 Giulia Biondani
Giulia Biondani Jean-François Peyron
Jean-François Peyron
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 10 August 2018
Sec. Cellular Endocrinology
Volume 9 - 2018 | https://doi.org/10.3389/fendo.2018.00446
This article is part of the Research TopicMetformin: Beyond DiabetesView all 12 articles
Metformin, a widely used anti-diabetic molecule, has attracted a strong interest in the last 10 years as a possible new anti-cancer molecule. Metformin acts by interfering with mitochondrial respiration, leading to an activation of the AMPK tumor-suppressive pathway to promote catabolic-energy saving reactions and block anabolic ones that are associated with abnormal cell proliferation. Metformin also acts at the organism level. In type 2 diabetes patients, metformin reduces hyperglycemia and increases insulin sensitivity by enhancing insulin-stimulated glucose uptake in muscles, liver, and adipose tissue and by reducing glucose output by the liver. Lowering insulin and insulin-like growth factor 1 (IGF-1) levels that stimulate cancer growth could be important features of metformin's mode of action. Despite continuous progress in treatments with the use of targeted therapies and now immunotherapies, acute leukemias are still of very poor prognosis for relapse patients, demonstrating an important need for new treatments deriving from the identification of their pathological supportive mechanisms. In the last decade, it has been realized that if cancer cells modify and reprogram their metabolism to feed their intense biochemical needs associated with their runaway proliferation, they develop metabolic addictions that could represent attractive targets for new therapeutic strategies that intend to starve and kill cancer cells. This Mini Review explores the anti-leukemic potential of metformin and its mode of action on leukemia metabolism.
Metformin is an active biguanide derivative extracted from the French Lilac (Galega officinalis), a plant discovered during the Middle Age for its healing effects on the diabetic condition. Metformin/Glucophage® was first prescribed in Europe in 1979, then in the United States by 1994 and is now the first-line treatment for type 2 diabetes (T2D) as more than 120 million patients are treated worldwide (1).
In 2001 metformin appeared on the cancer scene when it was observed that in hepatocytes it stimulated the AMP-activated serine threonine protein kinase (AMPK) (2), a sensor of the energetic cellular status and an important tumor suppressor pathway (3, 4).
This discovery prompted clinicians and researchers to measure cancer frequency in T2D patients under metformin. It was first shown in 2005 that metformin significantly reduced cancer incidence in a cohort of 983 T2D patients (5). Other studies confirmed that metformin was associated with a lower risk of cancer in treated diabetic patients (6–8).
These striking results led the renowned cancer researcher Lewis Cantley to consider that “Metformin may have saved more people from cancer deaths than any drug in history” (9).
Numerous investigations worldwide rapidly demonstrated direct anti-cancer effects of metformin on various models (10–12). In vitro, metformin exhibits a strong anti-proliferative action on cancer cell lines derived from breast, colon, ovaries, pancreas, lung, and prostate (13–15). These results were strengthened by pre-clinical in vivo experiments using xenografts or transgenic mice and chemically-induced cancers. As an example, in a tobacco-induced lung carcinogenesis mouse model, metformin decreases tumor burden by 72% (16). Evidences show that metformin can act through an AMPK dependent (17, 18) or independent (19) way. However, despite metformin is widely used in clinic, its molecular mechanism of action is still under debate.
From the different reports it appears that metformin exerts a double action at both organism and molecular levels.
Within the organism, metformin has an anti-hyperglycemic action but as it does not decrease insulin secretion there is no risk of hypoglycemia in normal subjects (20). In muscles, metformin reduces hyperglycemia through different mechanisms: by enhancing insulin-stimulated glucose uptake and reducing hepatic glucose output (21). It lowers the production of glucose by the liver, and increases glucose utilization by muscles and adipocytes. This results in a decreased insulinemia and an amelioration of insulin sensitivity, likely counteracting the increased glucose uptake by insulin, which facilitates tumor initiation and progression (22). It was thus envisioned that the anti-cancer effects of metformin could be due to its ability to reduce circulating levels of glucose and consequently of insulin and insulin-like growth factor 1(IGF-1) that are suspected to feed different cancers expressing the receptors for these growth factors on their surface (23–26).
Diabetes, in particular T2D, and obesity are clearly associated with an increased risk to develop various cancers (27). However, no increased incidence was observed for hematologic malignancies (28) suggesting at first that the systemic effects of metformin may not apply to leukemia. Nevertheless, a metabolic syndrome with insulin resistance has been reported in leukemic patients exposed to high dose glucocorticoids (29). This could favor a therapy-induced obesity with hyperinsulinemia that supports leukemic cell survival and worsens patient's outcome. Insulin and IGF-1 receptors were found expressed on acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) (30, 31) and insulin stimulates in vitro the proliferation of ALL cell lines and primary cells that were sensitive to metformin (32). At the molecular level, an IGF1-IGF-1R autocrine loop is responsible for activation of a leukemia-supportive PI3K/Akt/mTOR pathway (33). Pharmacological interference with the insulin receptor and/or IGF1R autocrine loops affects leukemic proliferation (34) and potentiates the apoptotic action of etoposide (31). Similarly, targeting IGF-1R interferes with the growth of chronic lymphocytic leukemia (CLL) (35).
If insulin/IGF-1 do not appear to be strong oncogenic drivers for acute leukemias, they are likely trophic factors, supporting the rational use of metformin to decrease hyperinsulinemia and to indirectly affect leukemic cells.
