 Iván E. Alfaro1*
Iván E. Alfaro1* Amelina Albornoz1
Amelina Albornoz1 Alfredo Molina2
Alfredo Molina2 José Moreno2Karina Cordero2
José Moreno2Karina Cordero2 Alfredo Criollo2,3,4
Alfredo Criollo2,3,4 Mauricio Budini2,3*
Mauricio Budini2,3*- 1Fundación Ciencia & Vida, Santiago, Chile
- 2Dentistry Faculty, Institute in Dentistry Sciences, University of Chile, Santiago, Chile
- 3Autophagy Research Center (ARC), Santiago, Chile
- 4Advanced Center for Chronic Diseases (ACCDiS), University of Chile, Santiago, Chile
Chaperone Mediated Autophagy (CMA) is a lysosomal-dependent protein degradation pathway. At least 30% of cytosolic proteins can be degraded by this process. The two major protein players of CMA are LAMP-2A and HSC70. While LAMP-2A works as a receptor for protein substrates at the lysosomal membrane, HSC70 specifically binds protein targets and takes them for CMA degradation. Because of the broad spectrum of proteins able to be degraded by CMA, this pathway has been involved in physiological and pathological processes such as lipid and carbohydrate metabolism, and neurodegenerative diseases, respectively. Both, CMA, and the mentioned processes, are affected by aging and by inadequate nutritional habits such as a high fat diet or a high carbohydrate diet. Little is known regarding about CMA, which is considered a common regulation factor that links metabolism with neurodegenerative disorders. This review summarizes what is known about CMA, focusing on its molecular mechanism, its role in protein, lipid and carbohydrate metabolism. In addition, the review will discuss how CMA could be linked to protein, lipids and carbohydrate metabolism within neurodegenerative diseases. Furthermore, it will be discussed how aging and inadequate nutritional habits can have an impact on both CMA activity and neurodegenerative disorders.
Introduction
Autophagy
Autophagy is a cellular process in which proteins and organelles become degraded through lysosomes in order to maintain an adequate cellular homeostasis. In mammals, three different types of autophagy pathways have been described, (i) macroautophagy (mostly known as “autophagy”), (ii) microautophagy, and (iii) chaperone-mediated autophagy (CMA).
Macroautophagy and CMA, together with proteasome, are involved in the recycling of cellular proteins, including those that can compromise the normal physiology of the cell by adopting aberrant folded forms which are able to generate aggregates or inclusions (1). Macroautophagy is a multistep pathway that starts with the formation of an autophagosome, a double membrane vesicular structure that includes the cargo to be degraded. Autophagosome formation involves the participation of different factors like the Atg protein family, LC3-II, p62, Beclin, among others.After its formation, the autophagosome fuses to the lysosome where it releases its proteolytic material to form an autolysosome structure that finally degrades the cargo (1).
In addition to proteins, macroautophagy targets can also include damaged and/or oxidized organelles such as mitochondria (mitophagy), endoplasmic reticulum (ERphagy), peroxisomes (pexophagy), lipid droplets (lipophagy), ferritin (ferritinophagy) and zymogen granules (zymophagy) (2–5).
CMA, which will be further described below, involves the participation of HSC70 (heat shock-cognate chaperone 70 KDa) and LAMP-2A (lysosomal associated-membrane protein 2A) proteins. With respect to macro- and microautophagy, CMA has been reported only to degrade cytosolic proteins previously bound to HSC70-specific sequence (see below) (6, 7). In the case of microautophagy, it carries the degradation of proteins and organelles out, by their direct engulfment of the lysosome. In microautophagy, whereas some of the proteins can be incorporated into the lysosome through an “unspecific way”, others, like those in CMA, also need the interaction with the Hsc70 chaperone. The similarities and differences between CMA and microautophagy have recently been discussed in the review of Tekiradg and Cuervo (8).
Chaperone Mediated Autophagy (CMA)
CMA Overview
Chaperone-mediated autophagy (CMA) is a specific lysosomal-dependent protein degradation pathway (9). It differs from macroautophagy because, (i) it does not involve the formation of autophagosomes and autolysosomes, (ii) its targets are cellular proteins and not organelles, (iii) the protein cargo is directly delivered into the lysosomal lumen through the interaction with HSC70 and LAMP-2A (10). HSC70 belongs to the Hsp70 protein family and it has a constitutive expression, participating principally in the CMA pathway but also in microautophagy (8). LAMP-2A protein is the A isoform of Lamp-2 (lysosome associated-membrane protein type 2) and restricts the CMA degradation process. Importantly, in CMA, selectivity resides in the fact that all CMA substrate proteins contain at least one amino acidic motif biochemically related to the penta-peptide KFERQ (11, 12). In the cell cytoplasm, HSC70 recognizes the KFERQ sequence in target proteins to form the complex HSC70-substrate. Although the temporality of this process is not well known, the complex HSC70-substrate interacts with the cytosolic tail of LAMP-2A that, in turns, drives the translocation of the target protein into the lysosome lumen (13). All cells have a basal CMA activity which helps to maintain the homeostasis of many cellular proteins. However, under certain stimulus, like nutrient starvation, serum deprivation or cellular stress (e.g., protein aggregation), CMA activity increases. This up-regulation condition can be visualized by mRNA LAMP-2A overexpression, HSC70 and LAMP-2A co-localization and LAMP-2A positive lysosomes with increased perinuclear distribution (14–16).
CMA Target Recognition
In 1985 Dice et al. found a pentapeptide in the Ribonuclease A (RNase A) which was necessary for the degradation of this protein by lysosomes in fibroblasts (11, 17–20). When different proteins were analyzed, it was found that the best aminoacidic sequence, to be present in a protein that is going to be degraded through CMA, corresponds to the pentapeptide KFERQ (11). The general rule, is that this sequence is composed of an amino acid Glutamine (Q), that must contain in one side (right of left) a tetrapeptide, including basic-, acidic- and hydrophobic- amino acids (11). Target proteins for CMA should contain at least one KFERQ-like domain, however some of them include more than one (21). Proteins containing a KFERQ-like domain do not enter the lysosomes by themselves, they first need to be recognized and bound by the chaperone HSC70 (22). This protein, also known as HspA8, is a 73 kDa protein belonging to the Hsp70 protein family (23). Thus, HSC70 specifically recognize the KFERQ-like motive in cytosolic proteins to target them for lysosomal degradation through CMA (22). In some cases, other co-factors, or covalent modifications have been found to be necessary for the binding between the complex HSC70-protein substrate and LAMP-2A. For example, STUB1/CHIP (a ubiquitin E3 ligase) assists the degradation of HIF1A through CMA by the ubiquitination of Lysin 63 (K63) (24) and also by supporting the interaction of LAMP-2A through its chaperon binding domain (24). In addition, covalent modifications, like acetylation and phosphorylation have been described to create KFERQ-like domains capable of targeting proteins for CMA degradation (25–29).
CMA Substrate Translocation and Degradation
In the lysosome membrane, the HSC70-substrate complex interacts with the lysosome receptor LAMP-2A. The protein LAMP-2A was first described by Dr. Ana María Cuervo and Dr. J. Fred Dice (30). It was identified as the LGP96 (lysosome membrane glycoprotein 96 kDa) and it was found to be a necessary receptor for the specific degradation of GAPDH (Glyceraldehyde- 3-phosphate dehydrogenase) and RNase I (Ribonuclease I) through lysosomes (30). The authors showed that both GAPDH and RNase I, were able to bind the LGP96, which were isolated from rat liver (30). The Lysosome-associated membrane glycoprotein 2 (LAMP-2) is able to generate three variants through its pre-mRNA alternative splicing, (LAMP-2A, Lamp-2B, and Lamp-2C) but, to date, it is well established that only the variant A is responsible for the CMA activity (31). Compared to variants B and C, LAMP-2A has four positively charged residues in its C-terminal domain that specifically allow its interaction with target proteins (31). Characterization of LAMP-2A showed that multimerization of this variant (and not the others) in the lysosomal membrane is directly correlated with CMA activity (13, 31). The substrate up-take by the lysosome is a step that depends on LAMP-2A (32). Monomeric LAMP-2A is able to interact with substrates, however, substrate translocation is dependent on the formation of LAMP-2A oligomers (32). Further evidence showed that LAMP-2A arranges in a stable homotrimer, with helical transmembrane domains bound by a coiled-coil conformation, and with the cytosolic tails interacting with the complex HSC70-substrate protein (13). In addition, LAMP-2A oligomerization was shown to be regulated by different proteins. Glial fibrillary acidic protein (GFAP) helps to stabilize the translocation complex in an EF1α dependent manner. The stabilizing effect of GFAP is disrupted by the association of EF1α to GTP, which in turn is released from the translocation complex and allows the self-association between GFAP molecules. The self-interaction between GFAP molecules have a negative impact on the stabilization of the translocation complex. Thus, GTP acts as an inhibitor of CMA activity (33). Phosphorylating and dephosphorylating signals also regulate CMA activity at the level of the translocation complex (34). For example, mTORC2 (mammalian target of rapamycin complex 2) has been shown to activate, through phosphorylation, Akt kinase, which in turn inhibits the CMA activity. The mechanism by which active Akt decreases the CMA activity is not yet well established. However, it has been postulated that active Akt phosphorylates GFAP, destabilizing its binding to the LAMP-2A translocation complex. In this scenario, both, mTORC2 and Akt act as negative regulators of CMA activity. On the contrary, Pleckstrin homology (PH) domain and leucine-rich repeat protein phosphatase 1 (PHLPP1), has been shown to be recruited to the lysosome membrane in a Rac1 dependent manner. On the lysosomal membrane, PHLPP1 induces CMA activation by dephosphorylating Akt at the same residues previously phosphorylated by mTORC2. Thus, PHLPP1 is a positive regulator of CMA activity (34). In addition, the binding and up-take of the CMA substrates through the translocation complex is assisted by HSC70 and Hsp90 (heat shock protein 90) chaperones. In the case of Hsp90, it was shown that this protein stabilizes the binding of LAMP-2A at the lysosomal membrane (32). HSC70, apart from its important role in binding to the KFERQ domain, participates in two additional steps of the CMA process. One step is related to the recycling of LAMP-2A in the absence of substrates, where HSC70 has been proposed to support the destabilization of LAMP-2A from the translocation complex (32). The other step occurs at the lumen of the lysosome, where a lysosomal HSC70 (lys-HSC70) is also involved in the up-take process of the substrate. The blockage of lys-HSC70, by a specific antibody, has been directly correlated with an inhibition of the CMA activity (35). Figure 1 summarizes the main aspects of the CMA mechanism.
