 Cathleen John1,2Jana Grune2,3
Cathleen John1,2Jana Grune2,3 Christiane Ott1,2Kerstin Nowotny1Stefanie Deubel1Arne Kühne4Carola Schubert2,4
Christiane Ott1,2Kerstin Nowotny1Stefanie Deubel1Arne Kühne4Carola Schubert2,4 Ulrich Kintscher2,4,5
Ulrich Kintscher2,4,5 Vera Regitz-Zagrosek2,5,6
Vera Regitz-Zagrosek2,5,6 Tilman Grune1,2,7,8*
Tilman Grune1,2,7,8*- 1Department of Molecular Toxicology, German Institute of Human Nutrition Potsdam-Rehbruecke, Potsdam, Germany
- 2German Center for Cardiovascular Research (DZHK), Partner Site Berlin, Berlin, Germany
- 3Institute of Physiology, Charité Universitätsmedizin Berlin, Berlin, Germany
- 4Institute of Pharmacology, Center for Cardiovascular Research, Charité -Universitätsmedizin Berlin, Berlin, Germany
- 5Center for Cardiovascular Research, Charité Universitätsmedizin Berlin, Berlin, Germany
- 6Institute for Gender in Medicine, Charité Universitätsmedizin Berlin, Berlin, Germany
- 7Institute of Nutritional Science, University of Potsdam, Potsdam, Germany
- 8German Center for Diabetes Research, Oberschleißheim, Germany
Background: Obesity is a risk factor for diseases including type 2 diabetes mellitus (T2DM) and cardiovascular disorders. Diabetes itself contributes to cardiac damage. Thus, studying cardiovascular events and establishing therapeutic intervention in the period of type T2DM onset and manifestation are of highest importance. Mitochondrial dysfunction is one of the pathophysiological mechanisms leading to impaired cardiac function.
Methods: An adequate animal model for studying pathophysiology of T2DM is the New Zealand Obese (NZO) mouse. These mice were maintained on a high-fat diet (HFD) without carbohydrates for 13 weeks followed by 4 week HFD with carbohydrates. NZO mice developed severe obesity and only male mice developed manifest T2DM. We determined cardiac phenotypes and mitochondrial function as well as cardiomyocyte signaling in this model.
Results: The development of an obese phenotype and T2DM in male mice was accompanied by an impaired systolic function as judged by echocardiography and MyH6/7 expression. Moreover, the mitochondrial function only in male NZO hearts was significantly reduced and ERK1/2 and AMPK protein levels were altered.
Conclusions: This is the first report demonstrating that the cardiac phenotype in male diabetic NZO mice is associated with impaired cardiac energy function and signaling events.
Introduction
Diabetes as Cardiovascular Risk Factor
Obesity and its related diseases, such as type 2 diabetes mellitus (T2DM) and cardiovascular diseases (CVD), developed through increasing caloric intake and/or reduced energy expenditure, are a present critical global health problem. The number of T2DM patients is expanding every year (1). Diabetes is characterized by high blood glucose, either because the body cannot produce enough insulin or is unable to use it effectively, consequently leading to insulin resistance or deficiency (2). It has been shown that T2DM is more common in men than in women (3). Moreover, men develop diabetes at a lower body mass index and are predestined to be more insulin-resistant (4). A recent review by Harreiter and Kautzky-Willer pointed out the importance of the investigation of sex differences in prevention of T2DM (5). The authors addressed that trials investigating lifestyle or pharmacological intervention in males and females at risk were promising so far, but that there is more need to analyze biological and psychosocial differences among women and men. In addition, T2DM increases the risk of CVD, known to be a major cause of morbidity and mortality in diabetic men and women (6–8).
Sex Differences in Heart Metabolism
Interestingly, marked sex differences of healthy individuals regarding mitochondrial function in the heart have been reported. In fact, female rat heart mitochondria produce less reactive oxygen species and had a greater antioxidant capacity than those from males (9), resulting in a better protection of heart function in females (10). Cardiac metabolic response is regulated by several intracellular signaling pathways, including the mitogen activated protein kinase (MAPK) signaling. One class of the MAPKs is the extracellular signal regulated kinases 1/2 (ERK1/2), mediating heart development, metabolism and function. Cumulative evidence confirms that MAPKs influence cardiac compensation and decompensation partly through mitochondria interactions (11). Previous MAPKs studies demonstrated a direct interaction with the outer mitochondrial membrane, translocation into mitochondria (12–14) and indirect effects between these kinases and mitochondria (15–20). Additionally, a key player in the modulation of metabolism is AMP-activated protein kinase (AMPK) (21). Recently, AMPK was shown to be required for the fragmentation of mitochondria and is sufficient to induce mitochondrial fission (22). Two other very important functionally distinct cardiac proteins, responsible for contractility, are the myosin heavy chain isoforms alpha (Myh6) and beta (Myh7). Relative expression levels are altered in cardiac disease (23). However, there is a gap regarding our knowledge on sex dimorphism in diabetes associated cardiovascular research (24). In addition, most preclinical research is done using male animals or cells with undefined/unmentioned sex (25, 26).
The New Zealand Obese Mouse as a Model for Diabetes
The New Zealand Obese (NZO) mouse is an appropriate animal model to examine sex differences in diabetes and related cardiac function. The NZO mouse represents a model of morbid obesity, insulin resistance, hypertension, and hypercholesterolemia which resembles the human metabolic syndrome (27). Obesity in the NZO mouse is the consequence of a moderately increased food intake and reduced energy expenditure. It is accompanied by a marked hyperglycemia and hyperinsulinemia at earlier age, followed by associated beta-cell destruction (27). Phenotypically, overt diabetes in NZO mice is defined by a threshold of 16.6 mM plasma glucose (27). Interestingly, NZO mice possess sex-dependent characteristics. While female and male NZO mice become obese on a high-fat diet, females are protected from becoming diabetic. In males instead, the diabetes prevalence is usually 50–75% at the age of 22 weeks (28, 29). However, it has been shown that female NZO mice can develop diabetes on a high-fat-diet (HFD) (30). Surprisingly, cardiovascular parameters considering heart functions in the NZO mice have not yet been investigated in relation to metabolic hear function.
In this study, we investigated whether changes in the diabetic state are associated with sex, and this correlates with changes in cardiac mitochondrial function or expression of metabolically-associated proteins and contractile proteins and cardiac function in the NZO mouse.
