
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 21 June 2018
Sec. Molecular and Structural Endocrinology
Volume 9 - 2018 | https://doi.org/10.3389/fendo.2018.00333
This article is part of the Research Topic Endocrine and Paracrine Role of FGF23 and Klotho in Health and Disease View all 13 articles
 Maren Leifheit-Nestler1*
Maren Leifheit-Nestler1* Beatrice Richter1,2
Beatrice Richter1,2 Melis Basaran1Julia Nespor1Isabel Vogt1Ioana Alesutan3,4
Melis Basaran1Julia Nespor1Isabel Vogt1Ioana Alesutan3,4 Jakob Voelkl3
Jakob Voelkl3 Florian Lang5Joerg Heineke6
Florian Lang5Joerg Heineke6 Stefanie Krick7Dieter Haffner1
Stefanie Krick7Dieter Haffner1
Clinical and experimental studies indicate a possible link between high serum levels of fibroblast growth factor 23 (FGF23), phosphate, and parathyroid hormone (PTH), deficiency of active vitamin D (1,25D) and klotho with the development of pathological cardiac remodeling, i.e., left ventricular hypertrophy and myocardial fibrosis, but a causal link has not been established so far. Here, we investigated the cardiac phenotype in klotho hypomorphic (kl/kl) mice and Hyp mice, two mouse models of elevated FGF23 levels and klotho deficiency, but differing in parameters of mineral metabolism, by using histology, quantitative real-time PCR, immunoblot analysis, and serum and urine biochemistry. Additionally, the specific impact of calcium, phosphate, PTH, and 1,25D on hypertrophic growth of isolated neonatal rat cardiac myocytes was investigated in vitro. Kl/kl mice displayed high serum Fgf23 levels, increased relative heart weight, enhanced cross-sectional area of individual cardiac myocytes, activated cardiac Fgf23/Fgf receptor (Fgfr) 4/calcineurin/nuclear factor of activated T cell (NFAT) signaling, and induction of pro-hypertrophic NFAT target genes including Rcan1, bMHC, brain natriuretic peptide (BNP), and atrial natriuretic peptide (ANP) as compared to corresponding wild-type (WT) mice. Investigation of fibrosis-related molecules characteristic for pathological cardiac remodeling processes demonstrated ERK1/2 activation and enhanced expression of Tgf-β1, collagen I, and Mmp2 in kl/kl mice than in WT mice. In contrast, despite significantly elevation of serum and cardiac Fgf23, and reduced renal klotho expression, Hyp mice showed no signs of pathological cardiac remodeling. Kl/kl mice showed enhanced serum calcium and phosphate levels, while Hyp mice showed unchanged serum calcium levels, lower serum phosphate, and elevated serum iPTH concentrations compared to corresponding WT mice. In cultured cardiac myocytes, treatment with both calcium or phosphate significantly upregulated endogenous Fgf23 mRNA expression and stimulated hypertrophic cell growth and expression of pro-hypertrophic genes. The treatment with PTH induced hypertrophic cell growth only, and stimulation with 1,25D had no significant effects. In conclusion, our data indicate that Hyp mice, in contrast to kl/kl mice appear to be protected from pathological cardiac remodeling during conditions of high FGF23 levels and klotho deficiency, which may be due, at least in part, to differences in mineral metabolism alterations, i.e., hypophosphatemia and lack of hypercalcemia.
Patients with chronic kidney disease (CKD) (1–3) and patients with heart failure (4–8) display abnormalities in mineral metabolism, which are associated with cardiovascular diseases (CVD). The phosphaturic hormone fibroblast growth factor (FGF) 23 is synthesized and secreted by osteocytes in response to various stimuli (9) to increase renal phosphate excretion by inhibiting the expression of sodium–phosphate co-transporters NaPi-2a and NaPi-2c (10, 11). Moreover, FGF23 suppresses parathyroid hormone (PTH) expression and secretion (12, 13) and inhibits renal formation of 1,25(OH)2D3 (1,25D, calcitriol) by downregulating 1α-hydroxylase (Cyp27b1) and upregulating 24-hydroxylase (Cyp24a1) (11, 14, 15). Klotho is an endocrine hormone primarily produced in the kidney (16) and functions as co-receptor for FGF23, whereby it increases the binding affinity of FGF23 to its FGF receptors (FGFRs) and, therefore, mediates different signaling pathways including renal phosphate and 1,25D metabolism (17–19).
With declining kidney function, the endocrine network of mineral metabolism becomes altered, which leads to elevated serum levels of FGF23, phosphate, and PTH in addition to a deficiency of 1,25D and klotho (20). All these parameter represent key risk factors for the development of endothelial dysfunction, left ventricular hypertrophy (LVH), myocardial fibrosis, and contribute to the overall cardiovascular mortality in CKD and non-CKD patients (1, 6, 21–26). We previously showed that FGF23 directly targets the heart and promotes LVH by binding to FGFR4 on cardiac myocytes activating phospholipase C gamma (PLCγ)/calcineurin/nuclear factor of activated T cells (NFAT) to induce pro-hypertrophic gene expression independently of its co-receptor klotho that is not expressed in the heart (16, 27–29). Moreover, we demonstrated that FGF23 contributes to pathologic cardiac remodeling and promotes the pro-fibrotic crosstalk between cardiac myocytes and fibroblasts resulting in enhanced cardiac hypertrophy and fibrosis in the absence of klotho (30).
Besides high FGF23 levels, klotho deficiency also seems to be associated with cardiac dysfunction in humans and rodents (25, 29, 31), and rescuing the availability of klotho by genetic overexpression or intravenous delivery of soluble klotho shows beneficial outcomes including amelioration of cardiac hypertrophy in klotho-deficient uremic mice and suppression of cardiac fibroblast activation and collagen synthesis (31–33). In addition, klotho ameliorates FGF23-mediated oxidative stress by inducing nitric oxide synthesis and degrading reactive oxygen species in endothelial cells (34). Furthermore, klotho protects against myocardial hypertrophy by reducing indoxyl sulfate-mediated oxidative stress in vitro and in vivo (35).
To date, the impact of high phosphate levels for the development of cardiomyopathy in the setting of high FGF23 levels is controversially discussed. Increased serum phosphate is associated with cardiovascular mortality in CKD (25, 36) and FGF23 mediates renal phosphate excretion only in the presence of klotho (19). Normalization of serum phosphate and FGF23 levels by dietary phosphate restriction in 5/6 nephrectomized klotho-deficient mice did not abrogate the development of cardiac hypertrophy suggesting that reduced klotho contributes to uremic cardiomyopathy independent of phosphate and FGF23 (32). In contrast, cardiac hypertrophy and fibrosis correlated with high phosphate levels in uremic and non-uremic klotho-deficient mice, and genetically induced elevation of soluble klotho ameliorated phosphate-induced hypertrophic growth of cardiac myocytes in vivo (31). The association between FGF23 and cardiac remodeling in klotho-deficient animals suggests that FGF23 affects cardiac hypertrophy and fibrosis only in states of high phosphate and klotho deficiency. Thus, the interplay between increased FGF23, phosphate, and reduced klotho participates in the development of CVD. However, the direct impact of each single factor on pathologic cardiac remodeling is still not clear. Here, we test whether elevated FGF23 levels in klotho deficiency invariably leads to cardiac hypertrophy and fibrosis and whether different alterations in mineral metabolism play a role in these effects. Therefore, we compared two mouse models with genetically elevated serum FGF23 levels and reduced renal klotho availability, Hyp mice, and klotho hypomorphic (kl/kl) mice, with respect to the development of a pathologic cardiac phenotype. The Hyp mouse is a murine homolog to the human disease of X-linked dominant hypophosphatemia resembling elevated circulating FGF23 concentrations resulting in renal phosphate wasting, hypophosphatemia, decreased renal 1,25D synthesis, and defects in bone mineralization (37, 38). Klotho hypomorphic mice present with high plasma concentrations of FGF23, 1,25D, and phosphate, resulting in severe soft tissue calcification and premature aging (16). In addition, we used neonatal rat ventricular myocytes (NRVM) to study the role of parameters of altered mineral metabolism including calcium, phosphate, PTH, and 1,25D for the induction of cardiac hypertrophy.
All experimental procedures were approved by the State Office committee on animal welfare Lower Saxony for Hyp mice and Baden-Württemberg for kl/kl mice and performed in accordance with national animal protection guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
Hemizygous male B6.Cg-PhexHyp/J (Hyp) mice (strain no. 000528; Jackson Laboratory, Bar Harbor, ME, USA) with X-linked semidominant mutation in the Phex gene causing defects in phosphate metabolism and male wild-type (WT) littermates produced from breeding of heterozygous females (Hyp/+) with C57BL/6J WT males were used in this study. Mice were fed normal rodent chow containing 600 IU/kg cholecalciferol, 0.7% calcium, and 0.5% phosphate (#1324, Altromin, Lage, Germany) ad lib. The origin of homozygous klotho hypomorphic mice (kl/kl), breeding, and genotyping were described previously (16). Five to 13 mice per group were used in this study and sacrificed at 6–8 weeks of age. Blood was collected via cardiac puncture, and hearts were isolated and prepared for histological and biochemical analyses. For serology, blood was centrifuged at 4°C and 13,000 rcf for 20 min. Serum supernatant was collected, stored at −80°C, and subsequently analyzed via ELISA techniques for C-term FGF23, intact FGF23, and 1-84 PTH (each from Immutopics, San Clemente, CA, USA), and spectrophotometrically for calcium and phosphate (each from DiaSys Diagnostic Systems GmbH, Holzheim, Germany).
