 Matthew S. Thimgan
Matthew S. Thimgan Natalie Kress
Natalie Kress Josh Lisse
Josh Lisse Courtney Fiebelman1
Courtney Fiebelman1 Thomas Hilderbrand
Thomas Hilderbrand- 1Department of Biological Sciences, Missouri University of Science and Technology, Rolla, MO, United States
- 2Department of Neuroscience, Washington University School of Medicine in St. Louis, St. Louis, MO, United States
The regulation of sleep and the response to sleep deprivation rely on multiple biochemical pathways. A critical connection is the link between sleep and metabolism. Metabolic changes can disrupt sleep, and conversely decreased sleep can alter the metabolic environment. There is building evidence that lipid metabolism, in particular, is a critical part of mounting the homeostatic response to sleep deprivation. We have evaluated an acyl-CoA synthetase, pudgy (pdgy), for its role in sleep and response to sleep deprivation. When pdgy transcript levels are decreased through transposable element disruption of the gene, mutant flies showed lower total sleep times and increased sleep fragmentation at night compared to genetic controls. Consistent with disrupted sleep, mutant flies had a decreased lifespan compared to controls. pdgy disrupted fatty acid handling as pdgy mutants showed increased sensitivity to starvation and exhibited lower fat stores. Moreover, the response to sleep deprivation is reduced when compared to a control flies. When we decreased the transcript levels for pdgy using RNAi, the response to sleep deprivation was decreased compared to background controls. In addition, when the pdgy transcription is rescued throughout the fly, the response to sleep deprivation is restored. These data demonstrate that the regulation and function of acyl-CoA synthetase plays a critical role in regulating sleep and the response to sleep deprivation. Endocrine and metabolic signals that alter transcript levels of pdgy impact sleep regulation or interfere with the homeostatic response to sleep deprivation.
Introduction
There is an increasing recognition that lipid metabolism contributes to sleep and wakefulness regulation and the response to sleep deprivation. The involvement of lipids supports the energy hypothesis of sleep regulation, in which energy is required to perform the restorative actions of sleep as well as to carry out waking activities (1). In addition, mobilized lipids can act as signaling molecules and change the composition of membranes within the cell (2). Mutations that alter free fatty acid mobilization reduce sleep rebound and protect the animal from the cognitive deficits usually seen with sleep deprivation (3). In addition, knockdown of two presumptive acyl-CoA synthetases reduces the homeostatic response to sleep deprivation and had metabolic phenotypes (4). Mutations in fatty acid binding protein result in reductions of sleep in flies and mammals (5, 6). In mammals, TRIB1 has been linked with population differences in sleep regulation (7) and mutations in fatty acid metabolism alters REM sleep (8). Plasma levels of lipid-related molecules are altered through sleep deprivation (9), and free fatty acids are elevated during sleep restriction (10). Lipid metabolism genes are found as genes that are the most changed in response to sleep deprivation in unbiased array or network analyses (11, 12). Thus lipid metabolism, and in particular free fatty acid handling plays an important, yet ill-defined, role in sleep regulation.
Acyl-CoA synthetases (ACSs) are enzymes that attach a CoA moiety to fatty acids to activate them. The resulting acyl-CoA molecule is an “activated” fatty acid that is retained in the cell, and the acyl-CoA can go on to fulfill energy needs, carry out signaling properties, or other functions within the cell or in other cells (2). Each organism has multiple ACSs with specific organ and cellular expression patterns (13). ACSs also have substrate specificity, in which different chain length fatty acids are conjugated to CoA by different ACSs (13). Knockout and knockdown data from mice reveal increases and decreases in lipid storage likely resulting from defects in the dynamics of lipid mobilization (14–16). In flies, mutations in the ACS, bubblegum (bgm), result in a severe neurodegeneration phenotype that can be rescued by fatty acid consumption (17). In flies, knockdown of two ACSs reduces the homeostatic response to sleep deprivation (4). Thus, ACSs play crucial role in energy and metabolic management and are responsive to changing physiologic conditions.
The ACS pudgy (pdgy) is upregulated under starvation conditions to continue to fulfill energy needs in the face of decreased energy consumption (18). Transcript levels of pdgy also vary between sleep and wake states in the fly (11). In this manuscript, we used a behavioral genetics approach to assess whether pdgy impacts the response to sleep deprivation and whether there were accompanying metabolic changes. We found that decreasing the expression of pdgy eliminated the homeostatic response to sleep deprivation. In contrast to the other ACSs observed before, pdgy, reduced baseline sleep and increased the fragmentation of the animal. Rescue of pdgy in a mutant background restored the sleep phenotypes. These results suggest that pdgy regulates sleep through the activation of fatty acids. Given that endocrine factors can alter the transcript levels of ACSs, our data suggest a mechanism by which endocrine factors can alter sleep regulation and lipid mobilization in response to sleep deprivation.
Materials and Methods
Flies and Husbandry
Flies were reared in standard laboratory conditions, such light:dark schedule, standard food (yeast, sucrose, corn syrup, molasses, and agar), 25°C and 50% humidity. The UAS-PdgyRNAi line was obtained from the Vienna Drosophila Resource Center (19). Actin-GAL4/CyO (Act-GAL4), pdgyBG02662, pdgyEY02124 (20), Mi{PT-GFSTF.0}pdgyMI04730−GFSTF.0, and flies to mobilize the transposon were obtained from the Bloomington Stock Center (Bloomington, Indiana). pdgyBG02662 has a 8,447 bp transposon inserted in the 5′ untranslated region and pdgyEY02124 has a 10,908 bp transposable element within the coding region of the pdgy gene (21). Background controls were generated by mobilizing the P-element using the Δ2-3 version of the transposase to extract the transposable element, termed “revertants.” The Mi{PT-GFSTF.0}pdgyMI04730−GFSTF.0 uses a Minos transposable element that integrates into an intronic region with a splice acceptor such that the GFP tag is expressed in the protein under the control of endogenous enhancers and repressors (22). The Fat body-GAL4 (FB-GAL4) was generously provided by Ronald Kuhnlein and the UAS-pdgywt was generously provided by Aurelio Teleman.
Sleep Measurements, Sleep Deprivation, and Starvation
Sleep in flies was measured using the Trikinetics Drosophila Activity Monitor (DAM) system. Activity counts were converted into sleep using the protocol developed previously (23) in which data was collected in 1 min bins and 5 min of inactivity as the empirically-derived definition of sleep (23, 24). Sleep architecture and sleep metrics were calculated using an in-house program (23–25).
Sleep deprivation was accomplished using the geotaxis method as described in (3). Briefly, 3-5 day old female flies were loaded into 65 mm tubes with food on one end and an air permeable plug on the other end. After 2 full days of baseline, flies were sleep deprived using the SNAP device. Flies were deprived of sleep for 12 h between ZT12 (lights out) to ZT0 (lights on) at which point flies were released into recovery where they remained unperturbed for 48 h. Increases in sleep were calculated from the second day of baseline and presented as a percentage of sleep lost.
Starvation experiments were carried out similarly, in which flies were allowed 2 days of baseline sleep on normal food and then put onto 1% agar and water during the starvation period. For starvation tolerance assays, flies were loaded into the DAMS system for continuous monitoring and which provided activity resolution to the nearest hour to identify when the fly died.
Real-time Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated from 20 fly heads using Trizol (Invitrogen, Carlsbad, CA) and cleaned of contaminating DNA using DNAse I. cDNA synthesis was performed in quadruplicate using Superscript (Invitrogen, Carlsbad, CA), according to manufacturer protocol. Equal amounts of cDNA were used as a starting material to amplify RP49. cDNA from reverse transcription reactions with comparable RP49 levels were compared. Expression values for RP49 were used to normalize results between groups. For flies maintained on an LD schedule, both experimental and untreated controls, were collected at the same circadian time, ZT0-1 for sleep deprived or starved animals. Relative levels of transcript were determined using the ΔΔ method as described (26) to determine the relative levels of transcript between genotype or experimental flies.
Longevity Assay
Three-day-old flies were randomly assigned to one of 3 vials of 10 flies. Flies were counted and transferred onto new food every 3 times per week. Flies alive were expressed as a percentage of the original starting number of flies. Lifespan curves were analyzed using Kaplan-Meier analysis to determine significant differences.
For peroxide tolerance, individual flies were loaded into the DAMS system. After 2 days on normal food, the flies were put onto normal food containing 1% hydrogen peroxide (27) and monitored continuously until the animal died. Deaths were visually confirmed.
Triglyceride Measurements
For each genotype, 10 female flies were frozen and stored at −80°C. Lipid measurements were carried similar to (3). Briefly, flies were weighed and homogenized in a 2:1 (methanol:chloroform) solution to extract the lipids (28). The MeOH:chloroform is evaporated using the speed vac, and the lipids were re-suspended in the starting reagent for Infinity (ThermoElectron, Waltham, MA) triglyceride reagent and triglyceride levels detected using the colorometric detection according to the manufacturer's specifications. Lipid levels are quantified using a standard curve of known triglyceride run in parallel.