Materials and Methods
Experimental Design
Mice were kept in agreement with the National Institutes of Health guidelines for care and use of laboratory animals. All animal procedures were performed in accordance with the guidelines of the German Law on the Protection of Animals and were approved by the local authorities (Landesamt für Gesundheit und Soziales, Berlin, Germany).
Male and female NZO/HIBomDife (German Institute of Human Nutrition Potsdam-Rehbruecke [DIfE], Nuthetal, Germany) and C57BL/6JRj (B6, control group) mice from Janvier were housed under identical conditions (12 light/dark cycle, 21°C room temperature and free access to food and water). The number of animals is indicated in the legends of the figures. At 5 weeks of age, mice were placed on a HFD with a low carbohydrate content (Altromin, custom made by the manufacturer, Lage, Germany: C 1057-89; fat: 30.5%, protein: 32%, carbohydrates: <0.1%) and at an age of 18 weeks, the animals obtained a high carbohydrate diet (HCD) (Altromin, custom made by the manufacturer: C 1090-60; fat: 35%, protein: 21.4%, carbohydrates: 29%) for 4 weeks. Body weight and blood glucose levels were weekly controlled. Body weight was measured with an electronic scale and blood glucose was determined with a Contour XT glucose meter (Bayer Health Care, Leverkusen, Germany). Blood samples were collected before sacrificing the mice and were stored after centrifugation at −80°C. For the oxygen consumption experiment hearts were cut longitudinally and fiber bundles were isolated. The remaining heart tissue was frozen in liquid nitrogen and stored at −80°C until protein or mRNA isolation.
Serum Insulin- and Proinsulin-ELISA
The concentration of insulin and proinsulin in murine serum was determined by using the Mouse High Range Insulin ELISA (ALPCO, Salem, USA) and carried out according to the manufacturer's instructions.
Mitochondrial Respiration Function
The mitochondrial function of heart fibers was measured by oxygraph chambers and Clarke electrodes from Hansatech. A heart-fiber bundle was removed and fibrillated to form individual fibers that were still connected at the ends. These fibers were permeabilized under stirring for 30 min in saponin buffer and then washed twice for 10 min. From the permeabilized fibers 2 × 2 mm pieces (about 3–5 mg wet weight) were separated and placed in 1 ml potassium chloride buffer in the oxygraph chambers. The chambers were closed airtight and the basal respiration of the fibers was determined. Thereafter, the oxygen consumption was recorded and presented as a graph during adding the following substrates and metabolites in an interval of about 2 min: 2.5 mM adenosine diphosphate (ADP), 4 μM rotenone, 10 mM succinate, 8 μM cytochrome c, 4 μM Antimycin A, 0.5 mM tetramethyl-p-phenylenediamine (TMPD), and 7.5 mM sodium azide. At the end, the respiratory control index (RCI), the ratio of maximum respiration (state 3) and respiration in the absence of ADP (state 4), was determined by the software program.
RNA and Protein Analysis
The mRNA isolation from 5 mg heart tissue was done with the Dynabeads™ mRNA Purification Kit (Thermo Fisher Scientific # 61006). According to the manufacturer's instructions mRNA samples were reverse transcribed with the SensiFAST cDNA Synthesis Kit (Bioline # BIO-65053) and quantitative real time-PCR reactions in the presence of Dream-Taq-Hot Start- DNA Polymerase (Thermo Fisher Scientific # EP1703) and SYBR Green (Life Sciences) was performed. Following murine primers were used: myosine heavy chain 6 (Myh6) (fwd 5′-AGAAGCCCAGCGCTCCCTCA-3′, rev 5′- TGCCTCGGGTCAGCTGGGAA-3′, myosine heavy chain 7 (Myh7) (fwd 5′-TTCCTTACTTGCTACCCTC-3′, rev 5′-CTTCTCAGACTTCCGCAG-3′). Relative abundance of mRNA was calculated after normalization to ribosomal protein L13a (Rpl13a) reference (primer sequence: fwd 5′-GTTCGGCTGAAGCCTACCAG-3′, rev 5′-TTCCGTAACCTCAAGATCTGCT-3′.
Protein isolation of approximately 10 mg of tissue was performed in 500 μl of RIPA buffer (including protease inhibitors) using ceramic spheres in a homogenizer (FastPrep-24, MP Biomedicals). The concentration of the protein samples was determined by a BCA Protein Assay Kit (Pierce # 23225). Lysates were analyzed by immunoblotting using primary antibodies raised against phosphor-p44/42 MAPK (pERK) (Cell Signaling #4370), p44/42 MAPK (ERK1/2) (Cell Signaling #4695), phospho-AMPKα (Thr172) (Cell Signaling #2535), AMPKα (Cell Signaling #2532), Myh6 (K-13) (Santa Cruz #168676), Myh7 (Santa Cruz #71575), α-tubulin (Sigma #T9026), and secondary antibodies, such as donkey anti-mouse IgG, donkey anti-rabbit-IgG or donkey anti-goat-IgG antibodies (Jackson Immuno Research Laboratories). For the investigation of mitochondrial respiration chain complexes a total OXPHOS Rodent WB Antibody Cocktail (abcam #110413) was taken. For detection, enhanced chemiluminescent reagents (ECL kit; Thermo Scientific) and a BioRad Chemidoc MP System were used.
Echocardiography
Echocardiographic analysis regarding cardiac morphology and function was carried out at the age of 22 weeks. Echocardiography was performed as previously described, with the exception that the isofluran dose needed to be adapted to the higher weight of animals (3–4%) (31, 32). Echocardiography was performed using a MX400 ultra-high frequency linear array transducer (18–38 MHz, center transmit: 30 MHz, axial resolution: 50 μm) together with a Vevo® 3100 high-resolution Imaging System (both FUJIFILM VisualSonics, Toronto, ON, Canada). M-Mode images of the maximum dimension of the LV in parasternal long axis view were used to analyze cardiac dimensions and calculate LV mass (LVM). B-Mode pictures were obtained in order to analyze cardiac volumes and LV function parameters. All data sets were acquired prospectively and analyzed for this study in a retrospective manner.
Statistics
All analyses were performed using GraphPad Prism 7. Results represent mean values ± standard deviation (SD). Unpaired t-test or Mann-Whitney test and for comparison of the multiple groups, two-way ANOVA followed by Tukey-B-Posthoc-test was used. Differences with p ≤ 0.05 were considered statistically significant.