Neonatal rat ventricular myocytes (NRVM) were isolated using a standard isolation system (Worthington Biochemical Corporation) (39). In brief, hearts from 1- to 2-days-old Sprague Dawley rats were harvested, minced in calcium- and magnesium-free Hank’s Balanced Salt Solution (HBSS) followed by tissue digestion with 50 µg/mL trypsin at 4°C for 20–24 h. Soybean trypsin inhibitor in HBSS was added and the tissue was further digested with collagenase (in Leibovitz L-15 medium) under slow rotation (15 rpm) at 37°C for 45 min. Cells were homogenized and resuspended 20 times with a standard 10 mL serological pipette and filtered twice through a 70-µm cell strainer (BD Falcon). After incubation at room temperature for 20 min, cells were centrifuged at 100 × g for 5 min and cell pellet was re-suspended in plating medium Dulbecco’s Modified Eagle Medium (DMEM) with 20% M199 (Invitrogen), 15% fetal bovine serum (FBS; Invitrogen), and 1% penicillin/streptomycin solution (P/S; Invitrogen).
Cells were plated on glass and plastic surfaces pre-coated with laminin (Invitrogen; 10 µg/mL in PBS) at room temperature for 1 h. For immunofluorescence analysis, 3 × 105 cells were seeded per well on pre-coated glass coverslips in 24-well plates and for RNA isolations, 8 × 105 cells were seeded in 6 cm-culture dishes. Cells were left in plating medium at 37°C over night. After starvation in maintenance medium DMEM with 20% M199, 1% insulin-transferrin-sodium selenite solution (ITS; Sigma-Aldrich), and 1% P/S, isolated NRVM were stimulated in duplicates in maintenance medium in the presence of 3 mM calcium, 1 mM phosphate, 10 nM PTH, 10 nM calcitriol, or vehicle, respectively, for 48 h. At least six independent cell isolations were used for all experiments.
Formalin-fixed paraffin-embedded heart tissue samples were deparaffinized in xylene, hydrated through a series of graded alcohols. For the quantification of cardiac myocyte size, fixed cardiac mid-chamber (MC) (33) sections were incubated with wheat germ agglutinin (WGA) Alexa Fluor555 (Invitrogen) at 5 µg/mL in PBS for 1 h to visualize cellular borders of individual cardiac myocytes. 4′,6-diamidino-2-phenylindole (DAPI; 0.2 µg/mL) was used for nuclear staining in the dark for 15 min. Representative immunofluorescence images of cardiac tissue were taken on a Zeiss AxioObserver Z1 microscope (Carl Zeiss) with a Plan-Apo 63×/N.A. 1.4 oil objective. ZEN Software (Carl Zeiss) was used to measure myocardial cross-sectional area in square micrometer of 100 cardiac myocytes in average.
For the detection of myocardial fibrosis and visualization of fibrillar collagen fibers, MC sections were stained with picrosirius red (Sigma-Aldrich) as described previously (28). In brief, deparaffinized MC sections were incubated with picrosirius red for 60 min followed by mounting in non-aqueous mounting medium and analyzed by bright field and polarized microcopy using a Keyence BZ-9000® microscope with a 20× objective. In addition, for polarized light microscopy, two polarization filters were used in a rectangular orientation, positioned above and below the sample.
To analyze hypertrophic growth of isolated NRVM on laminin-coated glass coverslips after 48 h of treatment with calcium, phosphate, PTH, and 1,25D, NRVM were fixed in 2% PFA in 5 mg/mL sucrose for 5 min and permeabilized in 1% Triton X-100 in PBS for 10 min followed by incubation with mouse monoclonal antibody against sarcomeric α-actinin (1:1,000 dilution; EA-53; Sigma-Aldrich). Cy3-conjugated goat-anti mouse (Jackson Immuno Research) was used as secondary antibody at 1:300 dilution. To visualize nuclei, fixed cells were incubated with DAPI (400 ng/mL in PBS) for 10 min. Immunofluorescence images were taken on a Zeiss AxioObserver Z1 microscope (Carl Zeiss) with a 40× objective. Myocyte cross-sectional area was measured based on α-actinin-positive staining using Carl Zeiss ZEN software. At least 100 cells per stimulation were quantified for the determination of cardiac myocyte cross-sectional area.
For RNA isolation of snap-frozen mouse myocardial tissue or NRVM, RNeasy Mini Kit (Qiagen) was used according to the manufacturer protocol. Total RNA (500 ng) was transcribed into cDNA using QuantiTect Reverse Transcription Kit (Qiagen) and qRT-PCR was performed in triplicates (20 ng cDNA per reaction) with appropriate primers in 5′–3′orientation (Table 1) using QuantiFAST SYBR Green PCR Kit including ROX dye (Qiagen). Forty-five cycles (95°C, 10 s; 60°C, 30 s) were performed on an ABI prism 7900HT Fast system (Applied Biosystems). Relative gene expression values, adjusted for the same CT-threshold and baseline settings, were calculated according to the 2−ΔΔCT method (40) using Gapdh as housekeeping gene (SDS Software v2.3, Applied Biosystems).

Table 1. The following oligonucleotides shown in 5′–3′orientation were used as primers for quantitative RT-PCR analyses.
For protein extraction from snap-frozen myocardial specimens of the left ventricle of Hyp and kl/kl mice, 30 mg tissue was homogenized in 250 µL RIPA extraction buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% Na-desoxycholate, 1 mM EDTA) with protease (P8340, Sigma-Aldrich) and phosphatase (S65208, Sigma-Aldrich) inhibitors, sonicated two times and incubated on ice for 30 min. Cell lysates were centrifuged at 13,000 rpm and 4°C for 10 min, protein concentration was quantified using BCA test, 100 µg total protein was boiled in sample buffer and analyzed by SDS-PAGE and immunoblotting. Antibodies to FGF23 (1:1,000; ab98000, Abcam), Calcineurin A (1:10,000; ab52761, Abcam), NFATc4 (1:200; sc-13036, Santa Crus Biotechnology Inc.), Rcan1 (1:1,000; SAB2101967, Sigma), pERK1/2 and ERK1/2 (each 1:1,000; #4370 and #9107, Cell Signaling Technology), connective tissue growth factor (Ctgf) (1:1,000; ab6992, Abcam), and GAPDH (1:1,000; #2118, Cell Signaling Technology) were used as primary antibodies in 5% BSA in LI-COR blocking buffer/TBS (1:2) over night, and IRDye® secondary goat-anti-mouse and goat-anti-rabbit (LI-COR Biosciences) were used as secondary antibodies. Primary antibodies to collagen 1 (1:1,000; ab34710, Abcam), MMP2 (1:500; ab92536, Abcam), and TGF-β1 (1:500; ab179695, Abcam) were used in 5% milk in TBS-T over night followed by using HRP-labeled secondary antibodies and enhanced chemiluminescent substrate. The Odyssey Imager (LI-COR Biosciences) was used for protein detection and quantification.
Data are presented as mean ± SEM if not indicated otherwise. Comparison between groups of Hyp or kl/kl mice and its respective WT littermates were done by unpaired t-test in case of normally distributed data or Mann–Whitney U test in case of non-Gaussian distributions, respectively (GraphPad Prism Software version 6.0). Different groups of NRVM were compared by one-way ANOVA and Bonferroni’s multiple comparison post hoc tests. Two-tailed P values of <0.05 were considered statistically significant.
First, we investigated circulating and cardiac levels of Fgf23 in Hyp and kl/kl mice. Hyp mice presented with 10-fold higher levels of both serum C-term and intact Fgf23 when compared to WT littermates. In kl/kl mice, circulating C-term and intact Fgf23 concentrations were 2,000- and 3,300-fold higher compared to respective WT controls (Figures 1A,B). In the heart tissue of Hyp and kl/kl mice, endogenous cardiac Fgf23 mRNA expression and full-length biological active Fgf23 protein were significantly upregulated compared to respective WT controls (Figures 1C,D). Thus, both mouse models showed elevated circulating and cardiac Fgf23 levels, although serum Fgf23 concentrations appeared to be higher in kl/kl compared to Hyp mice.
