:247–60. doi: 10.1016/j.cell.2006.01.051
12. Thurston RC, Sowers MR, Sternfeld B, Gold EB, Bromberger J, Chang Y, et al. Gains in body fat and vasomotor symptom reporting over the menopausal transition: the study of women's health across the nation. Am J Epidemiol. (2009) 170:766–74. doi: 10.1093/aje/kwp203
13. Senapati S, Gracia CR, Freeman EW, Sammel MD, Lin H, Kim C, et al. Hormone variations associated with quantitative fat measures in the menopausal transition. Climacteric. (2014) 17:183–90. doi: 10.3109/13697137.2013.845876
14. Randolph JF Jr, Sowers M, Gold EB, Mohr BA, Luborsky J, Santoro N, et al. Reproductive hormones in the early menopausal transition: relationship to ethnicity, body size, and menopausal status. J Clin Endocrinol Metab. (2003) 88:1516–22. doi: 10.1210/jc.2002-020777
15. Celestino Catao Da Silva D, Nogueira De Almeida Vasconcelos A, Cleto Maria Cerqueira J, De Oliveira Cipriano Torres D, Oliveira Dos Santos AC, De Lima Ferreira Fernandes Costa H, et al. Endogenous sex hormones are not associated with subclinical atherosclerosis in menopausal women. Minerva Ginecol. (2013) 65:297–302.
16. Munir JA, Wu H, Bauer K, Bindeman J, Byrd C, Feuerstein IM, et al. The perimenopausal atherosclerosis transition: relationships between calcified and noncalcified coronary, aortic, and carotid atherosclerosis and risk factors and hormone levels. Menopause. (2012) 19:10–5. doi: 10.1097/gme.0b013e318221bc8d
17. El Khoudary SR, Santoro N, Chen HY, Tepper PG, Brooks MM, Thurston RC, et al. Trajectories of estradiol and follicle-stimulating hormone over the menopause transition and early markers of atherosclerosis after menopause. Eur J Prev Cardiol. (2016) 23:694–703. doi: 10.1177/2047487315607044
18. Crawford ED, Schally AV, Pinthus JH, Block NL, Rick FG, Garnick MB, et al. The potential role of follicle-stimulating hormone in the cardiovascular, metabolic, skeletal, and cognitive effects associated with androgen deprivation therapy. Urol Oncol. (2017) 35:183–91. doi: 10.1016/j.urolonc.2017.01.025
19. Ghinea N. Vascular Endothelial FSH Receptor, a Target of Interest for Cancer Therapy. Endocrinology. (2018) 159:3268–74. doi: 10.1210/en.2018-00466
20. Fox KM, Dias JA, Van Roey P. Three-dimensional structure of human follicle-stimulating hormone. Mol Endocrinol. (2001) 15:378–89. doi: 10.1210/mend.15.3.0603
21. Demyashkin GA. Inhibin B in seminiferous tubules of human testes in normal spermatogenesis and in idiopathic infertility. Syst Biol Reprod Med. (2018) 65:1–9. doi: 10.1080/19396368.2018.1478470
22. Ying SY. Inhibins, activins, and follistatins: gonadal proteins modulating the secretion of follicle-stimulating hormone. Endocr Rev. (1988) 9:267–93. doi: 10.1210/edrv-9-2-267
23. Christensen A, Bentley GE, Cabrera R, Ortega HH, Perfito N, Wu TJ, et al. Hormonal regulation of female reproduction. Horm Metab Res. (2012) 44:587–91. doi: 10.1055/s-0032-1306301
24. Simoni M, Gromoll J, Nieschlag E. The follicle-stimulating hormone receptor: biochemistry, molecular biology, physiology, and pathophysiology. Endocr Rev. (1997) 18:739–73. doi: 10.1210/edrv.18.6.0320
25. Bruser A, Schulz A, Rothemund S, Ricken A, Calebiro D, Kleinau G, et al. The Activation Mechanism of Glycoprotein Hormone Receptors with Implications in the Cause and Therapy of Endocrine Diseases. J Biol Chem. (2016) 291:508–20. doi: 10.1074/jbc.M115.701102
26. Herndon MK, Law NC, Donaubauer EM, Kyriss B, Hunzicker-Dunn M. Forkhead box O member FOXO1 regulates the majority of follicle-stimulating hormone responsive genes in ovarian granulosa cells. Mol Cell Endocrinol. (2016) 434:116–26. doi: 10.1016/j.mce.2016.06.020
27. Crepieux P, Marion S, Martinat N, Fafeur V, Vern YL, Kerboeuf D, et al. The ERK-dependent signalling is stage-specifically modulated by FSH, during primary Sertoli cell maturation. Oncogene. (2001) 20:4696–709. doi: 10.1038/sj.onc.1204632
28. Escamilla-Hernandez R, Little-Ihrig L, Zeleznik AJ. Inhibition of rat granulosa cell differentiation by overexpression of Galphaq. Endocrine. (2008) 33:21–31. doi: 10.1007/s12020-008-9064-z
29. Tranchant T, Durand G, Gauthier C, Crepieux P, Ulloa-Aguirre A, Royere D, et al. Preferential beta-arrestin signalling at low receptor density revealed by functional characterization of the human FSH receptor A189 V mutation. Mol Cell Endocrinol. (2011) 331:109–18. doi: 10.1016/j.mce.2010.08.016
30. Nechamen CA, Thomas RM, Cohen BD, Acevedo G, Poulikakos PI, Testa JR, et al. Human follicle-stimulating hormone (FSH) receptor interacts with the adaptor protein APPL1 in HEK 293 cells: potential involvement of the PI3K pathway in FSH signaling. Biol Reprod. (2004) 71:629–36. doi: 10.1095/biolreprod.103.025833
31. McGee EA, Hsueh AJ. Initial and cyclic recruitment of ovarian follicles. Endocr Rev. (2000) 21:200–14. doi: 10.1210/edrv.21.2.0394
32. Howles CM. Role of LH and FSH in ovarian function. Mol Cell Endocrinol. (2000) 161:25–30. doi: 10.1016/S0303-7207(99)00219-1
33. Chan WK, Tan CH. Induction of aromatase activity in porcine granulosa cells by FSH and cyclic AMP. Endocr Res. (1987) 13:285–99.