Results
Manifestation of Obesity and Diabetes
All mice received HFD without carbohydrates for 13 weeks followed by 4 weeks HCD. Male and female NZO mice developed severe obesity until the end of the study compared to the B6-control group (Figure 1A). Blood glucose levels of NZO males and females were also increased in comparison to B6 mice, but only if mice were treated for 4 weeks with HCD (Figure 1B). Comparing the final body weight of male and female NZO with the corresponding B6 at 22 weeks, body weight of both sexes was significantly increased (Figure 1C). However, exclusively the male NZO mice showed significantly higher blood glucose levels compared to their B6 counterparts (Figure 1D). Development of T2DM in male NZO mice was accompanied by significant higher serum proinsulin (Figure 1E) and serum insulin levels compared to B6 controls, while values of female NZO mice did not reach statistical significance (Figure 1F).
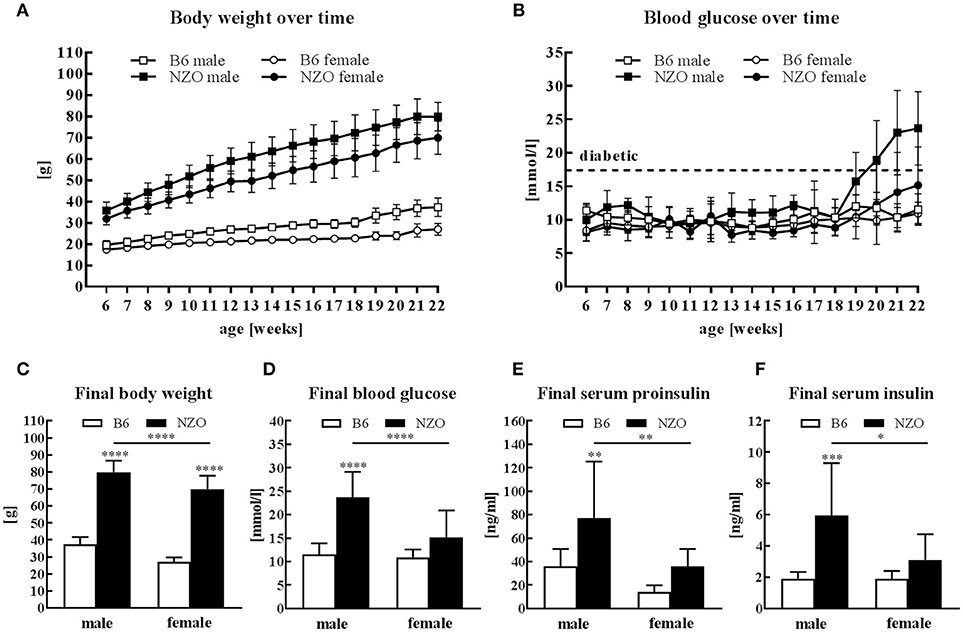
Figure 1. Body weight, glucose and insulin in male and female NZO and B6 mice. Body weight (A) and blood glucose (B) course during the whole study. Body weight (C), blood glucose (D), serum proinsulin (E) and serum insulin (F) at the end of the study (week 22). Mean ± SD. Two-way ANOVA with Tukey's posthoc test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; B6 male, n = 10–12; NZO male, n = 10–18; B6 female, n = 10–12; NZO female, n = 10–19.
Characterization of Cardiac Phenotype by Echocardiography
Echocardiography was used to phenotype the cardiac performance of HCD-fed NZO mice and B6 controls. First, we determined the cardiac phenotype, i.e., heart rate, LV wall thicknesses, and LVM, and measures of systolic cardiac performance (Table 1, first part). NZO males had a dramatically lowered heart rate when compared to B6 control males. The LV anterior and posterior walls were significantly thicker in NZO males than in B6 males. In female NZO mice exclusively the LV posterior wall demonstrated moderately increased wall thickening compared to female B6 mice. The LVM was significantly increased in both NZO sexes in comparison to corresponding B6 mice, and was also significantly increased in NZO males when compared to NZO females, which was not the case when looking at B6 males compared to B6 females. The inner diameters of the LV were significantly increased in both NZO sexes in comparison to corresponding B6 mice. No differences were observed, when comparing ESV and EDV of NZO mice with their sex-matched B6-controls. NZO mice of both sexes tended to have greater ESVs but smaller EDVs compared to sex-matched B6 controls, however reaching no statistical significance (Table 1, second part). In both genotypes, female mice showed lower absolute ESV and EDV compared to corresponding male controls. SV and EF were significantly decreased in both sexes of NZO mice when compared to sex-matched B6 controls.
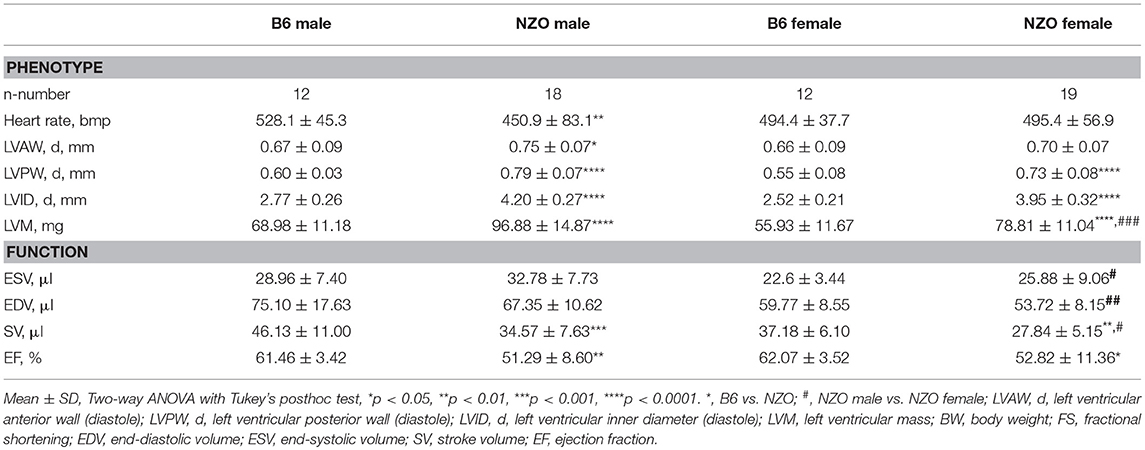
Table 1. Echocardiographic-assessed characterization of cardiac phenotypes after 22 weeks.