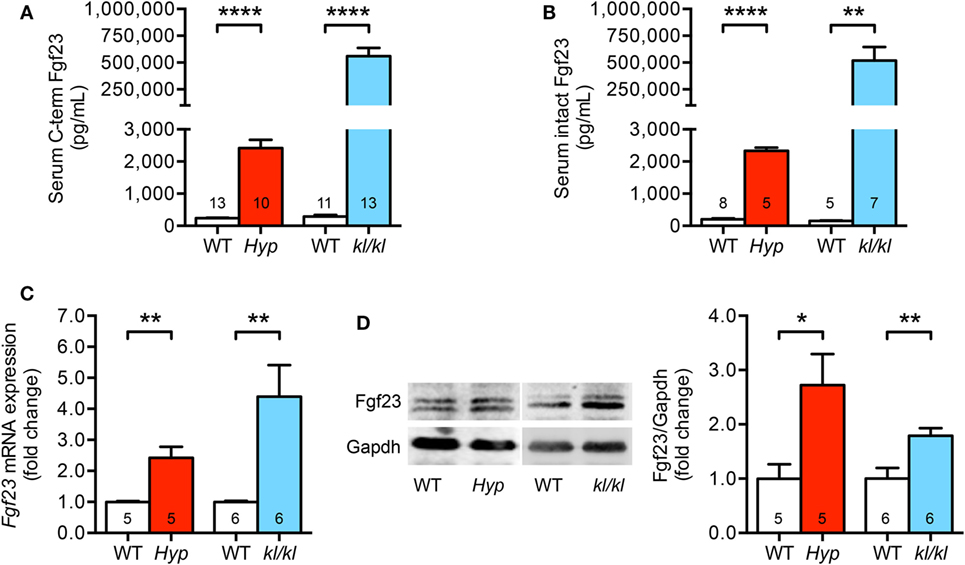
Figure 1. Serum Fgf23 levels and cardiac Fgf23 expression are elevated in Hyp and kl/kl mice. (A) Increased serum C-term Fgf23 levels in both Hyp and kl/kl mice as compared to respective wild-type mice. (B) Elevation of intact Fgf23 concentrations in the serum of Hyp and kl/kl mice than in respective control mice. (C) Fgf23 mRNA levels are increased in cardiac tissue of both mouse lines demonstrated by quantitative real-time PCR after normalization to Gapdh. (D) Representative immunoblot and respective quantification show the elevation of Fgf23 protein levels in total heart lysates of Hyp and kl/kl animals (Gapdh served as loading control). Values are presented as mean ± SEM; numbers in each bar graph represent the n-values used for the respective measurement; *P < 0.05, **P < 0.01, ****P < 0.0001.
Next, we investigated the development of cardiac hypertrophy in both mouse models. Due to growth retardation caused by abnormal mineral metabolism in Hyp mice, we calculated the relative heart weight on the basis of heart weight to body weight ratio. When compared to WT controls, the relative heart weight of kl/kl mice was 0.84 ± 0.15 mg/g higher (Figure 2A). In addition, kl/kl mice presented with 282 ± 13 μm2 cross-sectional area of individual cardiac myocytes compared to 189 ± 14 μm2 in WT littermates demonstrated by WGA staining of the myocyte cell borders in heart tissue sections. In contrast, Hyp mice showed unaltered cardiac myocytes cell size compared to WT littermates (202 ± 19 versus 178 ± 13 μm2; P = 0.287) (Figure 2B). Thus, it appears that only kl/kl mice display the development of cardiac hypertrophy.
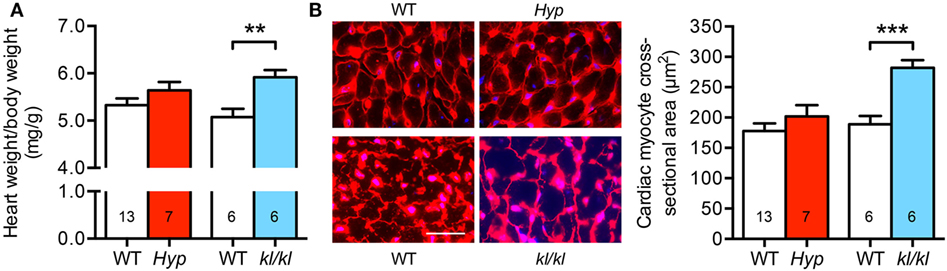
Figure 2. Development of cardiac hypertrophy in kl/kl mice. (A) The relative heart weight to body weight ratio is increased only in kl/kl mice. (B) Immunofluorescence co-staining for wheat germ agglutinin (WGA) Alexa Fluor555 (red) and cell nuclei (blue) demonstrates enlarged size of cardiac myocytes in kl/kl mice but not in Hyp mice (magnification, 63×; scale bar, 25 µm). Values are presented as mean ± SEM; numbers in each bar graph represent the n-values used for the respective measurement; **P < 0.01, ***P < 0.001.
Since Fgfr1 and Fgfr4 are the main Fgfrs expressed in the myocardial tissue of humans and rodents (27, 29), and each induces pathological cardiac remodeling through a different pathway, we next wanted to know whether both receptors were altered in the heart of Hyp and kl/kl mice. Cardiac Fgfr1 mRNA levels were unchanged in kl/kl mice, but tended to be lower in Hyp mice; however, the difference did not reach statistical significance (P = 0.184) (Figure 3A). In contrast, cardiac Fgfr4 mRNA expression was 3.8 ± 0.3-fold upregulated in kl/kl mice compared to controls. In Hyp mice, a trend of Fgfr4 elevation was recognized when compared to their respective WT controls (2.4 ± 0.7-fold; P = 0.097) (Figure 3B). Calcineurin protein levels, activated by Fgfr4 via PLCγ, were 1.6 ± 0.1-fold enhanced in heart tissue lysates from kl/kl mice (P = 0.017), but not from Hyp mice (Figure 3C). Moreover, the transcription factor Nfat was clearly de-phosphorylated (0.41 ± 0.16-fold; P = 0.027) and thereby activated in heart tissue only from kl/kl mice compared to WT littermates (Figure 3D). Taken together, the pro-hypertrophic Fgfr4/calcineurin/NFAT signaling pathway is induced in kl/kl, but not in Hyp mice.
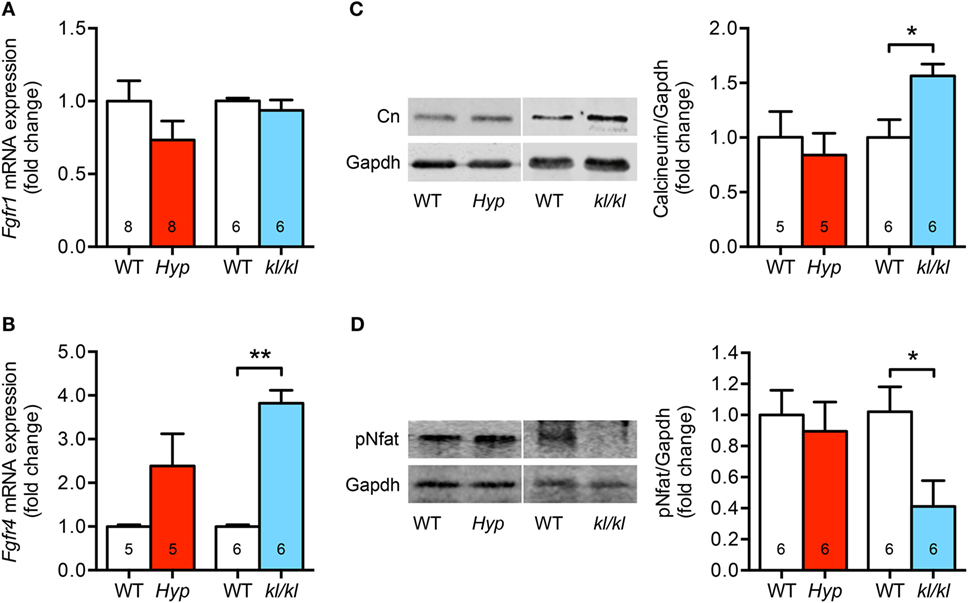
Figure 3. The cardiac Fgfr4/calcineurin/nuclear factor of activated T cell signaling is activated in kl/kl mice. (A) Quantitative real-time PCR analysis reveals no significant differences in Fgfr1 mRNA expression in cardiac tissue from both mouse lines compared to their respective wild-type controls. (B) Fgfr4 mRNA expression is elevated in hearts from kl/kl mice as well as Hyp mice; however, the differences are statistically significant only for the kl/kl animals. (C) Representative immunoblot and quantification indicate an upregulation of cardiac calcineurin protein expression only for kl/kl mice (Gapdh served as loading control). (D) Representative immunoblot and quantification of Nfat activation show reduced Nfat phosphorylation in cardiac tissue from kl/kl mice. Values are presented as mean ± SEM; numbers in each bar graph represent the n-values used for the respective measurement; *P < 0.05, **P < 0.01.
