 Jirapas Sripetchwandee
Jirapas Sripetchwandee Nipon Chattipakorn
Nipon Chattipakorn Siriporn C. Chattipakorn
Siriporn C. Chattipakorn- 1Neurophysiology Unit, Cardiac Electrophysiology Research and Training Center, Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand
- 2Cardiac Electrophysiology Unit, Department of Physiology, Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand
- 3Department of Oral Biology and Diagnostic Sciences, Faculty of Dentistry, Chiang Mai University, Chiang Mai, Thailand
It is widely recognized that obesity and associated metabolic changes are considered a risk factor to age-associated cognitive decline. Inflammation and increased oxidative stress in peripheral areas, following obesity, are patently the major contributory factors to the degree of the severity of brain insulin resistance as well as the progression of cognitive impairment in the obese condition. Numerous studies have demonstrated that the alterations in brain mitochondria, including both functional and morphological changes, occurred following obesity. Several studies also suggested that brain mitochondrial dysfunction may be one of underlying mechanism contributing to brain insulin resistance and cognitive impairment in the obese condition. Thus, this review aimed to comprehensively summarize and discuss the current evidence from various in vitro, in vivo, and clinical studies that are associated with obesity, brain insulin resistance, brain mitochondrial dysfunction, and cognition. Contradictory findings and the mechanistic insights about the roles of obesity, brain insulin resistance, and brain mitochondrial dysfunction on cognition are also presented and discussed. In addition, the potential therapies for obese-insulin resistance are reported as the therapeutic strategies which exert the neuroprotective effects in the obese-insulin resistant condition.
Introduction
Obesity has been of interest in several fields of medical research. It has been demonstrated that obesity can lead to the development of several complications, including cardiovascular diseases, diabetes, and neurodegeneration (1, 2). Several reports from both in vivo and clinical studies also showed that obesity is associated with the development of cognitive impairment by several proposed mechanisms including the impairment of leptin signaling and the induction of Alzheimer's-like pathologies which include β-amyloid accumulation and hyperphosphorylation of tau protein (1). Another pathological condition that commonly occurs following obesity (3) is peripheral insulin resistance. This is a pathologic state in which target tissues cannot respond to insulin at the physiological level, leading to the development of hyperinsulinemia with euglycemia. Hyperinsulinemia can disturb the physiological function of several vital organs via the impairment of insulin signaling and the disturbance of intracellular signaling transduction.
The brain is one of the vital organs that can be affected as a result of peripheral insulin resistance. Several previous studies from our group and others have demonstrated that obesity not only induces peripheral insulin resistance, but can also lead to the development of brain insulin resistance, as shown by an impairment of insulin-induced long-term depression (LTD) and a reduction in the activation of brain insulin signaling pathway (4–19). One possible explanation for the occurrence of brain insulin resistance due to peripheral insulin resistance may be the production of ceramide from high lipid generation in the liver (20, 21). Ceramide, a compound of sphingosine and a fatty acid, can enter circulation and cross the blood-brain barrier (BBB). Once in the brain, ceramide can induce brain oxidative stress, brain inflammation, and brain insulin resistance, leading to neurodegeneration (22, 23).
Mitochondria are the vital organelles that provide cellular energy. They play a pivotal role in insulin signaling (24). Normally, insulin binds with its receptor, mediating the activation of cellular glucose uptake through glucose transporters. Following uptake, glucose is converted to pyruvate by the glycolytic process and these pyruvates are then converted to Acetyl-CoA, a substrate of the Krebs cycle, by glucose oxidation (25, 26). In addition, insulin stimulates the uptake of cellular fatty acids into the cells and the fatty acids are further converted to fatty acyl-CoA (25). Fatty acyl-CoA can either be converted into several lipid products, including diacylglycerol (DAG), triacylglycerol (TAG) and ceramide or be directly transported to mitochondria to induce mitochondrial β-oxidation, resulting in the production of acetyl-CoA for the Krebs cycle (25, 26). A diagram illustrating the association between insulin signaling, glycolysis and beta oxidation is summarized in Figure 1.
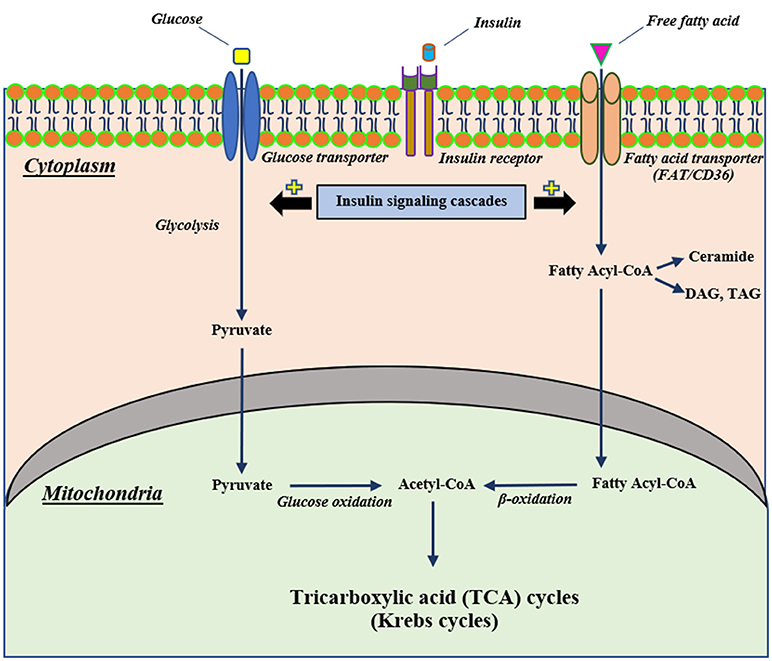
Figure 1. The proposed mechanism of cellular insulin signaling on glucose and fatty acid metabolisms. Extracellular insulin can bind with its receptor, resulting in stimulating insulin signaling cascades. Stimulation of insulin signaling cascades can activate cellular glucose uptake through glucose transporters that intracellular glucose further can be converted to pyruvate by glycolysis and subsequently pass into the mitochondria to change to be Acetyl-CoA for the Krebs cycle. In addition to glucose metabolism, an activation of an insulin signaling cascade can also induce intracellular uptake of free fatty acid via a fatty acid transporter (FAT/CD36) and this free fatty acid can convert to fatty acyl-CoA that translocates to mitochondria and changes to Acetyl-CoA as well as glucose metabolism. CD36, cluster of differentiation-36; DAG, diacylglycerol; FAT, fatty acid translocase; TAG, triacylglycerol.
Previous studies reported that mitochondrial dysfunction has been related to the development of insulin resistance (26, 27). Interestingly, it has been shown that brain mitochondrial dysfunction, as indicated by the overproduction of mitochondrial reactive oxygen species (ROS), mitochondrial depolarization and mitochondrial swelling, has occurred in association with brain insulin resistance and all of these events could lead to the development of cognitive decline and Alzheimer's disease (6, 8, 11, 13–15, 19, 28–31).
Several studies have indicated a relationship between insulin resistance and mitochondrial dysfunction in cognitive-impaired rats (32, 33). Barhwal and colleagues demonstrated that an increased activation of insulin receptor subunit A (IRA) phosphorylation, subsequently stimulating the α subunit of AMP-activated protein kinase (AMPK), leading to improved brain mitochondrial biogenesis (32). Furthermore, it has been demonstrated that intranasal insulin treatment restores cognitive function in methamphetamine-induced cognitive impairment by improving brain insulin signaling via the PI3K/Akt/GSK3β pathway and improving brain mitochondrial function via key-regulatory genes related to mitochondrial biogenesis (33). Beirami and colleagues found that insulin treatment could improve insulin signaling, particularly in the PI3K/Akt/GSK3β pathway and also increase key-regulatory genes related to mitochondrial biogenesis (33). Our previous reports also demonstrated that peripheral insulin resistance develops before impaired cognition in obese-insulin resistant rat model (4, 6, 28). Taken together, all of these findings suggested that (1) The stimulation of insulin receptor plays the important roles in improving brain mitochondrial biogenesis and preserving cognitive function and (2) insulin resistance is associated with mitochondrial dysfunction and the cognitive impairment.
However, the association between obesity and brain insulin resistance, brain mitochondrial function and cognition are still not clearly understood. In this review, the current evidence regarding the associations among obesity, brain insulin resistance, brain mitochondrial dysfunction, and cognition are comprehensively summarized in this review. In addition, controversial findings in these association are presented and discussed.
Obesity Induced Peripheral Insulin Resistance and Metabolic Disturbance Via the Induction of Inflammation and Increased Oxidative Stress
Obesity can induce peripheral insulin resistance which is an impairment of cellular insulin signaling via increased inflammatory cytokines, oxidative stress, and mitochondrial dysfunction of several tissues and organs, including adipose tissue, skeletal muscle, and the liver (34–36). Adipose tissue has commonly been acknowledged to be primarily associated with insulin resistance during obesity (37, 38). In addition to white and brown adipose tissue, beige adipose tissue also plays a possible role in the development of peripheral insulin resistance since it has ability to accumulate energy as white-type and produce heat as brown-type (39, 40).
Under peripheral insulin resistant condition, the overproduction of free fatty acids (FFAs) was occurred, resulting in a release of pro-inflammatory cytokines into the blood circulation. These cytokines can further activate several types of serine kinase such as IκB kinase (IKK) and c-Jun N-terminal kinase (JNK) (41–44). As reported previously, obese-mediated activation of these serine kinases resulted in inhibiting cellular insulin signaling by activating the phosphorylation of insulin receptor substrate-1 (IRS-1) at serine sites including serine-302 (pS302) and serine-307 (pS307), instead of its normal phosphorylated site at tyrosine residue (45–49). In addition, these cytokines activated other inflammatory-related negative regulators of IRS proteins, particularly in Suppressor of cytokine signaling (Socs) protein (50–52). This protein can bind with the phosphorylated insulin receptor (IR), consequently blocking an activation of the IRS proteins (50–52). Additionally, the Socs proteins promoted IRS proteins for ubiquitination, resulting in IRS degradation through the proteasomal complex. Those findings suggest that inflammation following obesity can lead to impaired insulin signaling or insulin resistance in the target organs.
Although numerous studies only found the association between brain mitochondrial dysfunction and brain insulin resistance in obese condition, it remains unclear whether brain mitochondrial dysfunction found in obese condition is a primary cause of brain insulin resistance. However, four previous studies suggest the possibility that mitochondrial dysfunction may be responsible for insulin resistance. The first study demonstrated that mitochondrial dysfunction induced by hyperglycemic condition impaired the AMPK-Akt pathway which is a downstream signaling cascade of the insulin signaling pathway and contributed to insulin resistance (53). Peng and colleagues suggest that mitochondrial dysfunction could lead to neuronal insulin resistance. In the second study, mice with liver-specific ablation of mitofusin-2 (Mfn2) developed glucose intolerance, enhanced hepatic gluconeogenesis as well as impaired insulin signaling in the liver and muscles (54). In the third and fourth studies, the depletion of mitochondrial DNA (mtDNA) resulted in an impaired glucose utilization and induced insulin resistance in the myotubes (55, 56). All these findings suggest that brain mitochondrial dysfunction under obesity could be the cause of brain insulin resistance.
Excess levels of FFAs induce not only systemic inflammation, but also increase the level of oxidative stress which is the major contributory factor in the development of several co-morbidities in the obese-condition (3). Oxidative stress, involving an overproduction of free-radicals, is one of these pathological conditions. Increased oxidative stress can damage several cellular components, including mitochondria, lysosomes, endoplasmic reticulum, nuclei and DNA (57). High levels of oxidative stress can also destroy the cellular membrane, resulting in an overproduction of cytotoxic aldehyde byproducts such as malondialdehyde (MDA) and 4-hydroxylnonenal (HNE) (58). Recent studies have demonstrated that excessive FFAs in WAT also increased oxidative stress, as indicated by increasing activity of NADPH oxidase, reduced activity of antioxidative enzymes such as glutathione peroxidase (GPX), superoxide dismutase (SOD), and catalase (CAT), and decreased glutathione (GSH) synthesis (59). Excessive oxidative stress itself also induces cellular inflammation by an activation of NF-κB in association with an alteration in nuclear processes, including acetylation and deacetylation of histones. It has been shown that oxidative stress itself results in increased promotion of gene expression of pro-inflammatory mediators such as IL-1β and TNF-α in several organs (60). All those findings suggest that obesity could induce peripheral insulin resistance by the induction of inflammation and oxidative stress. The proposed mechanism around insulin resistance in cases of obesity as a possible outcome of inflammation and oxidative stress is shown in Figure 2.