34. Simoni M, Weinbauer GF, Gromoll J, Nieschlag E. Role of FSH in male gonadal function. Ann Endocrinol. (1999) 60:102–6.
35. Vu Hai MT, Lescop P, Loosfelt H, Ghinea N. Receptor-mediated transcytosis of follicle-stimulating hormone through the rat testicular microvasculature. Biol Cell. (2004) 96:133–44. doi: 10.1016/j.biolcel.2003.11.008
36. Lindsay R. Hormones and bone health in postmenopausal women. Endocrine. (2004) 24:223–30. doi: 10.1385/ENDO:24:3:223
37. Colaianni G, Cuscito C, Colucci S. FSH and TSH in the regulation of bone mass: the pituitary/immune/bone axis. Clin Dev Immunol. (2013) 2013:382698. doi: 10.1155/2013/382698
38. Davis SR, Lambrinoudaki I, Lumsden M, Mishra GD, Pal L, Rees M, et al. Menopause. Nat Rev Dis Primers. (2015) 1:15004. doi: 10.1038/nrdp.2015.4
39. Sowers MR, Greendale GA, Bondarenko I, Finkelstein JS, Cauley JA, Neer RM, et al. Endogenous hormones and bone turnover markers in pre- and perimenopausal women: SWAN. Osteoporos Int. (2003) 14:191–7. doi: 10.1007/s00198-002-1329-4
40. Crandall CJ, Tseng CH, Karlamangla AS, Finkelstein JS, Randolph JF Jr, Thurston RC, et al. Serum sex steroid levels and longitudinal changes in bone density in relation to the final menstrual period. J Clin Endocrinol Metab. (2013) 98:E654–63. doi: 10.1210/jc.2012-3651
41. Gallagher CM, Moonga BS, Kovach JS. Cadmium, follicle-stimulating hormone, and effects on bone in women age 42-60 years, NHANES III. Environ Res. (2010) 110:105–11. doi: 10.1016/j.envres.2009.09.012
42. Cannon JG, Cortez-Cooper M, Meaders E, Stallings J, Haddow S, Kraj B, et al. Follicle-stimulating hormone, interleukin-1, and bone density in adult women. Am J Physiol Regul Integr Comp Physiol. (2010) 298:R790–8. doi: 10.1152/ajpregu.00728.2009
43. Adami S, Bianchi G, Brandi ML, Giannini S, Ortolani S, DiMunno O, et al. Determinants of bone turnover markers in healthy premenopausal women. Calcif Tissue Int. (2008) 82:341–7. doi: 10.1007/s00223-008-9126-5
44. Garcia-Martin A, Reyes-Garcia R, Garcia-Castro JM, Rozas-Moreno P, Escobar-Jimenez F, Munoz-Torres M. Role of serum FSH measurement on bone resorption in postmenopausal women. Endocrine. (2012) 41:302–8. doi: 10.1007/s12020-011-9541-7
45. Xu ZR, Wang AH, Wu XP, Zhang H, Sheng ZF, Wu XY, et al. Relationship of age-related concentrations of serum FSH and LH with bone mineral density, prevalence of osteoporosis in native Chinese women. Clin Chim Acta. (2009) 400:8–13. doi: 10.1016/j.cca.2008.09.027
46. Wu XY, Wu XP, Xie H, Zhang H, Peng YQ, Yuan LQ, et al. Age-related changes in biochemical markers of bone turnover and gonadotropin levels and their relationship among Chinese adult women. Osteoporos Int. (2010) 21:275–85. doi: 10.1007/s00198-009-0943-9
47. Wang B, Song Y, Chen Y, Wang ES, Zheng D, Qu F, et al. Correlation analysis for follicle-stimulating hormone and C-terminal cross-linked telopetides of type i collagen in menopausal transition women with osteoporosis. Int J Clin Exp Med. (2015) 8:2417–22.