In order to evaluate pro-hypertrophic genes targeted by activated calcineurin/NFAT pathway, we analyzed the expression of regulator of calcineurin 1 (Rcan1), brain natriuretic peptide (BNP), atrial natriuretic peptide (ANP), and both alpha- and beta-myosin heavy chain (aMHC, bMHC) in myocardial tissue of these two mouse models. In kl/kl mice, cardiac Rcan1 expression was 2.1-fold upregulated on both mRNA and protein level when compared to WT controls but not in Hyp mice (Figures 4A,B). The switch to a fetal cardiac gene expression pattern was only detected in kl/kl mice demonstrated by an enhanced bMHC to aMHC ratio when compared to respective WT littermates (Figure 4C). Moreover, the mRNA expression of the pro-hypertrophic factors ANP and BNP was clearly induced in kl/kl mice (5.3 ± 1.1-fold, P = 0.0035; 2.0 ± 0.3-fold, P = 0.0094) (Figures 4D,E). Interestingly, BNP levels were even 0.6-fold lower in Hyp mice in comparison to their WT controls (P = 0.010).
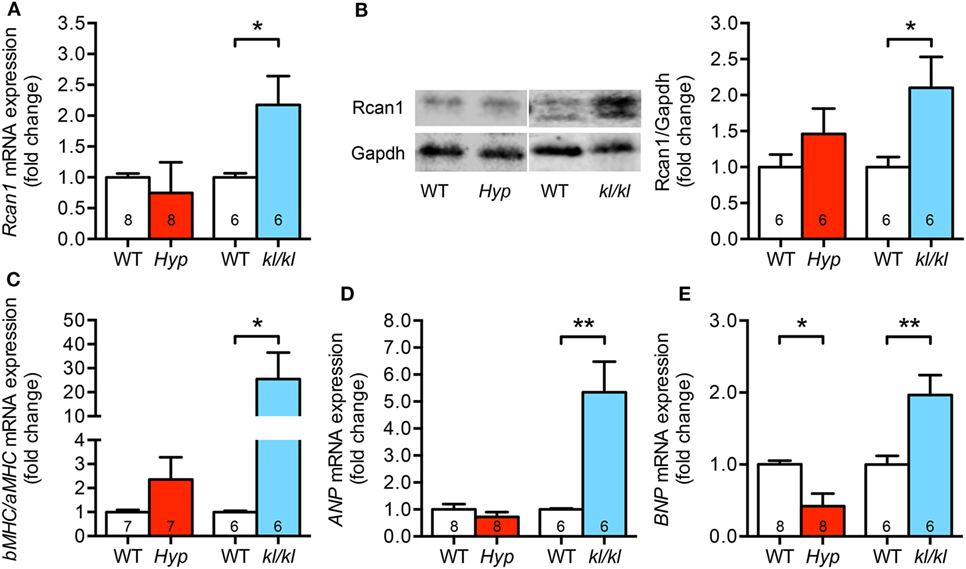
Figure 4. Kl/kl mice show an upregulation of cardiac pro-hypertrophic nuclear factor of activated T cell target genes. (A,B) Rcan1 expression is increased only in cardiac tissue from kl/kl mice demonstrated by quantification of mRNA levels by real-time PCR and protein levels by western blotting. (C) Quantification of bMHC and aMHC mRNA expression shows the induction of fetal gene program in heart tissue from kl/kl mice but not from Hyp mice. (D) ANP mRNA expression is induced in the myocardium of kl/kl mice in relation to respective wild-type animals. (E) Cardiac BNP mRNA expression is reduced in Hyp mice, but upregulated in kl/kl mice. Values are presented as mean ± SEM; numbers in each bar graph represent the n-values used for the respective measurement; *P < 0.05, **P < 0.01.
One major characteristic of pathologic cardiac hypertrophy is the concomitant development of myocardial fibrosis (3, 41). Therefore, we next investigated collagen synthesis and remodeling as well as the expression levels of fibrosis-related molecules involved in the transforming growth factor-beta (Tgf-β) signaling cascade. Demonstrated by picrosirius red staining of myocardial tissue sections, the accumulation of fibrillar collagens was clearly increased in kl/kl mice but not in Hyp mice (Figure 5A). This was confirmed by a 2.0-fold enhanced mRNA expression of both collagen 1 (Col1) and matrix metallopeptidase 2 (Mmp2) only in kl/kl mice compared to WT littermates (Figures 5B,C). One major pro-fibrotic pathway in cardiomyopathy among others is the induction of connective tissue growth factor (Ctgf) via Tgf-β1-mediated activation of extracellular signal-regulated kinases (ERK)1/2 (41–43). Only in kl/kl mice, Tgfb1 mRNA levels were 3.0-fold upregulated compared to respective WT controls (Figure 5D). The enhanced pro-fibrotic gene expressions in kl/kl mice were confirmed by higher Col1, Mmp2, and Tgf-β1 protein expressions in myocardial tissue of kl/kl mice but not in Hyp mice (Figures 5E–G). Moreover, ERK1/2 was activated in kl/kl mice demonstrated by a 2.6-fold enhanced phosphorylation in total heart tissue lysates (Figures 5H,I). Finally, Ctgf protein expression was 1.5-fold induced in kl/kl mice but not in Hyp mice (Figures 5H,J). In summary, interstitial cardiac fibrosis was only present in kl/kl mice.
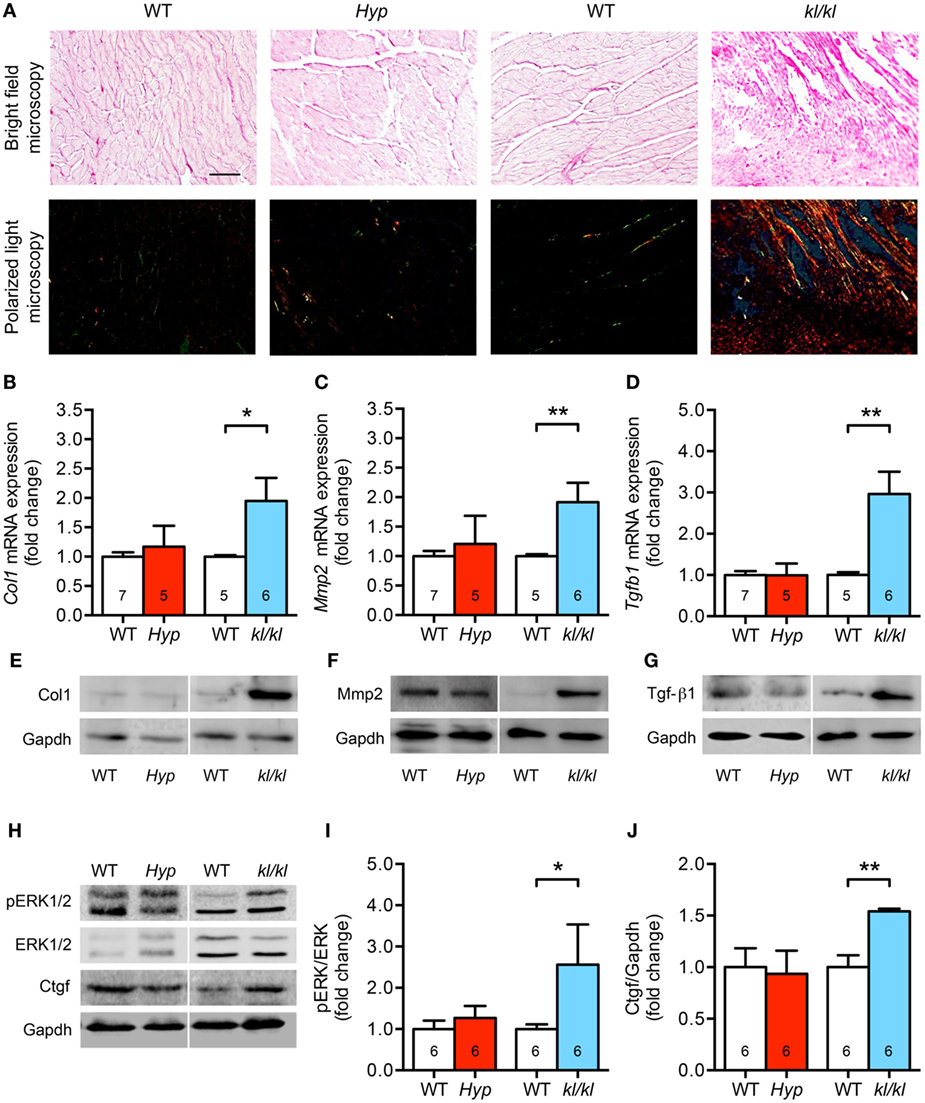
Figure 5. Development of cardiac fibrosis in kl/kl mice but not in Hyp mice. (A) Representative images of murine myocardial sections stained for fibrillar collagens by picrosirius red demonstrate increased accumulation of fibrillar collagens in kl/kl mice but not in Hyp mice (magnification, 20×; scale bar, 50 µm). (B) The mRNA levels of Col1 are higher in heart tissue of kl/kl mice and unchanged in Hyp animals. (C) Likewise, kl/kl mice show an enhanced Mmp2 mRNA expression. (D) The mRNA expression of Tgfb1 is significantly different between both mouse lines whereby it is upregulated in kl/kl animals. The protein expression of (E) Col1, (F) Mmp2, and (G) Tgf-β1 in heart tissue lysates is markedly enhanced in kl/kl mice but not in Hyp mice. (H) ERK1/2 is only activated in kl/kl mice demonstrated by increased phosphorylated protein levels in immunoblot analysis followed by (I) densitometric quantification. (J) The levels of Ctgf protein are increased in kl/kl mice but not in Hyp mice. Values are presented as mean ± SEM; numbers in each bar graph represent the n-values used for the respective measurement; *P < 0.05, **P < 0.01.
