 Ilze Mentoor
Ilze Mentoor Anna-Mart Engelbrecht
Anna-Mart Engelbrecht Paul J. van Jaarsveld
Paul J. van Jaarsveld Theo Nell
Theo Nell- 1Department of Physiological Sciences, Faculty of Science, Stellenbosch University, Stellenbosch, South Africa
- 2Non-Communicable Diseases Research Unit, South African Medical Research Council, Cape Town, South Africa
- 3Division of Medical Physiology, Faculty of Medicine and Health Sciences, Stellenbosch University, Stellenbosch, South Africa
Excess adipose tissue is a hallmark of an overweight and/or obese state as well as a primary risk factor for breast cancer development and progression. In an overweight/obese state adipose tissue becomes dysfunctional due to rapid hypertrophy, hyperplasia, and immune cell infiltration which is associated with sustained low-grade inflammation originating from dysfunctional adipokine synthesis. Evidence also supports the role of excess adipose tissue (overweight/obesity) as a casual factor for the development of chemotherapeutic drug resistance. Obesity-mediated effects/modifications may contribute to chemotherapeutic drug resistance by altering drug pharmacokinetics, inducing chronic inflammation, as well as altering tumor-associated adipocyte adipokine secretion. Adipocytes in the breast tumor microenvironment enhance breast tumor cell survival and decrease the efficacy of chemotherapeutic agents, resulting in chemotherapeutic resistance. A well-know chemotherapeutic agent, doxorubicin, has shown to negatively impact adipose tissue homeostasis, affecting adipose tissue/adipocyte functionality and storage. Here, it is implied that doxorubicin disrupts adipose tissue homeostasis affecting the functionality of adipose tissue/adipocytes. Although evidence on the effects of doxorubicin on adipose tissue/adipocytes under obesogenic conditions are lacking, this narrative review explores the potential role of obesity in breast cancer progression and treatment resistance with inflammation as an underlying mechanism.
Introduction
Breast cancer continues to be a major health risk for women globally (1, 2). Lifestyle-related risk factors including overweight and obesity (adiposity) have reached epidemic proportions (3, 4), and are considered major risk factors for breast cancer development and progression (5).
Adipose tissue plays an important physiological role as a metabolically active storage compartment and endocrine organ due to its diverse ability to secrete various adipokines (6). Adipose tissue dysfunction in relation to obesity has been linked to accelerated growth and the survival of breast cancer cells (7, 8).
Adipose tissue dysfunction is mainly characterized by inflammation which is primarily mediated by rapid adipose tissue remodeling (hypertrophy and hyperplasia) (9). This results in dysfunctional synthesis of several adipokines in coordination with immune cell infiltration leading to a state of sustained low-grade inflammation, which activates downstream signaling pathways favoring cancer cell survival (increased proliferation and decreased apoptosis) and hence contributing to cancer progression and metastasis (10–12).
Furthermore, adipose tissue and/or adipocytes in the tumor microenvironment serve as an exogenous energy source for the survival of breast cancer cells (13, 14), especially since adipose tissue is abundant in the breast (15). It is further proposed that breast cancer cells modulate lipid metabolism by altering the secretion of adipokines through adipocytes, resulting in the release of free fatty acid (FFA) providing energy substrates, that cancer cells need to sustain its high proliferation demand (13).
Pre-clinical evidence highlights obesity as a key player in breast cancer chemotherapeutic drug resistance (16–19). This finding bear's great clinical significance for overweight/obese breast cancer patients being treated with chemotherapeutic agents such as doxorubicin (20), since, obese and normal weight patients receive the same treatment regimen (21). Studies have also confirmed this in showing that obesity is associated with poor clinical outcomes in breast cancer patients treated with chemotherapeutic agents including doxorubicin (22, 23).
Doxorubicin is a highly sensitive alkylating antineoplastic agent used as a first line adjuvant regimen for breast cancer patients (24), despite its high sensitivity as a chemotherapeutic agent, it is also associated with a diverse range of cellular toxicities and the development of treatment resistance (25). Additionally, doxorubicin also negatively impacts on adipose tissue function (26–29). This is of clinical significance since obesity is associated with an increased risk for various types of cancers being treated with doxorubicin (20). However, few studies exist in which the effects of doxorubicin on adipose tissue in the context of obesity and dysfunctional adipose tissue is investigated. We proposed that using doxorubicin treatment on dysfunctional adipose tissue and/or adipocytes, may exacerbate the negative effects of obesity per se, and further dysregulate adipokine secretion.
It is imperative to explore and understand the cellular mechanisms whereby obesity negatively affects chemotherapy outcomes. Identifying molecular mechanisms in which doxorubicin affect adipose tissue could contribute in describing molecular mechanisms and identifying potential novel pharmacologic targets and development of the appropriate management protocols of doxorubicin related toxicities in order to improve over-all survival of these cancer patients. This narrative review will mainly focus on (i) the pathological links between adiposity and breast cancer in the context of inflammation as an underlying mechanism and, (ii) the role of adiposity in breast cancer treatment (doxorubicin) resistance and the possible mechanisms that contribute to treatment resistance.
Adiposity and Breast Cancer
Globally, the increasing burden of breast cancer is considered the second most prevalent cancer diagnosed amongst women (1, 30) in both developed and developing countries (2, 31). Estimations rank breast cancer as the fifth leading cause of death globally at 626,679 deaths per annum (1, 30).
Despite many efforts to reduce cancer mortality by implementing lifestyle-related modifications, limited progress has been made due to the very complicated interplay between dietary behaviors and other lifestyle modifications (32, 33). This is especially problematic since recent epidemiological studies strongly suggested that adiposity (excess adipose tissue) is considered a significant risk factor in many lifestyle-associated cancers including breast cancer (34–37).
Adipose Tissue Is a Complex Functional Tissue
Fundamentally, adipose tissue is a complex and important endocrine organ impacting various physiological systems (38). It functions as both an energy storage compartment and a metabolic active endocrine organ (6), secreting various bioactive substances (pro- and anti-inflammatory) known as adipokines (39), including but not limited to leptin, adiponectin (Apn), tumor necrosis factor-alpha (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), resistin and macrophage chemoattractant protein-1 (MCP-1) (40, 41). Additionally, adipose tissue also plays a functional role in steroid sex hormone and growth factor production, and is integral in the development of insulin resistance, hyperglycaemia and breast cancer (42, 43). Epidemiological and experimental models support the role of a dysregulation in adipokine synthesis and their actions in relation to adiposity and adipose tissue dysfunction, to the development of various disease states, including breast cancer (5, 44–51).
Dysfunctional Adipose Tissue, Inflammation, and Breast Cancer
Dysfunctional adipose tissue is characterized by low grade inflammation, primarily mediated by rapid hypertrophy/hyperplasia (adipose tissue remodeling) as well as immune cell infiltration (52), resulting in the deregulated synthesis of several adipokines i.e., IL-1β, IL-6, interleukin-8 (IL-8), resistin, leptin and MCP-1 (7, 8, 15, 53–55) (Figure 1). These inflammatory mediators attract monocytes (differentiated into macrophages) and T-lymphocytes, stimulating the synthesis of both pro-inflammatory and pro-angiogenic factors (56), collectively contributing to a chronic cycle that sustains an inflammatory milieu. Increased IL-6 and leptin levels has been shown to supress 5′ adenosine monophosphate-activated protein kinase (AMPK), well-known for its' anti-inflammatory effects in adipose tissue (57). Additionally, adipose tissue-induced inflammation also attenuates the suppression of nuclear factor kappa B (NFκB), p65 phosphorylation and also induces M1 to M2 macrophage phenotype switching. The latter is due to the pro-inflammatory state in which saturated fatty acids bind to the toll-like receptors on macrophages (58) and upregulate the secretion of various pro-inflammatory mediators (IL-1β and TNF-α) in adipose tissue (59, 60), creating a state of chronic systemic low-grade inflammation.
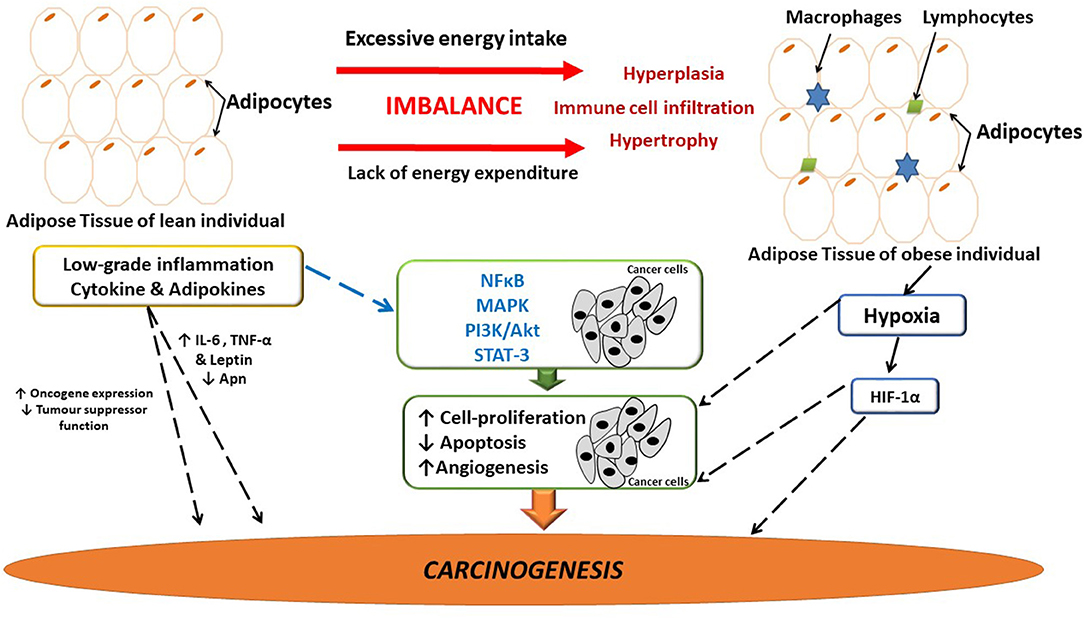
Figure 1. The link between adipose-induced inflammation and cancer. Adipose tissue dysfunction is associated with sustained low-grade inflammation and it may be linked to breast cancer development and progression. Several inflammatory mediators are implicated in tumor development and progression. Possibly as a result of the sustained inflammatory signaling having downstream effects on major pathways involved in angiogenesis, cell-proliferation and apoptosis, thus having the ability to influence carcinogenesis. Hypoxia in adipose tissue also induces the release of inflammatory mediators, thus further exacerbating inflammation. Apn, adiponectin; IL-6, interleukin-6; HIF-1α, hypoxia inducible factor-1α; MAPK, mitogen activated protein kinase; NFκB, nuclear factor kappa B; PI3K/Akt, phosphoinositide-3-kinase; STAT-3, Signal transducer and activator of transcription-3; TNF-α, tumor necrosis factor-α.
Inflammation is a well-known predisposing risk factor for tumorigenesis and a hallmark of cancer (61, 62). Pre-existing pro-inflammatory microenvironments are associated with an increased risk for cancer, as in the case for inflammatory breast cancer (63). Remarkable similarities exist between dysfunctional adipose tissue and the tumor microenvironment where infiltration of immune cells initiate the secretion of pro-inflammatory molecules, thereby sustaining and promoting the progression of both obesity and breast cancer (64) (Figure 1).
The complex pathophysiology that exists between adipose tissue, inflammation and breast cancer involves inflammatory mediators (i.e., IL-6 and TNF-α) that enhances tumor progression and survival (65). Persistent inflammatory signaling (intracellular NFκB) induces downstream effects on major biochemical pathways affecting carcinogenesis (Figure 1). For example, the mitogen activated protein kinase (MAPK) family modulates cellular proliferation via the phosphoinositide-3-kinase (PI3K/Akt), and the MAPK pathways which regulates and affects mitogenic, anti-apoptotic as well as pro-angiogenic effects (60). Moreover, IL-6 secreted by adipose tissue binds to IL-6 receptor on breast cancer cells and activates the Janus family of kinases that phosphorylates signal transducer and activator of transcription-3 (STAT-3) (66). These events induce the expression of pro-survival genes (i.e., bcl-x) (67), characteristic of a pro-carcinogenic state promoting breast cancer cell survival and proliferation (Figure 1).
Obesity-induced cytokine secretions are detected in local adipose tissue and serum (68). These elevated circulating cytokines (IL-6, IL-8, TNF-α, and vascular endothelial growth factor; VEGF), exert effects at distant sites (69), that can promote breast cancer development through upregulation of inflammatory mediator synthesis and increased immune cell infiltration as well as angiogenesis (70, 71). Additionally, overweight/obese patients display a large number of crown-like-structures (necrotic adipocytes surrounded by immune cells) in mammary adipose tissue compared to normal weight breast cancer patients. These crown-like structures are characteristic of local inflammation (66, 72), and associated with an upregulation of pro-inflammatory cytokines and aromatase expression (73). Although the role of cytokines in obesity and breast cancer development have been reported, the effects of other adipokines should be considered as a possible relationship between obesity and the development of breast cancer.