48. Cheung E, Tsang S, Bow C, Soong C, Yeung S, Loong C, et al. Bone loss during menopausal transition among southern Chinese women. Maturitas. (2011) 69:50–6. doi: 10.1016/j.maturitas.2011.01.010
49. Devleta B, Adem B, Senada S. Hypergonadotropic amenorrhea and bone density: new approach to an old problem. J Bone Miner Metab. (2004) 22:360–4. doi: 10.1007/s00774-004-0495-1
50. Podfigurna-Stopa A, Pludowski P, Jaworski M, Lorenc R, Genazzani AR, Meczekalski B. Skeletal status and body composition in young women with functional hypothalamic amenorrhea. Gynecol Endocrinol. (2012) 28:299–304. doi: 10.3109/09513590.2011.613972
51. Ozbek MN, Demirbilek H, Baran RT, Baran A. Bone Mineral Density in Adolescent Girls with Hypogonadotropic and Hypergonadotropic Hypogonadism. J Clin Res Pediatr Endocrinol. (2016) 8:163–9. doi: 10.4274/jcrpe.2228
52. Rendina D, Gianfrancesco F, De Filippo G, Merlotti D, Esposito T, Mingione A, et al. FSHR gene polymorphisms influence bone mineral density and bone turnover in postmenopausal women. Eur J Endocrinol. (2010) 163:165–72. doi: 10.1530/EJE-10-0043
53. Mendoza N, Quereda F, Presa J, Salamanca A, Sanchez-Borrego R, Vazquez F, et al. Estrogen-related genes and postmenopausal osteoporosis risk. Climacteric. (2012) 15:587–93. doi: 10.3109/13697137.2012.656160
54. Drake MT, McCready LK, Hoey KA, Atkinson EJ, Khosla S. Effects of suppression of follicle-stimulating hormone secretion on bone resorption markers in postmenopausal women. J Clin Endocrinol Metab. (2010) 95:5063–8. doi: 10.1210/jc.2010-1103
55. Uihlein AV, Finkelstein JS, Lee H, Leder BZ. FSH suppression does not affect bone turnover in eugonadal men. J Clin Endocrinol Metab. (2014) 99:2510–5. doi: 10.1210/jc.2013-3246
56. Sun L, Zhang Z, Zhu LL, Peng Y, Liu X, Li J, et al. Further evidence for direct pro-resorptive actions of FSH. Biochem Biophys Res Commun. (2010) 394:6–11. doi: 10.1016/j.bbrc.2010.02.113
57. Wu Y, Torchia J, Yao W, Lane NE, Lanier LL, Nakamura MC, et al. Bone microenvironment specific roles of ITAM adapter signaling during bone remodeling induced by acute estrogen-deficiency. PLoS ONE. (2007) 2:e586. doi: 10.1371/journal.pone.0000586
58. Wang J, Zhang W, Yu C, Zhang X, Zhang H, Guan Q, et al. Follicle-Stimulating Hormone Increases the Risk of Postmenopausal Osteoporosis by Stimulating Osteoclast Differentiation. PLoS ONE. (2015) 10:e0134986. doi: 10.1371/journal.pone.0134986
59. Ritter V, Thuering B, Saint Mezard P, Luong-Nguyen NH, Seltenmeyer Y, Junker U, et al. Follicle-stimulating hormone does not impact male bone mass in vivo or human male osteoclasts in vitro. Calcif Tissue Int. (2008) 82:383–91. doi: 10.1007/s00223-008-9134-5
60. Allan CM, Kalak R, Dunstan CR, McTavish KJ, Zhou H, Handelsman DJ, et al. Follicle-stimulating hormone increases bone mass in female mice. Proc Natl Acad Sci USA. (2010) 107:22629–34. doi: 10.1073/pnas.1012141108
61. Feng Y, Zhu S, Antaris AL, Chen H, Xiao Y, Lu X, et al. Live imaging of follicle stimulating hormone receptors in gonads and bones using near infrared II fluorophore. Chem Sci. (2017) 8:3703–11. doi: 10.1039/c6sc04897h
62. Meher BR, Dixit A, Bousfield GR, Lushington GH. Glycosylation Effects on FSH-FSHR Interaction Dynamics: a case study of different fsh glycoforms by molecular dynamics simulations. PLoS ONE. (2015) 10:e0137897. doi: 10.1371/journal.pone.0137897
63. Jiang C, Hou X, Wang C, May JV, Butnev VY, Bousfield GR, et al. Hypoglycosylated hFSH has greater bioactivity than fully glycosylated recombinant hfsh in human granulosa cells. J Clin Endocrinol Metab. (2015) 100:E852–60. doi: 10.1210/jc.2015-1317
64. Zaidi S, Zhu LL, Mali R, Iqbal J, Yang G, Zaidi M, et al. Regulation of FSH receptor promoter activation in the osteoclast. Biochem Biophys Res Commun. (2007) 361:910–5. doi: 10.1016/j.bbrc.2007.07.081
65. Blair HC, Zaidi M. Osteoclastic differentiation and function regulated by old and new pathways. Rev Endocr Metab Disord. (2006) 7:23–32. doi: 10.1007/s11154-006-9010-4
66. Liu S, Cheng Y, Fan M, Chen D, Bian Z. FSH aggravates periodontitis-related bone loss in ovariectomized rats. J Dent Res. (2010) 89:366–71. doi: 10.1177/0022034509358822