As we presented in this study, circulating Fgf23 levels and cardiac Fgf23 synthesis were significantly enhanced in both Hyp and kl/kl mice. However, only kl/kl mice developed cardiac hypertrophy and fibrosis. To identify additional causes mediating pathologic cardiac remodeling in kl/kl mice and to evaluate why Hyp mice might be protected from cardiovascular disease, we next investigated parameters of mineral metabolism. Serum calcium levels were normal to slightly reduced in Hyp mice (8.7 ± 0.8 versus 9.6 ± 0.5 mg/dL; P = 0.327) but significantly enhanced in kl/kl mice compared to WT littermates (11.3 ± 0.5 versus 9.5 ± 0.3 mg/dL; P = 0.007) (Figure 6A). In addition, Hyp mice displayed hypophosphatemia (5.6 ± 0.5 versus 8.7 ± 0.3 mg/dL; P < 0.0001) while kl/kl mice were hyperphosphatemic (9.9 ± 0.6 versus 7.4 ± 0.2 mg/dL; P = 0.0012) (Figure 6B). Furthermore, Hyp mice showed 6.5-fold enhanced serum PTH concentrations compared to WT animals (Figure 6C), whereas previous reports describe suppressed PTH levels in kl/kl mice (44, 45). Others and we showed previously that Hyp mice are deficient for 1,25D, whereas kl/kl mice have significantly higher serum 1,25D levels (37, 44–46). Finally, Hyp mice showed markedly reduced renal Klotho mRNA expression levels (0.6 ± 0.07-fold; P = 0.0012), which were barely detectable in renal tissue of kl/kl mice (Figure 6D). Taken together, despite enhanced circulating Fgf23 levels in both mouse models, serum calcium, phosphate, and 1,25D levels were increased in kl/kl mice. In Hyp mice, serum calcium was unchanged and serum phosphate and 1,25D levels were decreased compared to their WT littermates. In contrast, serum PTH levels were increased in Hyp mice, but suppressed in kl/kl mice.
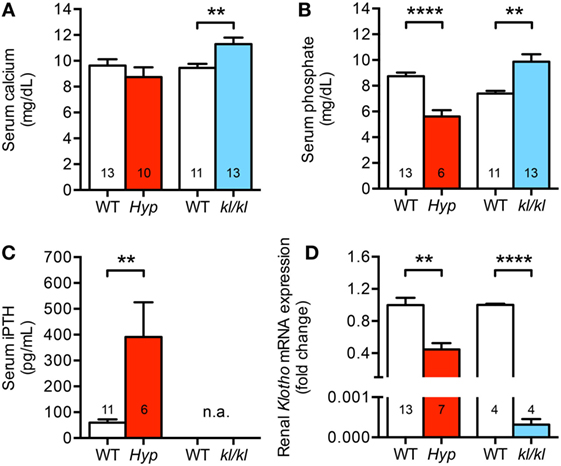
Figure 6. Different mineral metabolism between Hyp and kl/kl mice. (A) Serum calcium levels are normal to slightly reduced in Hyp mice and elevated in kl/kl animals. (B) Hyp mice are hypophosphatemic and kl/kl mice display hyperphosphatemia. (C) Serum parathyroid hormone (PTH) levels in Hyp mice are extensively elevated. For kl/kl mice, the PTH levels in the serum were not available (n.a.). (D) Renal Klotho mRNA expression is reduced in Hyp mice than in respective wild-type mice and barely detectable in kl/kl mice. Values are presented as mean ± SEM; numbers in each bar graph represent the n-values used for the respective measurement; **P < 0.01, ****P < 0.0001.
Since both animal models significantly differ with respect to serum calcium, phosphate, PTH, and 1,25D levels but only kl/kl mice developed cardiac remodeling processes (Table 2), we next investigated whether each single parameter was able to affect the endogenous Fgf23 expression and to promote cardiac hypertrophy in vitro. Therefore, we stimulated NRVM, which do not express klotho, with calcium, phosphate, PTH, and 1,25D and evaluated hypertrophic growth of individual cardiac myocytes, changes of endogenous Fgf23 expression, and finally induction of pro-hypertrophic markers. Treatment of NRVM with calcium, phosphate, and PTH but not with 1,25D significantly enhanced cardiac myocytes cross-sectional area demonstrated by immunocytochemical staining with anti-α-actinin antibody followed by quantification of the cell size (Figures 7A,B). Interestingly, Fgf23 mRNA levels of NRVM were significantly induced by calcium and phosphate treatment but neither by PTH nor by 1,25D (Figure 7C), which is in contrast to their Fgf23-stimulating properties reported for bone (9, 47). Rcan1 mRNA expression was significantly induced by treatment of NRVM with phosphate and tended to be increased by calcium treatment, a difference, however, not reaching statistical significance (Figure 7D). Moreover, the mRNA levels of bMHC, BNP, and ANP were significantly induced by both calcium and phosphate but not significantly modified after stimulation with PTH or 1,25D (Figures 7E–G). Taken together, among the altered parameters of mineral metabolism differing between Hyp and kl/kl mice, only calcium and phosphate clearly induced cardiac Fgf23 expression and promoted myocytes hypertrophy in vitro, suggesting that elevated phosphate and calcium levels may promote Fgf23-mediated cardiac hypertrophy in states of klotho deficiency.
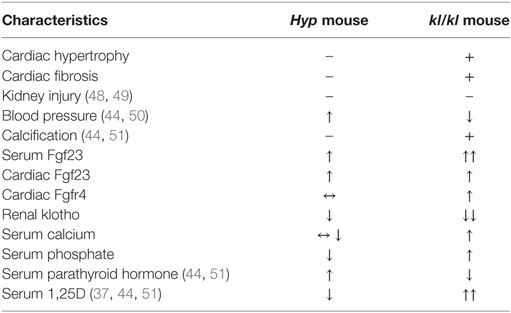
Table 2. Differences between Hyp and kl/kl mice regarding characteristics of cardiovascular and kidney disease, cardiac FGF23/FGFR4 system, and parameters of mineral metabolism.