As shown in Figure 1, metformine inhibits oxidative respiration by acting on the complex I of the mitochondrial respiratory chain (17, 18), leading to a drop in ATP synthesis, tilting the AMP/ATP balance toward AMP, with the consequent stimulation of AMPK. It is well known that the LKB1/AMPK pathway also regulates the protein synthesis rate through the control of mTOR. Activated AMPK stimulates tuberous sclerosis complex 1/2 (TSC1/2) through phosphorylation and its GTPase-activating protein (GAP) function toward the small G-protein Rheb (Ras homolog enriched in brain), thus determining the switching off of Rheb and resulting in the inhibition of mTOR activity (36–38). AMPK activation requires binding and phosphorylation by the tumor suppressor liver kinase B1 (LKB1) (39, 40). Therefore, the absence of LKB1 impedes an AMPK-negative regulation of cancerous cell metabolism.
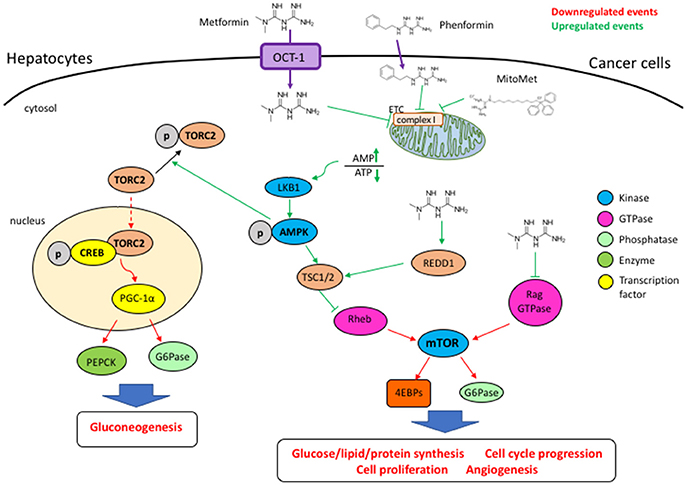
Figure 1. The entrance of metformin is mediated by the OCT1 transporter. By blocking the mitochondrial respiratory chain complex I, metformin and phenformin determine an increase of the AMP/ATP ratio, a condition that activates AMPK through phosphorylation by LKB1. Metformin-activated AMPK counteracts the activation of the mTORC1 complex, impairing cell cycle progression and proliferation, angiogenesis, as well as lipid and protein syntheses. Metformin can induce REDD1 and inhibit Rag GTPases, thus leading to the blocking of mTORC1 through an AMPK-independent way. In an AMPK-dependent way metformin promotes TORC2 phosphorylation and blocks its nuclear translocation, its association with phospho-CREB, impairing the transcription of genes such as PGC-1α, G6Pase, and PEPCK whose products promotes gluconeogenesis.
The AMPK pathway is a major repressor of the mTOR pathway that uses energy and nutrients to stimulate ATP-consuming anabolic reactions, favoring growth and proliferation (41). Activation of the PI3K/Akt pathway, a major upstream activator of mTOR, is restrained by the lipid phosphatase and tumor suppressor PTEN (phosphatase and tensin homolog), frequently inactivated in cancer (42, 43). Defects in control by PTEN lead to a constitutive activation of the Akt pathway that is involved in the etiology of various pathological conditions such as diabetes, aging, and cancer (44).
The mTOR serine/threonine kinase is the active central component of the mTORC1 and mTORC2 cellular complexes that function to coordinately stimulate cell growth (44). mTORC1 is crucial for the synthesis of proteins, lipids, and nucleic acids while mTORC2 phosphorylates Akt to stimulate proliferation and survival (45, 46). Furthermore, AMPK promotes phosphorylation of TORC2 (transducer of regulated CREB activity 2) to block its nuclear translocation and association with phospho-CREB (CRE binding protein), thus impairing the transcription of genes involved in gluconeogenesis such as peroxisome-proliferator-activated receptor-γ co-activator-1α (PGC-1α), glucose-6-phosphatase (G6Pase), and phosphoenolpyruvate carboxykinase (PEPCK) (38, 47).
Through AMPK stimulation, metformin interferes with mTORC1 activation. In addition, AMPK inhibits ATP formation through fatty acid oxidation (FAO) (48) and stimulates glycolysis by phosphorylation-induced activation of phosphofructo-2-kinase (PFK2) (49). Also, AMPK modulates gene expression for important metabolic enzymes (50) and induces a metabolic cell cycle checkpoint through p53 activation (51). Therefore, AMPK agonists as well as indirect activators such as metformin can be envisioned as promising anti-cancer compounds.
Not all actions of metformin are mediated by AMPK (19). Metformin together with hexokinase 2 (HK2) depletion synergistically interferes with mTORC1 activation through the induction of the mTORC1 inhibitor REDD1 (regulated in development and DNA damage) in hepatocellular carcinoma cells, even upon depletion of AMPKα1 and AMPKα2 (52). Repression of G6Pase and of hepatic glucose production by metformin still occurs in both AMPK and LKB1-deficient hepatocytes (53).
In AML cells metformin can block proliferation at either G0/G1 or S-G2/M, depending on the cell line analyzed. Furthermore, by using a siRNA of AMPKα1/2, Scotland and colleagues showed that metformin-induced cell death is not dependent by AMPK activation in AML cells (54). In prostate cancer cell lines, metformin has AMPK-independent anti-proliferative effects through induction of REDD1 (55). In breast cancer cells metformin interferes with purine/pyrimidine and glutathione synthesis upstream of AMPK (56).
Metformin and resveratrol synergistically block pancreatic cancer cell proliferation in vitro and in vivo by inhibiting vascular endothelial growth factor B (VEGF-B) signaling pathway (57).
Leukemia represent 2.8% of all cancers and 3.4% of deaths from cancer worldwide, with 351,000 new cases/year. Leukemia results from the transformation of hematopoietic stem-progenitor cells (HSPCs). Acute lymphoid or myeloid leukemia (ALL/AML) show an intense proliferation of immature leukemic blasts arrested at various stages of differentiation (58, 59). Despite important progress in treatments, the 5-year survival for T-ALL is 70–75% for children and only 35–40% for adults (59). New therapeutic strategies should therefore be identified to eradicate leukemia.
