 Sarah N. Framnes
Sarah N. Framnes Deanna M. Arble
Deanna M. Arble
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 06 August 2018
Sec. Translational and Clinical Endocrinology
Volume 9 - 2018 | https://doi.org/10.3389/fendo.2018.00440
This article is part of the Research Topic Metabolic Health in Normal and Abnormal Sleep View all 15 articles
Obstructive sleep apnea (OSA) is a common sleep disorder, effecting 17% of the total population and 40–70% of the obese population (1, 2). Multiple studies have identified OSA as a critical risk factor for the development of obesity, diabetes, and cardiovascular diseases (3–5). Moreover, emerging evidence indicates that metabolic disorders can exacerbate OSA, creating a bidirectional relationship between OSA and metabolic physiology. In this review, we explore the relationship between glycemic control, insulin, and leptin as both contributing factors and products of OSA. We conclude that while insulin and leptin action may contribute to the development of OSA, further research is required to determine the mechanistic actions and relative contributions independent of body weight. In addition to increasing our understanding of the etiology, further research into the physiological mechanisms underlying OSA can lead to the development of improved treatment options for individuals with OSA.
Obstructive sleep apnea (OSA) is a common sleep disorder classically characterized by apneic events leading to intermittent hypoxia and sleep fragmentation. OSA is most commonly found in obese, middle age men (6). Obesity is strongly associated with OSA (7) with approximately 40–70% of the obese population diagnosed with OSA (1, 2). Unfortunately, OSA has broad detrimental effects on health ranging from increased daytime sleepiness to a 4-fold increase in mortality (1). As the name implies, OSA derives from obstruction of the airway. While the cause of obstruction varies between individuals, common obstructions occur due to abnormal anatomy [e.g., narrow airway, enlarged tonsils (8)], obese anatomy [e.g., increased fat storage in pharyngeal tissue (9, 10)], and/or decreased neuromuscular tone (11). During a polysomnography evaluation in the sleep laboratory, an individual with OSA experiences periods of breathing reduction (hypopnea) or cessation (apnea) coincident with respiratory effort. The severity of an individuals' apnea and hypopnea is defined by the apnea-hypopnea index (AHI). An individual with mild OSA experiences 5–15 apnea-hypopnea events per hour, whereas those with moderate or severe OSA experience 15–30 or >30 events/h, respectively (12). Apneic events lead to reductions in blood oxygen saturation, and over the course of the night, present as intermittent hypoxia (IH) (13). It is estimated that an individual with severe OSA may reach blood oxygen saturation levels as low as ~76% (14) and it is widely regarded that these drops in oxygen play a key role in many of the downstream disease states associated with OSA. Reduction in blood oxygen and elevations in blood carbon dioxide are sensed by chemoreceptors in the brain and carotid bodies, which trigger brief microarousals and result in sleep fragmentation (15). These repeated microarousals are believed to contribute to Excessive Daytime Sleepiness (EDS), another characteristic of OSA. EDS, as scored by the Epworth Sleepiness Scale, measures an individuals' perceived sleepiness. Higher levels of EDS are associated with an increased risk of falling asleep at work or driving, and is associated with decreased life satisfaction (15). In a large sleep study, 76% of individuals with severe OSA exhibited EDS, and 56% of individuals with mild or moderate OSA exhibited EDS (16). In addition to apneic events, an individual with OSA exhibits a blunted hypercapnic ventilatory response (HCVR) and a blunted hypoxic ventilatory response (HVR) (17), demonstrating impaired chemosensitivity. Interestingly, blunted HCVR (18) and HVR (19, 20) are observed in some obese patients without OSA, most often those with obesity hypoventilation syndrome, suggesting that impaired chemosensitivity may occur before the onset of apneic events.
In contrast to OSA, central sleep apnea (CSA) is defined by the cessation of air flow without perceived respiratory effort (21). Like OSA, individuals with CSA may exhibit multiple apneas throughout the night. While CSA effects <5% of individuals referred to the sleep clinic (22), an increased risk for CSA is observed in individuals with compromised chemoreception. For example, CSA is found in ~24% of chronic opioid users (23) due to opioid-induced impairments to the carotid bodies and hypoglossal nerve signaling (23). Interestingly, ~13–20% of individuals diagnosed with OSA exhibit central apneas as well (24, 25). In particular, individuals with type 2 diabetes (T2D) have an increased chance of experiencing both OSA and CSA (i.e., mixed apnea) (26). Increased recognition of mixed apneic events has led to an emerging hypothesis which postulates that OSA and CSA share common mechanisms of action (22).
Currently, continuous positive airflow pressure, or CPAP, is the most effective and widely used treatment for OSA (27). By delivering a continuous flow of air, CPAP actively keeps the airway open and can improve the AHI of OSA patients an average of ~13 events/h (28). Despite the dramatic improvement in AHI from CPAP treatment, compliance is low. Only 39–50% of users will use CPAP (29) for the recommended minimum of at least 4 h per night for 5 days per week (30). Thus, improved treatment strategies for OSA are needed.
Yet, despite the prevalence of OSA, the serious health risks, and the inadequate treatment options, we have a poor understanding of how sleep apnea develops. While clinical studies have been instrumental in laying the foundation of OSA research, basic science approaches using rodent models have enabled investigators to explore the etiology of OSA. Initially, the English bulldog was used as a naturally occurring model of OSA which exhibited snoring, sleep disordered breathing, and daytime sleepiness (31). While it was first believed that the apnea of the English bulldog was occurring solely due to abnormalities of the upper airway (e.g., narrow nares and enlarged soft palate), these anatomical features only accounted for a subset of apneic events. Indeed, during sleep studies, English bulldogs displayed apneic events without respiratory effort, representative of central sleep apnea (31). While the English bulldog was a good initial model, and mirrored humans by exhibiting naturally occurring OSA (23), it also experienced apnea in a lean state. To better account for the obesity observed in many OSA individuals, lean and obese Yucatan miniature pigs were utilized as another naturally occurring model of OSA (32). Similar to the English bulldog, obese pigs experienced mixed apneic events, however, lean pigs did not experience any apneic events (32). These data suggested that obesity may be a key factor contributing to sleep apnea. While Yucatan miniature pigs were a naturally occurring model of OSA, further mechanistic studies were difficult owing to sheer size of the animals and lack of available genetic tools.
Currently, much of the mechanistic hypotheses involving OSA are tested in rodent models. The rodent offers superior capabilities in behavioral and genetic manipulation, allowing more detailed investigation into the mechanisms leading to the metabolic consequences of OSA. To examine sleep apnea in rodent models, researchers have modeled two main characteristics of OSA: sleep fragmentation and intermittent hypoxia (IH). In general, sleep loss and decreased sleep quality without the presence of OSA is associated with obesity, impairments in glucose regulation, and reductions in insulin sensitivity (33). While IH is associated with many of these same outcomes, IH often leads to weight loss instead of weight gain, perhaps due to the observed increases in circulating leptin (see leptin section below). Since IH mirrors both the oxygen desaturation as well as the microarousals associated with OSA (15), many of the mechanistic hypotheses on OSA and metabolism have sprouted from IH studies. A wealth of data indicates that chronic IH results in profound impairments in cardiometabolism similar to those experienced by individuals with OSA, including hypertension (34), ventricular hypertrophy (35), insulin resistance, and hyperlipidemia (36, 37). Using whole-body plethysmography, researchers have also observed similarities between the chemosensitivity of obese rodents (measured via the ventilatory responses to hypercapnia and hypoxia, with and without IH) to that of individuals with OSA (38, 39). In this way, the ventilatory responses of rodents presents itself as another measure analogous to the physiology of individuals with OSA. Diet-induced obese rodents can also be used alongside lean controls to determine the effect of obesity on ventilation parameters and IH-induced outcomes. Indeed, just as in humans (18), diet-induced obesity leads to a depressed ventilatory response in rodent models (38, 40). Using a combination of clinical and rodent studies, investigators can significantly increase our understanding of the etiology of sleep apnea.
In some individuals, OSA etiology is clearly associated with anatomical obstruction. For example, most OSA diagnosed in children is due to enlarged tonsils and is treated with tonsillectomy (41). However, many clinical evaluations for OSA do not reveal any obvious anatomical obstructions (42, 43). In the absence of a clear anatomical obstruction, much of the etiological theory on OSA has focused on one of its most profoundly associated factors: obesity.
Multiple studies have shown a clear, positive association between obesity and AHI (7). More specifically, increased visceral obesity (44) and neck circumference (45) have been linked to OSA. While it is generally accepted that obesity is an important prerequisite for OSA, the hypothesized mechanisms by which obesity contributes to OSA vary widely. Indeed, the relative contributions of an individuals' physical weight vs. an individuals' metabolic physiology in the development of OSA is an active area of debate (46–48).
Traditionally, the strong association between obesity and OSA led many to conclude that OSA occurred due to increased fat mass mechanically restricting airflow. Specifically, increased fat deposits in the tongue and/or larger pharyngeal tissue (9, 10, 49) were hypothesized to be too heavy for the reduced muscular tone normally experienced during rapid-eye movement (REM) sleep and thus, the tissue's increased physical weight lead to an obstruction the airway and apnea or hypopnea. Increased physical mass also affects lung mechanics, reducing functional residual capacity and tidal volume (50). For the remainder of this review, we refer to mechanisms supporting this hypothesis as weight-dependent (Figure 1). However, focusing only on physical body weight as an underlying mechanism to OSA does not explain why only a subset of obese individuals have sleep apnea (51). Nor does physical body weight alone explain why lean individuals develop sleep apnea (52). Nevertheless, the association between obesity and OSA suggests that these variables may be related to each other in ways that go beyond the physical mechanical weight of fat.
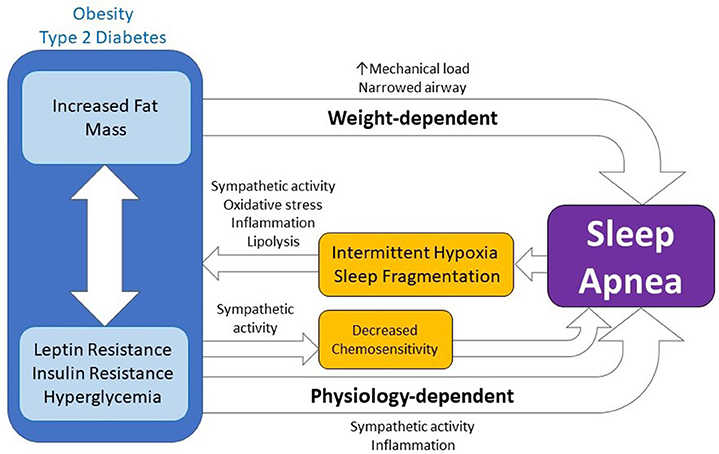
Figure 1. The bidirectional relationship between obstructive sleep apnea and metabolic disease. Sleep apnea results in intermittent hypoxia and sleep fragmentation which lead to and exacerbate obesity and type 2 diabetes by increasing sympathetic activity, oxidative stress, inflammation, and lipolysis. Moreover, metabolic disease can lead to, or exacerbate, sleep apnea through weight-dependent and physiology-dependent mechanisms. While weight-dependent mechanisms are a function of the physical increase in body mass or fat mass (e.g. increased mechanical load, narrowed airway), physiology-dependent mechanisms are physiological changes coincident with obesity or diabetes which go on to influence chemosensitivity and sleep apnea either directly or via action on sympathetic activity, inflammation, or other mechanisms.