Changes of Cardiac Contractile Proteins
Next, we investigated expression levels of contractile proteins Myh6 and Myh7. Male NZO mice showed a tendency to a lower Myh6 mRNA and protein expression level compared to B6 males. Female NZO mice had no changes in the Myh6 mRNA expression, while theMyh6 protein levels also have a tendency to be lower compared to B6 females (Figures 2A,B). In contrast, in male NZO mice, the Myh7 mRNA expression was significantly increased compared to female NZO and male B6 mice (Figures 2C,D). However, Myh7 mRNA and protein levels were not elevated in female NZO mice, compared to the B6 controls (Figures 2C,D).
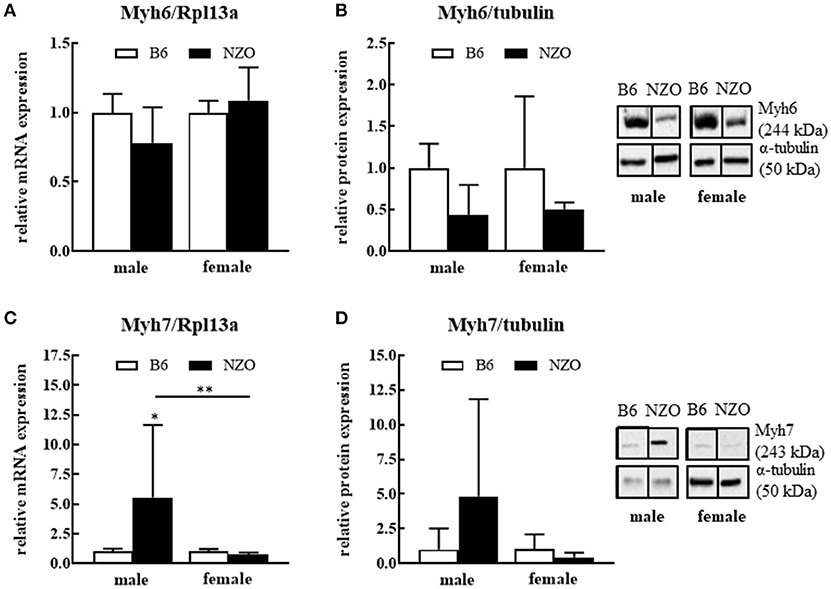
Figure 2. Protein and mRNA expression differences of cardiac myosine heavy chain isoforms 6 and 7. Myh6 mRNA expression level (A), Myh6 protein expression (B), Myh7 mRNA expression (C) and Myh7 protein expression (D). mRNA was normalized to Rpl13a and as protein expression reference α-tubulin was used. Mean ± SD. Two-way ANOVA with Tukey's posthoc test, *p < 0.05, **p < 0.01; B6 male: n = 5–10, NZO male: n = 7–10, B6 female: n = 5–10, NZO female: n = 7–10.
Mitochondrial Function
Since the major goal of our study was to investigate the influence of a sex-related diabetic phenotype on the energy function of the heart, we next investigated whether changes in mitochondrial function or expression of metabolism-associated proteins in the heart explain the observed sex differences in cardiac phenotype. After sacrificing the mice, single heart fibers were isolated and mitochondrial function was measured. The RCI was significantly reduced in NZO males vs. B6 controls. State 3 was significantly decreased, whereas state 4 showed no changes in male NZO mice compared to B6 males. In contrast to male NZOs, the mitochondrial respiration of female NZO mice was not significantly changed in comparison to B6 mice (Figure 3A). Furthermore, the relative protein expression of all mitochondrial respiration chain complexes, excluding complex IV, seemed to be reduced in NZO males vs. B6 males, although no significant differences were found. In female NZO mice no protein expression change of the complexes was seen (Figure 3B). In general, the protein expression levels of most mitochondrial complexes had a tendency to be higher in females compared to the corresponding males.
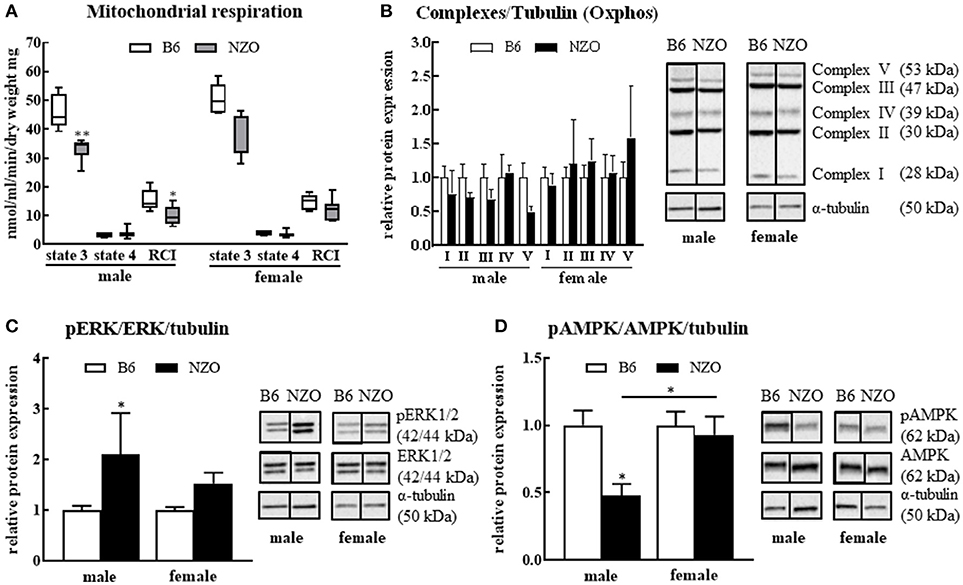
Figure 3. Changes in mitochondrial function and signaling pathways. Mitochondrial respiration (A) and protein levels of mitochondrial respiration chain complexes (B), phosphorylated ERK1/2 (C) and phosphorylated AMPK (D). α-tubulin was used as reference protein. Mean ± SD. Two-way ANOVA with Tukey's posthoc test, *p < 0.05, **p < 0.01; B6 male: n = 5, NZO male: n = 7, B6 female: n = 5, NZO female: n = 7.