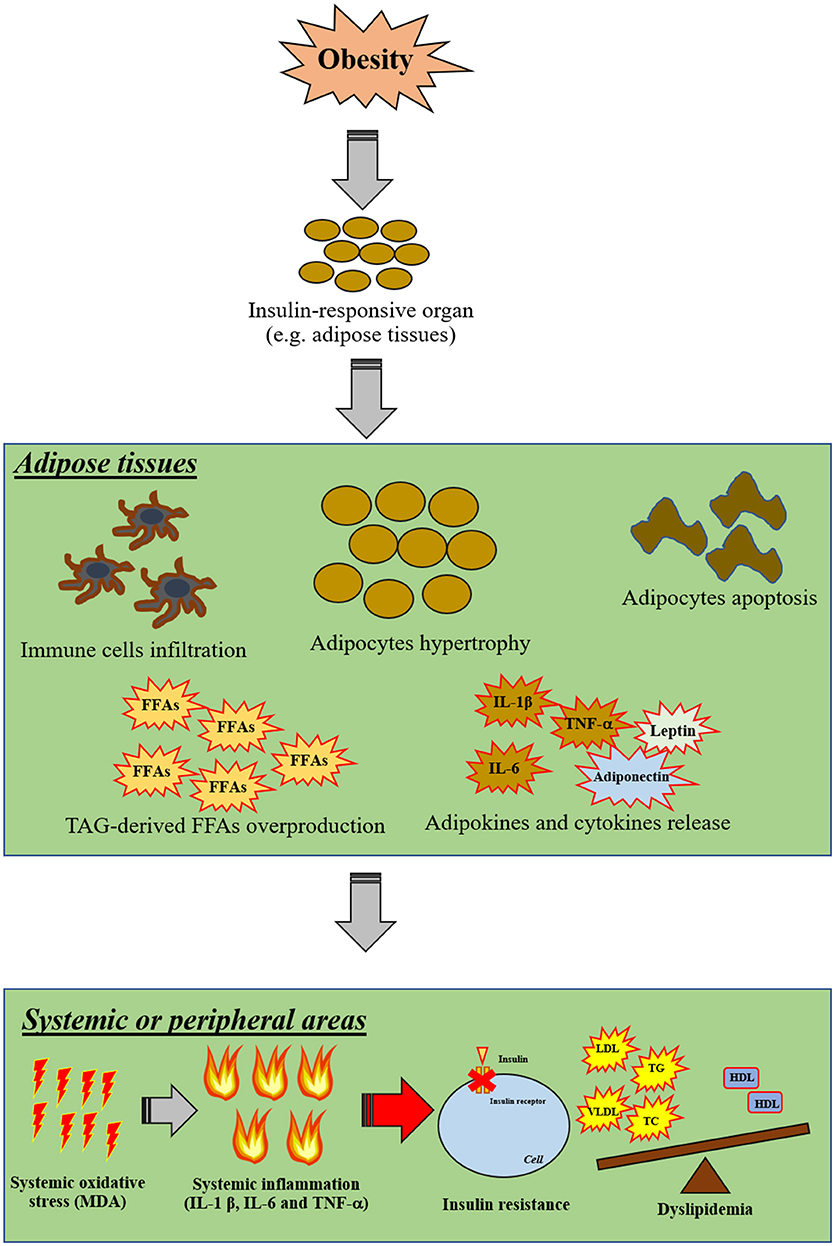
Figure 2. The proposed mechanisms of insulin resistance in obesity via inflammation and oxidative stress. Obesity primarily affects insulin-responsive organs particularly in adipose tissue by inducing adipocyte hypertrophy, adipocyte apoptosis, infiltration of immune cells, TAG-derived FFA overproduction and promoting the release of pro-inflammatory cytokines. Moreover, excessive FFA can also induce oxidative stress in adipose tissue. After that, cellular oxidative stress can induce systemic oxidative stress and further promote systemic inflammation. As a result, these systemic pathological conditions can impair cellular insulin signaling in other organs in association with the induction of dyslipidemia by excessive FFA production from adipose tissues. FFAs, free-fatty acids; HDL, high-density lipoprotein; IL, interleukin; LDL, low-density lipoprotein; MDA, malondialdehyde; TAG, triacylglycerol; TC, total cholesterol; TG, triglyceride; TNF, tumor necrosis factor; T2DM, type-2 diabetic mellitus; VLDL, very low-density lipoprotein; WAT, white-adipose tissue.
The Physiological Role of Insulin in the Brain
Under the physiological condition, insulin can be found in different brain areas, particularly in the hypothalamus, cortex and hippocampus. The level of insulin in those brain areas is higher than the plasma insulin level (61–66). In addition, Hersom and colleagues reported that the insulin level in cerebrospinal fluid has been found to be correlated with the plasma insulin level in a non-linear manner (67).
Several studies also showed that the insulin receptors (IRs) have been detected widely in different brain areas, particularly in brain areas that regulate olfaction, appetite, autonomic activity, and cognitive function (61, 68, 69). Those findings suggest that insulin in the brain not only comes from beta cells in the pancreas, but also can be synthesized from cells in the brain.
Unlike peripheral organs, several studies have shown the physiological roles of insulin in the brain are not primarily involved with the cellular glucose uptake but associated with cognitive function (70, 71). Normally, the glucose transporters (GLUT), particularly GLUT-4, play an important role in cellular glucose uptake and are regulated by insulin (72). In the brain, it has been reported that the expression of GLUT-4 is very low and GLUT-4 has not been regulated by insulin level (70). Moreover, brain GLUT-4 was in only a few areas such as the olfactory bulb, hippocampus and hypothalamus (73, 74). Along with GLUT-4, there are several types of GLUT which could require glucose uptake including GLUT-3 for neuronal glucose uptake and GLUT-1 for astrocytic glucose handling (75–77). Regarding the role of insulin in the brain, a recent study found that insulin failed to induce cellular glucose uptake into the neurons as well as the activation of insulin receptors because insulin has no effect on GLUT-4 translocation (78). Consistent with this concept, the GLUT-3-mediated neuronal glucose uptake occurs in an insulin-independent manner (79). It is possible that insulin does not play an essential role in neuronal glucose uptake, but it might have a role in the regulation of normal brain function. However, there is a recent controversial finding indicating the role of insulin-mediated glucose utilization in the brain via GLUT-4 which might affect the brain spatial working memory (80). In that study, they demonstrated that GLUT-4 inhibition, using a specific GLUT-4 inhibitor; indinavir sulfate (IND), alone did not impair neuronal glucose utilization and spatial memory in the hippocampus, whereas exogeneous insulin-mediated memory enhancement could be blocked by IND as well as impaired neuronal glucose utilization. These suggested that supra-basal insulin level might require functional GLUT-4 for neuronal glucose utilization and enhancement of brain spatial working memory (GLUT-4 dependent manner), but not for a basal insulin level which is GLUT-4 independent manner.
There is increasing evidence to show that the actions of insulin in the brain impact several aspects such as the neuromodulator, neuroprotective effects and also have a role in cognition and memory (70). Regarding the role of insulin as the neuromodulator, Kovacs and Hajnal demonstrated that insulin acts as one of the neuromodulators, as indicated by modulating GABAergic activity in the cerebellum (81). That study demonstrated that insulin exerted the GABA-dependent neuronal inhibitory effects in cerebellum, and that the effect of insulin was abolished when co-administrated with insulin receptor (IR) inhibitor. Those findings suggest that insulin in the brain plays a role as the neuromodulator. Regarding the role of insulin as the neuroprotective agent, several studies reported that insulin exerted neuroprotective effects via anti-apoptosis, β-amyloid inhibition and antioxidant agency (82–85).
Several studies regarding the benefits on brain cognition, both in in vivo and clinical situations have demonstrated that either peripheral or central administration of insulin showed positive effects on learning and memory (86, 87). Moreover, this effect has been shown to be related to activation via insulin receptors and downstream signaling (70, 88). The modification of learning and memory following insulin administration has been assessed by the improvement in brain synaptic plasticity, including long-term potentiation (LTP) and long-term depression (LTD) (89). Insulin could modulate neurotransmitter release at the synaptic areas particularly in the releasing of glutamate, a pivotal neurotransmitter required for the preservation of synaptic transmission (89, 90). Therefore, all those findings suggest that insulin in the brain is involved in several physiological functions, including the neuromodulation, neuroprotection, and cognition.
Excessive Free Fatty Acid and Neuronal Insulin Resistance (in vitro Studies)
As mentioned in the previous topics that insulin plays an important role in brain function, several studies demonstrated that neuronal insulin resistance can be found under obese condition (4, 6, 7, 9–14, 16–19, 28, 30, 31, 91–94). Moreover, the excessive free fatty acid (FFA) has been found under an obese condition. Growing evidence from in vitro studies has demonstrated the association between excessive FFA and neuronal insulin resistance. It has been shown that excessive FFA could activate the inflammation via increased release of pro-inflammatory cytokines (42, 44). Those inflammatory cytokines stimulate the activity of their downstream signaling, including the serine kinases, Ikkb, and JNK1, which subsequently inhibit the pathway of insulin receptor substrate 1 (IRS1) signaling via promoting the phosphorylation of IRS at serine sites, instead of tyrosine site (45–49). In addition, FFA can activate other inflammation-related negative regulators of IRS protein, particularly Socs proteins, and promote IRS degradation (51, 52). Those findings suggest that excessive FFA under obesity can impair insulin signaling, leading to an insulin resistant condition, via the activation of inflammation.
Several studies reported that high levels of circulating saturated free fatty acids (sFFAs) caused pathological conditions in several tissues and organs such as adipose tissue, the liver, and skeletal muscle as well as the brain, as a result of the induction of inflammation (95–100). For example, the administration of palmitate, a common sFFA, onto adipocytes, endothelial cells and myotubes led to the activation of inflammation via nuclear factor-κB (NF-κB) pathways (101–105). It has been shown that inflammation in adipose tissues triggers the release of several cytokines, resulting in aggravation of the severity of inflammation in other tissues (97, 100, 106).
A recent study established that sFFAs induced insulin resistance in the brain (107). Diaz and colleagues pointed out that the incubation of palmitate with hypothalamic cells also increased neuronal toxicity by raising ROS production, resulting in the induction of inflammation and ER stress, as well as mitochondrial dynamic impairment and reduced phosphorylation of insulin signaling protein (107).
In contrast to the previous study, Choi and colleagues demonstrated that exposure to either palmitate alone or a sFFA mixture with cultured hypothalamic neuronal cell lines induced neither neuronal inflammation nor neuronal insulin resistance (108). A possible explanation is that the type of hypothalamic neuronal cell line in Choi's study might be less sensitive to sFFA exposure, because inflammation and insulin resistance were still observed, when those cells were exposed to lipopolysaccharide (LPS). In addition, use of different types of hypothalamic cell lines might result in the different findings as shown in previous reports. All these findings from the in vitro models are summarized in Supplementary Table 1.