There are two alternative explanations to the strong association between obesity and OSA. The first of which is that OSA is leading to obesity and metabolic dysfunction (Table 1). Indeed, OSA-associated IH and sleep fragmentation have been repeatedly found to induce and exacerbate cardiometabolic disease (91). This directional hypothesis is generally accepted and well-reviewed [see (53, 77, 92)]. Therefore, we only highlight key studies supporting this hypothesis in this review. Instead, we focus on a second intriguing possibility, that obese physiology and not physical weight per se leads to the development of OSA (Table 2).
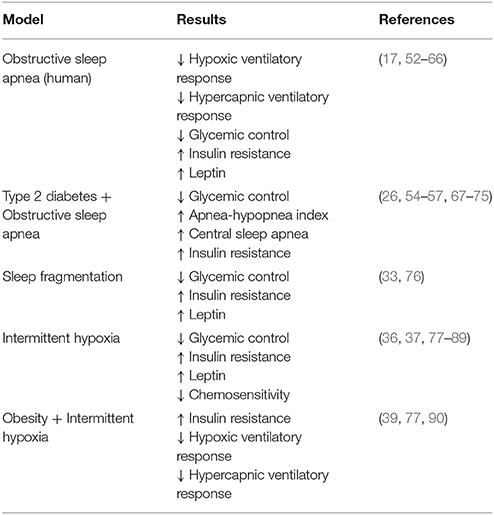
Table 1. Summary of presented evidence that obstructive sleep apnea and its components are associated with decreased glycemic control, insulin resistance, increased leptin, and decreased chemosensitivity.
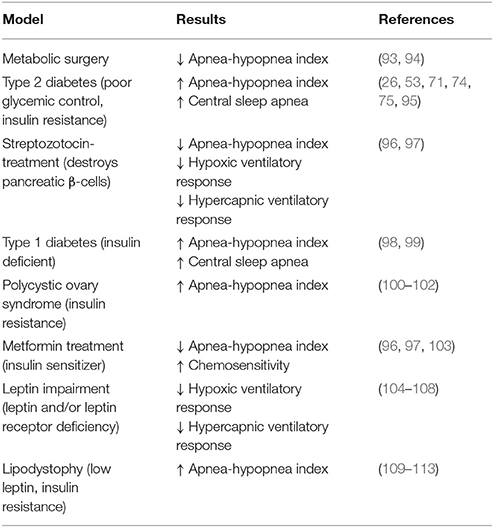
Table 2. Summary of presented evidence that the manipulation of glycemic control, insulin, and leptin are associated with increased apneic events and decreased chemosensitivity.
Emerging hypotheses postulate that physiological components of obesity, including glycemic control, insulin action, and leptin signaling, contribute to the development of OSA. It's possible that obese physiology leads to greater reductions in pharyngeal dilator muscle tone and results in increased chance of obstruction during sleep (114). This greater reduction in muscle tone may be due to chronically increased muscle activity, due to increased autonomic response, and/or histological changes to the muscle tissue itself via inflammatory pathways (49). Alternatively, obese physiology may be leading to disordered breathing and increased central sleep apnea via decreased chemosensitivity (18–20). Given the increased risk of mixed apneic events observed within type 2 diabetics, this latter observation is particularly interesting. Research on this front is on-going and it's possible that other mechanisms by which obese physiology impacts sleep apnea may soon be defined. Collectively, we refer to mechanisms that support these hypotheses as physiology-dependent or weight-independent (Figure 1).
Teasing apart the relative contribution of physical, weight-dependent mechanisms from physiological-dependent mechanisms is inherently difficult due to the close relationship between obesity and its associated changes in glycemic control, insulin action, and leptin signaling. For example, obesity is strongly associated with glucose dysregulation and weight loss alone can substantially improve fasting glucose and glucose tolerance within individuals with T2D (115). In the context of OSA, weight loss through dieting can also substantially improve AHI (116). However, it is unclear if these dramatic improvements in apneic symptoms are from weight-dependent or physiological-dependent mechanisms, as dieting both reduces physical body weight and improves glucose metabolism.
A unique way to partition the effect of weight loss from substantial changes in metabolic physiology has utilized data from bariatric surgical procedures. Bariatric surgical procedures, such as the Roux-en-Y Gastric Bypass (RYGB), the vertical sleeve gastrectomy (VSG), and the laparoscopic adjustable gastric band (LAGB) lead to significant, sustained weight loss and improvements in glucose regulation (117). However, RYGB and VSG are unique among bariatric surgical procedures in that glucose metabolism is improved through both weight-dependent and weight-independent mechanisms. In fact, due to their ability to improve glucose regulation in part through weight-independent mechanisms, RYGB and VSG are sometimes referred to as metabolic surgeries (118). This contrasts with the metabolic improvements following LAGB which parallel total weight loss without additional improvements from weight-independent means (117, 118). Following metabolic surgeries, improvements in glucose tolerance can occur quickly, before significant weight loss occurs (117, 118). In some cases, individuals can discontinue their diabetic medication before being discharged from the hospital (117). To determine how OSA may be affected by metabolic improvements independent of weight loss, it would be ideal to quantify OSA on a time scale before significant weight loss occurs. However, most polysomnography following bariatric surgical procedures occurs 6 months to 1 year post-operatively and thus after significant weight loss is achieved. However, quantifying EDS, closely related to OSA, can be done without polysomnography. In one study, individuals undergoing RYGB showed resolution of EDS symptoms within 1 month, accompanied by only marginal weight loss (119). While it is tempting to speculate that sleep apnea too may be improved on a time scale indicative of weight-independent mechanisms, this question remains unanswered. While EDS is associated with OSA, there is also an independent relationship between obesity and sleep. Overweight individuals are more likely to exhibit increased sleepiness during the day independent of OSA (120, 121). Moreover, decreased sleep duration and sleep quality has been linked to increases in BMI and metabolic dysfunction (122). Therefore, improvements in EDS following bariatric surgery could be a result of small to moderate changes in body weight and/or improvements in metabolic physiology independent of sleep apnea. Alternatively, directly comparing OSA outcomes following metabolic surgeries such as RYGB and VSG vs. weight-loss surgeries such as LAGB can provide insight into the relative contributions of weight-dependent and physiology-dependent mechanisms in the etiology of OSA. A number of comparative studies have reported that OSA resolution 1-year after RYGB or VSG is approximately double that of individuals undergoing LAGB (93, 94). Furthermore, other studies have shown that LAGB has no better OSA resolution compared to diet-induced weight loss, despite more weight loss attained via LAGB (123). Given the added weight-independent metabolic benefits following RYGB and VSG, these data suggest that some component of obese physiology and not body weight itself, may be involved in the etiology of OSA.
To better address how obese physiology may impact disordered breathing, investigators have incorporated preclinical animal models. Indeed, the preclinical setting allows researchers to systemically manipulate glycemic control, insulin sensitivity, and/or leptin and examine their specific contributions to disordered breathing. While we address each of these variables in detail in the sections below, a commonality among these experiments is the use of high-fat diets to induce obesity within the animal models. Similar to humans, diet-induced obesity leads to a depressed hypercapnic ventilatory response (40) and a restrictive ventilatory pattern (39) in mice. Importantly, since diet-induced obesity alone leads to both increased physical weight and metabolic syndrome, a more detailed approach (such as including weight as a covariate or using weight-matched controls) must be used to specifically determine how obese physiology contributes to disordered breathing. Moreover, the addition of high-fat diets has also been found to exacerbate the metabolic consequences of IH. Obese, high-fat fed mice exposed to chronic IH demonstrate further detriments in insulin resistance (39, 90), suggesting that obesity itself or obese physiology may exacerbate OSA disease outcomes.
A prominent characteristic of obese physiology is an impairment in glycemic control. Clinical association studies and randomized control trials have evaluated the relationship between OSA and glycemic control with mixed results. In support of an association between apnea and glycemic control, a recent pilot study found that the combination of respiratory events and nocturnal awakenings could predict variability of fasting blood glucose in T2D patients (67). Nocturnal hypoxemia has also been independently associated with the development of impaired glycemic control (54) and T2D in healthy individuals (55) and worsened glycemic control in individuals with T2D (68). Moreover, with the use of continuous glucose monitoring, T2D individuals with OSA have been shown to exhibit peaks in circulating glucose levels temporally following blood oxygen desaturation (69). Taken together, these studies demonstrate that OSA, and in particular nocturnal hypoxemia, likely leads to elevated glucose levels. In non-diabetic individuals, daily, 24-h rhythms in circulating glucose variability have been associated with OSA severity (56), suggesting that the association between OSA and improper glucose control may precede T2D. Whether circulating glucose levels directly impact disordered breathing or OSA is less clear. It would be informative to explore if individuals with recurrent hypoglycemia or nocturnal hypoglycemia are at increased risk for OSA and/or have reduced chemosensitivity (124). While unexplored, this information could advance our understanding of the involvement of glycemic control and/or glucose sensing in the development of sleep apnea.
In animal models, simulation of OSA using chronic IH has greatly advanced our knowledge of how OSA may impact disease states via cyclic drops in blood oxygen. Rodents exposed to chronic IH have increased gluconeogenesis in the liver (78–80), fasting hyperglycemia, and decreased glucose tolerance (81). Acute, 3-h, exposures of IH in healthy humans also leads to an increase in circulating glucose levels before noticeable changes to insulin sensitivity (125). Indeed, much of the effects on glycemic control from OSA may be attributed to IH (126). Moreover, altering metabolic state prior to IH impacts the outcome, indicating a bidirectional relationship between glycemic control and IH. For example, fasting can mitigate some cardiovascular consequences of IH, including the activation of glycogen synthase in the myocardium (127). Additionally, treatment with a lipolysis inhibitor ameliorates hyperglycemia and glucose intolerance induced by IH in mice (81), highlighting an important role for the adipose tissue and lipolysis in many of the downstream consequences of IH and perhaps OSA (77, 128). Taken together, it is likely that circulating and fasting glucose is increased by OSA and that elevated glucose before the theoretical onset of OSA is likely to exacerbate the cardiometabolic outcomes of OSA.
Another way to explore the relationship between glycemic control and OSA is by intervention and treatment studies. One would hypothesize that if alterations in glucose were downstream of OSA, then treatment of OSA alone would improve glycemic control. While there are randomized controlled studies which support this hypothesis (129), others report no improvement in glycemic control with CPAP use (130). One possibility for these conflicting results is the presence of existing glycemic impairment. For example, in a recent study, higher glycemic variability was associated with sleep disordered breathing in both T2D and non-diabetic individuals, however CPAP treatment only improved glycemic variability in those without T2D (57). Similarly, a meta-analysis concludes that CPAP may prevent the development of T2D in non-diabetic individuals (131), again pointing to the effectiveness of CPAP on glycemic control before T2D develops. However, withdraw from CPAP in both obese T2D and non-diabetics leads to an increase in nocturnal glucose without affecting glucose tolerance, production, or insulin (132), suggesting that CPAP use is leading to a reduction in glucose. Together, these data point to the likelihood that glucose impairment is downstream of OSA in non-diabetic individuals, but it remains to be elucidated the relationship between glycemic control and OSA within those with T2D.