In addition, the amount of the phosphorylated ERK1/2 was significantly elevated in male NZO mice compared to male B6 animals (Figure 3C). Inversely, the phosphorylated AMPK protein expression was significantly lower in male NZO mice than in B6 male controls and as well in comparison to female NZO mice (Figure 3D). No significant changes were seen in female NZO mice regarding the protein expression levels of phosphorylated ERK1/2 or AMPK (Figures 3C,D).
Discussion
Here we were able to show that the development of an diabetic phenotype in male NZO mice was accompanied by changes in the heart function, as judged by the echocardiography, by changes in the Myh6/7 expression and moreover by impairment of mitochondrial function. A reduced state 3 respiration and RCI was accompanied by significantly reduced ERK1/2 and AMPK protein levels.
Male and female NZO mice developed a severe obesity when they were maintained on a HFD until the age of 22 weeks which is in agreement with the study of Kluge et al. (33). After the feeding of carbohydrates from the 18th week of age only male NZO mice revealed a manifest T2DM status whereas female NZO mice seem to be protected from T2DM. Male NZO mice generated a marked hyperglycemia and hyperinsulinemia reflecting pronounced insulin resistance which is consistent with data from Joost and Schürmann (27). In females, the final blood glucose and the associated serum proinsulin as well as serum insulin levels were significantly lower than in male NZO mice and there were no changes to the B6-control group which indicates no overt diabetes development (Figure 1). Interestingly, a previous study showed that an ovariectomy of NZO females modulates diabetic insulin resistance and produced a phenotype which was comparable with that of NZO males (34).
Nevertheless, until now it is not known how this effect influences the onset and progression of cardiovascular events. To our knowledge, we describe here for the first time the cardiovascular damage events in relation to metabolic functions in the NZO mouse model in a sex-sensitive manner. In general a decrease in insulin sensitivity leads to less glucose uptake which perhaps can exert feedback on a molecular regulation leading to an increase in mitochondrial dysfunction in the heart (35). Today, no extensive knowledge exists about the energy metabolism in the diabetic human heart, but it is known that the energy supply through glucose in diabetic hearts is disturbed (36). The influence of sex regarding glucose utilization in obese and diabetic patients seems to be pronounced (37). The exact underlying mechanisms, impairing cardiac function and promoting cardiomyocyte injury, have not yet been demonstrated. Besides lipotoxicity, oxidative stress, intramyocardial inflammation and altered insulin and calcium signaling, mitochondrial dysfunction plays an important role (38). Moreover, hyperlipidemia has been shown to increase stress on mitochondria as they attempt to generate sufficient ATP, leading to increased ROS production (39). In fact, all this leads to further complications in the diabetic heart (40, 41). In our study, the mitochondrial respiration, represented by RCI, was significantly lower in male NZOs than in females where no significant changes were documented compared to the control group. One explanation for the decreased mitochondrial respiration could be that the mitochondrial respiration chain complexes are less expressed in the male NZO hearts (Figure 3, part one). Decreased expression level of mitochondrial chain complexes could either be due to a reduced number of mitochondria per cardiomyocyte or a low expression level of the proteins per mitochondrial unit. We could not study mitochondrial numbers but found a trend toward a lower expression of respiratory proteins in the male NZOs. Interestingly, investigations in other obesity mouse models related to T2DM, such as the ob/ob and the db/db mice, showed likewise decreased cardiac mitochondrial respiration capacity and lower expression of respiration chain complexes (42, 43).
Mitochondrial dysfunction activates various signaling pathways so we tested the phosphorylation level of ERK1/2 and AMPK two markers of mitochondrial metabolism. In our study, the level of pERK was significantly upregulated only in male NZO mice, but there were no differences in female NZO mice vs. the control group (Figure 3C). The activation of ERK1/2 was also shown in other studies with diabetic hearts (44) and it is also known that activated ERK1/2 contributes to apoptosis in cardiomyocytes under diabetic conditions (45, 46). In addition, it was reported that mitochondrial dysfunction was associated with increased activation and protein expression of ERK1/2 and an attenuated mitochondrial respiration and, therefore, ATP production, wherein a decreased complex I activity and substrate oxidation was observed as well (47, 48). A recent study with diabetic db/db mice showed that levels of phosphorylated ERK1/2 were increased and an inhibition of ERK1/2 can prevent cell death under diabetic conditions. The authors concluded a pathological cardiac change in diabetes through ERK1/2 (49). Therefore, these findings are a clear indication of a metabolic failure and stress in diabetic male hearts.
In contrast, pAMPK was significantly decreased in male NZO mice compared to the B6 controls and NZO females. AMPK has been identified in the past as a central actor of mitochondrial homeostasis. The cardiac AMPK downregulation has been reported in some T2DM animal models as an important intervention target (50, 51). Our data are supported by a study in db/db mice in which a significant decrease of AMPK phosphorylation in murine cardiac tissue was observed (52), but oppositely in ob/ob mice an increased cardiac AMPK phosphorylation was observed (53). If this cardiac benefit is secondary to systemic improvement or to intrinsic cardiomyocyte mechanisms remains to be clarified. Impaired cardiac glucose uptake and utilization are evident in T2DM male patients but not in females (54). Moreover, Boudina et al. conclude that the reduced mitochondrial oxidative capacity may contribute to cardiac dysfunction in their obese mouse model (42, 43).
In the present study, we observed as well differences of contractile proteins on sex-specific cardiac phenotypes. Two important heart contractile proteins, Myh6 and Myh7 were altered in the NZO mouse model (Figure 2). While the RNA- and protein expression of Myh6 was decreased in the NZO males, the Myh7 increased, demonstrating clear shift of these two contraction actors. In the non-diabetic female NZO mice there was no change or shift seen. Interestingly, in impaired adult mouse hearts, a shift from the predominant Myh6 toward Myh7 is often present (55). A study from Krenz et al., using transgenic mice, which predominantly expressed Myh7 instead of Myh6 in the heart, showed a significantly reduced cardiac contractility (56). Further, in cardiac hypertrophy an elevation of Myh7 can serve as an early and sensitive marker and recently downregulation of Myh6 expression in human hearts was observed, too (57–59). Transcriptional reprogramming of MHC gene expression has been described to be characteristic for the development of hypertrophy-induced heart failure. Hang et al. have well described that resulting cardiac stress triggers adult or stressed hearts to undergo a shift from α-MHC (Myh6) to β-MHC expression (Myh7) by reactivation of chromatin remodeling protein Brg1. Complexation of Brg1 with its partners PARP and HDAC induce the pathological Myh6 to Myh7 switch (60), that we could also show for the NZO males.
