 Zhi-Ping Tang1,2,3
Zhi-Ping Tang1,2,3 Jian-kui Du
Jian-kui Du Xiao-Yan Zhu
Xiao-Yan Zhu Jian-Qiang Lu
Jian-Qiang Lu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 14 May 2018
Sec. Experimental Endocrinology
Volume 9 - 2018 | https://doi.org/10.3389/fendo.2018.00215
This article is part of the Research Topic MicroRNAs as New Players in Endocrinology View all 9 articles
Oxidative stress plays a pivotal role in the initiation and progression of cardiac diseases. Estrogens have been demonstrated to exert pleiotropic cardioprotective effects, among which antioxidative stress is one of the key effects linking estrogens to cardioprotection. By using a microRNAs (miRs) microarray screening approach, we discovered an increase in miR-494, which is known to exert cardioprotective effects, in estrogen-treated cardiomyocytes. We hypothesized that the upregulation of miR-494 might contribute to estrogen-mediated cardioprotection against oxidative stress. We found that E2 stimulates miR-494 expression via ERα in both cardiomyocytes and the myocardium of female mice. The miR-494 inhibitor attenuated the protective effect of 17β-estradiol (E2) against oxidative stress-induced injury in cardiomyocytes. By contrast, the miR-494 mimic protected cardiomyocytes against oxidative stress-induced cardiomyocyte injury. Using real-time PCR, western blot and dual-luciferase reporter gene analyses, we identified nuclear factor kappa B (NF-κB) repressing factor (NKRF) as the miR-494 target in cardiomyocytes. E2 was found to inhibit NKRF, thus activating NF-κB through a miR-494-dependent mechanism. In addition, the protective effects of E2 and miR-494 against oxidative stress in cardiomyocytes were eliminated by the NF-κB inhibitor. In summary, this study demonstrates for the first time that estrogen inhibits NKRF expression through ERα-mediated upregulation of miR-494 in cardiomyocytes, leading to the activation of NF-κB, which in turn results in an increase in antioxidative defense. ERα-mediated upregulation of miR-494 may contribute to estrogen protection of cardiomyocytes against oxidative stress.
Cardiac diseases are a growing public health problem (1). Intriguingly, observational studies provide strong support for significant gender differences in the incidence and prognosis of cardiac diseases (2). Premenopausal women face a lower incidence of myocardial dysfunction, ventricular hypertrophy, heart failure, and myocardial ischemia than age-matched men. This gender advantage is lost, however, once women become postmenopausal (3, 4). These findings have attracted great attention on the cardioprotective role of estrogens, which is supported by a number of clinical and animal studies (5–7).
Estrogens have been demonstrated to exert pleiotropic cardioprotective effects, among which antioxidative stress is one of the key effects linking estrogens to cardioprotection (8). Oxidative stress plays a pivotal role in the initiation and progression of cardiac diseases including myocardial ischemia/reperfusion (I/R) injury, ventricular hypertrophy, and heart failure. Estrogens have been reported to prevent oxidative stress-induced apoptosis in cardiomyocytes by counteracting mitochondrial reactive oxygen species generation (9). Prior studies including ours also demonstrate that estrogens may exert cardioprotection via increasing myocardial antioxidants and suppressing myocardial oxidative stress in animal models of estrogen deprivation (10, 11), chronic volume overload (12), and pressure overload-induced hypertrophy (13). Thus far, the mechanisms by which estrogens protect cardiomyocytes against oxidative stress remain largely unknown. We speculate that estrogen-regulated microRNAs (miRs) may be involved.
MicroRNAs are a class of endogenous small noncoding RNAs that negatively regulate the stability and translation of target protein-coding mRNAs at the 3′ untranslated region (UTR) (14). The dysregulation of miRs has been reported in a variety of cardiac diseases (15, 16). In particular, Queirós et al. reported that estrogen regulates a miRs network including miR-21, -24, -27a, -27b, -106a, and -106b in female cardiac fibroblasts, thereby modulating a spectrum of genes involved in cardiac fibrosis and remodeling (17). However, the estrogen-responsive miRs in cardiomyocytes still remain to be elucidated.
In the present study, we first used a miRs microarray screening approach to address miRs expression profiling in estrogen-treated cardiomyocytes. We discovered an increase in miR-494, which is known to exert cardioprotective effects against I/R-induced injury (18). We hypothesized that the upregulation of miR-494 might contribute to estrogen-mediated cardioprotection against oxidative stress.
The preparation of the rat neonatal cardiomyocyte culture was as described previously (10, 11). Briefly, ventricle tissues were collected from rats that were up to 3 days old, and then minced in a dissociation buffer (in mmol/L: 116 NaCl, 20 HEPES, 0.8 Na2HPO4, 5.6 glucose, 5.4 KCl, 0.8 MgSO4, pH of 7.35) into 1 mm3 particles. Serial digestions were performed in a dissociation buffer containing 0.1% trypsin and 0.05% collagenase type II (Worthington Biochemical, Freehold, NJ, USA) at 37°C. Cell pellets were resuspended in DMEM containing 10% fetal bovine serum (FBS) and placed in culture dishes at 37°C for 1 h to allow for the selective attachment of nonmyocytes (primarily cardiac fibroblasts). The cardiomyocyte-enriched fraction (>95% cardiomyocytes as determined by immunocytochemistry staining) was then seeded onto a 12-well culture plate (Corning, Inc., Cambridge, MA, USA) at a density of 1 × 105 cells/cm2 and cultured in DMEM containing 15 mmol/L HEPES, 10% FBS, 0.1 mmol/L bromodeoxyuridine (BrdU), and antibiotics (100 U/mL penicillin and 100 mg/mL streptomycin) for 48 h. The culture medium was then exchanged for serum-free DMEM containing the same additives with the exception of BrdU.