Heart Rate
Heart rate was measured according to the methods laid out in (29). We anesthetized flies with 1 mL of FlyNap (Carolina Biological Supply) for 10 min (30). FlyNap does not affect heart function (31). Flies were attached to a slide with the dorsal side up by affixing their extended wings using double-stick tape. Flies were lit from below to see the heart contraction under 200X magnification. Contractions were counted over a 15 s period. The average of 5 separate observations per fly were used. There was a 15 s interval in between observation periods. The experimenter was blinded to genotype and counted the beats per 15 s in the fly.
Resting Metabolic Rate
Resting metabolic rates were determined using a Sable Systems International CA-10 (Las Vegas, NV USA). Virgin female flies per genotype were collected using light CO2 anesthesia and put into vial until use. Metabolic rates were measured by using groups of 30, 5–7 day old flies at rest within flow-through respirometry chambers. Dry, CO2-free air was passed through the 10-ml glass respirometry chambers at 50 mL/min and then dried again and passed through a Li-Cor 6251 carbon dioxide analyzer (Lincoln, NE). Metabolic rates were measured for 5 min using constant respiromentry. The metabolic rate was the stable rate established. Within each run, seven experimental chambers containing flies were sampled in a sequential fashion by using a computer-controlled valve system. Genotypes were assigned randomly to the chambers for each run. One additional chamber was kept empty and sampled before and after each experimental chamber to remove any baseline drift. Temperature was measured by using a thermocouple within the empty chamber. Analog signals from the flow meter, carbon dioxide analyzer, and thermocouple were converted to digital and recorded on a computer (Sable Systems). After CO2 levels were measured, the weight of the flies was measured on a microbalance to normalize levels to tissue weight.
Immunolocalization
Tissue from the Mi{Mic} flies inserted in pdgy were dissected and fixed in 4% paraformaldehyde in PBS for 1 h. Tissues was washed, blocked with normal goat serum, and then incubated overnight in 1:100 anti-GFP primary antibody (R&D Biosystems, Minneapolis, MN) at +4°C. After washing in PBS-T, the samples were incubated overnight in 1:100 Alexa Fluor 488 secondary antibody (Thermo Fisher, Eugene, Oregon) overnight at +4°C, and then washed and mounted in Vectashield. Images were taken using an Olympus IX51 inverted microscope at 600X total magnification using a UPLFLN 60X NA 1.25 objective and an MPlanFLN 10X NA0.30. FITC (EX 482/35 506DM EM 536/40) filter were used (Brightline). Images were captured with a Hamamatsu ORCA285 CCD camera. Shutters, filters, and camera were controlled using SlideBook software (Intelligent Imaging Innovations, Denver, CO).
Data Analysis and Statistics
Where not otherwise stated, groups were analyzed by Student's t-test, in the case of comparing 2 samples and an ANOVA analysis for more than 2 groups with a post hoc Student's t-test to determine which groups are different.
Results
We identified 2 independent P-element insertions within or near the first exon of the pdgy gene that could disrupt transcription or function, pdgyBG02662 (pdgyBG−P) and pdgyEY02124 (pdgyEY−P). We chose these two particular P-elements because they were created in two different backgrounds and were unlikely to have common mutations that might result in aberrant phenotypes (20). We first measured the RNA levels in the P-element compared to the revertant control. The revertants, pdgyBG02662rev (pdgyBGrev) and pdgyEY02124rev (pdgyEYrev) were generated by precisely excising the P-element to restore function in the same background as the P-element mutant. Both the pdgyBG−P (Figure 1A) and the pdgyEY−P (Figure 1D) had lower pdgy transcript levels compared to the revertants. To determine if pdgy impacts the response to sleep deprivation, we deprived flies of sleep for the 12 h primary sleep period from ZT13-ZT0. After sleep deprivation, both pdgy P-element alleles did not exhibit a sleep rebound after losing a full night's sleep. The background controls, pdgyBG2rev (Figure 1B) and pdgyEYrev (Figure 1E), had a large compensatory sleep rebound that were in the range of normal (23). Both the pdgyBGrev (Figure 1C) and the pdgyEYrev (Figure 1F) had increased average maximum day bout from ZT0-ZT12, an indicator of increased consolidation of sleep (32). The metric is compared the value from the baseline day before sleep deprivation to the same period the day after sleep deprivation (the 12 h post sleep deprivation). The increase in the maximum day bout indicates greater consolidation of sleep in the post-deprivation period, which is a property of the homeostatic response to sleep deprivation. Thus, the P-element mutations have an impaired ability to carry out a sleep rebound.
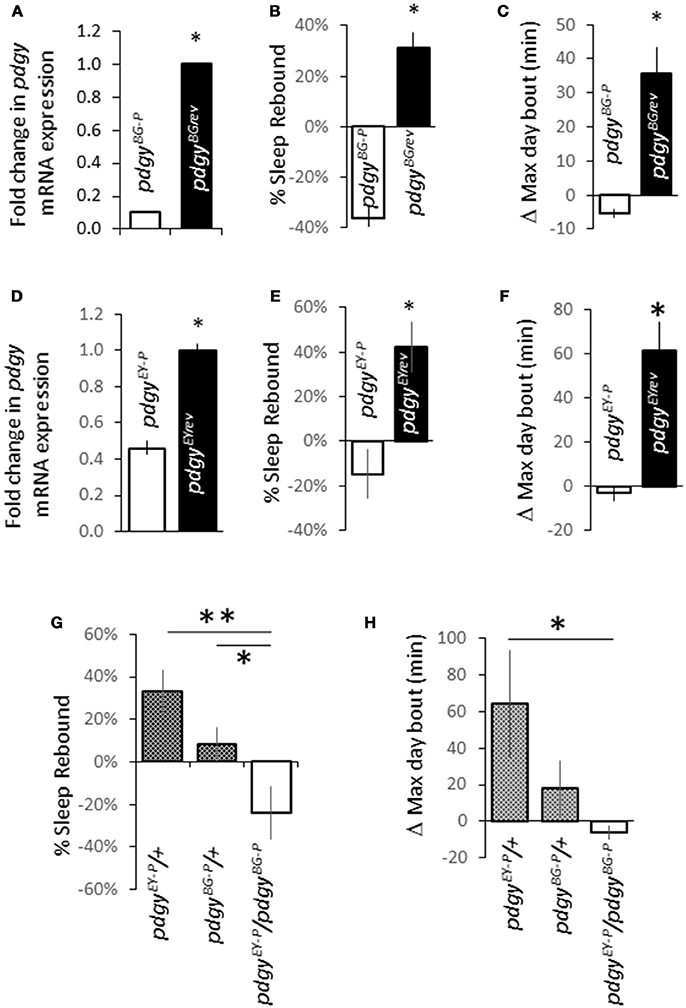
Figure 1. Disruption in pdgy expression alters the response to sleep deprivation. (A) Levels of pdgy mRNA are decreased in pdgyBG02662 (pdgyBG−P) flies compared to the background control, pdgyBGrev (n = 3 groups of 5 pooled flies for each genotype, p < 0.05 by Students t-test). (B) The amount of sleep recaptured after sleep deprivation is significantly decreased when pdgy is disrupted (n = 49 pdgyBG−P and n = 39 pdgyBGrev). (C) The individual change in the maximum day bout was increased in the pdgyBGrev compared to the pdgyBG−P. (D) A second P-element near the pudgyBG−P insertion and in a different background, pdgyEY02124 (pdgyEY−P) has decreased levels of pdgy RNA levels compared to the background controls, pdgyEYrev. (E) The sleep homeostatic response is muted in pdgyEY−P (n = 40) compared to the background control pdgyEYrev (n = 43). (F) The average increase in the maximum day bout is increased between the pdgyEY−P and the pdgyEYrev. (G,H) To determine if there was something in the background responsible for the sleep phenotype in the background of the two strains of flies, we ran a complementation test. pdgyEY−P/+ (n = 16) and pdgyBG−P/+ (n = 14) showed a positive response to sleep deprivation while the pdgyEY−P/pdgyBG−P (n = 13) heterozygote has a significantly lower sleep rebound (G) and maximum day bout on the day following sleep deprivation (H). *p < 0.05, **p < 0.01.
We then determined if the 2 P-elements or backgrounds complemented one another to rescue the phenotype. Heterozygous expression of each of the P-elements exhibited a rebound whereas the pdgyBG−P/pdgyEY−P heterozygous animal exhibited a significantly negative rebound (Figure 1G). The change in the maximum day bout was increased between the pdgyEY02124/+ and the pdgyBG02662/pdgyEY02124 but not a statistical difference between the pdgyBG02662/+, though the outcross showed a positive change and the heterozygote had a decrease in the max day bout (Figure 1H). Thus the sleep rebound phenotype is due to the pdgy locus and genetically supports the conclusion that both alleles have decreased levels of pdgy that do not compensate for each other.