This suggests a cardiac dysfunction in male NZO mice (61–63), which was further verified by echocardiography. Indeed, we observed a lower heart rate in NZO male mice in comparison to the control group, but no changes in female ones. In addition, male NZO mice had significant decreased EF and dramatically increased LVM, indicating more severe cardiac dysfunction than in females. Surprisingly, although in female NZO mice, only developing obesity, not manifest diabetes, we observed a significantly reduced EF and enhanced LVM in comparison with B6 controls, corresponding to a non-significant trend for a decrease in mitochondrial function (Table 1). Nevertheless, it seems that the diabetic status of the male NZO mice aggravated the cardiac dysfunction in males. Males had a lower mitochondrial respiration, increased ERK1/2 and decreased AMPK activity as well as an increased Myh7 expression level in the heart, which was not seen in female NZO mice.
In our NZO diabetic animal model, the cardiac function was severely decreased, particularly in the male NZO mice. It is possible that sex differences in mitochondrial function potentially contribute to a better cardiac performance in females. Thus, from T2DM protected female NZO mice show a better mitochondrial respiration rate than diabetic male NZO mice. Sex-specific changes in the related mitochondrial function and cellular stress signaling let us propose a functional impairment of the heart, which was confirmed by echocardiography.
Author Contributions
CJ: performed most of the animal treatment, analyzed the data and wrote the manuscript; JG: executed echocardiographic data acquisition and –analysis; CO and KN: helped to prepare the animal tissues; SD: conducted the analysis to serum proinsulin- and insulin levels and RT-PCR; AK: determined the mitochondrial respiration assay and western blots; CS: analyzed the data and proofed the statistics; CO, UK, VR-Z, and TG: designed and supervised the project and enabled the experiments. All authors reviewed the manuscript.
Funding
This work was supported by a grant from the German Ministry of Education and Research (BMBF) and the State of Brandenburg (DZD grant 82DZD00302). Additionally, the work was supported by the DZHK grant project NZOcardio (100290384) and DZHK core facility (echocardiographic system), the Margarete-Ammon Stiftung, and by the Gesundheitscampus Brandenburg.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Vanessa Riese for her excellent technical assistance in separating the single heart fibers and determination of the mitochondrial respiration.
References
1. Atlas IFDID. Atlas IFDID (7th edition 2015 and 8th edition 2017) Available online at: www.diabetesatlas.org/
2. Pulgaron ER, Delamater AM. Obesity and type 2 diabetes in children: epidemiology and treatment. Curr Diab Rep. (2014) 14:508. doi: 10.1007/s11892-014-0508-y
3. Emerging Risk Factors C, Sarwar N, Gao P, Seshasai SR, Gobin R, Kaptoge S, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet (2010) 375:2215–22. doi: 10.1016/S0140-6736(10)60484-9
4. Sattar N. Gender aspects in type 2 diabetes mellitus and cardiometabolic risk. Best Pract Res Clin Endocrinol Metab. (2013) 27:501–7. doi: 10.1016/j.beem.2013.05.006
5. Harreiter J, Kautzky-Willer A. Sex and gender differences in prevention of type 2 diabetes. Front Endocrinol. (2018) 9:220. doi: 10.3389/fendo.2018.00220
6. Gregg EW, Gu Q, Cheng YJ, Narayan KM, Cowie CC. Mortality trends in men and women with diabetes, 1971 to 2000. Ann Intern Med. (2007) 147:149–55. doi: 10.7326/0003-4819-147-3-200708070-00167
7. Mascarenhas-Melo F, Marado D, Palavra F, Sereno J, Coelho Á, Pinto R, et al. Diabetes abrogates sex differences and aggravates cardiometabolic risk in postmenopausal women. Cardiovasc Diabetol. (2013) 12:61. doi: 10.1186/1475-2840-12-61
8. Steinberg HO, Paradisi G, Cronin J, Crowde K, Hempfling A, Hook G, et al. Type II diabetes abrogates sex differences in endothelial function in premenopausal women. Circulation (2000) 101:2040–6. doi: 10.1161/01.CIR.101.17.2040
9. Lagranha CJ, Deschamps A, Aponte A, Steenbergen C, Murphy E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ Res. (2010) 106:1681–91. doi: 10.1161/CIRCRESAHA.109.213645
10. Ventura-Clapier R, Moulin M, Piquereau J, Lemaire C, Mericskay M, Veksler V, et al. Mitochondria: a central target for sex differences in pathologies. Clin Sci. (2017) 131:803–22. doi: 10.1042/CS20160485
11. Javadov S, Jang S, Agostini B. Crosstalk between mitogen-activated protein kinases and mitochondria in cardiac diseases: therapeutic perspectives. Pharmacol Ther. (2014) 144:202–25. doi: 10.1016/j.pharmthera.2014.05.013
12. Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, et al. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J Biol Chem. (2000) 275:322–7. doi: 10.1074/jbc.275.1.322
13. Baines CP, Zhang J, Wang GW, Zheng YT, Xiu JX, Cardwell EM, et al. Mitochondrial PKCepsilon and MAPK form signaling modules in the murine heart: enhanced mitochondrial PKCepsilon-MAPK interactions and differential MAPK activation in PKCepsilon-induced cardioprotection. Circ Res. (2002) 90:390–7. doi: 10.1161/01.RES.0000012702.90501.8D
14. Ballard-Croft C, Kristo G, Yoshimura Y, Reid E, Keith BJ, Mentzer RM, et al. Acute adenosine preconditioning is mediated by p38 MAPK activation in discrete subcellular compartments. Am J Physiol Heart Circ Physiol. (2005) 288:H1359–1366. doi: 10.1152/ajpheart.01006.2004