Targeting the energetic metabolism of cancer cells is emerging as an attractive option (60) as cancer/leukemic cells reprogram their metabolism to fulfill their intense metabolic needs. Consequently, they develop metabolic addictions that can be used as new targeting options to starve and kill them (60).
The control of the PI3K/Akt/mTOR axis by PTEN is fundamental for the self-renewal of HSCs and PTEN knock-out generates leukemia in mice (61). A common biochemical feature among acute leukemia is the abnormal and constitutive activation of the PI3K/Akt/mTOR pathway (62, 63). Separated or combined pharmacological targeting of PI3K, Akt, or mTOR triggers leukemic cell death in AML and ALL (64, 65). PI-103, a dual inhibitor of PI3K and mTOR displays anti-leukemic properties (66). Unfortunately, the immunosuppressant rapalogs (Temsirolimus, Everolimus) that target mTORC1 activation, showed a limited anti-cancer activity as they failed to inhibit mTORC2 activity and reactivated the tumor supportive Akt pathway (41, 67, 68). Torkinibs, ATP-competitive inhibitors of the mTOR kinase activity, target both mTOR complexes (69, 70) and have already displayed promising anti-cancer properties on leukemia models (71–73).
Metformin represents an interesting opportunity to target leukemia through inhibition of constitutive mTOR, a pathological hallmark in leukemogenesis. In 2010 metformin was shown to interfere with AML proliferation and clonogenic activity and to induce apoptosis in human immortalized cell lines and primary samples while it did not affect normal CD34+ HSCs (37). Metformin, after blocking mTORC1 activation, prevents initiation of translation, in particular of c-myc, cyclin D1, and Bcl-xL that are crucial for cancer proliferation (37). Metformin induces apoptosis of leukemic megakaryoblasts from acute megakaryoblastic leukemia (AMKL) which is a rare type of leukemia with poor prognosis (74). Metformin could be an option for the DNA repair defective Fanconi Anemia pre-leukemic disorder as it is toxic after inhibiting the respiratory chain (75).
In T-ALL cells, metformin stimulates AMPK to inhibit mTOR and trigger an autophagic response that precedes apoptosis. By affecting protein synthesis, metformin strongly decreases c-myc and Bcl-xL levels (76). This apoptotic action of metformin in T-ALL also involves an AMPK-dependent activation of the ER stress/unfolded protein response (UPR) (77). In this model, metformin induces a compensatory, anti-apoptotic activation of Akt and of PIM-2, that could be reversed by inhibitors, synergizing with metformin for cell death induction.
Genetic defects in the PTEN tumor suppressor gene are leading to the constitutive activation of the PI3K/Akt/mTOR pathway in T-ALL (78) and are associated with a poor outcome in pediatric T-ALL (79). Tumor cells from a mouse T-ALL model generated by the T-cell specific deletion of PTEN display a constitutive activation of PI3K/Akt/mTOR that could be inhibited by metformin through AMPK activation and by torkinibs (80). Deletion of LKB1 in mice with a PTEN+/- background increases lymphoma incidence that appeared with a shorter latency and were sensitive to metformin (81).
Metformin counteracted the activation of the PI3K/Akt/mTOR pathway triggered by several oncogenes such as the Bcr-abl fusion tyrosine kinase in CML and Phi+ T-ALL and B-ALL and the Tax oncoprotein in HTLV-1-induced ATL (human T-lymphotropic virus type 1-induced adult-T-cell leukemia). Through AMPK activation, metformin suppresses proliferation and clonogenic activity of various CML lines, including those expressing the imatinib-resistant T315I Bcr-abl mutant (82). In ATL, LKB1/AMPK activation by metformin inhibits leukemic proliferation by reducing Tax expression (83). In CLL, metformin prevents cell cycle entry of leukemic cells in vitro after engagement of a CD40-CD40L proliferative stimulus (84). CLL cells that are sensitive to the tyrosine kinase inhibitor dasatinib appears to be selectively killed by metformin (85).
Leukemic stem cells (LSCs) are the rare cells at the origin of leukemia and also of relapse because of their intrinsic mechanisms of resistance to chemotherapies (86). Interestingly, in T-ALL metformin targets the Hoescht 33342low side population and the CD34+CD7-CD4- subset that are known to be enriched in LSCs (76). Similarly, cancer stem cells (CSCs) in different solid tumors appears to be highly sensitive to low doses of metformin (87, 88).
The eradication of cancer will require the combination of multiple therapeutic strategies in a personalized manner. Anti-cancer clinical protocols and drug cocktails would need to be adapted to the specific genetic defects of each patient. Nevertheless, targeting a common dysregulated cellular function such as the reprogrammed cancer metabolism with a metabolic disruptor such as metformin is likely to be an interesting adjuvant approach.
Metformin has already been associated to several classical chemotherapeutic drugs with promising results. Metformin shows additive effects with anthracyclines (doxorubicin, daunorubicin) to reduce growth and survival of lymphoma cells (80), T-ALL cells (89), and ALL (32). The use of metformin could help to reduce the dose of doxorubicin necessary to prolong remission (88) and consequently to reduce cardiac toxicity of anthracyclines.
In T-ALLs metformin synergizes with dexamethasone, the glucocorticoid used as first line treatment for acute leukemias (80), and also potentiates the effect of the microtubule-disrupting agent vincristine (90) and of the topoisomerase II inhibitor etoposide (32).
All-trans retinoic acid (ATRA) is used in acute promyeloid leukemia (APL) to overcome the differentiation block induced by the PML-RAR fusion oncoprotein. By inducing PML-RAR degradation, metformin synergized with ATRA to induce APL cell death (91).