67. Liu S, Cheng Y, Xu W, Bian Z. Protective effects of follicle-stimulating hormone inhibitor on alveolar bone loss resulting from experimental periapical lesions in ovariectomized rats. J Endod. (2010) 36:658–63. doi: 10.1016/j.joen.2010.01.011
68. Zaidi M, Sun L, Kumar TR, Sairam MR, Blair HC. Response: both FSH and sex steroids influence bone mass. Cell. (2006) 127:1080–1. doi: 10.1016/j.cell.2006.12.003
69. Oz OK, Hirasawa G, Lawson J, Nanu L, Constantinescu A, Antich PP, et al. Bone phenotype of the aromatase deficient mouse. J Steroid Biochem Mol Biol. (2001) 79:49–59. doi: 10.1016/S0960-0760(01)00130-3
70. Zhu LL, Blair H, Cao J, Yuen T, Latif R, Guo L, et al. Blocking antibody to the beta-subunit of FSH prevents bone loss by inhibiting bone resorption and stimulating bone synthesis. Proc Natl Acad Sci USA. (2012) 109:14574–9. doi: 10.1073/pnas.1212806109
71. Zhu LL, Tourkova I, Yuen T, Robinson LJ, Bian Z, Zaidi M, et al. Blocking FSH action attenuates osteoclastogenesis. Biochem Biophys Res Commun. (2012) 422:54–8. doi: 10.1016/j.bbrc.2012.04.104
72. Sowers M, Zheng H, Tomey K, Karvonen-Gutierrez C, Jannausch M, Li X, et al. Changes in body composition in women over six years at midlife: ovarian and chronological aging. J Clin Endocrinol Metab. (2007) 92:895–901. doi: 10.1210/jc.2006-1393
73. Gavaler JS, Rosenblum E. Predictors of postmenopausal body mass index and waist hip ratio in the oklahoma postmenopausal health disparities study. J Am Coll Nutr. (2003) 22:269–76. doi: 10.1080/07315724.2003.10719303
74. Seth B, Arora S, Singh R. Association of obesity with hormonal imbalance in infertility: a cross-sectional study in north Indian women. Indian J Clin Biochem. (2013) 28:342–7. doi: 10.1007/s12291-013-0301-8
75. Gourlay ML, Specker BL, Li C, Hammett-Stabler CA, Renner JB, Rubin JE. Follicle-stimulating hormone is independently associated with lean mass but not BMD in younger postmenopausal women. Bone. (2012) 50:311–6. doi: 10.1016/j.bone.2011.11.001
76. Jaff NG, Norris SA, Snyman T, Toman M, Crowther NJ. Body composition in the Study of Women Entering and in Endocrine Transition (SWEET): A perspective of African women who have a high prevalence of obesity and HIV infection. Metabolism. (2015) 64:1031–41. doi: 10.1016/j.metabol.2015.05.009
77. Ecochard R, Marret H, Barbato M, Boehringer H. Gonadotropin and body mass index: high FSH levels in lean, normally cycling women. Obstet Gynecol. (2000) 96:8–12. doi: 10.1016/S0029-7844(00)00842-5
78. Caillon H, Freour T, Bach-Ngohou K, Colombel A, Denis MG, Barriere P, et al. Effects of female increased body mass index on in vitro fertilization cycles outcome. Obes Res Clin Pract. (2015) 9:382–8. doi: 10.1016/j.orcp.2015.02.009
79. De Pergola G, Maldera S, Tartagni M, Pannacciulli N, Loverro G, Giorgino R. Inhibitory effect of obesity on gonadotropin, estradiol, and inhibin B levels in fertile women. Obesity. (2006) 14:1954–60. doi: 10.1038/oby.2006.228
80. Tepper PG, Randolph JF Jr, McConnell DS, Crawford SL, El Khoudary SR, Joffe H, et al. Trajectory clustering of estradiol and follicle-stimulating hormone during the menopausal transition among women in the Study of Women's Health across the Nation (SWAN). J Clin Endocrinol Metab. (2012) 97:2872–80. doi: 10.1210/jc.2012-1422
81. Ausmanas MK, Tan DA, Jaisamrarn U, Tian XW, Holinka CF. Estradiol, FSH and LH profiles in nine ethnic groups of postmenopausal Asian women: the Pan-Asia Menopause (PAM) study. Climacteric. (2007) 10:427–37. doi: 10.1080/13697130701610780
82. Simoncig Netjasov A, Tancic-Gajic M, Ivovic M, Marina L, Arizanovic Z, Vujovic S. Influence of obesity and hormone disturbances on sexuality of women in the menopause. Gynecol Endocrinol. (2016) 32:762–6. doi: 10.3109/09513590.2016.1161746
83. Sorensen K, Juul A. BMI percentile-for-age overestimates adiposity in early compared with late maturing pubertal children. Eur J Endocrinol. (2015) 173:227–35. doi: 10.1530/EJE-15-0239
84. Bouvattier C, Lahlou N, Roger M, Bougneres P. Hyperleptinaemia is associated with impaired gonadotrophin response to GnRH during late puberty in obese girls, not boys. Eur J Endocrinol. (1998) 138:653–8.