Leptin and adiponectin (Apn) have been antagonistically implicated for their roles in inflammation and tumorigenesis (Figure 1) (74). Leptin increases the synthesis of pro-inflammatory cytokines and plays a role in breast cancer development by increasing cellular proliferation and angiogenesis (75). Elevated serum leptin levels and increased expression of leptin receptors is also reported in breast cancer patients that is often associated with higher pathological grade tumors and cancer treatment resistance (76, 77). Adiponectin is decreased in obese patients, the metabolic syndrome as well as in breast cancer patients thus lowering the risk of cancer development due to an upregulation of apoptosis and its' anti-inflammatory properties (78–81).
Excess adipose tissue (overweight/obesity) is also associated with increased secretion of insulin-like growth factor-1 (IGF-1) in breast, colon, lung and prostate cancer patients (82). The over expression of insulin-like growth factor-1-receptor (IGF-1R) was observed in both breast and pancreatic tumor tissue (83), where inhibition of apoptosis and stimulation of cellular proliferation via the PI3K-AKT-mTOR and RAS/Raf/MEK pathways are implicated (83).
Additionally, loss of tumor suppressor function, increased cell cycling and stimulation of oncogenes also promote inflammation and exacerbates inflammatory related signaling pathways (69, 84, 85) (Figure 1). For example, the p53 gene mutation promotes inflammation in the tumor microenvironment by inducing the synthesis of IL-1, IL-6, TNF-α, and activates NFκB (86, 87), which maintains inflammation in the tumor microenvironment and enhances genomic instability (88, 89). In addition, p53 has also been shown to induce the PI3k/Akt/mTOR pathway, which can induce the synthesis of pro-inflammatory mediators (90). As a result of rapid hypertrophy and hyperplasia (91) (Figure 1), hypoxia inducible factor-1α (HIF-1α) is upregulated (92), which binds to transcription factors on VEGF and angiopoietin-2 target genes, stimulating angiogenesis in the microenvironment, which is also known to exacerbate local inflammation (92).
Others report that obesity-induced inflammation may also play a role in breast cancer tumor invasion and metastasis. Here, epithelial mesenchymal transition (EMT) can be induced by various pro-inflammatory markers, i.e., IL-6, IL-8, TNF-α, and CCL2 derived from cancer-associated adipocytes (93–95).
It is evident from these findings that obesity is a factor casual in the development of breast cancer, involving molecular mechanisms in relation to inflammation, immune cell infiltration and adipokine dysfunction. Supporting evidence includes obesity as a negative prognostic factor for breast cancer independent of menopausal status, tumor stage, and tumor hormone–binding characteristics (96, 97).
Breast Cancer Treatment
Chemotherapy still remains one of the conventional treatment options in addition to radiotherapy and surgery, which significantly improves cancer patients' overall-survival (98, 99). Several chemotherapeutic drug classes exist which are associated with beneficial clinical outcomes for cancer patients (100).
Doxorubicin, also known as Adriamycin or hydroxyl daunorubicin (101), is classified as an anthracycline antibiotic, exhibiting broad-spectrum anti-neoplastic activity (24, 101), and is used to treat a range of malignancies of the breast (used as first line adjuvant chemotherapeutic agent), bladder, stomach, lung, ovaries, thyroid as well as multiple myeloma, Hodgkin- and non-Hodgkin's lymphoma, due to poor tumor selectivity (102).
Doxorubicin interacts with deoxyribonucleic acid (DNA) by intercalation, thereby inhibiting macromolecule biosynthesis (103). This inhibits topoisomerase II (DNA repair function), which relaxes DNA transcription supercoils (104, 105). Secondly, doxorubicin generates reactive oxygen species (ROS) damaging cell membranes, DNA and proteins (103, 104) through stimulation of p53-DNA binding; subsequently it initiates caspase signaling and DNA cross-linking (106). Doxorubicin treatment efficacy is often associated with adverse side effects such as nephrotoxicity, hepatotoxicity, sarcopenia, cardiotoxicity (102), and changes in body composition (decreased body weight and lipoatrophy, discussed in section Doxorubicin Toxicity on Adipose Tissue/Adipocytes) (107, 108). These effects contribute toward recurrence as well as metastasis in breast cancer patients, making doxorubicin treatment protocols ineffective and prone to develop treatment resistance (109, 110).
Obesity and Treatment Resistance
Experimental animal models showed that diet-induced obesity increases tumor development, progression and metastasis with decreased chemotherapeutic efficacy (7, 8, 55, 111–113), specifically in the case of breast cancer (16–18). Additionally, obesity is associated with larger tumor sizes and positive lymph node involvement compared to non-obese breast cancer patients (114, 115).
Human studies also show that obesity is linked to poor clinical outcomes in breast cancer patients treated with chemotherapeutic, hormonal-based chemotherapy agents and radiotherapy (22, 116). Obesity was also associated with lower pathological complete response, disease free survival, clinical benefit rate and worse overall-survival (22). Iwase et al. reported that a high visceral fat area is associated with poor clinical outcomes for patients receiving neo-adjuvant chemotherapy (anthracycline followed by taxane) treatment regimens (19). In fact, treatment protocols for overweight and obese cancer patients includes prescribed lower doses of chemotherapeutic agents to avoid co-morbidities, side effects and adverse toxicities (117). This could also compromise drug efficacy and contribute to the development of treatment resistance and added cytotoxicity (118). However, alterations in dosages cannot clarify all occurrences of treatment resistance in relation to obesity (96, 119, 120).
Resistance Mechanisms
Drug resistance can either be classified as intrinsic (pre-treatment), or acquired (post treatment) (121). Currently, known drug resistance mechanisms include, the evasion of therapy-induced apoptosis, activation of drug transporter proteins and enhanced DNA repair mechanisms, which describe cellular mechanisms (109). Drug resistance can additionally ensue as a result of alterations in pharmacokinetics, drug inactivation and metabolism (109, 121), which can be induced and/or exacerbated in obese states.
Cellular mechanisms
Treatment resistance can develop due to the evasion of apoptotic pathways by increased anti-apoptotic protein (blc-2) and decreased pro-apoptotic protein (bax) expression (122). Adipocytes protect cancer cells from chemotherapeutic agents (i.e., vincristine and daunorubicin) by upregulating anti-apoptotic bcl-2, and downregulation of pro-apoptotic bad and pim-2 family members (an oncogene which phosphorylates bad) (123). Although the mechanisms by which adipocytes achieved this ‘protection of cancer cells' was not assessed, a recent in vitro study identified resistin (mainly secreted from adipose tissue) (124) as a causal factor for acquiring resistance to doxorubicin treatment in both the MCF-7 human breast cancer cell line (estrogen receptor positive) as well as in the MDA-MB-231 human triple negative cell line. Here, doxorubicin induced apoptosis (increased cytochrome-c concentration, cleaved caspase-9, cleaved PARP) in a time and dose dependant manner. Addition of recombinant resistin to the treatment protocol downregulated apoptosis by inducing autophagy (a self-degradation process which cancer cells can utilize to eliminate toxic materials to avoid cell death) (25). Although resistin receptor expression was not assessed, and no supporting evidence for animal or human models were provided, it would be plausible to motivate for more experimental research to investigate potential mechanisms and causal factors involved in acquiring doxorubicin treatment resistance.
Additionally, treatment resistance can also be the result of gene mutations coding for apoptotic proteins (125), for example the mutation in p53 has been associated with acquired resistance to doxorubicin in breast cancer patients, possibly due to inhibition of apoptosis by activating Bax/Bak (pro-apoptotic factors) (125). General-, and central obesity showed a positive association with mutations in p53 of tumor tissue, which was further associated with less favorable tumor characteristics including poorly differentiated and higher nuclear grade tumors (126).
Moreover, modifications in the activation and expression of drug transporter proteins alters drug responses by reducing intracellular drug concentration, which promotes treatment resistance (127). Examples are (i) P-glycoprotein (P-gp), (ii) multi-drug resistance protein-1 (MDR-1), (iii) multi-drug resistance associated protein-1 (MDRP-1), and (iv) breast cancer resistance protein (BCRP), which are ATP-binding cassette (ABC) transmembrane pumps responsible for the elimination of toxic compounds from cells (128, 129). Although normally expressed in healthy tissue, overexpression of P-gp, MDR-1, MDRP-1 and BCRP are present in breast cancer cells in relation to doxorubicin resistance (130–132). P-glycoprotein expression can also be upregulated by inflammation (NFκB), resulting in an altered expression of MDRP-1, which increases the expression of P-gp and consequently modify drug responses. (110, 133).
Adipose tissue is also a source of mesenchymal stem cells which share similar characteristics to tumor-initiating stem cells (134), which can be recruited to the tumor microenvironment to support breast tumor growth and proliferation (135). Tumor-initiating stem cells has the ability to self-renew and/or differentiate, tolerate high levels of DNA damage, increase ABC transmembrane transporter protein expression and induce the synthesis of various cytokines and growth factors (increased IL-6 and C-C motif ligand 5 (CXCL5) levels) (63, 135–137), and therefore may be an alternative treatment resistance mechanism. Elevated leptin concentrations and leptin receptor expression (increased in adiposity) is associated with the promotion of cancer stem cells survival and self-renewal, by inducing JAK2/STAT3 signaling pathways, that increase stem cell renewal transcription factors (NANOG, OCT-4, and SOX-2) expression in breast cancer cells (77, 138).
Leptin, well-known for its role in inflammation and tumorigenesis (74, 139), increases cellular proliferation and angiogenesis (75), and is also associated with higher pathological grade breast cancer tumors (76) and breast cancer treatment resistance (136, 140). However, in contrast leptin also shows anti-cancer effects, by enhancing the anti-proliferative effects of 3′-5′-cyclic adenosine monophosphate (cAMP) elevating agents in breast cancer cells (141). 3′-5′-cyclic adenosine monophosphate is an intracellular second messenger, generated from ATP by adenylate cyclase's (142, 143) and plays a regulatory role in cellular proliferation, apoptosis as well as differentiation, proposed to be induced via protein kinase A (144, 145). The utilization of cAMP elevating agents has been explored in pre-clinical models and shows anti-cancer effects i.e., inducing apoptosis (downregulation of Bcl-2 which leads to caspase-3 mediated apoptosis) (146) and cell cycle arrest (146, 147) in breast cancer cells. Additionally, cAMP elevating agents inhibit both cellular proliferation (148), and angiogenesis (decreased VEGF) (149) as well as sensitize breast cancer cells to chemotherapeutic drug treatments (150). Naviglio et al. showed that co-treatment of triple negative breast cancer cells (MDA-MB-231 cells) with leptin and a cAMP elevating agent (forskolin) decreased breast cancer cell proliferation by inhibiting the activation of the ERK signaling pathway (141), which is well-known to be over active in breast cancer cells (151, 152). Interestingly, the authors also showed that leptin enhanced the anti-proliferative effects of cAMP elevating agents, by inducing both apoptosis and cell cycle arrest (141). Additionally, Spina et al. showed that an increase in cAMP levels inhibits leptin-induced migration of breast cancer cells (MDA-MB-231) (153). Recently, Illiano et al. demonstrated that forskolin treatment (cAMP elevating agent) inhibited ERK1/2 activity via protein kinase A-mediated inhibition, which induced apoptosis and increased the sensitivity of breast cancer cells (MDA-MB-231 and MDA-MB-468) to doxorubicin treatment (154). This finding is significant since, doxorubicin treatment resistance in breast cancer cells involves the activation of the RAS/RAF/ERK signaling pathway (152, 155). The anti-proliferative interaction between leptin and cAMP elevating drugs might provide potentially new strategies for therapeutic intervention in overweight/obese breast cancer patients (since leptin levels are elevated in overweight/obese patients), who are at risk/prone to develop treatment resistance. However, treatment of breast cancer cells with cAMP elevating agents under obesogenic conditions, is yet to be explored, especially considering cAMP can stimulate lipolysis in adipose tissue (156).
Additional growth factors secreted by adipocytes also implicated in treatment resistance include IGF-1 and IGF-1R (increased systemic bioavailability in adiposity and adipocytes also secrete IGF-1). These growth factors are linked to decreased apoptosis, increased cancer cell proliferation and pro-inflammatory mediator secretion which are directly associated with breast cancer risk and progression (157–160). Upregulation of IGF-1R was associated with poor disease prognosis and chemotherapy resistance through increased expression of MDR-1 and MDRP-1 affecting drug transportation and delivery in cancer cells (161).