Triggering leukemia apoptosis at the mitochondrial level with the bcl-2 inhibitor ABT-737 is a promising therapy which was shown to be enhanced by metformin-induced mitochondrial membrane depolarization (92). The anti-leukemic activity of the Flt3 inhibitor sorafenib, that was developed to target poor prognosis-ITD Flt3 AML cells, could be enhanced by metformin, thus inducing a strong decrease in the expression of several components of the mTOR pathway (93).
In several studies metformin displays strong potentiating effects when combined with molecules affecting metabolism, in particular glycolysis, such as ritonavir in multiple myeloma (MM) (94) and CLL (95). In silico, metformin was predicted to combine with an inhibitor of the Glut4 glucose transporter to affect MM (96).
Disruption of the mitochondrial respiratory complex I by metformin is followed by a compensatory upregulation of glucose uptake and glycolysis (97, 98). As a consequence metformin was shown to synergize with the non-metabolizable glucose analog and hexokinase inhibitor 2-deoxy-glucose (2-DG) in T-ALL (80) and in CML (99) and with the glycolysis inhibitor sodium dichloroacetate (DCA) in B-CLL (100). Similar effects have been observed in Flt3-positive AML when metformin was associated with the metabolic inhibitor 6-BT (101). Cell death induction of MM cells after disrupting protein homeostasis with the proteasome inhibitor bortezomib can be enhanced by metformin, preventing a protective autophagic response (102). ALL cells display a metabolic dependency on asparagine that can be targeted with L-asparaginase, an effect further amplified by metformin (80).
In the 1920s, Otto Warburg and colleagues observed for the first time that cancer tissues were taking up enormous amounts of glucose compared to the surrounding tissue. Later, in 1956 Otto Warburg proposed that cancer cells have defective mitochondria because they utilize glucose through aerobic glycolysis, unlike normal cells which use glucose to produce ATP through oxidative phosphorylation (OXPHOS) in mitochondria (103). It was realized a couple of years ago that despite a far less efficient ATP production, this metabolic reprogramming represents an adaptation to optimize the utilization of nutrients to produce the biomass necessary for the generation of new proliferating cancer cells (104–106). Nevertheless, cancer and leukemic cells need active mitochondria for their fitness. Targeting the mitochondria respiratory function by inhibiting the electron transport chain (80), mitochondrial translation (107), or the FAO (108), are all new efficient approaches to kill leukemic cells. Recently, a mitochondrial transfer from stromal cells toward leukemic AML cells provided them with a survival advantage toward chemotherapy (109, 110). An important metabolic plasticity appears to take place as the environment of leukemic cells is changing (111). AML cells can become more sensitive to metformin when cultured in low-glucose medium or after downregulating glycolysis with 2-DG or an Akt inhibitor (54). Similarly, pharmacological approaches to inhibit OXPHOS markedly enhanced the anti-leukemic effects of cytarabine (112).
There are at least two important questions pending about the use of metformin in cancer.
First, will the ability of metformin to control hyperinsulinemia and glycemia in T2D patients stand for non-diabetic people? The 306 registered clinical trials on metformin and cancer will provide important answers. In relation to this review, metformin is tested (NCT01324180) in relapsed childhood ALL in association with vincristine, dexamethasone, doxorubicin, and PEG-asparaginase that are classical drugs for these leukemias. Metformin will be evaluated as a monotherapy for untreated or relapsed CLL patients in a phase 2 pilot study (NCT01750567).
Second, the doses of metformin that are efficient in vitro on cancer models are in the mM range, far above those obtained in treated T2D patients (6–30 μM) (113, 114). The cellular entry of the highly hydrophilic metformin is limited by expression of the organic cation transporter (OCT) (115). Interesting areas of research aim at facilitating metformin uptake through specific encapsulation, use of nanocarriers, or after chemical modifications. Coupling a mitochondrial vector to metformin (MitoMet) increases its ability to interfere with OXPHOS and consequently its efficiency to affect proliferation and to trigger ROS-dependent apoptosis in pancreatic cancer in vitro and in vivo, without affecting normal fibroblasts (116).
Phenformin, a hydrophobic metformin derivative is more active than metformin (81, 117) but was rapidly withdrawn from the market in the late 1970s because of numerous deadly cases of lactic acidosis. We now believe that phenformin could be worth testing as an adjuvant molecule for cancer patients with a monitoring of lactic acidosis. A clinical trial (NCT03026517) will evaluate phenformin in combination with dabrafenib and trametinib for patients with BRAF-mutated melanoma.
Recently Higurashi et al demonstrated the important role of metformin in chemoprevention of colorectal cancer (118). Other clinical trials are ongoing for coloncancer and other tumor types (e.g., NCT03047837; NCT02581137; NCT01312467; NCT01579812; NCT02581137).
Many studies support to use metformin and derivatives like phenformin as global adjuvants for classical anti-leukemic drugs. Improving metformin entry and access to its cellular target(s) through chemical modifications or the use of nanocarriers could be important means to increase the potential of this interesting anti-metabolic molecule.
GB and J-FP equally wrote the article. GB did the illustration work.
This article is supported by an institutional grant from Inserm. GB is supported by a postdoctoral fellowship from INCa.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, though no other collaboration, with the authors at time of review.