85. Bieniek JM, Kashanian JA, Deibert CM, Grober ED, Lo KC, Brannigan RE, et al. Influence of increasing body mass index on semen and reproductive hormonal parameters in a multi-institutional cohort of subfertile men. Fertil Steril. (2016) 106:1070–5. doi: 10.1016/j.fertnstert.2016.06.041
86. Foresta C, Di Mambro A, Pagano C, Garolla A, Vettor R, Ferlin A. Insulin-like factor 3 as a marker of testicular function in obese men. Clin Endocrinol. (2009) 71:722–6. doi: 10.1111/j.1365-2265.2009.03549.x
87. Casimirri F, Pasquali R, Cantobelli S, Melchionda N, Barbara L. [Obesity and adipose tissue distribution in men: relation to sex steroids and insulin]. Minerva Endocrinol. (1991) 16:31–5.
88. Yamacake KG, Cocuzza M, Torricelli FC, Tiseo BC, Frati R, Freire GC, et al. Impact of body mass index, age and varicocele on reproductive hormone profile from elderly men. Int Braz J Urol. (2016) 42:365–72. doi: 10.1590/S1677-5538.IBJU.2014.0594
89. Pauli EM, Legro RS, Demers LM, Kunselman AR, Dodson WC, Lee PA. Diminished paternity and gonadal function with increasing obesity in men. Fertil Steril. (2008) 90:346–51. doi: 10.1016/j.fertnstert.2007.06.046
90. Ostergren PB, Kistorp C, Fode M, Bennedbaek FN, Faber J, Sonksen J. Metabolic consequences of gonadotropin-releasing hormone agonists vs. orchiectomy: a randomized clinical study. BJU Int. (2018) doi: 10.1111/bju.14609. [Epub ahead of print].
91. Ostergren PB, Kistorp C, Fode M, Henderson J, Bennedbaek FN, Faber J, et al. Luteinizing Hormone-Releasing Hormone Agonists are Superior to Subcapsular Orchiectomy in Lowering Testosterone Levels of Men with Prostate Cancer: Results from a Randomized Clinical Trial. J Urol. (2017) 197:1441–7. doi: 10.1016/j.juro.2016.12.003
92. Stefanska A, Sypniewska G, Ponikowska I, Cwiklinska-Jurkowska M. Association of follicle-stimulating hormone and sex hormone binding globulin with the metabolic syndrome in postmenopausal women. Clin Biochem. (2012) 45:703–6. doi: 10.1016/j.clinbiochem.2012.03.011
93. Stefanska A, Ponikowska I, Cwiklinska-Jurkowska M, Sypniewska G. Association of FSH with metabolic syndrome in postmenopausal women: a comparison with CRP, adiponectin and leptin. Biomark Med. (2014) 8:921–30. doi: 10.2217/bmm.14.49
94. Wang N, Li Q, Han B, Chen Y, Zhu C, Chen Y, et al. Follicle-stimulating hormone is associated with non-alcoholic fatty liver disease in Chinese women over 55 years old. J Gastroenterol Hepatol. (2016) 31:1196–202. doi: 10.1111/jgh.13271
95. Li X, Jing L, Lin F, Huang H, Chen Z, Chen Y, et al. Diurnal rhythm of follicle-stimulating hormone is associated with nonalcoholic fatty liver disease in a Chinese elderly population. Eur J Obstet Gynecol Reprod Biol. (2018) 222:166–70. doi: 10.1016/j.ejogrb.2018.01.034
96. Cui H, Zhao G, Liu R, Zheng M, Chen J, Wen J. FSH stimulates lipid biosynthesis in chicken adipose tissue by upregulating the expression of its receptor FSHR. J Lipid Res. (2012) 53:909–17. doi: 10.1194/jlr.M025403
97. Liu XM, Chan HC, Ding GL, Cai J, Song Y, Wang TT, et al. FSH regulates fat accumulation and redistribution in aging through the Galphai/Ca(2+)/CREB pathway. Aging Cell. (2015) 14:409–20. doi: 10.1111/acel.12331
98. Cohen P, Spiegelman BM. Brown and beige fat: molecular parts of a thermogenic machine. Diabetes. (2015) 64:2346–51. doi: 10.2337/db15-0318
99. Wu J, Bostrom P, Sparks LM, Ye L, Choi JH, Giang AH, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. (2012) 150:366–76. doi: 10.1016/j.cell.2012.05.016
100. Rosen CJ, Zaidi M. Contemporaneous reproduction of preclinical science: a case study of FSH and fat. Ann N Y Acad Sci. (2017) 1404:17–9. doi: 10.1111/nyas.13457
101. Rosenwald M, Perdikari A, Rulicke T, Wolfrum C. Bi-directional interconversion of brite and white adipocytes. Nat Cell Biol. (2013) 15:659–67. doi: 10.1038/ncb2740
102. Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med. (2013) 19:1338–44. doi: 10.1038/nm.3324
103. Song Y, Wang ES, Xing LL, Shi S, Qu F, Zhang D, et al. Follicle-Stimulating Hormone Induces postmenopausal dyslipidemia through inhibiting hepatic cholesterol metabolism. J Clin Endocrinol Metab. (2016) 101:254–63. doi: 10.1210/jc.2015-2724
104. Ma L, Song Y, Li C, Wang E, Zheng D, Qu F, et al. Bone turnover alterations across the menopausal transition in south-eastern Chinese women [corrected]. Climacteric. (2016) 19:400–5. doi: 10.1080/13697137.2016.1180677
105. Bosco C, Bosnyak Z, Malmberg A, Adolfsson J, Keating NL, Van Hemelrijck M. Quantifying observational evidence for risk of fatal and nonfatal cardiovascular disease following androgen deprivation therapy for prostate cancer: a meta-analysis. Eur Urol. (2015) 68:386–96. doi: 10.1016/j.eururo.2014.11.039
106. Tsai HK, D'Amico AV, Sadetsky N, Chen MH, Carroll PR. Androgen deprivation therapy for localized prostate cancer and the risk of cardiovascular mortality. J Natl Cancer Inst. (2007) 99:1516–24. doi: 10.1093/jnci/djm168
107. Zhao J, Zhu S, Sun L, Meng F, Zhao L, Zhao Y, et al. Androgen deprivation therapy for prostate cancer is associated with cardiovascular morbidity and mortality: a meta-analysis of population-based observational studies. PLoS ONE. (2014) 9:e107516. doi: 10.1371/journal.pone.0107516
108. Wang N, Shao H, Chen Y, Xia F, Chi C, Li Q, et al. Follicle-Stimulating Hormone, Its association with cardiometabolic risk factors, and 10-year risk of cardiovascular disease in postmenopausal women. J Am Heart Assoc. (2017) 6:05918. doi: 10.1161/JAHA.117.005918
109. Hopmans SN, Duivenvoorden WC, Werstuck GH, Klotz L, Pinthus JH. GnRH antagonist associates with less adiposity and reduced characteristics of metabolic syndrome and atherosclerosis compared with orchiectomy and GnRH agonist in a preclinical mouse model. Urol Oncol. (2014) 32:1126–34. doi: 10.1016/j.urolonc.2014.06.018
110. Moulton KS, Heller E, Konerding MA, Flynn E, Palinski W, Folkman J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation. (1999) 99:1726–32.
111. Albertsen PC, Klotz L, Tombal B, Grady J, Olesen TK, Nilsson J. Cardiovascular morbidity associated with gonadotropin releasing hormone agonists and an antagonist. Eur Urol. (2014) 65:565–73. doi: 10.1016/j.eururo.2013.10.032
112. Stilley JA, Guan R, Duffy DM, Segaloff DL. Signaling through FSH receptors on human umbilical vein endothelial cells promotes angiogenesis. J Clin Endocrinol Metab. (2014) 99:E813–20. doi: 10.1210/jc.2013-3186
113. El Khoudary SR, Wildman RP, Matthews K, Thurston RC, Bromberger JT, Sutton-Tyrrell K. Endogenous sex hormones impact the progression of subclinical atherosclerosis in women during the menopausal transition. Atherosclerosis. (2012) 225:180–6. doi: 10.1016/j.atherosclerosis.2012.07.025
114. Choi SH, Hong ES, Lim S. Clinical implications of adipocytokines and newly emerging metabolic factors with relation to insulin resistance and cardiovascular health. Front Endocrinol. (2013) 4:97. doi: 10.3389/fendo.2013.00097
115. Iqbal J, Sun L, Kumar TR, Blair HC, Zaidi M. Follicle-stimulating hormone stimulates TNF production from immune cells to enhance osteoblast and osteoclast formation. Proc Natl Acad Sci USA. (2006) 103:14925–30. doi: 10.1073/pnas.0606805103
116. Mertens-Walker I, Baxter RC, Marsh DJ. Gonadotropin signalling in epithelial ovarian cancer. Cancer Lett. (2012) 324:152–9. doi: 10.1016/j.canlet.2012.05.017
117. Rzepka-Gorska I, Chudecka-Glaz A, Kosmowska B. FSH and LH serum/tumor fluid ratios and malignant tumors of the ovary. Endocr Relat Cancer. (2004) 11:315–21. doi: 10.1677/erc.0.0110315
118. Ben-Josef E, Yang SY, Ji TH, Bidart JM, Garde SV, Chopra DP, et al. Hormone-refractory prostate cancer cells express functional follicle-stimulating hormone receptor (FSHR). J Urol. (1999) 161:970–6.
