 Lauren Winter1,2
Lauren Winter1,2 Lydia A. Wong1George Jerums1Jas-mine Seah1Michele Clarke1
Lydia A. Wong1George Jerums1Jas-mine Seah1Michele Clarke1 Sih Min Tan3Melinda T. Coughlan3Richard J. MacIsaac4,5
Sih Min Tan3Melinda T. Coughlan3Richard J. MacIsaac4,5 Elif I. Ekinci1,2*
Elif I. Ekinci1,2*
- 1Endocrine Centre of Excellence, Austin Health, Melbourne, VIC, Australia
- 2Department of Medicine, Austin Health, University of Melbourne, Melbourne, VIC, Australia
- 3Department of Diabetes, Central Clinical School, Monash University, Melbourne, VIC, Australia
- 4Department of Endocrinology and Diabetes, St Vincent’s Health, Melbourne, VIC, Australia
- 5Department of Medicine, St Vincent’s Health, University of Melbourne, Melbourne, VIC, Australia
Diabetic kidney disease is a common complication of type 1 and type 2 diabetes and is the primary cause of end-stage renal disease in developed countries. Early detection of diabetic kidney disease will facilitate early intervention aimed at reducing the rate of progression to end-stage renal disease. Diabetic kidney disease has been traditionally classified based on the presence of albuminuria. More recently estimated glomerular filtration rate has also been incorporated into the staging of diabetic kidney disease. While albuminuric diabetic kidney disease is well described, the phenotype of non-albuminuric diabetic kidney disease is now widely accepted. An association between markers of inflammation and diabetic kidney disease has previously been demonstrated. Effector molecules of the innate immune system including C-reactive protein, interleukin-6, and tumor necrosis factor-α are increased in patients with diabetic kidney disease. Furthermore, renal infiltration of neutrophils, macrophages, and lymphocytes are observed in renal biopsies of patients with diabetic kidney disease. Similarly high serum neutrophil and low serum lymphocyte counts have been shown to be associated with diabetic kidney disease. The neutrophil–lymphocyte ratio is considered a robust measure of systemic inflammation and is associated with the presence of inflammatory conditions including the metabolic syndrome and insulin resistance. Cross-sectional studies have demonstrated a link between high levels of the above inflammatory biomarkers and diabetic kidney disease. Further longitudinal studies will be required to determine if these readily available inflammatory biomarkers can accurately predict the presence and prognosis of diabetic kidney disease, above and beyond albuminuria, and estimated glomerular filtration rate.
Introduction
In patients with type 1 and type 2 diabetes mellitus, methods to assist in the early identification of kidney disease are a priority so that measures to prevent its progression can be put in place. Newer medications to prevent diabetic kidney disease are increasingly becoming available that may offer benefits in addition to good glycemic and blood pressure control and inhibition of the renin–angiotensin system. These newer medications include sodium glucose co-transporter-linked blockers (1, 2), and glucagon-like peptide-1 analogs (3, 4) are already being used in clinical practice, and the anticipated release of novel antifibrotic agents and anti-monocyte chemoattractant protein-1 agents is likely to help prevent the onset of diabetic kidney disease (5).
Currently, diabetic kidney disease accounts for the majority of cases of end-stage renal disease in developed countries (6) and is of increasing concern in developing countries (7). It is estimated that 40% of individuals with either type 1 or type 2 diabetes will go on to develop diabetic kidney disease (8). Rates of type 2 diabetes alone are estimated to double within the next 25 years to 352 million worldwide (7). Increasing numbers of patients with diabetes and subsequently diabetic kidney disease will place a significant financial burden on the healthcare system (9).
The full pathogenesis of diabetic kidney disease is still being elicited (10); however, early detection and subsequent intervention may slow rates of disease progression (11). Current diagnosis of diabetic kidney disease is based on the presence of reduced estimated glomerular filtration rate and/or an increased urinary albumin excretion in absence of other renal disease. The phenotype of normoalbuminuric renal insufficiency is also becoming increasingly recognized (12). The limitations of using albuminuria alone as a marker of diabetic kidney disease are well recognized, emphasizing the importance of developing novel, cheap, and accessible ways to detect early renal disease which will be useful in the context of the increasing burden of diabetes (13, 14). Improving our understanding of the pathogenesis of diabetic kidney disease is likely to assist in the management of patients (15).
Activation of immune and inflammatory pathways is increasingly recognized as key mediators for the development and progression of renal damage in diabetes with diabetic kidney disease (16). In this review, we focus on the potential role of readily accessible indicators of activation of the innate and adaptive immune system as markers of diabetic kidney disease. In particular, we review evidence for the immune cells neutrophils, macrophages, and monocytes which reflect innate immunity and lymphocytes which reflect adaptive immunity as indicators for the presence of diabetic kidney disease.
Discussion
Overview of the Immune System
The role of the immune system is to defend the human body against injury from foreign pathogens, toxins, and allergens (17) (Figure 1). There are two components of the immune system; the innate immune system and the adaptive immune system (18). The innate immune system provides the first line of defense when a foreign molecule is detected. It involves physical barriers such as the epidermis (19), the complement system (19), a cellular component including both neutrophils and macrophages and the acute-phase response (18). Physical barriers act to stop foreign pathogens from entering the body (17). These include the epithelial cell layers that form tight cell–cell contacts, the secreted mucus layer lining the epithelium of the respiratory, gastrointestinal, and genitourinary tracts, and the epithelial cilia that help to move particles out of the body (17). The complement system, which involves plasma and cell surface proteins, marks foreign pathogens for destruction (17). In addition to this, it activates the cellular component of the innate immune system (17). This includes neutrophils, macrophages, and CD4+ T cells, members of the adaptive immune system (17). On and in, these cells are a number of pattern recognition receptors, including toll-like receptors and the receptor for advanced glycation end products that recognize danger-associated molecular patterns. In diabetic kidneys, toll-like receptors 1 and 2 as well as the receptor for advanced glycation end products are thought to be the main receptors for danger-associated molecular patterns in promoting immune-mediated injury (16). Increased expression of these receptors in diabetic kidney disease leads to the increased activation of myeloid differentiation primary response 88 or nuclear factor-κB and production of pro-inflammatory cytokines (16), which are the main mediators of inflammation and the acute-phase response (20).
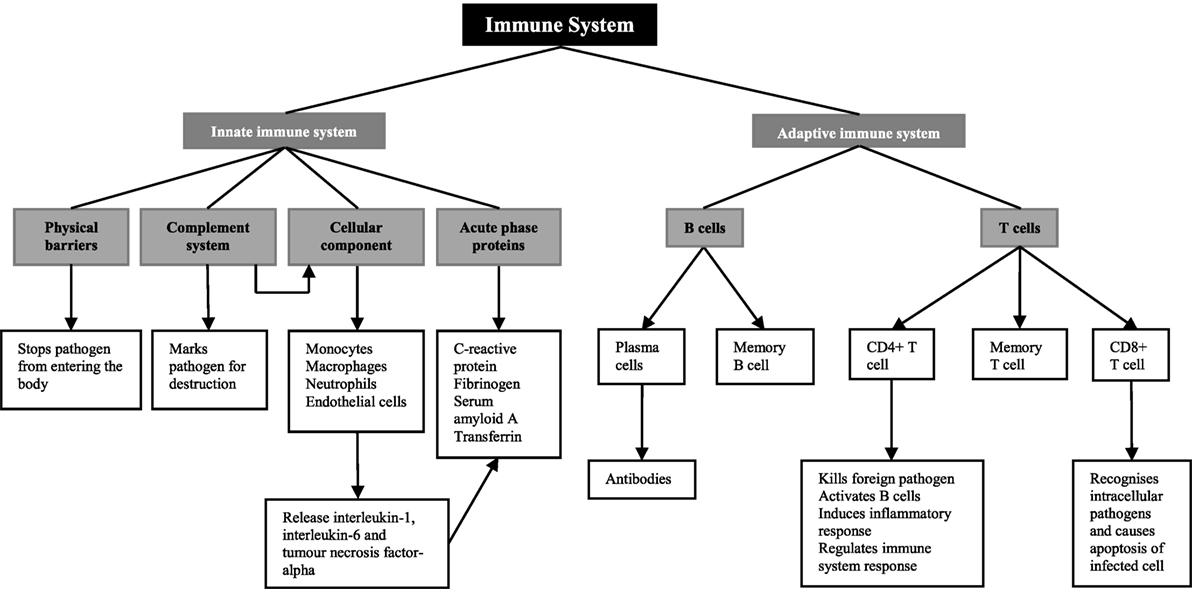
Figure 1. Overview of the immune system.
Pro-inflammatory cytokines, including interleukin-6 and tumor necrosis factor-alpha released from macrophages, monocytes, and endothelial cells stimulate the liver to synthesize acute-phase proteins (21), such as C-reactive protein, fibrinogen, and complement (20). C-reactive protein acts as opsonin for bacteria, parasites, and immune complexes and can activate the classical pathway of complement (22, 23). Furthermore, it induces the release of pro-inflammatory cytokines and tissue factors in monocytes (24, 25). Increase in the plasma concentration of complement cascade components can lead to the accumulation of neutrophils, macrophages, and plasma proteins, participating in the killing of infectious agents, the clearance of foreign and host cellular debris, and the repair of damaged tissue (23). Fibrinogen, which is a component of the coagulation cascade, plays an essential role in promoting wound healing (23).
While the innate immune system is the host’s non-specific resistance to pathogens and is poised to act rapidly, the adaptive immune system acts in a highly specific nature involving antigen specificity and immunologic memory (26). The adaptive immune system involves lymphocytes known as T cells and B cells, and both express highly specific antigen receptors on their surface (27). T cells are important mediators of the cell-mediated immune response, whereas B cells are critical for the humoral immune response.
There are two main types of T cells; CD4+ and CD8+ T cells (27). CD4+ T cells function as both helper and regulatory cells and are activated by antigen presenting cells such as macrophages and influence the nature of the immune response (27). Cytotoxic CD8+ T cells recognize intracellular pathogens and activate apoptosis of the infected cell (27). B cells are defined by their production of immunoglobulin, or antibody, against a specific antigen, also known as humoral immunity (17). Once a B cell encounters an antigen and is activated, it will become either a memory cell to be activated once again in the future or it will become a plasma cell, producing large amounts of antibody (27).
Inflammation in Type 2 Diabetes Mellitus
Chronic systemic inflammation has been hypothesized to contribute to the development of type 2 diabetes (28) as well as its complications (29). Activation of inflammatory pathways is common to type 2 diabetes, atherosclerosis, and cardiovascular disease (30). Furthermore, the metabolic syndrome, which includes insulin resistance, type 2 diabetes, hypertension, dyslipidemia, and central obesity (31), is a condition of chronic low-grade inflammation (32). Importantly, anti-inflammatory therapies such as canakinumab, a monoclonal antibody against interleukin-1β, has been shown to reduce the risk of cardiovascular disease by reducing inflammation without lowering baseline lipid levels (33).
Acute-Phase Proteins and Pro-Inflammatory Cytokines in the Development of Type 2 Diabetes Mellitus
It is thought that innate immune system activation may play a role in the development and progression of type 2 diabetes and its complications (18). There have been a small number of cross-sectional analyses performed regarding the relationship between circulating C-reactive protein, interleukin-6, fibrinogen, and type 2 diabetes (34–36) (Table 1). There have been many more longitudinal studies examining whether baseline measurements can predict the development of type 2 diabetes within a given follow-up period (37–50) (Table 2). The majority of these studies showed a positive relationship between C-reactive protein and the risk of developing type 2 diabetes. The strength of these associations was often lessened when adjusting for measures of adiposity, some to the point of losing significance. In general, these studies reported no significance between C-reactive protein and the risk of developing type 2 diabetes after adjusting for body mass index (51), waist–hip ratio in addition to body mass index (50) and without adjusting for any patient characteristics (52). Overall, however, these results suggest that having markers of chronic low-grade inflammation is indeed associated with an increased risk of developing type 2 diabetes.