1. Bailey CJ, Turner RC. Metformin. N Engl J Med. (1996) 334:574–9. doi: 10.1056/NEJM199602293340906
2. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. (2001) 108:1167–74. doi: 10.1172/JCI13505
3. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer (2009) 9:563–75. doi: 10.1038/nrc2676
4. Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol. (2015) 33:1–7. doi: 10.1016/j.ceb.2014.09.004
5. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ (2005) 330:1304–5. doi: 10.1136/bmj.38415.708634.F7
6. Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, et al. Diabetes and cancer: a consensus report. CA Cancer J Clin. (2010) 60:207–21. doi: 10.3322/caac.20078
7. Zhang P, Li H, Tan X, Chen L, Wang S. Association of metformin use with cancer incidence and mortality: a meta-analysis. Cancer Epidemiol. (2013) 37:207–18. doi: 10.1016/j.canep.2012.12.009
8. Singh S, Singh H, Singh PP, Murad MH, Limburg PJ. Antidiabetic medications and the risk of colorectal cancer in patients with diabetes mellitus: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. (2013) 22:2258–68. doi: 10.1158/1055-9965.EPI-13-0429
9. Taubes G. Cancer research. Cancer prevention with a diabetes pill. Science (2012) 335: 29. doi: 10.1126/science.335.6064.29
10. Ben Sahra I, Le Marchand-Brustel Y, Tanti JF, Bost F. Metformin in cancer therapy: a new perspective for an old antidiabetic drug Mol Cancer Ther. (2010) 9:1092–9. doi: 10.1158/1535-7163.MCT-09-1186
11. Dowling RJ, Niraula S, Stambolic V, Goodwin PJ. Metformin in cancer: translational challenges. J Mol Endocrinol. (2012) 48:R31–43. doi: 10.1530/JME-12-0007
12. Pollak M. Potential applications for biguanides in oncology. J Clin Invest. (2013) 123:3693–700. doi: 10.1172/JCI67232
13. Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. (2007) 67:6745–52. doi: 10.1158/0008-5472.CAN-06-4447
14. Wang LW, Li ZS, Zou DW, Jin ZD, Gao J, Xu GM. Metformin induces apoptosis of pancreatic cancer cells. World J Gastroenterol. (2008) 14:7192–8. doi: 10.3748/wjg.v14.i47.7192
15. Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin Dl level. Oncogene (2008) 27:3576–86. doi: 10.1038/sj.onc.1211024
16. Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen–induced lung tumorigenesis. Cancer Prev Res. (2010) 3:1066–76. doi: 10.1158/1940-6207.CAPR-10-0055
17. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex of the mitochondrial respiratory chain. Biochem J. (2000) 348(Pt 3):607–14. doi: 10.1042/bj3480607
18. El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. (2000) 275:223–8. doi: 10.1074/jbc.275.1.223
19. Miller RA, Birnbaum MJ. An energetic tale of AMPK-independent effects of metformin. J Clin Invest. (2010) 120:2267–70. doi: 10.1172/JCI43661
20. Barnett AH. Avoiding hypoglycaemia while achieving good glycaemic control in type diabetes through optimal use of oral agent therapy. Curr Med Res Opin. (2010) 26:1333–42. doi: 10.1185/03007991003738063
22. Yu X, Mao W, Zhai Y, Tong C, Liu M, Ma L, et al. Anti-tumor activity of metformin: from metabolic and epigenetic perspectives. Oncotarget (2017) 8:5619–28. doi: 10.18632/oncotarget.13639
23. Taubes G. Cancer research. Unraveling the obesity-cancer connection. Science (2012) 335:28, 30–2. doi: 10.1126/science.335.6064.28
24. Cohen DH, LeRoith D. Obesity, type diabetes, and cancer: the insulin and IGF connection. Endocr Relat Cancer (2012) 19:F27–45. doi: 10.1530/ERC-11-0374
25. Gallagher EJ, LeRoith D. The proliferating role of insulin and insulin-like growth factors in cancer. Trends Endocrinol Metab. (2010) 21:610–8. doi: 10.1016/j.tem.20
26. Frasca F, Pandini G, Sciacca L, Pezzino V, Squatrito S, Belfiore A, et al. The role of insulin receptors and IGF-I receptors in cancer and other diseases. Arch Physiol Biochem. (2008) 114:23–37. doi: 10.1080/13813450801969715
28. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. (2003) 348:1625–38. doi: 10.1056/NEJMoa021423
29. Chow EJ, Pihoker C, Friedman DL, Lee SJ, McCune JS, Wharton C, et al. Glucocorticoids and insulin resistance in children with acute lymphoblastic leukemia. Pediatr Blood Cancer (2013) 60:621–6. doi: 10.1002/pbc.24364