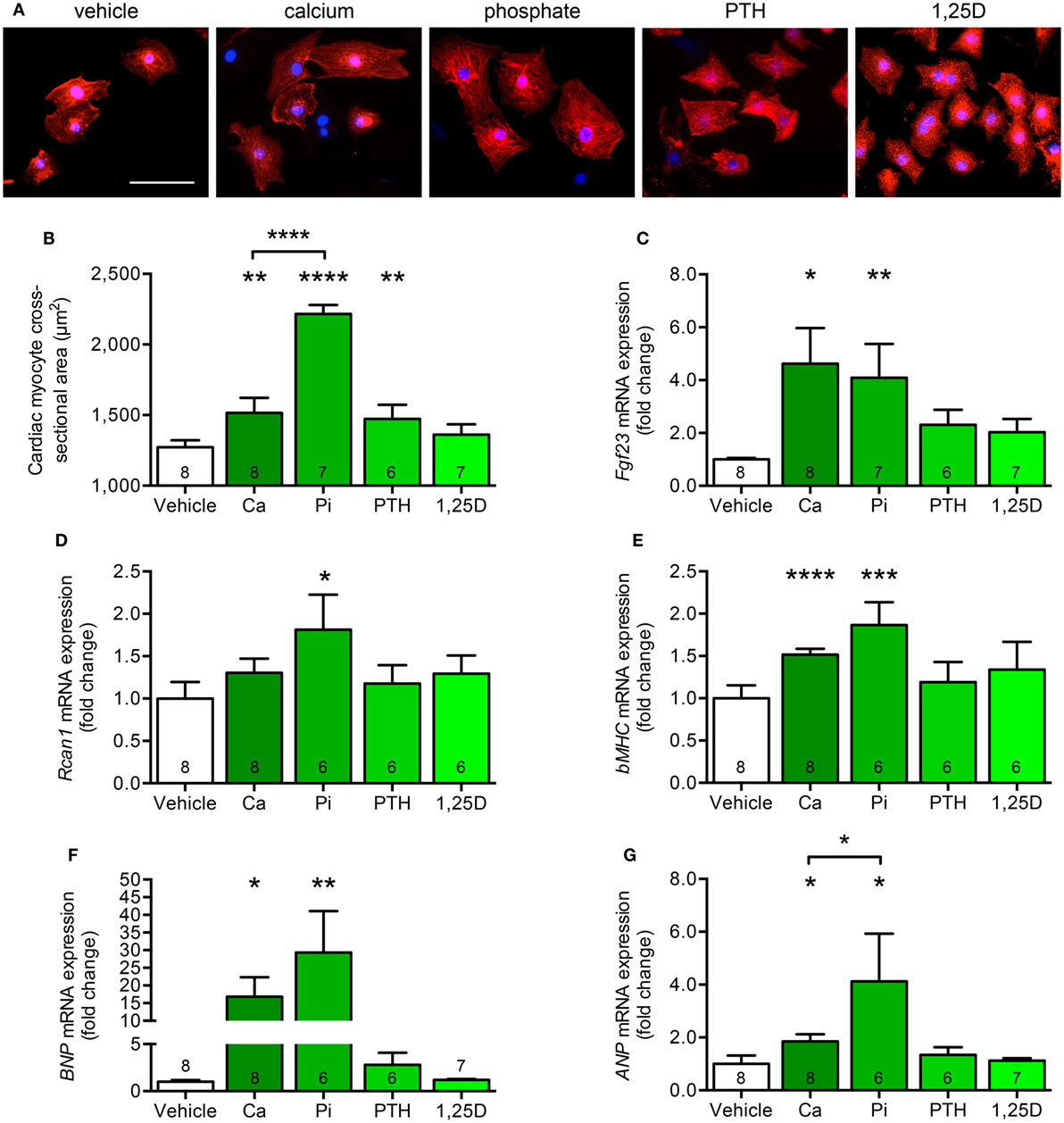
Figure 7. Calcium and phosphate treatment induces cell growth and pro-hypertrophic gene expression in cultured neonatal rat ventricular myocytes (NRVM). (A,B) Immunofluorescence staining for sarcomeric α-actinin followed by quantification demonstrates enhanced cardiac myocyte cross-sectional area after treatment of NRVM with calcium, phosphate, and PTH, but not 1,25D (magnification, 40×; scale bar, 50 µm). (C) Calcium and phosphate stimulation significantly increases Fgf23 mRNA expression in NRVM. (D) Rcan1 mRNA levels are significantly enhanced by treatment of NRVM with phosphate. The mRNA levels of (E) bMHC, (F) BNP, and (G) ANP are significantly upregulated after stimulation of NRVM with calcium and phosphate. Values are presented as mean ± SEM; numbers in each bar graph represent the n-values used for the respective measurement; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Abbreviations: Ca, calcium; Pi, phosphate; PTH, parathyroid hormone; 1,25D, 1,25(OH)2D3.
High FGF23 levels and klotho deficiency are postulated as key risk factors for the development of uremic cardiomyopathy (2, 23, 25, 31, 52) and increase of FGF23 is further associated with poor outcome in patients with heart failure, cardiogenic shock, or arrhythmia at normal kidney function (4–8). Even though FGF23 directly targets the heart and contributes to hypertrophy and fibrosis independent of klotho (27, 28), it is not known whether the increase of FGF23 in parallel with reduction of klotho alone is sufficient to lead to CVD. Here, we show that enhanced FGF23 synthesis in kl/kl mice led to the development of cardiac hypertrophy and fibrosis while Hyp mice, despite of high circulating and cardiac FGF23 levels and reduced renal klotho expression, did not show any cardiac phenotype. This suggests that elevated FGF23 levels in the presence of klotho deficiency do not necessarily promote cardiac toxicity of FGF23 in vivo per se. Among additional key factors of altered mineral metabolism in Hyp and kl/kl mice, only calcium and phosphate stimulated endogenous FGF23 expression in cultured cardiac myocytes and induced pro-hypertrophic gene expression resulting in hypertrophic cell growth in vitro. The present results suggest that the differences in serum calcium and phosphate levels between the two mouse models may contribute to the effects on cardiac hypertrophy and fibrosis at high FGF23 levels. Concomitant normal serum calcium levels and hypophosphatemia may prevent Hyp mice from the development of LVH, whereas concomitant hypercalcemia and hyperphosphatemia may have further promoted pathological cardiac remodeling in kl/kl mice.
In the present study, both Hyp and kl/kl mice presented with enhanced circulating levels of C-term and intact FGF23 as well as upregulated cardiac FGF23 synthesis in addition to reduced renal Klotho expression, although both alterations were more pronounced in kl/kl mice. However, kl/kl mice, but not Hyp mice, developed cardiac hypertrophy, demonstrated by an increase in relative heart weight and cardiac myocyte cross-sectional area. In addition, we demonstrated for the first time that FGFR4 is upregulated in heart tissue of kl/kl mice and this resulted in activation of calcineurin/NFAT signaling and finally induction of pro-hypertrophic NFAT target genes Rcan1, bMHC, ANP, and BNP. Furthermore, kl/kl mice further displayed enhanced myocardial fibrosis with concomitant induction of collagen 1, Mmp2 and Tgf-β1 expression, activated ERK1/2 and Ctgf protein. Thereby, we support the findings of Hu et al. showing that pERK1/2 is elevated and myocardial fibrosis is present in heart tissue of homozygous kl/kl mice as well (31). Moreover, this group investigated the Tgf-β1-mediated activation of ERK1/2 in neonatal rat cardiac myocytes and fibroblasts in vitro, which was only present in the absence of klotho suggesting that there is a strong connection between klotho deficiency and the induction of pro-fibrotic pathways. In addition, treatment of neonatal mouse cardiac fibroblasts with the 65 kDa soluble klotho isoform was shown to suppress myofibroblast proliferation and collagen synthesis (33). Taken together, our data suggest that the well-established cardiac FGF23/FGFR4 signaling involved in the development of pathologic cardiac hypertrophy with the parallel appearance of cardiac fibrosis is induced in kl/kl mice with high FGF23 levels and klotho deficiency.
Our results in homozygous kl/kl mice are well in line with Yang and colleagues who showed that heterozygous kl/+ mice had elevated relative heart weight and enhanced left ventricular posterior wall thickness with reduced left ventricular internal diastolic diameter (35). In another study by Hu et al., heterozygous kl/+ mice showed reduced ejection fraction, stroke volume, and cardiac output in addition to enlarged septum and posterior wall thickness within the diastole (31). In contrast, neither kl/+ nor kl/kl mice with comparable age had baseline cardiac abnormalities published in other studies by Xie and colleagues (32, 53). Interestingly, according to this study, serum FGF23 and phosphate levels were similar in WT and kl/+ mice. The different cardiac outcome in klotho-deficient mice within these studies might be, at least partially, due to differences in dietary phosphate content ranging from 0.35 to 0.6% (31, 32, 35, 53).
The development of pathologic cardiac remodeling including hypertrophy and fibrosis is less clear in Hyp mice. Recently, it was described that 8-week-old Hyp mice showed increased relative heart weight, and administration of an FGFR1 activating antibody resulted in normalization of relative heart weight, left ventricular wall thickness, and blood pressure. Treatment of Hyp mice with soluble klotho ameliorated systolic, diastolic, and mean arterial blood pressure (MAP) (54). Andrukhova and colleagues showed enhanced FGF23 serum levels in 3-month-old Hyp mice with enhanced MAP in addition to significantly higher relative heart weight compared to WT littermates (50). Thus, both studies suggest that Hyp mice on a regular phosphate diet present with enhanced blood pressure and cardiac hypertrophy. However, a detailed evaluation of the cardiac phenotype in Hyp mice, i.e., investigation of cardiac myocyte cross-sectional area, pro-hypertrophic signaling pathways, and concomitant fibrosis, in order to evalute pathological cardiac hypertrophy in-depth, was not yet been performed in Hyp mice. In the present study, we investigated histological and molecular biological analyses of heart tissue in 6- to 8-week-old Hyp mice on a regular phosphate diet and compared our findings to their WT littermates. In contrast to the abovementioned studies (50, 54), relative heart weight was not significantly altered in Hyp mice in the present study, and additionally, cardiac myocyte cross-sectional area was similar to controls. Although, Hyp mice showed comparably increased circulating FGF23 levels and the presence of enhanced cardiac FGF23 synthesis in the present study, FGFR4 and its respective downstream signaling pathway, i.e., calcineurin/NFAT, Rcan1, bMHC, ANP, and BNP, were not induced at all. Neither accumulation of fibrillar collagens nor fibrosis-related pathways, including collagen 1 expression, Tgf-β1-mediated activation of ERK1/2, or Ctgf were induced in Hyp mice compared to controls. Thus, our data point out that high FGF23 levels in addition to klotho deficiency does not necessarily result in pathological cardiac remodeling in vivo. The fact that blood pressure is elevated in Hyp mice (50) while low in kl/kl mice (44) further supported the hypothesis that FGF23-mediated cardiac hypertrophy might be blood pressure independent.