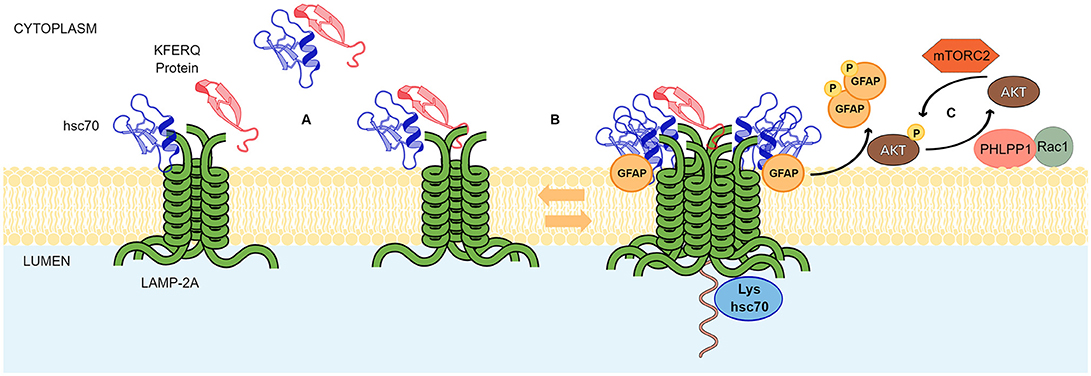
Figure 1. Chaperone mediated autophagy mechanism. The scheme shows the principal events in CMA mediated translocation of proteins through the lysosomal membrane and known regulatory mechanisms. (A) LAMP-2A exist as an inactive oligomer at the lysosomal membrane. CMA protein substrates containing KFERQ motifs and HSC70 can bind to the cytosolic tail of LAMP-2A with similar affinity. The short C-terminal tail of LAMP-2A exposed to the cytoplasm and conformed by 12 amino acids probably binds to the chaperone and the substrate in a competitive manner. However, on the substrate, binding sites for HSC70 and LAMP-2A do not overlap allowing the HSC70-substrate complexes to interact with LAMP-2A oligomer in a substrate dependent manner. (B) In a dynamic fashion, GFAP favors the formation of high molecular weight aggregates of LAMP2A. Somehow, coupled and simultaneous binding of the substrate to HSC70 and LAMP-2A is sensed by the transmembrane domains of the LAMP-2A for rearrangements in the supramolecular complexes. Next, substrate unfolding and translocation to the lysosomal lumen occurs. A lysosomal-HSC70 (Lys-hsc70) collaborates in this process. (C) A signaling pathway involving phosphorylating and dephosphorylating signals, where AKT, MTORC2, PHLPP1, RAC1, and GFAP, regulate the stabilization of the LAMP-2A complex at lysosomal membranes.
CMA Activation
Koga et al. (36) showed that basal CMA activity is present in a variety of cell types. However, an up-regulation in this pathway can be observed under different stimulus or conditions. The most common stimulus for CMA activation is nutrient deprivation (or starvation). Starvation activates CMA both in vitro and in vivo (11, 20, 36) and, although the exact mechanism has not been described yet, at least in vivo, it has been proposed that it depends on the circulating ketone bodies (37). CMA over-activation has also been observed under DNA damage. Under this condition, CMA activity is up-regulated with the purpose to degrade the checkpoint protein kinase 1 (Chk1). The accelerated degradation of Chk1 by CMA reduces its nuclear entrance and consequently decreases the phosphorylation and destabilization of the MRN (Mre11–Rad50–Nbs1), a complex that participates in the early steps of particular DNA repair pathways (38). In line with this result, DNA irradiation of a hepatocellular carcinoma cell line also over-activates CMA. In this cell line, CMA up-regulation was responsible for the degradation of HMGB1 (high-mobility group box 1 protein) degradation, which in turn, provoked the down-regulation of the p53 protein and the consequent decrease in the apoptotic rate. In this case, CMA activation could be considered a mechanism by which this type of carcinoma is able to resist the irradiation treatment (39). Oxidative stress also activates CMA constitutively. Both, mRNA LAMP-2A levels and the recruitment of LAMP-2A protein to the lysosomal membrane (40, 41) were augmented under oxidative stress. The reasons by which CMA is activated under oxidative stress are unknown. However, a possibility could be that CMA acts as a degradative backup system for an inhibited macroautophagy pathway (42). With respect to the molecular mechanism, recent work showed that under hydrogen peroxide treatment, the transcription factor NFE2L2/NRF2 (nuclear factor, erythroid derived 2, like 2) can drive the expression of the lamp2 gene. Pharmacological activation of NFE2L2/NRF2 further supported the role of this transcription factor on lamp-2A gene overexpression (43). However, under oxidative stress, CMA up-regulation could also have a toxic effect. It was shown that CMA over-activity strongly reduces the levels of MEF2A (myocyte enhancer factor 2a), a protein necessary for the adaptive response of the cells to oxidative stress (41). Another stressor that up-regulates CMA activity is hypoxia. Over-activation of CMA was directly correlated with the survival of neuronal cells exposed to different hypoxic conditions and, on the contrary, the down-regulation of this pathway sensitized the cells to the stress (44). On the other hand, it was reported that HIF-1α, a protein that activates HIF-1 (Hypoxia-inducible factor-1) and mediates an adaptive response to hypoxia, is degraded by CMA in cells exposed to hypoxic conditions (45). Therefore, the exact mechanisms by which CMA protects cells against hypoxia, needs to be further elucidated. A constitutive activation of CMA has also been observed in different models of neurodegenerative diseases. For example, in cellular and mouse models of Huntington Disease, CMA was activated as a compensatory mechanism for the inhibition of macroautophagy. In this case, both lys-HSC70 and LAMP-2A protein levels were markedly augmented. In particular, there was an observed stability in LAMP-2A proteins at the lysosomal membrane and a transcriptional upregulation of this splice variant (46). Up-regulation of CMA was also observed in molecular mechanisms related with Parkinson's disease. For example, recent studies performed with a dopaminergic neuronal cell line, using an endoplasmic reticulum (ER) stress mouse model, showed that ER stressors are able to induce CMA activation. The mechanism for such activation involved the recruitment of MKK4 protein kinase to the lysosomal membrane and the subsequent phosphorylation of LAMP-2A by p38 MAPK protein kinase (47). This regulatory mechanism for CMA up-regulation is relevant as neurotoxins associated with PD are directly correlated with an activation of the ER-p38 MAPK–CMA response pathway (48). Additionally, in PD models, LAMP-2A and HSC70 were observed to be up-regulated when α-syn was over-expressed in vivo, indicating that a CMA over-activation is triggered in response to a α-syn pathological condition (49)
CMA and Metabolism
Participation of CMA in the Regulation of General Protein Metabolism
Protein homeostasis is regulated by a series of interconnected signaling pathways that sense amino acid availability, among them, the integrated signaling pathway (ISR) through the general control nonderepressible-2 protein (GCN2) and the V-ATPase/Ragulator/mTOR pathway located in lysosomes (50, 51). GCN2 is a protein kinase that is activated in the presence of accumulated deacetylated tRNAs, sensing amino acid depletion. The main target of GCN2 is the eukaryotic initiation factor 2 (eIF2), whose phosphorylation induces the inhibition of general translation and favors the synthesis of a specific set of mRNAs that regulates cell survival and promotion of macroautophagy (52). On the other hand, mTORC1 kinase is activated in response to amino acid availability, promoting translation and inhibiting macroautophagy. The mechanism of amino acid sensing, coupled with mTOR activation, involves the lysosomal amino acid transporter SLC38A9, and mTOR-binding proteins, Sestrin2 and Castor1 (50). These factors that regulate mTOR activation or inhibition, also modulate mTOR activity and thus mTOR-downstream targets involved in promotion of protein translation, cell growth and anabolism, and inhibition of macroautophagy initiation. In response to amino acid deprivation, mTOR activity is attenuated, protein translation is inhibited and macroautophagy is maximally activated to restore amino acid levels.
These non-selective mechanisms of protein homeostasis, added to a variety of mechanisms of stabilization and destabilization of protein pools, define the average life time of every protein in the cell, in nutrient rich conditions and in response to nutritional stress. Selective mechanisms of protein degradation are in contrast, generally linked to the modulation of the function of specific proteins or to the degradation of misfolded proteins that have been targeted for efficient elimination. Selective and non-selective mechanisms of proteasomal and macroautophagy protein degradation pathways have been extensively reviewed in several articles (53–56). Whereas proteins degraded by CMA need to be recognized specifically by HSC70 protein through its KFERQ domain, CMA can be considered a selective protein degradation process. An initial analysis indicated that at least 30% of cytosolic proteins contain CMA KFERQ-like motifs (7). These proteins mainly correspond to a fraction of long-lived proteins that were more rapidly degraded by lysosomes in response to serum starvation or amino acids withdrawal. Indeed, at least 90% of enhanced proteolysis observed during serum withdrawal in fibroblast cells is thought to be part of the protein pools targeted for degradation by CMA (20). In comparison to macroautophagy, the increase in degradation rates of the pool of proteins targeted to CMA is observed in response to prolonged periods of serum or nutrient deprivation (7, 12, 57). Indeed, macroautophagy activity decreases in activity after 6–12 h of nutrient removal, but CMA progressively increases in response to prolonged periods of serum or nutrient deprivation (57). Thus, CMA is a protein degradation pathway specifically activated during long term starvation. The pool of proteins degraded by CMA is very specific and different to the protein pools degraded by other protein catabolic neutral or acid proteolytic pathways. Additionally, CMA-targeted proteins accumulate in lysosomal membranes under starvation and, probably, because of this reason, this pool is excluded for degradation by proteasome or macroautophagy.