One possible mechanism linking glucose dysregulation and OSA is via autonomic dysfunction. T2D leads to autonomic dysfunction and this directly affects respiratory control and cardiac outcomes consistent with the presentation of OSA (133). This mechanism is supported by impaired autonomic activity observed in individuals with central hypoventilation syndrome which exhibit sleep disordered breathing, hypoglycemia and hyperinsulinemia (134). Sympathetic activity is also directly involved in modulating fasting hyperglycemia following exposure to IH (135), pointing to the ability of the sympathetic system to modulate glucose metabolism in addition to respiratory outcomes (133). Chronic IH has also been observed to increase tonic and reactive afferent chemoreceptor outputs from the carotid body which in turn effects catecholamine to modulate the autonomic nervous system (82–84) and leads to fasting hyperglycemia (136) and hypertension (84). An interesting area of research positions the carotid bodies as key integrators of glucose metabolism, OSA, and autonomic function. Glomus cells in the carotid bodies sense oxygen, carbon dioxide, and glucose. Interestingly, oxygen and glucose signals can potentiate one another, leading to scenarios where dysregulation of glucose may lead to a dysregulation of O2 and CO2 sensing which in turn may affect breathing (137). Addition of 2-deoxy-d-glucose (2DG; a glucoprivic agent) in the drinking water of rats can prevent phrenic long-term facilitation, a form of respiratory motor plasticity, suggesting that alterations in glucose sensing can directly alter breathing (138). Work in this field is ongoing and shows great potential in elucidating bidirectional pathways between glucose control and disordered breathing via the sympathetic system.
A key player in glucose metabolism and tightly linked to obesity, insulin action has also been investigated in the context of OSA (95, 139). OSA is correlated with an increased risk of T2D (53) and within the diagnosed OSA population, approximately 15–30% exhibit symptoms of T2D (140, 141). Moreover, a meta-analysis of longitudinal studies concludes that the relative risk ratio of an individual with moderate/severe OSA developing T2D is 1.63 (95% CI: 1.09–2.45) compared to an individual without significant apneic events (58). Within the T2D population, reportedly 58–86% of individuals also present with OSA (70–72). Moreover, in individuals with existing T2D, a dose-dependent relationship is found between worsening glycemic control and the severity of OSA independent of obesity (68, 73, 74). If autonomic neuropathy is present alongside T2D, the individual is at an increased risk for mixed apneic events due to the degradation of respiratory neurons resulting in overall decreases in chemoreception and increased HCVR (75). While these statistics may suggest that T2D precedes the development of OSA, this hypothesis has not been supported by clinical longitudinal studies (142) or meta-analysis (143). Instead, the clinical data point to the likelihood that OSA exacerbates existing T2D through an insulin-related mechanism.
Within non-diabetic and T2D individuals, insulin resistance appears to be more closely tied with OSA than fasting hyperglycemia or glucose variability. Clinical association studies have generally found that insulin resistance is independently associated with OSA (52, 59–64), however data undermining this association, particularly from early clinical studies are present (144, 145). Much of the research exploring the relationship between insulin and OSA has been pioneered in rodent models utilizing IH. Indeed, chronic IH exposure leads to insulin resistance in lean rodents and exacerbates insulin resistance in diet-induced obese models (39, 77, 80, 90). IH can also affect β cell function, leading to augmented basal secretion and reduced glucose-stimulated insulin secretion (80, 146). Much of insulin resistance induced by IH has been attributed to elevated sympathetic activity (147–149), as pharmacological or surgical methods used to block the sympathetic response prevent the development of IH-induced insulin resistance (136, 150). Indeed, individuals with OSA demonstrate increased sympathetic nerve activity (151). Additionally, IH is observed to increase pancreatic oxidative stress and reduce β3-adrenergic receptor mediated insulin secretion (152). A possible mediator between elevated sympathetic activity and insulin resistance following IH may be increased lipolysis. Increased sympathetic outflow contributes to lipolysis, which in turn leads to elevated free-fatty acids and finally insulin resistance (153). This hypothesis is supported by data from animal models where pharmacological inhibition of lipolysis prevents IH-induced decreases in insulin sensitivity (81). A recent clinical study further demonstrated that lipoprotein abnormalities observed in OSA individuals are more directly related to insulin resistance than OSA severity itself (154). Upstream of lipolysis, hypoxia-inducible factor-mediated transcription (e.g., HIF-1α, HIF-2α) may play an important role in linking oxygen desaturation induced by IH with lipolysis (155–157) and/or insulin resistance (158).
While mounting evidence supports the conclusion that IH leads to insulin resistance, research on how decreased insulin sensitivity may lead to the development of OSA is scant due in part to the challenging experimental designs. One such way to specifically manipulate insulin is with the drug streptozotocin (STZ). STZ leads to apoptosis of pancreatic beta cells and, when given in low to moderate doses, is used as a model of T2D, reflecting insufficient insulin action and hyperglycemia. Interestingly, STZ-induced T2D (e.g., STZ-T2D) rats have marked reductions in ventilatory control, including reductions in the HCVR and the HVR, as well as increased incidents of apnea (96, 97). Insulin or metformin treatment can substantially improve disordered breathing in STZ-T2D rats (96, 97), suggesting that insufficient insulin action may contribute to the development of sleep apnea. However, it is possible that observed changes in chemoreception and disordered breathing are secondary to STZ-induced decreases in peripheral sympathetic activity (159) as opposed to insulin action per se. Along these lines, STZ-T2D rats exposed to chronic IH exhibit an attenuation in fasting hyperglycemia and mitigated (160) or improved (161) insulin resistance, perhaps reflecting the inability of IH to stimulate a sympathetic system dampened by STZ treatment. Notably, this effect in STZ-T2D rodents is distinct from IH's effect in diet-induced obese T2D animals, which experience an exacerbation in insulin resistance (39, 77, 80, 90).
If insulin action were central to the pathogenesis of sleep apnea, one might expect insulin deficient, Type 1 Diabetic (T1D) individuals to have a higher incident of disordered breathing. In support of this conclusion, children with T1D exhibit more total apneic events and increased CSA, associated with hyperglycemia and autonomic dysfunction (98, 99). Conversely, individuals with T1D are also at risk for a rare syndrome presenting with disordered breathing and hypoglycemia. Dead-in-bed syndrome is believed to occur due to initial bouts of nocturnal hypoglycemia associated with excessive hypotonia of the airway followed by IH, breathing depression, and finally cardiac arrhythmia (162). While these two conditions are distinct in insulin action, they share a common result on sympathetic function. Indeed, chemoreceptors at the carotid bodies are known to respond to elevated insulin with sympathetic activation (163, 164) while hyperglycemic events also cause autonomic dysfunction (99). These data suggest that it may not be insulin action per se associated with disordered breathing, but insulin's effect on the autonomic system. Beyond T1D and T2D individuals, other disease states associated with insulin resistance have increased risk of exhibiting disordered breathing. Women with polycystic ovary syndrome (PCOS) exhibit insulin resistance and are 30 times more likely to exhibit OSA compared to women without PCOS (100). Moreover, the insulin resistance displayed by PCOS individuals predicts OSA independent of obesity (101, 102). Hyperinsulinemia and hypoglycemia is also present in individuals with congenital central hypoventilation syndrome (CCHS), a syndrome associated with impairments in chemosensitivity and sleep disordered breathing due to a mutation in the PHOX2B gene (134). Individuals with CCHS also exhibit dysregulation to their autonomic nervous system which likely contributes to both their metabolic and disordered breathing phenotype (134). Taken together, these clinical studies suggest that insulin resistance may be an important contributing factor in OSA pathogenesis. However, it is difficult to determine the isolated role of insulin action as alterations in autonomic nervous system activity and/or chemosensitivity are often occurring simultaneously.
In most randomized clinical trials, CPAP treatment improves short-term insulin resistance (165), however the impact of CPAP on long-term insulin resistance is unknown (77). Long-term improvements in insulin action due to CPAP would support the hypothesis that OSA leads to or exacerbates insulin resistance and undermine the hypothesis that insulin resistance itself was leading to OSA. Echoing the latter, a recent randomized, placebo-controlled pilot study reported that manipulating insulin sensitivity via treatment with pioglitazone did not affect OSA (145). However, data from rodent models complicate these findings. In non-obese, high-fat diet fed rats, metformin treatment increased insulin sensitivity and prevented the development of sleep apnea independently of body weight (103). This discrepancy may be due to the specific type of apnea being studied. In rats, central apneic events occurring with relatively higher frequency than in the general human population. If this is true, further research into differentiating between obstructive, central, and mixed apneic events may yield differential contributions of insulin resistance.
Overall, ample evidence demonstrates that insulin resistance is associated with OSA independent of obesity, and that the cyclic bouts of hypoxia experienced by OSA individuals may be key to exacerbating insulin resistance. However, evidence demonstrating that insulin action alone leads to or exacerbates OSA is limited. One possibility is that insulin resistance is one of many factors affecting sleep disordered breathing and requires coincident impairments in the autonomic nervous system, glycemic control, or others (see leptin in the following section) to generate the conditions necessary for promoting OSA.
Leptin is a satiety hormone released by and in proportion to adipose tissue stores. The robustly positive relationship between leptin and body fat makes leptin an obvious confound when speculating on the root cause of OSA. In general, as leptin increases with fat mass, it acts as an anti-obesity hormone. However, too much leptin can lead to leptin resistance wherein the anti-obesity properties are no longer triggered. Indeed, treating obese individuals with peripheral leptin fails to reduce body weight (166). However, leptin resistance may not impact all of leptin actions. For example, even in obese individuals, leptin's action on sympathoexcitatory actions is maintained (167). It is possible that elevated leptin and/or leptin resistance observed in obesity may be contributing to OSA.
In non-T2D individuals, clinical studies have identified a positive association between OSA and leptin independent of body fat (61, 65). Though a causal relationship has not been defined, there is also evidence that both leptin resistance (168, 169) and OSA increase with aging (144). Healthy pre-menopausal women have significantly higher circulating leptin levels compared to men independent of body weight (168) and are also significantly less affected by OSA (0.6% of pre-menopausal females vs. 3.9% of males) (170), suggesting that increased leptin signaling or elevated leptin may be protective of OSA. However, this effect appears to be absent in post-menopausal women (171). Based on these association studies like these, if leptin action is involved in OSA, then the involvement of other endocrine systems including sex-hormones and insulin resistance may be important co-contributors to OSA.
Recently, accumulating evidence points to leptin action upstream of disordered breathing. Clinical data from individuals with obesity hypoventilation syndrome suggest that leptin resistance contributes to a reduction in HCVR and HVR likely via an impaired chemosensitivity (172). Leptin deficient ob/ob mice exhibit a disordered breathing phenotype (104), including a reduction in HCVR (105), and treating ob/ob mice with leptin improves ventilation within 3 days, before significant weight loss occurs (105). The obese Zucker rat, which lacks leptin receptors, also exhibit a decreased HVR (106) however maintain a stable upper airway during sleep (173). Leptin resistant New Zealand Obese mice exhibit inspiratory flow limitation, suggestive of sleep disordered breathing (107). These rodent data are partially recapitulated in individuals with lipodystrophy which exhibit chronically low levels of leptin (109, 110) and are at a greater risk to the development of OSA (111), suggesting that insufficient leptin action may lead to OSA in humans. However, lipodystrophic individuals also have increased fat deposits around the neck and exhibit characteristic insulin resistance (112, 113), making it difficult to determine the individual contribution of leptin on apneic events independent of physical body weight or other physiological variables such as insulin.