Therefore, the question arises if alterations in the mineral metabolism may modulate the cardiac phenotype in these two mouse models. In line with previous published studies by others (37, 55) and us (46), Hyp mice showed normal to reduced serum calcium levels, presented with hypophosphatemia, secondary hyperparathyroidism, and vitamin D deficiency. In contrast, kl/kl mice were hypercalcemic and hyperphosphatemic, had elevated 1,25D levels, and suppressed serum PTH levels (44, 45). Thus, the observed differences in calcium and phosphate serum concentrations may at least partly explain the different cardiac phenotypes in the two mouse models. The importance of phosphate as a risk factor for cardiomyopathy was previously demonstrated in a variety of experimental studies showing that high-phosphate diet induced cardiac hypertrophy and fibrosis in rodents (25, 28, 56). Moreover, stimulation with phosphate induced pERK1/2, Ctgf, and collagen 1 in isolated neonatal rat cardiac fibroblasts and in addition pERK1/2, Ctgf, and pSmad2/3 in cardiac myocytes in vitro (31). Interestingly, co-treatment with soluble klotho only ameliorated Ctgf and collagen 1 levels in cardiac fibroblasts and Smad2/3 phosphorylation in myocytes suggesting that klotho primarily targets phosphate-induced pro-fibrotic signaling pathways. The impact of phosphate on the development of cardiac hypertrophy on cellular level was not investigated so far. Here, we present that both calcium and phosphate stimulated hypertrophic growth of cultured cardiac myocytes with upregulation of Rcan1, bMHC, ANP, and BNP, respectively. Interestingly, among the known parameters of mineral metabolism that induce FGF23 synthesis in bone (9), only calcium and phosphate upregulated endogenous Fgf23 expression in NRVM.
Circulating FGF23 levels were considerably higher and the degree of klotho deficiency was lower in kl/kl mice compared to Hyp mice in the present study. Therefore, we cannot exclude that further stimulation of circulating FGF23, e.g., by increased phosphate load may result in pathological cardiac remodeling in Hyp mice despite the presence of hypophosphatemia. In addition, kl/kl mice presented with hypercalcemia, which was shown to stimulate cardiac myocyte hypertrophy in vitro, although the effects of calcium were lower compared to phosphate. However, concomitant hypercalcemia may have further promoted the cardiac phenotype in kl/kl mice. Furthermore, in kl/kl mice, the regulation of cardiac hypertrophy and fibrosis by other pathological factors cannot be ruled out (57–59). Clearly, further studies are needed to confirm an association of altered mineral metabolism at high FGF23 levels and pathological cardiac remodeling.
In extension to previously published studies, our results support the hypothesis that both altered parameters of mineral metabolism and elevated FGF23 levels contribute to cardiac hypertrophy and fibrosis in the setting of klotho deficiency, and consequently, Hyp mice might be protected from pathologic cardiac remodeling by the presence of concomitant hypophosphatemia and lack of hypercalcemia. In addition, high FGF23 levels are only cardiotoxic in the presence of high calcium and/or high phosphate and independent of klotho deficiency. The latter supports the concept of early initiation of phosphate lowering treatment in states of elevated FGF23 and klotho deficiency, e.g., uremia, in order to prevent pathological cardiac remodeling.
All experimental procedures were approved by the State Office committee on animal welfare Lower Saxony for Hyp mice and Baden-Württemberg for kl/kl mice, and performed in accordance with national animal protection guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
Design: ML-N, BR, JV, and DH; experiments: BR, MB, JN, and IV; experimental support and critical revision of the manuscript: IA, JV, FL, JH, and SK; data evaluation: ML-N, BR, MB, JN, and IV; preparation of manuscript: ML-N, BR, and DH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Anja Ziolek for technical assistance and Anna Gigina for isolation of neonatal rat ventricular myocytes.
This work was supported by the Deutsche Forschungsgemein-schaft (La315-15).
1. Himmelfarb J, Ikizler TA. Hemodialysis. N Engl J Med (2010) 363(19):1833–45. doi:10.1056/NEJMra0902710
2. Gutierrez OM, Januzzi JL, Isakova T, Laliberte K, Smith K, Collerone G, et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation (2009) 119(19):2545–52. doi:10.1161/CIRCULATIONAHA.108.844506
3. Gross ML, Ritz E. Hypertrophy and fibrosis in the cardiomyopathy of uremia – beyond coronary heart disease. Semin Dial (2008) 21(4):308–18. doi:10.1111/j.1525-139X.2008.00454.x
4. Andersen IA, Huntley BK, Sandberg SS, Heublein DM, Burnett JC Jr. Elevation of circulating but not myocardial FGF23 in human acute decompensated heart failure. Nephrol Dial Transplant (2016) 31(5):767–72. doi:10.1093/ndt/gfv398
5. Wohlfahrt P, Melenovsky V, Kotrc M, Benes J, Jabor A, Franekova J, et al. Association of fibroblast growth factor-23 levels and angiotensin-converting enzyme inhibition in chronic systolic heart failure. JACC Heart Fail (2015) 3(10):829–39. doi:10.1016/j.jchf.2015.05.012
6. Seiler S, Cremers B, Rebling NM, Hornof F, Jeken J, Kersting S, et al. The phosphatonin fibroblast growth factor 23 links calcium-phosphate metabolism with left-ventricular dysfunction and atrial fibrillation. Eur Heart J (2011) 32(21):2688–96. doi:10.1093/eurheartj/ehr215
7. Mathew JS, Sachs MC, Katz R, Patton KK, Heckbert SR, Hoofnagle AN, et al. Fibroblast growth factor-23 and incident atrial fibrillation: the Multi-Ethnic Study of Atherosclerosis (MESA) and the Cardiovascular Health Study (CHS). Circulation (2014) 130(4):298–307. doi:10.1161/CIRCULATIONAHA.113.005499
8. Fuernau G, Poss J, Denks D, Desch S, Heine GH, Eitel I, et al. Fibroblast growth factor 23 in acute myocardial infarction complicated by cardiogenic shock: a biomarker substudy of the Intraaortic Balloon Pump in Cardiogenic Shock II (IABP-SHOCK II) trial. Crit Care (2014) 18(6):713. doi:10.1186/s13054-014-0713-8
9. Haffner D, Leifheit-Nestler M. Extrarenal effects of FGF23. Pediatr Nephrol (2017) 32(5):753–65. doi:10.1007/s00467-016-3505-3
10. Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, et al. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol (2009) 297(2):F282–91. doi:10.1152/ajprenal.90742.2008
11. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res (2004) 19(3):429–35. doi:10.1359/JBMR.0301264
12. Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest (2007) 117(12):4003–8. doi:10.1172/JCI32409
13. Quarles LD. Endocrine functions of bone in mineral metabolism regulation. J Clin Invest (2008) 118(12):3820–8. doi:10.1172/JCI36479
14. Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol (2006) 17(5):1305–15. doi:10.1681/ASN.2005111185
15. Erben RG, Andrukhova O. FGF23-Klotho signaling axis in the kidney. Bone (2017) 100:10062–8. doi:10.1016/j.bone.2016.09.010
16. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature (1997) 390(6655):45–51. doi:10.1038/36285
17. Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem (2006) 281(10):6120–3. doi:10.1074/jbc.C500457200
18. Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature (2006) 444(7120):770–4. doi:10.1038/nature05315
19. Razzaque MS. The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol (2009) 5(11):611–9. doi:10.1038/nrendo.2009.196
20. Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol (2013) 75:503–33. doi:10.1146/annurev-physiol-030212-183727
21. Richter M, Lautze HJ, Walther T, Braun T, Kostin S, Kubin T. The failing heart is a major source of circulating FGF23 via oncostatin M receptor activation. J Heart Lung Transplant (2015) 34(9):1211–4. doi:10.1016/j.healun.2015.06.007
22. Hasegawa H, Nagano N, Urakawa I, Yamazaki Y, Iijima K, Fujita T, et al. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int (2010) 78(10):975–80. doi:10.1038/ki.2010.313
23. Heine GH, Seiler S, Fliser D. FGF-23: the rise of a novel cardiovascular risk marker in CKD. Nephrol Dial Transplant (2012) 27(8):3072–81. doi:10.1093/ndt/gfs259