In the course of the sleep deprivation experiments, we observed that the flies with the transposon insertion sleep considerably less. In fact they had a decreased overall duration (Figures 2A,D), with a striking decreased in night duration (Figures 2B,E), the fly's most consolidated sleep period. In addition, these flies were significantly more fragmented than their background controls (Figures 2C,F). Unlike mutations in other lipid metabolism genes in Drosophila, pdgy reduces baseline duration and consolidation in the primary sleep period of both the pdgyBG−P and pdgyEY−P mutants.
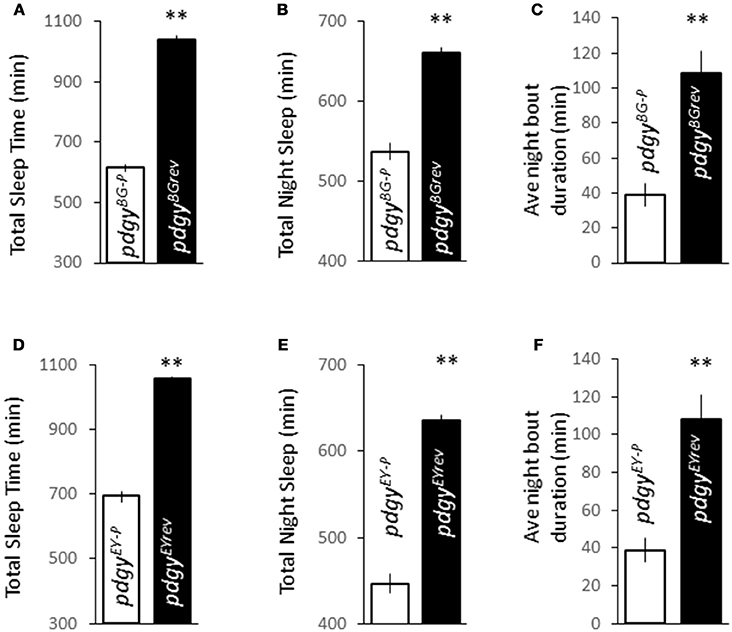
Figure 2. Disruption in pdgy expression alters baseline sleep characteristics. (A–C) Flies with decreased pdgy expression, pdgyBG−P (n = 84), show reductions in sleep with sleep times over 24 h sleep duration throughout the night (B), (A) and sleep consolidation as measured by average night bout duration (C) compared to background control pdgyBGrev (n = 63). (D–F) Total sleep time (D) nighttime sleep duration (E), and night and consolidation (F) are decreased in flies with decreased pdgy (pdgyEY−P) (n = 89) compared to background controls (pdgyEYrev) in a second background (n = 68). **p < 0.01 by Student's t-test.
We then confirmed whether our pdgy hypomorphs showed metabolic phenotypes compared to the background controls. We started by determining the fly's starvation sensitivity. After 48 h of starvation, ~75% of the pdgyBGrev were still alive while the pdgyBG−P mutant flies had completely died after approximately 27 h (Figure 3A). The same relationship was observed with the pdgyEYrevand pdgyEY−P flies (Figure 3C). These results suggest that there is a decreased levels of energy stores or a decreased ability to mobilize those resources under starvation conditions. We directly measured the triglyceride levels. In our hands, the pdgyBG−P had decreased triglyceride levels compared to the pdgyBGrev (Figure 3B) and the relationship was similar between the EY mutant and background control as well (Figure 3D). Thus, it appears that both the pdgyBG−P and pdgyEY−P had decreased triglyceride levels compared to the revertants and less available resources to mobilize. Though these results do not preclude an inability to properly mobilize or utilize liberated free fatty acids.
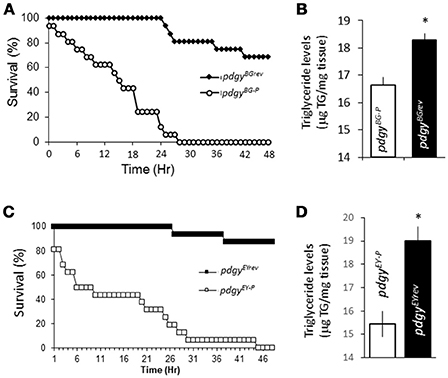
Figure 3. pdgy mutants display metabolic phenotypes. (A) Under starvation conditions, the pdgyBG−P displayed decreased survival compared to background controls, pdgyBGrev. (B) pdgyBG−P had decreased lipid stores compared to background controls. (C,D) Starvation phenotypes compared to its background control (pdgyEYrev) as the pdgyBG−P (D) and pdgyEY−P shows a similar triglyceride phenotypes. *p < 0.05.
Both decreased sleep durations and fragmented sleep have been associated with adverse consequences related to inadequate sleep. We assessed whether lifespan was decreased in the mutants. pdgyBG−P had a 50% survival at ~33 days and a final lifespan to 59 days while the pdgyBGrev had a 50% survival at 42 days and final survival at 81 days (Figure 4A). A similar phenotype was observed in pdgyEY−P as the 50% survival was at 38 days and the genotype survived till 59 days while pdgyEYrev had a 50% survival at day 50 and a final survival for 95 days (Figure 4B). These results are consistent with previous results from inadequate sleep.
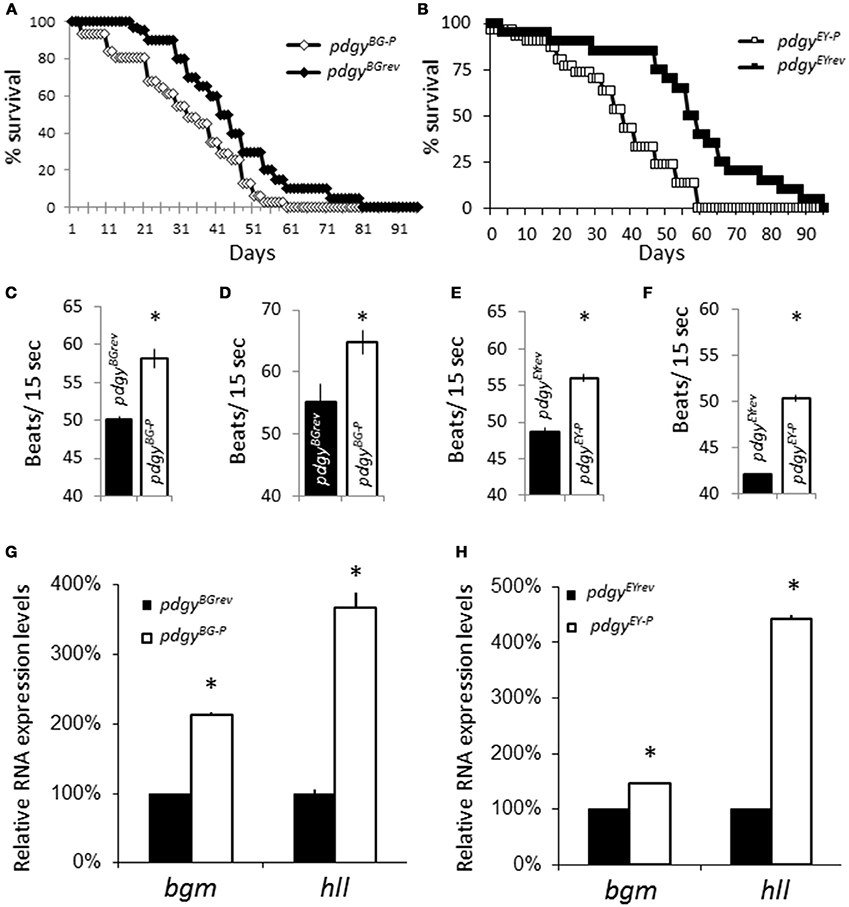
Figure 4. Consequences of the pdgy mutants. (A) Lifespan measure for the pdgyBG−P (n = 30) compared to the revertant control, pdgyBGrev (n = 30). (B) Lifespan measure for the pdgyEY−P (n = 30) compared to the revertant control, pdgyEYrev (n = 30). pdgyBG−P has increased heart rates compared to background controls at (C) 9–11 d.o. and (D) 22–25 d.o. A similar phenotype is observed in pdgyEY−P compared to background controls at (E) 9–11 d.o. and (F) 22–25 d.o. (G) Levels of the ACSs bubblegum (bgm) and heimdall (hll) are elevated in the pdgy hypomorph (pdgyBG−P in G) and (pdgyEY−P in H) compared to the revertant control. *p < 0.01.