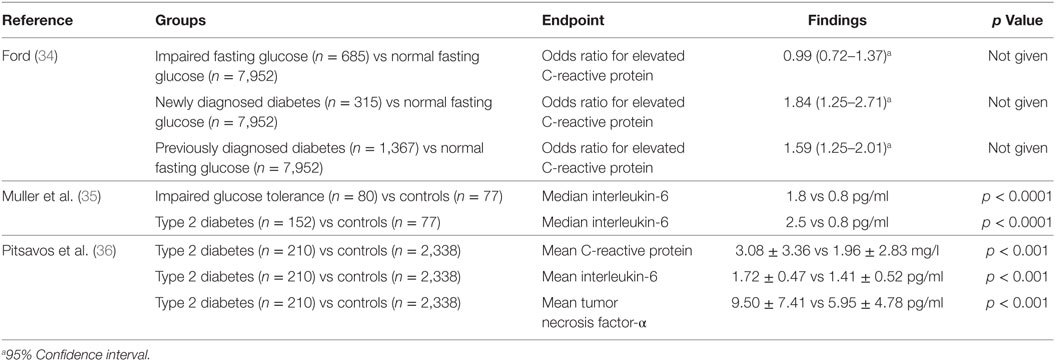
Table 1. Cross-sectional studies of C-reactive protein, interleukin-6, and tumor necrosis factor-α in type 2 diabetes.
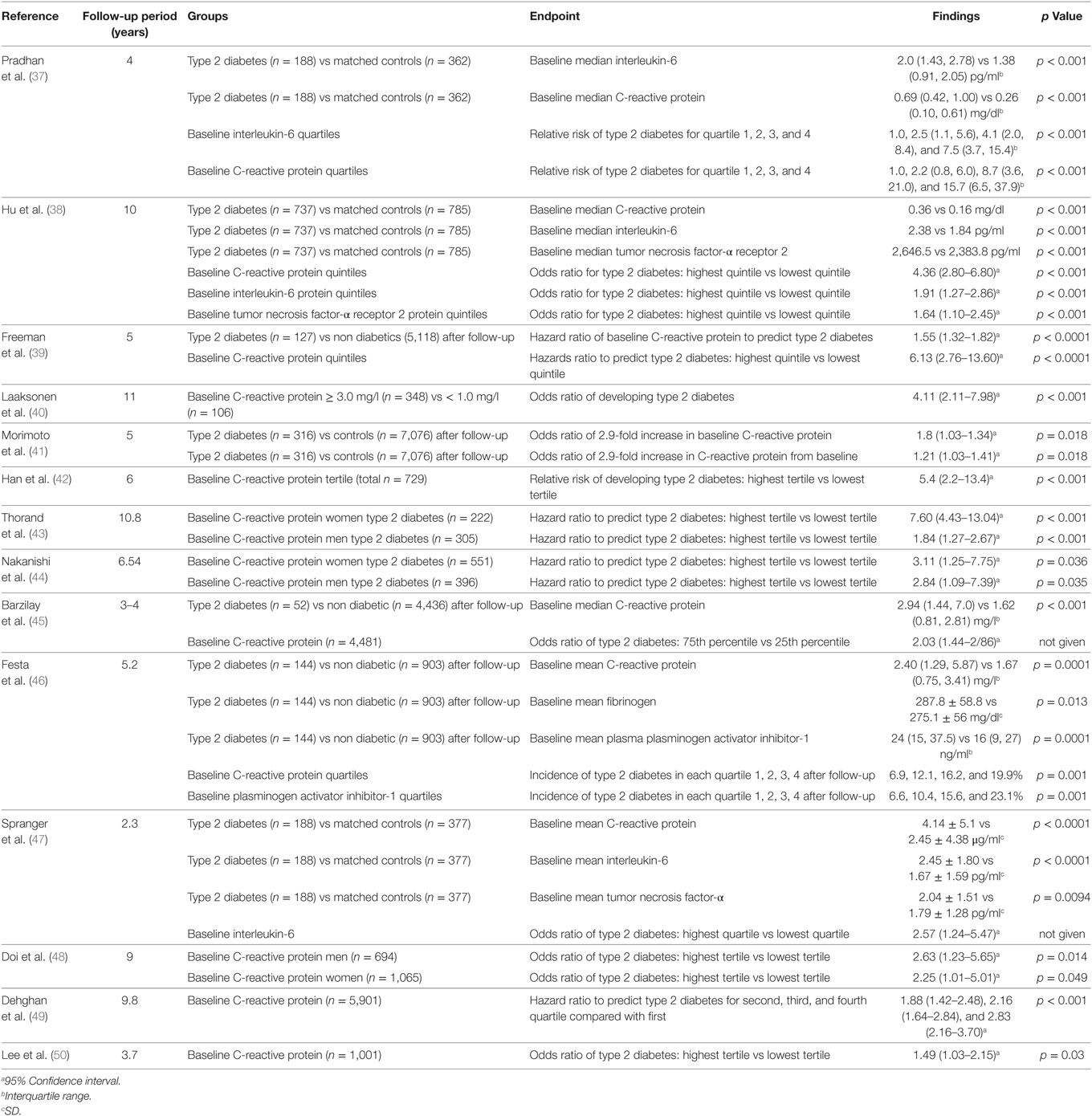
Table 2. Longitudinal studies of C-reactive protein, interleukin-6, tumor necrosis factor-α, fibrinogen, and plasminogen activator inhibitor-1 in type 2 diabetes.
White Cell Count in the Development of Type 2 Diabetes Mellitus
Several studies have examined the relationship between total white cell count and type 2 diabetes. The white cell count includes neutrophils, lymphocytes, monocytes, eosinophils, and basophils (53). Both cross-sectional studies (54, 55) (Table 3) and longitudinal studies (56, 57) (Table 4) have assessed this relationship between leukocytes and type 2 diabetes. These studies suggest particular white cells may be implicated in the development of type 2 diabetes and diabetic kidney disease based on albuminuria status.
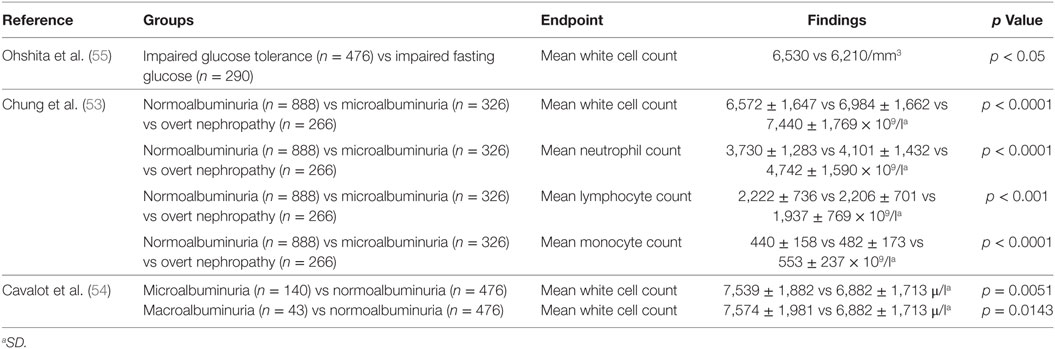
Table 3. Cross-sectional studies of the white cell count in type 2 diabetes.
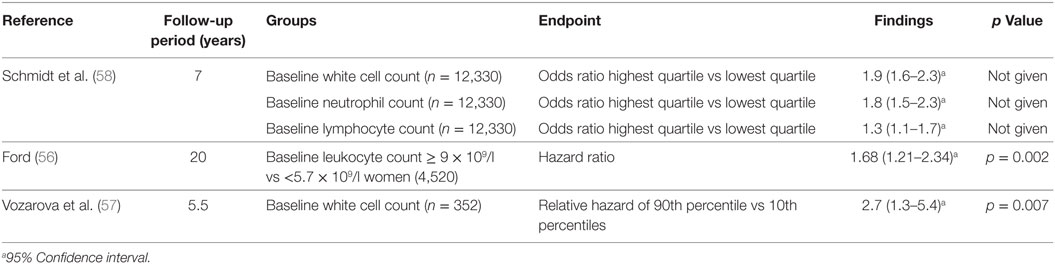
Table 4. Longitudinal studies of the white cell count in type 2 diabetes.
Innate Immune System and the Development of Diabetic Kidney Disease
Acute-Phase Proteins and Pro-Inflammatory Cytokines in the Development of Diabetic Kidney Disease in Type 2 Diabetes Mellitus
Acute-phase proteins and pro-inflammatory cytokines have been implicated in the development of diabetic kidney disease. Albuminuria is often present in patients with diabetic kidney disease but there is also a subset of patients who develop diabetic kidney disease, as determined by declining glomerular filtration rate, without the presence of an elevated albumin excretion rate (59–61). Despite this, many studies continue to use urinary albumin excretion alone to determine the presence of diabetic kidney disease. In addition to this, microalbuminuria is thought to represent widespread endothelial dysfunction and vascular damage (62) and therefore may not always represent the presence of diabetic kidney disease (63). Microalbuminuria in type 2 diabetes is known to predict cardiovascular mortality (64) which may suggest a common underlying process such as inflammation (65).
Multiple studies have examined a relationship between microalbuminuria and acute-phase proteins, without necessarily using microalbuminuria to infer the presence of diabetic kidney disease. In a cross-sectional study of 467 patients and 1,014 controls, a significant positive correlation between microalbuminuria and both C-reactive protein and fibrinogen was demonstrated (66). In 64 patients, C-reactive protein and fibrinogen were significantly higher in those with microalbuminuria compared with those with normoalbuminuria (65). Elevated C-reactive protein and microalbuminuria at baseline resulted in an increased risk of death in 328 patients followed for 10 years (62). C-reactive protein was also found to be strongly associated with increasing albuminuria with annual follow-up.
Other studies have been designed to determine the relationship between circulating acute-phase proteins, pro-inflammatory cytokines, and diabetic kidney disease (Figure 2). In a small sample of 32 patients, there was a positive correlation between urinary albumin excretion and circulating fibrinogen (67). There was no significant relationship observed, however, with C-reactive protein and interleukin-6. In 151 patients compared with 80 matched controls, there was no significant relationship between interleukin-6 and albuminuria (68). There was, however, a significant difference in interleukin-6 between patients with either microalbuminuria or macroalbuminuria when compared with those with normoalbuminuria. Tumor necrosis factor-α levels were significantly different between patients with microalbuminuria and those with macroalbuminuria. Similarly circulating tumor necrosis factor-α was also significantly higher in all patients compared with controls.