Leptin action may also be instrumental in downstream signaling of OSA. IH has been shown to lead to a significant increase in leptin levels in both rodents and humans (37, 85–89). Similar increases in leptin are observed in OSA patients (66) and in those with shortened sleep (76). CPAP treatment in OSA individuals tend to decrease leptin levels independent of body weight, however this is not consistently observed in all studies (128). As many patients lose weight with CPAP, noting changes in body fat specifically (174), is particularly important to consider when reflecting on leptin action. When exposed to IH, rodents with deficient leptin signaling have exacerbated insulin resistance (175) and increased cardiovascular impairments including endothelial dysfunction (176). Leptin treatment prior to IH reduces insulin resistance and hyperlipidemia and improves endothelial relaxation and vascular stiffness in ob/ob mice (175, 177). Most intriguingly, leptin treatment can mitigate IH-induced hyperlipidemia and cardiovascular outcomes in lean, wild type (177) suggesting that a boost in leptin signaling may prevent downstream cardiometabolic consequences of IH. As these studies focus on peripheral leptin treatment, it is unclear if leptin is acting primarily on peripheral or central targets. However, recent evidence that manipulation of specific neuronal leptin receptors can lead to tachypnea and a decreased HCVR (108) supports the hypothesis that neuronal leptin signaling may contribute to disordered breathing.
Given the role of leptin in ventilatory drive and the increases observed following IH, leptin may be acting by way of a counterregulatory mechanism in an attempt to improve disordered breathing. Some have proposed leptin is directly controlled by hypoxia (86). However, leptin's tight relationship with other key players in OSA, including obesity and insulin sensitivity (178), especially in T2D individuals (179), make it difficult to draw specific conclusions about the role of leptin in OSA. Key areas of leptin's involvement in OSA require further exploration, including leptin's action in chemosensitive regions, and the synergistic role of leptin, insulin, and other hormones on downstream cardiometabolic outcomes associated with OSA.
The strong association between obesity, OSA, and T2D has led many to speculate about the bidirectional relationship between metabolic disease and OSA. A wealth of clinical studies suggests that OSA can exacerbate T2D, and animal studies have echoed this conclusion demonstrating that rodents exposed to IH show impairments in glycemic control, insulin resistance, and altered leptin levels. With the aid of these animal models, a number of mechanistic hypotheses have been posed which link OSA to the metabolic syndrome, including an elevation in sympathetic tone, increased lipolysis, inflammation (180), and reductions in chemosensitivity. A more debated hypothesis positions the physiological components of obesity, including glucose, insulin, and leptin signaling as key contributors to the etiology of OSA. While it is becoming clear that elements beyond the physical weight of body fat may be leading to OSA, the field is largely undecided on which factor(s) are critical to OSA's etiology. Novel hypotheses on this aspect of the directional relationship would do well to consider the synergistic relationship between insulin and leptin at the foundation for healthy and disordered breathing. Other new avenues of research show great promise in increasing our understanding of OSA and the relationship to cardiometabolic diseases. Emerging evidence that the gut microbiota is altered following IH (181), for example, elucidates a novel, potential link between OSA, glucose metabolism, and the gut (182). The involvement of the circadian biology with OSA and sleep disordered breathing also shows great promise (183). OSA individuals exhibit a circadian dysregulation of cortisol (184), and treatment with melatonin has been found to mitigate IH-induced hyperglycemia (185), insulin resistance, and microvascular damage (186). Research in these fields are on-going and may revel exciting new information about OSA etiology.
Advancing our knowledge on the etiology of OSA may lead to novel treatment strategies. Currently CPAP is the most effective and widely used treatment for individuals with OSA (27). Despite its low compliance (29), CPAP treatment modestly improves blood pressure (151), attenuates heart failure (187), and improves cardiac function (188, 189) and can significantly reduce mortality due to cardiovascular diseases (190). CPAP can also improve AHI (191) and blood oxygenation in individuals presenting predominantly with CSA (192). CPAP is unique in that it not only targets physical obstructions but also alleviates a brain-central failure to breathe. Indeed, the success of CPAP reflects the heterogenous nature of sleep apnea with both anatomical and neuronal underpinnings. For comparison, surgical treatments such as the Uvulopalatopharyngoplasty (UPPP) target anatomical obstructions and success rates are heavily dependent on degree of anatomical obstruction (193). Whereas drugs targeting the brain improve OSA (194), but not to the extent as CPAP. For example, fluoxetine (Prozac), a selective serotonin reuptake inhibitor commonly used to treat depression, in combination with ondansetron, improves apneic events by ~40% (195). Similarly, acetazolamide, a carbonic anhydrase inhibitor used to treat glaucoma and other conditions, has been shown to improve central sleep apnea and oxygen saturation (196). Taken together, the current treatment data supports a growing hypothesis that OSA involves more than physical anatomical obstructions and implicates a physiological component in the development of apneic events. Especially in the cases of mixed apneic events, more common in those with T2D (26), it becomes critical to understand the etiology of sleep apnea in order to effectively treat it beyond physical and anatomical obstructions.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
DA receives laboratory research funds from the American Heart Association (17SDG33660108) and the Department of Biological Sciences at Marquette University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer RA and handling Editor declared their shared affiliation.
1. Young T, Finn L, Peppard PE, Szklo-Coxe M, Austin D, Nieto FJ, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin sleep cohort. Sleep (2008) 31:1071–8. doi: 10.5665/sleep/31.8.1071
2. Lopez PP, Stefan B, Schulman CI, Byers PM. Prevalence of sleep apnea in morbidly obese patients who presented for weight loss surgery evaluation: more evidence for routine screening for obstructive sleep apnea before weight loss surgery. Am Surg. (2008) 74:834–8.
3. Coughlin SR, Mawdsley L, Mugarza JA, Calverley PM, Wilding JP. Obstructive sleep apnoea is independently associated with an increased prevalence of metabolic syndrome. Eur Heart J. (2004) 25:735–41. doi: 10.1016/j.ehj.2004.02.021
4. Somers VK, White DP, Amin R, Abraham WT, Costa F, Culebras A, et al. Sleep apnea and cardiovascular disease: an American Heart Association/American College of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council on Cardiovascular Nursing. J Am Coll Cardiol. (2008) 52:686–717. doi: 10.1016/j.jacc.2008.05.002
5. Somers VK, White DP, Amin R, Abraham WT, Costa F, Culebras A, et al. American Heart Association Council for High Blood Pressure Research Professional Education Committee, C. American Heart Association Stroke, N. American Heart Association Council on Cardiovascular, and F. American College of Cardiology, Sleep apnea and cardiovascular disease: an American Heart Association/american College Of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council On Cardiovascular Nursing. In collaboration with the National Heart, Lung, and Blood Institute National Center on Sleep Disorders Research (National Institutes of Health). Circulation (2008) 118:1080–111. doi: 10.1161/CIRCULATIONAHA.107.189375
6. Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc. (2008) 5:136–43. doi: 10.1513/pats.200709-155MG
7. Gami AS, Caples SM, Somers VK. Obesity and obstructive sleep apnea. Endocrinol Metab Clin North Am. (2003) 32:869–94. doi: 10.1016/S0889-8529(03)00069-0
8. Friedman M, Ibrahim H, Joseph NJ. Staging of obstructive sleep apnea/hypopnea syndrome: a guide to appropriate treatment. Laryngoscope (2004) 114:454–9. doi: 10.1097/00005537-200403000-00013
9. Kim AM, Keenan BT, Jackson N, Chan EL, Staley B, Poptani H, et al. Tongue fat and its relationship to obstructive sleep apnea. Sleep (2014) 37:1639–48. doi: 10.5665/sleep.4072
10. Shelton KE, Woodson H, Gay S, Suratt PM. Pharyngeal fat in obstructive sleep apnea. Am Rev Respir Dis. (1993) 148:462–6. doi: 10.1164/ajrccm/148.2.462
11. Jordan AS, McSharry DG, Malhotra A. Adult obstructive sleep apnoea. Lancet (2014) 383:736–47. doi: 10.1016/S0140-6736(13)60734-5
12. Sleep-related breathing disorders in adults: recommendations for syndrome definition and measurement techniques in clinical research. The Report of an American Academy of Sleep Medicine Task Force. Sleep (1999) 22:667–89. doi: 10.1093/sleep/22.5.667
13. Dempsey JA, Veasey SC, Morgan BJ, O'Donnell CP. Pathophysiology of sleep apnea. Physiol Rev. (2010) 90:47–112. doi: 10.1152/physrev.00043.2008
14. Chung F, Liao P, Elsaid H, Islam S, Shapiro CM, Sun Y. Oxygen desaturation index from nocturnal oximetry: a sensitive and specific tool to detect sleep-disordered breathing in surgical patients. Anesth Analg. (2012) 114:993–1000. doi: 10.1213/ANE.0b013e318248f4f5
15. McNicholas WT. Diagnosis of obstructive sleep apnea in adults. Proc Am Thorac Soc. (2008) 5:154–60. doi: 10.1513/pats.200708-118MG
16. Chen R, Xiong KP, Lian YX, Huang JY, Zhao MY, Li JX, et al. Daytime sleepiness and its determining factors in Chinese obstructive sleep apnea patients. Sleep Breath (2011) 15:129–35. doi: 10.1007/s11325-010-0337-4
17. Javaheri S, Colangelo G, Lacey W, Gartside PS. Chronic hypercapnia in obstructive sleep apnea-hypopnea syndrome. Sleep (1994) 17:416–23.
18. Piper AJ, Grunstein RR. Big breathing: the complex interaction of obesity, hypoventilation, weight loss, and respiratory function. J Appl Physiol. (2010) 108:199–205. doi: 10.1152/japplphysiol.00713.2009
20. Ge RL, Stone JA, Levine BD, Babb TG. Exaggerated respiratory chemosensitivity and association with SaO2 level at 3,568 m in obesity. Respir Physiol Neurobiol. (2005) 146:47–54. doi: 10.1016/j.resp.2004.11.009
22. Eckert DJ, Jordan AS, Merchia P, Malhotra A. Central sleep apnea: pathophysiology and treatment. Chest (2007) 131:595–607. doi: 10.1378/chest.06.2287
23. Correa D, Farney RJ, Chung F, Prasad A, Lam D, Wong J. Chronic opioid use and central sleep apnea: a review of the prevalence, mechanisms, and perioperative considerations. Anesth Analg. (2015) 120:1273–85. doi: 10.1213/ANE.0000000000000672
24. Lehman S, Antic NA, Thompson C, Catcheside PG, Mercer J, McEvoy RD. Central sleep apnea on commencement of continuous positive airway pressure in patients with a primary diagnosis of obstructive sleep apnea-hypopnea. J Clin Sleep Med. (2007) 3:462–6.