Another consequence of poor sleep is that there are increases in heart rate. We evaluated heart rate in the flies. Heart rate was elevated in both sets of mutants compared to their revertant controls at 9-11 days old (Figures 4C,E). We were interested if the phenotype would progress as the flies aged because the consequences of low sleep duration and sleep fragmentation would continue to build up. In flies that were 22–25 days old, both sets of mutants still had an elevated heart rate, but the phenotype did not appear to get worse with increased time (Figure 4D,F). The increased heart rate is consistent with phenotypes associated with inadequate sleep. We reasoned that the similar phenotypes might have similar compensatory responses to in other ACSs. Both hypomorphic lines showed increased transcript levels of bgm and hll compared to revertant controls, indicating that there are modifications in lipid processing enzymes in response to decreased levels of pdgy (Figures 4G,H).
Since the flies displayed a decreased lifespan, we wanted to determine if the flies were generally weaker than the background flies. When the flies were challenged with hydrogen peroxide, flies with the P-element integrated into the genome were able to tolerate the infusion of a compound known to increase cellular stress throughout the animal (33). We evaluated young mutant flies and the background revertants (Figure 5A). To our surprise, the P-element mutants appeared to be more resilient to the infusion of peroxide compared to the background revertants at 5 days old. It is possible that with age, the P-elements would have shown an increasing susceptibility with age. We then tested middle age and older flies as well. In each case, the P-element flies were more resilient to peroxide treatment than the background controls (Figures 5B,C). Interestingly, the peroxide had the intended result as the number of hours that the population survived on peroxide decreased with age but the relationship between the genotypes was not changed. These results indicate that the P-element flies are not inherently weaker than the background controls.
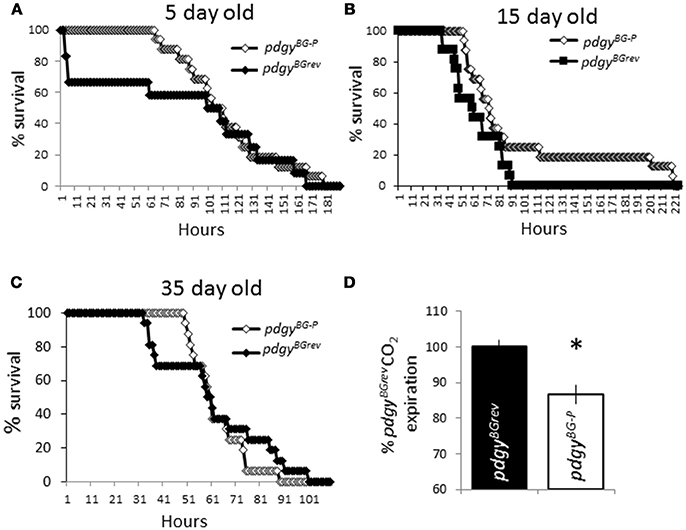
Figure 5. Pdgy mutants are not generally unfit flies. pdgyBG−P and pdgyBGrev were put on peroxide in their food different ages, 5 d.o. (A), 15 d.o. (B), and 35 d.o. (C). n = 16 for each condition. (D) We measured levels of CO2 expired as an indicator of respiration. Levels of CO2 are displayed as percent of control. *p < 0.05.
We evaluated whether the resting metabolic rate was different between the pdgy mutants and background controls (Figure 5D). Interestingly, the metabolic rate was lower in the pdgy mutants, perhaps reflecting the decreased ability to appropriately process fatty acids. In addition, these data suggest that the respiration rate is not higher in the mutant flies.
The P-element mutations suggested a decreased level of pdgy was responsible for the decreased duration and increased sleep fragmentation. We used an independent knockdown technique guided by the bipartite GAL4-UAS system to decrease levels of pdgy (34). We knocked down pdgy throughout the animal using the ubiquitous driver Actin-GAL4 (Act-GAL4). We confirmed knockdown as levels of pdgy transcript were about 20% in the Act-GAL4>UAS-pdgyRNAi of what they were in the background controls (Figure 6A). When the animal was sleep deprived, the control lines, Act-GAL4/+ and the UAS-pdgyRNAi/+ flies showed a significantly higher rebound in sleep rebound compared to the experimental line, Act-GAL4>UAS-pdgyRNAi (Figure 6B). The maximum day bout was lower in the knockdown flies compared to both controls (Figure 6C). We then wanted to determine if a more localized knockdown in the fat body would alter the rebound using the Fat body-GAL4 (FB-GAL4), an enhancer trap that has been shown to express in the adult fly fat body (35). When pdgy is reduced using FB-GAL4, the rebound to sleep deprivation was again reduced below the background lines, indicating that the knockdown of pdgy in the fat bodies may contributes to the sleep rebound phenotype (Figure 6D). When max day bout was assessed, it was lower in the knockdown flies compared to the FB-GAL4/+ background flies but did not reach significance in compared to UAS-pdgyRNAi/+ flies (Figure 6E). Though, the UAS-pdgyRNAi/+ flies showed an increase while the knockdown flies showed a decrease in max day bout, which is consistent with how the sleep rebound was expressed. With both drivers, we did not observe the baseline sleep phenotype, possibly because the knockdown was not strong enough or in the appropriate cells.
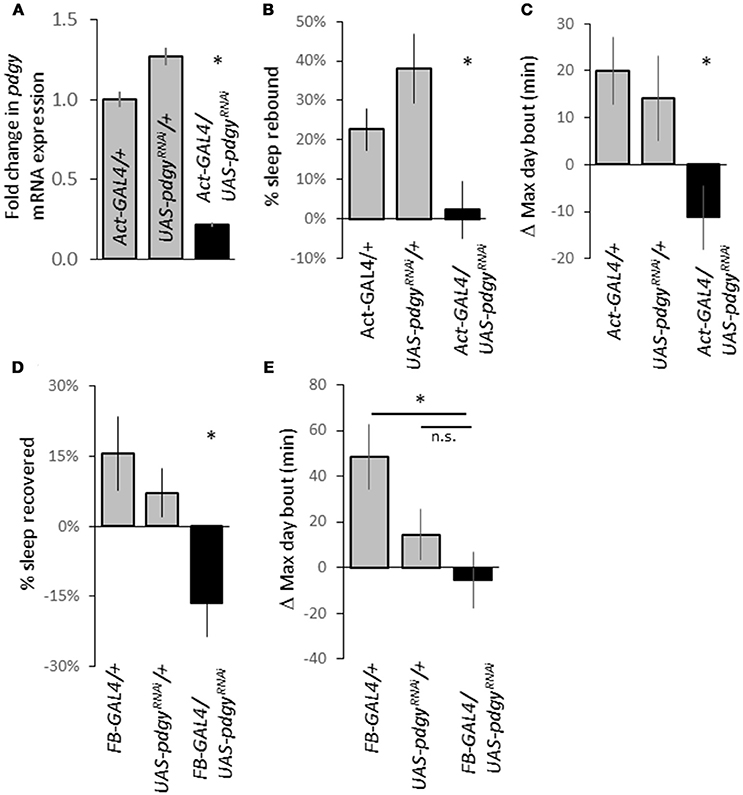
Figure 6. Pdgy knockdown results in a decreased rebound. (A) We confirmed knockdown using real-time qPCR as RNA levels were decreased in Act-GAL4>UAS-pdgyRNAi compared to background controls. (B) Ubiquitous pdgy RNAi knock down (Act-GAL4>UAS-pdgyRNAi) reduced the response to sleep deprivation compared to the background controls, Act-GAL4/+ and UAS-pdgyRNAi/+. N = Act 50, UAS = 38, Act/UAS = 40). (C) An increase in the maximum day bout was suppressed when pdgy was knocked down ubiquitiously. (D) Knockdown in the fat body (FB-GAL4>UAS-pdgyRNAi) also decreased the response to sleep deprivation compared to background controls FB-GAL4/+ and UAS-pdgyRNAi/+. (E) The change in the maximum day bout in FB-GAL4>UAS-pdgyRNAi compared to background controls. For (D,E), N = FB-GAL4/+ = 29, UAS-pdgyRNAi/+ = 53, FB-GAL4/UAS-pdgyRNAi = 32). *p < 0.05.
As a final confirmation that pdgy was a critical part of the sleep rebound, we employed a rescue strategy in which the wild-type pdgy transcript was expressed ubiquitously in an otherwise pdgy hypomorphic animal. We put both Act-GAL4 and UAS-pdgywt, generously provided by Aurelio Teleman (18), into the pdgyBG−P background. The rescue flies exhibited increase night sleep (Figure 7A) and increased night time consolidation (Figure 7B) compared to flies that did not carry the rescue construct. In addition, rescue flies had a homeostatic response to sleep deprivation whereas mutant flies did not (Figures 7C,D). Thus, these data indicate that rescuing pdgy restores more wild-type sleep traits.