Peripheral Insulin Resistance Induced Brain Insulin Resistance and Brain Dysfunction
A previous report has shown that peripheral insulin resistance was detected at the end of 8 weeks consumption of a HFD as characterized by an increase in body weight, an increase in visceral fat, an elevation in plasma metabolic parameters (fasting plasma glucose, fasting plasma insulin and HOMA index) and an increase in hepatic triglycerides (4). Several reports also stated that there is an association between peripheral insulin-resistance and liver-brain connection (22, 23, 109). Regarding the association between peripheral insulin resistance and brain connection, a previous study demonstrated that the development of brain insulin resistance as indicated by an impairment in both brain insulin signaling pathways and brain insulin sensitivity occurred after 12 weeks of HFD consumption (4). The possible explanation could be that peripheral insulin resistance led to increased hepatic lipid production, particularly in ceramides. Ceramide is generated from fatty acids and sphingosines (22). Ceramide is commonly known to have lipid soluble properties. Thus, it is possible to easily cross the blood-brain barrier. In addition, several studies demonstrated that ceramide induced brain insulin resistance via an impaired brain insulin signaling pathway (110), and caused neurodegeneration (22, 23, 111). Although there was no evidence showing that ceramide directly induced blood-brain barrier disruption, it is possible that a large amount of ceramide under obesity condition may be one risk factor to cause the disruption of the blood-brain barrier. Moreover, there was no direct evidence demonstrating that local insulin production in the brain can be related to the impaired insulin response.
There are a few studies reporting the timeline events of peripheral insulin resistance and the occurrence of brain insulin resistance. According to those recent studies in a rat model, chronic HFD consumption for 8 weeks only induced peripheral insulin resistance but did not initially cause brain insulin resistance. However, the extension of HFD consumption to 12 weeks could lead to the development of brain insulin resistance as shown by a reduction in insulin-related protein expression along with the impairment of insulin-induced long-term depression (LTD). These findings suggested that peripheral insulin resistance occurred prior to the brain insulin resistance.
In contrast to those reports, Filippi and colleagues reported that 3 days of lard oil-enriched HFD consumption in male Sprague-Dawley (SD) rats demonstrated brain insulin resistance (92). In addition, a study by Pathan and colleagues has been reported that metabolic disturbance including the increased plasma levels of glucose, insulin, total cholesterol, and triglyceride occurred in 5-weeks of HFD consumption (112). These different finding might be due to different compositions of HFD and different strains of animals that were used in their studies.
Supporting evidence shows that obese condition as a result of chronic consumption of a high-fat diet (HFD) or genetic-induced obesity and in obese-T2DM animals indicated the development of peripheral insulin resistance subsequently caused brain insulin resistance and could be associated with cognitive impairment (4, 6, 7, 9–14, 16–19, 28–31, 91–94, 113). Interestingly, a recent study also showed that maternal obesity can lead to the development of metabolic disturbance, hippocampal insulin resistance, and further altered the distribution of markers of neurogenesis, neuronal synaptic plasticity and function of the offspring (94). This suggest that maternal obesity might affect the offspring's neurocognitive outcome.
Systemic inflammation and oxidative stress following obesity could be the causes leading to disruption of the brain defense mechanistic structures, known as the blood-brain barrier (114). Bank and colleagues observed that under conditions of obese-insulin resistance, either circulating inflammation or oxidative stress decreased the expression of tight junction proteins in the BBB, resulting in an increase in permeability to the brain (114). That study suggested that the systemic pathological conditions occurring in the obese-insulin resistant condition, could induce brain pathological conditions, such as the induction of brain insulin resistance.
Several previous studies demonstrated that chronic HFD consumption induced obese-insulin resistance prior to causing brain insulin resistance, as indicated by decreasing phosphorylation of insulin receptors (IR), insulin receptor substrate (IRS) and downstream signaling, including PI3K-Akt, GSK3β, and AMPK pathways (4, 6, 7, 9–14, 16–19, 28–31, 91–94). Impaired brain insulin signaling in the obese-insulin resistant condition also resulted in a loss of inhibitory function to FoxO transcription factors, which normally modulate cellular metabolism and autophagy (115).
Several studies also demonstrated that obese-insulin resistance was caused by the impairment of mitochondrial function in the brain, as indicated by increased mitochondrial ROS production, induced mitochondrial membrane depolarization, and mitochondrial swelling, demonstrated by the observation of unfolded cristae in brain mitochondria (6, 8, 11–16, 19, 28–31, 92, 116). Decrease in the functional processes of mitochondria and reduced ATP content were observed in brain mitochondria of obese insulin resistant mice and Zucker diabetic rats (12, 116). Moreover, this mitochondrial change was observed, along with brain cell apoptosis, by the alteration in apoptotic related proteins including bax, bad and bcl-2 (10, 15, 19).
Although numerous studies found the association between brain mitochondrial dysfunction and brain insulin resistance which occurred in an obese condition, it remains unclear whether brain mitochondrial dysfunction in an obese condition is a primary cause of brain insulin resistance. According to Peng's study, hyperglycemic condition by high-glucose exposed to neurons led to mitochondrial dysfunction followed by an impaired AMPK-Akt signaling pathway, which is the downstream cascade of the insulin signaling pathway, and possibly contributing to insulin resistance. That finding suggested that mitochondrial dysfunction could lead to neuronal insulin resistance (53).
However, only one study from Peng and colleagues reported the idea that mitochondrial dysfunction is one causative factor for the development of brain insulin resistance. In contrast, several lines of evidence demonstrated that insulin can control mitochondrial function in pancreatic β-cells, cardiomyocytes and hepatocytes (117–119). In addition, the use of the anti-diabetic drug, thiazolidinediones, regulated the mitochondrial biogenesis suggesting that insulin can directly regulate mitochondria (120–122). All those findings imply that brain mitochondrial activity and brain insulin signaling have bidirectional communication to regulate normal brain function.
Regarding to the links among obesity-related insulin resistance, brain mitochondrial dysfunction and dementia, several studies reported that brain mitochondrial dysfunction has been observed in obese-insulin resistant condition with cognitive decline (6, 8, 11–16, 19, 28–31, 116). In addition, several pharmacological interventions that preserved brain mitochondrial function in obese-insulin resistant models could improve cognitive function (6, 8, 11–16, 19, 28–31). Those findings suggested that mitochondrial dysfunction and insulin resistance would be underlying mechanisms leading to cognitive decline.
Obese-insulin resistance caused by HFD consumption not only induced systemic inflammation, but also caused brain inflammation, as shown by increased brain pro-inflammatory cytokine; TNF-α and a transcriptional factor; NF-κB (13, 15, 19, 116). Furthermore, brain oxidative stress was shown by an elevation in cytotoxic aldehyde products; MDA was also found in association with the brain inflammation following obese-insulin resistance (8, 11, 13–15, 19, 28–31).
All of these findings suggested that obesity can increase both systemic inflammation and oxidative stress, and both events can lead to the development of peripheral insulin resistance as well as brain dysfunction.
Several studies in chronic HFD consumption-induced obese-insulin resistant condition showed an association between cognitive decline and obese-insulin resistance, occurring in association with impaired brain insulin signaling, brain mitochondrial dysfunction, brain apoptosis, increased brain inflammation, increased brain oxidative stress, and synaptic dysplasticity (8, 11–16, 19, 28–31, 123, 124). Cognitive decline was determined by several methods, including the Morris Water Maze (MWM) test (8, 11, 13–16, 19, 28–31), and a Novel Object Recognition (NOR) test (8, 12). Brain synaptic plasticity plays a pivotal role in the learning and memory process (125). Several studies reported synaptic dysfunction in the chronic HFD consumption-induced obese-insulin resistant condition, as indicated by a loss of the long-term potentiation (LTP) process, the reduction in synaptic proteins, or a decrease in dendritic spine density (9, 11, 13–15, 19, 31, 124, 126). In addition, Alzheimer's like pathologies, such as β-amyloid accumulation and tau-hyperphosphorylation have been found in obese-insulin resistant mice and diabetic rats (10, 16). All those findings suggest that obese-insulin resistance can lead to brain damage, resulting in cognitive decline and neurodegeneration.
According to several reports, the cafeteria diet could induce metabolic syndromes by altering metabolic profiles and causing induced hyperlipidemia, hyperinsulinemia and increased body weight. Although there are numerous studies that demonstrated that the cafeteria diet caused metabolic changes along with alterations in brain functions such as anxiety, depression, stress and impairment of cognition (127–135), there was no direct evidence indicating that the cafeteria diet induced brain insulin resistance.
In contrast to the HFD-induced obese-insulin resistant model, the genetically induced obese-insulin resistant models, such as ob/ob mice and Zucker diabetic fatty rats, developed peripheral insulin resistance without brain insulin resistance (116, 126). However, other brain dysfunction, including increased brain oxidative stress, brain mitochondrial dysfunction and brain synaptic dysplasticity were observed in those genetically-induced obese models. Those findings suggested that brain insulin resistance might not be the main cause of brain dysfunction in the genetically-induced obese-insulin resistant condition (116, 125).
Regarding the studies from type 2 diabetes mellitus (T2DM) animal models, the findings in these examples were similar to the previous findings from the model of HFD- induced obese-insulin resistance, in which peripheral insulin resistance occurred before the development of cognitive impairment, impaired brain insulin sensitivity, and brain mitochondrial dysfunction (16). The key findings of the effect of obese-insulin resistance on the brain are summarized in Supplementary Table 2.
Evidence of Brain Mitochondrial Dysfunction Occurring in Association With the Development of Cognitive Impairment in the Obese-Insulin Resistant Condition
Mitochondria are commonly known as a cellular power house (136). Mitochondria play roles in the regulation of ATP production, Ca2+-storage, and the control of cell survival or death (137). As regards brain activity, a previous study has shown that mitochondria play a key role in brain synaptic transmission and age-associated cognitive function (138–141). That study suggested that changes in shape of mitochondria in presynaptic neurons affected the synaptic transmission. Donut-shape mitochondria has been reported as being a hallmark of mitochondrial stress (142, 143) and also may be related to a deficient working memory in an old-monkey model (140). In addition, the presence of donut-shaped mitochondria showed a correlation to the reduction of synapses as indicated by the smaller size of the active zone (140). All these findings indicated that brain mitochondria have a significant impact on cognitive function.
To support the concept that mitochondrial dysfunction could lead to cognitive decline, several studies reported the impact of brain mitochondria on cognitive function (144, 145). In an AD-mouse model, brain mitochondrial dysfunction and the accumulation of mitochondrial Aβ has been observed and these impairments depended on a degree of cognitive impairment in AD transgenic mice (144). In addition, Baek and colleagues demonstrated that an inhibition of mitochondrial fission (Drp-1) inhibitor ameliorated the synaptic depression, Aβ accumulation, and subsequently attenuated cognitive impairment in an AD-mouse model (145).
In addition, in cases of obese-insulin resistance, mitochondrial dysfunction was also observed in the brain (146). Several studies showed that brain mitochondrial dysfunction occurred in cases of HFD consumption, the genetically-induced obese-insulin resistant condition, and in cases of T2DM, indicated by the excessive production of mitochondrial ROS, mitochondrial membrane potential changes, and swollen mitochondria with evidence of unfolded cristae in mitochondria (6, 8, 11–16, 19, 28–31, 116). The reduction of ATP level in association with the malfunction of brain mitochondria, including decreased O2 consumption and CO2 production, has been found in brain tissue from rats with obese-insulin resistant condition (12, 116, 123, 126).
There was no evidence to directly indicate that brain mitochondrial dysfunction, particularly brain mitochondrial membrane potential change, led to brain insulin resistance. There was only an association between brain mitochondrial dysfunction and brain insulin resistance since most of studies demonstrated that brain mitochondrial dysfunction was promptly observed in obese-insulin resistant animals with brain insulin resistance as indicated by the reduction in brain insulin-related proteins expression and impairment of insulin-induced long-term depression in the hippocampal areas (5, 6, 8, 11, 14, 15, 19, 29–31).
Also, several studies have demonstrated that brain apoptosis occurs in association with brain mitochondrial dysfunction (10, 15, 19). One possible explanation is that cytochrome C is released following mitochondrial swelling, leading to the formation of a complex with APAF-1. These complexes become the apoptosomes which activate the caspase cascades and finally induce cell death (147). Previous studies showed that brain mitochondrial dysfunction increased the levels of pro-apoptotic proteins (bax and bad), and decreased levels of the anti-apoptotic protein (Bcl-2) were observed in brain tissue from rats with the obese-insulin resistant conditions (10, 15, 19). An increase in pro-apoptotic proteins can induce cytochrome C release, resulting in brain apoptosis (148). In addition, apoptotic-mediated neuronal death has been known to be one underlying mechanism for cognitive impairment and other neurodegenerative diseases (149).