The pool of proteins targeted by CMA depends, most likely, on the periods of deprivation, type of deprivation (glucose, amino acids, serum or specific growth factors) and the cell type in the study (12). In addition, the selective pool of proteins degraded by CMA would also be associated with certain functions capable of overcoming metabolic changes in response to starvation. This has been observed in the regulation of glycolytic flux, through the degradation of glycolytic enzymes by CMA, in order to protect cells from apoptosis (7, 21). On the other hand, proteins that do not contain KFERQ-like regions would most likely be protected against degradation, to sustain the critical functions of cells under nutritional stress. It has also been suggested that activation of non-selective macroautophagy in the first stages of starvation, help to provide metabolite building blocks (as amino acids) for the continuous synthesis of macromolecules. However, at prolonged times of starvation, a relief of macroautophagy by selective CMA-dependent protein degradation, would be necessary to avoid the elimination of essential proteins for cell survival (57). It is unknown whether nutrient deprivation directly controls the CMA activity, as with macroautophagy. The main hypothesis is that an indirect regulation of CMA by nutrient deprivation would be carried out by translational and transcriptional up-regulation of lysosomal proteins and genes involved in lysosome biogenesis (58–60). However, a recent report indicates that upregulation of CMA was responsible for mTOR activation by a mechanism that sensed the augmentation of the free amino acid content dependent on CMA activation (61). In addition, increased CMA activity, in response to LAMP2-A overexpression, was also shown to down-regulate macroautophagy through mTOR activation (62). Overall, this latter evidence reveals that CMA activation functions as a negative feedback loop for mTOR and, thus, would regulate the general amino acid and protein homeostasis signaling.
As mentioned before, basal CMA activity can be detected in different tissues, suggesting that this pathway has a constant role in the regulation of certain proteins, even under conditions of nutrient or growth factor availability (57). However, whether the degradation of these target proteins increases under CMA activation (e.g., starvation), or another specific cell response, remains to be clarified. Most likely, the latter will depend on the different affinities that these proteins would have for HSC70 and LAMP-2A, and/or on the availability of HSC70 and LAMP-2A (63).
As a consequence of its important role in controlling protein homeostasis, CMA is able to regulate metabolic pathways that are crucial to maintain balance in cellular and systemic physiology.
CMA and Regulation of Lipid Metabolism
Neutral cytosolic lipolysis and lysosome-associated acid lipolysis were classically classified in two different pathways. However, both are now recognized as synergistic, cooperative and interconnected mechanisms that contribute to lipid catabolism (64). Lysosomal lipolysis involves the degradation of lipids derived from endocytosis, as well as from cytosolic lipid stores (lipid droplets) through an autophagic process known as Lipophagy (65). Lipid droplets (LDs) are specialized organelles that store neutral lipids during fatty acid availability, mainly triglycerides (TG) and cholesterol esters, for their posterior use as energy or precursors for lipids and steroids (66). LDs consist of a core of neutral lipids separated from the aqueous cytoplasm by a phospholipid monolayer decorated by a set of proteins. These proteins are enzymes involved in lipid synthesis and lipid hydrolysis, membrane-trafficking proteins and a set of specialized LDs-associated structural proteins called perilipins (PLINs). PLINs cover a family of 5 major proteins and some additional splice variants expressed in lower amounts. Perilipin 1 (PLIN1) and 2 (PLIN2) are exclusively associated with LDs whereas perilipins 3–5 are found in the cytoplasm, associated with LDs, as well as microdroplets and lipoprotein particles (67). Perilipins 1, 2 and 5 control, at the surface of the lipid droplets, the access of cytosolic neutral lipases. For example, phosphorylation of PLIN1 and PLIN5 by protein kinase A (PKA), in response to hormones or growth factors, trigger the recruitment of neutral lipases like hormone sensitive lipase (HSL) and triacyl-glycerol lipase (ATGL) (68, 69). In contrast to PLN1 and PLIN5, PLN2 is not phosphorylated by PKA and does not recruit lipases at LDs (67). Similar to PLIN2, PLIN3 and PLIN4 protect LDs from degradation and they seem to have a tissue-specific function in the regulation of LDs size, maturation and mitochondrial association (70–72).
The involvement of autophagy in lipid turnover was first demonstrated for macroautophagy (65). Electron-microscopy studies indicated that autophagosomes engulf LDs or small portions of large LDs for degradation, forming autolipophagosomes in a process now known as macrolipophagy. In hepatocytes, macrolipophagy and the presence of lipid-containing autophagolysosomes, is stimulated by nutrient starvation and low fatty acid treatments (65). Members of the Rab GTPase family, RAB7 and RAB10 have been involved in the early steps of autophagosome recruitment to LDs (73). Moreover, the Rab effectors, EHBP1 and EH2, have been suggested to help in the elongation of autophagosomal membranes around LDs (74, 75). Neutral lipases ATGL and HSL contain LIR motifs (LC3-interaction region) and interact with the cytosolic face of autophagosomes through LC3-II, being recruited to LDs in response to macrolipophagy activation (64, 76).
In addition to macroautophagy, CMA also plays a central role in lipid homeostasis. In a mouse model, where LAMP-2A was specifically down-regulated in liver by deleting the exon 9 of lamp2 gene, it was shown that the constitutive blockade of CMA activity induces pronounced steatosis without a substantial increase in TG synthesis (77). In addition, LAMP-2A knock-out cells were insensitive to lipolysis induced by starvation and displayed increased density, size and occupancy area of LDs (78). Interestingly, in this model, non-selective macroautophagy was intact, indicating a specific and new role of CMA in lipophagy (77). In line with this evidence, further studies found the presence of KFERQ-like motifs in PLIN2 and PLIN3 proteins, and identified these two proteins as substrates for CMA lysosomal degradation. In addition, lysosomes actives in CMA isolated from rat liver, were enriched in PLIN2 and PLIN3, supporting the fact that these proteins are CMA substrates. This evidence was further complemented by experiments indicating that PLIN2 and PLIN3 degradation was reduced in LAMP-2A knockout cells (78). With regards to the mechanism, the degradation of PLIN2 through CMA in response to nutrient starvation and lipid challenges, was correlated with an increment in the association between HSC70 and perilipins (78). Reduced degradation of PLIN2 in cells that are deficient for the CMA pathway, leads to reduced lipolysis, likely due to an impairment in the association of ATGL with LDs as well as due to the defective recruitment of autophagosomal proteins and Rab7 to LDs (25, 78). Moreover, PLIN2 phosphorylation by AMPK was found to be a necessary modification for PLIN2 association with HSC70 in a form dependent on CMA activation (25, 78). Therefore, the role of CMA in lipophagy not only relays in the regulation of the recruitment of neutral lipases to LDs trough perlipin degradation, but also in the integration of a regulatory step that involves a post-transcriptional modification guided by AMPK (25). Thus, CMA itself could be considered a nutrient sensing pathway supporting the fact that lysosomes can play a role as initial sensors and integrators of lipid homeostasis (79). Additional research, however, is needed to determine how cellular pathways involved in cellular lipid homeostasis communicate with CMA and lipophagy, and how CMA-dependent lipolysis affects different tissues.
CMA and Regulation of Carbohydrate Metabolism
As mentioned before, glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was one of the first characterized as a CMA substrate (80). Additionally, at least 8 of 10 glycolytic enzymes contain CMA-targeting motifs (77). Substrates of CMA that participate in glycolysis, validated by LAMP2-A loss of function or lysosome-transport assays, also include hexokinase-2 (HK-2) (28), the M2 isoform of pyruvate kinase (81), aldolase-A, enolase-1, 3-phosphoglyceric phosphokinase, phosphoglycerate mutase, glucose-6-phosphate-dehydrogenase and the mitochondrial proteins glutamate dehydrogenase, ornithine transcarbamoylase and malate dehydrogenase (77). Other related substrates that have been suggested to be regulated by CMA in their function include phosphofructokinase-2 (82) and aldolase B (83). Glycolytic flux regulation by CMA has been confirmed in vivo in a mouse model with specific down-regulation of LAMP-2A in hepatocytes (77). These mice displayed higher protein levels of glycolytic enzymes and enzymes from the tricarboxylic acid cycle (TCA), a reduction in hepatic gluconeogenesis, lower glycogen synthesis and an increase in lactate production and TCA intermediates (77). This metabolic profile suggests a switch in hepatic metabolism to carbohydrate consumption as a source of energy vs. glucose biosynthesis in response to low CMA activity (77). On the other hand, classical inhibition of hepatic glycolysis caused by serum starvation (84) was not observed in mice with liver-specific CMA down-regulation (77). These results suggest that CMA activity would be necessary for a metabolic adaptive mechanism that triggers glucose production in liver to support peripheral organs under nutritional stress conditions.
The mechanism regulating CMA in response to changes in glucose availability are not fully understood. Pointing to a central role of the lysosome in sensing glucose homeostasis, new evidence indicates that glucose starvation induces changes in lysosomal acidification in an AMPK activity dependent-manner (84, 85). The mechanisms implicated in this regulation may involve a glucose-dependent regulation of the lysosome biogenesis through the transcription factor EB (TFEB) (86). Additional research is needed to elucidate how these lysosomal changes, induced by carbohydrate availability, regulate the CMA activity and, in turn, how this affects the cellular glycolytic flux.