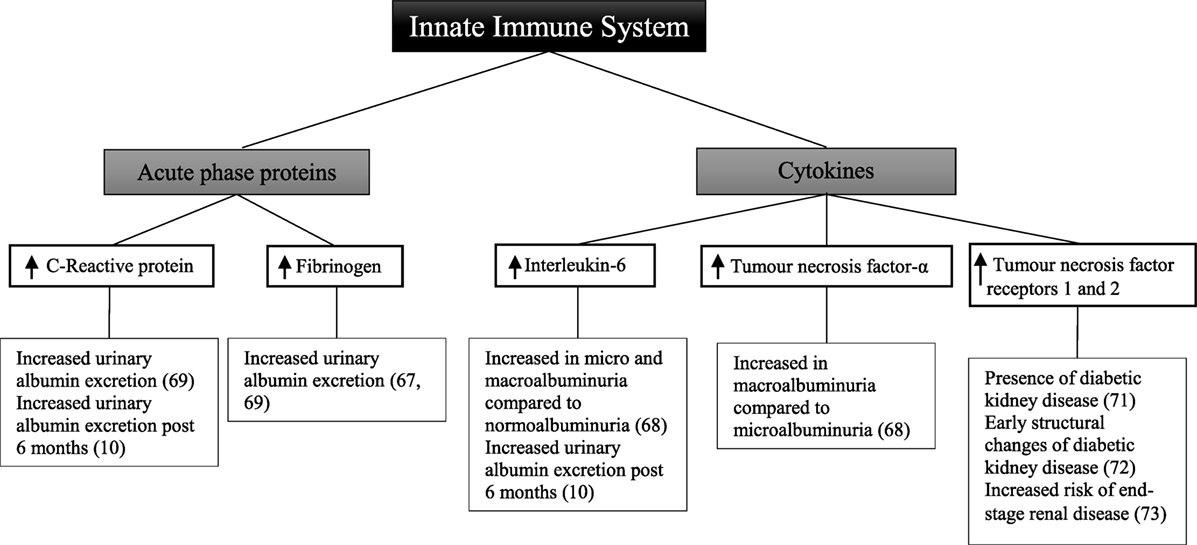
Figure 2. Inflammatory components of the innate immune system and diabetic kidney disease in type 2 diabetes.
A cross-sectional study of 74 patients with type 2 diabetes demonstrated that there was no significant relationship between fibrinogen, total leukocyte count, and glomerular filtration rate (69). Similarly in a subset of 66 patients, there was no association seen with C-reactive protein. Fibrinogen, total leukocyte count, and C-reactive protein were, however, positively correlated with increasing levels of albuminuria, with the highest levels observed in patients with macroalbuminuria.
In a cohort study of 80 patients followed for 6 months, there was no change in degree of albuminuria from baseline (10). This was attributed to both the short follow-up time in addition to the decision not to exclude patients who had been prescribed either angiotensin-converting enzyme inhibitors or angiotensin receptor blockers, with their well-known anti-albuminuric effects (70). Nevertheless, at baseline and at 6 months, interleukin-6 and C-reactive protein were significantly correlated with elevated urinary albumin excretion.
The receptors for tumor necrosis factor have also been explored as markers of renal injury. In a cross-sectional analysis of 607 patients, the level of soluble tumor necrosis factor receptors 1 and 2 is positively correlated with the presence of diabetic kidney disease (71). Similarly in a study of 83 patients, tumor necrosis factor receptors 1 and 2 were associated with early structural changes seen in diabetic kidney disease such as glomerular basement membrane width and podocyte foot process width (72). Increased risk of end-stage renal disease was correlated with tumor necrosis factor receptors 1 and 2 in 193 American Indian patients followed for a mean of 9.5 years (73).
Although only observational in nature, these studies demonstrate that in patients with type 2 diabetes and diabetic kidney disease, circulating levels of C-reactive protein, fibrinogen, interleukin-6, and tumor necrosis factor are significantly increased. This may suggest that an inflammatory response involving the innate immune system plays a role in the development of albuminuria in these patients.
Acute-Phase Proteins and Pro-Inflammatory Cytokines in the Development of Diabetic Kidney Disease in Type 1 Diabetes Mellitus
There have been a smaller number of studies looking at components of the innate immune system in patients with type 1 diabetes (Figure 3). In 909 patients with type 1 diabetes, there was a difference in the relationship between urinary albumin excretion and circulating fibrinogen in males and females (74). In males, there was a persistent positive correlation at all stages of albuminuria. By contrast, in females, a difference was only seen in those with macroalbuminuria compared with those without. In this study, however, urine and blood to measure urinary albumin excretion and circulating fibrinogen levels were collected within 1 year of each other; hence results may not reflect the true relationship. After 3 years of follow-up, there was no significant difference in C-reactive protein between 49 patients with microalbuminuria and 49 matched controls (75). When analyzing the controls who developed microalbuminuria during follow-up, there was a significant difference in C-reactive protein before and after development of microalbuminuria.
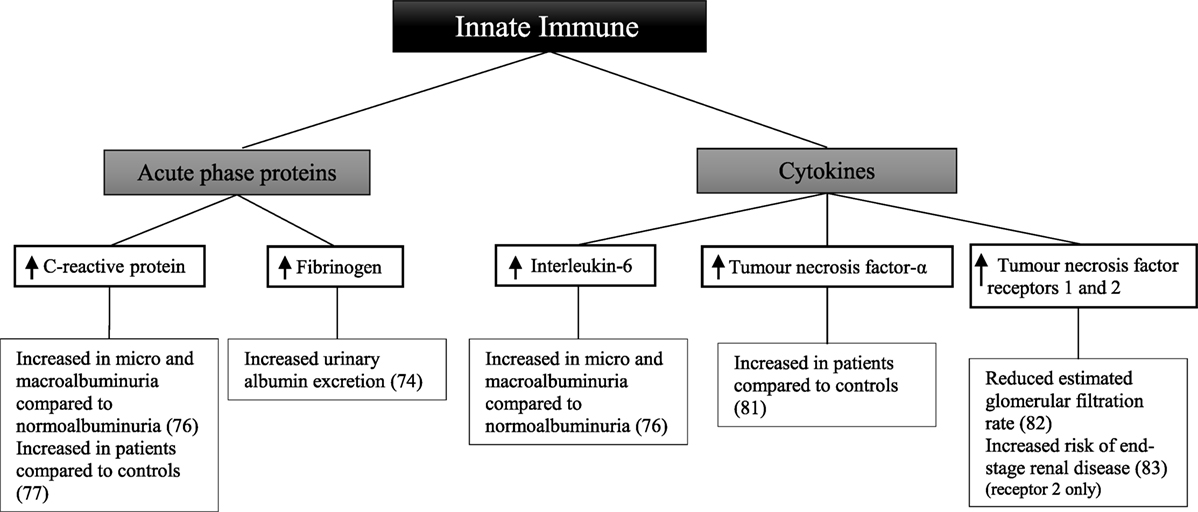
Figure 3. Inflammatory components of the innate immune system and diabetic kidney disease in type 1 diabetes.
Other studies have shown C-reactive protein and interleukin-6 to be significantly greater in 194 patients with normoalbuminuria compared with 66 healthy controls (76). Furthermore, patients with microalbuminuria and macroalbuminuria had significantly greater levels of C-reactive protein and interleukin-6 than those with normoalbuminuria. Similarly, C-reactive protein was significantly higher in 40 patients compared with 40 matched controls irrespective of albuminuria status (77). In patients with known diabetic kidney disease low-grade inflammation, calculated based on C-reactive protein, interleukin-6, soluble intercellular adhesion molecule-1, and secreted phospholipase A2, was shown to be associated with an increased risk of all-cause mortality after 10 years of follow-up (78). Similarly the risk of the combined cardiovascular endpoint; taking into account cardiovascular-related death, non-fatal event or need for intervention was significantly higher in patients with diabetic kidney disease at baseline. Soluble intercellular adhesion molecule-1, which is associated with vascular wall inflammation (79), has been shown to be significantly associated with the progression from normoalbuminuria or microalbuminuria to macroalbuminuria after a follow-up period of 6 years in an African-American population of 725 patients with type 1 diabetes (80).
A comparison of 44 patients and 24 healthy controls showed increased circulating tumor necrosis factor-α in the patient group (81). Furthermore, tumor necrosis factor-α and tumor necrosis factor receptors 1 and 2 were associated with a lower estimated glomerular filtration rate in 667 patients (82). In a follow-up study of 349 patients with diabetic kidney disease, followed for 5–18 years, baseline tumor necrosis factor receptor 2 was associated with an increased risk of end-stage renal disease (83).
Some studies have examined whether these markers are related to diabetic complications in patients with type 1 diabetes. In 543 patients divided into two groups based on the presence or absence of any complication of type 1 diabetes, circulating levels of C-reactive protein, interleukin-6, and tumor necrosis factor-α were increased in individuals with complications compared with those without (84). Similarly C-reactive protein and fibrinogen were higher in 88 patients without evidence of macrovascular complications compared with 40 controls (85). The patient group was also stratified based on the presence of microvascular complications. When comparing patients with advanced complications to both those without complications and with early-stage complications, a significant difference in the above markers was seen.
Such studies suggest there is an underlying systemic inflammatory process occurring in patients with type 1 diabetes and its complications including diabetic kidney disease. Whether the inflammation occurring in individuals with type 1 diabetes is comparable those with type 2 diabetes is not known and requires further investigation.
The Complement Cascade in the Development of Diabetic Kidney Disease
The complement cascade is a highly sophisticated network of immune proteins that are activated in response to invading pathogens or tissue injury. Under normal conditions complement is tightly regulated by a number of fluid-phase and cell surface proteins (86); however, when complement is hyperactivated it drives a severe inflammatory response (87). The complement system comprises over 40 proteins that are present either in soluble form (mostly in blood) or in a bound form at local inflammatory sites and in cell membranes. The soluble proteins normally exist in an inactive form that can be rapidly activated and amplified by a series of tightly regulated sequentially acting proteases. Complement comprises four major pathways that are initiated through antibody/antigen complexes (classical pathway), carbohydrate moieties (lectin pathway), foreign surfaces (alternative pathway), and activated neutral proteases that can cleave C5, especially during inflammatory conditions independent of C3 (extrinsic pathway). All pathways result in the formation of C5a, a major effector molecule which, via ligation with its receptor C5aR1, initiates pathology in a number of inflammatory diseases (88).
The kidney appears to be particularly vulnerable to complement-mediated injury (86). Injury may arise from deposition of circulating active complement fragments in glomeruli. Alternatively, local production and activation of complement in the kidney may lead to inflammation. Several renal disorders have been associated with abnormal complement activation (86, 89) including immune complex-mediated glomerulonephritis and tubulointerstitial injury associated with progressive proteinuric diseases (86) in addition to the more recently introduced pathological entity C3 glomerulopathy (89). Excessive complement activation is also involved in two rare but severe kidney diseases that often culminate in end-stage renal failure, membranoproliferative glomerulonephritis type II (dense-deposit disease), and atypical hemolytic uremic syndrome.
It is increasingly appreciated that the complement pathway is activated in diabetic kidney disease (90). Deposition of the membrane attack complex (C5b-9) has been shown in the kidney of patients with diabetic kidney disease (91). A known inhibitor of the membrane attack complex, anti-glycated human CD59-specific antibody, has reduced activity in erythrocytes of diabetic patients (92). This may help to explain the increased membrane attack complex deposition in patients with diabetes (92). In addition to membrane attack complex, deposition in the kidney of diabetic patients with moderate and severe glomerulosclerosis is correlated with medial smooth muscle injury in intrarenal arteries (93).
Serum concentrations of mannose-binding lectin, a key protein involved in the lectin pathway, are increased in patients with diabetic kidney disease (94) and are predictive of the subsequent development of albuminuria (95). Ostergaard et al. found that serum H-ficolin, an activator of the lectin pathway, was associated with an increased risk of future progression to microalbuminuria in patients with newly diagnosed type 1 diabetes (96). In a mouse model of type 1 diabetes, circulating levels of mannan-binding lectin significantly increased after induction (97). Similarly in humans, mannan-binding lectin is significantly higher in diabetic patients compared with healthy controls (98). Furthermore, mannan-binding lectin positively correlates with urinary albumin excretion in patients with type 1 diabetes (98–101) as well as type 2 diabetes (102). Streptozotocin-induced diabetic mice with a genetic deletion in mannose-binding lectin had significantly lower rates of urinary albumin excretion compared with diabetic wild-type mice (103).