25. Wang J, Wang Y, Feng J, Chen BY, Cao J. Complex sleep apnea syndrome. Patient Prefer Adherence (2013) 7:633–41. doi: 10.2147/PPA.S46626
26. Iftikhar IH, Hoyos CM, Phillips CL, Magalang UJ. Meta-analyses of the Association of Sleep Apnea with insulin resistance, and the effects of CPAP on HOMA-IR, adiponectin, and visceral adipose fat. J Clin Sleep Med. (2015) 11:475–85. doi: 10.5664/jcsm.4610
27. Collard P, Pieters T, Aubert G, Delguste P, Rodenstein DO. Compliance with nasal CPAP in obstructive sleep apnea patients. Sleep Med Rev. (1997) 1:33–44. doi: 10.1016/S1087-0792(97)90004-6
28. Giles TL, Lasserson TJ, Smith BH, White J, Wright J, Cates CJ. Continuous positive airways pressure for obstructive sleep apnoea in adults. Cochrane Database Syst Rev. (2006):CD001106. doi: 10.1002/14651858.CD001106.pub2
29. Hong SO, Chen YF, Jung J, Kwon YD, Liu SYC. Hypoglossal nerve stimulation for treatment of obstructive sleep apnea (OSA): a primer for oral and maxillofacial surgeons. Maxillofac Plast Reconstr Surg. (2017) 39:27. doi: 10.1186/s40902-017-0126-0
30. Richard W, Venker J, den Herder C, Kox D, van den Berg B, Laman M, et al. Acceptance and long-term compliance of nCPAP in obstructive sleep apnea. Eur Arch Otorhinolaryngol. (2007) 264:1081–6. doi: 10.1007/s00405-007-0311-3
31. Hendricks JC, Kline LR, Kovalski RJ, O'Brien JA, Morrison AR, Pack AI. The English bulldog: a natural model of sleep-disordered breathing. J Appl Physiol. (1987) 63:1344–50.
32. Lonergan RP, III, Ware JC, Atkinson RL, Winter WC, Suratt PM, Sleep apnea in obese miniature pigs. J Appl Physiol. (1998) 84:531–6.
33. Spiegel K, Tasali E, Leproult R, Van Cauter E. Effects of poor and short sleep on glucose metabolism and obesity risk. Nat Rev Endocrinol. (2009) 5:253–61. doi: 10.1038/nrendo.2009.23
34. Lesske J, Fletcher EC, Bao G, Unger T. Hypertension caused by chronic intermittent hypoxia–influence of chemoreceptors and sympathetic nervous system. J Hypertens. (1997) 15:1593–603.
35. Verdecchia P, Schillaci G, Guerrieri M, Gatteschi C, Benemio G, Boldrini F, et al. Circadian blood pressure changes and left ventricular hypertrophy in essential hypertension. Circulation (1990) 81:528–36.
36. Campen MJ, Shimoda LA, O'Donnell CP. Acute and chronic cardiovascular effects of intermittent hypoxia in C57BL/6J mice. J Appl Physiol. (2005) 99:2028–35. doi: 10.1152/japplphysiol.00411.2005
37. Li J, Thorne LN, Punjabi NM, Sun CK, Schwartz AR, Smith PL, et al. Intermittent hypoxia induces hyperlipidemia in lean mice. Circ Res. (2005) 97:698–706. doi: 10.1161/01.RES.0000183879.60089.a9
38. Davis EM, O'Donnell CP. Rodent models of sleep apnea. Respir Physiol Neurobiol. (2013) 188:355–61. doi: 10.1016/j.resp.2013.05.022
39. Olea E, Agapito MT, Gallego-Martin T, Rocher A, Gomez-Nino A, Obeso A, et al. Intermittent hypoxia and diet-induced obesity: effects on oxidative status, sympathetic tone, plasma glucose and insulin levels, and arterial pressure. J Appl Physiol. (2014) 117:706–19. doi: 10.1152/japplphysiol.00454.2014
40. Polotsky VY, Wilson JA, Haines AS, Scharf MT, Soutiere SE, Tankersley CG, et al. The impact of insulin-dependent diabetes on ventilatory control in the mouse. Am J Respir Crit Care Med. (2001) 163:624–32. doi: 10.1164/ajrccm.163.3.2007120
41. Todd CA, Bareiss AK, McCoul ED, Rodriguez KH. Adenotonsillectomy for obstructive sleep apnea and quality of life: systematic review and meta-analysis. Otolaryngol Head Neck Surg. (2017) 157:767–73. doi: 10.1177/0194599817717480
42. Kuna ST, Sant'Ambrogio G. Pathophysiology of upper airway closure during sleep. JAMA (1991) 266:1384–9. doi: 10.1001/jama.1991.03470100076036
43. Wellman A, Jordan AS, Malhotra A, Fogel RB, Katz ES, Schory K, et al. Ventilatory control and airway anatomy in obstructive sleep apnea. Am J Respir Crit Care Med. (2004) 170:1225–32. doi: 10.1164/rccm.200404-510OC
45. Davies RJ, Ali NJ, Stradling JR. Neck circumference and other clinical features in the diagnosis of the obstructive sleep apnoea syndrome. Thorax (1992) 47:101–5.
46. Motamedi GK. Obstructive sleep apnea; is it the anatomy or physiology? Clin Neurophysiol. (2014) 125:1717–8. doi: 10.1016/j.clinph.2014.02.004
47. Phillips CL, Hoyos CM, Yee BJ, Grunstein RR. CrossTalk opposing view: sleep apnoea causes metabolic syndrome. J Physiol. (2016) 594:4691–4. doi: 10.1113/JP272115
48. Vgontzas AN, Gaines J, Ryan S, McNicholas WT. CrossTalk proposal: metabolic syndrome causes sleep apnoea. J Physiol. (2016) 594:4687–90. doi: 10.1113/JP272114
49. Ryan CM, Bradley TD. Pathogenesis of obstructive sleep apnea. J Appl Physiol (1985) (2005) 99:2440–50. doi: 10.1152/japplphysiol.00772.2005
50. Salome CM, King GG, Berend N. Physiology of obesity and effects on lung function. J Appl Physiol (1985) (2010) 108:206–11. doi: 10.1152/japplphysiol.00694.2009
51. Mortimore IL, Marshall I, Wraith PK, Sellar RJ, Douglas NJ. Neck and total body fat deposition in nonobese and obese patients with sleep apnea compared with that in control subjects. Am J Respir Crit Care Med. (1998) 157:280–3.
52. Pamidi S, Wroblewski K, Broussard J, Day A, Hanlon EC, Abraham V, et al. Obstructive sleep apnea in young lean men: impact on insulin sensitivity and secretion. Diabetes Care (2012) 35:2384–9. doi: 10.2337/dc12-0841
53. Martinez Ceron E, Casitas Mateos R, Garcia-Rio F. Sleep apnea-hypopnea syndrome and type 2 diabetes. A reciprocal relationship? Arch Bronconeumol. (2015) 51:128–39. doi: 10.1016/j.arbres.2014.06.017
54. Appleton SL, Vakulin A, Wittert GA, Martin SA, Grant JF, Taylor AW. The association of obstructive sleep apnea (OSA) and nocturnal hypoxemia with the development of abnormal HbA1c in a population cohort of men without diabetes. Diabetes Res Clin Pract. (2016) 114:151–9. doi: 10.1016/j.diabres.2015.12.007
55. Appleton SL, Vakulin A, McEvoy RD, Wittert GA, Martin SA, Grant JF, et al. Nocturnal hypoxemia and severe obstructive sleep apnea are associated with incident type 2 diabetes in a population cohort of men. J Clin Sleep Med. (2015) 11:609–14. doi: 10.5664/jcsm.4768
56. Kallianos A, Trakada G, Papaioannou T, Nikolopouloss I, Mitrakou A, Manios E, et al. Glucose and arterial blood pressure variability in obstructive sleep apnea syndrome. Eur Rev Med Pharmacol Sci. (2013) 17:1932–7.
57. Nakata K, Miki T, Tanno M, Ohnishi H, Yano T, Muranaka A, et al. Distinct impacts of sleep-disordered breathing on glycemic variability in patients with and without diabetes mellitus. PLoS ONE (2017) 12:e0188689. doi: 10.1371/journal.pone.0188689
58. Wang X, Bi Y, Zhang Q, Pan F. Obstructive sleep apnoea and the risk of type 2 diabetes: a meta-analysis of prospective cohort studies. Respirology (2013) 18:140–6. doi: 10.1111/j.1440-1843.2012.02267.x
59. Brusik M, Strbova Z, Petrasova D, Pobeha P, Kuklisova Z, Tkacova R, et al. Increased resting energy expenditure and insulin resistance in male patients with moderate-to severe obstructive sleep apnoea. Physiol Res. (2016) 65:969–77.
60. Araujo Lda S, Fernandes JF, Klein MR, Sanjuliani AF, Obstructive sleep apnea is independently associated with inflammation and insulin resistance, but not with blood pressure, plasma catecholamines, and endothelial function in obese subjects. Nutrition (2015) 31:1351–7. doi: 10.1016/j.nut.2015.05.017
61. Manzella D, Parillo M, Razzino T, Gnasso P, Buonanno S, Gargiulo A, et al. Soluble leptin receptor and insulin resistance as determinant of sleep apnea. Int J Obes Relat Metab Disord. (2002) 26:370–5. doi: 10.1038/sj.ijo.0801939
62. Punjabi NM, Shahar E, Redline S, Gottlieb DJ, Givelber R, Resnick HE, Sleep Heart Health Study Investigators. Sleep-disordered breathing, glucose intolerance, and insulin resistance: the Sleep Heart Health Study. Am J Epidemiol. (2004) 160:521–30. doi: 10.1093/aje/kwh261
63. Makino S, Handa H, Suzukawa K, Fujiwara M, Nakamura M, Muraoka S, et al. Obstructive sleep apnoea syndrome, plasma adiponectin levels, and insulin resistance. Clin Endocrinol (Oxf). (2006) 64:12–9. doi: 10.1111/j.1365-2265.2005.02407.x
64. Ip MS, Lam B, Ng MM, Lam WK, Tsang KW, Lam KS. Obstructive sleep apnea is independently associated with insulin resistance. Am J Respir Crit Care Med. (2002) 165:670–6. doi: 10.1164/ajrccm.165.5.2103001
65. Ip MS, Lam KS, Ho C, Tsang KW, Lam W. Serum leptin and vascular risk factors in obstructive sleep apnea. Chest (2000) 118:580–6. doi: 10.1378/chest.118.3.580
66. Phillips BG, Kato M, Narkiewicz K, Choe I, Somers VK. Increases in leptin levels, sympathetic drive, and weight gain in obstructive sleep apnea. Am J Physiol Heart Circ Physiol. (2000) 279:H234–7. doi: 10.1152/ajpheart.2000.279.1.H234
67. Tatti P, Strollo F, Passali D. Sleep apnea, sleep disturbance, and fasting glucose variability: a pilot study. J Diabetes Sci Technol. (2013) 7:743–8. doi: 10.1177/193229681300700320
68. Torrella M, Castells I, Gimenez-Perez G, Recasens A, Miquel M, Simo O, et al. Intermittent hypoxia is an independent marker of poorer glycaemic control in patients with uncontrolled type 2 diabetes. Diabetes Metab. (2015) 41:312–8. doi: 10.1016/j.diabet.2015.01.002
69. Hui P, Zhao L, Xie Y, Wei X, Ma W, Wang J, et al. Nocturnal hypoxemia causes hyperglycemia in patients with obstructive sleep apnea and type 2 diabetes mellitus. Am J Med Sci. (2016) 351:160–8. doi: 10.1016/j.amjms.2015.12.002
70. Drager LF, Lopes HF, Maki-Nunes C, Trombetta IC, Toschi-Dias E, Alves MJ, et al. The impact of obstructive sleep apnea on metabolic and inflammatory markers in consecutive patients with metabolic syndrome. PLoS ONE (2010) 5:e12065. doi: 10.1371/journal.pone.0012065
71. Foster GD, Sanders MH, Millman R, Zammit G, Borradaile KE, Newman AB, et al. Obstructive sleep apnea among obese patients with type 2 diabetes. Diabetes Care (2009) 32:1017–9. doi: 10.2337/dc08-1776
72. Mokhlesi B, Ham SA, Gozal D. The effect of sex and age on the comorbidity burden of OSA: an observational analysis from a large nationwide US health claims database. Eur Respir J. (2016) 47:1162–9. doi: 10.1183/13993003.01618-2015
73. Aronsohn RS, Whitmore H, Van Cauter E, Tasali E. Impact of untreated obstructive sleep apnea on glucose control in type 2 diabetes. Am J Respir Crit Care Med. (2010) 181:507–13. doi: 10.1164/rccm.200909-1423OC
74. Grimaldi D, Beccuti G, Touma C, Van Cauter E, Mokhlesi B. Association of obstructive sleep apnea in rapid eye movement sleep with reduced glycemic control in type 2 diabetes: therapeutic implications. Diabetes Care (2014) 37:355–63. doi: 10.2337/dc13-0933
75. Rasche K, Keller T, Tautz B, Hader C, Hergenc G, Antosiewicz J, et al. Obstructive sleep apnea and type 2 diabetes. Eur J Med Res. (2010) 15:152–6. doi: 10.1186/2047-783X-15-S2-152