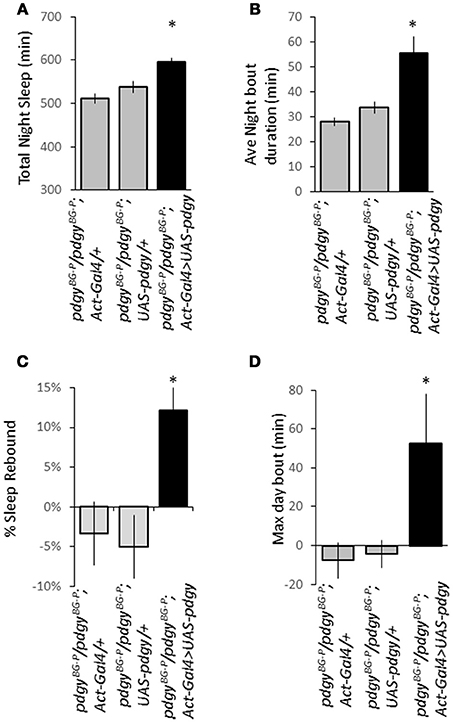
Figure 7. Rescue of pdgy in the pdgyBG−P background restores sleep homeostasis, duration, and consolidation. Re-introduction of the wild-type version of pdgy (dark bars) restores the duration of night time sleep (A) and increases the average night bout duration as a measure of sleep consolidation (B) compared to the driver (Act-GAL4/+) and the responder (UAS-pdgy) both in gray. (For both A,B, n = 63 for Act-GAL4/+, 64 for UAS-pdgy/+, and 61 for Act-GAL4>UAS-pdgy). (C) Rescue also restored a significant rebound after sleep deprivation. (D) The maximum day bout is increased on the first recovery day after sleep deprivation. For (C,D), n = 29 for Act-GAL4/+, 26 for UAS-pdgy/+, and 30 for Act-GAL4>UAS-pdgy. *p < 0.05.
Since knockdown using FB-GAL4 did not phenocopy the pdgy hypomorphs, we wanted to determine if there was pdgy expression elsewhere in the animal. We took advantage of a set of flies in which an EGFP tag has been integrated into the pdgy gene through random integration. This allows us to visualize where the pdgy gene is expressed endogenously. This technique comes with inherent caveats, but it has also revealed novel expression patterns and generated interesting hypotheses when an antibody is unavailable, as it is with pdgy. Using this technique, we observed faint pdgy expression in the fat bodies. We did observe expression in the Malpighian tubules (Figures 8A–D) as well as throughout the gut with particular localization in the cardia of the gut (Figures 8E–F). In addition, we observed expression in the Malpighian tubules (Figure 8B). Thus, the expression of pdgy may be more widespread in the fly and it is possible these organs could play a role in the response to sleep deprivation.
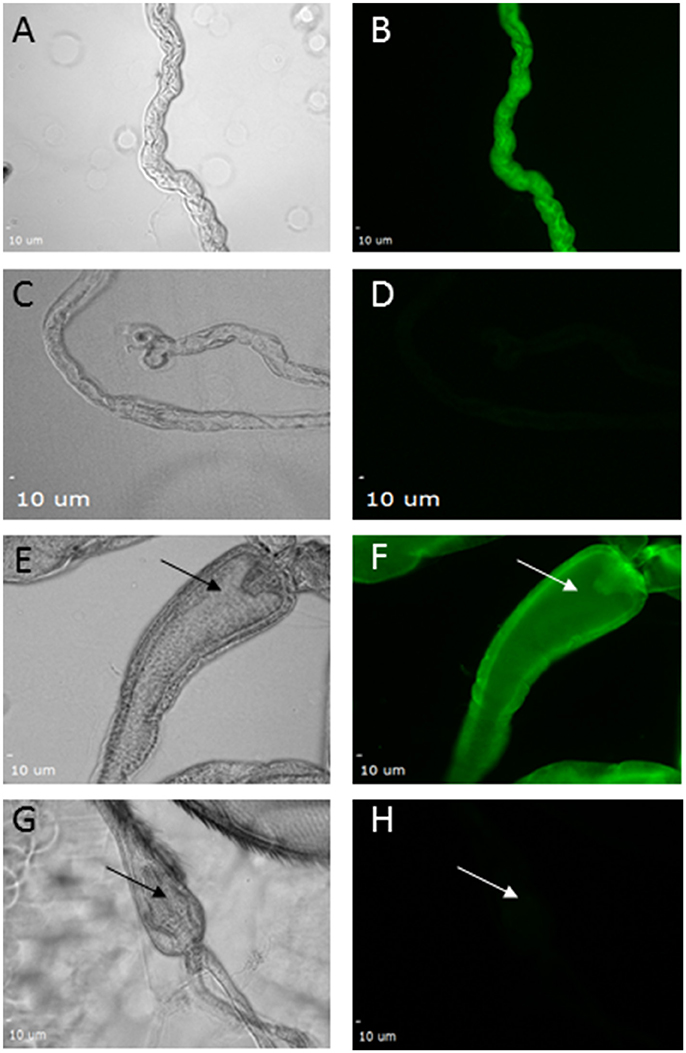
Figure 8. Expression of pdgy was detected in the gut and Malpighian tubules using an enhancer trap line. GFP expression was detected in the Malpighian tubules (A–D) and in the gut (E–H). Bright field images show Malpighian tubules (A,C). Fluorescence expression was detected in (B) when the secondary antibody was excited by wavelengths in the green wavelengths as opposed to a control (D) with a secondary antibody not excited or emitting in this same wavelength. GFP expression was also expressed in the gut. Shown here is the recognizable portion of the gut, the cardia. Expression was seen in (F) but not in (H) with the same conditions as above. Bright field images showing the field (E,G). Arrows point to the cardia in each image.
Discussion
Lipid metabolism has become a critical regulator of sleep and the response to sleep deprivation. The role of metabolism is taking a more prominent role in governing sleep regulation and the response to sleep deprivation (1, 36–38). During sleep there is very small drop in energy expenditure (39), suggesting that there is a substantial energy requirement, even during sleep. In microarray studies, lipid metabolism genes have elevated transcription in the brain in both mammals and flies (11, 40). In humans, the brain is active during sleep, which will consume energy, especially during rapid eye movement (REM) sleep (41, 42). In addition, energy consumption continues throughout the body during sleep, resulting in only a modest decrease in energy consumption compared to waking (39). Energy may play a critical role because both waking activities and the restorative activities of sleep require energy to carry out their functions (1). Thus, molecules and enzymes that control energy metabolism may regulate sleep and wake cycles and disruptions or changes in metabolic handling and the signals, such as endocrine signals, may have an impact on sleep behavior.
We have been evaluating the role that lipid metabolism plays in regulating the response to sleep deprivation (3). Through a microarray that compared waking that induced damage to the organism compared to waking that does not result in performance and health decrements, we identified several lipid metabolism genes, including the ACSs bgm and heimdall (hll) (4). One lipid metabolism gene, the ACS pdgy, was not identified, whereas pdgy (identified as CG9009) had been identified as a sleep regulatory gene (11). It remains unclear if pdgy plays a role in sleep regulation or the response to sleep deprivation.
Here, we have demonstrated that the acyl-CoA synthetase, pdgy, plays a critical role in the response to sleep deprivation. This is consistent with the role that other lipid metabolism genes in the fly (3, 4). For example, mutations in ACSs, such as bgm and hll, result in decreases in the sleep rebound. Knockdown of hll also has a lipid phenotype as lipid levels were lower but retained the distribution in mutant flies as measured by lipidomics (4). In contrast with other ACSs, we also demonstrated that pdgy also reduced the night sleep duration and increased fragmentation, both of which decrease the restorative ability of sleep. Pdgy (or CG9009 as it was previously designated) had been identified as a gene that increased its transcription levels with sleep deprivation. (11). Thus, we provide strong evidence that pdgy is involved in sleep regulation and the response to sleep deprivation.
Pdgy has been identified as an ACS (21), metabolic enzymes that activate fatty acids. These fatty acids can act as signaling molecules, fulfill cellular functions or transferred into the mitochondria for oxidative phosphorylation to generate energy (Figure 9). In that study, the authors hypothesized that the pdgyBG−P had a difficulty utilizing stored triglycerides. The results presented here agree with those conclusions. In the starvation experiments, pdgyBG−P and pdgyEY−P died in about 24 h without food, whereas the majority of the revertants were still alive after 48 h of starvation. In addition, we found a triglyceride phenotype, suggesting that pdgy is involved in lipid metabolism. Where the manuscripts do not match is in the precise metabolic outcomes. In Xu et al., the pdgyBG−P had elevated triglycerides compared to the controls, and in a starvation assay, the pdgyBG−P flies exhibited increased starvation resistance. In our study, we also examined another P-element mutation pdgyEY−P also show a metabolic phenotype similar to the pdgyBG−P. Since these mutations were generated in two different backgrounds, the BG lines were made in an isogenized line while the EY lines were made in a different background that were more conducive to P-element integration (20), these two lines do not carry the same background mutations and gene expression levels. In addition, when the two P-element lines were crossed to each other, there was no complementation, again suggesting that pdgy was responsible for the sleep phenotype. Therefore, the phenotypes observed in the pdgyBG−P and pdgyEY−P are likely due to the pdgy gene and not second-site mutations for 2 different lines.