30. Esber EC, Buell DN, Leikin SL. Insulin binding of acute lymphocytic leukemia cells. Blood (1976) 48:33–9.
31. Doepfner KT, Spertini O, Arcaro A. Autocrine insulin-like growth factor-I signaling promotes growth and survival of human acute myeloid leukemia cells via the phosphoinositide 3-kinase/Akt pathway. Leukemia (2007) 21:1921–30. doi: 10.1038/sj.leu.2404813
32. Pan J, Chen C, Jin Y, Fuentes-Mattei E, Velazquez-Tores G, Benito JM, et al. Differential impact of structurally different anti-diabetic drugs on proliferation and chemosensitivity of acute lymphoblastic leukemia cells. Cell Cycle (2012) 11:2314–26. doi: 10.4161/cc.20770
33. Chapuis N, Tamburini J, Cornillet-Lefebvre P, Gillot L, Bardet V, Willems L, et al. Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica (2010) 95:415–23. doi: 10.3324/haematol.2009.010785
34. Tazzari PL, Tabellini G, Bortul R, Papa V, Evangelisti C, Grafone T, et al. The insulin-like growth factor-I receptor kinase inhibitor NVP-AEW5 induces apoptosis in acute myeloid leukemia cells exhibiting autocrine insulin-like growth factor-I secretion. Leukemia (2007) 21:886–96. doi: 10.1038/sj.leu.2404643
35. Yaktapour N, Übelhart R, Schüler J, Aumann K, Dierks C, Burger M, et al. Insulin-like growth factor-receptor (IGF1R) as a novel target in chronic lymphocytic leukemia. Blood (2013) 122:1621–33. doi: 10.1182/blood-2013-02-484386
36. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell (2003) 115:577–90. doi: 10.1016/S0092-8674(03)00929-2
37. Green AS, Chapuis N, Maciel TT, Willems L, Lambert M, Arnoult C, et al. The LKB1/AMPK signaling pathway has tumor suppressor activity in acute myeloid leukemia through the repression of mTOR-dependent oncogenic mRNA translation. Blood (2010) 116:4262–73. doi: 10.1182/blood-2010-02-269837
38. Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci. (2012) 122:253–70. doi: 10.1042/CS20110386
39. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. (2004) 101:3329–35. doi: 10.1073/pnas.0308061100
40. Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. (2003) 13:2004–8. doi: 10.1016/j.cub.2003.10.031
41. Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem. (2010) 285:14071–7. doi: 10.1074/jbc.R109.094003
42. Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.that is mutated in multiple advanced cancers. Nat Genet. (1997) 15: 356–62. doi: 10.1038/ng0497-356
43. Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression Cell (2008) 133:403–14. doi: 10.1016/j.cell.2008.04.013
44. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. (2011) 12:21–35. doi: 10.1038/nrm3025
45. Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell (2010) 40:310–22. doi: 10.1016/j.molcel.2010.09.026
46. Ben-Sahra I, Manning BD. mTORCsignaling and the metabolic control of cell growth. Curr Opin Cell Biol. (2017) 45:72–82. doi: 10.1016/j.ceb.2017.02.012
47. Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, et al. The CREB coactivator TORCis a key regulator of fasting glucose metabolism. Nature (2005) 437:1109–11. doi: 10.1038/nature03967
48. Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. (1987) 223:217–22.
49. Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, et al. Phosphorylation and activation of heart PFK-by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. (2000) 10:1247–55. doi: 10.1016/S0960-9822(00)00742-9
50. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. (2011) 13:1016–23. doi: 10.1038/ncb2329
51. Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell (2005) 18:283–93. doi: 10.1016/j.molcel.2005.03.027
52. DeWaal D, Nogueira V, Terry AR, Patra KC, Jeon SM, Guzman G, et al. Hexokinase-depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun. (2018) 9:446. doi: 10.1038/s41467-017-02733-4
53. Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. (2010) 120:2355–69. doi: 10.1172/JCI40671
54. Scotland S, Saland E, Skuli N, de Toni F, Boutzen H, Micklow E, et al. Mitochondrial energetic and AKT status mediate metabolic effects and apoptosis of metformin in human leukemic cells. Leukemia (2013) 27:2129–38. doi: 10.1038/leu.2013.107
55. Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. (2011) 71:4366–72. doi: 10.1158/0008-5472.CAN-10-1769
56. Corominas-Faja B, Quirantes-Piné R, Oliveras-Ferraros C, Vazquez-Martin A, Cufí S, et al. Metabolomic fingerprint reveals that metformin impairs one-carbon metabolism in a manner similar to the antifolate class of chemotherapy drugs. Aging (2012) 4:480–98. doi: 10.18632/aging.100472
57. Zhu M, Zhang Q, Wang X, Kang L, Yang Y, Liu Y, et al. Metformin potentiates anti-tumor effect of resveratrol on pancreatic cancer by down-regulation of VEGF-B signaling pathway. Oncotarget (2016) 7:84190–200. doi: 10.18632/oncotarget.12391
58. Löwenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. (1999) 341:1051–62. doi: 10.1056/NEJM199909303411407
59. Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. (2006) 354:166–78. doi: 10.1056/NEJMra052603
60. Vander Heiden MG. Exploiting tumor metabolism: challenges for clinical translation J Clin Invest. (2013) 123:3648–51. doi: 10.1172/JCI72391
61. Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature (2006) 441: 475-82. doi: 10.1038/nature04703
62. Barrett D, Brown VI, Grupp SA, Teachey DT. Targeting the PI3K/AKT/mTOR signaling axis in children with hematologic malignancies. Paediatr Drugs (2012) 14:299–316. doi: 10.2165/11594740-000000000-00000
63. Polak R, Buitenhuis M. The PI3K/PKB signaling module as key regulator of hematopoiesis: implications for therapeutic strategies in leukemia. Blood (2012) 119:911–23. doi: 10.1182/blood-2011-07-366203