During a median of 5.8 years follow-up, baseline mannose-binding lectin was associated with increased urinary albumin excretion in patients with type 1 diabetes (104). Mannose-binding lectin was also significantly associated with progression from macroalbuminuria to end-stage renal disease (104). Similarly after a median of 18 years follow-up of newly diagnosed type 1 diabetic patients’ mannose-binding lectin measured after 3 years was associated with the development of microalbuminuria and macroalbuminuria (95). After 15 years of follow-up, the mortality of patients with type 2 diabetes was found to be significantly associated with baseline mannose-binding lectin (105). A recent study using genome-wide transcriptome analysis has shown upregulation of the complement pathway in micro-dissected human renal glomerular and tubule samples from patients with diabetic kidney disease (106). Although these studies indicate an association between activation of the complement pathway and diabetic kidney disease, the role of specific complement factors in mediating cellular injury in the development and progression of diabetic kidney disease has not yet been determined. It remains to be determined if complement proteins are useful markers for chronic kidney disease progression.
Role of Leukocytes in Diabetes Mellitus and Diabetic Kidney Disease
Studies at the cellular level have demonstrated that not only do leukocytes infiltrate the kidneys in patients with diabetic kidney disease but that functional changes in these cells also occur (Figure 4).

Figure 4. Changes to leukocytes in diabetic kidney disease.
Lymphocytes
To explore the role of lymphocytes in the development of diabetic nephropathy, Lim et al. studied recombination activating gene-1-deficient mice made diabetic with streptozotocin (107). Recombination activating gene-1-deficient mice lack mature T and B cells and hence allowing the study of the development of diabetic kidney disease in absence of lymphocytes. Diabetic recombination activating gene-1-deficient mice showed a reduction in albuminuria compared with diabetic wild-type mice in addition to preserved podocytes and reduced glomerular macrophage accumulation. These features suggest that lymphocytes play a role in the development of albuminuria. However, it should be noted that both groups showed equal levels of decline in creatinine clearance and other characteristic histological features of diabetic kidney disease.
Another study using non-obese diabetic mice (NOD mice, a model of autoimmune diabetes) showed that diabetic mice had T cell and B cell infiltration into the glomeruli, as well as CD11c+ dendritic cells (108). There was also deposition of glomerular IgG and complement C3. A similar study showed at both 1 and 8 months post diabetes induction (STZ) in rats, there was an increased infiltration of CD4+ T cells, CD8+ T cells, and macrophages into the glomeruli (109). The change in CD4+ cells appeared to be more significant at 1 month, whereas CD8+ T cells become more significant at later stages of the disease when tissue loss was evident.
Clinical studies have also shown lymphocyte activation in diabetes. In patients with type 2 diabetes and proteinuria, there was no difference in absolute numbers of circulating lymphocytes compared with age-, sex-, and duration-of-diabetes-matched controls with type 2 diabetes and without proteinuria (110). There was, however, a higher proportion of activated lymphocytes, as measured by an antihuman leukocyte antigen D-related antigen, in diabetic patients with non-nephrotic range proteinuria compared with non-proteinuric diabetic controls.
Otton et al. performed a study investigating apoptosis of lymphocytes obtained from diabetic subjects or from rats with alloxan-induced diabetes (111). Lymphocytes harvested from rat mesenteric lymph nodes or human circulating lymphocytes showed significantly higher rates of fragmented DNA, chromatin condensation, and blebbing. The high occurrence of apoptosis in lymphocytes was accompanied by a reduced number of blood-circulating lymphocytes in diabetic patients. Interestingly, in rats with insulin therapy, the proportion of lymphocytes with fragmented DNA was decreased.
Furthermore, Moon et al. have found increased infiltration of interstitial CD4+ and CD8+ T cells in 20 weeks diabetic mice kidney when compared with non-diabetic controls (112). Using flow cytometry, intrarenal CD4+ and CD8+ T cells were also significantly increased in proteinuric diabetic mice. However, the population of T and B cells in diabetic spleen was not changed, indicating the changes in lymphocytes in diabetes occurred predominantly in the kidney and not within the circulation. More importantly, in the same study, there was a significant increase in the infiltration of renal interstitial T (CD4+, CD8+) and B cells (CD20+) in type 2 diabetic human kidney, and the increase in CD4+ T cells and CD20+ B cell correlated with the amount of proteinuria in these patients (112).
Neutrophils
In 11 patients with type 1 diabetes and albuminuria, there was a significant downregulation of the adhesion molecule CD11b on neutrophils following activation by phorbol 12-myristate 13-acetate (113). The pathways controlling granule exocytosis in neutrophils from diabetic patients were shown to be abnormal. In type 2 diabetes, neutrophils display enhanced adherence to fetal-calf serum coated surfaces (114). In 22 renal biopsies from patients with biopsy proven diabetic kidney disease, there was increased infiltration of neutrophils and macrophages into interstitial, peritubular, and capillary regions (115). Furthermore, the number of interstitial and peritubular renal polymorphic neutrophils in diabetic nephropathy was proportional to estimated glomerular filtration rate at the time of the biopsy. The study did not report whether patients had type 1 diabetes or type 2 diabetes.
Macrophages
Kidney macrophage accumulation is associated with the progression of type 2 diabetic nephropathy. Chow et al. performed a time course of renal disease progression and kidney macrophage infiltration in the db/db mouse model, which exhibits obesity and type 2 diabetes (116). In db/db mice at 8 months of age, there was an increased macrophage infiltration in glomerular and interstitial regions of the kidney in mice which developed hyperglycemia compared with those that did not. Macrophage accumulation and activation in db/db mice correlated with HbA1c, glomerular and tubular damage, albuminuria, elevated plasma creatinine, renal fibrosis, and kidney expression of macrophage chemokines. In another study looking at 20 patients with diabetic kidney disease, macrophage accumulation in both glomerular and interstitial regions of renal biopsies was demonstrated (117). However, only infiltration into the interstitium was found to be correlated with progression of disease. Total macrophage count was shown to correlate with renal impairment but not albuminuria. In renal biopsies of patients with histologically proven diabetic kidney disease, compared with controls with no diabetic kidney disease, the presence of glomerular anti-inflammatory CD163+ macrophages were significantly associated with the presence of glomerulosclerosis, interstitial fibrosis, and tubular atrophy (118). Similarly both estimated glomerular filtration rate and level of albuminuria were associated with the presence of interstitial CD68+ macrophages.
In streptozotocin-induced diabetic mice, there was increased macrophage infiltration in both glomeruli and the interstitium when compared with controls (119). In the same study, genetic knockout of macrophage scavenger receptor A, which results in a reduction of macrophage infiltration into the kidneys, afforded renoprotection from STZ diabetes as shown by decreased albuminuria, glomerular hypertrophy, and renal fibrosis. It has been shown that this infiltration occurs early in diabetic kidney disease development, with macrophage infiltration being seen by immunohistochemical labeling 8 days after diabetes induction in streptozotocin-induced diabetic rats (120). However, streptozotocin may have acute effects. Nonetheless, in one study using another model of type 1 diabetes, the spontaneously diabetic Ins2Akita mouse, increased accumulation of macrophages was also observed in the glomeruli of diabetic Ins2Akita mice after 14–15 weeks of diabetes (121), thus suggesting that the recruitment of macrophages into the diabetic kidneys is not an exclusive feature of streptozotocin in animal models of type 1 diabetes.
A key mediator in the infiltration of macrophages is monocyte chemoattractant protein-1 (122). In response to pro-inflammatory cytokines interleukin-1, tumor necrosis factor-α, and interferon-γ, monocyte chemoattractant protein-1 is released from kidney tubular cells, smooth muscle cells, mesangial cells, and podocytes (122). In addition to macrophage accumulation, monocyte chemoattractant protein-1 is thought to be indirectly involved in the recruitment of neutrophils through an intermediate mediator leukotriene B4 (123). Monocyte chemoattractant protein-1 deficient diabetic mice do not develop albuminuria and are protected from an increase in plasma creatinine (124). The P2X7 receptor is expressed on macrophages and mediates pro-inflammatory signaling pathways (125). In human mesangial cells exposed to a high glucose environment, P2X7 receptors have increased release of monocyte chemoattractant protein-1 (125). Furthermore, mice with a knockdown of the P2X7 receptor had reduced glomerular macrophage recruitment and collagen IV deposition (125).
The use of NOX-E36 (empaticap pegol) which binds and inhibits MCP-1 resulted in a reduction of albumin–creatinine ratio in patients with type 2 diabetes (126). Furthermore, treatment with CCX140-B, an antagonist to C–C chemokine receptor 2 which monocyte chemoattractant protein-1 binds to, resulted in a reduction in albumin–creatinine ratio (127), glomerular hypertrophy, and increased podocyte density in experimental diabetes (127). Treatment with CCX140-B in patients with type 2 diabetes and nephropathy resulted in a reduced albumin–creatinine ratio (5).
Emergence of Neutrophil–Lymphocyte Ratio as a Potential Marker of Diabetic Kidney Disease
Both neutrophils and lymphocytes have been shown to have altered function in patients with type 1 diabetes and type 2 diabetes. Neutrophils are positively correlated with urinary albumin excretion in patients with type 2 diabetes while lymphocytes are negatively correlated (53).
Neutrophil–lymphocyte ratio, calculated by dividing the total neutrophil count by the total lymphocyte count (128), has been shown to be associated with coronary artery disease (7, 129, 130), atherosclerosis formation (131), adverse cardiac outcomes (132–134), and the metabolic syndrome (135, 136) (Figure 5). The neutrophil–lymphocyte ratio is a cheap, commonly available (137), and stable marker which provides better predictive value than individual leukocyte counts (138, 139).
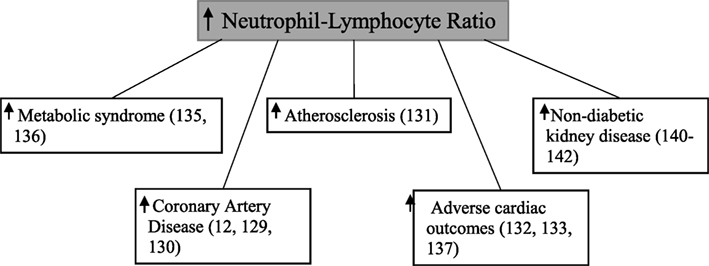
Figure 5. Elevated neutrophil–lymphocyte ratio and various disease states.
There have been a small number of studies relating neutrophil–lymphocyte ratio to non-diabetic kidney disease. Sixty one dialysis patients with higher neutrophil–lymphocyte ratio were found to also have significantly elevated levels of tumor necrosis factor-α (140). Neutrophil–lymphocyte ratio has also been demonstrated to be significantly higher in patients with chronic kidney disease when compared with healthy controls (141). Finally in 105 patients with stage 4 kidney disease, a higher baseline neutrophil–lymphocyte ratio was significantly associated with a rapid decline in estimated glomerular filtration rate, which was defined as >5 ml/min/1.73 m2/year (142).
Use of Neutrophil–Lymphocyte Ratio in Diabetes Mellitus
A relationship between neutrophil–lymphocyte ratio and the metabolic syndrome has been established (122, 123). Furthermore, there is a common inflammatory pathway observed between cardiovascular disease and type 2 diabetes (30). Several studies have examined the relationship between neutrophil–lymphocyte ratio and complications of type 2 diabetes (Figure 6).
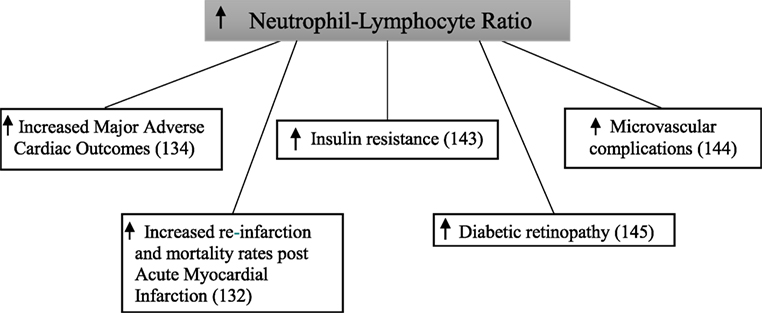
Figure 6. Increased neutrophil–lymphocyte ratio and complications in diabetes mellitus.