In addition to brain mitochondrial function, mitochondrial dynamics, including fusion and fission, play a critical role in cell survival or death (150). It has been demonstrated that an imbalance in mitochondrial dynamics as well as malfunction of brain mitochondria has been associated with neurodegeneration, and cognitive impairment (151, 152). Previous studies demonstrated that the imbalance of mitochondrial dynamics as indicated by an increase in the mitochondrial fission process in association with a decrease in the mitochondrial fusion process, resulted in cell death and the development of cognitive decline (152, 153). Consistent with these reports, obese-insulin resistance led to a development in an imbalance in this dynamic process in the brain, specifically an increase in mitochondrial fission protein and a reduction in mitochondrial fusion protein (10, 92, 107).
All the findings pertaining to brain mitochondria suggested that brain mitochondrial dysfunction or an imbalance in mitochondrial dynamics in the brain would be the underlying mechanisms responsible for cognitive impairment associated with the obese- insulin resistant condition. The findings associated with brain mitochondria and obesity are summarized in Supplementary Table 2.
Pharmacological Interventions Exerted Peripheral Benefits and Neuroprotection Against Obese-Insulin Resistance
Obesity not only induces peripheral insulin resistance, but it also leads to brain insulin resistance, brain mitochondrial dysfunction, resulting in cognitive decline. However, several pharmacological interventions such as anti-diabetic drugs, hormone therapy, and alternative medicine, which exert beneficial effects on peripheral insulin sensitivity, have also been reported to provide neuroprotection in the brain. Despite medication, non-pharmacological intervention as well as subcellular targeting interventions also provide benefits in the brain under obese-insulin resistant condition. All these findings are summarized and discussed in the following section.
Neuroprotective Effects of Therapeutic Interventions Using Unsaturated Free Fatty Acid in vitro Studies
Recent in vitro study found unsaturated free fatty acids provided beneficial effects in neuronal insulin-resistance caused by saturated free fatty acid (sFFA) exposure through improving cell insulin-related signaling and mitochondrial function in association with reducing neuronal inflammation, oxidative stress, and apoptosis (107). This finding suggested that neuronal insulin resistance by sFFA induction occurred through the impairment of neuronal inulin-related signaling, mitochondrial dysfunction, and inflammation-oxidative stress and that intervention using unsaturated free fatty acids could ameliorate this neurotoxicity.
Beneficial Effects of Anti-Diabetic Drugs on the Brain in the Obese-Insulin Resistant Condition
Therapeutic interventions including anti-diabetic drugs and hormone therapy are required to relieve the deleterious effects occurring as a result of obese-insulin resistance (154, 155). Of these interventions, anti-diabetic drugs are commonly known to reduce body weight, decrease dyslipidemia, and improve insulin sensitivity in the obese-insulin resistant condition (155).
Several classes of anti-diabetic drugs, including a peroxisome proliferator-activated receptor gamma (PPARγ) agonist, biguanide, dipeptidyl peptidase-4 (DPP-4) inhibitors, and sodium-glucose co-transporter 2 (SGLT-2) inhibitors, have been used in obese-insulin resistant models. These drugs not only provided beneficial effects in attenuating metabolic disturbance, but also exerted neuroprotective effects, as indicated by improved brain insulin sensitivity, brain mitochondrial function and hippocampal synaptic plasticity, as well as reducing brain inflammation, brain oxidative stress, brain apoptosis, and dendritic spine loss. They also led to improved cognitive function (6, 17, 19, 28–31, 112, 113, 124, 156, 157).
The neuroprotective effects of pharmacological interventions particularly anti-diabetic drugs and hormone therapy could possibly be due to direct effects in the brain since some types of these drugs can pass the blood-brain barrier (158, 159) and improve the periphery.
Beneficial Effects of Hormone Therapy in the Brain in the Obese Insulin-Resistant Condition
Not only the use of anti-diabetic drugs, but also the addition of incretin hormone therapy is needed in some cases to ameliorate the obese-insulin resistant condition (160). Incretin hormones are a group of metabolic hormones that play a role in decreased blood glucose levels via stimulating insulin secretion in response to meals (161). Incretin hormones include glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) (162). Both GLP-1 and GIP are secreted from enteroendocrine cells in the intestinal epithelium. Recently, incretin hormones have been considered as a potential intervention for diabetic therapies since they exert blood-glucose lowering effects (161). In addition, several previous studies reported that incretin hormones not only reduced blood glucose levels, but also exerted other beneficial effects such as anti-oxidant, anti-inflammation, anti-apoptotic effects and an enhancement of cell proliferation (163–165).
However, the use of incretin hormone therapy showed controversial effects as regards neuroprotection in conditions of obese-insulin resistance (123, 126, 166, 167). Examples of the findings include: (1) Use of an incretin mimetic drug, exendin-4, resulted in neuroprotective effects by improving hippocampal synaptic plasticity and cognitive function along with improvement of metabolic parameters in an obese-insulin resistant condition (166). (2) Another incretin mimetic drug, liraglutide, in cases of either HFD or genetically induced obese insulin resistance demonstrated that the improvement in metabolic parameters along with a preservation in the neurogenesis and neuronal survival resulted in improved hippocampal synaptic plasticity and cognitive function (126, 167). (3) Lixisenatide, known as GLP-1 receptor agonist, could improve cognitive performance by attnueating peripheral insulin resistance and enhancing cells proliferation in brain of HFD-fed animal model by Lennox et al. (168). (4) However, use of incretin metabolites [GLP-1(9-36), GIP (3-42) and exendin (9-39)] did not lead to the attenuation of either metabolic or brain parameters including hippocampal synaptic plasticity, brain locomotor activity and cognitive function in the HFD-fed mice model (123). The possible explanation for the differences could be due to the differences in pharmacokinetic structures between incretin metabolites and incretin mimetic drugs. This possibility requires further investigation to explore these controversial findings.
In addition, the administration of fibroblast growth factor-21, a starvation hormone, also exerted neuroprotection in the obese-insulin resistant condition (15, 113). To support these findings, FGF21 administration has been reported to improve peripheral parameters including (1) increased an energy expenditure, resulting in body weight reduction (169, 170), (2) improved peripheral insulin sensitivity through an increased adipocyte glucose uptake rate, increased insulin synthesis and decreased hepatic gluconeogenesis (171–174), (3) exerted anti-inflammatory effect (175), and (4) increased adiponectin biosynthesis (176, 177). In addition, FGF21 has also demonstrated the neuroprotective effects by several underlying mechanisms including (1) improving mitochondrial biogenesis and function, by increasing mitochondrial respiratory capacity and mitochondrial anti-oxidant levels (178) and (2) decreasing brain cell damage (179).
Beneficial Effects of Alternative Medicine in the Brain in the Obese Insulin-Resistant Condition
Several studies have shown that other alternative interventions, including herbal extracts or recipes, also exerted neuroprotective effects in the obese-insulin resistant condition (8, 12, 13, 15, 16, 180). Interestingly, the beneficial effects of these interventions were not only indicated in the peripheral area, but also provided neuroprotective effects against cognitive impairment in the obese insulin-resistant condition (8, 12, 13, 15, 16, 180).
Herbal extracts, including cinnamon extract, garlic extract, naringin, a ZiBu PiYin recipe, Thymol and Ginsenoside Re have recently been reported as leading to improvements in brain cognition and locomotor activity in obese insulin-resistant animals with or without the improvement of metabolic parameters (8, 12, 16, 17, 23, 93).
Beneficial Effects of Subcellular-Targeting Intervention in Brain in the Obese-Insulin Resistant Condition
Interestingly, application of a N-methyl D-aspartate receptor (NMDARs) antagonist in a genetically-induced obese-model also demonstrated neuroprotective effects through rescuing dendritic spine arborization and synaptic density (180). Moreover, the inhibition of mitochondrial fission by MDIVI-1 could prevent brain insulin resistance through the attenuation of ER stress and iNOS expression (92).
Beneficial Effects of Non-Pharmacological Intervention in Brain in the Obese-Insulin Resistant Condition
In addition, non-pharmacological interventions, including energy restriction and vagus nerve stimulation, also exerted neuroprotective effects in the obese-insulin resistant condition (13, 31). Regarding obese-insulin resistant models, energy restriction attenuated metabolic disturbance and preserved dendritic spine density (31), while the vagus nerve stimulation led to attenuated peripheral insulin resistance as well as improved brain function, as indicated by improved brain insulin sensitivity/brain mitochondrial function/cognitive function, and reduced brain inflammation, /brain oxidative stress/ brain apoptosis/dendritic spine loss (13).
Taken together, all those findings from therapeutic interventions suggested that all interventions in the treatment of obese-insulin resistance not only improved the metabolic parameters, but also protected the brain against cognitive decline. All these findings are summarized in Supplementary Table 3.
According to previous reports, pharmacological interventions such as anti-diabetic drugs can attenuate cognitive impairment via the improvement of peripheral insulin sensitivity (181, 182), the provision of antioxidative effects (17, 19, 28–31, 157, 183), a decrease in inflammation and apoptosis (17, 19, 113, 157, 183), as well as the improvement of mitochondrial function in the brain (6, 19, 28–31, 113). Another factor associated with cognitive impairment is an imbalance in brain mitochondrial dynamics (92). Only one previous study has demonstrated that a modulation of brain mitochondrial dynamics attenuated neuronal insulin resistance (92). The beneficial effects of this modulation on cognitive function in obese models have not yet been investigated. Therefore, further investigation is needed to prove this mechanism.
Conclusion
Several studies demonstrated that obese-insulin resistance contributed to cognitive impairment through several proposed mechanisms, including inflammation and oxidative stress. Brain mitochondria are also damaged as a result of an obese-insulin resistant condition with resulting cognitive decline. Therapeutic approaches for obese-insulin resistance not only had beneficial effects to the metabolic parameters, but also led to improvement in brain function, brain mitochondrial function, and cognitive function. Therefore, it is possible that mitochondria may play an important role in cognitive decline in conditions pertaining to obesity. A summary of the possible mechanisms involved in the impact of obese-insulin resistance on the brain is shown in Figure 3. Although many studies have reported an association between peripheral insulin resistance and brain pathologies such as brain insulin resistance, brain mitochondrial dysfunction, and cognitive impairment, these studies could not conclude the causative relationship between insulin resistance and brain mitochondrial dysfunction. All of these studies imply that an improvement in peripheral insulin sensitivity along with an improvement in brain mitochondrial function could preserve cognitive function under obese condition. Further investigations are needed to better understand the underlying mechanisms of cognitive impairment in obese condition. This information is necessary to provide specific molecular targets for drug development programs.