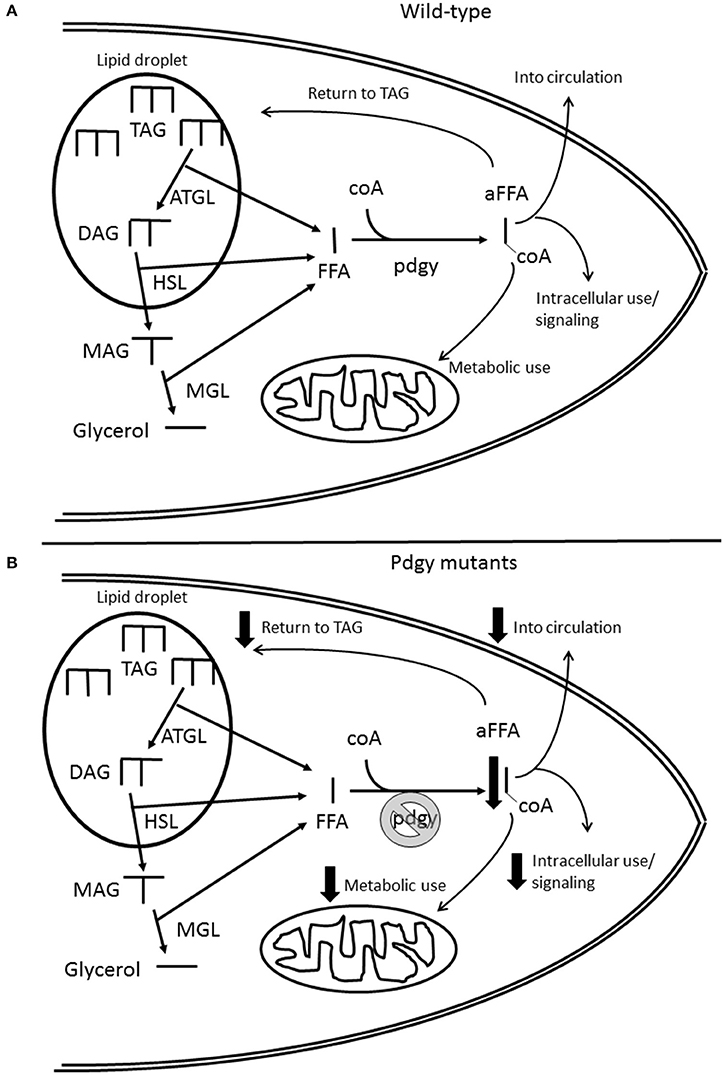
Figure 9. Schematic of the role of pdgy in cellular lipid metabolism. (A) Under normal conditions, the adipose triglyceride lipase (ATGL) releases free fatty acids (FFA) and diacylglycerides (DAG) from the lipid droplet. Another FFA is released from DAG by hormone sensitive lipase (HSL) and another FFA is released from the Monacylglyceride (MAG) by monoglyceride lipase (MGL) leaving glycerol. Pdgy then adds a coA molecule to the FFA to activate it (aFFA). The aFFA then is used for metabolic, may be reinserted into the lipid droplet, cell signaling or protein post-translational modifications, or distribution to other tissues through the circulation. (B) The absence or reduction of pdgy would result in less FFA coverted to aFFA. This mutation would potentially reduce the FFA available to carry out necessary functions within the cell and throughout the organism. Another consequence may be the increase in the levels of FFA within the cell.
On its face these results may seem incompatible, but the relative differences may be due to how each of the mutants were generated. In Xu et al., they outcrossed the BG mutant five times to a white mutant, which in theory exchanges the background around the P-element for the background on the white-eyed flies. The white-eyed mutant is then the comparator genotype. This method standardizes the background and make it easier to compare between different mutants. The white-eyed mutant background is not a preferable background, just a standardized background with all of the mutations and differences that occur in the white background. Our approach was to precisely excise the P-element to restore pdgy transcript levels in the same background that the original mutation was in. This is a common method to restore function in flies. Therefore, our comparator group is different than their comparator group and triglyceride levels would be relatively different. The metabolic system is complex and may have complex interactions. Therefore, in the Xu et al. experiments, the background may allow enough of the lipids to be processed to permit starvation resistance whereas in the pdgy revertant background may not allow lipid utilization that supports survival under starvation conditions.
These results highlight how the difference in genetic context can lead to serious consequences or not so serious consequences. One example of where this has occurred is with the CLOCK mutant mouse. In one case, the impact of the mutation was increased weight gain, lipids dysregulation and cholesterol possibly due to the lack of circadian rhythms (43). Yet when that same mutation was moved into a second background, the opposite phenotype was observed (44). In both cases, circadian rhythms were disrupted, but the consequences were different in the different circumstances. These results may have a clinical implication as shift workers, who often have misaligned circadian rhythms, have an increased likelihood to be obese (45). Interestingly, understanding the differences between the two backgrounds may provide insights into possible treatment options to prevent lipid dysregulation in people with circadian disruptions.
We have gone on to demonstrate using other genetic methods that pdgy plays a role in the response to sleep deprivation. The sleep deprivation phenotype was replicated using both knockdown and rescue indicating that pdgy is necessary and sufficient to play a role in this process. The baseline sleep phenotype was replicated in the rescue experiments. We hypothesize that the knockdown was not strong enough in the appropriate cells to impact the sleep duration and fragmentation phenotypes, but that presents an opportunity to identify cells responsible for these phenotypes. Moreover, the phenotypes were observed in 2 different P-element lines with 2 different backgrounds. In sum, the genetics strongly implicates the pdgy in sleep regulatory pathways.
In the fly disruptions in the metabolic system have been shown to alter the response to sleep deprivation. Presumably, the energy demand is created to carry out the restorative aspects of sleep. At a cellular level, the restorative role that sleep plays will require energy to convert and/or dispose of the damage generated during waking activities. Under most normal circumstances that energy is limited (1). Therefore during sleep deprivation the body cannot satisfy the energy demands of both restoration and waking functions and must leave some things undone. Consistent with this hypothesis, levels of circulating free fatty acids are increased with sleep deprivation, consistent with an increase in available energy with sleep deprivation (10). These incomplete functions may underlie the deleterious consequences of waking. Mutations that decrease triglyceride lipase activity inhibit the response to sleep deprivation. Moreover, a mutation in the gene Lipid storage droplet-2 (LSD-2), results in increased triglyceride lipase activity and a blunted sleep rebound (3). Interestingly, starved flies (both the cycle01 mutant and the w1118 line) and LSD-2 mutant flies were able to learn after the same amount of sleep loss as sleep deprived flies that showed learning impairments. What these two conditions have in common is that they both have elevated levels of lipolysis that could sustain increased energy needs. In other animals there are temporary periods in which sleep is disrupted, but function is maintained. Female orcas and dolphins that have just given birth remain awake with their calves for approximately a month without a subsequent increase in sleep to compensate (46). White-crowned sparrows retain cognitive function during the migratory season, despite substantial sleep deprivation (47). Cognitive function is not retained if the sleep loss is outside of the migratory period. Sandpipers are able to maintain reproductive fitness in the face of sleep deprivation (48). This possibility of increased lipolysis is consistent with all of the natural conditions in which sleep deprivation is not accompanied by impairments from sleep loss. These data support the need for lipid metabolism to support sleep and the response to sleep deprivation though the effects are a complex process and may depend on the genetic and environmental context (49). The pdgy sleep phenotypes observed here are consistent with other lipid metabolism mutants that do not permit a homeostatic response to sleep deprivation.
We evaluated potential consequences of short sleep duration and increased fragmentation sleep in the P-elements. In other situations, short sleep increases cardiovascular effects, including heart rate potentially due to increases in the sympathetic system (50, 51). Moreover, short sleep duration has been associated with shorter lifespan in flies and humans (52–54). The phenotypes observed, elevate heart rate and decreased longevity, were consistent with the consequences of inadequate sleep. These occurred in both P-element mutants, but these flies were not more sensitive to stresses as measured by exposure to hydrogen peroxide. These consequences were not observed in either the RNAi knockdown experiment or the rescue experiments in the pdgyBG−P background. The GAL4/+ in the BG background showed the decreased lifespan, but the UAS/+ did not shown the decrements in lifespan on the same timeline as the rescue experimental line. This despite the fact that sleep duration was decreased and sleep was fragmented in the background lines, but was normal in the rescue line. This result argues against that sleep loss and fragmentation result in a reduced lifespan phenotype. Alternatively, the rescue expression of pdgy may provide protection under these circumstance. Another possibility is that in the P-element background, the lack of proper fatty acid utilization results in the observed phenotypes.