76. Hayes AL, Xu F, Babineau D, Patel SR. Sleep duration and circulating adipokine levels. Sleep (2011) 34:147–52.
77. Gileles-Hillel A, Kheirandish-Gozal L, Gozal D. Biological plausibility linking sleep apnoea and metabolic dysfunction. Nat Rev Endocrinol. (2016) 12:290–8. doi: 10.1038/nrendo.2016.22
78. Savransky V, Bevans S, Nanayakkara A, Li J, Smith PL, Torbenson MS, et al. Chronic intermittent hypoxia causes hepatitis in a mouse model of diet-induced fatty liver. Am J Physiol Gastrointest Liver Physiol. (2007) 293:G871–7. doi: 10.1152/ajpgi.00145.2007
79. Savransky V, Nanayakkara A, Vivero A, Li J, Bevans S, Smith PL, et al. Chronic intermittent hypoxia predisposes to liver injury. Hepatology (2007) 45:1007–13. doi: 10.1002/hep.21593
80. Polak J, Shimoda LA, Drager LF, Undem C, McHugh H, Polotsky VY, et al. Intermittent hypoxia impairs glucose homeostasis in C57BL6/J mice: partial improvement with cessation of the exposure. Sleep (2013) 36:1483–90; 1490A−1490B. doi: 10.5665/sleep.3040
81. Weiszenstein M, Shimoda LA, Koc M, Seda O, Polak J. Inhibition of lipolysis ameliorates diabetic phenotype in a mouse model of obstructive sleep apnea. Am J Respir Cell Mol Biol. (2016) 55:299–307. doi: 10.1165/rcmb.2015-0315OC
82. Semenza GL, Prabhakar NR. Neural regulation of hypoxia-inducible factors and redox state drives the pathogenesis of hypertension in a rodent model of sleep apnea. J Appl Physiol. (2015) 119:1152–6. doi: 10.1152/japplphysiol.00162.2015
83. Jacintho JD, Kovacic P. Neurotransmission and neurotoxicity by nitric oxide, catecholamines, and glutamate: unifying themes of reactive oxygen species and electron transfer. Curr Med Chem. (2003) 10:2693–703. doi: 10.2174/0929867033456404
84. Iturriaga R, Oyarce MP, Dias ACR. Role of carotid body in intermittent hypoxia-related hypertension. Curr Hypertens Rep. (2017) 19:38. doi: 10.1007/s11906-017-0735-0
85. Grosfeld A, Zilberfarb V, Turban S, Andre J, Guerre-Millo M, Issad T. Hypoxia increases leptin expression in human PAZ6 adipose cells. Diabetologia (2002) 45:527–30. doi: 10.1007/s00125-002-0804-y
86. Guerre-Millo M, Grosfeld A, Issad T. Leptin is a hypoxia-inducible gene. Obes Res. (2002) 10:856; author reply 857–8. doi: 10.1038/oby.2002.116
87. Reinke C, Bevans-Fonti S, Drager LF, Shin MK, Polotsky VY. Effects of different acute hypoxic regimens on tissue oxygen profiles and metabolic outcomes. J Appl Physiol. (2011) 111:881–90. doi: 10.1152/japplphysiol.00492.2011
88. Trzepizur W, Gaceb A, Arnaud C, Ribuot C, Levy P, Martinez MC, et al. Vascular and hepatic impact of short-term intermittent hypoxia in a mouse model of metabolic syndrome. PLoS ONE (2015) 10:e0124637. doi: 10.1371/journal.pone.0124637
89. Ling Q, Sailan W, Ran J, Zhi S, Cen L, Yang X, et al. The effect of intermittent hypoxia on bodyweight, serum glucose and cholesterol in obesity mice. Pak J Biol Sci. (2008) 11:869–75. doi: 10.3923/pjbs.2008.869.875
90. Drager LF, Li J, Reinke C, Bevans-Fonti S, Jun JC, Polotsky VY. Intermittent hypoxia exacerbates metabolic effects of diet-induced obesity. Obesity (Silver Spring) (2011) 19:2167–74. doi: 10.1038/oby.2011.240
91. Drager LF, Togeiro SM, Polotsky VY, Lorenzi-Filho G. Obstructive sleep apnea: a cardiometabolic risk in obesity and the metabolic syndrome. J Am Coll Cardiol. (2013) 62:569–76. doi: 10.1016/j.jacc.2013.05.045
92. Drager LF, Polotsky VY, O'Donnell CP, Cravo SL, Lorenzi-Filho G, Machado BH. Translational approaches to understanding metabolic dysfunction and cardiovascular consequences of obstructive sleep apnea. Am J Physiol Heart Circ Physiol. (2015) 309:H1101–11. doi: 10.1152/ajpheart.00094.2015
93. Hutter MM, Schirmer BD, Jones DB, Ko CY, Cohen ME, Merkow RP, et al. First report from the American College of Surgeons Bariatric Surgery Center Network: laparoscopic sleeve gastrectomy has morbidity and effectiveness positioned between the band and the bypass. Ann Surg. (2011) 254:410–20; discussion 420–2. doi: 10.1097/SLA.0b013e31822c9dac
94. Carlin AM, Zeni TM, English WJ, Hawasli AA, Genaw JA, Krause KR, et al., Michigan Bariatric Surgery C. The comparative effectiveness of sleeve gastrectomy, gastric bypass, and adjustable gastric banding procedures for the treatment of morbid obesity. Ann Surg. (2013) 257:791–7. doi: 10.1097/SLA.0b013e3182879ded
95. Brooks B, Cistulli PA, Borkman M, Ross G, McGhee S, Grunstein RR, et al. Obstructive sleep apnea in obese noninsulin-dependent diabetic patients: effect of continuous positive airway pressure treatment on insulin responsiveness. J Clin Endocrinol Metab. (1994) 79:1681–5.
96. Hein MS, Schlenker EH, Patel KP. Altered control of ventilation in streptozotocin-induced diabetic rats. Proc Soc Exp Biol Med. (1994) 207:213–9. doi: 10.3181/00379727-207-43809
97. Ramadan W, Petitjean M, Loos N, Geloen A, Vardon G, Delanaud S. Effect of high-fat diet and metformin treatment on ventilation and sleep apnea in non-obese rats. Respir Physiol Neurobiol. (2006) 150:52–65. doi: 10.1016/j.resp.2005.02.011
98. Perfect MM, Patel PG, Scott RE, Wheeler MD, Patel C, Griffin K, et al. Sleep, glucose, and daytime functioning in youth with type 1 diabetes. Sleep (2012) 35:81–8. doi: 10.5665/sleep.1590
99. Villa MP, Multari G, Montesano M, Pagani J, Cervoni M, Midulla F, et al. Sleep apnoea in children with diabetes mellitus: effect of glycaemic control. Diabetologia (2000) 43:696–702. doi: 10.1007/s001250051365
100. Vgontzas AN, Legro RS, Bixler EO, Grayev A, Kales A, Chrousos GP. Polycystic ovary syndrome is associated with obstructive sleep apnea and daytime sleepiness: role of insulin resistance. J Clin Endocrinol Metab. (2001) 86:517–20. doi: 10.1210/jcem.86.2.7185
101. Gopal M, Duntley S, Uhles M, Attarian H. The role of obesity in the increased prevalence of obstructive sleep apnea syndrome in patients with polycystic ovarian syndrome. Sleep Med. (2002) 3:401–4. doi: 10.1016/S1389-9457(02)00033-3
102. Vgontzas AN, Bixler EO, Chrousos GP. Metabolic disturbances in obesity versus sleep apnoea: the importance of visceral obesity and insulin resistance. J Intern Med. (2003) 254:32–44. doi: 10.1046/j.1365-2796.2003.01177.x
103. Ramadan W, Dewasmes G, Petitjean M, Wiernsperger N, Delanaud S, Geloen A, et al. Sleep apnea is induced by a high-fat diet and reversed and prevented by metformin in non-obese rats. Obesity (Silver Spring) (2007) 15:1409–18. doi: 10.1038/oby.2007.169
104. Polotsky M, Elsayed-Ahmed AS, Pichard L, Harris CC, Smith PL, Schneider H, et al. Effects of leptin and obesity on the upper airway function. J Appl Physiol. (2012) 112:1637–43. doi: 10.1152/japplphysiol.01222.2011
105. O'Donnell CP, Schaub CD, Haines AS, Berkowitz DE, Tankersley CG, Schwartz AR, et al. Leptin prevents respiratory depression in obesity. Am J Respir Crit Care Med. (1999) 159:1477–84. doi: 10.1164/ajrccm.159.5.9809025
106. Farkas GA, Schlenker EH. Pulmonary ventilation and mechanics in morbidly obese Zucker rats. Am J Respir Crit Care Med. (1994) 150:356–62. doi: 10.1164/ajrccm.150.2.8049815
107. Hernandez AB, Kirkness JP, Smith PL, Schneider H, Polotsky M, Richardson RA, et al. Novel whole body plethysmography system for the continuous characterization of sleep and breathing in a mouse. J Appl Physiol. (2012) 112:671–80. doi: 10.1152/japplphysiol.00818.2011
108. Flak JN, Arble D, Pan W, Patterson C, Lanigan T, Goforth PB, et al. A leptin-regulated circuit controls glucose mobilization during noxious stimuli. J Clin Invest. (2017) 127:3103–13. doi: 10.1172/JCI90147
109. Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. (2002) 346:570–8. doi: 10.1056/NEJMoa012437
110. Carr A. HIV lipodystrophy: risk factors, pathogenesis, diagnosis and management. AIDS (2003) 17:S141–8. doi: 10.1097/00002030-200301030-00027
111. Lo Re V, III, Schutte-Rodin S, Kostman JR. Obstructive sleep apnoea among HIV patients. Int J STD AIDS (2006) 17:614–20. doi: 10.1258/095646206778113078
112. Dorey-Stein Z, Amorosa VK, Kostman JR, Lo Re V, III, and Shannon RP. Severe weight gain, lipodystrophy, dyslipidemia, and obstructive sleep apnea in a human immunodeficiency virus-infected patient following highly active antiretroviral therapy. J Cardiometab Syndr. (2008) 3:111–4. doi: 10.1111/j.1559-4572.2008.07552.x
113. Schulz R, Lohmeyer J, Seeger W. Obstructive sleep apnea due to HIV-associated lipodystrophy. Clin Infect Dis. (2003) 37:1398–9. doi: 10.1086/379131
114. Mezzanotte WS, Tangel DJ, White DP. Influence of sleep onset on upper-airway muscle activity in apnea patients versus normal controls. Am J Respir Crit Care Med. (1996) 153:1880–7.