Baseline neutrophil–lymphocyte ratio was significant higher in patients with type 2 diabetes compared with those without in a study following 2,559 patients for 1 year after being admitted to hospital with an acute myocardial infarction (132). After follow-up, the composite endpoint of re-infarction and mortality increased in a step-wise manner across quartiles of neutrophil–lymphocyte ratio in patients with type 2 diabetes. This did not occur in patients without type 2 diabetes. In a retrospective cohort study of 338 patients with type 2 diabetes, those in the highest tertile of baseline neutrophil–lymphocyte ratio had significantly increased rates of major adverse cardiac events and mortality compared with those in the lowest tertile (134).
In a cross-sectional analysis of 586 patients, neutrophil–lymphocyte ratio was significantly higher in patients with type 2 diabetes when compared with those with normal glucose tolerance (143). This was similarly shown when comparing patients with type 2 diabetes to those with impaired glucose tolerance. Furthermore, patients with impaired glucose tolerance had a significantly higher neutrophil–lymphocyte ratio than those with normal glucose tolerance. Neutrophil–lymphocyte ratio was significantly higher in 146 patients with type 2 diabetes with microvascular complications compared with 97 patients without microvascular complications (144). Patients with microvascular complications also had a significantly higher neutrophil–lymphocyte ratio when compared with 218 healthy controls. Interestingly, no difference was seen between the control group and the patients with type 2 diabetes without microvascular complications. In this study, diabetic kidney disease was diagnosed based on urinary albumin excretion alone. In 58 type 2 diabetic patients, neutrophil–lymphocyte ratio was significantly higher when compared with 52 controls (145). In addition to this, neutrophil–lymphocyte ratio was also significantly higher in participants who also had diabetic retinopathy.
These studies demonstrate that inflammation, as measured by neutrophil–lymphocyte ratio, is associated with both macrovascular and microvascular diabetic complications.
Use of Neutrophil–Lymphocyte Ratio in Diabetic Kidney Disease
To date, only a handful of studies have looked at the association between neutrophil–lymphocyte ratio and diabetic kidney disease (Figure 7).

Figure 7. Increased neutrophil–lymphocyte ratio and diabetic kidney disease.
In 338 type 2 diabetic patients followed for 3 years, those with a higher baseline neutrophil–lymphocyte ratio were found to have increased rates of the primary endpoint of >12 ml/min loss in estimated glomerular filtration rate over the 4-year study period with the last estimated glomerular filtration rate being <60 ml/min (11). Similarly, a high baseline neutrophil–lymphocyte ratio correlated with a low baseline estimated glomerular filtration rate in this study. In 114 patients with type 2 diabetes, neutrophil–lymphocyte ratio was significantly higher in those with diabetic kidney disease compared with those without—with the presence of diabetic kidney disease being defined on the basis of both albuminuria and estimated glomerular filtration rate criteria (137). In a multiple logistic regression analysis, neutrophil–lymphocyte ratio was independently predictive of diabetic kidney disease. In this study, there was no association seen between neutrophil–lymphocyte ratio and diabetic retinopathy.
In a cross-sectional analysis of 200 patients with type 2 diabetes, elevated neutrophil–lymphocyte ratio was significantly associated with microalbuminuria, macroalbuminuria, and reduced estimated glomerular filtration rate (146). Neutrophil–lymphocyte ratio was significantly higher in 115 patients with type 2 diabetes and early-stage diabetic kidney disease when compared with 138 type 2 diabetics with no diabetic kidney disease (147). However, early-stage diabetic kidney disease was defined as urinary albumin excretion within the microalbuminuria range only. Furthermore, patients with type 2 diabetes who had declining estimated glomerular filtration rate without evidence of albuminuria were excluded.
These studies demonstrate that changes in both the innate immune system by way of the total neutrophil count and the adaptive immune system in terms of the total lymphocyte count are associated with the development of diabetic kidney disease.
Conclusion
Although diagnosis of diabetic kidney disease is well established, there is ongoing research to identify cheap and accessible ways to detect disease at an earlier stage and thus facilitate early intervention. It is hoped that through such research, reliable ways of predicting future risk may also emerge. Given the link between diabetes and inflammation, the use of common blood tests including the white cell count which is used to calculate the neutrophil–lymphocyte ratio may help provide this early detection.
Previous studies have traditionally used urinary albumin excretion as an indicator of the presence of diabetic kidney disease, along with estimates of glomerular filtration rate. It is important to recognize the limitations of using urinary albumin excretion alone in patients with type 2 diabetes to determine presence of diabetic kidney disease.
Observational studies have shown positive associations between inflammatory mediators, such as C-reactive protein, and the development of diabetic kidney disease. This has been demonstrated in both patients with type 1 and type 2 diabetes and suggests that activity of the innate immune system is implicated in this development. Furthermore, diabetic kidney disease is positively associated with neutrophils and negatively associated with lymphocytes. This suggests both the innate and adaptive immune systems play a role in the pathogenesis of diabetic kidney disease. This is further supported by the association between diabetic kidney disease and the neutrophil–lymphocyte ratio. Due to the lack of specificity of the neutrophil–lymphocyte ratio, its ability to detect diabetic kidney disease is not superior to that of albuminuria. However, it does provide important insight into the mechanisms that drive diabetic kidney disease development.
Further research is required to understand in detail how this interaction occurs as it may lead to the development of interventions. Treatments that are able to stop the key mediators of inflammation in these patients could not only reduce rates of diabetic kidney disease but also help to stop progression to end-stage renal disease. Renoprotective effects of anti-inflammatory agents which block the effects of monocyte chemoattractant protein-1 reducing albuminuria have recently been demonstrated. Other medications that affect inflammation are being developed to target diabetic kidney disease.
However, longitudinal studies with long-term follow-up periods are required to determine the exact role of inflammation and key mediators of the immune system in the development of diabetic kidney disease. Furthermore, treatments which block known inflammatory mediators in the pathogenesis of diabetic nephropathy need further research to determine if these are viable targets for treatment in patients with diabetes.
Author Contributions
LW—primary author of review. LAW—contributed to background research and references. GJ and RM—co-supervisor. J-mS and MC—helped with editing manuscript. ST and MTC—primary author of complement subsection. EE—main supervisor of the manuscript- supervised the research, and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer FP and handling Editor declared their shared affiliation.
Funding
EE was supported by an NHMRC Early Career Research Fellowship (#1054312), Viertel Clinical Investigatorship, Sir Edward Weary Dunlop Medical Research Foundation grant, RACP fellowship, and Diabetes Australia Research Program Grant. MTC is supported by a Career Development Award from the JDRF Type 1 Diabetes Clinical Research Network. ST is supported by a JDRF Advanced Postdoctoral Fellowship. RM was supported by an Australian Diabetes Society-Servier Diabetes Research Grant, St Vincent’s Health Australia’s Inclusive Innovation Fund, and the Rebecca Cooper Medical Research Foundation.
References
1. Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med (2016) 375(4):323–34. doi:10.1056/NEJMoa1515920
2. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med (2017) 377(7):644–57. doi:10.1056/NEJMoa1611925
3. Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JFE, Nauck MA, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med (2016) 375(4):311–22. doi:10.1056/NEJMoa1603827
4. Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jódar E, Leiter LA, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med (2016) 375(19):1834–44. doi:10.1056/NEJMoa1607141
5. de Zeeuw D, Bekker P, Henkel E, Hasslacher C, Gouni-Berthold I, Mehling H, et al. The effect of CCR2 inhibitor CCX140-B on residual albuminuria in patients with type 2 diabetes and nephropathy: a randomised trial. Lancet Diabetes Endocrinol (2015) 3(9):687–96. doi:10.1016/S2213-8587(15)00261-2
6. Packham DK, Alves TP, Dwyer JP, Atkins R, de Zeeuw D, Cooper M, et al. Relative incidence of ESRD versus cardiovascular mortality in proteinuric type 2 diabetes and nephropathy: results from the DIAMETRIC (Diabetes Mellitus Treatment for Renal Insufficiency Consortium) database. Am J Kidney Dis (2012) 59(1):75–83. doi:10.1053/j.ajkd.2011.09.017
7. Aygun F, Efe D. Association of neutrophil/lymphocyte ratio with obstructive coronary artery disease and coronary artery calcium score detected by multislice computed tomography in type 2 diabetes mellitus patients. Patient Prefer Adherence (2015) 9:1023–31. doi:10.2147/PPA.S85577
8. Gross JL, de Azevedo MJ, Silveiro SP, Canani LH, Caramori ML, Zelmanovitz T. Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care (2005) 28(1):164–76. doi:10.2337/diacare.28.1.164
9. Zhang P, Zhang X, Brown J, Vistisen D, Sicree R, Shaw J, et al. Global healthcare expenditure on diabetes for 2010 and 2030. Diabetes Res Clin Pract (2010) 87(3):293–301. doi:10.1016/j.diabres.2010.01.026
10. Choudhary N, Ahlawat RS. Interleukin-6 and C-reactive protein in pathogenesis of diabetic nephropathy: new evidence linking inflammation, glycemic control, and microalbuminuria. Iran J Kidney Dis (2008) 2(2):72–9.
11. Azab B, Daoud J, Naeem FB, Nasr R, Ross J, Ghimire P, et al. Neutrophil-to-lymphocyte ratio as a predictor of worsening renal function in diabetic patients (3-year follow-up study). Ren Fail (2012) 34(5):571–6. doi:10.3109/0886022X.2012.668741
12. MacIsaac RJ, Jerums G. Diabetic kidney disease with and without albuminuria. Curr Opin Nephrol Hypertens (2011) 20(3):246–57. doi:10.1097/MNH.0b013e3283456546
13. MacIsaac R, Ekinci E, Jerums G. Progressive diabetic nephropathy. How useful is microalbuminuria? contra. Kidney Int (2014) 86(1):50–7. doi:10.1038/ki.2014.98
14. Watts AK, Dwyer K, Ekinci E, Jerums G, Macisaac R. The Shifting Paradigm of Albuminuria in Diabetic Kidney Disease. Neutral Bay: Medicine Today (2015). p. 34–8.
15. Tolouian R, T Hernandez G. Prediction of diabetic nephropathy: the need for a sweet biomarker. J Nephropathol (2013) 2(1):4–5. doi:10.5812/nephropathol.8966
16. Tesch GH. Diabetic nephropathy – is this an immune disorder? Clin Sci (2017) 131(16):2183–99. doi:10.1042/CS20160636
17. Chaplin DD. Overview of the immune response. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S3–23. doi:10.1016/j.jaci.2009.12.980
18. Pickup JC, Crook MA. Is type II diabetes mellitus a disease of the innate immune system? Diabetologia (1998) 41(10):1241–8. doi:10.1007/s001250051058
19. Alberts B, Johnson A, Lewis J, Morgan D, Raff M, Roberts K, et al. Molecular Biology of the Cell. New York: Garland Science (2015).