We used an enhancer trap line to localize pdgy within the fly. Enhancer traps integrate an exogenous piece of DNA into the animal's DNA. Once present, expression of the GFP falls under the control of local enhancer and repressors to guide expression. For pdgy, the MI{MIC} line is integrated within the pdgy gene and thus likely reflects endogenous expression. Our results demonstrated strong expression in the cardia and throughout the gut as well as expression in the Malpighian tubules, the kidney-like organ in the fly. We didn't see strong expression in the adipose tissue, though we did not test it under starvation conditions where there is likely a large upregulation of pdgy in the fat bodies (18). In mammals, ACSs have been found in the intestinal epithelial cells (55), supporting the interpretation that MI{MIC} expression mimics endogenous expression. Another possibility is that the MI{MIC} expression responds to only part of the enhancer control. As we did not see complete phenocopy between the RNAi knockdown in the fat body and the P-element hypomorphs. Therefore it is possible that this expression pattern may contribute to sleep regulation.
The fact that lipid metabolic enzyme pdgy can alter sleep regulation has a two broader implications. First, this is further evidence that lipid metabolism plays a critical role in sleep regulation under baseline conditions and after sleep deprivation. Energy management appears to be an obvious route to impact sleep regulation, but this does not rule out a contribution by lipid signaling. Secondly at the physiological level, endocrine signals change the expression of metabolic enzymes. For example, both growth hormone and insulin alter the transcription for lipogenesis (56). Given that there are numerous ACSs that likely have specificity for chain lengths of fatty acids, not all of these changes would have the same impact on sleep regulation as pdgy it will alter the cellular metabolic environment. In fact, starvation alters the metabolic environment and increases wakefulness (3, 57–59). Therefore, when the hormonal environment changes because of dietary conditions or because of disease, one of the consequences could be that the fatty acid mobilization and activation enzymes are changed and that those changes may impact sleep regulation and the response to sleep deprivation. In addition, inadequate sleep can alter the endocrine environment (37, 60–62). Thus, there is a direct connection between the endocrine system to sleep regulation through metabolic enzymes.
Author Contributions
MT designed experiments, ran experiments, analyzed data, and wrote the manuscript. NK, JL, CF, and TH all designed and performed experiments, and analyzed the subsequent data.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all of the students that helped maintain the flies and the other work that keeps a lab working. We would like to thank Yasuko Suzuki and Christie Koch for completing experiments related to his project. We would like to thank the funding sources from the University of Missouri Research Board and the NIH 1R15GM117507 to MT and P40OD018537 to the Bloomington Drosophila Stock Center (BDSC).
References
1. Schmidt MH, The energy allocation function of sleep: a unifying theory of sleep torpor and continuous wakefulness. Neurosci Biobehav Rev. (2014) 47:122–53. doi: 10.1016/j.neubiorev.2014.08.001
2. Miyamoto J, Hasegawa S, Kasubuchi M, Ichimura A, Nakajima A, Kimura I. Nutritional signaling via free fatty acid receptors. Int J Mol Sci. (2016) 17:450. doi: 10.3390/ijms17040450
3. Thimgan MS, Suzuki Y, Seugnet L, Gottschalk L, Shaw PJ. The perilipin homologue, lipid storage droplet 2, regulates sleep homeostasis and prevents learning impairments following sleep loss. PLoS Biol. (2010) 8:e1000466. doi: 10.1371/journal.pbio.1000466
4. Thimgan MS, Seugnet L, Turk J, Shaw PJ. Identification of genes associated with resilience/vulnerability to sleep deprivation and starvation in Drosophila. Sleep (2014) 38:801–14. doi: 10.5665/sleep.4680
5. Gerstner JR, Vanderheyden WM, Shaw PJ, Landry CF, Yin JC. Fatty-acid binding proteins modulate sleep and enhance long-term memory consolidation in Drosophila. PLoS ONE (2011) 6:e15890. doi: 10.1371/journal.pone.0015890
6. Gerstner JR, Perron IJ, Riedy SM, Yoshikawa T, Kadotani H, Owada Y, et al. Normal sleep requires the astrocyte brain-type fatty acid binding protein FABP7. Sci Adv. (2017) 3:e1602663. doi: 10.1126/sciadv.1602663
7. Ollila HM, Utge S, Kronholm E, Aho V, Van Leeuwen W, Silander K, et al. TRIB1 constitutes a molecular link between regulation of sleep and lipid metabolism in humans. Transl Psychiatry (2012) 2:e97. doi: 10.1038/tp.2012.20
8. Tafti M, Petit B, Chollet D, Neidhart E, de Bilbao F, Kiss JZ, et al. Deficiency in short-chain fatty acid beta-oxidation affects theta oscillations during sleep. Nat Genetics (2003) 34:320–5. doi: 10.1038/ng1174
9. Weljie AM, Meerlo P, Goel N, Sengupta A, Kayser MS, et al. Oxalic acid and diacylglycerol 36:3 are cross-species markers of sleep debt. Proc Natl Acad Sci USA. (2015) 112:2569–74. doi: 10.1073/pnas.1417432112
10. Broussard JL, Chapotot F, Abraham V, Day A, Delebecque F, Whitmore HRet al. Sleep restriction increases free fatty acids in healthy men. Diabetologia (2015) 58:791–8. doi: 10.1007/s00125-015-3500-4
11. Cirelli C, LaVaute TM, Tononi G. Sleep and wakefulness modulate gene expression in Drosophila. J Neurochem. (2005) 94:1411–9. doi: 10.1111/j.1471-4159.2005.03291.x
12. Harbison ST, Sehgal A. Quantitative genetic analysis of sleep in Drosophila melanogaster. Genetics (2008) 178:2341–60. doi: 10.1534/genetics.107.081232
13. Grevengoed TJ, Klett EL, Coleman RA. Acyl-CoA metabolism and partitioning. Ann Rev Nutr. (2014) 34:1–30. doi: 10.1146/annurev-nutr-071813-105541
14. Ellis JM, Li LO, Wu PC, Koves TR, Ilkayeva O, Stevens RD, et al. Adipose acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell Metab. (2010) 12:53–64. doi: 10.1016/j.cmet.2010.05.012
15. Bu SY, Mashek DG. Hepatic long-chain acyl-CoA synthetase 5 mediates fatty acid channeling between anabolic and catabolic pathways. J Lipid Res. (2010) 51:3270–80. doi: 10.1194/jlr.M009407
16. Meller N, Morgan ME, Wong WP, Altemus JB, Sehayek E. Targeting of Acyl-CoA synthetase 5 decreases jejunal fatty acid activation with no effect on dietary long-chain fatty acid absorption. Lipids Health Dis. (2013) 12:88. doi: 10.1186/1476-511X-12-88
17. Min KT, Benzer S. Preventing neurodegeneration in the Drosophila mutant bubblegum. Science (1999) 284:1985–8. doi: 10.1126/science.284.5422.1985
18. Xu X, Gopalacharyulu P, Seppanen-Laakso T, Ruskeepaa AL, Aye CC, Carson BP, et al. Insulin signaling regulates fatty acid catabolism at the level of CoA activation. PLoS Genet. (2012) 8:e1002478. doi: 10.1371/journal.pgen.1002478
19. Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature (2007) 448:151–6. doi: 10.1038/nature05954
20. Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G, et al. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics (2004) 167:761–81. doi: 10.1534/genetics.104.026427
21. Teleman AA, Ratzenbock I, Oldham S. Drosophila: a model for understanding obesity and diabetic complications. Exp Clin Endocrinol Diabetes (2012) 120:184–5. doi: 10.1055/s-0032-1304566
22. Nagarkar-Jaiswal S, Lee PT, Campbell ME, Chen K, Anguiano-Zarate S, Gutierrez MC, et al. A library of MiMICs allows tagging of genes and reversible, spatial and temporal knockdown of proteins in Drosophila. eLife (2015) 4:e05338. doi: 10.7554/eLife.05338
23. Shaw PJ, Cirelli C, Greenspan RJ, Tononi G. Correlates of sleep and waking in Drosophila melanogaster. Science (2000) 287:1834–7. doi: 10.1126/science.287.5459.1834
24. Andretic R, Shaw PJ. Essentials of sleep recordings in Drosophila: moving beyond sleep time. Methods Enzymol. (2005) 393:759–72. doi: 10.1016/S0076-6879(05)93040-1
25. Shaw PJ, Tononi G, Greenspan RJ, Robinson DF. Stress response genes protect against lethal effects of sleep deprivation in Drosophila. Nature (2002) 417:287–91. doi: 10.1038/417287a
26. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (2001) 25:402–8. doi: 10.1006/meth.2001.1262
27. Runko AP, Griswold AJ, Min KT. Overexpression of frataxin in the mitochondria increases resistance to oxidative stress and extends lifespan in Drosophila. FEBS Lett. (2008) 582:715–9. doi: 10.1016/j.febslet.2008.01.046
28. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. (1959) 37:911–7. doi: 10.1139/y59-099
29. Neckameyer WS, Matsuo H. Distinct neural circuits reflect sex, sexual maturity, and reproductive status in response to stress in Drosophila melanogaster. Neuroscience (2008) 156:841–56. doi: 10.1016/j.neuroscience.2008.08.020
30. Wessells RJ, Bodmer R. Screening assays for heart function mutants in Drosophila. Biotechniques (2004) 37:58–60, 62, 64 passim. doi: 10.2144/04371ST01
31. Paternostro G, Vignola C, Bartsch DU, Omens JH, McCulloch AD, Reed JC. Age-associated cardiac dysfunction in Drosophila melanogaster. Circ Res. (2001) 88:1053–8. doi: 10.1161/hh1001.090857
32. Donlea JM, Thimgan MS, Suzuki Y, Gottschalk L, Shaw PJ. Inducing sleep by remote control facilitates memory consolidation in Drosophila. Science (2011) 332:1571–6. doi: 10.1126/science.1202249
33. Sohal RS, Effect of hydrogen peroxide administration on life span, superoxide dismutase catalase and glutathione in the adult housefly, Musca domestica. Exp Gerontol. (1988) 23:211–6. doi: 10.1016/0531-5565(88)90008-3
34. Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development (1993) 118:401–15.