115. Henry RR, Wallace P, Olefsky JM. Effects of weight loss on mechanisms of hyperglycemia in obese non-insulin-dependent diabetes mellitus. Diabetes (1986) 35:990–8.
116. Anandam A, Akinnusi M, Kufel T, Porhomayon J, El-Solh AA. Effects of dietary weight loss on obstructive sleep apnea: a meta-analysis. Sleep Breath (2013) 17:227–34. doi: 10.1007/s11325-012-0677-3
117. Sandoval D. Bariatric surgeries: beyond restriction and malabsorption. Int J Obes (Lond). (2011) 35:S45–9. doi: 10.1038/ijo.2011.148
118. Arble DM, Sandoval DA, Seeley RJ. Mechanisms underlying weight loss and metabolic improvements in rodent models of bariatric surgery. Diabetologia (2015) 58:211–20. doi: 10.1007/s00125-014-3433-3
119. Varela JE, Hinojosa MW, Nguyen NT. Resolution of obstructive sleep apnea after laparoscopic gastric bypass. Obes Surg. (2007) 17:1279–82. doi: 10.1007/s11695-007-9228-6
120. Fernandez-Mendoza J, Calhoun SL. Excessive Daytime Sleepiness: Age, Sleep, Mood, and Metabolic Modulation. London; San Diego, CA; Waltham, MA; Oxford: Academic Press (2015).
121. Panossian LA, Veasey SC. Daytime sleepiness in obesity: mechanisms beyond obstructive sleep apnea–a review. Sleep (2012) 35:605–15. doi: 10.5665/sleep.1812
122. Reutrakul S, Van Cauter E. Sleep influences on obesity, insulin resistance, and risk of type 2 diabetes. Metabolism (2018) 84:56–66. doi: 10.1016/j.metabol.2018.02.010
123. Dixon JB, Schachter LM, O'Brien PE, Jones K, Grima M, Lambert G, et al. Surgical vs. conventional therapy for weight loss treatment of obstructive sleep apnea: a randomized controlled trial. JAMA (2012) 308:1142–9. doi: 10.1001/2012.jama.11580
124. Jennum P, Stender-Petersen K, Rabol R, Jorgensen NR, Chu PL, Madsbad S. The impact of nocturnal hypoglycemia on sleep in subjects with type 2 diabetes. Diabetes Care (2015) 38:2151–7. doi: 10.2337/dc15-0907
125. Newhouse LP, Joyner MJ, Curry TB, Laurenti MC, Man CD, Cobelli C, et al. Three hours of intermittent hypoxia increases circulating glucose levels in healthy adults. Physiol Rep. (2017) 5:e13106. doi: 10.14814/phy2.13106
126. Mesarwi OA, Sharma EV, Jun JC, Polotsky VY. Metabolic dysfunction in obstructive sleep apnea: a critical examination of underlying mechanisms. Sleep Biol Rhythms (2015) 13:2–17. doi: 10.1111/sbr.12078
127. Wu Y, Wang H, Brautigan DL, Liu Z. Activation of glycogen synthase in myocardium induced by intermittent hypoxia is much lower in fasted than in fed rats. Am J Physiol Endocrinol Metab. (2007) 292:E469–75. doi: 10.1152/ajpendo.00486.2006
128. Lam DC, Lam KS, Ip MS. Obstructive sleep apnoea, insulin resistance and adipocytokines. Clin Endocrinol (Oxf). (2015) 82:165–77. doi: 10.1111/cen.12597
129. Martinez-Ceron E, Barquiel B, Bezos AM, Casitas R, Galera R, Garcia-Benito C, et al. Effect of continuous positive airway pressure on glycemic control in patients with obstructive sleep apnea and type 2 diabetes. A randomized clinical trial. Am J Respir Crit Care Med. (2016) 194:476–85. doi: 10.1164/rccm.201510-1942OC
130. Shaw JE, Punjabi NM, Naughton MT, Willes L, Bergenstal RM, Cistulli PA, et al. The effect of treatment of obstructive sleep apnea on glycemic control in type 2 diabetes. Am J Respir Crit Care Med. (2016) 194:486–92. doi: 10.1164/rccm.201511-2260OC
131. Chen L, Kuang J, Pei JH, Chen HM, Chen Z, Li ZW, et al. Continuous positive airway pressure and diabetes risk in sleep apnea patients: a systemic review and meta-analysis. Eur J Intern Med. (2017) 39:39–50. doi: 10.1016/j.ejim.2016.11.010
132. Chopra S, Rathore A, Younas H, Pham LV, Gu C, Beselman A, et al. Obstructive sleep apnea dynamically increases nocturnal plasma free fatty acids, glucose, and cortisol during sleep. J Clin Endocrinol Metab. (2017) 102:3172–81. doi: 10.1210/jc.2017-00619
133. Bernardi L, Bianchi L. Integrated cardio-respiratory control: insight in diabetes. Curr Diab Rep. (2016) 16:107. doi: 10.1007/s11892-016-0804-9
134. Farina MI, Scarani R, Po C, Agosto C, Ottonello G, Benini F. Congenital central hypoventilation syndrome and hypoglycaemia. Acta Paediatr. (2012) 101:e92–6. doi: 10.1111/j.1651-2227.2011.02533.x
135. Shin MK, Han W, Joo H, Bevans-Fonti S, Shiota M, Stefanovski D, et al. Effect of adrenal medullectomy on metabolic responses to chronic intermittent hypoxia in the frequently sampled intravenous glucose tolerance test. J Appl Physiol. (2017) 122:767–74. doi: 10.1152/japplphysiol.00975.2016
136. Shin MK, Yao Q, Jun JC, Bevans-Fonti S, Yoo DY, Han W, et al. Carotid body denervation prevents fasting hyperglycemia during chronic intermittent hypoxia. J Appl Physiol. (2014) 117:765–76. doi: 10.1152/japplphysiol.01133.2013
137. Gao L, Ortega-Saenz P, Garcia-Fernandez M, Gonzalez-Rodriguez P, Caballero-Eraso C, Lopez-Barneo J. Glucose sensing by carotid body glomus cells: potential implications in disease. Front Physiol. (2014) 5:398. doi: 10.3389/fphys.2014.00398
138. MacFarlane PM, Vinit S, Mitchell GS. Enhancement of phrenic long-term facilitation following repetitive acute intermittent hypoxia is blocked by the glycolytic inhibitor 2-deoxyglucose. Am J Physiol Regul Integr Comp Physiol (2018) 314:R135–44. doi: 10.1152/ajpregu.00306.2017
139. Vgontzas AN, Papanicolaou DA, Bixler EO, Hopper K, Lotsikas A, Lin HM, et al. Sleep apnea and daytime sleepiness and fatigue: relation to visceral obesity, insulin resistance, and hypercytokinemia. J Clin Endocrinol Metab. (2000) 85:1151–8. doi: 10.1210/jcem.85.3.6484
140. Rajan P, Greenberg H. Obstructive sleep apnea as a risk factor for type 2 diabetes mellitus. Nat Sci Sleep (2015) 7:113–25. doi: 10.1016/j.amjmed.2009.04.026
141. Nagayoshi M, Punjabi NM, Selvin E, Pankow JS, Shahar E, Iso H, et al. Obstructive sleep apnea and incident type 2 diabetes. Sleep Med. (2016) 25:156–61. doi: 10.1016/j.sleep.2016.05.009
142. Liu CL, Wu CS. Assessing whether the association between sleep apnea and diabetes is bidirectional. Can J Diabetes (2017) 41:197–203. doi: 10.1016/j.jcjd.2016.09.009
143. Beck MK, Westergaard D, Jensen AB, Groop L, Brunak S. Temporal order of disease pairs affects subsequent disease trajectories: the case of diabetes and sleep apnea. Pac Symp Biocomput. (2017) 22:380–9. doi: 10.1142/9789813207813_0036
144. Vgontzas AN, Bixler EO, Chrousos GP. Sleep apnea is a manifestation of the metabolic syndrome. Sleep Med Rev. (2005) 9:211–24. doi: 10.1016/j.smrv.2005.01.006
145. Liu A, Kim SH, Ariel D, Abbasi F, Lamendola C, Cardell J, et al. Does enhanced insulin sensitivity improve sleep measures in patients with obstructive sleep apnea: a randomized, placebo-controlled pilot study. Sleep Med. (2016) 22:57–60. doi: 10.1016/j.sleep.2016.06.005
146. Wang N, Khan SA, Prabhakar NR, Nanduri J. Impairment of pancreatic beta-cell function by chronic intermittent hypoxia. Exp Physiol. (2013) 98:1376–85. doi: 10.1113/expphysiol.2013.072454
147. Lin M, Ai J, Li L, Huang C, Chapleau MW, Liu R, et al. Structural remodeling of nucleus ambiguus projections to cardiac ganglia following chronic intermittent hypoxia in C57BL/6J mice. J Comp Neurol. (2008) 509:103–17. doi: 10.1002/cne.21732
148. Gu H, Lin M, Liu J, Gozal D, Scrogin KE, Wurster R, et al. Selective impairment of central mediation of baroreflex in anesthetized young adult Fischer 344 rats after chronic intermittent hypoxia. Am J Physiol Heart Circ Physiol. (2007) 293:H2809–18. doi: 10.1152/ajpheart.00358.2007
149. Chalacheva P, Thum J, Yokoe T, O'Donnell CP, Khoo MC. Development of autonomic dysfunction with intermittent hypoxia in a lean murine model. Respir Physiol Neurobiol. (2013) 188:143–51. doi: 10.1016/j.resp.2013.06.002
150. Jun JC, Shin MK, Devera R, Yao Q, Mesarwi O, Bevans-Fonti S, et al. Intermittent hypoxia-induced glucose intolerance is abolished by alpha-adrenergic blockade or adrenal medullectomy. Am J Physiol Endocrinol Metab. (2014) 307:E1073–83. doi: 10.1152/ajpendo.00373.2014
151. Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest. (1995) 96:1897–904. doi: 10.1172/JCI118235
152. Sherwani SI, Aldana C, Usmani S, Adin C, Kotha S, Khan M, et al. Intermittent hypoxia exacerbates pancreatic beta-cell dysfunction in a mouse model of diabetes mellitus. Sleep (2013) 36:1849–58. doi: 10.5665/sleep.3214
153. Delarue J, Magnan C. Free fatty acids and insulin resistance. Curr Opin Clin Nutr Metab Care (2007) 10:142–8. doi: 10.1097/MCO.0b013e328042ba90
154. Liu A, Cardell J, Ariel D, Lamendola C, Abbasi F, Kim SH, et al. Abnormalities of lipoprotein concentrations in obstructive sleep apnea are related to insulin resistance. Sleep (2015) 38:793–9. doi: 10.5665/sleep.4678
155. Trayhurn P. Hypoxia and adipocyte physiology: implications for adipose tissue dysfunction in obesity. Annu Rev Nutr. (2014) 34:207–36. doi: 10.1146/annurev-nutr-071812-161156
156. Pasarica M, Sereda OR, Redman LM, Albarado DC, Hymel DT, Roan LE, et al. Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes (2009) 58:718–25. doi: 10.2337/db08-1098
157. Li J, Bosch-Marce M, Nanayakkara A, Savransky V, Fried SK, Semenza GL, et al. Altered metabolic responses to intermittent hypoxia in mice with partial deficiency of hypoxia-inducible factor-1alpha. Physiol Genomics (2006) 25:450–7. doi: 10.1152/physiolgenomics.00293.2005
158. Carreras A, Zhang SX, Almendros I, Wang Y, Peris E, Qiao Z, et al. Resveratrol attenuates intermittent hypoxia-induced macrophage migration to visceral white adipose tissue and insulin resistance in male mice. Endocrinology (2015) 156:437–43. doi: 10.1210/en.2014-1706
159. Dall'Ago P, Fernandes TG, Machado UF, Bello AA, Irigoyen MC. Baroreflex and chemoreflex dysfunction in streptozotocin-diabetic rats. Braz J Med Biol Res. (1997) 30:119–24.