20. Pickup JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care (2004) 27(3):813–23. doi:10.2337/diacare.27.3.813
21. Crook M. Type 2 diabetes mellitus: a disease of the innate immune system? An update. Diabet Med (2004) 21(3):203–7. doi:10.1046/j.1464-5491.2003.01030.x
22. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med (1999) 340(6):448–54. doi:10.1056/NEJM199902113400607
23. Steel DM, Whitehead AS. The major acute phase reactants: C-reactive protein, serum amyloid P component and serum amyloid A protein. Immunol Today (1994) 15(2):81–8. doi:10.1016/0167-5699(94)90138-4
24. Ballou SP, Lozanski G. Induction of inflammatory cytokine release from cultured human monocytes by C-reactive protein. Cytokine (1992) 4(5):361–8. doi:10.1016/1043-4666(92)90079-7
25. Cermak J, Key NS, Bach RR, Balla J, Jacob HS, Vercellotti GM. C-reactive protein induces human peripheral blood monocytes to synthesize tissue factor. Blood (1993) 82(2):513–20.
26. Rabb H. The T cell as a bridge between innate and adaptive immune systems: implications for the kidney. Kidney Int (2002) 61(6):1935–46. doi:10.1046/j.1523-1755.2002.00378.x
27. Bonilla FA, Oettgen HC. Adaptive immunity. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S33–40. doi:10.1016/j.jaci.2009.09.017
28. Pickup JC, Mattock MB, Chusney GD, Burt D. NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia (1997) 40(11):1286–92. doi:10.1007/s001250050822
29. Macisaac RJ, Ekinci EI, Jerums G. Markers of and risk factors for the development and progression of diabetic kidney disease. Am J Kidney Dis (2014) 63(2 Suppl 2):S39–62. doi:10.1053/j.ajkd.2013.10.048
30. Pradhan AD, Ridker PM. Do atherosclerosis and type 2 diabetes share a common inflammatory basis? Eur Heart J (2002) 23(11):831–4. doi:10.1053/euhj.2001.3052
31. Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation (2009) 120(16):1640–5. doi:10.1161/CIRCULATIONAHA.109.192644
32. Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract (2014) 105(2):141–50. doi:10.1016/j.diabres.2014.04.006
33. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med (2017) 377(12):1119–31. doi:10.1056/NEJMoa1707914
34. Ford ES. Body mass index, diabetes, and C-reactive protein among U.S. adults. Diabetes Care (1999) 22(12):1971–7. doi:10.2337/diacare.22.12.1971
35. Muller S, Martin S, Koenig W, Hanifi-Moghaddam P, Rathmann W, Haastert B, et al. Impaired glucose tolerance is associated with increased serum concentrations of interleukin 6 and co-regulated acute-phase proteins but not TNF-alpha or its receptors. Diabetologia (2002) 45(6):805–12. doi:10.1007/s00125-002-0829-2
36. Pitsavos C, Tampourlou M, Panagiotakos DB, Skoumas Y, Chrysohoou C, Nomikos T, et al. Association between low-grade systemic inflammation and type 2 diabetes mellitus among men and women from the ATTICA Study. Rev Diabet Stud (2007) 4(2):98–104. doi:10.1900/RDS.2007.4.98
37. Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA (2001) 286(3):327–34. doi:10.1001/jama.286.3.327
38. Hu FB, Meigs JB, Li TY, Rifai N, Manson JE. Inflammatory markers and risk of developing type 2 diabetes in women. Diabetes (2004) 53(3):693–700. doi:10.2337/diabetes.53.3.693
39. Freeman DJ, Norrie J, Caslake MJ, Gaw A, Ford I, Lowe GD, et al. C-reactive protein is an independent predictor of risk for the development of diabetes in the West of Scotland Coronary Prevention Study. Diabetes (2002) 51(5):1596–600. doi:10.2337/diabetes.51.5.1596
40. Laaksonen DE, Niskanen L, Nyyssonen K, Punnonen K, Tuomainen TP, Valkonen VP, et al. C-reactive protein and the development of the metabolic syndrome and diabetes in middle-aged men. Diabetologia (2004) 47(8):1403–10. doi:10.1007/s00125-004-1472-x
41. Morimoto H, Sakata K, Oishi M, Tanaka K, Nakada S, Nogawa K, et al. Effect of high-sensitivity C-reactive protein on the development of diabetes as demonstrated by pooled logistic-regression analysis of annual health-screening information from male Japanese workers. Diabetes Metab (2013) 39(1):27–33. doi:10.1016/j.diabet.2012.03.004
42. Han TS, Sattar N, Williams K, Gonzalez-Villalpando C, Lean ME, Haffner SM. Prospective study of C-reactive protein in relation to the development of diabetes and metabolic syndrome in the Mexico City Diabetes Study. Diabetes Care (2002) 25(11):2016–21. doi:10.2337/diacare.25.11.2016
43. Thorand B, Baumert J, Kolb H, Meisinger C, Chambless L, Koenig W, et al. Sex differences in the prediction of type 2 diabetes by inflammatory markers: results from the MONICA/KORA Augsburg case-cohort study, 1984-2002. Diabetes Care (2007) 30(4):854–60. doi:10.2337/dc06-1693
44. Nakanishi S, Yamane K, Kamei N, Okubo M, Kohno N. Elevated C-reactive protein is a risk factor for the development of type 2 diabetes in Japanese Americans. Diabetes Care (2003) 26(10):2754–7. doi:10.2337/diacare.26.10.2754
45. Barzilay JI, Abraham L, Heckbert SR, Cushman M, Kuller LH, Resnick HE, et al. The relation of markers of inflammation to the development of glucose disorders in the elderly: the Cardiovascular Health Study. Diabetes (2001) 50(10):2384–9. doi:10.2337/diabetes.50.10.2384
46. Festa A, D’Agostino R Jr, Tracy RP, Haffner SM; Insulin Resistance Atherosclerosis Study. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: the insulin resistance atherosclerosis study. Diabetes (2002) 51(4):1131–7. doi:10.2337/diabetes.51.4.1131
47. Spranger J, Kroke A, Mohlig M, Hoffmann K, Bergmann MM, Ristow M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes (2003) 52(3):812–7. doi:10.2337/diabetes.52.3.812
48. Doi Y, Kiyohara Y, Kubo M, Ninomiya T, Wakugawa Y, Yonemoto K, et al. Elevated C-reactive protein is a predictor of the development of diabetes in a general Japanese population: the Hisayama Study. Diabetes Care (2005) 28(10):2497–500. doi:10.2337/diacare.28.10.2497
49. Dehghan A, Kardys I, de Maat MP, Uitterlinden AG, Sijbrands EJ, Bootsma AH, et al. Genetic variation, C-reactive protein levels, and incidence of diabetes. Diabetes (2007) 56(3):872–8. doi:10.2337/db06-0922
50. Lee CC, Adler AI, Sandhu MS, Sharp SJ, Forouhi NG, Erqou S, et al. Association of C-reactive protein with type 2 diabetes: prospective analysis and meta-analysis. Diabetologia (2009) 52(6):1040–7. doi:10.1007/s00125-009-1338-3
51. Thorand B, Lowel H, Schneider A, Kolb H, Meisinger C, Frohlich M, et al. C-reactive protein as a predictor for incident diabetes mellitus among middle-aged men: results from the MONICA Augsburg cohort study, 1984-1998. Arch Intern Med (2003) 163(1):93–9. doi:10.1001/archinte.163.1.93
52. Krakoff J, Funahashi T, Stehouwer CD, Schalkwijk CG, Tanaka S, Matsuzawa Y, et al. Inflammatory markers, adiponectin, and risk of type 2 diabetes in the Pima Indian. Diabetes Care (2003) 26(6):1745–51. doi:10.2337/diacare.26.6.1745
53. Chung FM, Tsai JC, Chang DM, Shin SJ, Lee YJ. Peripheral total and differential leukocyte count in diabetic nephropathy: the relationship of plasma leptin to leukocytosis. Diabetes Care (2005) 28(7):1710–7. doi:10.2337/diacare.28.7.1710
54. Cavalot F, Massucco P, Perna P, Traversa M, Anfossi G, Trovati M. White blood cell count is positively correlated with albumin excretion rate in subjects with type 2 diabetes. Diabetes Care (2002) 25(12):2354–5. doi:10.2337/diacare.25.12.2354-a
55. Ohshita K, Yamane K, Hanafusa M, Mori H, Mito K, Okubo M, et al. Elevated white blood cell count in subjects with impaired glucose tolerance. Diabetes Care (2004) 27(2):491–6. doi:10.2337/diacare.27.2.491
56. Ford ES. Leukocyte count, erythrocyte sedimentation rate, and diabetes incidence in a national sample of US adults. Am J Epidemiol (2002) 155(1):57–64. doi:10.1093/aje/155.1.57
57. Vozarova B, Weyer C, Lindsay RS, Pratley RE, Bogardus C, Tataranni PA. High white blood cell count is associated with a worsening of insulin sensitivity and predicts the development of type 2 diabetes. Diabetes (2002) 51(2):455–61. doi:10.2337/diabetes.51.2.455
58. Schmidt MI, Duncan BB, Sharrett AR, Lindberg G, Savage PJ, Offenbacher S, et al. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): a cohort study. Lancet (1999) 353(9165):1649–52. doi:10.1016/S0140-6736(99)01046-6
59. Ekinci EI, Jerums G, Skene A, Crammer P, Power D, Cheong KY, et al. Renal structure in normoalbuminuric and albuminuric patients with type 2 diabetes and impaired renal function. Diabetes Care (2013) 36(11):3620–6. doi:10.2337/dc12-2572
60. MacIsaac RJ, Tsalamandris C, Panagiotopoulos S, Smith TJ, McNeil KJ, Jerums G. Nonalbuminuric renal insufficiency in type 2 diabetes. Diabetes Care (2004) 27(1):195–200. doi:10.2337/diacare.27.1.195
61. Tsalamandris C, Allen TJ, Gilbert RE, Sinha A, Panagiotopoulos S, Cooper ME, et al. Progressive decline in renal function in diabetic patients with and without albuminuria. Diabetes (1994) 43(5):649–55. doi:10.2337/diabetes.43.5.649
62. Stehouwer CD, Gall MA, Twisk JW, Knudsen E, Emeis JJ, Parving HH. Increased urinary albumin excretion, endothelial dysfunction, and chronic low-grade inflammation in type 2 diabetes: progressive, interrelated, and independently associated with risk of death. Diabetes (2002) 51(4):1157–65. doi:10.2337/diabetes.51.4.1157
63. Bakris GL, Molitch M. Microalbuminuria as a risk predictor in diabetes: the continuing saga. Diabetes Care (2014) 37(3):867–75. doi:10.2337/dc13-1870
64. MacLeod JM, Lutale J, Marshall SM. Albumin excretion and vascular deaths in NIDDM. Diabetologia (1995) 38(5):610–6. doi:10.1007/BF00400732
65. Gomes MB, Nogueira VG. Acute-phase proteins and microalbuminuria among patients with type 2 diabetes. Diabetes Res Clin Pract (2004) 66(1):31–9. doi:10.1016/j.diabres.2004.02.009
66. Festa A, D’Agostino R, Howard G, Mykkänen L, Tracy RP, Haffner SM. Inflammation and microalbuminuria in nondiabetic and type 2 diabetic subjects: the Insulin Resistance Atherosclerosis Study. Kidney Int (2000) 58(4):1703–10. doi:10.1046/j.1523-1755.2000.00331.x