Acquired resistance to doxorubicin and docetaxel in breast cancer cells was also attributed to the transfer of microRNA present in exosomes (nanovesicles which mediates cell-cell transfer of DNA, mRNA, microRNA, proteins and lipids) (162). Adipocyte derived exosomes has been associated with increased migration in breast cancer cells (163), immune cell recruitment of macrophages and chronic inflammation (164, 165). Resistance to paclitaxel in ovarian cancer cells was attributed to the transfer of microRNA (miR21) present in adipocyte derived exosomes (166, 167), which downregulated the expression of apoptotic protease activating factor-1, a key protein involved in apoptosome formation (166). In addition, adipocyte derived exosomes increased the invasion of melanoma cancer cells and induced metabolic reprogramming by transferring proteins (ECHA (subunit of mitochondrial trifunctional protein) and hydroxyacyl-coenzyme A dehydrogenase), involved in fatty acid oxidation to these cancer cells. In addition, these effects were found to be worsened by obese adipocytes (168). However, evidence on the role of adipocyte and/or obese adipocytes derived exosomes in treatment resistance on breast cancer cells are lacking and therefore motivates experimental models to investigate potential mechanisms and causal factors involved in acquiring doxorubicin treatment resistance.
Drug metabolism mechanisms
Adiposity alters chemotherapeutic pharmacokinetics by; (i) increasing drug distribution, (ii) altering drug clearance, and (iii) modifying the drug-protein binding process (169). For example, obesity increases the distribution volume of lipolytic drugs by increasing its' accumulation in excess adipose tissue (169), thereby decreasing exposure of cancer cells to treatment agents. Behan et al. showed that excess adipose tissue could act as a “shelter” for protection against treatment toxicity, as cancer cells migrate into adipose tissue (123).
Obesity affects drug clearance via the liver, which primarily metabolizes, detoxifies and clears drugs from circulation (170). Hepatosteatosis decreases hepatic microcirculation, whereas the glomerular filtration and tubular secretion, and reabsorption in the kidneys, leads to increased drug clearance (117). Ghose et al. reported that mice fed a high fat diet, showed a decreased expression of key hepatic drug metabolizing enzymes (i.e., CYP3A11, CYP2B10 and CYP2A4), which could be the result of the high levels of pro-inflammatory mediators (IL-1β, IL-6 and TNF-α), increased phosphorylation of JNK and increased activation of NFκB (171). CYP34 activity, has also been found to be increased in a leptin knockout obesity model (172). In addition, the elimination, or the half-life of a drug may also be altered in obese individuals (173). Lastly, obesity is also associated with an increase in alpha-1 acid glycoprotein concentration, which could increase the binding of drugs in the plasma, thereby decreasing its bioavailability (174).
Furthermore, cytarabine, a treatment agent used in acute myeloid leukemia, is only toxic to cancer cells in its phosphorylated form (cytarabine triphosphate) (129). Cancer cells disrupt the phosphorylation reactions by altering the expression of enzymes involved in the metabolic activation of cytarabine i.e., aldo-keto reductase (AKR) and carbonyl reductase (CBR) (122). Sheng et al. showed that adipocytes metabolized daunorubicin (by increasing the expression of daunorubicin-metabolizing enzymes i.e., AKR-1C1, AKR-1C2, AKR-1C3 and CBR-1), which lead to the inactivation of daunorubicin and acquired resistance (175). This could implicate adipocytes/adipose tissue as a co-factor to decrease certain drug concentration in lipid-enriched tumor microenvironments (175). Evidence now also suggest that cancer cells “manipulate” adipocytes in the tumor microenvironment, in order to survive, but also alter drug pharmacokinetics and induce drug resistance by disrupting lipid storage and metabolism (13, 176).
Several mechanisms exist which can result in the modification of drug metabolism, drug transport and the failure of tumor cells to respond to chemotherapeutic drugs, due to overexpression of drug export proteins in cancer cells (169). It should be emphasized that limited evidence, investigating the role of overweight/obesity on pharmacokinetics of the majority of anti-cancer drugs in clinical trials, exists (170, 177). This is mainly attributed to participant inclusion criteria into phase I clinical trials and pharmacokinetic analyses, that exclude patients with co-morbidities, which is highly prevalent in overweight and obese cases (177).
Adipocytes in the Tumor Microenvironment: Lipid-Related Mechanisms
Breast cancer cells co-exist in a sophisticated microenvironment with various adjacent cell types including adipocytes, macrophages, fibroblast and endothelial cells (178). Evidence exist on the beneficial roles of fibroblasts, endothelial cells and macrophages in the tumor microenvironment (179–181). The exact role of adipocytes in the breast tumor microenvironment in treatment resistance remains unclear.
The presence of adipocytes in the tumor microenvironment revealed that breast tumor cells utilize adipocytes to their advantage to promote its survival, growth as well as proliferation and metastasis (13, 176). In addition, the presence of adipocytes in the tumor microenvironment also reduces the toxic effects of breast cancer treatment agents (18). For example, Trastuzumab® treatment (a monoclonal antibody targeting human epidermal growth factor-2) inhibited breast cancer cell growth in the absence of a lipoma. However, this inhibition was hindered in the presence of a lipoma suggesting that adipose tissue/adipocytes may have an impact on resistance to cancer therapy (176).
Adipocytes in the breast tumor microenvironment is characterized by both morphological and phenotypical changes. Histological analysis of human mammary tumor biopsies shows no, or very few adipocytes present (174), with characteristic smaller cell size (14). Adipocytes in the breast tumor microenvironment also display a more fibroblast like morphology known as cancer-associated adipocytes (127, 182). These phenotypical and morphological alterations induce functional changes in adipocytes to yield free fatty acids (FFA) from triglycerides (TG) stored in lipid droplets (Figure 2) (73). This is proposed to be as a result of tumor growth inducing lipolysis in adipocytes, which can result in adipose tissue mass reduction (183).
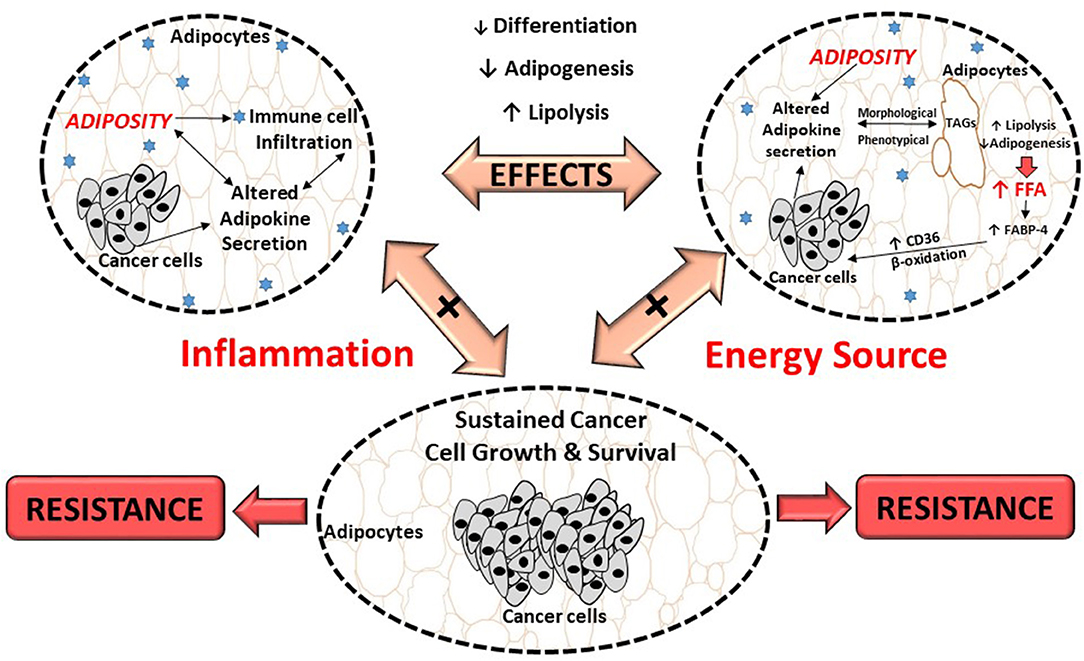
Figure 2. Proposed effects of breast cancer cells on adipocytes and its role in treatment resistance. Breast cancer cells dysregulate metabolic pathways, by altering the secretion of adipokines from adipocytes which results in inflammation. This, could result in morphological and phenotypical changes (delipidation) and thereby increase the release of FFA. These FFA provide energy substrates for cancer cells to sustain its high proliferation demand, contributing to cancer treatment resistance. FABP-4, fatty acid binding protein-4; FFA, free fatty acids; TAGs, triglycerides; CD36, fatty acid translocase.
Previously it was shown that breast cancer cells induce lipolysis by increasing the expression of hormone sensitive lipase (HSL) and adipose triglyceride lipase enzymes in adipocytes (13). It is proposed that adipocyte-derived fatty acids are either used as metabolic substrates for energy (β-oxidation) (184), or stored in lipid droplets and/or membranes within tumors (185), to sustain survival. Fatty acids and its derivatives serve as building blocks for various membrane lipids (i.e., phospholipids and sterol esters) and signaling molecules, both implicated in carcinogenesis and treatment resistance (186–188).
Additional supporting evidence include breast cancer cells increasing exogenous fatty acid uptake and utilization (FFA derived from adipocytes), by altering the expression of various enzymes in fatty acid uptake (i.e., increased fatty acid binding protein-4 (FABP-4) and fatty acid translocase (CD36) expression) (189–191) and β-oxidation (i.e., increased carnitine palmitoyltransferase I expression) (13, 192, 193). In addition, “obese” adipocytes provided higher concentrations of FFA to breast cancer cells to sustain survival and migration (13), however treatment resistance was not assessed in this obese breast cancer model.
Furthermore, adipocytes also provide FFA to breast cancer cells by dedifferentiation and/or inhibition of adipogenesis (13) (Figure 2), evident by adipocytes showing decreased expression of adipogenic markers including peroxisome proliferator-activated receptor-γ (PPAR-γ), FABP-4 and cytosine-cytosine-adenosine-adenosine-thymidine (CCAAT) enhancer binding protein-α (CEBP-α) (14). Breast cancer cells can also alter fatty acid metabolism by increasing de novo synthesis of fatty acids, by altering the expression of fatty acid synthase (FAS), acetyl-CoA carboxylase (ACC), and stearoyl CoA-desaturase-1 (SCD-1) enzymes (194–198). The result of this alteration is lipid saturation of cancer cell membranes, which protects against the cytotoxic effects of chemotherapeutic anti-cancer drugs (199). Increased FFA are also stored in tumors in the form of lipid droplets in order to avert lipotoxicity and/or to serve as an energy reserve (200). This is also supported by lipid depositions found in tumors (185), including breast tumors which is considered a characteristic of cancer aggressiveness (201).
It is proposed that the dysregulation of cytokines (increased IL-6, TNF- α and IL−1β), adipokines (increased leptin and decreased Apn and resistin), chemokines, as well as extracellular matrix proteins (collagen IV) from adipocytes (12, 64, 202, 203) (Figure 2), affect the expression of transcription factors involved in lipid metabolism. Examples include, HSL, FABP-4 and CEBP-α (14, 72). The outcome is altered adipocyte-signaling pathways and gene expression in tumor cells which induce stromal cells to produce adipokines (204).
Evidence points toward adipocytes being stimulated by breast tumor cells to increase expression of matrix metalloproteinase-11, a negative regulator of adipogenesis by decreasing pre-adipocyte differentiation and reversing mature adipocyte differentiation (11). Macrophage chemoattractant protein-1 and CCLC-5 may also be active in the host microenvironment promoting survival, invasion metastasis, and unfavorable drug responses. Here, it is proposed that the recruitment of immune cells to the tumor microenvironment, promotes inflammation, stimulating the secretion of matrix metalloproteinase-9 (role in matrix degradation) and evading the host's immune responses (202). Additionally, MCP-1 benefits vascular endothelial cell survival and activate the JAK2/STAT5 and p38 MAPK pathways, inducing angiogenesis (205).
Several groups support the role of cytokines in drug resistance. Estrogen receptor positive sensitive breast cancer cell line, MCF-7, did not express IL-6, however in a drug resistant breast cancer cell line (MCF-7/R) IL-6 was expressed (206, 207). Elevated IL-6 is linked to doxorubicin resistance in breast cancer cells, by increasing cytosine-cytosine-adenosine-adenosine-thymidine (CCAAT) enhancer binding protein (CEBP) activity, which leads to an increased expression of MDRP-1 (206, 208). Additionally, ex vivo mature adipocytes significantly increased the proliferation of both mammary cancer cells (MCF-7) and normal mammary cells (184B5) (209). Adipocytes derived from obese patient's diminished Tamoxifen treatment efficacy compared to adipocytes derived from normal weight patients. The authors identified IL-6, TNF-α and leptin as potential mediators (209). In agreement, Incio et al. demonstrated that obesity decreased the efficacy of anti-VEGF treatment in both breast cancer patients and in diet-induced obese mice. The authors proposed that inflammation (increased IL-6) and angiogenesis (increased fibroblast-growth factor-2) in adipocyte dense hypoxic microenvironments within tumors, which can sustain tumor survival (18). However, additional studies are needed to target IL-6 i.e., anti-IL-6R (tocilizumab), in order to identify the exact role of these biomarkers.
To summarize, morphological and phenotypic changes (delipidation), as a result of increased pro-inflammatory cytokines and a deregulated adipokine profile in adipose tissue/adipocytes, may be responsible for breast tumor enhancing effects of adipocytes, providing a potential mechanism for cancer treatment resistance. The role and contribution of adipose tissue/adipocytes in the tumor microenvironment and pathogenesis of breast cancer remains unclear. The exact molecular mechanisms in which breast cancer cells in an obesogenic environment use adipocyte to their physiological advantage to induce treatment resistance, needs to be explored extensively.