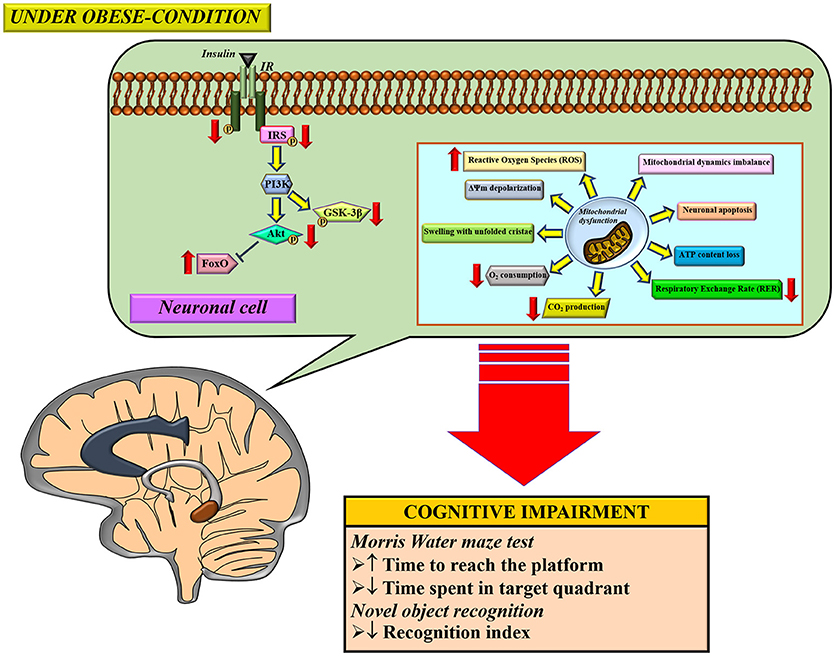
Figure 3. The proposed mechanisms of obese-insulin resistance and brain mitochondrial dysfunction induced brain cognitive decline. Under obese-condition, brain insulin signaling was initially impaired by a reduction of insulin receptor (IR) phosphorylation which led to further its downstream signaling cascade including decreasing phosphorylation of the insulin receptor substrates (IRS), Akt and GSK3β. On the other hand, the reduction in Akt phosphorylation subsequently decreased an inhibitory response to FoxO protein, resulting in an increased FoxO expression. In parallel with impaired brain insulin signaling pathways, brain mitochondrial dysfunction was also observed as indicated by several anomalies in function changes (increased ROS production, ΔΨm depolarization, in association with decreased O2 consumption, CO2 production and respiratory exchange rate (RER), and thus induced loss of ATP content), morphological changes (mitochondrial swelling and unfolded cristae), dynamics changes (imbalance of mitochondrial fusion and fission proteins expression), and causing neuronal apoptosis. Eventually, all these pathological changes can lead to cognitive impairment, determined by learning and memory behavior tests. ΔΨm; mitochondrial membrane potential, ATP; adenosine triphosphate, FoxO, forkhead box O; GSK-3β, glycogen synthase kinase-3-beta; IR, insulin receptor; IRS, insulin receptor substrate.
Author Contributions
Conception and design of the study: JS, NC, and SC. Analyzed data: JS, NC, and SC. Initial draft of manuscript: JS and SC. Manuscript editing: JS, NC, and SC. All authors approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Thailand Research Fund Grants TRF-RTA6080003 (SC) and MRG6080226 (JS), The National Research Council of Thailand (SC), a NSTDA Research Chair Grant from the National Science and Technology Development Agency Thailand (NC), and a Chiang Mai University Center of Excellence Award (NC).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2018.00496/full#supplementary-material
References
1. Frisardi V, Solfrizzi V, Seripa D, Capurso C, Santamato A, Sancarlo D, et al. Metabolic-cognitive syndrome: a cross-talk between metabolic syndrome and Alzheimer's disease. Ageing Res Rev. (2010) 9:399–417. doi: 10.1016/j.arr.2010.04.007
2. Bastien M, Poirier P, Lemieux I, Despres JP. Overview of epidemiology and contribution of obesity to cardiovascular disease. Prog Cardiovasc Dis. (2014) 56:369–81. doi: 10.1016/j.pcad.2013.10.016
3. Boden G. Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes. (2011) 18:139–43. doi: 10.1097/MED.0b013e3283444b09
4. Pratchayasakul W, Kerdphoo S, Petsophonsakul P, Pongchaidecha A, Chattipakorn N, Chattipakorn SC. Effects of high-fat diet on insulin receptor function in rat hippocampus and the level of neuronal corticosterone. Life Sci. (2011) 88:619–27. doi: 10.1016/j.lfs.2011.02.003
5. de la Monte SM. Therapeutic targets of brain insulin resistance in sporadic Alzheimer's disease. Front Biosci. (2012) 4:1582–605. doi: 10.2741/e482
6. Pipatpiboon N, Pratchayasakul W, Chattipakorn N, Chattipakorn SC. PPARgamma agonist improves neuronal insulin receptor function in hippocampus and brain mitochondria function in rats with insulin resistance induced by long term high-fat diets. Endocrinology (2012) 153:329–38. doi: 10.1210/en.2011-1502
7. Arnold SE, Lucki I, Brookshire BR, Carlson GC, Browne CA, Kazi H, et al. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol Dis. (2014) 67:79–87. doi: 10.1016/j.nbd.2014.03.011
8. Pintana H, Sripetchwandee J, Supakul L, Apaijai N, Chattipakorn N, Chattipakorn S. Garlic extract attenuates brain mitochondrial dysfunction and cognitive deficit in obese-insulin resistant rats. Appl Physiol Nutr Metab. (2014) 39:1373–9. doi: 10.1139/apnm-2014-0255
9. Liu Z, Patil IY, Jiang T, Sancheti H, Walsh JP, Stiles BL, et al. High-fat diet induces hepatic insulin resistance and impairment of synaptic plasticity. PLoS ONE (2015) 10:e0128274. doi: 10.1371/journal.pone.0128274
10. Nuzzo D, Picone P, Baldassano S, Caruana L, Messina E, Marino Gammazza A, et al. Insulin resistance as common molecular denominator linking obesity to Alzheimer's disease. Curr Alzheimer Res. (2015) 12:723–35. doi: 10.2174/1567205012666150710115506
11. Pratchayasakul W, Sa-Nguanmoo P, Sivasinprasasn S, Pintana H, Tawinvisan R, Sripetchwandee J, et al. Obesity accelerates cognitive decline by aggravating mitochondrial dysfunction, insulin resistance and synaptic dysfunction under estrogen-deprived conditions. Horm Behav. (2015) 72:68–77. doi: 10.1016/j.yhbeh.2015.04.023
12. Wang D, Yan J, Chen J, Wu W, Zhu X, Wang Y. Naringin improves neuronal insulin signaling, brain mitochondrial function, and cognitive function in high-fat diet-induced obese mice. Cell Mol Neurobiol. (2015) 35:1061–71. doi: 10.1007/s10571-015-0201-y
13. Chunchai T, Samniang B, Sripetchwandee J, Pintana H, Pongkan W, Kumfu S, et al. Vagus nerve stimulation exerts the neuroprotective effects in obese-insulin resistant rats, leading to the improvement of cognitive function. Sci Rep. (2016) 6:26866. doi: 10.1038/srep26866
14. Pintana H, Pratchayasakul W, Sa-nguanmoo P, Pongkan W, Tawinvisan R, Chattipakorn N, et al. Testosterone deprivation has neither additive nor synergistic effects with obesity on the cognitive impairment in orchiectomized and/or obese male rats. Metabolism (2016) 65:54–67. doi: 10.1016/j.metabol.2015.10.015
15. Sa-Nguanmoo P, Tanajak P, Kerdphoo S, Satjaritanun P, Wang X, Liang G, et al. FGF21 improves cognition by restored synaptic plasticity, dendritic spine density, brain mitochondrial function and cell apoptosis in obese-insulin resistant male rats. Horm Behav. (2016) 85:86–95. doi: 10.1016/j.yhbeh.2016.08.006
16. Sun Z, Zhan L, Liang L, Sui H, Zheng L, Sun X, et al. ZiBu PiYin recipe prevents diabetes-associated cognitive decline in rats: possible involvement of ameliorating mitochondrial dysfunction, insulin resistance pathway and histopathological changes. BMC Complement Altern Med. (2016) 16:200. doi: 10.1186/s12906-016-1177-y
17. Li H, Qin T, Li M, Ma S. Thymol improves high-fat diet-induced cognitive deficits in mice via ameliorating brain insulin resistance and upregulating NRF2/HO-1 pathway. Metab Brain Dis. (2017) 32, 385–393. doi: 10.1007/s11011-016-9921-z
18. Kothari V, Luo Y, Tornabene T, O'Neill AM, Greene MW, Geetha T, et al. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim Biophys Acta (2017) 1863:499–508. doi: 10.1016/j.bbadis.2016.10.006
19. Sa-Nguanmoo P, Tanajak P, Kerdphoo S, Jaiwongkam T, Pratchayasakul W, Chattipakorn N, et al. SGLT2-inhibitor and DPP-4 inhibitor improve brain function via attenuating mitochondrial dysfunction, insulin resistance, inflammation, and apoptosis in HFD-induced obese rats. Toxicol Appl Pharmacol. (2017) 333:43–50. doi: 10.1016/j.taap.2017.08.005
20. Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. (2006) 45:42–72. doi: 10.1016/j.plipres.2005.11.002
21. Gorski J. Ceramide and insulin resistance: how should the issue be approached? Diabetes (2012) 61:3081–3. doi: 10.2337/db12-1157
22. Tong M, de la Monte SM. Mechanisms of ceramide-mediated neurodegeneration. J Alzheimers Dis. (2009) 16:705–14. doi: 10.3233/JAD-2009-0983
23. Sartorius T, Peter A, Schulz N, Drescher A, Bergheim I, Machann J, et al. Cinnamon extract improves insulin sensitivity in the brain and lowers liver fat in mouse models of obesity. PLoS ONE (2014) 9:e92358. doi: 10.1371/journal.pone.0092358
24. Cheng Z, Tseng Y, White MF. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab. (2010) 21:589–98. doi: 10.1016/j.tem.2010.06.005
25. Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. (2008) 118:2992–3002. doi: 10.1172/JCI34260
26. Montgomery MK, Turner N. Mitochondrial dysfunction and insulin resistance: an update. Endocr Connect. (2015) 4:R1–15. doi: 10.1530/EC-14-0092
27. Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. (2008) 102:401–14. doi: 10.1161/CIRCRESAHA.107.165472
28. Pintana H, Apaijai N, Pratchayasakul W, Chattipakorn N, Chattipakorn SC. Effects of metformin on learning and memory behaviors and brain mitochondrial functions in high fat diet induced insulin resistant rats. Life Sci. (2012) 91:409–14. doi: 10.1016/j.lfs.2012.08.017
29. Pintana H, Apaijai N, Chattipakorn N, Chattipakorn SC. DPP-4 inhibitors improve cognition and brain mitochondrial function of insulin-resistant rats. J Endocrinol. (2013) 218:1–11. doi: 10.1530/JOE-12-0521
30. Pipatpiboon N, Pintana H, Pratchayasakul W, Chattipakorn N, Chattipakorn SC. DPP4-inhibitor improves neuronal insulin receptor function, brain mitochondrial function and cognitive function in rats with insulin resistance induced by high-fat diet consumption. Eur J Neurosci. (2013) 37:839–49. doi: 10.1111/ejn.12088
31. Pintana H, Tanajak P, Pratchayasakul W, Sa-Nguanmoo P, Chunchai T, Satjaritanun P, et al. Energy restriction combined with dipeptidyl peptidase-4 inhibitor exerts neuroprotection in obese male rats. Br J Nutr. (2016) 1–9. doi: 10.1017/S0007114516003871
32. Barhwal K, Das SK, Kumar A, Hota SK, Srivastava RB. Insulin receptor A and Sirtuin 1 synergistically improve learning and spatial memory following chronic salidroside treatment during hypoxia. J Neurochem. (2015) 135:332–46. doi: 10.1111/jnc.13225
33. Beirami E, Oryan S, Seyedhosseini Tamijani SM, Ahmadiani, A, Dargahi L. Intranasal insulin treatment restores cognitive deficits and insulin signaling impairment induced by repeated methamphetamine exposure. J Cell Biochem. (2018) 119:2345–55. doi: 10.1002/jcb.26398
34. Schinner S, Scherbaum WA, Bornstein SR, Barthel A. Molecular mechanisms of insulin resistance. Diabet Med. (2005) 22:674–82. doi: 10.1111/j.1464-5491.2005.01566.x
35. Martyn JA, Kaneki M, Yasuhara S. Obesity-induced insulin resistance and hyperglycemia: etiologic factors and molecular mechanisms. Anesthesiology (2008) 109:137–48. doi: 10.1097/ALN.0b013e3181799d45
36. Gutierrez-Rodelo C, Roura-Guiberna A, Olivares-Reyes JA. [Molecular Mechanisms of Insulin Resistance: an Update]. Gac Med Mex. (2017) 153:214–28.