67. Otto C, Engelschalk C, Fraunberger P, Laubach E, Schwandt P. Lack of an association of urinary albumin excretion with interleukin-6 or C-reactive protein in patients with type 2 diabetes. Acta Diabetol (2001) 38(4):153–5. doi:10.1007/s592-001-8072-5
68. Moriwaki Y, Yamamoto T, Shibutani Y, Aoki E, Tsutsumi Z, Takahashi S, et al. Elevated levels of interleukin-18 and tumor necrosis factor-α in serum of patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Metabolism (2003) 52(5):605–8. doi:10.1053/meta.2003.50096
69. Dalla Vestra M, Mussap M, Gallina P, Bruseghin M, Cernigoi AM, Saller A, et al. Acute-phase markers of inflammation and glomerular structure in patients with type 2 diabetes. J Am Soc Nephrol (2005) 16(Suppl 1):S78–82. doi:10.1681/ASN.2004110961
70. Cao Z, Bonnet F, Davis B, Allen TJ, Cooper ME. Additive hypotensive and anti-albuminuric effects of angiotensin-converting enzyme inhibition and angiotensin receptor antagonism in diabetic spontaneously hypertensive rats. Clin Sci (Lond) (2001) 100(6):591–9. doi:10.1042/cs1000591
71. Carlsson AC, Östgren CJ, Nystrom FH, Länne T, Jennersjö P, Larsson A, et al. Association of soluble tumor necrosis factor receptors 1 and 2 with nephropathy, cardiovascular events, and total mortality in type 2 diabetes. Cardiovasc Diabetol (2016) 15:40. doi:10.1186/s12933-016-0359-8
72. Pavkov ME, Weil EJ, Fufaa GD, Nelson RG, Lemley KV, Knowler WC, et al. Tumor necrosis factor receptors 1 and 2 are associated with early glomerular lesions in type 2 diabetes. Kidney Int (2016) 89(1):226–34. doi:10.1038/ki.2015.278
73. Pavkov ME, Nelson RG, Knowler WC, Cheng Y, Krolewski AS, Niewczas MA. Elevation of circulating TNF receptors 1 and 2 increases the risk of end-stage renal disease in American Indians with type 2 diabetes. Kidney Int (2015) 87(4):812–9. doi:10.1038/ki.2014.330
74. Klein RL, Hunter SJ, Jenkins AJ, Zheng D, Semler AJ, Clore J, et al. Fibrinogen is a marker for nephropathy and peripheral vascular disease in type 1 diabetes: studies of plasma fibrinogen and fibrinogen gene polymorphism in the DCCT/EDIC cohort. Diabetes Care (2003) 26(5):1439–48. doi:10.2337/diacare.26.5.1439
75. Marcovecchio ML, Giannini C, Widmer B, Dalton RN, Martinotti S, Chiarelli F, et al. C-reactive protein in relation to the development of microalbuminuria in type 1 diabetes: the Oxford Regional Prospective Study. Diabetes Care (2008) 31(5):974–6. doi:10.2337/dc07-2101
76. Saraheimo M, Teppo AM, Forsblom C, Fagerudd J, Groop PH. Diabetic nephropathy is associated with low-grade inflammation in type 1 diabetic patients. Diabetologia (2003) 46(10):1402–7. doi:10.1007/s00125-003-1194-5
77. Schalkwijk CG, Poland DC, van Dijk W, Kok A, Emeis JJ, Drager AM, et al. Plasma concentration of C-reactive protein is increased in type I diabetic patients without clinical macroangiopathy and correlates with markers of endothelial dysfunction: evidence for chronic inflammation. Diabetologia (1999) 42(3):351–7. doi:10.1007/s001250051162
78. Astrup AS, Tarnow L, Pietraszek L, Schalkwijk CG, Stehouwer CD, Parving HH, et al. Markers of endothelial dysfunction and inflammation in type 1 diabetic patients with or without diabetic nephropathy followed for 10 years: association with mortality and decline of glomerular filtration rate. Diabetes Care (2008) 31(6):1170–6. doi:10.2337/dc07-1960
79. Witkowska AM, Borawska MH. Soluble intercellular adhesion molecule-1 (sICAM-1): an overview. Eur Cytokine Netw (2004) 15(2):91–8.
80. Roy MS, Janal MN, Crosby J, Donnelly R. Markers of endothelial dysfunction and inflammation predict progression of diabetic nephropathy in African Americans with type 1 diabetes. Kidney Int (2015) 87(2):427–33. doi:10.1038/ki.2014.212
81. Lechleitner M, Koch T, Herold M, Dzien A, Hoppichler F. Tumour necrosis factor-alpha plasma level in patients with type 1 diabetes mellitus and its association with glycaemic control and cardiovascular risk factors. J Intern Med (2000) 248(1):67–76. doi:10.1046/j.1365-2796.2000.00705.x
82. Niewczas MA, Ficociello LH, Johnson AC, Walker W, Rosolowsky ET, Roshan B, et al. Serum concentrations of markers of TNFalpha and Fas-mediated pathways and renal function in nonproteinuric patients with type 1 diabetes. Clin J Am Soc Nephrol (2009) 4(1):62–70. doi:10.2215/CJN.03010608
83. Skupien J, Warram JH, Niewczas MA, Gohda T, Malecki M, Mychaleckyj JC, et al. Synergism between circulating tumor necrosis factor receptor 2 and HbA(1c) in determining renal decline during 5-18 years of follow-up in patients with type 1 diabetes and proteinuria. Diabetes Care (2014) 37(9):2601–8. doi:10.2337/dc13-1983
84. Schram MT, Chaturvedi N, Schalkwijk C, Giorgino F, Ebeling P, Fuller JH, et al. Vascular risk factors and markers of endothelial function as determinants of inflammatory markers in type 1 diabetes: the EURODIAB Prospective Complications Study. Diabetes Care (2003) 26(7):2165–73. doi:10.2337/diacare.26.7.2165
85. Targher G, Bertolini L, Zoppini G, Zenari L, Falezza G. Increased plasma markers of inflammation and endothelial dysfunction and their association with microvascular complications in Type 1 diabetic patients without clinically manifest macroangiopathy. Diabet Med (2005) 22(8):999–1004. doi:10.1111/j.1464-5491.2005.01562.x
86. Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol (2013) 33(6):479–92. doi:10.1016/j.semnephrol.2013.08.001
87. Walport MJ. Complement. First of two parts. N Engl J Med (2001) 344(14):1058–66. doi:10.1056/NEJM200104053441406
88. Woodruff TM, Nandakumar KS, Tedesco F. Inhibiting the C5-C5a receptor axis. Mol Immunol (2011) 48(14):1631–42. doi:10.1016/j.molimm.2011.04.014
89. Pickering MC, D’Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, et al. C3 glomerulopathy: consensus report. Kidney Int (2013) 84(6):1079–89. doi:10.1038/ki.2013.377
90. Flyvbjerg A. The role of the complement system in diabetic nephropathy. Nat Rev Nephrol (2017) 13(5):311–8. doi:10.1038/nrneph.2017.31
91. Falk RJ, Sisson SP, Dalmasso AP, Kim Y, Michael AF, Vernier RL. Ultrastructural localization of the membrane attack complex of complement in human renal tissues. Am J Kidney Dis (1987) 9(2):121–8. doi:10.1016/S0272-6386(87)80089-6
92. Qin X, Goldfine A, Krumrei N, Grubissich L, Acosta J, Chorev M, et al. Glycation inactivation of the complement regulatory protein CD59: a possible role in the pathogenesis of the vascular complications of human diabetes. Diabetes (2004) 53(10):2653–61. doi:10.2337/diabetes.53.10.2653
93. Uesugi N, Sakata N, Nangaku M, Abe M, Horiuchi S, Hisano S, et al. Possible mechanism for medial smooth muscle cell injury in diabetic nephropathy: glycoxidation-mediated local complement activation. Am J Kidney Dis (2004) 44(2):224–38. doi:10.1053/j.ajkd.2004.04.027
94. Hansen TK, Tarnow L, Thiel S, Steffensen R, Stehouwer CD, Schalkwijk CG, et al. Association between mannose-binding lectin and vascular complications in type 1 diabetes. Diabetes (2004) 53(6):1570–6. doi:10.2337/diabetes.53.6.1570
95. Hovind P, Hansen TK, Tarnow L, Thiel S, Steffensen R, Flyvbjerg A, et al. Mannose-binding lectin as a predictor of microalbuminuria in type 1 diabetes: an inception cohort study. Diabetes (2005) 54(5):1523–7. doi:10.2337/diabetes.54.5.1523
96. Ostergaard JA, Thiel S, Hovind P, Holt CB, Parving HH, Flyvbjerg A, et al. Association of the pattern recognition molecule H-ficolin with incident microalbuminuria in an inception cohort of newly diagnosed type 1 diabetic patients: an 18 year follow-up study. Diabetologia (2014) 57(10):2201–7. doi:10.1007/s00125-014-3332-7
97. Ostergaard JA, Bjerre M, Dagnaes-Hansen F, Hansen TK, Thiel S, Flyvbjerg A. Diabetes-induced changes in mannan-binding lectin levels and complement activation in a mouse model of type 1 diabetes. Scand J Immunol (2013) 77(3):187–94. doi:10.1111/sji.12027
98. Hansen TK, Thiel S, Knudsen ST, Gravholt CH, Christiansen JS, Mogensen CE, et al. Elevated levels of mannan-binding lectin in patients with type 1 diabetes. J Clin Endocrinol Metab (2003) 88(10):4857–61. doi:10.1210/jc.2003-030742
99. Saraheimo M, Forsblom C, Hansen TK, Teppo AM, Fagerudd J, Pettersson-Fernholm K, et al. Increased levels of mannan-binding lectin in type 1 diabetic patients with incipient and overt nephropathy. Diabetologia (2005) 48(1):198–202. doi:10.1007/s00125-004-1594-1
100. Kaunisto MA, Sjölind L, Sallinen R, Pettersson-Fernholm K, Saraheimo M, Fröjdö S, et al. Elevated MBL concentrations are not an indication of association between the MBL2 gene and type 1 diabetes or diabetic nephropathy. Diabetes (2009) 58(7):1710–4. doi:10.2337/db08-1495
101. Zhao SQ, Hu Z. Mannose-binding lectin and diabetic nephropathy in type 1 diabetes. J Clin Lab Anal (2016) 30(4):345–50. doi:10.1002/jcla.21861
102. Guan LZ, Tong Q, Xu J. Elevated serum levels of mannose-binding lectin and diabetic nephropathy in type 2 diabetes. PLoS One (2015) 10(3):e0119699. doi:10.1371/journal.pone.0119699
103. Østergaard J, Thiel S, Gadjeva M, Hansen TK, Rasch R, Flyvbjerg A. Mannose-binding lectin deficiency attenuates renal changes in a streptozotocin-induced model of type 1 diabetes in mice. Diabetologia (2007) 50(7):1541–9. doi:10.1007/s00125-007-0686-0
104. Hansen TK, Forsblom C, Saraheimo M, Thorn L, Waden J, Hoyem P, et al. Association between mannose-binding lectin, high-sensitivity C-reactive protein and the progression of diabetic nephropathy in type 1 diabetes. Diabetologia (2010) 53(7):1517–24. doi:10.1007/s00125-010-1742-8
105. Hansen TK, Gall MA, Tarnow L, Thiel S, Stehouwer CD, Schalkwijk CG, et al. Mannose-binding lectin and mortality in type 2 diabetes. Arch Intern Med (2006) 166(18):2007–13. doi:10.1001/archinte.166.18.2007
106. Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, Susztak K. Transcriptome analysis of human diabetic kidney disease. Diabetes (2011) 60(9):2354–69. doi:10.2337/db10-1181