CMA and Neurodegenerative Diseases
There is increasing evidence supporting the idea that dysregulation in the CMA pathway plays a crucial role in neurodegeneration.
Parkinson's Disease (PD)
Evidence indicates that a dysregulation in CMA could impact on the onset or progression of Parkinson's Disease (PD). As mentioned above, the main protein associated with this neurogenerative disorder, alpha-synuclein protein (α-syn), has been identified as a CMA substrate (87). More specifically, reduced α-syn degradation was observed when its KFERQ motif was mutated and the expression of LAMP-2A was knocked-down. The involvement of CMA in α-syn degradation was confirmed in different neuronal cell lines (PC12 and SH-SY5Y) and primary cultures of cortical and midbrain neurons (87). One of the hallmarks of PD is the neurotoxicity caused by the abnormal aggregation of α-syn. In this context, mutations in the protein impair its degradation through a CMA pathway, causing the accumulation α-syn oligomers that are unable to be degraded by the lysosome. This event blocks the entire CMA pathway, enhancing the oligomers formation and compromising the degradation of other CMA substrates (88, 89). As mentioned above, LAMP-2A and HSC70 were observed to be up-regulated when α-syn was over-expressed in vivo (49). In line with these results, it was shown that the down-regulation of LAMP-2A in adult rat substantia nigra, via an adeno-associated virus, induced intracellular accumulation of α-syn puncta. In addition, LAMP-2A down-regulation was also correlated with a progressive loss of dopaminergic neurons, severe reduction in striatal dopamine levels/terminals, increased astro- and microgliosis and relevant motor deficits (90). Furthermore, studies using the Drosophila melanogaster model, showed that the overexpression of human LAMP-2A protein protected the flies from progressive locomotor and oxidative defects induced by neuronal expression of a human pathological form of α-syn (91). Also, other PD related proteins seem to be regulated by CMA activity. This is the case of PARK7/DJ-1 and MEF2D. PARK7/DJ-1 is an autosomal recessive familial PD gene that plays a critical role in the antioxidative response and its dysfunction leads to mitochondrial defects. CMA was shown to degrade, preferentially, an oxidized and altered form of PARK7, providing protection from mitochondrial damage and impairing cell viability (92). For MEF2D (myocyte-specific enhancer factor 2D protein), it was shown that an inhibition of CMA provoked the accumulation of this protein in the cell cytoplasm (93). These results are in line with the observation that MEF2D levels are increased in brains of α-syn transgenic mice and in samples from patients with PD (93). Thus, an impairment in CMA activity, caused by the α-syn oligomers, could be the reason for MEF2D accumulation in PD. Beyond the studies performed in cell cultures and in vivo models, strong evidence indicates that CMA could also be dysregulated in individuals affected by PD. In this regard, LAMP-2A and HSC70 were observed to be down-regulated in PD patients, suggesting that a CMA dysregulation can occur before the appearance of α-syn aggregation and other PD associated disorders (94, 95). Although the mechanisms that lead to a reduction in LAMP-2A and HSC70 levels in PD remain unknown, some studies suggest that genetic variations in the promoter of the lamp-2A gene and the up-regulation of different microRNAs that target both LAMP-2A and HSC70, could be implicated in their down-regulation (96, 97).
Alzheimer Disease (AD)
The association between CMA and Alzheimer's Disease (AD) can be described by the degradation of the RCAN1 protein (Regulator of calcineurin 1) through this pathway. Two KFERQ-like motifs identified in RCAN1 were responsible for its degradation by the CMA pathway (98). In addition, the inhibition of CMA pathway increased RCAN1 protein levels and consequently reduced the NFAT transcription factor activity (98). Interestingly, NFAT has been involved in the transcriptional regulation of the lamp2 gene (99). Thus, inhibition of CMA activity could be further enhanced by increased levels of RCAN1, that will subsequently impair NFAT-dependent transcription of lamp2. On the other hand, the Tau protein, one of the principal factors associated with AD, has also shown to be a CMA substrate (100, 101). The Tau protein is degraded by the autophagy–lysosomal system producing different fragments that, in turn, bind to HSC70 and become CMA substrates. Although these fragments are able to reach the lysosomes, they remain bound to the lysosomal membrane, causing the formation of pathological Tau aggregates that cause lysosomal damage and block the degradation of other CMA targets (100). Additionally, the amyloid precursor protein (APP), can be processed to produce another pathogenic molecule associated with AD, the β-amyloid peptide (Aβ), which contains a KFERQ related motif (102). However, deletion of KFERQ did not affect its binding to HSC70, but did somehow keep the APP and its C-terminal fragments (CTFs) away from lysosomes. Thus, the KFERQ-like domain of APP is relevant to preclude the accumulation of APP and its CTFs but, in this case, in an CMA degradation independent manner (102).
Huntington Disease (HD)
As previously mentioned, a constitutive activation of CMA has been observed in cell lines and mouse models of HD (46, 103). In a mouse model for this disease, a strong co-localization between LAMP-2A and HSC70 was correlated with augmented lysosomal degradation of the native and aberrant huntingtin protein (Htt) (46). Furthermore, additional studies performed in vivo also showed that Htt aggregates could be forced to be degraded through CMA, by targeting them with a polyQ binding protein (QBP1) including a KFERQ domain in its amino acidic sequence (104). The data indicated that modulation of the CMA pathway can be a plausible strategy for HD treatment.
Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Lobar Degeneration (FTLD)
Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Lobar Degeneration (FTLD) are two important neurodegenerative diseases where the implication of CMA has been poorly studied. So far, only two studies have connected these disorders with CMA and both focus on the ribonuclear protein TDP-43. Huang et al. described an KFERQ-like domain in TDP-43 that was responsible for the interaction of this protein with HSC70 under ubiquitination condition. Mutation of the KFERQ-like domain disrupted the ubiquitin-dependent binding of TDP-43 with HSC70. In addition, the down-regulation LAMP-2A by siRNA treatment, seems to increase the level of the pathologically-related 25-KDa and 35-KDa TDP-43 C-terminal fragments, but not the full-length protein (105). On the other hand, in a recent study, Tamaki, Y., et al designed an antibody able to recognize abnormal (unfolded or mislocated) TDP-43. The addition of the KFERQ sequence to this antibody, was able to promote the degradation of abnormal TDP-43 through the CMA pathway. Although in this case the connection of TDP-43 with CMA was not in a physiological context, this work indicates that a forced CMA target degradation of TDP-43 can be used as a strategy to ameliorate neurodegenerative diseases associated with this protein (106).
CMA in the Crosstalk With Metabolic Disorders and Neurodegeneration
Reduced CMA Activity With Aging
Aging can be considered as a physiological process that is associated with metabolic and neurodegenerative disorders via mechanisms that in many cases are completely unknown. With regards to autophagy, it has been observed that this lysosomal degradative mechanism also declines with age (107). In the case of CMA, this was determined by isolating lysosomes from the liver of young and old rats (108). Lysosomes from old rats had reduced LAMP-2A protein levels at both, total and lysosomal levels. The decrease of the LAMP-2A protein in lysosomes is associated with changes in lipid composition of these organelles that accelerate the degradation rate of the protein (108). With respect to the HSC70 protein, although the total protein levels remained unchanged, lysosomal HSC70 levels were augmented, probably to compensate the decline observed in the CMA activity (108). Further experiments demonstrated that, although the levels of Lamp2A mRNA were unchanged, there was a problem in the localization of LAMP-2A at the lysosomal membrane in old rats, consequently reducing the CMA activity (109). Furthermore, by using an animal model with specific down-regulation of CMA in the liver, it was possible to demonstrate that reduced CMA activity in old animals had an impact on the ability to overcome proteostasis induced by different stressors like oxidative stress, lipid challenging and aging (110). The mentioned evidences clearly demonstrate that CMA activity is reduced in older individuals and that this protein degradative pathway helps to stabilize the different disorders associated with aging.
Thus, considering that CMA activity is reduced with age, it is possible to argue that a decline in CMA activity during aging can be a risk factor for the development of neurodegenerative disorders associated with adult and senior people. In fact, beyond of what it can be concluded from in vitro and in vivo models, more evidence is coming out revealing that a dysregulation in CMA activity can be present in human neurodegeneration. Most of the evidence is a result of studies on PD. For example, a study performed with peripheral blood mononuclear cells from PD patients, showed that affected people had reduced HSC70 protein levels (111). Another study, analyzing CSF (cerebrospinal fluid) of PD patients with mutations in the repeat kinase 2 (LRRK2) gene, confirmed that LAMP-2A protein levels were reduced in affected females, compared to healthy people (112). However, studies focused on analyzing the levels of LAMP-2A and HSC70 in healthy people during normal aging, failed to find any positive association (113). Thus, future studies need to focus on whether changes in CMA activity (or CMA protein players) are altered during normal aging in healthy people, or if they are only associated with patients affected by neurodegeneration. The latter will help to elucidate whether observed changes in CMA protein players are a cause or a consequence of these disorders.
CMA Activity, Metabolic Disorders and Neurodegenerative Diseases
In vivo models strongly support the hypothesis that CMA activity decreases with aging. Considering this evidence, which of these could be the consequence of CMA activity in people with a high fat diet (HFD)? Rodriguez-Navarro et al. showed that chronic exposure of rats to an HFD or cholesterol-enriched diet, provoked a decrease in CMA activity (114). Further characterization of these animals showed that isolated lysosomes from liver have reduced LAMP-2A protein at lysosomal membranes. The latter was triggered by an accelerated degradation of lysosomal LAMP-2A, something also observed in older animals. Interestingly, authors also found that lysosomal membranes from animals with a HFD presented changes in lipid composition, in a similar way than that was observed with age (114). Thus, a dysregulation of CMA activity produced by aging, could impact on the LDs composition, affecting the aggregation of some proteins involved in neurodegenerative diseases.