37. Chondronikola M, Volpi E, Borsheim E, Porter C, Annamalai P, Enerback S, et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes (2014) 63:4089–99. doi: 10.2337/db14-0746
38. Zhou L, Park SY, Xu L, Xia X, Ye J, Su L, et al. Insulin resistance and white adipose tissue inflammation are uncoupled in energetically challenged Fsp27-deficient mice. Nat Commun. (2015) 6:5949. doi: 10.1038/ncomms6949
39. Lizcano F, Vargas D. Biology of beige adipocyte and possible therapy for type 2 diabetes and obesity. Int J Endocrinol. (2016) 2016:9542061. doi: 10.1155/2016/9542061
40. Min SY, Kady J, Nam M, Rojas-Rodriguez R, Berkenwald A, Kim JH, et al. Human 'brite/beige' adipocytes develop from capillary networks, and their implantation improves metabolic homeostasis in mice. Nat Med. (2016) 22:312–8. doi: 10.1038/nm.4031
41. Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie (2005) 87:99–109. doi: 10.1016/j.biochi.2004.10.019
42. Trayhurn P, Wood IS. Signalling role of adipose tissue: adipokines and inflammation in obesity. Biochem Soc Trans. (2005) 33(Pt 5):1078–81. doi: 10.1042/BST0331078
43. Sethi JK, Vidal-Puig AJ. Thematic review series: adipocyte biology. Adipose tissue function and plasticity orchestrate nutritional adaptation J Lipid Res. (2007) 48:1253–62. doi: 10.1194/jlr.R700005-JLR200
44. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. (2012) 18:363–74. doi: 10.1038/nm.2627
45. Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem. (2002) 277:1531–7. doi: 10.1074/jbc.M101521200
46. Gao Z, Zhang X, Zuberi A, Hwang D, Quon MJ, Lefevre M, et al. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3-L1 adipocytes. Mol Endocrinol. (2004) 18:2024–34. doi: 10.1210/me.2003-0383
47. Werner ED, Lee J, Hansen L, Yuan M, Shoelson SE. Insulin resistance due to phosphorylation of insulin receptor substrate-1 at serine 302. J Biol Chem. (2004) 279:35298–305. doi: 10.1074/jbc.M405203200
48. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. (2006) 116:1793–801. doi: 10.1172/JCI29069
49. Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol. (2014) 6:a009191. doi: 10.1101/cshperspect.a009191
50. Ronn SG, Billestrup N, Mandrup-Poulsen T. Diabetes and suppressors of cytokine signaling proteins. Diabetes (2007) 56:541–8. doi: 10.2337/db06-1068
51. de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett. (2008) 582:97–105. doi: 10.1016/j.febslet.2007.11.057
52. Johnson AM, Olefsky JM. The origins and drivers of insulin resistance. Cell (2013) 152:673–84. doi: 10.1016/j.cell.2013.01.041
53. Peng Y, Liu J, Shi L, Tang Y, Gao D, Long J, et al. Mitochondrial dysfunction precedes depression of AMPK/AKT signaling in insulin resistance induced by high glucose in primary cortical neurons. J Neurochem. (2016) 137:701–13. doi: 10.1111/jnc.13563
54. Sebastian D, Hernandez-Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA. (2012) 109:5523–8. doi: 10.1073/pnas.1108220109
55. Park SY, Choi GH, Choi HI, Ryu J, Jung CY, Lee W. Depletion of mitochondrial DNA causes impaired glucose utilization and insulin resistance in L6 GLUT4myc myocytes. J Biol Chem. (2005) 280:9855–64. doi: 10.1074/jbc.M409399200
56. Park SY, Lee W. The depletion of cellular mitochondrial DNA causes insulin resistance through the alteration of insulin receptor substrate-1 in rat myocytes. Diabetes Res Clin Pract. (2007) 77 (Suppl. 1):S165–71. doi: 10.1016/j.diabres.2007.01.051
57. Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. (2002) 192:1–15. doi: 10.1002/jcp.10119
58. Marnett LJ. Oxy radicals, lipid peroxidation and DNA damage. Toxicology (2002) 181–2, 219–22. doi: 10.1016/S0300-483X(02)00448-1
59. Le Lay S, Simard G, Martinez MC, Andriantsitohaina R. Oxidative stress and metabolic pathologies: from an adipocentric point of view. Oxid Med Cell Longev. (2014) 2014:908539. doi: 10.1155/2014/908539
60. Rahman I. Oxidative stress, transcription factors and chromatin remodelling in lung inflammation. Biochem Pharmacol. (2002) 64:935–42. doi: 10.1016/S0006-2952(02)01153-X
61. Havrankova J, Schmechel D, Roth J, Brownstein M. Identification of insulin in rat brain. Proc Natl Acad Sci USA. (1978) 75:5737–41. doi: 10.1073/pnas.75.11.5737
62. Dorn A, Rinne A, Bernstein HG, Hahn HJ, Ziegler M. Insulin and C-peptide in human brain neurons (insulin/C-peptide/brain peptides/immunohistochemistry/radioimmunoassay). J Hirnforsch. (1983) 24:495–9.
63. Devaskar SU, Giddings SJ, Rajakumar PA, Carnaghi LR, Menon RK, Zahm DS. Insulin gene expression and insulin synthesis in mammalian neuronal cells. J Biol Chem. (1994) 269:8445–54.
64. Kuwabara T, Kagalwala MN, Onuma Y, Ito Y, Warashina M, Terashima K, et al. Insulin biosynthesis in neuronal progenitors derived from adult hippocampus and the olfactory bulb. EMBO Mol Med. (2011) 3:742–54. doi: 10.1002/emmm.201100177
65. Luo J, Becnel J, Nichols CD, Nassel DR. Insulin-producing cells in the brain of adult Drosophila are regulated by the serotonin 5-HT1A receptor. Cell Mol Life Sci. (2012) 69:471–84. doi: 10.1007/s00018-011-0789-0
66. Molnar G, Farago N, Kocsis AK, Rozsa M, Lovas S, Boldog E, et al. GABAergic neurogliaform cells represent local sources of insulin in the cerebral cortex. J Neurosci. (2014) 34:1133–7. doi: 10.1523/JNEUROSCI.4082-13.2014
67. Hersom M, Helms HC, Schmalz C, Pedersen TA, Buckley ST, Brodin B. The insulin receptor is expressed and functional in cultured blood-brain barrier endothelial cells, but does not mediate insulin entry from blood-to-brain. Am J Physiol Endocrinol Metab. (2018). doi: 10.1152/ajpendo.00350.2016. [Epub ahead of print].
68. Werther GA, Hogg A, Oldfield BJ, McKinley MJ, Figdor R, Allen AM, et al. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology (1987) 121:1562–70. doi: 10.1210/endo-121-4-1562
69. Marks JL, Porte DJr, Stahl WL, Baskin DG. Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology (1990) 127:3234–6. doi: 10.1210/endo-127-6-3234
70. Blazquez E, Velazquez E, Hurtado-Carneiro V, Ruiz-Albusac JM. Insulin in the brain: its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer's disease. Front Endocrinol. (2014) 5:161. doi: 10.3389/fendo.2014.00161
71. Gray SM, Meijer RI, Barrett EJ. Insulin regulates brain function, but how does it get there? Diabetes (2014) 63:3992–7. doi: 10.2337/db14-0340
72. Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. (2007) 5:237–52. doi: 10.1016/j.cmet.2007.03.006
73. El Messari S, Leloup C, Quignon M, Brisorgueil MJ, Penicaud L, Arluison M. Immunocytochemical localization of the insulin-responsive glucose transporter 4 (Glut4) in the rat central nervous system. J Comp Neurol. (1998) 399:492–512.
74. Vannucci SJ, Koehler-Stec EM, Li K, Reynolds TH, Clark R, Simpson IA. GLUT4 glucose transporter expression in rodent brain: effect of diabetes. Brain Res. (1998) 797:1–11. doi: 10.1016/S0006-8993(98)00103-6
75. Uemura E, Greenlee HW. Insulin regulates neuronal glucose uptake by promoting translocation of glucose transporter GLUT3. Exp Neurol. (2006) 198:48–53. doi: 10.1016/j.expneurol.2005.10.035
76. Benarroch EE. Brain glucose transporters: implications for neurologic disease. Neurology (2014) 82:1374–9. doi: 10.1212/WNL.0000000000000328
77. Fernandez AM, Hernandez-Garzon E, Perez-Domper P, Perez-Alvarez A, Mederos S, Matsui T, et al. Insulin regulates astrocytic glucose handling through cooperation with IGF-I. Diabetes (2017) 66:64–74. doi: 10.2337/db16-0861
78. Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. (2012) 122:1316–38. doi: 10.1172/JCI59903
79. Gould GW, Brant AM, Kahn BB, Shepherd PR, McCoid SC, Gibbs EM. Expression of the brain-type glucose transporter is restricted to brain and neuronal cells in mice. Diabetologia (1992) 35:304–9. doi: 10.1007/BF00401196
80. Pearson-Leary J, Jahagirdar V, Sage J, McNay EC. Insulin modulates hippocampally-mediated spatial working memory via glucose transporter-4. Behav Brain Res. (2018) 338:32–9. doi: 10.1016/j.bbr.2017.09.033
81. Kovacs P, Hajnal A. In vivo electrophysiological effects of insulin in the rat brain. Neuropeptides (2009) 43:283–93. doi: 10.1016/j.npep.2009.05.006
82. Sevanian A, Davies KJ, Hochstein P. Serum urate as an antioxidant for ascorbic acid. Am J Clin Nutr. (1991) 54(Suppl. 6):1129S−34S. doi: 10.1093/ajcn/54.6.1129s
83. Ryu BR, Ko HW, Jou I, Noh JS, Gwag BJ. Phosphatidylinositol 3-kinase-mediated regulation of neuronal apoptosis and necrosis by insulin and IGF-I. J Neurobiol. (1999) 39:536–46.