35. Gronke S, Mildner A, Fellert S, Tennagels N, Petry S, Muller G, et al. Brummer lipase is an evolutionary conserved fat storage regulator in Drosophila. Cell Metab. (2005) 1:323–30. doi: 10.1016/j.cmet.2005.04.003
36. Thimgan MS, Schilli K. The role of metabolic genes in sleep regulation. In: Shaw PJ, Tafti M, and Thorpy M editors. The Genetic Basis of Sleep and Sleep Disorders. Cambridge: Cambridge University Press (2013). pp. 91–103.
37. Markwald RR, Melanson EL, Smith MR, Higgins J, Perreault L, Eckel RH, et al. Impact of insufficient sleep on total daily energy expenditure, food intake, and weight gain. Proc Natl Acad Sci USA. (2013) 110:5695–700. doi: 10.1073/pnas.1216951110
38. Benington JH, Heller HC. Restoration of brain energy metabolism as the function of sleep. Prog Neurobiol. (1995) 45:347–60. doi: 10.1016/0301-0082(94)00057-O
39. Jung CM, Melanson EL, Frydendall EJ, Perreault L, Eckel RH, Wright KP. Energy expenditure during sleep, sleep deprivation and sleep following sleep deprivation in adult humans. J Physiol. (2011) 589:235–44. doi: 10.1113/jphysiol.2010.197517
40. Cirelli C, Gutierrez CM, Tononi G. Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron (2004) 41:35–43. doi: 10.1016/S0896-6273(03)00814-6
41. Kaufmann C, Wehrle R, Wetter TC, Holsboer F, Auer DP, Pollmacher T, et al. Brain activation and hypothalamic functional connectivity during human non-rapid eye movement sleep: an EEG/fMRI study. Brain (2006) 129:655–67. doi: 10.1093/brain/awh686
42. Nofzinger EA, Neuroimaging of sleep and sleep disorders. Curr Neurol Neurosci Rep. (2006) 6:149–55. doi: 10.1007/s11910-996-0038-3
43. Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science (2005) 308:1043–5. doi: 10.1126/science.1108750
44. Oishi K, Atsumi G, Sugiyama S, Kodomari I, Kasamatsu M, Machida K, et al. Disrupted fat absorption attenuates obesity induced by a high-fat diet in Clock mutant mice. FEBS Lett. (2006) 580:127–30. doi: 10.1016/j.febslet.2005.11.063
45. Sun M, Feng W, Wang F, Li P, Li Z, Li M, et al. Meta-analysis on shift work and risks of specific obesity types. Obes Rev. (2018) 19:28–40. doi: 10.1111/obr.12621
46. Lyamin O, Pryaslova J, Lance V, Siegel J. Animal behaviour: continuous activity in cetaceans after birth. Nature (2005) 435:1177. doi: 10.1038/4351177a
47. Rattenborg NC, Mandt BH, Obermeyer WH, Winsauer PJ, Huber R, Wikelski M, et al. Migratory sleeplessness in the white-crowned sparrow (Zonotrichia leucophrys gambelii). PLoS Biol. (2004) 2:E212. doi: 10.1371/journal.pbio.0020212
48. Lesku JA, Rattenborg NC, Valcu M, Vyssotski AL, Kuhn S, Kuemmeth F, et al. Adaptive sleep loss in polygynous pectoral sandpipers. Science (2012) 337:1654–8. doi: 10.1126/science.1220939
49. St-Onge MP. The role of sleep duration in the regulation of energy balance: effects on energy intakes and expenditure. J Clin Sleep Med. (2013) 9:73–80. doi: 10.5664/jcsm.2348
50. Tobaldini E, Pecis M, Montano N. Effects of acute and chronic sleep deprivation on cardiovascular regulation. Arch Ital Biol. (2014) 152:103–10. doi: 10.12871/000298292014235
51. Virtanen I, Kalleinen N, Urrila AS, Leppanen C, Polo-Kantola P. Cardiac autonomic changes after 40 hours of total sleep deprivation in women. Sleep Med. (2015) 16:250–7. doi: 10.1016/j.sleep.2014.10.012
52. Cirelli C, Bushey D, Hill S, Huber R, Kreber R, Ganetzky B, et al. Reduced sleep in Drosophila Shaker mutants. Nature (2005) 434:1087–92. doi: 10.1038/nature03486
53. Seugnet L, Suzuki Y, Thimgan M, Donlea J, Gimbel SI, Gottschalk L, et al. Identifying sleep regulatory genes using a Drosophila model of insomnia. J Neurosci. (2009) 29:7148–57. doi: 10.1523/JNEUROSCI.5629-08.2009
54. Kripke DF, Garfinkel L, Wingard DL, Klauber MR, Marler MR. Mortality associated with sleep duration and insomnia. Arch Gen Psychiatry (2002) 59:131–6. doi: 10.1001/archpsyc.59.2.131
55. Oikawa E, Iijima H, Suzuki T, Sasano H, Sato H, Kamataki A, et al. A novel acyl-CoA synthetase, ACS5, expressed in intestinal epithelial cells and proliferating preadipocytes. J Biochem. (1998) 124:679–85. doi: 10.1093/oxfordjournals.jbchem.a022165
56. Kersten S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep. (2001) 2:282–6. doi: 10.1093/embo-reports/kve071
57. Keene AC, Duboue ER, McDonald DM, Dus M, Suh GS, Waddell S, et al. Clock and cycle limit starvation-induced sleep loss in Drosophila. Curr Biol. (2010) 20:1209–15. doi: 10.1016/j.cub.2010.05.029
58. Borbely AA. Sleep in the rat during food deprivation and subsequent restitution of food. Brain Res. (1977) 124:457–71. doi: 10.1016/0006-8993(77)90947-7
59. Danguir J, Nicolaidis S. Dependence of sleep on nutrients' availability. Physiol Behav. (1979) 22:735–40. doi: 10.1016/0031-9384(79)90240-3
60. Sassin JF, Parker DC, Mace JW, Gotlin RW, Johnson LC, Rossman LG. Human growth hormone release: relation to slow-wave sleep and sleep-walking cycles. Science (1969) 165:513–5. doi: 10.1126/science.165.3892.513
61. Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet (1999) 354:1435–9. doi: 10.1016/S0140-6736(99)01376-8
Keywords: acyl-CoA synthetase, sleep deprivation, sleep regulation, lipid metabolism, sleep fragmentation, Drosophila, lifespan
Citation: Thimgan MS, Kress N, Lisse J, Fiebelman C and Hilderbrand T (2018) The acyl-CoA Synthetase, pudgy, Promotes Sleep and Is Required for the Homeostatic Response to Sleep Deprivation. Front. Endocrinol. 9:464. doi: 10.3389/fendo.2018.00464
Received: 27 April 2018; Accepted: 27 July 2018;
Published: 21 August 2018.
Edited by:
Sushmita Pamidi, McGill University, CanadaReviewed by:
Tamas Kozicz, Mayo Clinic, United StatesLicio A. Velloso, Universidade Estadual de Campinas, Brazil
Copyright © 2018 Thimgan, Kress, Lisse, Fiebelman and Hilderbrand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew S. Thimgan, dGhpbWdhbkBtc3QuZWR1