Doxorubicin Toxicity on Adipose Tissue/Adipocytes
Doxorubicin treatment has been shown to negatively impact adipose tissue/adipocytes (26, 29), ranging from metabolic dysfunction to phenotypical changes (27, 106, 210–212), which contribute toward the disruption of adipose tissue homeostasis and lipid storage (Table 1).
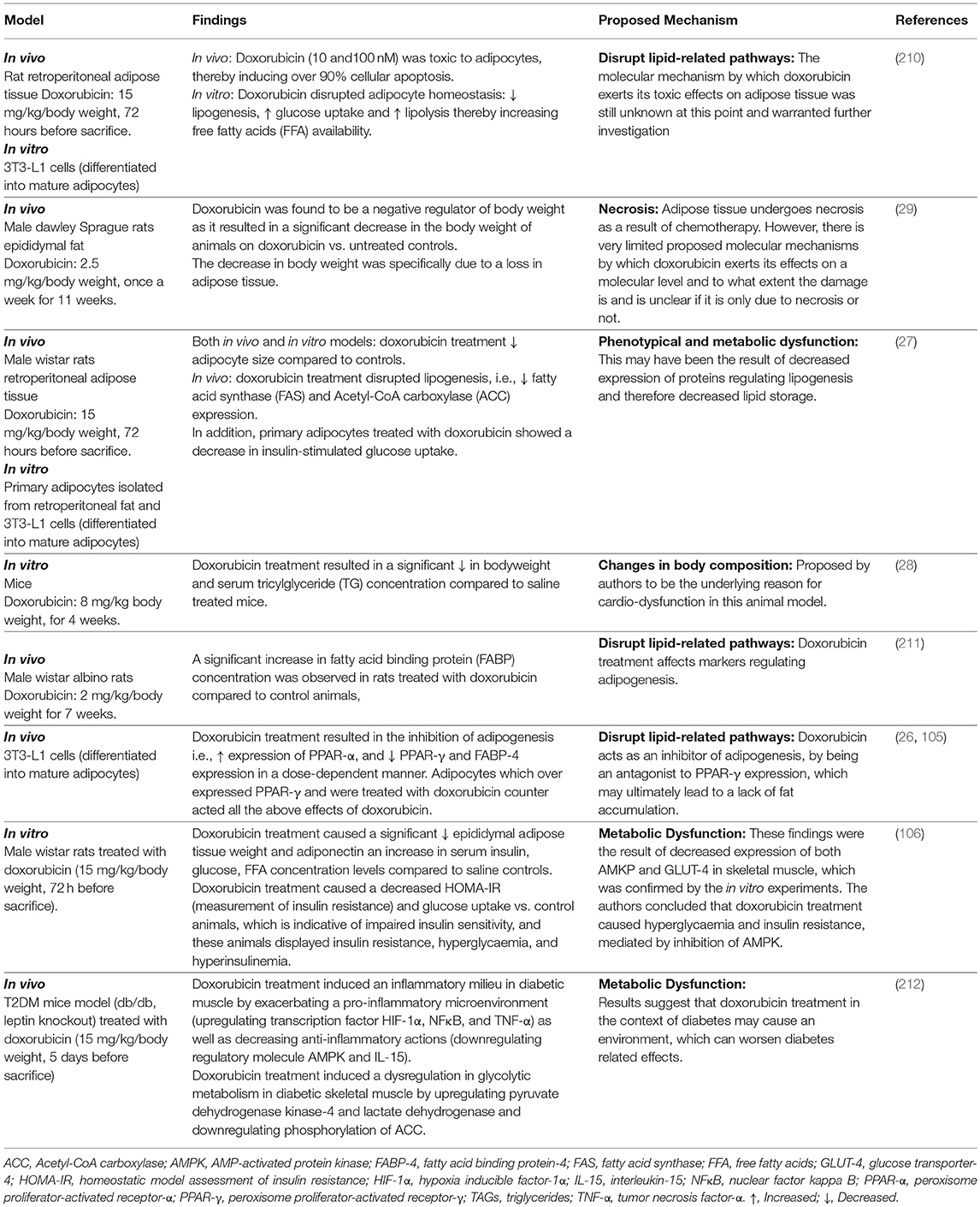
Table 1. Effect of doxorubicin on adipose tissue and/or adipocytes.
The molecular mechanisms underlying doxorubicin's negative effects on adipose tissue/adipocytes is proposed to involve adipokine dysregulation, which in turn affects factors regulating lipid metabolism pathways. For example, decreasing and/or inhibition of adipogenesis (decreased PPAR-γ and FABP expression) and lipogenesis (decreased FAS expression) as well as the induction of lipolysis (increased HSL expression) (27, 29) (Figure 3). This in turn induces an increase in FFA release as the result of the phenotypical changes (27, 29), thereby disrupting lipid storage. Doxorubicin induced metabolic dysfunction (increased FFA levels), could potentially increase the availability of energy substrates (FFA) for cancer cells to utilize to sustain both its' survival and proliferation demands (26, 27, 29, 106), and thereby indirectly contribute to breast cancer treatment resistance itself. However, it should be stressed that evidence on the effects of doxorubicin on adipose tissue/adipocytes (Table 1) is based on normal functioning adipose tissue/adipocytes, and not on an obesity model, where adipose tissue is dysfunctional.
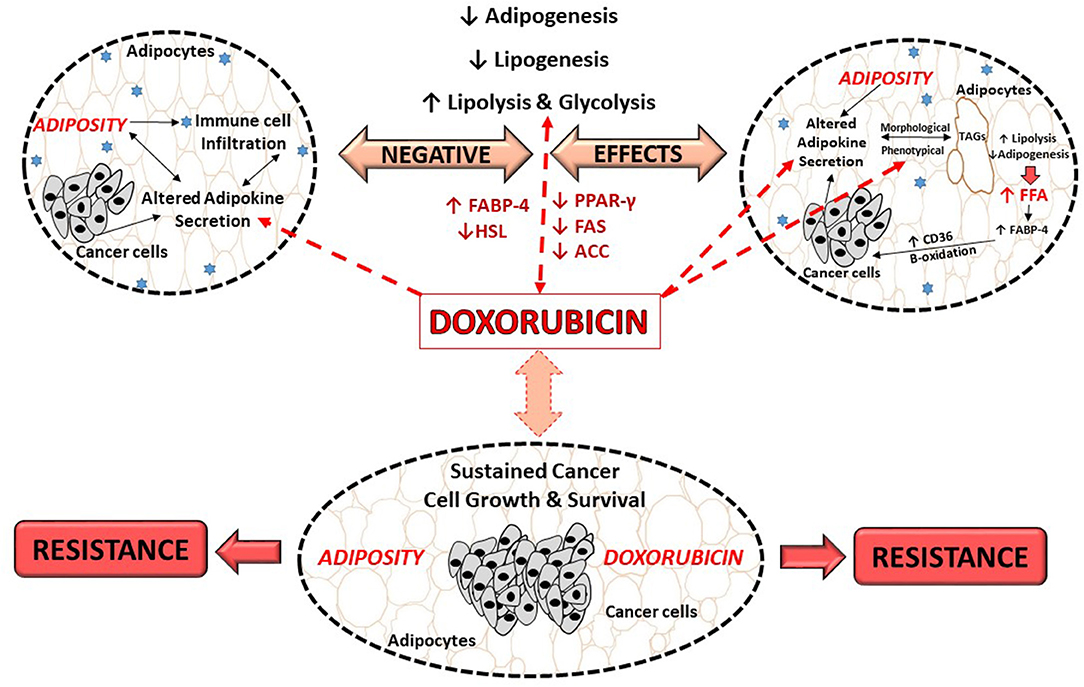
Figure 3. Proposed role of doxorubicin in an obesogenic breast cancer model. ACC, Acetyl-CoA carboxylase; CD36, fatty acid translocase; FFA, free fatty acids; FABP, fatty acid binding protein; FABP-4, fatty acid binding protein-4; FAS, fatty acid synthase; HSL, hormone sensitive lipase; PPAR-γ, peroxisome proliferator-activated receptor-γ; TAGs, triglycerides.
Evidence on the effects of doxorubicin on adipose tissue/adipocytes in the context of obesity, where adipose tissue is dysfunctional is lacking. In light of this, we proposed that doxorubicin treatment on dysfunctional adipose tissue and/or adipocytes, may further exacerbate the negative effects of obesity itself, toward cancer treatment by further dysregulating adipokines secretion, which in turn affects the factors regulates lipogenesis, adipogenesis and lipolysis, thereby further implicating obesity in the context of breast cancer treatment (Figure 3).
Future Research and Conclusion
Adipose tissue plays an important physiological role as a metabolically active storage compartment and endocrine organ. A disruption in adipose tissue homeostasis results in potentially serious health and clinical-related outcomes. Obesity induced adipose-dysfunction is associated with an increased risk for breast tumor development and progression.
Obesity is associated with chronic low grade inflammation as a result of adipokine secretion (immune cell infiltration), which results in a sustained inflammatory milieu. These inflammatory mediators activate downstream signaling pathways (MAPK and PI3K) in breast cancer cells that favors cancer cell survival (increased proliferation and decreased apoptosis), and contribute to breast cancer development and progression (Figure 3).
Recent evidence also implicate obesity as a causal factor for reduced chemotherapy efficacy, resulting in treatment resistance. Obesity-driven changes may contribute to chemotherapy resistance by altering drug pharmacokinetics, impairing drug metabolism and delivery, and inducing chronic inflammation as well as altering tumor-associated adipocyte adipokine secretion. However, the exact underlying mechanisms by which obesity achieves this remains unclear.
It is suggested that adipose tissue/adipocytes, serve as a potential energy source for cancer cells to sustain their survival thereby promoting cell growth and proliferation (Figure 3). This is especially significant in the case of breast cancer; as adipose tissue is the most abundant tissue type in the breast. Breast cancer cells dysregulate lipid related metabolic pathways i.e., lipolysis, adipogenesis, de novo fatty acid synthesis and exogenous lipid uptake by altering the secretion of adipokines by adipocytes, which in turn results in the release of FFA (Figure 3). These fatty acids can then serve as energy substrates for breast cancer cells to sustain its high proliferation rates or can be stored in tumors in the form of lipid droplets and/or in membrane lipids in order to avoid lipotoxicity, which protects against the cytotoxic effects of anti-cancer drugs.
Additionally, doxorubicin treatment itself has also been shown to modify adipose tissue/adipocytes through inhibition of adipogenesis, downregulating lipogenesis, inducing lipolysis, and subsequently disrupting lipid storage. Resulting in phenotypical changes in adipocytes (Figure 3), which in turn produces more “bioavailable” energy substrates (increased FFA), which cancer cells can potentially utilize to sustain survival and proliferation demands and thereby could indirectly contribute to chemotherapeutic treatment resistance (Figure 3).
It should be stressed that studies investigating the effects of doxorubicin on adipose tissue/adipocytes and lipid metabolism in the context of obesity, where adipose tissue is dysfunctional are lacking. We thus propose that doxorubicin treatment in patients with dysfunctional adipose tissue and/or adipocytes, may further exacerbate the tumor promoting effects of obesity itself. This may be achieved by further dysregulating adipokine secretion, which in turn affects lipogenesis, adipogenesis and lipolysis, linking adiposity to breast cancer treatment resistance (Figure 3). It is thus of importance to investigate the effect of doxorubicin in the context of obesity, and how obesity may aggregate factors playing a role in the development of doxorubicin treatment resistance, as there is an increase in the prevalence of breast cancer patients who are either overweight or obese, treated with doxorubicin. Specifically, since, obese and normal weight patients receive the same treatment regimens. Therefore, extensive investigation is needed to elucidate the underlying mechanism by which obesity contributes to treatment resistance.
The role of lipid metabolism in breast cancer also remains understudied as well as the cytotoxic effects of chemotherapeutic drugs on adipose tissue/adipocytes, both of which may contribute to the promotion of breast cancer cell survival and treatment resistance. Therefore, the identification of molecular mechanisms underlying both the effects of a neoplastic state and doxorubicin treatment on adipose tissue, will promote the identification of novel pharmacologic targets as well as the development of appropriate management protocols for adipose tissue driven chemotherapeutic drug resistance as well as doxorubicin related toxicities in order to improve over-all survival of breast cancer patients.
Author Contributions
IM wrote the first draft of the manuscript. TN, A-ME, and PvJ contributed to critical revision and intellectual input of the manuscript. All authors read and approved the final manuscript.