In addition, a systemic decrease in CMA activity, and the onset or progression of neurodegenerative disorders, could be enhanced by an HFD. Different works demonstrate that a HFD can be a risk factor for the development of neurodegenerative diseases. For example, in a mouse model of PD, it was observed that HFD provoked a reduction in Parkin protein levels (115). In addition, similar results were obtained with a α-syn transgenic mouse model of PD. Compared with wild type mice, an HFD causes obesity and glucose intolerance in transgenic mice. Furthermore, transgenic mice also had an accelerated onset of PD disease and premature movement phenotype and death (116). The same situation was observed in transgenic mice models for AD. The model with an HFD, presented exacerbated neuropathology, defects in synaptic stability/plasticity, apoptotic neuronal cell death (117) and increased levels of insoluble amyloid-β (AB) and Tau (118). On the other hand, LDs have been found to play a role in neurodegenerative diseases. For example, as reviewed by Pennetta et al. LDs seem to be important for the progression of motor neuron diseases (MND) (119). The mechanisms by which LDs are affecting neurodegeneration processes are unknown, however it has been observed that they can affect protein aggregation. For example, in cells treated with an HFD, α-syn accumulated in triglyceride-riche LDs (120). In a similar way, amyloidal fibrils from amyloidotic polyneuropathy (FAP), were observed to be colocalizing with a high density lipoprotein (HDL) (121). Altogether, these data support the idea that LDs could be involved with the aggregation process of neurodegenerative disease associated proteins.
On the other hand, there is increasing evidence which now supports the idea that a high carbohydrate diet (HCD) is a risk factor for certain neurodegenerative diseases (122). Probably, because of a dysregulation in protein glycation, which can control the balance between protein solubility and aggregation (123, 124). Moreover, many reports indicate that there is a strong association between diabetes and people affected by AD (125, 126), suggesting that a dysregulation in glucose metabolism could impact on the onset of this and other neurodegenerative diseases (127). Although the mechanisms are not clear, evidence has shown that there is an increase in the activity of glycolytic enzymes in AD patients (128). In addition, α-syn was observed to bind GAPDH, increasing the activity of this enzyme (129). Also, analyses performed with postmortem human brain tissue, reported a reduction of glucose-6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase in AD and PD (127).
Carbohydrate metabolism dysregulation has also been found in different models of HD. Gouarné et al. found that a rat model of HD had a deficit in glycolysis in striatal neurons (130). In a mouse model of HD, it was observed that glycolysis inhibition decreases the levels of glutamate transport and provoked neurotoxicity (131). On the contrary, in two different models of HD, it was found that the activity of some glycolytic enzymes was higher. The latter results were not correlated with the enhanced conversion of glucose to lactate and increased ATP in the brain and tissue, respectively. Altogether, this evidence indicates that in neurodegenerative diseases, an imbalance in a glycolytic pathway could play an important role in the onset or progression of the disease. Considering that CMA regulates the homeostasis of glycolytic enzymes, changes in the activity of this pathway along with aging could strongly impact on the risk to develop a neurodegenerative disease. In addition, the risk can also be augmented by aging and by a continuous intake of a high carbohydrate diet. Figure 2 shows the main mechanisms that can influence CMA down-regulation and its impact in neurodegenerative diseases.
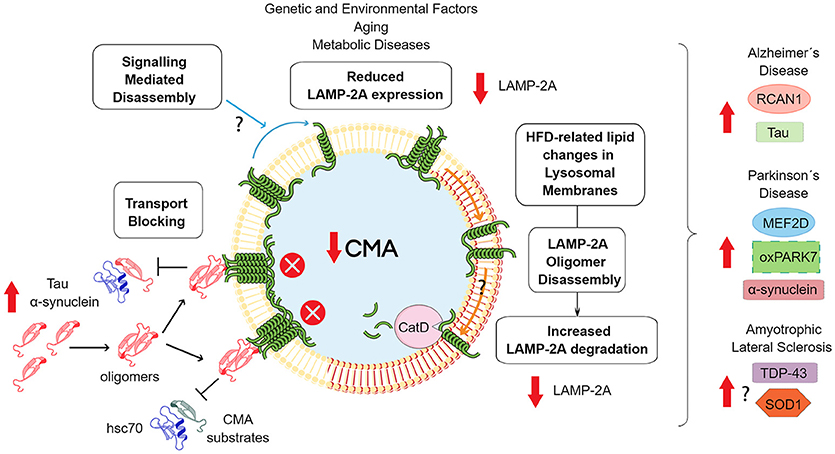
Figure 2. Consequences of downregulated CMA activity. The scheme shows the principal mechanisms of CMA downregulation. Reduced expression of the lamp2 gene has been related with genetic polymorphisms (96), aging (108) and metabolic disorders (132). In relation to the latter situation, HFD has been shown to modify the lipid composition of lysosomal membranes, including increased levels of cholesterol and ceramide (132). These modifications induces the formation of organized lipid microdomains, which prevent LAMP-2A multimerization and stimulate subsequent degradation through proteolysis by Cathepsin D (CatD) (133). Independently of the mechanism, a reduction in CMA activity can induce an increase in the levels of some proteins related with the development of neurodegenerative diseases. In the case of tau, α-synuclein or other less characterized CMA protein substrates that are able to aggregate, increases the total mass that these proteins would contribute to protein oligomerization, an event associated with a blockade of CMA transport. In addition, oxidative damage, would enhance the aggregation process. Additionally, changes in signaling pathways that control LAMP2A oligomerization and membrane stabilization can alter LAMP-2A membrane dynamics and CMA activity. Finally, CMA down-regulation can alter a series of other specific proteins related with neuronal survival or neuroinflammation, that participate in the development of neurodegenerative diseases.
Concluding Remarks
As part of the autophagic pathways, CMA has been involved in regulating the metabolism of different proteins. Many of these proteins are strongly associated with neurodegenerative diseases affecting humans, while others are involved in the regulation of metabolic pathways such as lipids and carbohydrate metabolism. In addition, neurodegenerative diseases and metabolic disorders are common features of aged humans and many reports associate the dysregulation in lipids or carbohydrate metabolism, with the risk of developing some type of neurodegenerative disease. It has also been reported that a high fat, or high carbohydrate diet (or glucose reach diet), can also increase neurodegeneration prevalence or progression. On the other hand, CMA activity, has been observed to decline with age and with a non-balanced dietary intake such as a high fat diet. Overall, CMA can be considered a common factor in the regulation of metabolic and neurodegenerative pathways and a dysregulation in CMA, provoked by normal aging, or by metabolic disorders induced by deficient nutritional habits, which could tilt the balance toward a pathological situation. Figure 3 shows an overview of how CMA could be connected with metabolic dysfunctions and neurodegenerative disorders.
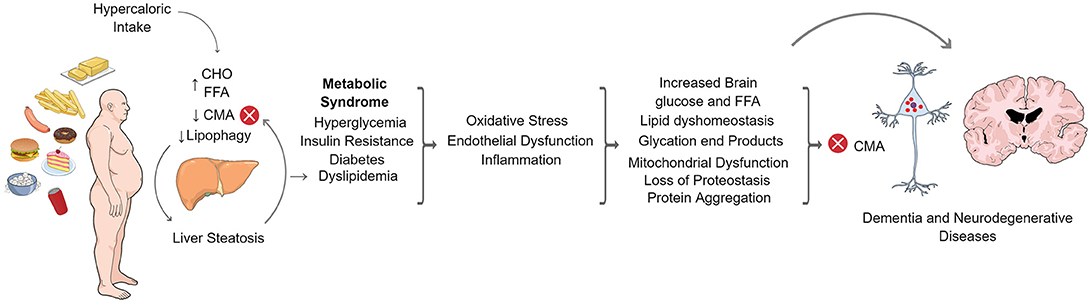
Figure 3. CMA in the crosstalk of metabolic dysfunction and neurodegenerative diseases. Increased caloric intake in humans produces elevated circulating levels of carbohydrates (CHO) and free fatty acids (FFA). In the liver, FFAs accumulate in lipid droplets leading to hepatic steatosis. FFAs also inhibit CMA and lipophagy. The latter contributes to hepatic fat accumulation and systemic dyslipidemia that, together with increased levels of glucose, leads to metabolic syndrome characterized by insulin resistance and diabetes. Metabolic syndrome is a general status also characterized by oxidative stress, inflammation and endothelial dysfunction. This condition is present throughout the body and is also capable of affecting the nervous system, resulting in lipid dyshomeostasis, mitochondrial dysfunction, loss of proteostasis and protein aggregation in the brain. All of these processes have been shown to alter proteostatic mechanisms, including CMA. Loss of neuronal proteostasis finally alters neuronal function which leads to dementia or the development of neurodegenerative diseases.
Author Contributions
AA and MB wrote the article. IEA wrote the article and created the figures. AM, JM, KC, and AC made important contributions, participated in discussions and provided corrections to the manuscript.