84. Rensink AA, Otte-Holler I, de Boer R, Bosch RR, ten Donkelaar HJ, de Waal RM, et al. Insulin inhibits amyloid beta-induced cell death in cultured human brain pericytes. Neurobiol Aging (2004) 25:93–103. doi: 10.1016/S0197-4580(03)00039-3
85. Duarte AI, Proenca T, Oliveira CR, Santos MS, Rego AC. Insulin restores metabolic function in cultured cortical neurons subjected to oxidative stress. Diabetes (2006) 55:2863–70. doi: 10.2337/db06-0030
86. Park CR, Seeley RJ, Craft S, Woods SC. Intracerebroventricular insulin enhances memory in a passive-avoidance task. Physiol Behav. (2000) 68:509–14. doi: 10.1016/S0031-9384(99)00220-6
87. Kern W, Peters A, Fruehwald-Schultes B, Deininger E, Born J, Fehm HL. Improving influence of insulin on cognitive functions in humans. Neuroendocrinology (2001) 74:270–80. doi: 10.1159/000054694
88. Zhao W, Chen H, Xu H, Moore E, Meiri N, Quon MJ, et al. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats J Biol Chem. (1999) 274:34893–902. doi: 10.1074/jbc.274.49.34893
89. Huang CC, Lee CC, Hsu KS. An investigation into signal transduction mechanisms involved in insulin-induced long-term depression in the CA1 region of the hippocampus. J Neurochem. (2004) 89:217–31. doi: 10.1111/j.1471-4159.2003.02307.x
90. Ahmadian G, Ju W, Liu L, Wyszynski M, Lee SH, Dunah AW, et al. Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J. (2004) 23:1040–50. doi: 10.1038/sj.emboj.7600126
91. Pratchayasakul W, Chattipakorn N, Chattipakorn SC. Effects of estrogen in preventing neuronal insulin resistance in hippocampus of obese rats are different between genders. Life Sci. (2011) 89:702–7. doi: 10.1016/j.lfs.2011.08.011
92. Filippi BM, Abraham MA, Silva PN, Rasti M, LaPierre MP, Bauer PV, et al. Dynamin-related protein 1-dependent mitochondrial fission changes in the dorsal vagal complex regulate insulin action. Cell Rep. (2017) 18:2301–9. doi: 10.1016/j.celrep.2017.02.035
93. Kim JM, Park CH, Park SK, Seung TW, Kang JY, Ha JS, et al. Ginsenoside re ameliorates brain insulin resistance and cognitive dysfunction in high fat diet-induced C57BL/6 mice. J Agric Food Chem. (2017) 65:2719–29. doi: 10.1021/acs.jafc.7b00297
94. Schmitz L, Kuglin R, Bae-Gartz I, Janoschek R, Appel S, Mesaros A, et al. Hippocampal insulin resistance links maternal obesity with impaired neuronal plasticity in adult offspring. Psychoneuroendocrinology (2018) 89:46–52. doi: 10.1016/j.psyneuen.2017.12.023
95. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. (2003) 112:1821–30. doi: 10.1172/JCI200319451
96. De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology (2005) 146:4192–9. doi: 10.1210/en.2004-1520
97. Boden G. Fatty acid-induced inflammation and insulin resistance in skeletal muscle and liver. Curr Diab Rep. (2006) 6:177–81. doi: 10.1007/s11892-006-0031-x
98. Funaki M. Saturated fatty acids and insulin resistance. J Med Invest. (2009) 56:88–92. doi: 10.2152/jmi.56.88
99. Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab. (2009) 296:E1003–12. doi: 10.1152/ajpendo.90377.2008
100. Zeyda M, Stulnig TM. Obesity, inflammation, and insulin resistance–a mini-review. Gerontology (2009) 55:379–86. doi: 10.1159/000212758
101. Ajuwon KM, Spurlock ME. Palmitate activates the NF-kappaB transcription factor and induces IL-6 and TNFalpha expression in 3T3-L1 adipocytes. J Nutr. (2005) 135:1841–6. doi: 10.1093/jn/135.8.1841
102. Coll T, Jove M, Rodriguez-Calvo R, Eyre E, Palomer X, Sanchez RM, et al. Palmitate-mediated downregulation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha in skeletal muscle cells involves MEK1/2 and nuclear factor-kappaB activation. Diabetes (2006) 55:2779–87. doi: 10.2337/db05-1494
103. Jove M, Planavila A, Sanchez RM, Merlos M, Laguna JC, Vazquez-Carrera M. Palmitate induces tumor necrosis factor-alpha expression in C2C12 skeletal muscle cells by a mechanism involving protein kinase C and nuclear factor-kappaB activation. Endocrinology (2006) 147:552–61. doi: 10.1210/en.2005-0440
104. Maloney E, Sweet IR, Hockenbery DM, Pham M, Rizzo NO, Tateya S, et al. Activation of NF-kappaB by palmitate in endothelial cells: a key role for NADPH oxidase-derived superoxide in response to TLR4 activation. Arterioscler Thromb Vasc Biol. (2009) 29:1370–5. doi: 10.1161/ATVBAHA.109.188813
105. Green CJ, Macrae K, Fogarty S, Hardie DG, Sakamoto K, Hundal HS. Counter-modulation of fatty acid-induced pro-inflammatory nuclear factor kappaB signalling in rat skeletal muscle cells by AMP-activated protein kinase. Biochem J. (2011) 435:463–74. doi: 10.1042/BJ20101517
106. Cai D. NFkappaB-mediated metabolic inflammation in peripheral tissues versus central nervous system. Cell Cycle (2009) 8:2542–8. doi: 10.4161/cc.8.16.9386
107. Diaz B, Fuentes-Mera L, Tovar A, Montiel T, Massieu L, Martinez-Rodriguez HG, et al. Saturated lipids decrease mitofusin 2 leading to endoplasmic reticulum stress activation and insulin resistance in hypothalamic cells. Brain Res. (2015) 1627:80–9. doi: 10.1016/j.brainres.2015.09.014
108. Choi SJ, Kim F, Schwartz MW, Wisse BE. Cultured hypothalamic neurons are resistant to inflammation and insulin resistance induced by saturated fatty acids. Am J Physiol Endocrinol Metab. (2010) 298:E1122–30. doi: 10.1152/ajpendo.00006.2010
109. de la Monte S, Derdak Z, Wands JR. Alcohol, insulin resistance and the liver-brain axis. J Gastroenterol Hepatol. (2012) 27(Suppl. 2):33–41. doi: 10.1111/j.1440-1746.2011.07023.x
110. Arboleda G, Huang TJ, Waters C, Verkhratsky A, Fernyhough P, Gibson RM. Insulin-like growth factor-1-dependent maintenance of neuronal metabolism through the phosphatidylinositol 3-kinase-Akt pathway is inhibited by C2-ceramide in CAD cells. Eur J Neurosci. (2007) 25:3030–8. doi: 10.1111/j.1460-9568.2007.05557.x
111. Arboleda G, Morales LC, Benitez B, Arboleda H. Regulation of ceramide-induced neuronal death: cell metabolism meets neurodegeneration. Brain Res Rev. (2009) 59:333–46. doi: 10.1016/j.brainresrev.2008.10.001
112. Pathan AR, Gaikwad AB, Viswanad B, Ramarao P. Rosiglitazone attenuates the cognitive deficits induced by high fat diet feeding in rats. Eur J Pharmacol. (2008) 589:176–9. doi: 10.1016/j.ejphar.2008.06.016
113. Sa-Nguanmoo P, Tanajak P, Kerdphoo S, Jaiwongkam T, Wang X, Liang G, et al. FGF21 and DPP-4 inhibitor equally prevents cognitive decline in obese rats. Biomed Pharmacother. (2018) 97:1663–72. doi: 10.1016/j.biopha.2017.12.021
114. Banks WA, Gray AM, Erickson MA, Salameh TS, Damodarasamy M, Sheibani N, et al. Lipopolysaccharide-induced blood-brain barrier disruption: roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J Neuroinflammation (2015) 12:223. doi: 10.1186/s12974-015-0434-1
115. Guo S. Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J Endocrinol. (2014) 220:T1–23. doi: 10.1530/JOE-13-0327
116. Raza H, John A, Howarth FC. Increased oxidative stress and mitochondrial dysfunction in zucker diabetic rat liver and brain. Cell Physiol Biochem. (2015) 35:1241–51. doi: 10.1159/000373947
117. Boudina S, Bugger H, Sena S, O'Neill BT, Zaha VG, Ilkun O, et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation (2009) 119:1272–83. doi: 10.1161/CIRCULATIONAHA.108.792101
118. Liu HY, Yehuda-Shnaidman E, Hong T, Han J, Pi J, Liu Z, et al. Prolonged exposure to insulin suppresses mitochondrial production in primary hepatocytes. J Biol Chem. (2009) 284:14087–95. doi: 10.1074/jbc.M807992200
119. Liu S, Okada T, Assmann A, Soto J, Liew CW, Bugger H, et al. Insulin signaling regulates mitochondrial function in pancreatic beta-cells. PLoS ONE (2009) 4:e7983. doi: 10.1371/journal.pone.0007983
120. Bogacka I, Xie H, Bray GA, Smith SR. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes (2005) 54:1392–9. doi: 10.2337/diabetes.54.5.1392
121. Ghosh S, Patel N, Rahn D, McAllister J, Sadeghi S, Horwitz G, et al. The thiazolidinedione pioglitazone alters mitochondrial function in human neuron-like cells. Mol Pharmacol. (2007) 71:1695–702. doi: 10.1124/mol.106.033845
122. Strum JC, Shehee R, Virley D, Richardson J, Mattie M, Selley P, et al. Rosiglitazone induces mitochondrial biogenesis in mouse brain. J Alzheimers Dis. (2007) 11:45–51. doi: 10.3233/JAD-2007-11108
123. Porter D, Faivre E, Flatt PR, Holscher C, Gault VA. Actions of incretin metabolites on locomotor activity, cognitive function and in vivo hippocampal synaptic plasticity in high fat fed mice. Peptides (2012) 35:1–8. doi: 10.1016/j.peptides.2012.03.014
124. Sripetchwandee J, Pipatpiboon N, Pratchayasakul W, Chattipakorn N, Chattipakorn SC. DPP-4 inhibitor and PPARgamma agonist restore the loss of CA1 dendritic spines in obese insulin-resistant rats. Arch Med Res. (2014) 45:547–52. doi: 10.1016/j.arcmed.2014.09.002
125. Neves G, Cooke SF, Bliss TV. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci. (2008) 9:65–75. doi: 10.1038/nrn2303
126. Porter WD, Flatt PR, Holscher C, Gault VA. Liraglutide improves hippocampal synaptic plasticity associated with increased expression of Mash1 in ob/ob mice. Int J Obes. (2013) 37:678–84. doi: 10.1038/ijo.2012.91
127. Maniam J, Morris MJ. Palatable cafeteria diet ameliorates anxiety and depression-like symptoms following an adverse early environment. Psychoneuroendocrinology (2010) 35:717–28. doi: 10.1016/j.psyneuen.2009.10.013
128. Beilharz JE, Maniam J, Morris MJ. Short exposure to a diet rich in both fat and sugar or sugar alone impairs place, but not object recognition memory in rats. Brain Behav Immun. (2014) 37:134–41. doi: 10.1016/j.bbi.2013.11.016
129. Martire SI, Maniam J, South T, Holmes N, Westbrook RF, Morris MJ. Extended exposure to a palatable cafeteria diet alters gene expression in brain regions implicated in reward and withdrawal from this diet alters gene expression in brain regions associated with stress. Behav Brain Res. (2014) 265:132–41. doi: 10.1016/j.bbr.2014.02.027
130. Leffa DD, Valvassori SS, Varela RB, Lopes-Borges J, Daumann F, Longaretti LM, et al. Effects of palatable cafeteria diet on cognitive and noncognitive behaviors and brain neurotrophins' levels in mice. Metab Brain Dis. (2015) 30:1073–82. doi: 10.1007/s11011-015-9682-0
131. Pini RTB, Ferreira do Vales LDM, Braga Costa TM, Almeida SS. Effects of cafeteria diet and high fat diet intake on anxiety, learning and memory in adult male rats. Nutr Neurosci. (2017) 20:396–408. doi: 10.1080/1028415X.2016.1149294
132. Sack M, Lenz JN, Jakovcevski M, Biedermann SV, Falfan-Melgoza C, Deussing J, et al. Early effects of a high-caloric diet and physical exercise on brain volumetry and behavior: a combined MRI and histology study in mice. Brain Imaging Behav. (2017) 11:1385–96. doi: 10.1007/s11682-016-9638-y
133. Soares MB, Ramalho JB, Izaguirry AP, Pavin NF, Spiazzi CC, Schimidt HL, et al. Comparative effect of Camellia sinensis teas on object recognition test deficit and metabolic changes induced by cafeteria diet. Nutr Neurosci. (2017). doi: 10.1080/1028415X.2017.1418726. [Epub ahead of print].
134. Teixeira D, Cecconello AL, Partata WA, de Fraga LS, Ribeiro, MFM, Guedes RP. The metabolic and neuroinflammatory changes induced by consuming a cafeteria diet are age-dependent. Nutr Neurosci (2017) doi: 10.1080/1028415X.2017.1380892. [Epub ahead of print].