Funding
Work in this laboratory is supported by research grants from the Cancer Association of South Africa (CANSA), the South African Medical Research Council (SAMRC) and the National Research Foundation (NRF) of South Africa. Funding bodies had no role in the preparation of this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer (2015) 136:E359–386. doi: 10.1002/ijc.29210
3. Nagrani R, Mhatre S, Rajaraman P, Soerjomataram I, Boffetta P, Gupta S, et al. Central obesity increases risk of breast cancer irrespective of menopausal and hormonal receptor status in women of South Asian Ethnicity. Eur J Cancer (2016) 66:153–161. doi: 10.1016/j.ejca.2016.07.022
4. Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, et al. Global, regional and national prevalence of overweight and obesity in children and adults 1980–2013: A systematic analysis. Lancet (2014) 384:766–81. doi: 10.1016/S0140-6736(14)60460-8
5. Sparano JA, Wang M, Zhao F, Stearns V, Martino S, Ligibel JA, et al. Obesity at diagnosis is associated with inferior outcomes in hormone receptor-positive operable breast cancer. Cancer (2012) 118:5937–46. doi: 10.1002/cncr.27527
6. Rezaee F, Dashty M. Role of adipose tissue in metabolic system disorders: adipose tissue is the initiator of Metabolic diseases. J Diabetes Meta (2013) S13:008. doi: 10.4172/2155-6156.S13-008
7. Cowen S, McLaughlin S, Hobbs G, Coad J, Martin KH, Olfert IM, et al. High-fat, high-calorie diet enhances mammary carcinogenesis and local inflammation in MMTV-PyMT mouse model of breast cancer. Cancers (2015) 7:1125–42. doi: 10.3390/cancers7030828
8. Khalid S, Hwang D, Babichev Y, Kolli R, Altamentova S, Koren S, et al. Evidence for a tumor promoting effect of high-fat diet independent of insulin resistance in HER2/Neu mammary carcinogenesis. Breast Cancer Res Treat. (2010) 122:647–59. doi: 10.1007/s10549-009-0586-8
9. Choe SS, Huh JY, Hwang IJ, Kim JI, Kim JB. Adipose tissue remodeling: its role in energy metabolism and metabolic disorders. Front Endocrinol. (2016) 7:30. doi: 10.3389/fendo.2016.00030
10. Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer (2004) 4:579–91. doi: 10.1038/nrc1408
11. Toren P, Mora BC, Venkateswaran V. Diet, obesity, and cancer progression: are adipocytes the link. Lipid Insights (2013) 6:37–45. doi: 10.4137/LPI.S10871
12. Mahon KL, Lin H-M, Castillo L, Lee BY, Lee-Ng M, Chatfield MD, et al. Cytokine profiling of docetaxel-resistant castration-resistant prostate cancer. Br J Cancer (2015) 112:1340–8. doi: 10.1038/bjc.2015.74
13. Balaban S, Shearer RF, Lee LS, van Geldermalsen M, Schreuder M, Shtein HC, et al. Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. (2017) 5:1. doi: 10.1186/s40170-016-0163-7
14. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. (2011) 71:2455–65. doi: 10.1158/0008-5472.CAN-10-3323
15. Liu E, Samad F, Mueller BM. Local adipocytes enable estrogen-dependent breast cancer growth: role of leptin and aromatase. Adipocyte (2013) 2:165–9. doi: 10.4161/adip.23645
16. Bousquenaud M, Fico F, Solinas G, Rüegg C, Santamaria-Martínez A. Obesity promotes the expansion of metastasis-initiating cells in breast cancer. Breast Cancer Res. (2018) 20:104. doi: 10.1186/s13058-018-1029-4
17. Dong L, Yuan Y, Opansky C, Chen Y, Aguilera-Barrantes I, Wu S, et al. Diet-induced obesity links to ER positive breast cancer progression via LPA/PKD-1-CD36 signaling-mediated microvascular remodeling. Oncotarget (2017) 8:22550–62. doi: 10.18632/oncotarget.15123
18. Incio J, Ligibel JA, McManus DT, Suboj P, Jung K, Kawaguchi K, et al. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci Transl Med. (2018) 10:eaag0945. doi: 10.1126/scitranslmed.aag0945
19. Iwase T, Sangai T, Nagashima T, Sakakibara M, Sakakibara J, Hayama S, et al. Impact of body fat distribution on neoadjuvant chemotherapy outcomes in advanced breast cancer patients. Cancer Med. (2016) 5:41–48. doi: 10.1002/cam4.571
20. Hydock DS, Lien CY, Jensen BT Schneider CM, Hayward R. Switching to a low-fat diet attenuates the intensified doxorubicin cardiotoxicity associated with high-fat feeding. Cancer Chemother Pharmacol. (2013) 71:1551–60. doi: 10.1007/s00280-013-2154-5
21. Sirin O, Kolonin MG. Treatment of obesity as a potential complementary approach to cancer therapy. Drug Discov Today (2013) 18:567–73. doi: 10.1016/j.drudis.2012.05.008
22. Gevorgyan A, Bregni G, Galli G, Ganzinelli M, Martinetti A, Lo Vullo S, et al. Body mass index and clinical benefit of fulvestrant in postmenopausal women with advanced breast cancer. Tumori (2016) 102:e11–4. doi: 10.5301/tj.5000515
23. Karpinska A, Safranow K, Kładny J, Sulzyc-Bielicka V. The influence of obesity on results of AT (doxorubicin plus docetaxel) neoadjuvant chemotherapy in locally advanced breast cancer patients. Pol Przegl Chir. (2015) 87:231–7. doi: 10.1515/pjs-2015-0047
24. Guenancia C, Ladoire S, Ghiringelli F, Rochette L, Vergely C, Cottin Y. Implications of excess weight in the cardiotoxicity of anthracyclines and trastuzumab in breast cancer. Arch Cardiovasc Dis. (2017) 110:69–71. doi: 10.1016/j.acvd.2016.12.004
25. Liu Z, Shi A, Song D, Han B, Zhang Z, Ma L, et al. Resistin confers resistance to doxorubicin-induced apoptosis in human breast cancer cells through autophagy induction. Am J Cancer Res. (2017) 7:574–83. eCollection 2017.
26. Arunachalam S, Kim SY, Kim MS, Yi HK, Yun BS, Lee DY, et al. Adriamycin inhibits adipogenesis through the modulation of PPARγ and restoration of adriamycin-mediated inhibition of adipogenesis by PPARγ over-expression. Toxicol Mech Methods (2012) 22:540–6. doi: 10.3109/15376516.2012.692110
27. Biondo LA, Lima Junior EA, Souza CO, Cruz MM, Cunha R, Alonso-Vale MI, et al. Impact of doxorubicin treatment on the physiological functions of white adipose tissue. PLoS ONE (2016) 11:e0151548. doi: 10.1371/journal.pone.0151548
28. Nagendran J, Kienesberger PC, Pulinilkunnil T, Zordoky BN, Sung MM, Kim T, et al. Cardiomyocyte specific adipose triglyceride lipase overexpression prevents doxorubicin induced cardiac dysfunction in female mice. Heart (2013) 99:1041–7. doi: 10.1136/heartjnl-2013-303843
29. Xiang P, Deng HY, Li K Huang GY, Chen Y, Tu L, et al. Dexrazoxane protects against doxorubicin-induced cardiomyopathy: upregulation of Akt and Erk phosphorylation in a rat model. Cancer Chemother Pharmacol. (2009) 63:343–9. doi: 10.1007/s00280-008-0744-4
30. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global Cancer Statistics, 2012. CA Cancer J Clin. (2015) 65:87–108. doi: 10.3322/caac.21262
31. Moodley J, Cairncross L, Naiker T, Momberg M. Understanding pathways to breast cancer diagnosis among women in the Western Cape Province, South Africa: a qualitative study. BMJ Open (2016) 6:e009905. doi: 10.1136/bmjopen-2015-009905
32. Brinton LA, Figueroa JD, Awnuah B, Yarney J, Wiafe S, Wood SN, et al. Breast Cancer in Sub-Saharan Africa: Opportunities for Prevention. Breast Cancer Res Treat. (2014) 144:467–78. doi: 10.1007/s10549-014-2868-z
33. Ghoncheh M, Momenimovahed Z, Salehiniya H. epidemiology, incidence and mortality of breast cancer in Asia. Asian Pac J Cancer Prev. (2016) 17:47–52. doi: 10.7314/APJCP.2016.17.S3.47
34. Donohoe CL, Doyle SL, Reynolds JV. Visceral adiposity, insulin resistance and cancer risk. Diabetol Metab Syndr. (2011) 3:12. doi: 10.1186/1758-5996-3-12
35. O'Neill S, O'Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. (2015) 16:1–12. doi: 10.1111/obr.12229
36. White AJ, Nichols HB, Bradshaw PT, Sandler DP. Overall and central adiposity and breast cancer risk in the sister study. Cancer (2015) 121:3700–8. doi: 10.1002/cncr.29552
37. Zhu QL, Xu WH, Tao MH. Biomarkers of the metabolic syndrome and breast cancer prognosis. Cancers (2010) 2:721–739. doi: 10.3390/cancers2020721
38. Divella R, De Luca R, Abbate I, Naglieri E, Daniele A. 2016. Obesity and cancer: the role of adipose tissue and adipo-cytokines-induced chronic inflammation. J Cancer (2016) 7:2346–59. eCollection 2016. doi: 10.7150/jca.16884
39. Mendonça FM, de Sousa FR, Barbosa AL, Martins SC, Araújo RL, Soares R, et al. Metabolic syndrome and risk of cancer: which link? Metabolism (2015) 64:182–9. doi: 10.1016/j.metabol.2014.10.008
40. Fuentes E, Fuentes F, Vilahur G, Badimon L, Palomo I. Mechanisms of chronic state of inflammation as mediators that link obese adipose tissue and metabolic syndrome. Mediators Inflamm (2013) 2013:136584. doi: 10.1155/2013/136584
41. Gade W, Schmit J, Collins M, Gade J. Beyond obesity: the diagnosis and pathophysiology of metabolic syndrome. Clin Lab Sci. (2010) 23:51–61; quiz 62–5.