15. Bogoyevitch MA, Ng DC, Court NW, Draper KA, Dhillon A, Abas L. Intact mitochondrial electron transport function is essential for signalling by hydrogen peroxide in cardiac myocytes. J Mol Cell Cardiol. (2000) 32:1469–80. doi: 10.1006/jmcc.2000.1187
16. Zhao TC, Hines DS, Kukreja RC. Adenosine-induced late preconditioning in mouse hearts: role of p38 MAP kinase and mitochondrial K(ATP) channels. Am J Physiol Heart Circ Physiol. (2001) 280:H1278–1285. doi: 10.1152/ajpheart.2001.280.3.H1278
17. Yue Y, Qin Q, Cohen MV, Downey JM, Critz SD. The relative order of mK(ATP) channels, free radicals and p38 MAPK in preconditioning's protective pathway in rat heart. Cardiovasc Res. (2002) 55:681–9. doi: 10.1016/S0008-6363(02)00452-2
18. Kaiser RA, Bueno OF, Lips DJ, Doevendans PA, Jones F, Kimball TF, et al. Targeted inhibition of p38 mitogen-activated protein kinase antagonizes cardiac injury and cell death following ischemia-reperfusion in vivo. J Biol Chem. (2004) 279:15524–30. doi: 10.1074/jbc.M313717200
19. Kong JY, Klassen SS, Rabkin SW. Ceramide activates a mitochondrial p38 mitogen-activated protein kinase: a potential mechanism for loss of mitochondrial transmembrane potential and apoptosis. Mol Cell Biochem. (2005) 278:39–51. doi: 10.1007/s11010-005-1979-6
20. Wall JA, Wei J, Ly M, Belmont P, Martindale JJ, Tran D, et al. Alterations in oxidative phosphorylation complex proteins in the hearts of transgenic mice that overexpress the p38 MAP kinase activator, MAP kinase kinase 6. Am J Physiol Heart Circ Physiol. (2006) 291:H2462–72. doi: 10.1152/ajpheart.01311.2005
21. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. (2018) 19:121–35. doi: 10.1038/nrm.2017.95
22. Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Losón OC, Hellberg K, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science (2016) 351:275–81. doi: 10.1126/science.aab4138
23. Nadal-Ginard B, Mahdavi V. Molecular basis of cardiac performance. Plasticity of the myocardium generated through protein isoform switches. J Clin Invest. (1989) 84:1693–700. doi: 10.1172/JCI114351
24. Reichelt ME, Mellor KM, Bell JR, Chandramouli C, Headrick JP, Delbridge LM. Sex, sex steroids, and diabetic cardiomyopathy: making the case for experimental focus. Am J Physiol Heart Circ Physiol. (2013) 305:H779–792. doi: 10.1152/ajpheart.00141.2013
25. Zucker I, Beery AK. Males still dominate animal studies. Nature (2010) 465:690. doi: 10.1038/465690a
26. Yoon DY, Mansukhani NA, Stubbs VC, Helenowski IB, Woodruff TK, Kibbe MR. Sex bias exists in basic science and translational surgical research. Surgery (2014) 156:508–16. doi: 10.1016/j.surg.2014.07.001
27. Joost HG, Schürmann A. The genetic basis of obesity-associated type 2 diabetes (diabesity) in polygenic mouse models. Mamm Genome (2014) 25:401–12. doi: 10.1007/s00335-014-9514-2
28. Jürgens HS, Neschen S, Ortmann S, Scherneck S, Schmolz K, Schüler G, et al. Development of diabetes in obese, insulin-resistant mice: essential role of dietary carbohydrate in beta cell destruction. Diabetologia (2007) 50:1481–9. doi: 10.1007/s00125-007-0662-8
29. Ortlepp JR, Kluge R, Giesen K, Plum L, Radke P, Hanrath P, et al. A metabolic syndrome of hypertension, hyperinsulinaemia and hypercholesterolaemia in the New Zealand obese mouse. Eur J Clin Invest. (2000) 30:195–202. doi: 10.1046/j.1365-2362.2000.00611.x
30. Lubura M, Hesse D, Kraemer M, Hallahan N, Schupp M, von Löffelholz C, et al. Diabetes prevalence in NZO females depends on estrogen action on liver fat content. Am J Physiol Endocrinol Metab. (2015) 309:E968–980. doi: 10.1152/ajpendo.00338.2015
31. Grune J, Benz V, Brix S, Salatzki J, Blumrich A, Höft B, et al. Steroidal and nonsteroidal mineralocorticoid receptor antagonists cause differential cardiac gene expression in pressure overload-induced cardiac hypertrophy. J Cardiovasc Pharmacol. (2016) 67:402–11. doi: 10.1097/FJC.0000000000000366
32. Grune J, Blumrich A, Brix S, Jeuthe S, Drescher C, Grune T, et al. Evaluation of a commercial multi-dimensional echocardiography technique for ventricular volumetry in small animals. Cardiovasc Ultrasound (2018) 16:10. doi: 10.1186/s12947-018-0128-9
33. Kluge R, Scherneck S, Schürmann A, Joost HG. Pathophysiology and genetics of obesity and diabetes in the New Zealand obese mouse: a model of the human metabolic syndrome. Methods Mol Biol. (2012) 933:59–73. doi: 10.1007/978-1-62703-068-7_5
34. Vogel H, Mirhashemi F, Liehl B, Taugner F, Kluth O, Kluge R, et al. Estrogen deficiency aggravates insulin resistance and induces beta-cell loss and diabetes in female New Zealand obese mice. Horm Metab Res. (2013) 45:430–5. doi: 10.1055/s-0032-1331700
35. Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia (2014) 57:660–71. doi: 10.1007/s00125-014-3171-6
36. Peterson LR, Saeed IM, McGill JB, Herrero P, Schechtman KB, Gunawardena R, et al. Sex and type 2 diabetes: obesity-independent effects on left ventricular substrate metabolism and relaxation in humans. Obesity (2012) 20:802–10. doi: 10.1038/oby.2011.208
37. Lyons MR, Peterson LR, McGill JB, Herrero P, Coggan AR, Saeed IM, et al. Impact of sex on the heart's metabolic and functional responses to diabetic therapies. Am J Physiol Heart Circ Physiol. (2013) 305:H1584–91. doi: 10.1152/ajpheart.00420.2013
38. Reichelt ME, Mellor KM, Curl CL, Stapleton D, Delbridge LM. Myocardial glycophagy - a specific glycogen handling response to metabolic stress is accentuated in the female heart. J Mol Cell Cardiol. (2013) 65:67–75. doi: 10.1016/j.yjmcc.2013.09.014
39. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. (2005) 115:1111–9. doi: 10.1172/JCI25102
40. Nielsen R, Jorsal A, Iversen P, Tolbod L, Bouchelouche K, Sørensen J, et al. Heart failure patients with prediabetes and newly diagnosed diabetes display abnormalities in myocardial metabolism. J Nucl Cardiol. (2018) 25:169–76. doi: 10.1007/s12350-016-0622-0
41. Taegtmeyer H, Beauloye C, Harmancey R, Hue L. Insulin resistance protects the heart from fuel overload in dysregulated metabolic states. Am J Physiol Heart Circ Physiol. (2013) 305:H1693–97. doi: 10.1152/ajpheart.00854.2012
42. Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes (2007) 56:2457–66. doi: 10.2337/db07-0481
43. Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation (2005) 112:2686–95. doi: 10.1161/CIRCULATIONAHA.105.554360
44. Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res. (2006) 98:596–605. doi: 10.1161/01.RES.0000207406.94146.c2
45. Yu BC, Chang CK, Ou HY, Cheng KC, Cheng JT. Decrease of peroxisome proliferator-activated receptor delta expression in cardiomyopathy of streptozotocin-induced diabetic rats. Cardiovasc Res. (2008) 80:78–87. doi: 10.1093/cvr/cvn172
46. Thandavarayan RA, Watanabe K, Ma M, Gurusamy N, Veeraveedu PT, Konishi T, et al. Dominant-negative p38alpha mitogen-activated protein kinase prevents cardiac apoptosis and remodeling after streptozotocin-induced diabetes mellitus. Am J Physiol Heart Circ Physiol. (2009) 297:H911–9. doi: 10.1152/ajpheart.00124.2009
47. Nowak G. Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J Biol Chem. (2002) 277:43377–88. doi: 10.1074/jbc.M206373200
48. Nowak G, Clifton GL, Godwin ML, Bakajsova D. Activation of ERK1/2 pathway mediates oxidant-induced decreases in mitochondrial function in renal cells. Am J Physiol Renal Physiol. (2006) 291:F840–55. doi: 10.1152/ajprenal.00219.2005
49. Ni R, Cao T, Xiong S, Ma J, Fan GC, Lacefield JC, et al. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic Biol Med. (2016) 90:12–23. doi: 10.1016/j.freeradbiomed.2015.11.013
50. Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes (2011) 60:1770–8. doi: 10.2337/db10-0351
51. Daniels A, van Bilsen M, Janssen BJ, Brouns AE, Cleutjens JP, Roemen TH, et al. Impaired cardiac functional reserve in type 2 diabetic db/db mice is associated with metabolic, but not structural, remodelling. Acta Physiol. (2010) 200:11–22. doi: 10.1111/j.1748-1716.2010.02102.x
52. Li YJ, Wang PH, Chen C, Zou MH, Wang DW. Improvement of mechanical heart function by trimetazidine in db/db mice. Acta Pharmacol Sin. (2010) 31:560–9. doi: 10.1038/aps.2010.31
53. Tabbi-Anneni I, Buchanan J, Cooksey RC, Abel ED. Captopril normalizes insulin signaling and insulin-regulated substrate metabolism in obese (ob/ob) mouse hearts. Endocrinology (2008) 149:4043–50. doi: 10.1210/en.2007-1646
54. Peterson LR, Herrero P, Coggan AR, Kisrieva-Ware Z, Saeed I, Dence C, et al. Type 2 diabetes, obesity, and sex difference affect the fate of glucose in the human heart. Am J Physiol Heart Circ Physiol. (2015) 308:H1510–6. doi: 10.1152/ajpheart.00722.2014
55. Harada K, Sugaya T, Murakami K, Yazaki Y, Komuro I. Angiotensin II type 1A receptor knockout mice display less left ventricular remodeling and improved survival after myocardial infarction. Circulation (1999) 100:2093–9. doi: 10.1161/01.CIR.100.20.2093
56. Krenz M, Robbins J. Impact of beta-myosin heavy chain expression on cardiac function during stress. J Am Coll Cardiol. (2004) 44:2390–7. doi: 10.1016/j.jacc.2004.09.044
57. Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. (2000) 86:386–90. doi: 10.1161/01.RES.86.4.386
58. Lowes BD, Minobe W, Abraham WT, Rizeq MN, Bohlmeyer TJ, Quaife RA, et al. Changes in gene expression in the intact human heart. Downregulation of alpha-myosin heavy chain in hypertrophied, failing ventricular myocardium. J Clin Invest (1997) 100:2315–24. doi: 10.1172/JCI119770
59. Reiser PJ, Portman MA, Ning XH, Schomisch Moravec C. Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am J Physiol Heart Circ Physiol. (2001) 280:H1814–20. doi: 10.1152/ajpheart.2001.280.4.H1814
60. Hang CT, Yang J, Han P, Cheng HL, Shang C, Ashley E, et al. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature (2010) 466:62–7. doi: 10.1038/nature09130
61. Kiriazis H, Kranias EG. Genetically engineered models with alterations in cardiac membrane calcium-handling proteins. Annu Rev Physiol. (2000) 62:321–51. doi: 10.1146/annurev.physiol.62.1.321
62. Fatkin D, McConnell BK, Mudd JO, Semsarian C, Moskowitz IG, Schoen FJ, et al. An abnormal Ca(2+) response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. J Clin Invest. (2000) 106:1351–9. doi: 10.1172/JCI11093
Keywords: NZO, heart, obesity, mitochondrial function, echocardiography, systolic function
Citation: John C, Grune J, Ott C, Nowotny K, Deubel S, Kühne A, Schubert C, Kintscher U, Regitz-Zagrosek V and Grune T (2018) Sex Differences in Cardiac Mitochondria in the New Zealand Obese Mouse. Front. Endocrinol. 9:732. doi: 10.3389/fendo.2018.00732
Received: 10 September 2018; Accepted: 16 November 2018;
Published: 04 December 2018.
Edited by:
Alexandra Kautzky-Willer, Medical University of Vienna, AustriaReviewed by:
Serdar Farhan, Mount Sinai Hospital, United StatesBrian M. Shewchuk, The Brody School of Medicine at East Carolina University, United States
Copyright © 2018 John, Grune, Ott, Nowotny, Deubel, Kühne, Schubert, Kintscher, Regitz-Zagrosek and Grune. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tilman Grune, c2NpZW50aWZpYy5kaXJlY3RvckBkaWZlLmRl