64. Martelli AM, Evangelisti C, Chiarini F, Grimaldi C, Manzoli L, McCubrey JA. Targeting the PI3K/AKT/mTOR signaling network in acute myelogenous leukemia. Expert Opin Investig Drugs (2009) 18:1333–49. doi: 10.1517/14728220903136775
65. Simioni C, Neri LM, Tabellini G, Ricci F, Bressanin D, Chiarini F, et al. Cytotoxic activity of the novel Akt inhibitor, MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia (2012) 26:2336–42. doi: 10.1038/leu.2012.136
66. Park S, Chapuis N, Bardet V, Tamburini J, Gallay N, Willems L, et al. PI-103, a dual inhibitor of Class IA phosphatidylinositide 3-kinase and mTOR, has antileukemic activity in AML. Leukemia (2008) 22:1698–706. doi: 10.1038/leu.2008.144
67. Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. (2008) 118:3065–74. doi: 10.1172/JCI34739
68. Efeyan A, Sabatini DM. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. (2010) 22:169–76. doi: 10.1016/j.ceb.2009.10.007
69. García-Martínez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM, et al. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem J (2009) 421:29–42. doi: 10.1042/BJ20090489
70. Sun SY. mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett. (2013) 340:1–8. doi: 10.1016/j.canlet.2013.06.017
71. Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, et al. Effective and selective targeting of leukemia cells using a TORC1/kinase inhibitor. Nat Med. (2010) 16:205–13. doi: 10.1038/nm.2091
72. Evangelisti C, Ricci F, Tazzari P, Tabellini G, Battistelli M, Falcieri E, et al. Targeted inhibition of mTORC1 and mTORC2 by active-site mTOR inhibitors has cytotoxic effects in T-cell acute lymphoblastic leukemia. Leukemia (2011) 25:781–91. doi: 10.1038/leu.2011.20
73. Willems L, Chapuis N, Puissant A, Maciel TT, Green AS, Jacque N, et al. The dual mTORC1 and mTOR2 Cinhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia (2012) 26:1195–202. doi: 10.1038/leu.2011.339
74. Liang X, Kong P, Wang J, Xu Y, Gao C, Guo G. Effects of metformin on proliferation and apoptosis of human megakaryoblastic Dami and MEG-cells. J Pharmacol Sci. (2017) 135:14–21. doi: 10.1016/j.jphs.2017.08.003
75. Ravera S, Cossu V, Tappino B, Nicchia E, Dufour C, Cavani S, et al. Concentration-dependent metabolic effects of metformin in healthy and Fanconi anemia lymphoblast cells. J Cell Physiol. (2018) 233:1736–51. doi: 10.1002/jcp.26085
76. Grimaldi C, Chiarini F, Tabellini G, Ricci F, Tazzari PL, Battistelli M, et al. AMP-dependent kinase/mammalian target of rapamycin complex signaling in T-cell acute lymphoblastic leukemia: therapeutic implications. Leukemia (2012) 26:91–100. doi: 10.1038/leu.2011.269
77. Leclerc GM, Leclerc GJ, Kuznetsov JN, DeSalvo J, Barredo JC. Metformin induces apoptosis through AMPK-dependent inhibition of UPR signaling in ALL lymphoblasts. PLoS ONE (2013) 8:e74420. doi: 10.1371/journal.pone.0074420
78. Silva A, Yunes JA, Cardoso BA, Martins LR, Jotta PY, Abecasis M, et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest. (2008) 118:3762–74. doi: 10.1172/JCI34616
79. Jotta PY, Ganazza MA, Silva A, Viana MB, da Silva MJ, Zambaldi LJ, et al. Negative prognostic impact of PTEN mutation in pediatric T-cell acute lymphoblastic leukemia. Leukemia (2010) 24:239–42. doi: 10.1038/leu.2009.209
80. Rosilio C, Lounnas N, Nebout M, Imbert V, Hagenbeek T, Spits H, et al. The metabolic perturbators metformin, phenformin and AICAR interfere with the growth and survival of murine PTEN-deficient T cell lymphomas and human T-ALL/T-LL cancer cells. Cancer Lett. (2013) 336:114–26. doi: 10.1016/j.canlet.2013.04.015
81. Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, et al. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. (2008) 412:211–21. doi: 10.1042/BJ20080557
82. Vakana E, Altman JK, Glaser H, Donato NJ, Platanias LC. Antileukemic effects of AMPK activators on BCR-ABL-expressing cells. Blood (2011) 118:6399–402. doi: 10.1182/blood-2011-01-332783
83. Tang HM, Gao WW, Chan CP, Siu YT, Wong CM, Kok KH, et al. LKB1 tumor suppressor and salt-inducible kinases negatively regulate human T-cell leukemia virus type transcription. Retrovirology (2013) 10:40. doi: 10.1186/1742-4690-10-40
84. Bruno S, Ledda B, Tenca C, Ravera S, Orengo AM, Mazzarello AN, et al. Metformin inhibits cell cycle progression of B-cell chronic lymphocytic leukemia cells. Oncotarget (2015) 6:22624–40. doi: 10.18632/oncotarget.4168
85. Martinez Marignac VL, Smith S, Toban N, Bazile M, Aloyz R. Resistance to Dasatinib in primary chronic lymphocytic leukemia lymphocytes involves AMPK-mediated energetic re-programming. Oncotarget (2013) 4:2550–66. doi: 10.18632/oncotarget.1508
86. Huntly BJ, Gilliland DG. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer (2005) 5:311–21. doi: 10.1038/nrc1592
87. Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. (2009) 69:7507–11. doi: 10.1158/0008-5472.CAN-09-2994
88. Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. (2011) 71:3196–201. doi: 10.1158/0008-5472.CAN-10-3471
89. Shi WY, Xiao D, Wang L, Dong LH, Yan ZX, Shen ZX, et al. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. (2012) 3:e275. doi: 10.1038/cddis.2012.13
90. Yi Y, Gao L, Wu M, Ao J, Zhang C, Wang X, et al. Metformin Sensitizes Leukemia Cells to Vincristine via Activation of AMP-activated Protein Kinase. J Cancer (2017) 8:2636–42. doi: 10.7150/jca.19873
91. Huai L, Wang C, Zhang C, Li Q, Chen Y, Jia Y, et al. Metformin induces differentiation in acute promyelocytic leukemia by activating the MEK/ERK signaling pathway. Biochem Biophys Res Commun. (2012) 422:398–404. doi: 10.1016/j.bbrc.2012.05.001