42. Cefalu WT. Inflammation, insulin resistance, and type 2 diabetes: back to the future? Diabetes (2009) 58:307–8. doi: 10.2337/db08-1656
43. Friedrich N, Thuesen B, Jørgensen T, Juul A, Spielhagen C, Wallaschofksi H, et al. The association between IGF-I and insulin resistance: a general population study in Danish adults. Diabetes Care (2012) 35:768–73. doi: 10.2337/dc11-1833
44. Blüher M. Adipose tissue dysfunction contributes to obesity related metabolic diseases. Best Pract Res Clin Endocrinol Metab. (2013) 27:163–77. doi: 10.1016/j.beem.2013.02.005
45. Ewertz M, Jensen M-B, Gunnarsdóttir KÁ, Højris I, Jakobsen EH, Nielsen D, et al. Effect of Obesity on Prognosis after early-stage breast cancer. J Clin Oncol. (2011) 29:25–31. doi: 10.1200/JCO.2010.29.7614
46. Goossens GH, Blaak EE. Adipose tissue dysfunction and impaired metabolic health in human obesity: a matter of oxygen? Front Endocrinol. (2015) 6:55. doi: 10.3389/fendo.2015.00055
47. Witt PM, Christensen JH, Schmidt EB, Dethlefsen C, Tjønneland A, Overvad K, et al. Marine n-3 polyunsaturated fatty acids in adipose tissue and breast cancer risk : a case-cohort study from Denmark. Cancer Causes Control. (2009) 20:1715–21. doi: 10.1007/s10552-009-9423-y
48. Allott EH, Hursting SD. Obesity and cancer: mechanistic insights from transdisciplinary studies. Endocr Relat Cancer (2015) 22:R365–86. doi: 10.1530/ERC-15-0400
49. Azrad M, Demark-Wahnefried W. The association between adiposity and breast cancer recurrence and survival: a review of the recent literature. Curr Nutr Rep. (2014) 3:9–15. doi: 10.1007/s13668-013-0068-9
50. Kyrgiou M, Kalliala I, Markozannes G, Gunter MJ, Paraskevaidis E, Gabra H, et al. Adiposity and cancer at major anatomical sites: umbrella review of the literature. BMJ (2017) 356:j477. doi: 10.1136/bmj.j477
51. Sun X, Nichols HB, Robinson W, Sherman ME, Olshan AF, Troester MA. Post-diagnosis adiposity and survival among breast cancer patients: influence of breast cancer subtype. Cancer Causes Control. (2015) 26:1803–11. doi: 10.1007/s10552-015-0673-6
52. Maury E, Brichard SM. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol Cell Endocrinol. (2010) 314:1–16. doi: 10.1016/j.mce.2009.07.031
53. Lasselin J, Magne E, Beau C, Ledaguenel P, Dexpert S, Aubert A, et al. Adipose inflammation in obesity: relationship with circulating levels of inflammatory markers and association with surgery-induced weight loss. J Clin Endocrinol Metab. (2014) 99:E53–61. doi: 10.1210/jc.2013-2673
54. Lautenbach A, Budde A, Wrann CD, Teichmann B, Vieten G, Karl T, et al. Obesity and the associated mediators leptin, estrogen and IGF-I enhance the cell proliferation and early tumorigenesis of breast cancer cells, Nutr Cancer (2009) 61:484–91. doi: 10.1080/01635580802610115
55. Stemmer K, Perez-Tilve D, Ananthakrishnan G, Bort A, Seeley RJ, Tschöp MH, et al. High-fat-diet-induced obesity causes an inflammatory and tumor-promoting microenvironment in the rat kidney. Dis Model Mech. (2012) 5:627–635. doi: 10.1242/dmm.009407
56. Gilbert CA, Slingerland JM. Cytokines, obesity, and cancer: new insights on mechanisms linking obesity to cancer risk and progression. Annu Rev Med. (2013) 64:45–57. doi: 10.1146/annurev-med-121211-091527
57. Lindholm CR, Ertel RL, Bauwens JD, Schmuck EG, Mulligan JD, Saupe KW. A high-fat diet decreases AMPK activity in multiple tissues in the absence of hyperglycemia or systemic inflammation in rats. J Physiol Biochem. (2013) 69:165–75. doi: 10.1007/s13105-012-0199-2
58. Suganami T, Ogawa Y. Adipose tissue macrophages: their role in adipose tissue remodeling. J Leukoc Biol. (2010) 88:33–9. doi: 10.1189/jlb.0210072
59. Galic S, Fullerton MD, Schertzer JD, Sikkema S, Marcinko K, Walkley CR, et al. Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J Clin Invest. (2011) 121:4903–15. doi: 10.1172/JCI58577
60. Roberts DL, Dive C, Renehan AG. Biological mechanisms linking obesity and cancer risk: new perspectives. Annu Rev Med. (2010) 61:301–16. doi: 10.1146/annurev.med.080708.082713
61. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–899. doi: 10.1016/j.cell.2010.01.025
62. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
63. Yao F, Zhang M, Chen L. Adipose tissue-specialized immunologic features might be the potential therapeutic target of prospective medicines for obesity. J Diabetes Res. (2017) 2017:4504612. doi: 10.1155/2017/4504612
64. Nieman KM, Romero IL, Van Houten B, Lengyel E. Adipose tissue and adipocytes supports tumorigenesis and metastasis. Biochimica et Biophysica Acta (2013) 1831:1533–41. doi: 10.1016/j.bbalip.2013.02.010
65. Pérez-Hernández AI, Catalán V, Gómez-Ambrosi J, Rodríguez A, Frühbeck G. Mechanisms linking excess adiposity and carcinogenesis promotion. Front Endocrinol. (2014) 5:65. doi: 10.3389/fendo.2014.00065
66. Khan S, Shukla S, Sinha S, Meeran SM. Role of adipokines and cytokines in obesity-associated breast cancer: therapeutic targets. Cytokine Growth Factor Rev. (2013) 24:503–13. doi: 10.1016/j.cytogfr.2013.10.001
67. Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. (2007) 178:2623–9. doi: 10.4049/jimmunol.178.5.2623
68. Balistreri CR, Caruso C, Candore G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediators Inflamm. (2010) 2010:802078. doi: 10.1155/2010/802078
69. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA, et al. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. (2014) 2014:149185. doi: 10.1155/2014/149185
70. Guo D, Bell EH, Mischel P, Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharm Des. (2014) 20: 2619–26. doi: 10.2174/13816128113199990486
71. Gupta S, Roy A, Dwarakanath BS. Metabolic cooperation and competition in the tumor microenvironment: implications for therapy. Front Oncol. (2017) 7:68. doi: 10.3389/fonc.2017.00068
72. Vaysse C, Lømo J, Garred Ø, Fjeldheim F, Lofteroed T, Schlichting E, et al. Inflammation of mammary adipose tissue occurs in overweight and obese patients exhibiting early-stage breast cancer. NPJ Breast Cancer (2017) 3:35. doi: 10.1038/s41523-017-0030-x
73. Choi J, Cha YJ, Koo JS. Adipocyte biology in breast cancer: from silent bystander to active facilitator. Prog Lipid Res. (2018) 69:11–20. doi: 10.1016/j.plipres.2017.11.002
74. Surmacz E. Leptin and adiponectin: emerging therapeutic targets in breast cancer. J Mammary Gland Biol Neoplasia (2013) 18:321–32. doi: 10.1007/s10911-013-9302-8
75. Rodríguez AJ, Mastronardi C, Paz-Filho G. Leptin as a risk factor for the development of colorectal cancer. Trans Gastrointest Cancer (2013) 2:211–22. doi: 10.3978/j.issn.2224-4778.2013.10.04
76. Aleksandrova K, Boeing H, Jenab M, Bueno-de-Mesquita HB, Jansen E, van Duijnhoven FJ, et al. Leptin and soluble leptin receptor in risk of colorectal cancer in the European prospective investigation into cancer and nutrition cohort. Cancer Res. (2012) 72:5328–37. doi: 10.1158/0008-5472.CAN-12-0465
77. Sultana R, Kataki AC, Borthakur BB, Basumatary TK, Bose S, et al. Imbalance in leptin-adiponectin levels and leptin receptor expression as chief contributors to triple negative breast cancer progression in Northeast India. Gene (2017) 621:51–58. doi: 10.1016/j.gene.2017.04.021
78. Gnacinska M, Małgorzewicz S, Lysiak-Szydłowska W, Sworczak K. The serum profile of adipokines in overweight patients with metabolic syndrome. Endokrynol Pol. (2010) 61:36–41.
79. Otake S, Takeda H, Fujishima S, Fukui T, Orii T, Sato T, et al. Decreased levels of plasma adiponectin associated with increased risk of colorectal cancer. World J Gastroenterol. (2010) 16:1252–7. doi: 10.3748/wjg.v16.i10.1252
80. Zhao D, Liu H. Adipose tissue dysfunction and the pathogenesis of metabolic syndrome. World J Hypertens (2013) 3:18–26. doi: 10.5494/wjh.v3.i3.18
81. Nalabolu MR, Palasamudram K, Jamil K. Adiponectin and leptin molecular actions and clinical significance in breast cancer. Int J Hematol Oncol Stem Cell Res. (2014) 8:31–40.
82. Denduluri SK, Idowu O, Wang Z, Liao Z, Yan Z, Mohammed MK, et al. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. (2015) 2:13–25. doi: 10.1016/j.gendis.2014.10.004
83. Tan J, Buache E, Chenard MP, Dali-Youcef N, Rio MC. Adipocyte is a non-trivial, dynamic partner of breast cancer cells. Int J Dev Biol. (2011) 55:851–9. doi: 10.1387/ijdb.113365jt
84. Dibra D, Mishra L, Li S. Molecular of oncogene-induced inflammation and inflammation-sustained oncogene activation in gastrointestinal tumors: an underappreciated symbiotic relationship. Biochim biophys Acta (2014) 1846:152–60. doi: 10.1016/j.bbcan.2014.05.001
85. Sethi G, Shanmugam MK, Ramachandran L, Kumar AP, Tergaonkar V. Multifaceted link between cancer and inflammation. Biosci Rep. (2012) 32:1–15. doi: 10.1042/BSR20100136
86. Cooks T, Pateras IS, Tarcic O, Solomon H, Schetter AJ, Wilder S, et al. Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell (2013) 23:634–46. doi: 10.1016/j.ccr.2013.03.022
87. Komarova EA, Krivokrysenko V, Wang K, Neznanov N, Chernov MV, Komarov PG, et al. p53 is a suppressor of inflammatory response in mice. FASEB J. (2005) 19:1030–2. doi: 10.1096/fj.04-3213fje
88. Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer (2008) 8:180–92. doi: 10.1038/nrc2344
89. Yang L, Karin M. Roles of tumor suppressors in regulating tumor-associated inflammation. Cell Death Diff. (2014) 21:1677–86. doi: 10.1038/cdd.2014.131
90. Wang X, Simpson ER, Brown KA. p53: protection against tumor growth beyond effects on cell cycle and apoptosis. Cancer Res. (2015) 75:5001–7. doi: 10.1158/0008-5472.CAN-15-0563
91. Trayhurn P. Hypoxia and adipose tissue function and dysfunction in obesity. Phys Revs. (2013) 93:1–21. doi: 10.1152/physrev.00017.2012
92. Rausch LK, Netzer NC, Hoegel J, Pramsohler S. The linkage between breast cancer, hypoxia, and adipose tissue. Front Oncol. (2017) 7:211. doi: 10.3389/fonc.2017.00211
93. Hwang WL, Yang MH, Tsai ML, Lan HY, Su SH, Chang SC, et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology (2011) 141:279–91, 291.e1–5. doi: 10.1053/j.gastro.2011.04.008
94. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature (2011) 475:222–5. doi: 10.1038/nature10138
95. Xie G, Yao Q, Liu Y, Du S, Liu A, Guo Z, et al. IL-6-induced epithelial-mesenchymal transition promotes the generation of breast cancer stem-like cells analogous to mammosphere cultures. Int J Oncol. (2012) 40:1171–9. doi: 10.3892/ijo.2011.1275
96. Chen H, Ding A, Wang M. 2016. Impact of central obesity on prognostic outcome of triple negative breast cancer in Chinese women. Springer Plus (2016) 5:594. doi: 10.1186/s40064-016-2200-y
97. Pierobon M, Frankenfeld CL. Obesity as a risk factor for triple-negative breast cancers: a systematic review and meta-analysis. Breast Cancer Res Treat. (2013) 137:307–314. doi: 10.1007/s10549-012-2339-3
98. Kabel AM, Baali FH. Breast cancer : insights into risk factors, pathogenesis, diagnosis and management. J Can Res Treat. (2015) 3:28–33. doi: 10.12691/jcrt-3-2-3
99. Matsen CB, Neumayer LA. Breast Cancer. JAMA Surg. (2013) 148:971–9. doi: 10.1001/jamasurg.2013.3393
100. Hassan MS, Ansari J, Spooner D, Hussain SA. Chemotherapy for breast cancer (Review). Oncol Rep. (2010) 24:1121–31. doi: 10.3892/or_00000963
101. Meredith AM, Dass CR. Increasing role of the cancer chemotherapeutic doxorubicin in cellular metabolism. J Pharm Pharmacol. (2016) 68:729–41. doi: 10.1111/jphp.12539
102. Rivankar S. An overview of doxorubicin formulations in cancer therapy. J Cancer Res Ther. (2014) 10:853–8. doi: 10.4103/0973-1482.139267
103. Thorn CF, Oshiro C, Marsh S, Hernandez-Boussard T, McLeod H, Klein TE, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics (2011) 21:440–6. doi: 10.1097/FPC.0b013e32833ffb56
104. Anampa J, Makower D, Sparano JA. Progress in adjuvant chemotherapy for breast cancer: an overview. BMC Med. (2015) 13:195. doi: 10.1186/s12916-015-0439-8