135. Gutierrez-Martos M, Girard B, Mendonca-Netto S, Perroy J, Valjent E, Maldonado R, et al. Cafeteria diet induces neuroplastic modifications in the nucleus accumbens mediated by microglia activation. Addict Biol. (2018) 23:735–49. doi: 10.1111/adb.12541
136. Yin F, Boveris A, Cadenas E. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid. Redox Signal. (2014) 20:353–71. doi: 10.1089/ars.2012.4774
137. Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G. Mitochondrial control of cellular life, stress, and death. Circ Res. (2012) 111:1198–207. doi: 10.1161/CIRCRESAHA.112.268946
138. Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron (2008) 60:748–66. doi: 10.1016/j.neuron.2008.10.010
139. Cheng A, Hou Y, Mattson MP. Mitochondria and neuroplasticity. ASN Neuro (2010) 2:e00045. doi: 10.1042/AN20100019
140. Hara Y, Yuk F, Puri R, Janssen WG, Rapp PR, Morrison JH. Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment. Proc Natl Acad Sci USA (2014) 111:486–91. doi: 10.1073/pnas.1311310110
141. Raefsky SM, Mattson MP. Adaptive responses of neuronal mitochondria to bioenergetic challenges: roles in neuroplasticity and disease resistance. Free Radic Biol Med. (2017) 102:203–16. doi: 10.1016/j.freeradbiomed.2016.11.045
142. Liu X, Hajnoczky G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia-reoxygenation stress. Cell Death Differ. (2011) 18:1561–72. doi: 10.1038/cdd.2011.13
143. Ahmad T, Aggarwal K, Pattnaik B, Mukherjee S, Sethi T, Tiwari BK, et al. Computational classification of mitochondrial shapes reflects stress and redox state. Cell Death Dis. (2013) 4:e461. doi: 10.1038/cddis.2012.213
144. Dragicevic N, Mamcarz M, Zhu Y, Buzzeo R, Tan J, Arendash GW, et al. Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer's transgenic mice. J Alzheimers Dis. (2010) 20(Suppl. 2):S535–50. doi: 10.3233/JAD-2010-100342
145. Baek SH, Park SJ, Jeong JI, Kim SH, Han J, Kyung JW, et al. Inhibition of Drp1 ameliorates synaptic depression, abeta deposition, and cognitive impairment in an Alzheimer's disease model. J Neurosci. (2017) 37:5099–110. doi: 10.1523/JNEUROSCI.2385-16.2017
146. Koliaki C, Roden M. Alterations of mitochondrial function and insulin sensitivity in human obesity and diabetes mellitus. Annu Rev Nutr. (2016) 36:337–67. doi: 10.1146/annurev-nutr-071715-050656
147. Cain K, Bratton SB, Cohen GM. The Apaf-1 apoptosome: a large caspase-activating complex. Biochimie (2002) 84:203–14. doi: 10.1016/S0300-9084(02)01376-7
148. Gomez-Crisostomo NP, Lopez-Marure R, Zapata E, Zazueta C, Martinez-Abundis E. Bax induces cytochrome c release by multiple mechanisms in mitochondria from MCF7 cells. J Bioenerg Biomembr. (2013) 45:441–8. doi: 10.1007/s10863-013-9508-x
149. Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. (2014) 112:24–49. doi: 10.1016/j.pneurobio.2013.10.004
150. Perfettini JL, Roumier T, Kroemer G. Mitochondrial fusion and fission in the control of apoptosis. Trends Cell Biol. (2005) 15:179–83. doi: 10.1016/j.tcb.2005.02.005
151. Knott AB, Bossy-Wetzel E. Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Ann N Y Acad Sci. (2008) 1147:283–92. doi: 10.1196/annals.1427.030
152. Mancuso M, Calsolaro V, Orsucci D, Carlesi C, Choub A, Piazza S, et al. Mitochondria, cognitive impairment, and Alzheimer's disease. Int J Alzheimers Dis. (2009) 2009. doi: 10.4061/2009/951548
153. Cho DH, Nakamura T, Lipton SA. Mitochondrial dynamics in cell death and neurodegeneration. Cell Mol Life Sci. (2010) 67:3435–47. doi: 10.1007/s00018-010-0435-2
154. Zhang Y, Liu J, Yao J, Ji G, Qian L, Wang J, et al. Obesity: pathophysiology and intervention. Nutrients (2014) 6:5153–83. doi: 10.3390/nu6115153
155. Martin KA, Mani MV, Mani A. New targets to treat obesity and the metabolic syndrome. Eur J Pharmacol. (2015) 763(Pt A):64–74. doi: 10.1016/j.ejphar.2015.03.093
156. Lennox R, Porter DW, Flatt PR, Holscher C, Irwin N, Gault VA. Comparison of the independent and combined effects of sub-chronic therapy with metformin and a stable GLP-1 receptor agonist on cognitive function, hippocampal synaptic plasticity and metabolic control in high-fat fed mice. Neuropharmacology (2014) 86:22–30. doi: 10.1016/j.neuropharm.2014.06.026
157. Gault VA, Lennox R, Flatt PR. Sitagliptin, a dipeptidyl peptidase-4 inhibitor, improves recognition memory, oxidative stress and hippocampal neurogenesis and upregulates key genes involved in cognitive decline. Diabetes Obes Metab. (2015) 17:403–13. doi: 10.1111/dom.12432
158. Labuzek K, Suchy D, Gabryel B, Bielecka A, Liber S, Okopien B. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol Rep. (2010) 62:956–65. doi: 10.1016/S1734-1140(10)70357-1
159. Grommes C, Karlo JC, Caprariello A, Blankenship D, Dechant A, Landreth GE. The PPARgamma agonist pioglitazone crosses the blood-brain barrier and reduces tumor growth in a human xenograft model. Cancer Chemother Pharmacol. (2013) 71:929–36. doi: 10.1007/s00280-013-2084-2
160. de Mello AH, Pra M, Cardoso LC, de Bona Schraiber R, Rezin GT. Incretin-based therapies for obesity treatment. Metabolism (2015) 64:967–81. doi: 10.1016/j.metabol.2015.05.012
161. Kim W, Egan JM. The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol Rev. (2008) 60:470–512. doi: 10.1124/pr.108.000604
162. Flock G, Baggio LL, Longuet C, Drucker DJ. Incretin receptors for glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide are essential for the sustained metabolic actions of vildagliptin in mice. Diabetes (2007) 56:3006–13. doi: 10.2337/db07-0697
163. Guo C, Huang T, Chen A, Chen X, Wang L, Shen F, et al. Glucagon-like peptide 1 improves insulin resistance in vitro through anti-inflammation of macrophages. Braz J Med Biol Res. (2016) 49:e5826. doi: 10.1590/1414-431x20165826
164. Wu L, Liu X, Wang L, Wang Y, Wang L, Guan B, et al. Exendin-4 protects HUVECs from tunicamycin-induced apoptosis via inhibiting the IRE1a/JNK/caspase-3 pathway. Endocrine (2017) 55:764–72. doi: 10.1007/s12020-016-1190-4
165. Zhang SS, Wu Z, Zhang Z, Xiong ZY, Chen H, Huang QB. Glucagon-like peptide-1 inhibits the receptor for advanced glycation endproducts to prevent podocyte apoptosis induced by advanced oxidative protein products. Biochem Biophys Res Commun. (2017) 482:1413–9. doi: 10.1016/j.bbrc.2016.12.050
166. Gault VA, Porter WD, Flatt PR, Holscher C. Actions of exendin-4 therapy on cognitive function and hippocampal synaptic plasticity in mice fed a high-fat diet. Int J Obes. (2010) 34:1341–4. doi: 10.1038/ijo.2010.59
167. Porter DW, Kerr BD, Flatt PR, Holscher C, Gault VA. Four weeks administration of Liraglutide improves memory and learning as well as glycaemic control in mice with high fat dietary-induced obesity and insulin resistance. Diabetes Obes Metab. (2010) 12:891–9. doi: 10.1111/j.1463-1326.2010.01259.x
168. Lennox R, Flatt PR, Gault VA. Lixisenatide improves recognition memory and exerts neuroprotective actions in high-fat fed mice. Peptides (2014) 61:38–47. doi: 10.1016/j.peptides.2014.08.014
169. Owen BM, Ding X, Morgan DA, Coate KC, Bookout AL, Rahmouni K, et al. FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab. (2014) 20:670–7. doi: 10.1016/j.cmet.2014.07.012
170. Douris N, Stevanovic DM, Fisher FM, Cisu TI, Chee MJ, Nguyen NL, et al. Central fibroblast growth factor 21 browns white fat via sympathetic action in male mice. Endocrinology (2015) 156:2470–81. doi: 10.1210/en.2014-2001
171. Kharitonenkov A, Shanafelt AB. FGF21: a novel prospect for the treatment of metabolic diseases. Curr Opin Investig Drugs (2009) 10:359–64.
172. Xu J, Stanislaus S, Chinookoswong N, Lau YY, Hager T, Patel J, et al. Acute glucose-lowering and insulin-sensitizing action of FGF21 in insulin-resistant mouse models–association with liver and adipose tissue effects. Am J Physiol Endocrinol Metab. (2009) 297:E1105–14. doi: 10.1152/ajpendo.00348.2009
173. Kong LJ, Feng W, Wright M, Chen Y, Dallas-yang Q, Zhou YP, et al. FGF21 suppresses hepatic glucose production through the activation of atypical protein kinase Ciota/lambda. Eur J Pharmacol. (2013) 702:302–8. doi: 10.1016/j.ejphar.2012.11.065
174. Kralisch S, Tonjes A, Krause K, Richter J, Lossner U, Kovacs P, et al. Fibroblast growth factor-21 serum concentrations are associated with metabolic and hepatic markers in humans. J Endocrinol. (2013) 216:135–43. doi: 10.1530/JOE-12-0367
175. Wang Q, Yuan J, Yu Z, Lin L, Jiang Y, Cao Z, et al. FGF21 attenuates high-fat diet-induced cognitive impairment via metabolic regulation and anti-inflammation of obese mice. Mol Neurobiol. (2017) 55:4702–17. doi: 10.1007/s12035-017-0663-7
176. Kim HW, Lee JE, Cha JJ, Hyun YY, Kim JE, Lee MH, et al. Fibroblast growth factor 21 improves insulin resistance and ameliorates renal injury in db/db mice. Endocrinology (2013) 154:3366–76. doi: 10.1210/en.2012-2276
177. Lin Z, Tian H, Lam KS, Lin S, Hoo RC, Konishi M, et al. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. (2013) 17:779–89. doi: 10.1016/j.cmet.2013.04.005
178. Makela J, Tselykh TV, Maiorana F, Eriksson O, Do HT, Mudo G, et al. Fibroblast growth factor-21 enhances mitochondrial functions and increases the activity of PGC-1alpha in human dopaminergic neurons via Sirtuin-1. Springerplus (2014) 3:2. doi: 10.1186/2193-1801-3-2
179. Yu Y, Bai F, Wang W, Liu Y, Yuan Q, Qu S, et al. Fibroblast growth factor 21 protects mouse brain against D-galactose induced aging via suppression of oxidative stress response and advanced glycation end products formation. Pharmacol Biochem Behav. (2015) 133:122–31. doi: 10.1016/j.pbb.2015.03.020
180. Akhtar MW, Sanz-Blasco S, Dolatabadi N, Parker J, Chon K, Lee MS, et al. Elevated glucose and oligomeric beta-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation. Nat Commun. (2016) 7:10242. doi: 10.1038/ncomms10242
181. Alagiakrishnan K, Sankaralingam S, Ghosh M, Mereu L, Senior P. Antidiabetic drugs and their potential role in treating mild cognitive impairment and Alzheimer's disease. Discov Med. (2013) 16:277–86.
182. Palleria C, Leporini C, Maida F, Succurro E, De Sarro G, Arturi F, et al. Potential effects of current drug therapies on cognitive impairment in patients with type 2 diabetes. Front Neuroendocrinol. (2016) 42:76–92. doi: 10.1016/j.yfrne.2016.07.002
Keywords: obese-insulin resistance, oxidative stress, inflammation, brain mitochondria, cognitive decline
Citation: Sripetchwandee J, Chattipakorn N and Chattipakorn SC (2018) Links Between Obesity-Induced Brain Insulin Resistance, Brain Mitochondrial Dysfunction, and Dementia. Front. Endocrinol. 9:496. doi: 10.3389/fendo.2018.00496
Received: 02 February 2018; Accepted: 07 August 2018;
Published: 31 August 2018.
Edited by:
Andre Kleinridders, Deutsches Institut für Ernährungsforschung Potsdam-Rehbrücke (DIfE), GermanyReviewed by:
Aniko Korosi, University of Amsterdam, NetherlandsRalf Jockers, Université Paris-Sorbonne, France
Andréa Silva Torrão, Universidade de São Paulo, Brazil
Copyright © 2018 Sripetchwandee, Chattipakorn and Chattipakorn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Siriporn C. Chattipakorn, c2NjaGF0dGlwYWtvcm5AZ21haWwuY29t; c2lyaXBvcm4uY0BjbXUuYWMudGg=