92. Velez J, Pan R, Lee JT, Enciso L, Suarez M, Duque JE, et al. Biguanides sensitize leukemia cells to ABT-737-induced apoptosis by inhibiting mitochondrial electron transport. Oncotarget (2016) 7:51435–49. doi: 10.18632/oncotarget.9843
93. Wang F, Liu Z, Zeng J, Zhu H, Li J, Cheng X, et al. Metformin synergistically sensitizes FLT3-ITD-positive acute myeloid leukemia to sorafenib by promoting mTOR-mediated apoptosis and autophagy. Leuk Res. (2015) 39:1421–7. doi: 10.1016/j.leukres.2015.09.016
94. Dalva-Aydemir S, Bajpai R, Martinez M, Adekola KU, Kandela I, Wei C, et al. Targeting the metabolic plasticity of multiple myeloma with FDA-approved ritonavir and metformin. Clin Cancer Res. (2015) 21:1161–71. doi: 10.1158/1078-0432.CCR-14-1088
95. Adekola KU, Dalva Aydemir S, Ma S, Zhou Z, Rosen ST, Shanmugam M. Investigating and targeting chronic lymphocytic leukemia metabolism with the human immunodeficiency virus protease inhibitor ritonavir and metformin. Leuk Lymphoma (2015) 56:450–9. doi: 10.3109/10428194.2014.922180
96. Mishra RK, Wei C, Hresko RC, Bajpai R, Heitmeier M, Matulis SM, et al. In silico modeling-based identification of Glucose Transporter (GLUT4)-selective Inhibitors for Cancer Therapy. J Biol Chem. (2015) 290:14441–53. doi: 10.1074/jbc.M114.628826
97. Pryor PR, Liu SC, Clark AE, Yang J, Holman GD, Tosh D. Chronic insulin effects on insulin signalling and GLUT4 endocytosis are reversed by metformin. Biochem J. (2000) 3(Pt 1):83–91. doi: 10.1042/0264-6021:3480083
98. Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, et al. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. (2010) 70:2465–75. doi: 10.1158/0008-5472.CAN-09-2782
99. Xue C, Wang C, Liu Q, Meng Q, Sun H, Huo X, et al. Targeting P-glycoprotein expression and cancer cell energy metabolism: combination of metformin and 2-deoxyglucose reverses the multidrug resistance of K562/Dox cells to doxorubicin. Tumour Biol. (2016) 37:8587–97. doi: 10.1007/s13277-015-4478-8
100. Voltan R, Rimondi E, Melloni E, Gilli P, Bertolasi V, Casciano F, et al. Metformin combined with sodium dichloroacetate promotes B leukemic cell death by suppressing anti-apoptotic protein Mcl-1. Oncotarget (2016) 7:18965–77. doi: 10.18632/oncotarget.7879
101. Sabnis HS, Bradley HL, Tripathi S, Yu WM, Tse W, Qu CK, et al. Synergistic cell death in FLT3-ITD positive acute myeloid leukemia by combined treatment with metformin and 6-benzylthioinosine. Leuk Res. (2016) 50:132–40. doi: 10.1016/j.leukres.2016
102. Jagannathan S, Abdel-Malek MA, Malek E, Vad N, Latif T, Anderson KC, et al. Pharmacologic screens reveal metformin that suppresses GRP78-dependent autophagy to enhance the anti-myeloma effect of bortezomib. Leukemia (2015) 29:2184–91. doi: 10.1038/leu.2015.157
104. Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell (2008) 134:703–7. doi: 10.1016/j.cell.2008.08.021
105. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324:1029–33. doi: 10.1126/science.1160809
106. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell (2012) 21:297–308. doi: 10.1016/j.ccr.2012.02.014
107. Skrtić M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell (2011) 20:674–88. doi: 10.1016/j.ccr.2011.10.015
108. Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. (2010) 120:142–56. doi: 10.1172/JCI38942
109. Moschoi R, Imbert V, Nebout M, Chiche J, Mary D, et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood (2016) 128:253–64. doi: 10.1182/blood-2015-07-655860
110. Griessinger E, Moschoi R, Biondani G, Peyron JF. Mitochondrial transfer in the leukemia microenvironment. Trends Cancer (2017) 3:828–39. doi: 10.1016/j.trecan.2017.10.003
111. Suganuma K, Miwa H, Imai N, Shikami M, Gotou M, Goto M, et al. Energy metabolism of leukemia cells: glycolysis versus oxidative phosphorylation. Leuk Lymphoma (2010) 51:2112–9. doi: 10.3109/10428194.20
112. Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. (2017) 7:716–35. doi: 10.1158/2159-8290.CD-16-0441
114. Hundal RS, Inzucchi SE. Metformin: new understandings, new uses. Drugs (2003) 63:1879–94. doi: 10.2165/00003495-200363180-00001
115. Segal ED, Yasmeen A, Beauchamp MC, Rosenblatt J, Pollak M, Gotlieb WH. Relevance of the OCT1 transporter to the antineoplastic effect of biguanides. Biochem Biophys Res Commun. (2011) 414:694–9. doi: 10.1016/j.bbrc.2011.09.134
116. Boukalova S, Stursa J, Werner L, Ezrova Z, Cerny J, Bezawork-Geleta A, et al. Mitochondrial targeting of metformin enhances its activity against pancreatic cancer. Mol Cancer Ther. (2016) 15:2875–86. doi: 10.1158/1535-7163.MCT-15-1021
117. Caraci F, Chisari M, Frasca G, Chiechio S, Salomone S, Pinto A, et al. Effects of phenformin on the proliferation of human tumor cell lines. Life Sci. (2003) 74:643–50. doi: 10.1016/j.lfs.2003.07.015
118. Higurashi T, Hosono K, Takahashi H, Komiya Y, Umezawa S, Sakai E, et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: a multicentre double-blind, placebo-controlled, randomised phase trial. Lancet Oncol. (2016) 17:475–83. doi: 10.1016/S1470-2045(15)00565-3
Keywords: metformin, leukemia, chemotherapy, adjuvant, AMPK pathway, metabolism and bioenergetics
Citation: Biondani G and Peyron J-F (2018) Metformin, an Anti-diabetic Drug to Target Leukemia. Front. Endocrinol. 9:446. doi: 10.3389/fendo.2018.00446
Received: 02 May 2018; Accepted: 19 July 2018;
Published: 10 August 2018.
Edited by:
Benoit Viollet, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Giulia Cantini, Università degli Studi di Firenze, ItalyCopyright © 2018 Biondani and Peyron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-François Peyron, cGV5cm9uQHVuaWNlLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.