105. Arunachalam S, Tirupathi Pichiah PB, Achiraman S. Doxorubicin treatment inhibits PPARγ and may induce lipotoxicity by mimicking a type 2 diabetes-like condition in rodent models. FEBS Lett. (2013) 587:105–10. doi: 10.1016/j.febslet.2012.11.019
106. de Lima Junior EA, Yamashita AS, Pimentel GD, De Sousa LG, Santos RV, Gonçalves CL, et al. Doxorubicin caused severe hyperglycaemia and insulin resistance, mediated by inhibition in AMPk signalling in skeletal muscle. J Cachexia Sarcopenia Muscle (2016) 7:615–25. doi: 10.1002/jcsm.12104
107. Thivat E, Thérondel S, Lapirot O, Abrial C, Gimbergues P, Gadéa E, et al. Weight change during chemotherapy changes the prognosis in non metastatic breast cancer for the worse. BMC Cancer (2010) 10:648. doi: 10.1186/1471-2407-10-648
108. Vagenas D, DiSipio T, Battistutta D, Demark-Wahnefried W, Rye S, Bashford J, et al. Weight and weight change following breast cancer: evidence from a prospective, population-based, breast cancer cohort study. BMC Cancer (2015) 15:28. doi: 10.1186/s12885-015-1026-2
109. de Visser KE, Jonkers J. Towards understanding the role of cancer-associated inflammation in chemoresistance. Cur Pharm Des. (2009) 15:1844–53. doi: 10.2174/138161209788453239
110. Xu F, Wang F, Yang T, Sheng Y, Zhong T, Chen Y, et al. Differential drug resistance acquisition to doxorubicin and paclitaxel in breast cancer cells. Can Cell Int. (2014) 14:538. doi: 10.1186/s12935-014-0142-4
111. Guiu B, Petit JM, Bonnetain F, Ladoire S, Guiu S, Cercueil JP, et al. Visceral fat area is an independent predictive biomarker of outcome after first-line bevacizumab-based treatment in metastatic colorectal cancer. Gut (2010) 59:341–7. doi: 10.1136/gut.2009.188946
112. Lashinger LM, Rossi EL, Hursting SD. Obesity and resistance to cancer chemotherapy: interacting roles of inflammation and metabolic dysregulation. Clin Pharmacol Ther. (2014) 96:458–63. doi: 10.1038/clpt.2014.136
113. Thomas AP, Hoang J, Vongbunyong K, Nguyen A, Rakshit K, Matveyenko AV. Administration of melatonin and metformin prevents deleterious effects of circadian disruption and obesity in male rats. Endocrinology (2016) 157:4720–31. doi: 10.1210/en.2016-1309
114. De Azambuja E, McCaskill-Stevens W, Francis P, Quinaux E, Crown JP, Vicente M, et al. The effect of body mass index on overall and disease-free survival in node-positive breast cancer patients treated with docetaxel and doxorubicin-containing adjuvant chemotherapy: the experience of the BIG 02–98 trial. Breast Cancer Res Treat. (2010) 119:145–53. doi: 10.1007/s10549-009-0512-0
115. Haakinson DJ, Leeds SG, Dueck AC, Gray RJ, Wasif N, Stucky CC, et al. The impact of obesity on breast cancer: a retrospective review. Ann Surg Oncol. (2012) 19:3012–8. doi: 10.1245/s10434-012-2320-8
116. Bochet L, Meulle A, Imbert S, Salles B, Valet P, Muller C. Cancer-associated adipocytes promotes breast tumor radioresistance. Biochem Biophys Res Commun. (2011) 411:102–6. doi: 10.1016/j.bbrc.2011.06.101
117. Lyman GH, Sparreboom A. Chemotherapy dosing in overweight and obese patients with cancer. Nat Rev Clin Oncol. (2013) 10:451–9. doi: 10.1038/nrclinonc.2013.108
118. Ritzmo C, Söderhäll S, Karlén J, Nygren H, Eksborg S. Pharmacokinetics of doxorubicin and etoposide in a morbidly obese pediatric patient. Pediatr Hematol Oncol. (2007) 24:437–45. doi: 10.1080/08880010701451343
119. Fuentes-Mattei E. Obesity and cancer: jet fuel accelerating cancer hallmarks and increasing the economic burden of cancer. Adv Obes Weight Manag Control. (2015) 2:00032. doi: 10.15406/aowmc.2015.02.00032
120. Healy LA, Ryan AM, Carroll P, Ennis D, Crowley V, Boyle T, et al. Metabolic syndrome, central obesity and insulin resistance are associated with adverse pathological features in postmenopausal breast cancer. Clin Oncol. (2010) 22:281–8. doi: 10.1016/j.clon.2010.02.001
121. Zahreddine H, Borden KL. Mechanisms and insights into drug resistance in cancer. Front Pharmacol. (2013) 4:28. doi: 10.3389/fphar.2013.00028
122. Wind NS, Holen I. Multidrug resistance in breast cancer: from in vitro models to clinical studies. Int J Breast Cancer (2011) 2011:967419. doi: 10.4061/2011/967419
123. Behan JW, Yun JP, Proektor MP, Ehsanipour EA, Arutyunyan A, Moses AS, et al. Adipocytes impair leukemia treatment in mice. Can Res. (2009) 69:7867–74. doi: 10.1158/0008-5472.CAN-09-0800
124. Sadashiv Tiwari S, Paul BN, Kumar S, Chandra A, Dhananjai S, et al. Over expression of resistin in adipose tissue of the obese induces insulin resistance. World J Diabetes (2012) 3:135–41. doi: 10.4239/wjd.v3.i7.135
125. Oh KT, Baik HJ, Lee AH, Oh YT, Youn YS, Lee ES. The reversal of drug-resistance in tumors using a drug-carrying nanoparticular system. Int J Mol Sci. (2009) 10:3776–92. doi: 10.3390/ijms10093776
126. Ochs-Balcom HM, Marian C, Nie J, Brasky TM, Goerlitz DS, Trevisan M, et al. Adiposity is associated with p53 gene mutations in breast cancer. Breast Cancer Res. Treat. (2015) 153:635–45. doi: 10.1007/s10549-015-3570-5
127. Park J, Euhus DM, Scherer PE. Paracrine and endocrine effects of adipose tissue on cancer development and progression. Endocr Rev. (2011) 32:550–70. doi: 10.1210/er.2010-0030
128. Jones VS, Huang RY, Chen LP, Chen ZS, Fu L, Huang RP, et al. Cytokines in cancer drug resistance: cues to new therapeutic strategies. Biochim Biophys Acta (2016) 1865:255–65. doi: 10.1016/j.bbcan.2016.03.005
129. Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull. (2017) 7:339–48. doi: 10.15171/apb.2017.041
130. Hembruff SL, Laberge ML, Villeneuve DJ, Guo B, Veitch Z, Cecchetto M, et al. Role of drug transporters and drug accumulation in the temporal acquisition of drug resistance. BMC Cancer (2008) 8:318. doi: 10.1186/1471-2407-8-318
131. Pajic M, Iyer JK, Kersbergen A, van der Burg E, Nygren AO, Jonkers J, Borst P, et al. Moderate increase in Mdr1a/1b expression causes in vivo resistance to doxorubicin in a mouse model for hereditary breast cancer. Cancer Res. (2009) 69:6396–404. doi: 10.1158/0008-5472.CAN-09-0041
132. Rottenberg S, Nygren AO, Pajic M, van Leeuwen FW, van der Heijden I, van de Wetering K, et al. Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc Natl Acad Sci USA. (2007) 104:12117–22. doi: 10.1073/pnas.0702955104
133. Malvi P, Chaube B, Singh SV, Mohammad N, Pandey V, Vijayakumar MV, et al. Weight control interventions improve therapeutic efficacy of dacarbazine in melanoma by reversing obesity-induced drug resistance. Cancer Metab. (2016) 4:21. doi: 10.1186/s40170-016-0162-8
134. Onstad MA, Schmandt RE, Lu KH. Addressing the role of obesity in endometrial cancer risk, prevention, and treatment. J Clin Oncol. (2016) 34:4225–30. doi: 10.1200/JCO.2016.69.4638
135. Zhao Y, Zhang X, Zhao H, Wang J, Zhang Q. CXCL5 secreted from adipose tissue-derived stem cells promotes cancer cell proliferation. Oncol Lett. (2018) 15:1403–1410.
136. Candelaria PV, Rampoldi A, Harbuzariu A, Gonzalez-Perez RR. Leptin signaling and cancer chemoresistance: perspectives. World J Clin Oncol. (2017) 8:106–19. doi: 10.5306/wjco.v8.i2.106
137. Schweizer R, Tsuji W, Gorantla VS, Marra KG, Rubin JP, Plock JA. The role of adipose-derived stem cells in breast cancer progression and metastasis. Stem Cells Int. (2015) 2015:120949. doi: 10.1155/2015/120949
138. Zheng Q, Banaszak L, Fracci S, Basali D, Dunlap SM, Hursting SD, et al. Leptin receptor maintains cancer stem-like properties in triple negative breast cancer cells. Endocr Relat Cancer (2013) 20:797–808. doi: 10.1530/ERC-13-0329
139. Nyasani E, Munir I, Perez M, Payne K, Khan S. Linking obesity-induced leptin-signaling pathways to common endocrine-related cancers in women. Endocrine (2018). doi: 10.1007/s12020-018-1748-4. [Epub ahead of print].
140. Mishra AK, Parish CR, Wong M-L, Licinio J, Blackburn AC. Leptin signals via TGFB1 to promote metastatic potential and stemness in breast cancer. PLoS ONE (2017) 12:e0178454. doi: 10.1371/journal.pone.0178454
141. Naviglio S, Di Gesto D, Illiano F, Chiosi E, Giordano A, Illiano G, et al. Leptin potentiates antiproliferative action of cAMP elevation via protein kinase A down regulation in breast cancer cells. J Cell Physiol. (2010) 225:801–9. doi: 10.1002/jcp.22288
142. Bahreyni A, Samani SS, Rahmani F, Behnam-Rassouli R, Khazaei M, Ryzhikov M, et al. Role of adenosine signaling in the pathogenesis of breast cancer. J Cell Physiol. (2018) 233:1836–43. doi: 10.1002/jcp.25944
143. Naviglio S, Caraglia M, Abbruzzese A, Chiosi E, Di Gesto D, Marra M, et al. Protein kinase A as a biological target in cancer therapy. Expert Opin Ther Targets (2009) 13:83–92. doi: 10.1517/14728220802602349
144. Fatemi A, Kazemi A, Kashiri M, Safa M. Elevation of cAMP Levels Inhibits Doxorubicin-Induced Apoptosis in Pre- B ALL NALM- 6 Cells through Induction of BAD Phosphorylation and Inhibition of P53 Accumulation. Int J Mol Cell Med. (2015) 4:94–102.
145. Vitale G, Dicitore A, Mari D, Cavagnini F. A new therapeutic strategy against cancer: cAMP elevating drugs and leptin. Cancer Biol Ther. (2009) 8:1191–3. doi: 10.4161/cbt.8.12.8937
146. Naviglio S, Di Gesto D, Romano M, Sorrentino A, Illiano F, Sorvillo L, et al. Leptin enhances growth inhibition by cAMP elevating agents through apoptosis of MDA-MB-231 breast cancer cells. Cancer Biol Ther. (2009) 8:1183–90. doi: 10.4161/cbt.8.12.8562
147. Spina A, Di Maiolo F, Esposito A, D'Auria R, Di Gesto D, Chiosi E, et al. Integrating leptin and cAMP signalling pathways in triple-negative breast cancer cells. Front Biosci. (2013) 18:92. doi: 10.2741/4092
148. Oerlecke I, Bauer E, Dittmer A, Leyh B, Dittmer J. Cyclic AMP enhances TGFβ responses of breast cancer cells by upregulating TGFβ receptor i expression. Ahmad A, ed. PLoS ONE (2013) 8:e54261. doi: 10.1371/journal.pone.0054261
149. del Valle-Pérez B, Martínez-Estrada OM, Vilaró S, Ventura F, Viñals F. cAMP inhibits TGF beta1-induced in vitro angiogenesis. FEBS Lett. (2004) 569:105–11. doi: 10.1016/j.febslet.2004.05.058
150. Wang W, Li Y, Zhu JY, Fang D, Ding HF, Dong Z, et al. Triple negative breast cancer development can be selectively suppressed by sustaining an elevated level of cellular cyclic AMP through simultaneously blocking its efflux and decomposition. Oncotarget (2016) 7:87232–45. doi: 10.18632/oncotarget.13601
151. Eckert LB, Repasky GA, Ülkü AS, McFall A, Zhou H, Sartor CI, et al. Involvement of Ras activation in human breast cancer cell signaling, invasion, and anoikis. Cancer Res. (2004) 64:4585–92. doi: 10.1158/0008-5472.CAN-04-0396
152. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al., 2007. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta (2007) 1773:1263–84. doi: 10.1016/j.bbamcr.2006.10.001
153. Spina A, Di Maiolo F, Esposito A, Sapio L, Chiosi E, Sorvillo L, et al. cAMP elevation down-regulates β3 integrin and focal adhesion kinase and inhibits leptin-induced migration of MDA-MB-231 breast cancer cells. Biores. Open Access (2012) 1:324–32. doi: 10.1089/biores.2012.0270
154. Illiano M, Sapio L, Salzillo A, Capasso L, Caiafa I, Chiosi E, et al. Forskolin improves sensitivity to doxorubicin of triple negative breast cancer cells via Protein Kinase A-mediated ERK1/2 inhibition. Biochem Pharmacol. (2018) 152:104–13. doi: 10.1016/j.bcp.2018.03.023
155. Huang J, Luo Q, Xiao Y, Li H, Kong L, Ren G. The implication from RAS/RAF/ERK signalling pathway increased activation in epirubicin treated triple negative breast cancer. Oncotarget (2017) 8:108249–60. doi: 10.18632/oncotarget.22604
156. Madsen L, Kristiansen K. The importance of dietary modulation of cAMP and insulin signaling in adipose tissue and the development of obesity. Ann N Y Acad Sci. (2010) 1190:1–14. doi: 10.1111/j.1749-6632.2009.05262.x
157. Arcidiacono B, Iiritano S, Nocera A, Possidente K, Nevolo MT, Ventura V, et al. Insulin resistance and cancer risk: an overview of the pathogenetic mechanisms. Exp Diabetes Res. (2012) 2012:789174. doi: 10.1155/2012/789174