160. Chen X, Zhao T, Huang X, Wu L, Wu K, Fan M, et al. Intermittent hypoxia maintains glycemia in streptozotocin-induced diabetic rats. Cell Stress Chaperones (2016) 21:515–22. doi: 10.1007/s12192-016-0679-3
161. Tian YM, Liu Y, Wang S, Dong Y, Su T, Ma HJ, et al. Anti-diabetes effect of chronic intermittent hypobaric hypoxia through improving liver insulin resistance in diabetic rats. Life Sci. (2016) 150:1–7. doi: 10.1016/j.lfs.2016.02.053
162. Parekh B. The mechanism of dead-in-bed syndrome and other sudden unexplained nocturnal deaths. Curr Diabetes Rev. (2009) 5:210–5. doi: 10.2174/157339909789804387
163. Bin-Jaliah I, Maskell PD, Kumar P. Indirect sensing of insulin-induced hypoglycaemia by the carotid body in the rat. J Physiol. (2004) 556:255–66. doi: 10.1113/jphysiol.2003.058321
164. Ribeiro MJ, Sacramento JF, Gonzalez C, Guarino MP, Monteiro EC, Conde SV. Carotid body denervation prevents the development of insulin resistance and hypertension induced by hypercaloric diets. Diabetes (2013) 62:2905–16. doi: 10.2337/db12-1463
165. Salord N, Fortuna AM, Monasterio C, Gasa M, Perez A, Bonsignore MR, et al. A randomized controlled trial of continuous positive airway pressure on glucose tolerance in obese patients with obstructive sleep apnea. Sleep (2016) 39:35–41. doi: 10.5665/sleep.5312
166. Kalra SP. Circumventing leptin resistance for weight control. Proc Natl Acad Sci USA. (2001) 98:4279–81. doi: 10.1073/pnas.091101498
167. Correia ML, Rahmouni K. Role of leptin in the cardiovascular and endocrine complications of metabolic syndrome. Diabetes Obes Metab. (2006) 8:603–10. doi: 10.1111/j.1463-1326.2005.00562.x
168. Ostlund RE Jr., Yang JW, Klein S, Gingerich R. Relation between plasma leptin concentration and body fat, gender, diet, age, and metabolic covariates. J Clin Endocrinol Metab. (1996) 81:3909–13. doi: 10.1210/jcem.81.11.8923837
169. Gabriely I, Ma XH, Yang XM, Rossetti L, Barzilai N. Leptin resistance during aging is independent of fat mass. Diabetes (2002) 51:1016–21. doi: 10.2337/diabetes.51.4.1016
170. Bixler EO, Vgontzas AN, Lin HM, Ten Have T, Rein J, Vela-Bueno A, et al. Prevalence of sleep-disordered breathing in women: effects of gender. Am J Respir Crit Care Med. (2001) 163:608–13. doi: 10.1164/ajrccm.163.3.9911064
171. BaHammam AS, Pandi-Perumal SR, Piper A, Bahammam SA, Almeneessier AS, Olaish AH, et al. Gender differences in patients with obesity hypoventilation syndrome. J Sleep Res. (2016) 25:445–53. doi: 10.1111/jsr.12400
172. Pierce AM, Brown LK. Obesity hypoventilation syndrome: current theories of pathogenesis. Curr Opin Pulm Med. (2015) 21:557–62. doi: 10.1097/MCP.0000000000000210
173. Sood S, Liu X, Liu H, Horner RL. Genioglossus muscle activity and serotonergic modulation of hypoglossal motor output in obese Zucker rats. J Appl Physiol. (2007) 102:2240–50. doi: 10.1152/japplphysiol.01229.2006
174. Kostopoulos K, Alhanatis E, Pampoukas K, Georgiopoulos G, Zourla A, Panoutsopoulos A, et al. CPAP therapy induces favorable short-term changes in epicardial fat thickness and vascular and metabolic markers in apparently healthy subjects with obstructive sleep apnea-hypopnea syndrome (OSAHS). Sleep Breath (2016) 20:483–93. doi: 10.1007/s11325-015-1236-5
175. Polotsky VY, Li J, Punjabi NM, Rubin AE, Smith PL, Schwartz AR, et al. Intermittent hypoxia increases insulin resistance in genetically obese mice. J Physiol. (2003) 552:253–64. doi: 10.1113/jphysiol.2003.048173
176. Badran M, Abuyassin B, Golbidi S, Ayas N, Laher I. Uncoupling of vascular nitric oxide synthase caused by intermittent hypoxia. Oxid Med Cell Longev. (2016) 2016:2354870. doi: 10.1155/2016/2354870
177. Yang R, Sikka G, Larson J, Watts VL, Niu X, Ellis CL, et al. Restoring leptin signaling reduces hyperlipidemia and improves vascular stiffness induced by chronic intermittent hypoxia. Am J Physiol Heart Circ Physiol. (2011) 300:H1467–76. doi: 10.1152/ajpheart.00604.2009
178. Huang KC, Lin RC, Kormas N, Lee LT, Chen CY, Gill TP, et al. Plasma leptin is associated with insulin resistance independent of age, body mass index, fat mass, lipids, and pubertal development in nondiabetic adolescents. Int J Obes Relat Metab Disord. (2004) 28:470–5. doi: 10.1038/sj.ijo.0802531
179. Fischer S, Hanefeld M, Haffner SM, Fusch C, Schwanebeck U, Kohler C, et al. Insulin-resistant patients with type 2 diabetes mellitus have higher serum leptin levels independently of body fat mass. Acta Diabetol. (2002) 39:105–10. doi: 10.1007/s005920200027
180. Sapin E, Peyron C, Roche F, Gay N, Carcenac C, Savasta M, et al. Chronic intermittent hypoxia induces chronic low-grade neuroinflammation in the dorsal hippocampus of mice. Sleep (2015) 38:1537–46. doi: 10.5665/sleep.5042
181. Moreno-Indias I, Torres M, Montserrat JM, Sanchez-Alcoholado L, Cardona F, Tinahones FJ, et al. Intermittent hypoxia alters gut microbiota diversity in a mouse model of sleep apnoea. Eur Respir J. (2015) 45:1055–65. doi: 10.1183/09031936.00184314
182. Utzschneider KM, Kratz M, Damman CJ, Hullarg M. Mechanisms linking the gut microbiome and glucose metabolism. J Clin Endocrinol Metab. (2016) 101:1445–54. doi: 10.1210/jc.2015-4251
183. Reutrakul S, Van Cauter E. Interactions between sleep, circadian function, and glucose metabolism: implications for risk and severity of diabetes. Ann N Y Acad Sci. (2014) 1311:151–73. doi: 10.1111/nyas.12355
184. Bozic J, Galic T, Supe-Domic D, Ivkovic N, Ticinovic Kurir T, Valic Z, et al. Morning cortisol levels and glucose metabolism parameters in moderate and severe obstructive sleep apnea patients. Endocrine (2016) 53:730–9. doi: 10.1007/s12020-016-0925-6
185. Kaminski RS, Martinez D, Fagundes M, Martins EF, Montanari CC, Rosa DP, et al. Melatonin prevents hyperglycemia in a model of sleep apnea. Arch Endocrinol Metab. (2015) 59:66–70. doi: 10.1590/2359-3997000000012
186. Bertuglia S, Reiter RJ. Melatonin reduces microvascular damage and insulin resistance in hamsters due to chronic intermittent hypoxia. J Pineal Res. (2009) 46:307–13. doi: 10.1111/j.1600-079X.2008.00662.x
187. Arias MA, Garcia-Rio F, Alonso-Fernandez A, Mediano O, Martinez I, Villamor J. Obstructive sleep apnea syndrome affects left ventricular diastolic function: effects of nasal continuous positive airway pressure in men. Circulation (2005) 112:375–83. doi: 10.1161/CIRCULATIONAHA.104.501841
188. Otto ME, Belohlavek M, Romero-Corral A, Gami AS, Gilman G, Svatikova A, et al. Comparison of cardiac structural and functional changes in obese otherwise healthy adults with versus without obstructive sleep apnea. Am J Cardiol. (2007) 99:1298–302. doi: 10.1016/j.amjcard.2006.12.052
189. Drager LF, Bortolotto LA, Figueiredo AC, Silva BC, Krieger EM, Lorenzi-Filho G. Obstructive sleep apnea, hypertension, and their interaction on arterial stiffness and heart remodeling. Chest (2007) 131:1379–86. doi: 10.1378/chest.06-2703
190. Partinen M, Jamieson A, Guilleminault C. Long-term outcome for obstructive sleep apnea syndrome patients. Mortality. Chest (1988) 94:1200–4. doi: 10.1378/chest.94.6.1200
191. Issa FG, Sullivan CE. Reversal of central sleep apnea using nasal CPAP. Chest (1986) 90:165–71. doi: 10.1378/chest.90.2.165
192. Bradley TD, Logan AG, Kimoff RJ, Series F, Morrison D, Ferguson K, et al., Investigators C. Continuous positive airway pressure for central sleep apnea and heart failure. N Engl J Med. (2005) 353:2025–33. doi: 10.1056/NEJMoa051001
193. Rosvall BR, Chin CJ. Is uvulopalatopharyngoplasty effective in obstructive sleep apnea? Laryngoscope (2017) 127:2201–2. doi: 10.1002/lary.26631
194. Mason M, Welsh EJ, Smith I. Drug therapy for obstructive sleep apnoea in adults. Cochrane Database Syst Rev. (2013) 5:CD003002. doi: 10.1002/14651858.CD003002.pub3
195. Prasad B, Radulovacki M, Olopade C, Herdegen JJ, Logan T, Carley DW. Prospective trial of efficacy and safety of ondansetron and fluoxetine in patients with obstructive sleep apnea syndrome. Sleep (2010) 33:982–9.
Keywords: sleep apnea, leptin, glucose, diabetes, obesity, insulin, metabolism, disordered breathing
Citation: Framnes SN and Arble DM (2018) The Bidirectional Relationship Between Obstructive Sleep Apnea and Metabolic Disease. Front. Endocrinol. 9:440. doi: 10.3389/fendo.2018.00440
Received: 03 April 2018; Accepted: 17 July 2018;
Published: 06 August 2018.
Edited by:
Jonathan C. Jun, Johns Hopkins University, United StatesReviewed by:
Hariom Yadav, Wake Forest School of Medicine, United StatesCopyright © 2018 Framnes and Arble. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deanna M. Arble, ZGVhbm5hLmFyYmxlQG1hcnF1ZXR0ZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.