Funding
This work was supported by the Programa de Apoyo a Centros con Financiamiento Basal AFB 170004 to Fundación Ciencia & Vida. By CONICYT Fondo Nacional de Desarrollo Científico y Tecnológico de Chile, FONDECYT N°11161056 to IEA, FONDECYT N° 1161123 to MB, FONDECYT N° 1171075 to AC. By FONDAP-ACCDis N° 15130011 to AC. By the PEW Latin American Fellows Program in the Biomedical Science to AC. By the International Centre for Genetic Engineering and Biotechnology, ICGEB, N° CRP/CH113-04RG to MB. This work was also funded by the CONICYT PIA ACT172066 to MB and AC.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. (2018) 19:365–81. doi: 10.1038/s41580-018-0001-6
2. Iwata J, Ezaki J, Komatsu M, Yokota S, Ueno T, Tanida I, et al. Excess peroxisomes are degraded by autophagic machinery in mammals. J Biol Chem. (2006) 281:4035–41. doi: 10.1074/jbc.M512283200
3. Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. (2013) 14:759–74. doi: 10.1038/nrm3696
4. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. (2008) 183:795–803. doi: 10.1083/jcb.200809125
5. Grasso D, Ropolo A, Lo Re A, Boggio V, Molejon MI, Iovanna JL, et al. Zymophagy, a novel selective autophagy pathway mediated by VMP1-USP9x-p62, prevents pancreatic cell death. J Biol Chem. (2011) 286:8308–24. doi: 10.1074/jbc.M110.197301
6. Agarraberes FA, Dice JF. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci. (2001) 114:2491–9.
7. Cuervo AM, Knecht E, Terlecky SR, Dice JF. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol. (1995) 269:C1200–8. doi: 10.1152/ajpcell.1995.269.5.C1200
8. Tekirdag K, Cuervo AM. Chaperone-mediated autophagy and endosomal microautophagy: joint by a chaperone. J Biol Chem. (2018) 293:5414–24. doi: 10.1074/jbc.R117.818237
9. Isenman LD, Dice JF. Secretion of intact proteins and peptide fragments by lysosomal pathways of protein degradation. J Biol Chem. (1989) 264:21591–6.
10. Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol. (2011) 23:184–9. doi: 10.1016/j.ceb.2010.10.009
11. Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. (1990) 15:305–9. doi: 10.1016/0968-0004(90)90019-8
12. Wing SS, Chiang HL, Goldberg AL, Dice JF. Proteins containing peptide sequences related to Lys-Phe-Glu-Arg-Gln are selectively depleted in liver and heart, but not skeletal muscle, of fasted rats. Biochem J. (1991) 275(Pt 1):165–9. doi: 10.1042/bj2750165
13. Rout AK, Strub MP, Piszczek G, Tjandra N. Structure of transmembrane domain of lysosome-associated membrane protein type 2a (LAMP-2A) reveals key features for substrate specificity in chaperone-mediated autophagy. J Biol Chem. (2014) 289:35111–23. doi: 10.1074/jbc.M114.609446
14. Arias E. Methods to study chaperone-mediated autophagy. Methods Enzymol. (2017) 588:283–305. doi: 10.1016/bs.mie.2016.10.009
15. Kaushik S, Cuervo AM. Chaperone-mediated autophagy. Methods Mol Biol. (2008) 445:227–44. doi: 10.1007/978-1-59745-157-4_15
16. Patel B, Cuervo AM. Methods to study chaperone-mediated autophagy. Methods (2015) 75:133–40. doi: 10.1016/j.ymeth.2015.01.003
17. McElligott MA, Miao P, Dice JF. Lysosomal degradation of ribonuclease A and ribonuclease S-protein microinjected into the cytosol of human fibroblasts. J Biol Chem. (1985) 260:11986–93.
18. McElligott MA, Dice JF. Degradation of microinjected ribonuclease A and ribonuclease S-protein by lysosomal pathways. Prog Clin Biol Res. (1985) 180:471–3.
19. Dice JF, Backer JM, Miao P, Bourret L, McElligott MA. Regulation of catabolism of ribonuclease A microinjected into human fibroblasts. Prog Clin Biol Res. (1985) 180:385–94.
20. Chiang HL, Dice JF. Peptide sequences that target proteins for enhanced degradation during serum withdrawal. J Biol Chem. (1988) 263:6797–805.
21. Cuervo AM, Gomes AV, Barnes JA, Dice JF. Selective degradation of annexins by chaperone-mediated autophagy. J Biol Chem. (2000) 275:33329–35. doi: 10.1074/jbc.M005655200
22. Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science (1989) 246:382–5. doi: 10.1126/science.2799391
23. Stricher F, Macri C, Ruff M, Muller S. HSPA8/HSC70 chaperone protein: structure, function, and chemical targeting. Autophagy (2013) 9:1937–54. doi: 10.4161/auto.26448
24. Ferreira JV, Soares AR, Ramalho JS, Pereira P, Girao H. K63 linked ubiquitin chain formation is a signal for HIF1A degradation by Chaperone-Mediated Autophagy. Sci Rep. (2015) 5:10210. doi: 10.1038/srep10210
25. Kaushik S, Cuervo AM. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy (2016) 12:432–8. doi: 10.1080/15548627.2015.1124226
26. Quintavalle C, Di Costanzo S, Zanca C, Tasset I, Fraldi A, Incoronato M, et al. Phosphorylation-regulated degradation of the tumor-suppressor form of PED by chaperone-mediated autophagy in lung cancer cells. J Cell Physiol. (2014) 229:1359–68. doi: 10.1002/jcp.24569
27. Bonhoure A, Vallentin A, Martin M, Senff-Ribeiro A, Amson R, Telerman A, et al. Acetylation of translationally controlled tumor protein promotes its degradation through chaperone-mediated autophagy. Eur J Cell Biol (2017) 96:83–98. doi: 10.1016/j.ejcb.2016.12.002
28. Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell (2011) 42:719–30. doi: 10.1016/j.molcel.2011.04.025
29. Thompson LM, Aiken CT, Kaltenbach LS, Agrawal N, Illes K, Khoshnan A, et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. (2009) 187:1083–99. doi: 10.1083/jcb.200909067
30. Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science (1996) 273:501–3. doi: 10.1126/science.273.5274.501
31. Cuervo AM, Dice JF. Unique properties of lamp2a compared to other lamp2 isoforms. J Cell Sci. (2000) 113(Pt 24):4441–50.
32. Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol. (2008) 28:5747–63. doi: 10.1128/MCB.02070-07
33. Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R, Cuervo AM. Identification of regulators of chaperone-mediated autophagy. Mol Cell (2010) 39:535–47. doi: 10.1016/j.molcel.2010.08.004
34. Arias E, Koga H, Diaz A, Mocholi E, Patel B, Cuervo AM. Lysosomal mTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol Cell (2015) 59:270–84. doi: 10.1016/j.molcel.2015.05.030
35. Agarraberes FA, Terlecky SR, Dice JF. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol. (1997) 137:825–34. doi: 10.1083/jcb.137.4.825
36. Koga H, Martinez-Vicente M, Macian F, Verkhusha VV, Cuervo AM. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat Commun. (2011) 2:386. doi: 10.1038/ncomms1393
37. Finn PF, Dice JF. Ketone bodies stimulate chaperone-mediated autophagy. J Biol Chem. (2005) 280:25864–70. doi: 10.1074/jbc.M502456200
38. Park C, Suh Y, Cuervo AM. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun. (2015) 6:6823. doi: 10.1038/ncomms7823
39. Wu JH, Guo JP, Shi J, Wang H, Li LL, Guo B, et al. CMA down-regulates p53 expression through degradation of HMGB1 protein to inhibit irradiation-triggered apoptosis in hepatocellular carcinoma. World J Gastroenterol. (2017) 23:2308–17. doi: 10.3748/wjg.v23.i13.2308
40. Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell (2004) 15:4829–40. doi: 10.1091/mbc.e04-06-0477
41. Zhang L, Sun Y, Fei M, Tan C, Wu J, Zheng J, et al. Disruption of chaperone-mediated autophagy-dependent degradation of MEF2A by oxidative stress-induced lysosome destabilization. Autophagy (2014) 10:1015–35. doi: 10.4161/auto.28477
42. Wang Y, Singh R, Xiang Y, Czaja MJ. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology (2010) 52:266–77. doi: 10.1002/hep.23645
43. Pajares M, Rojo AI, Arias E, Diaz-Carretero A, Cuervo AM, Cuadrado A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy (2018) 14:1310–22. doi: 10.1080/15548627.2018.1474992
44. Dohi E, Tanaka S, Seki T, Miyagi T, Hide I, Takahashi T, et al. Hypoxic stress activates chaperone-mediated autophagy and modulates neuronal cell survival. Neurochem Int. (2012) 60:431–42. doi: 10.1016/j.neuint.2012.01.020
45. Hubbi ME, Hu H, Kshitiz, Ahmed I, Levchenko A, Semenza GL. Chaperone-mediated autophagy targets hypoxia-inducible factor-1alpha (HIF-1alpha) for lysosomal degradation. J Biol Chem. (2013) 288:10703–14. doi: 10.1074/jbc.M112.414771
46. Koga H, Martinez-Vicente M, Arias E, Kaushik S, Sulzer D, Cuervo AM. Constitutive upregulation of chaperone-mediated autophagy in Huntington's disease. J Neurosci. (2011) 31:18492–505. doi: 10.1523/JNEUROSCI.3219-11.2011
47. Li W, Yang Q, Mao Z. Signaling and induction of chaperone-mediated autophagy by the endoplasmic reticulum under stress conditions. Autophagy (2018) 14:1094–96. doi: 10.1080/15548627.2018.1444314
48. Li W, Zhu J, Dou J, She H, Tao K, Xu H, et al. Phosphorylation of LAMP2A by p38 MAPK couples ER stress to chaperone-mediated autophagy. Nat Commun. (2017) 8:1763. doi: 10.1038/s41467-017-01609-x
49. Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA. Lysosomal degradation of alpha-synuclein in vivo. J Biol Chem. (2010) 285:13621–9. doi: 10.1074/jbc.M109.074617
50. Wolfson RL, Sabatini DM. The dawn of the age of amino acid sensors for the mTORC1 pathway. Cell Metab. (2017) 26:301–9. doi: 10.1016/j.cmet.2017.07.001
51. Anthony TG. Homeostatic responses to amino acid insufficiency. Anim Nutr. (2015) 1:135–7. doi: 10.1016/j.aninu.2015.10.001
52. Castilho BA, Shanmugam R, Silva RC, Ramesh R, Himme BM, Sattlegger E. Keeping the eIF2 alpha kinase Gcn2 in check. Biochim Biophys Acta (2014) 1843:1948–68. doi: 10.1016/j.bbamcr.2014.04.006
53. Dikic I. Proteasomal and autophagic degradation systems. Annu Rev Biochem. (2017) 86:193–224. doi: 10.1146/annurev-biochem-061516-044908
54. Sorokin AV, Kim ER, Ovchinnikov LP. Proteasome system of protein degradation and processing. Biochemistry (2009) 74:1411–42. doi: 10.1134/S000629790913001X
55. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. (2014) 24:24–41. doi: 10.1038/cr.2013.168
56. Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. (2018) 20:233–242. doi: 10.1038/s41556-018-0037-z
57. Massey A, Kiffin R, Cuervo AM. Pathophysiology of chaperone-mediated autophagy. Int J Biochem Cell Biol. (2004) 36:2420–34. doi: 10.1016/j.biocel.2004.04.010
58. Lai MC, Chang CM, Sun HS. Hypoxia induces autophagy through translational up-regulation of lysosomal proteins in human colon cancer cells. PLoS ONE (2016) 11:e0153627. doi: 10.1371/journal.pone.0153627
59. Puertollano R, Ferguson SM, Brugarolas J, Ballabio A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. (2018) 37:e98804. doi: 10.15252/embj.201798804
60. Martina JA, Puertollano R. TFEB and TFE3: the art of multi-tasking under stress conditions. Transcription (2017) 8:48–54. doi: 10.1080/21541264.2016.1264353
61. Le Y, Zhang S, Ni J, You Y, Luo K, Yu Y, et al. Sorting nexin (2018) 10 controls mTOR activation through regulating amino-acid metabolism in colorectal cancer. Cell Death Dis. 9:666. doi: 10.1038/s41419-018-0719-2
62. Han Q, Deng Y, Chen S, Chen R, Yang M, Zhang Z, et al. Downregulation of ATG5-dependent macroautophagy by chaperone-mediated autophagy promotes breast cancer cell metastasis. Sci Rep. (2017) 7:4759. doi: 10.1038/s41598-017-04994-x
63. Cuervo AM, Terlecky SR, Dice JF, Knecht E. Selective binding and uptake of ribonuclease A and glyceraldehyde-3-phosphate dehydrogenase by isolated rat liver lysosomes. J Biol Chem. (1994) 269:26374–80.