158. Brahmkhatri VP, Prasanna C, Atreya HS. Insulin-like growth factor system in cancer: novel targeted therapies. Biomed Res Int. (2015) 2015:538019. doi: 10.1155/2015/538019
159. Lewitt MS, Dent MS, Hall K. 2014. The insulin-like growth factor system in obesity, insulin resistance and type 2 diabetes mellitus. J Clin Med. (2014) 3:1561–74. doi: 10.3390/jcm3041561
160. van Bunderen CC, Oosterwerff MM, van Schoor NM, Deeg DJ, Lips P, Drent ML. Serum IGF1, metabolic syndrome, and incident cardiovascular disease in older people: a population-based study. Eur J Endocrinol. (2013) 168:393–401. doi: 10.1530/EJE-12-0784
161. Vigneri PG, Tirrò E, Pennisi MS, Massimino M, Stella S, Romano C, et al. The Insulin/IGF System in Colorectal Cancer Development and Resistance to Therapy. Front Oncol. (2015) 5:230. doi: 10.3389/fonc.2015.00230
162. Chen WX, Liu XM, Lv MM, Chen L, Zhao JH, Zhong SL, et al. Exosomes from drug-resistant breast cancer cells transmit chemoresistance by a horizontal transfer of microRNAs. PLoS ONE (2014) 9:e95240. doi: 10.1371/journal.pone.0095240
163. Lin R, Wang S, Zhao RC. Exosomes from human adipose-derived mesenchymal stem cells promote migration through Wnt signaling pathway in a breast cancer cell model. Mol Cell Biochem. (2013) 383:13–20. doi: 10.1007/s11010-013-1746-z
164. Eguchi A, Mulya A, Lazic M, Radhakrishnan D, Berk MP, Povero D, et al. Microparticles release by adipocytes act as “find-me” signals to promote macrophage migration. PLoS ONE (2015) 10:e0123110. doi: 10.1371/journal.pone.0123110
165. Kranendonk ME, Visseren FL, van Balkom BW, Nolte-'t Hoen EN, van Herwaarden JA, de Jager W, et al. Human adipocyte extracellular vesicles in reciprocal signaling between adipocytes and macrophages. Obesity (2014) 22:1296–308. doi: 10.1002/oby.20679
166. Au Yeung CL, Co NN, Tsuruga T, Yeung TL, Kwan SY, Leung CS, et al., 2016. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat Commun. (2016) 7:11150. doi: 10.1038/ncomms11150
167. Jia Y, Chen Y, Wang Q, Jayasinghe U, Luo X, Wei Q, et al. Exosome: emerging biomarker in breast cancer. Oncotarget (2017) 8:41717–41733. doi: 10.18632/oncotarget.16684
168. Lazar I, Clement E, Dauvillier S, Milhas D, Ducoux-Petit M, LeGonidec S, et al. Adipocyte exosomes promote melanoma aggressiveness through fatty acid oxidation: a novel mechanism linking obesity and cancer. Cancer Res. (2016) 76:4051–7. doi: 10.1158/0008-5472.CAN-16-0651
169. Thompson PA, Rosner GL, Matthay KK, Moore TB, Bomgaars LR, Ellis KJ, et al. Impact of body composition on pharmacokinetics of doxorubicin in children: a Glaser Pediatric Research Network study. Cancer Chemother Pharmacol. (2009) 64:243–51. doi: 10.1007/s00280-008-0854-z
170. Griggs JJ, Mangu PB, Temin S, Balaban EP, Dignam JJ, Hryniuk WM, et al. Appropriate chemotherapy dosing for obese adult patients with cancer: American Society of clinical oncology clinical practice guideline. J Clin Oncol. (2012) 30:1553–61. doi: 10.1200/JCO.2011.39.9436
171. Ghose R, Omoluabi O, Gandhi A, Shah P, Strohacker K, Carpenter KC, et al. Role of high-fat diet in regulation of gene expression of drug metabolizing enzymes and transporters. Life Sci. (2011) 89:57–64. doi: 10.1016/j.lfs.2011.05.005
172. Behan JW, Avramis VI, Yun JP, Louie SG, Mittelman SD. Diet-induced obesity alters vincristine pharmacokinetics in blood and tissues of mice. Pharmacol Res. (2010) 61:385–90. doi: 10.1016/j.phrs.2010.01.007
173. Hanley MJ, Abernethy DR, Greenblatt DJ. Effect of obesity on the pharmacokinetics of drugs in humans. Clin Pharmacokinet. (2010) 49:71–87. doi: 10.2165/11318100-000000000-00000
174. Sheng X, Mittelman SD. The role of adipose tissue and obesity in causing treatment resistance of acute lymphoblastic leukaemia. Front Pediatr. (2014) 2:53. doi: 10.3389/fped.2014.00053
175. Sheng X, Parmentier JH, Tucci J, Pei H, Cortez-Toledo O, Dieli-Conwright CM, et al. Adipocytes Sequester and Metabolize the Chemotherapeutic Daunorubicin. Mol Cancer Res. (2017) 15:1704–13. doi: 10.1158/1541-7786.MCR-17-0338
176. Duong MN, Cleret A, Matera EL, Chettab K, Mathé D, Valsesia-Wittmann S, et al. Adipose cells promote resistance of breast cancer cells to trastuzumab-mediated antibody-dependent cellular cytotoxicity. Breast Cancer Res. (2015) 17:57. doi: 10.1186/s13058-015-0569-0
177. Horowitz NS, Wright AA. Impact of obesity on chemotherapy management and outcomes in women with gynecologic malignancies. Gynecol Oncol. (2015) 138:201–6. doi: 10.1016/j.ygyno.2015.04.002
178. Hefetz-Sela S, Scherer PE. Adipocytes: impact on tumor growth and potential sites for therapeutic intervention. Pharmacol Ther. (2013) 138:197–210. doi: 10.1016/j.pharmthera.2013.01.008
179. Hoy AJ, Balaban S, Saunders DN. Adipocyte-tumor cell metabolic crosstalk in breast cancer. Trends Mol Med. (2017) 23:381–392. doi: 10.1016/j.molmed.2017.02.009
180. Liao Z, Tan ZW, Zhu P, Tan NS. Cancer-associated fibroblasts in tumor microenvironment-Accomplices in tumor malignancy. Cell Immunol. (2018). doi: 10.1016/j.cellimm.2017.12.003. [Epub ahead of print].
181. Tao L, Huang G, Song H, Chen Y, Chen L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Onco Lett. (2017) 14:2611–20. doi: 10.3892/ol.2017.6497
182. Bochet L, Lehuédé C, Dauvillier S, Wang YY, Dirat B, Laurent V, et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. (2013) 73:5657–68. doi: 10.1158/0008-5472.CAN-13-0530
183. Arner P, Langin D. Lipolysis in lipid turnover, cancer cachexia, and obesity-induced insulin resistance. Trends Endocrinol Metab. (2014) 25:255–62. doi: 10.1016/j.tem.2014.03.002
184. Lengyel E, Makowski L, DiGiovanni J, Kolonin MG. Cancer as a matter of fat: the crosstalk between adipose tissue and tumors. Trends Cancer (2018) 4:374–84. doi: 10.1016/j.trecan.2018.03.004
185. Steuwe C, Patel II, Ul-Hasan M, Schreiner A, Boren J, Brindle KM, et al. CARS based label-free assay for assessment of drugs by monitoring lipid droplets in tumour cells. J Biophotonics (2014) 7:906–13. doi: 10.1002/jbio.201300110
186. Greene ER, Huang S, Serhan CN, Panigrahy D. Regulation of Inflammation in Cancer by Eicosanoids. Prostaglandins Other Lipid Mediat (2011) 96:27–36. doi: 10.1016/j.prostaglandins.2011.08.004
187. Nakajima M, Nagahashi M, Rashid OM, Takabe K, Wakai T. The role of sphingosine-1-phosphate in the tumor microenvironment and its clinical implications. Tumour Biol. (2017) 39:1010428317699133. doi: 10.1177/1010428317699133
188. Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. (2010) 70:8117–26. doi: 10.1158/0008-5472.CAN-09-3871
189. Guaita-Esteruelas S, Bosquet A, Saavedra P, Gumà J, Girona J, Lam EW, et al. Exogenous FABP4 increases breast cancer cell proliferation and activates the expression of fatty acid transport proteins. Mol Carcinog (2017) 56:208–17. doi: 10.1002/mc.22485
190. Nath A, Li I, Roberts LR, Chan C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci Rep. (2015) 5:14752. doi: 10.1038/srep14752
191. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy or rapid tumor growth. Nat Med. (2011) 17:1498–503. doi: 10.1038/nm.2492
192. Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, et al. Emerging roles of lipid metabolism in cancer metastasis. Molecular Cancer (2017) 16:76. doi: 10.1186/s12943-017-0646-3
193. Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, et al. JAK/STAT3-regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. (2018) 27:136–50.e5. doi: 10.1016/j.cmet.2017.11.001
194. Long J-P, Li X-Na, Zhang F. Targeting metabolism in breast cancer: how far we can go? World J Clin Oncol. (2016) 7:122. doi: 10.5306/wjco.v7.i1.122
195. Menendez JA, Lupu R. Fatty acid synthase regulates estrogen receptor-α signaling in breast cancer cells. Oncogenesis (2017) 6:e299. doi: 10.1038/oncsis.2017.4
196. Veigel D, Wagner R, Stübiger G, Wuczkowski M, Filipits M, Horvat R, et al. Fatty acid synthase is a metabolic marker of cell proliferation rather than malignancy in ovarian cancer and its precursor cells. Int J Cancer (2015) 136:2078–90. doi: 10.1002/ijc.29261
197. Yoon S, Lee MY, Park SW, Moon JS, Koh YK, Ahn YH, et al. Up-regulation of acetyl-CoA carboxylase alpha and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. J Biol Chem.(2007) 282:26122–31. doi: 10.1074/jbc.M702854200
198. Zhao J, Zhi Z, Wang C, Xing H, Song G, Yu X, et al. Exogenous lipids promote the growth of breast cancer cells via CD36. Oncol Rep. (2017) 38:2105–15. doi: 10.3892/or.2017.5864
199. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis (2016) 5:e189. doi: 10.1038/oncsis.2015.49
200. Scalfi-Happ C, Udart M, Hauser C, Rück A. Investigation of lipid bodies in a colon carcinoma cell line by confocal Raman microscopy. Med Laser Appl. (2011) 26:152–7. doi: 10.1016/j.mla.2011.08.002
201. de Gonzalo-Calvo D, López-Vilaró L, Nasarre L, Perez-Olabarria M, Vázquez T, Escuin D, et al. Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: a molecular and clinicopathological study. BMC Cancer (2015) 15:460. doi: 10.1186/s12885-015-1469-5
202. Koti M, Siu A, Clément I, Bidarimath M, Turashvili G, Edwards A, et al. A distinct pre-existing inflammatory tumour microenvironment is associated with chemotherapy resistance in high-grade serous epithelial ovarian cancer. Br J Cancer (2015) 112:1215–22. doi: 10.1038/bjc.2015.459
203. Vyas D, Laput G, Vyas AK. Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. Onco Targets Ther. (2014) 7:1015–23. doi: 10.2147/OTT.S60114
204. Pramanik R, Sheng X, Ichihara B, Heisterkamp N, Mittelman SD. Adipose tissue attracts and protects acute lymphoblastic leukemia cells from chemotherapy. Leuk Res. 37:503–9. doi: 10.1016/j.leukres.2012.12.013
205. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. (2017) 17:559–572. doi: 10.1038/nri.2017.49
206. Conze D, Weiss L, Regen PS, Bhushan A, Weaver D, Johnson P, et al. Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res, (2001) 61:8851–8.
207. Shi Z, Yang WM, Chen LP, Yang DH, Zhou Q, Zhu J, et al. Enhanced chemosensitization in multidrug-resistant human breast cancer cells by inhibition of IL-6 and IL-8 production. Breast Cancer Res Treat. (2012) 135:737–47. doi: 10.1007/s10549-012-2196-0
208. Tan C, Hu W, He Y, Zhang Y, Zhang G, Xu Y, Tang J. Cytokine-mediated therapeutic resistance in breast cancer. Cytokine (2018) 108:151–9. doi: 10.1016/j.cyto.2018.03.020
209. Bougaret L, Delort L, Billard H, Le Huede C, Boby C, De la Foye A, et al. Adipocyte/breast cancer cell crosstalk in obesity interferes with the anti-proliferative efficacy of tamoxifen. PLoS ONE (2018) 13:e0191571. doi: 10.1371/journal.pone.0191571
210. Batatinha H, Souza C, Lima E, Alonso-Vale MI, Cruz M, Da Cunha R, et al. Adipose tissue homeostasis is deeply disrupted by doxorubicin treatment. Cancer Metab (2014) 2(Suppl. 1):P5. doi: 10.1186/2049-3002-2-S1-P5
211. Dudka J, Gieroba R, Korga A, Burdan F, Matysiak W, Jodlowska-Jedrych B, et al. Different effects of resveratrol on dose-related Doxorubicin-induced heart and liver toxicity. Evid Based Complement Alternat Med. (2012) 2012:606183. doi: 10.1155/2012/606183
Keywords: obesity, adipose tissue, breast cancer, inflammation, treatment resistance
Citation: Mentoor I, Engelbrecht A-M, van Jaarsveld PJ and Nell T (2018) Chemoresistance: Intricate Interplay Between Breast Tumor Cells and Adipocytes in the Tumor Microenvironment. Front. Endocrinol. 9:758. doi: 10.3389/fendo.2018.00758
Received: 27 July 2018; Accepted: 29 November 2018;
Published: 11 December 2018.
Edited by:
Veronica Vella, Kore University of Enna, ItalyReviewed by:
Marco Falasca, Curtin University, AustraliaSilvio Naviglio, Università degli Studi della Campania Luigi Vanvitelli Caserta, Italy
Copyright © 2018 Mentoor, Engelbrecht, van Jaarsveld and Nell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Theo Nell, dG5lbGxAc3VuLmFjLnph