24. Gunta SS, Thadhani RI, Mak RH. The effect of vitamin D status on risk factors for cardiovascular disease. Nat Rev Nephrol (2013) 9(6):337–47. doi:10.1038/nrneph.2013.74
25. Scialla JJ, Wolf M. Roles of phosphate and fibroblast growth factor 23 in cardiovascular disease. Nat Rev Nephrol (2014) 10(5):268–78. doi:10.1038/nrneph.2014.49
26. Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int (2003) 64(6):2272–9. doi:10.1046/j.1523-1755.2003.00328.x
27. Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest (2011) 121(11):4393–408. doi:10.1172/JCI46122
28. Grabner A, Amaral AP, Schramm K, Singh S, Sloan A, Yanucil C, et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab (2015) 22(6):1020–32. doi:10.1016/j.cmet.2015.09.002
29. Leifheit-Nestler M, Grosse Siemer R, Flasbart K, Richter B, Kirchhoff F, Ziegler WH, et al. Induction of cardiac FGF23/FGFR4 expression is associated with left ventricular hypertrophy in patients with chronic kidney disease. Nephrol Dial Transplant (2016) 31(7):1088–99. doi:10.1093/ndt/gfv421
30. Leifheit-Nestler M, Kirchhoff F, Nespor J, Richter B, Soetje B, Klintschar M, et al. Fibroblast growth factor 23 is induced by an activated renin-angiotensin-aldosterone system in cardiac myocytes and promotes the pro-fibrotic crosstalk between cardiac myocytes and fibroblasts. Nephrol Dial Transplant (2018). doi:10.1093/ndt/gfy006
31. Hu MC, Shi M, Cho HJ, Adams-Huet B, Paek J, Hill K, et al. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J Am Soc Nephrol (2015) 26(6):1290–302. doi:10.1681/ASN.2014050465
32. Xie J, Yoon J, An SW, Kuro-o M, Huang CL. Soluble Klotho protects against uremic cardiomyopathy independently of fibroblast growth factor 23 and phosphate. J Am Soc Nephrol (2015) 26(5):1150–60. doi:10.1681/ASN.2014040325
33. Liu X, Chen Y, McCoy CW, Zhao T, Quarles DL, Pi M, et al. Differential regulatory role of soluble Klothos on cardiac fibrogenesis in hypertension. Am J Hypertens (2016) 29(10):1140–7. doi:10.1093/ajh/hpw062
34. Richter B, Haller J, Haffner D, Leifheit-Nestler M. Klotho modulates FGF23-mediated NO synthesis and oxidative stress in human coronary artery endothelial cells. Pflugers Arch (2016) 468(9):1621–35. doi:10.1007/s00424-016-1858-x
35. Yang K, Wang C, Nie L, Zhao X, Gu J, Guan X, et al. Klotho protects against indoxyl sulphate-induced myocardial hypertrophy. J Am Soc Nephrol (2015) 26(10):2434–46. doi:10.1681/ASN.2014060543
36. Kestenbaum B, Sampson JN, Rudser KD, Patterson DJ, Seliger SL, Young B, et al. Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol (2005) 16(2):520–8. doi:10.1681/ASN.2004070602
37. Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab (2006) 291(1):E38–49. doi:10.1152/ajpendo.00008.2006
38. Francis F, Hennig S, Korn B, Reinhardt R, de Jong P, Poustka A, et al. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet (1995) 11(2):130–6. doi:10.1038/ng1095-130
39. Zwadlo C, Schmidtmann E, Szaroszyk M, Kattih B, Froese N, Hinz H, et al. Antiandrogenic therapy with finasteride attenuates cardiac hypertrophy and left ventricular dysfunction. Circulation (2015) 131(12):1071–81. doi:10.1161/CIRCULATIONAHA.114.012066
40. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res (2001) 29(9):e45. doi:10.1093/nar/29.9.e45
41. Creemers EE, Pinto YM. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc Res (2011) 89(2):265–72. doi:10.1093/cvr/cvq308
42. Chen Y, Blom IE, Sa S, Goldschmeding R, Abraham DJ, Leask A. CTGF expression in mesangial cells: involvement of SMADs, MAP kinase, and PKC. Kidney Int (2002) 62(4):1149–59. doi:10.1111/j.1523-1755.2002.kid567.x
43. Phanish MK, Wahab NA, Hendry BM, Dockrell ME. TGF-beta1-induced connective tissue growth factor (CCN2) expression in human renal proximal tubule epithelial cells requires Ras/MEK/ERK and Smad signalling. Nephron Exp Nephrol (2005) 100(4):e156–65. doi:10.1159/000085445
44. Leibrock CB, Voelkl J, Kohlhofer U, Quintanilla-Martinez L, Kuro OM, Lang F. Bicarbonate-sensitive calcification and lifespan of klotho-deficient mice. Am J Physiol Renal Physiol (2016) 310(1):F102–8. doi:10.1152/ajprenal.00037.2015
45. Leibrock CB, Alesutan I, Voelkl J, Michael D, Castor T, Kohlhofer U, et al. Acetazolamide sensitive tissue calcification and aging of klotho-hypomorphic mice. J Mol Med (Berl) (2016) 94(1):95–106. doi:10.1007/s00109-015-1331-x
46. Leifheit-Nestler M, Kucka J, Yoshizawa E, Behets G, D’Haese P, Bergen C, et al. Comparison of calcimimetic R568 and calcitriol in mineral homeostasis in the Hyp mouse, a murine homolog of X-linked hypophosphatemia. Bone (2017) 103:103224–32. doi:10.1016/j.bone.2017.06.019
47. Lanske B, Razzaque MS. Molecular interactions of FGF23 and PTH in phosphate regulation. Kidney Int (2014) 86(6):1072–4. doi:10.1038/ki.2014.316
48. Baum M, Loleh S, Saini N, Seikaly M, Dwarakanath V, Quigley R. Correction of proximal tubule phosphate transport defect in Hyp mice in vivo and in vitro with indomethacin. Proc Natl Acad Sci U S A (2003) 100(19):11098–103. doi:10.1073/pnas.1834060100
49. Sugiura H, Yoshida T, Shiohira S, Kohei J, Mitobe M, Kurosu H, et al. Reduced Klotho expression level in kidney aggravates renal interstitial fibrosis. Am J Physiol Renal Physiol (2012) 302(10):F1252–64. doi:10.1152/ajprenal.00294.2011
50. Andrukhova O, Slavic S, Smorodchenko A, Zeitz U, Shalhoub V, Lanske B, et al. FGF23 regulates renal sodium handling and blood pressure. EMBO Mol Med (2014) 6(6):744–59. doi:10.1002/emmm.201303716
51. Leibrock CB, Feger M, Voelkl J, Kohlhofer U, Quintanilla-Martinez L, Kuro-o M, et al. Partial reversal of tissue calcification and extension of life span following ammonium nitrate treatment of Klotho-deficient mice. Kidney Blood Press Res (2016) 41(1):99–107. doi:10.1159/000443411
52. Gutierrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med (2008) 359(6):584–92. doi:10.1056/NEJMoa0706130
53. Xie J, Cha SK, An SW, Kuro OM, Birnbaumer L, Huang CL. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat Commun (2012) 3:1238. doi:10.1038/ncomms2240
54. Han X, Ross J, Kolumam G, Pi M, Sonoda J, King G, et al. Cardiovascular effects of renal distal tubule deletion of the FGF receptor 1 gene. J Am Soc Nephrol (2017) 29(1):69–80. doi:10.1681/ASN.2017040412
55. Li H, Martin A, David V, Quarles LD. Compound deletion of Fgfr3 and Fgfr4 partially rescues the Hyp mouse phenotype. Am J Physiol Endocrinol Metab (2011) 300(3):E508–17. doi:10.1152/ajpendo.00499.2010
56. Grabner A, Schramm K, Silswal N, Hendrix M, Yanucil C, Czaya B, et al. FGF23/FGFR4-mediated left ventricular hypertrophy is reversible. Sci Rep (2017) 7(1):1993. doi:10.1038/s41598-017-02068-6
57. Voelkl J, Alesutan I, Leibrock CB, Quintanilla-Martinez L, Kuhn V, Feger M, et al. Spironolactone ameliorates PIT1-dependent vascular osteoinduction in klotho-hypomorphic mice. J Clin Invest (2013) 123(2):812–22. doi:10.1172/JCI64093
58. Leibrock CB, Alesutan I, Voelkl J, Pakladok T, Michael D, Schleicher E, et al. NH4Cl treatment prevents tissue calcification in Klotho deficiency. J Am Soc Nephrol (2015) 26(10):2423–33. doi:10.1681/ASN.2014030230
Keywords: fibroblast growth factor 23, klotho deficiency, mineral metabolism, left ventricular hypertrophy, cardiac fibrosis, klotho hypomorphic mice, Hyp mice
Citation: Leifheit-Nestler M, Richter B, Basaran M, Nespor J, Vogt I, Alesutan I, Voelkl J, Lang F, Heineke J, Krick S and Haffner D (2018) Impact of Altered Mineral Metabolism on Pathological Cardiac Remodeling in Elevated Fibroblast Growth Factor 23. Front. Endocrinol. 9:333. doi: 10.3389/fendo.2018.00333
Received: 04 December 2017; Accepted: 31 May 2018;
Published: 21 June 2018
Edited by:
Reinhold Gottfried Erben, Veterinärmedizinische Universität Wien, AustriaReviewed by:
Chrishan S. Samuel, Monash University, AustraliaCopyright: © 2018 Leifheit-Nestler, Richter, Basaran, Nespor, Vogt, Alesutan, Voelkl, Lang, Heineke, Krick and Haffner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maren Leifheit-Nestler, bGVpZmhlaXQtbmVzdGxlci5tYXJlbkBtaC1oYW5ub3Zlci5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.