64. Zechner R, Madeo F, Kratky D. Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat Rev Mol Cell Biol. (2017) 18:671–84. doi: 10.1038/nrm.2017.76
65. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature (2009) 458:1131–5. doi: 10.1038/nature07976
66. Guo Y, Cordes KR, Farese RV Jr, Walther TC. Lipid droplets at a glance. J Cell Sci. (2009) 122:749–52. doi: 10.1242/jcs.037630
67. Sztalryd C, Brasaemle DL. The perilipin family of lipid droplet proteins: gatekeepers of intracellular lipolysis. Biochim Biophys Acta (2017) 1862:1221–32. doi: 10.1016/j.bbalip.2017.07.009
68. Cuervo AM, Mann L, Bonten EJ, A. d'Azzo, Dice JF. Cathepsin A regulates chaperone-mediated autophagy through cleavage of the lysosomal receptor. EMBO J. (2003) 22:47–59. doi: 10.1093/emboj/cdg002
69. Wang H, Sreenivasan U, Hu H, Saladino A, Polster BM, Lund LM, et al. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J Lipid Res. (2011) 52:2159–68. doi: 10.1194/jlr.M017939
70. Ramos SV, Turnbull PC, MacPherson RE, LeBlanc PJ, Ward WE, Peters SJ. Changes in mitochondrial perilipin 3 and perilipin 5 protein content in rat skeletal muscle following endurance training and acute stimulated contraction. Exp Physiol. (2015) 100:450–62. doi: 10.1113/expphysiol.2014.084434
71. Bell M, Wang H, Chen H, McLenithan JC, Gong DW, Yang RZ, et al. Consequences of lipid droplet coat protein downregulation in liver cells: abnormal lipid droplet metabolism and induction of insulin resistance. Diabetes (2008) 57:2037–45. doi: 10.2337/db07-1383
72. Chen W, Chang B, Wu X, Li L, Sleeman M, Chan L. Inactivation of Plin4 downregulates Plin5 and reduces cardiac lipid accumulation in mice. Am J Physiol Endocrinol Metab. (2013) 304:E770–9. doi: 10.1152/ajpendo.00523.2012
73. Li Z, Schulze RJ, Weller SG, Krueger EW, Schott MB, Zhang X, et al. A novel Rab10-EHBP1-EHD2 complex essential for the autophagic engulfment of lipid droplets. Sci Adv. (2016) 2:e1601470. doi: 10.1126/sciadv.1601470
74. Wang P, Liu H, Wang Y, Liu O, Zhang J, Gleason A, et al. RAB-10 Promotes EHBP-1 bridging of filamentous actin and tubular recycling endosomes. PLoS Genet. (2016) 12:e1006093. doi: 10.1371/journal.pgen.1006093
75. Cai B, Giridharan SS, Zhang J, Saxena S, Bahl K, Schmidt JA, et al. Differential roles of C-terminal Eps15 homology domain proteins as vesiculators and tubulators of recycling endosomes. J Biol Chem. (2013) 288:30172–80. doi: 10.1074/jbc.M113.488627
76. Martinez-Lopez N, Garcia-Macia M, Sahu S, Athonvarangkul D, Liebling E, Merlo P, et al. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab. (2016) 23:113–27. doi: 10.1016/j.cmet.2015.10.008
77. Schneider JL, Suh Y, Cuervo AM. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. (2014) 20:417–32. doi: 10.1016/j.cmet.2014.06.009
78. Kaushik S, Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol. (2015) 17:759–70. doi: 10.1038/ncb3166
79. Mony VK, Benjamin S, O'Rourke EJ. A lysosome-centered view of nutrient homeostasis. Autophagy (2016) 12:619–31. doi: 10.1080/15548627.2016.1147671
80. Aniento F, Roche E, Cuervo AM, Knecht E. Uptake and degradation of glyceraldehyde-3-phosphate dehydrogenase by rat liver lysosomes. J Biol Chem. (1993) 268:10463–70.
81. Xia HG, Najafov A, Geng J, Galan-Acosta L, Han X, Guo Y, et al. Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J Cell Biol. (2015) 210:705–16. doi: 10.1083/jcb.201503044
82. Bockus LB, Matsuzaki S, Vadvalkar SS, Young ZT, Giorgione JR, Newhardt MF, et al. Cardiac insulin signaling regulates glycolysis through phosphofructokinase 2 Content and Activity. J Am Heart Assoc. (2017) 6:e007159. doi: 10.1161/JAHA.117.007159
83. Susan PP, Dunn WA Jr. Starvation-induced lysosomal degradation of aldolase B requires glutamine 111 in a signal sequence for chaperone-mediated transport. J Cell Physiol. (2001) 187:48–58. doi: 10.1002/1097-4652(2001)9999:9999<00::AID-JCP1050>3.0.CO;2-I
84. McGuire CM, Forgac M. Glucose starvation increases V-ATPase assembly and activity in mammalian cells through AMP kinase and phosphatidylinositide 3-kinase/Akt signaling. J Biol Chem. (2018) 293:9113–23. doi: 10.1074/jbc.RA117.001327
85. Nwadike C, Williamson LE, Gallagher LE, Guan JL, Chan EYW. AMPK inhibits ULK1-dependent autophagosome formation and lysosomal acidification via distinct mechanisms. Mol Cell Biol. (2018) 38:e00023-18. doi: 10.1128/MCB.00023-18
86. Li L, Friedrichsen HJ, Andrews S, Picaud S, Volpon L, Ngeow K, et al. A TFEB nuclear export signal integrates amino acid supply and glucose availability. Nat Commun. (2018) 9:2685. doi: 10.1038/s41467-018-04849-7
87. Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. (2008) 283:23542–56. doi: 10.1074/jbc.M801992200
88. Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. (2008) 118:777–88. doi: 10.1172/JCI32806
89. Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE (2009) 4:e5515. doi: 10.1371/journal.pone.0005515
90. Xilouri M, Brekk OR, Polissidis A, Chrysanthou-Piterou M, Kloukina I, Stefanis L. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy (2016) 12:2230–47. doi: 10.1080/15548627.2016.1214777
91. Issa AR, Sun J, Petitgas C, Mesquita A, Dulac A, Robin M, et al. The lysosomal membrane protein LAMP2A promotes autophagic flux and prevents SNCA-induced Parkinson disease-like symptoms in the Drosophila brain. Autophagy (2018) 14:1898–910. doi: 10.1080/15548627.2018.1491489
92. Wang B, Cai Z, Tao K, Zeng W, Lu F, Yang R, et al. Essential control of mitochondrial morphology and function by chaperone-mediated autophagy through degradation of PARK7. Autophagy (2016) 12:1215–28. doi: 10.1080/15548627.2016.1179401
93. Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ, et al. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science (2009) 323:124–7. doi: 10.1126/science.1166088
94. Murphy KE, Gysbers AM, Abbott SK, Spiro AS, Furuta A, Cooper A, et al. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson's disease. Move Disord. (2015) 30:1639–47. doi: 10.1002/mds.26141
95. Sala G, Stefanoni G, Arosio A, Riva C, Melchionda L, Saracchi E, et al. Reduced expression of the chaperone-mediated autophagy carrier hsc70 protein in lymphomonocytes of patients with Parkinson's disease. Brain Res. (2014) 1546:46–52. doi: 10.1016/j.brainres.2013.12.017
96. Pang S, Chen D, Zhang A, Qin X, Yan B. Genetic analysis of the LAMP-2 gene promoter in patients with sporadic Parkinson's disease. Neurosci Lett. (2012) 526:63–7. doi: 10.1016/j.neulet.2012.07.044
97. Alvarez-Erviti L, Seow Y, Schapira AH, Rodriguez-Oroz MC, Obeso JA, Cooper JM. Influence of microRNA deregulation on chaperone-mediated autophagy and alpha-synuclein pathology in Parkinson's disease. Cell Death Dis. (2013) 4:e545. doi: 10.1038/cddis.2013.73
98. Liu H, Wang P, Song W, Sun X. Degradation of regulator of calcineurin 1 (RCAN1) is mediated by both chaperone-mediated autophagy and ubiquitin proteasome pathways. FASEB J. (2009) 23:3383–92. doi: 10.1096/fj.09-134296
99. Valdor R, Mocholi E, Botbol Y, Guerrero-Ros I, Chandra D, Koga H,., et al. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat Immunol. (2014) 15:1046–54. doi: 10.1038/ni.3003
100. Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Cuervo M, Wong E, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. (2009) 18:4153–70. doi: 10.1093/hmg/ddp367
101. Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, et al. Synergy and antagonism of macroautophagy and chaperone-mediated autophagy in a cell model of pathological tau aggregation. Autophagy (2010) 6:182–3. doi: 10.4161/auto.6.1.10815
102. Park JS, Kim DH, Yoon SY. Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep. (2016) 49:337–42. doi: 10.5483/BMBRep.2016.49.6.212
103. Qi L, Zhang XD, Wu JC, Lin F, Wang J, DiFiglia M, et al. The role of chaperone-mediated autophagy in huntingtin degradation. PLoS ONE (2012) 7:e46834. doi: 10.1371/journal.pone.0046834
104. Bauer PO, Goswami A, Wong HK, Okuno M, Kurosawa M, Yamada M, et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat Biotechnol. (2010) 28:256–63. doi: 10.1038/nbt.1608
105. Huang CC, Bose JK, Majumder P, Lee KH, Huang JT, Huang JK, et al. Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J Cell Sci. (2014) 127:3024–38. doi: 10.1242/jcs.136150
106. Tamaki Y, Shodai A, Morimura T, Hikiami R, Minamiyama S, Ayaki T, et al. Elimination of TDP-43 inclusions linked to amyotrophic lateral sclerosis by a misfolding-specific intrabody with dual proteolytic signals. Sci Rep. (2018) 8:6030. doi: 10.1038/s41598-018-24463-3
107. Marino G, Fernandez AF, Lopez-Otin C. Autophagy and aging: lessons from progeria models. Adv Exp Med Biol. (2010) 694:61–8. doi: 10.1007/978-1-4419-7002-2_6
108. Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. (2000) 275:31505–13. doi: 10.1074/jbc.M002102200
109. Kiffin R, Kaushik S, Zeng M, Bandyopadhyay U, Zhang C, Massey AC, et al. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J Cell Sci. (2007) 120:782–91. doi: 10.1242/jcs.001073
110. Schneider JL, Villarroya J, Diaz-Carretero A, Patel B, Urbanska AM, Thi M. M, et al. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell (2015) 14:249–64. doi: 10.1111/acel.12310
111. Papagiannakis N, Xilouri M, Koros C, Stamelou M, Antonelou R, Maniati M, et al. Lysosomal alterations in peripheral blood mononuclear cells of Parkinson's disease patients. Mov Disord. (2015) 30:1830–4. doi: 10.1002/mds.26433
112. Klaver AC, Coffey MP, Aasly JO, Loeffler DA. CSF lamp2 concentrations are decreased in female Parkinson's disease patients with LRRK2 mutations. Brain Res. (2018) 1683:12–16. doi: 10.1016/j.brainres.2018.01.016
113. Loeffler DA, Klaver AC, Coffey MP, Aasly JO. Cerebrospinal fluid concentration of key autophagy protein lamp2 changes little during normal aging. Front Aging Neurosci. (2018) 10:130. doi: 10.3389/fnagi.2018.00130
114. Rodriguez-Navarro JA, Kaushik S, Koga H, Dall'Armi C, Shui G, Wenk MR, et al. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci USA (2012) 109:E705–14. doi: 10.1073/pnas.1113036109
115. Khang R, Park C, Shin JH. Dysregulation of parkin in the substantia nigra of db/db and high-fat diet mice. Neuroscience (2015) 294:182–92. doi: 10.1016/j.neuroscience.2015.03.017
116. Rotermund C, Truckenmuller FM, Schell H, Kahle PJ. Diet-induced obesity accelerates the onset of terminal phenotypes in alpha-synuclein transgenic mice. J Neurochem. (2014) 131:848–58. doi: 10.1111/jnc.12813
117. Kim D, Cho J, Lee I, Jin Y, Kang H. Exercise attenuates high-fat diet-induced disease progression in 3xTg-AD Mice. Med Sci Sports Exerc. (2017) 49:676–86. doi: 10.1249/MSS.0000000000001166
118. Julien C, Tremblay C, Phivilay A, Berthiaume L, Emond V, Julien P, et al. High-fat diet aggravates amyloid-beta and tau pathologies in the (2010) 3xTg-AD mouse model. Neurobiol Aging 31:1516–31. doi: 10.1016/j.neurobiolaging.2008.08.022
119. Pennetta G, Welte MA. Emerging links between lipid droplets and motor neuron diseases. Dev Cell (2018) 45:427–32. doi: 10.1016/j.devcel.2018.05.002
120. Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL. Lipid droplet binding and oligomerization properties of the Parkinson's disease protein alpha-synuclein. J Biol Chem. (2002) 277:6344–52. doi: 10.1074/jbc.M108414200
121. Sun X, Ueda M, Yamashita T, Nakamura M, Bergstrom J, Zeledon Ramirez ME, et al. Lipid droplets are present in amyloid deposits in familial amyloidotic polyneuropathy and dialysis related amyloidosis. Amyloid (2006) 13:20–3. doi: 10.1080/13506120500537137
122. Taylor MK, Sullivan DK, Swerdlow RH, Vidoni ED, Morris JK, Mahnken JD, et al. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am J Clin Nutr. (2017) 106:1463–70. doi: 10.3945/ajcn.117.162263
123. Vicente Miranda H, Outeiro TF. The sour side of neurodegenerative disorders: the effects of protein glycation. J Pathol. (2010) 221:13–25. doi: 10.1002/path.2682
124. Liu F, Zaidi T, Iqbal K, Grundke-Iqbal I, Gong CX. Aberrant glycosylation modulates phosphorylation of tau by protein kinase A and dephosphorylation of tau by protein phosphatase 2A and 5. Neuroscience (2002) 115:829–37. doi: 10.1016/S0306-4522(02)00510-9
125. Valente T, Gella A, Fernandez-Busquets X, Unzeta M, Durany N. Immunohistochemical analysis of human brain suggests pathological synergism of Alzheimer's disease and diabetes mellitus. Neurobiol Dis. (2010) 37:67–76. doi: 10.1016/j.nbd.2009.09.008
126. Seneff S, Wainwright G, Mascitelli L. Nutrition and Alzheimer's disease: the detrimental role of a high carbohydrate diet. Eur J Intern Med. (2011) 22:134–40. doi: 10.1016/j.ejim.2010.12.017
127. Dunn L, Allen GF, Mamais A, Ling H, Li A, Duberley KE, et al. Dysregulation of glucose metabolism is an early event in sporadic Parkinson's disease. Neurobiol Aging (2014) 35:1111–5. doi: 10.1016/j.neurobiolaging.2013.11.001
128. Bigl M, Bruckner MK, Arendt T, Bigl V, Eschrich K. Activities of key glycolytic enzymes in the brains of patients with Alzheimer's disease. J Neural Transm (Vienna) (1999) 106:499–511. doi: 10.1007/s007020050174
129. Barinova K, Khomyakova E, Semenyuk P, Schmalhausen E, Muronetz V. Binding of alpha-synuclein to partially oxidized glyceraldehyde-3-phosphate dehydrogenase induces subsequent inactivation of the enzyme. Arch Biochem Biophys. (2018) 642:10–22. doi: 10.1016/j.abb.2018.02.002
130. Gouarne C, Tardif G, Tracz J, Latyszenok V, Michaud M, Clemens LE, et al. Early deficits in glycolysis are specific to striatal neurons from a rat model of huntington disease. PLoS ONE (2013) 8:e81528. doi: 10.1371/journal.pone.0081528
131. Estrada-Sanchez AM, Montiel T, Massieu L. Glycolysis inhibition decreases the levels of glutamate transporters and enhances glutamate neurotoxicity in the R6/2 Huntington's disease mice. Neurochem Res. (2010) 35:1156–63. doi: 10.1007/s11064-010-0168-5
132. Rodriguez-Navarro JA, Cuervo AM. Dietary lipids and aging compromise chaperone-mediated autophagy by similar mechanisms. Autophagy (2012) 8:1152–4. doi: 10.4161/auto.20649
Keywords: CMA, neurodegeneration, lipids, carbohydrates, metabolism
Citation: Alfaro IE, Albornoz A, Molina A, Moreno J, Cordero K, Criollo A and Budini M (2019) Chaperone Mediated Autophagy in the Crosstalk of Neurodegenerative Diseases and Metabolic Disorders. Front. Endocrinol. 9:778. doi: 10.3389/fendo.2018.00778
Received: 19 September 2018; Accepted: 11 December 2018;
Published: 31 January 2019.
Edited by:
Maria Ines Vaccaro, Universidad de Buenos Aires, ArgentinaReviewed by:
Angelo Poletti, University of Milan, ItalyCarsten Merkwirth, Ferring Research Institute, Inc., United States
Copyright © 2019 Alfaro, Albornoz, Molina, Moreno, Cordero, Criollo and Budini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Iván E. Alfaro, aWFsZmFyb0BjaWVuY2lhdmlkYS5vcmc=
Mauricio Budini, bWJ1ZGluaUB1LnVjaGlsZS5jbA==