119. Mariani S, Salvatori L, Basciani S, Arizzi M, Franco G, Petrangeli E, et al. Expression and cellular localization of follicle-stimulating hormone receptor in normal human prostate, benign prostatic hyperplasia and prostate cancer. J Urol. (2006) 175:2072–7; discussion: 7. doi: 10.1016/S0022-5347(06)00273-4
120. Zheng W, Lu JJ, Luo F, Zheng Y, Feng Y, Felix JC, et al. Ovarian epithelial tumor growth promotion by follicle-stimulating hormone and inhibition of the effect by luteinizing hormone. Gynecol Oncol. (2000) 76:80–8. doi: 10.1006/gyno.1999.5628
121. Wang J, Lin L, Parkash V, Schwartz PE, Lauchlan SC, Zheng W. Quantitative analysis of follicle-stimulating hormone receptor in ovarian epithelial tumors: a novel approach to explain the field effect of ovarian cancer development in secondary mullerian systems. Int J Cancer. (2003) 103:328–34. doi: 10.1002/ijc.10848
122. Perales-Puchalt A, Svoronos N, Rutkowski MR, Allegrezza MJ, Tesone AJ, Payne KK, et al. Follicle-stimulating hormone receptor is expressed by most ovarian cancer subtypes and is a safe and effective immunotherapeutic target. Clin Cancer Res. (2017) 23:441–53. doi: 10.1158/1078-0432.CCR-16-0492
123. Urbanska K, Stashwick C, Poussin M, Powell DJ Jr. Follicle-Stimulating hormone receptor as a target in the redirected t-cell therapy for cancer. Cancer Immunol Res. (2015) 3:1130–7. doi: 10.1158/2326-6066.CIR-15-0047
124. Modi DA, Sunoqrot S, Bugno J, Lantvit DD, Hong S, Burdette JE. Targeting of follicle stimulating hormone peptide-conjugated dendrimers to ovarian cancer cells. Nanoscale. (2014) 6:2812–20. doi: 10.1039/c3nr05042d
125. Choi JH, Choi KC, Auersperg N, Leung PC. Gonadotropins activate proteolysis and increase invasion through protein kinase A and phosphatidylinositol 3-kinase pathways in human epithelial ovarian cancer cells. Cancer Res. (2006) 66:3912–20. doi: 10.1158/0008-5472.CAN-05-1785
126. Hong H, Yan Y, Shi S, Graves SA, Krasteva LK, Nickles RJ, et al. PET of follicle-stimulating hormone receptor: broad applicability to cancer imaging. Mol Pharm. (2015) 12:403–10. doi: 10.1021/mp500766x
127. Sanchez AM, Flamini MI, Russo E, Casarosa E, Pacini S, Petrini M, et al. LH and FSH promote migration and invasion properties of a breast cancer cell line through regulatory actions on the actin cytoskeleton. Mol Cell Endocrinol. (2016) 437:22–34. doi: 10.1016/j.mce.2016.08.009
128. Liu L, Zhang J, Fang C, Zhang Z, Feng Y, Xi X. OCT4 mediates FSH-induced epithelial-mesenchymal transition and invasion through the ERK1/2 signaling pathway in epithelial ovarian cancer. Biochem Biophys Res Commun. (2015) 461:525–32. doi: 10.1016/j.bbrc.2015.04.061
129. Yang Y, Zhang J, Zhu Y, Zhang Z, Sun H, Feng Y. Follicle-stimulating hormone induced epithelial-mesenchymal transition of epithelial ovarian cancer cells through follicle-stimulating hormone receptor PI3K/Akt-Snail signaling pathway. Int J Gynecol Cancer. (2014) 24:1564–74. doi: 10.1097/IGC.0000000000000279
130. Babu PS, Krishnamurthy H, Chedrese PJ, Sairam MR. Activation of extracellular-regulated kinase pathways in ovarian granulosa cells by the novel growth factor type 1 follicle-stimulating hormone receptor. Role in hormone signaling and cell proliferation. J Biol Chem. (2000) 275:27615–26. doi: 10.1074/jbc.M003206200
131. Li Y, Ganta S, Cheng C, Craig R, Ganta RR, Freeman LC. FSH stimulates ovarian cancer cell growth by action on growth factor variant receptor. Mol Cell Endocrinol. (2007) 267:26–37. doi: 10.1016/j.mce.2006.11.010
132. Keereweer S, Van Driel PB, Robinson DJ, Lowik CW. Shifting focus in optical image-guided cancer therapy. Mol Imaging Biol. (2014) 16:1–9. doi: 10.1007/s11307-013-0688-x
133. Pento JT. Monoclonal antibodies for the treatment of cancer. Anticancer Res. (2017) 37:5935–9. doi: 10.21873/anticanres.12040