107. Lim AK, Ma FY, Nikolic-Paterson DJ, Kitching AR, Thomas MC, Tesch GH. Lymphocytes promote albuminuria, but not renal dysfunction or histological damage in a mouse model of diabetic renal injury. Diabetologia (2010) 53(8):1772–82. doi:10.1007/s00125-010-1757-1
108. Xiao X, Ma B, Dong B, Zhao P, Tai N, Chen L, et al. Cellular and humoral immune responses in the early stages of diabetic nephropathy in NOD mice. J Autoimmun (2009) 32(2):85–93. doi:10.1016/j.jaut.2008.12.003
109. Mensah-Brown EP, Obineche EN, Galadari S, Chandranath E, Shahin A, Ahmed I, et al. Streptozotocin-induced diabetic nephropathy in rats: the role of inflammatory cytokines. Cytokine (2005) 31(3):180–90. doi:10.1016/j.cyto.2005.04.006
110. Bending JJ, Lobo-Yeo A, Vergani D, Viberti GC. Proteinuria and activated T-lymphocytes in diabetic nephropathy. Diabetes (1988) 37(5):507–11. doi:10.2337/diabetes.37.5.507
111. Otton R, Soriano F, Verlengia R, Curi R. Diabetes induces apoptosis in lymphocytes. J Endocrinol (2004) 182(1):145–56. doi:10.1677/joe.0.1820145
112. Moon JY, Jeong KH, Lee TW, Ihm CG, Lim SJ, Lee SH. Aberrant recruitment and activation of T cells in diabetic nephropathy. Am J Nephrol (2012) 35(2):164–74. doi:10.1159/000334928
113. Fardon NJM, Wilkinson R, Thomas T. Abnormalities in primary granule exocytosis in neutrophils from type I diabetic patients with nephropathy. Clin Sci (2002) 102(1):69–75. doi:10.1042/cs1020069
114. Takahashi T, Hato F, Yamane T, Inaba M, Okuno Y, Nishizawa Y, et al. Increased spontaneous adherence of neutrophils from type 2 diabetic patients with overt proteinuria. Diabetes Care (2000) 23(3):417–8. doi:10.2337/diacare.23.3.417
115. Kelly KJ, Dominguez J. Rapid progression of diabetic nephropathy is linked to inflammation and episodes of acute renal failure. Am J Nephrol (2010) 32(5):469–75. doi:10.1159/000320749
116. Chow F, Ozols E, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int (2004) 65(1):116–28. doi:10.1111/j.1523-1755.2004.00367.x
117. Nguyen D, Ping F, Mu W, Hill P, Atkins RC, Chadban SJ. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology (Carlton) (2006) 11(3):226–31. doi:10.1111/j.1440-1797.2006.00576.x
118. Klessens CQF, Zandbergen M, Wolterbeek R, Bruijn JA, Rabelink TJ, Bajema IM, et al. Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol Dial Transplant (2017) 32(8):1322–9. doi:10.1093/ndt/gfw260
119. Usui HK, Shikata K, Sasaki M, Okada S, Matsuda M, Shikata Y, et al. Macrophage scavenger receptor-a-deficient mice are resistant against diabetic nephropathy through amelioration of microinflammation. Diabetes (2007) 56(2):363–72. doi:10.2337/db06-0359
120. Sassy-Prigent C, Heudes D, Mandet C, Belair MF, Michel O, Perdereau B, et al. Early glomerular macrophage recruitment in streptozotocin-induced diabetic rats. Diabetes (2000) 49(3):466–75. doi:10.2337/diabetes.49.3.466
121. Awad AS, Kinsey GR, Khutsishvili K, Gao T, Bolton WK, Okusa MD. Monocyte/macrophage chemokine receptor CCR2 mediates diabetic renal injury. Am J Physiol Renal Physiol (2011) 301(6):F1358–66. doi:10.1152/ajprenal.00332.2011
122. Haller H, Bertram A, Nadrowitz F, Menne J. Monocyte chemoattractant protein-1 and the kidney. Curr Opin Nephrol Hypertens (2016) 25(1):42–9. doi:10.1097/MNH.0000000000000186
123. Matsukawa A, Hogaboam CM, Lukacs NW, Lincoln PM, Strieter RM, Kunkel SL. Endogenous monocyte chemoattractant protein-1 (MCP-1) protects mice in a model of acute septic peritonitis: cross-talk between MCP-1 and leukotriene B4. J Immunol (1999) 163(11):6148–54.
124. Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Rollin BJ, Tesch GH. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int (2006) 69(1):73–80. doi:10.1038/sj.ki.5000014
125. Menzies RI, Booth JWR, Mullins JJ, Bailey MA, Tam FWK, Norman JT, et al. Hyperglycemia-induced renal P2X7 receptor activation enhances diabetes-related Injury. EBioMedicine (2017) 19:73–83. doi:10.1016/j.ebiom.2017.04.011
126. Menne J, Eulberg D, Beyer D, Baumann M, Saudek F, Valkusz Z, et al. C-C motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol Dial Transplant (2016) 32(2):307–15. doi:10.1093/ndt/gfv459
127. Sullivan T, Miao Z, Dairaghi DJ, Krasinski A, Wang Y, Zhao BN, et al. CCR2 antagonist CCX140-B provides renal and glycemic benefits in diabetic transgenic human CCR2 knockin mice. Am J Physiol Renal Physiol (2013) 305(9):F1288–97. doi:10.1152/ajprenal.00316.2013
128. Arbel Y, Finkelstein A, Halkin A, Birati EY, Revivo M, Zuzut M, et al. Neutrophil/lymphocyte ratio is related to the severity of coronary artery disease and clinical outcome in patients undergoing angiography. Atherosclerosis (2012) 225(2):456–60. doi:10.1016/j.atherosclerosis.2012.09.009
129. Balta S, Celik T, Mikhailidis DP, Ozturk C, Demirkol S, Aparci M, et al. The relation between atherosclerosis and the neutrophil-lymphocyte ratio. Clin Appl Thromb Hemost (2015) 22(5):405–11. doi:10.1177/1076029615569568
130. Verdoia M, Barbieri L, Giovine GD, Marino P, Suryapranata H, De Luca G. Neutrophil to lymphocyte ratio and the extent of coronary artery disease: results from a Large Cohort Study. Angiology (2015) 67(1):75–82. doi:10.1177/0003319715577529
131. Kalay N, Dogdu O, Koc F, Yarlioglues M, Ardic I, Akpek M, et al. Hematologic parameters and angiographic progression of coronary atherosclerosis. Angiology (2012) 63(3):213–7. doi:10.1177/0003319711412763
132. Lee G-K, Lee L-C, Chong E, Lee C-H, Teo S-G, Chia B-L, et al. The long-term predictive value of the neutrophil-to-lymphocyte ratio in type 2 diabetic patients presenting with acute myocardial infarction. QJM (2012) 105(11):1075–82. doi:10.1093/qjmed/hcs123
133. Arbel Y, Shacham Y, Ziv Baran T, Laufer Perl M, Finkelstein A, Halkin A, et al. Higher neutrophil/lymphocyte ratio is related to lower ejection fraction and higher long-term all-cause mortality in ST-elevation myocardial infarction patients. Can J Cardiol (2014) 30(10):1177–82. doi:10.1016/j.cjca.2014.05.010
134. Azab B, Chainani V, Shah N, McGinn JT. Neutrophil-lymphocyte ratio as a predictor of major adverse cardiac events among diabetic population: a 4-year follow-up study. Angiology (2013) 64(6):456–65. doi:10.1177/0003319712455216
135. Balta S, Cakar M, Demirkol S, Arslan Z, Akhan M. Higher neutrophil to lymhocyte ratio in patients with metabolic syndrome. Clin Appl Thromb Hemost (2013) 19(5):579. doi:10.1177/1076029612475023
136. Buyukkaya E, Karakas MF, Karakas E, Akcay AB, Tanboga IH, Kurt M, et al. Correlation of neutrophil to lymphocyte ratio with the presence and severity of metabolic syndrome. Clin Appl Thromb Hemost (2014) 20(2):159–63. doi:10.1177/1076029612459675
137. Ciray H, Aksoy AH, Ulu N, Cizmecioglu A, Gaipov A, Solak Y. Nephropathy, but not angiographically proven retinopathy, is associated with neutrophil to lymphocyte ratio in patients with type 2 diabetes. Exp Clin Endocrinol Diabetes (2015) 123(5):267–71. doi:10.1055/s-0035-1547257
138. Balta S, Kurtoglu E, Kucuk U, Demirkol S, Ozturk C. Neutrophil-lymphocyte ratio as an important assessment tool. Expert Rev Cardiovasc Ther (2014) 12(5):537–8. doi:10.1586/14779072.2014.902309
139. Horne B, Anderson J, John J, Weaver A, Bair T, Jensen K, et al. Which white blood cell subtypes predict increased cardiovascular risk? J Am Coll Cardiol (2005) 45(10):1638–43. doi:10.1016/j.jacc.2005.02.054
140. Turkmen K, Guney I, Yerlikaya FH, Tonbul HZ. The relationship between neutrophil-to-lymphocyte ratio and inflammation in end-stage renal disease patients. Ren Fail (2012) 34(2):155–9. doi:10.3109/0886022X.2011.641514
141. Okyay GU, İnal S, Öneç K, Er RE, Paşaoğlu Ö, Paşaoğlu H, et al. Neutrophil to lymphocyte ratio in evaluation of inflammation in patients with chronic kidney disease. Ren Fail (2013) 35(1):29–36. doi:10.3109/0886022X.2012.734429
142. Kocyigit I, Eroglu E, Unal A, Sipahioglu MH, Tokgoz B, Oymak O, et al. Role of neutrophil/lymphocyte ratio in prediction of disease progression in patients with stage-4 chronic kidney disease. J Nephrol (2013) 26(2):358–65. doi:10.5301/jn.5000152
143. Shiny A, Bibin Y, Shanthirani C, Regin B, Anjana R, Balasubramanyam M, et al. Association of neutrophil-lymphocyte ratio with glucose intolerance: an indicator of systemic inflammation in patients with type 2 diabetes. Diabetes Technol Ther (2014) 16(8):524–30. doi:10.1089/dia.2013.0264
144. Ozturk ZA, Kuyumcu ME, Yesil Y, Savas E, Yildiz H, Kepekci Y, et al. Is there a link between neutrophil-lymphocyte ratio and microvascular complications in geriatric diabetic patients? J Endocrinol Invest (2013) 36(8):593–9. doi:10.3275/8894
145. Ulu SM, Dogan M, Ahsen A, Altug A, Demir K, Acartürk G, et al. Neutrophil-to-lymphocyte ratio as a quick and reliable predictive marker to diagnose the severity of diabetic retinopathy. Diabetes Technol Ther (2013) 15(11):942–7. doi:10.1089/dia.2013.0097
146. Akbas EM, Demirtas L, Ozcicek A, Timuroglu A, Bakirci EM, Hamur H, et al. Association of epicardial adipose tissue, neutrophil-to-lymphocyte ratio and platelet-to-lymphocyte ratio with diabetic nephropathy. Int J Clin Exp Med (2014) 7(7):1794–801.
Keywords: diabetic kidney disease, neutrophil–lymphocyte ratio, inflammation, neutrophils, lymphocytes, diabetic nephropathy, atherosclerosis
Citation: Winter L, Wong LA, Jerums G, Seah J-m, Clarke M, Tan SM, Coughlan MT, MacIsaac RJ and Ekinci EI (2018) Use of Readily Accessible Inflammatory Markers to Predict Diabetic Kidney Disease. Front. Endocrinol. 9:225. doi: 10.3389/fendo.2018.00225
Received: 20 February 2018; Accepted: 20 April 2018;
Published: 22 May 2018
Edited by:
Tarunveer Singh Ahluwalia, Steno Diabetes Center Copenhagen (SDCC), DenmarkReviewed by:
Frederik Ivar Persson, Steno Diabetes Center Copenhagen (SDCC), DenmarkEusebio Chiefari, Università degli Studi Magna Graecia di Catanzaro, Italy
Copyright: © 2018 Winter, Wong, Jerums, Seah, Clarke, Tan, Coughlan, MacIsaac and Ekinci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elif I. Ekinci, ZWxpZi5la2luY2lAdW5pbWVsYi5lZHUuYXU=