134. Babiker HM, McBride A, Newton M, Boehmer LM, Drucker AG, Gowan M, et al. Cardiotoxic effects of chemotherapy: a review of both cytotoxic and molecular targeted oncology therapies and their effect on the cardiovascular system. Crit Rev Oncol Hematol. (2018) 126:186–200. doi: 10.1016/j.critrevonc.2018.03.014
135. Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov. (2010) 9:325–38. doi: 10.1038/nrd3003
136. Bouis D, Kusumanto Y, Meijer C, Mulder NH, Hospers GA. A review on pro- and anti-angiogenic factors as targets of clinical intervention. Pharmacol Res. (2006) 53:89–103. doi: 10.1016/j.phrs.2005.10.006
137. Raica M, Cimpean AM. Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals. (2010) 3:572–99. doi: 10.3390/ph3030572
138. Korc M, Friesel RE. The role of fibroblast growth factors in tumor growth. Curr Cancer Drug Targets. (2009) 9:639–51. doi: 10.2174/156800909789057006
139. Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, et al. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. (1999) 284:1994–8.
140. Barinaga M. Cancer research - Designing therapies that target tumor blood vessels. Science. (1997) 275:482–4. doi: 10.1126/science.275.5299.482
141. Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic therapy in oncology: current status and future directions. Lancet. (2016) 388:518–29. doi: 10.1016/S0140-6736(15)01088-0
142. Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat Rev Cancer. (2005) 5:423–35. doi: 10.1038/nrc1628
143. Thorpe PE. Vascular targeting agents as cancer therapeutics. Clin Cancer Res. (2004) 10:415–27. doi: 10.1158/1078-0432.Ccr-0642-03
144. Neri D, Bicknell R. Tumour vascular targeting. Nat Rev Cancer. (2005) 5:436–46. doi: 10.1038/nrc1627
145. Siemann DW, Chaplin DJ, Horsman MR. Realizing the Potential of Vascular targeted therapy: the rationale for combining vascular disrupting agents and anti-angiogenic agents to treat cancer. Cancer Invest. (2017) 35:519–34. doi: 10.1080/07357907.2017.1364745
146. Cesca M, Bizzaro F, Zucchetti M, Giavazzi R. Tumor delivery of chemotherapy combined with inhibitors of angiogenesis and vascular targeting agents. Front Oncol. (2013) 3:259. doi: 10.3389/fonc.2013.00259
147. Huang X, Molema G, King S, Watkins L, Edgington TS, Thorpe PE. Tumor infarction in mice by antibody-directed targeting of tissue factor to tumor vasculature. Science. (1997) 275:547–50.
148. Thorpe PE, Chaplin DJ, Blakey DC. The first international conference on vascular targeting: meeting overview. Cancer Res. (2003) 63:1144–7.
149. Renner M, Goeppert B, Siraj MA, Radu A, Penzel R, Wardelmann E, et al. Follicle-stimulating hormone receptor expression in soft tissue sarcomas. Histopathology. (2013) 63:29–35. doi: 10.1111/his.12135
150. Lizneva D, Yuen T, Sun L, Kim SM, Atabiekov I, Munshi LB, et al. Emerging concepts in the epidemiology, pathophysiology, and clinical care of osteoporosis across the menopausal transition. Matrix Biol. (2018) 71–72:70–81. doi: 10.1016/j.matbio.2018.05.001
152. Sponton CH, Kajimura S. Burning fat and building bone by FSH blockade. Cell Metab. (2017) 26:285–7. doi: 10.1016/j.cmet.2017.07.018
Keywords: FSH, FSHR, obesity, osteoporosis, BMI, cardiovascular risk
Citation: Lizneva D, Rahimova A, Kim S-M, Atabiekov I, Javaid S, Alamoush B, Taneja C, Khan A, Sun L, Azziz R, Yuen T and Zaidi M (2019) FSH Beyond Fertility. Front. Endocrinol. 10:136. doi: 10.3389/fendo.2019.00136
Received: 21 November 2018; Accepted: 13 February 2019;
Published: 19 March 2019.
Edited by:
Livio Casarini, University of Modena and Reggio Emilia, ItalyReviewed by:
Adolfo Rivero-Muller, Turku Centre for Biotechnology, FinlandCopyright © 2019 Lizneva, Rahimova, Kim, Atabiekov, Javaid, Alamoush, Taneja, Khan, Sun, Azziz, Yuen and Zaidi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mone Zaidi, bW9uZS56YWlkaUBtc3NtLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.