The H9c2 myocardial cell line was originally obtained from the American Type Culture Collection and kindly provided by the Shanghai Institute for Biological Sciences. The cells were cultured in a DMEM medium containing 10% FBS at 37°C in 5% CO2–95% air.
Rat neonatal cardiomyocytes treated with 17β-estradiol (E2) for 24 h were used for small RNA extraction with the miRcute miRNA isolation kit (TianGen) according to the manufacturer’s protocol. The miRNAs were dissolved in DEPC treated water and the concentration was determined by Nanodrop2000c (Thermo). miRNA microarrays were performed as previously described (19). Briefly, RNA samples were isolated, size fractionated, and labeled with Cy3 or Cy5. The samples were hybridized to a dual-channel microarray using the μParaflo microfluidics chips of LC Sciences (Houston, TX, USA). This array contained probes for rat, mouse, and human miRNAs listed in Sanger miRBase Release 11.0. The reverse transcription, cRNA synthesis, labeling, and hybridization with Affymetrix GeneChip Rat Genome 230 2.0 Array were conducted following the standard Affymetrix protocol. The background-subtracted data were normalized by the LOWESS and quantile normalization methods. The log2-fold change values were calculated for each miRNA by comparing their expression between any two epididymal regions. Meanwhile, the statistical significance of the fold change (P value) was inspected through a Student’s t-test. A volcano plot was used to portray the difference in miRNA expression between the two epididymal regions, in which the x-axis indicated the log2-fold change value and the y-axis indicated the negative log10 P value. The raw datasets of microarray results have been submitted to the NCBI GEO database (Accession number GSE106501. miRNA microarray results are available in Table S1 in Supplementary Material).
Total RNA from the heart tissue or cardiomyocytes was extracted by a TRIzol reagent (Invitrogen), and then 2 µg RNA was reverse transcribed to generate cDNA by using superscript reverse Transcriptase (Invitrogen) with a special stem-loop primer for miR-22 and oligodeoxythymidine for mRNAs. Quantitative real-time PCR was performed using a MiniOpticon real-time PCR detection system (Bio-Rad Laboratories). The primer sequences for nuclear factor kappa B (NF-κB) repressing factor (NKRF) were designed based on the cDNA sequences in GeneBank. The following primers were used: sense 5′- GTTCTGCCAAACACTGGACC-3′ and anti-sense 5′-CTGAGATAGGCTCCCGTATGCCC-3′. The reaction solution comprised 2.0 µl diluted cDNA, 0.2 µM/L of each paired primer, 200 µM/L deoxynucleotide triphosphates, 1 U Taq DNA polymerase (Qiagen, Beijing, China), and 1× PCR buffer. SYBRGreen (Roche Ltd, Basel, Switzerland) was used as the detection dye. The annealing temperature was set at 58–62°C and amplification was set at 40 cycles. The temperature range to detect the melting temperature of the PCR product was set from 60 to 95°C. To determine the relative quantification of gene expression, the comparative Ct (threshold cycle) method with arithmetic formulae (2−ΔΔCt) was used (20). The mRNA levels were normalized relative to the house-keeping gene β-actin.
Female C57BL/6 mice at 6–8 weeks of age (18–22 g) were obtained from Shanghai SLAC Laboratory Animal Co. (Shanghai, China) and housed at controlled room temperature with free access to food and water under a 12-h light/dark cycle. All animal protocols were approved by the Ethical Committee of Experimental Animals of Second Military Medical University. Bilateral OVX or a sham operation (Sham) was performed under anesthesia with sodium pentobarbital (60 mg/kg, ip). After 2 weeks, OVX mice were divided into four subgroups, and they were treated subcutaneously with the solvent sesame oil (30 μl/day, Sigma-Aldrich, St. Louis, MO, USA), E2 (30 μg/kg/day, Sigma-Aldrich), estrogen receptor ERα selective agonist PPT (30 μg/kg/day, Tocris bioscience, Bristol, UK) and ERβ selective agonist DPN (30 μg/kg/day, Tocris bioscience) for four weeks, as previously described (10, 11). All of the mice were then sacrificed for tissue collection. To avoid the influence of the estrus cycle, sham mice were sacrificed at the diestrus phase. Plasma E2 was determined with a commercial RIA kit (Sino-UK Institute of Biological Technology, Shanghai, China).
Cell viability was evaluated by an MTT assay based on the reduction of MTT (Sigma-Aldrich) by functional mitochondria to formazan, as described previously (21).
Lactate dehydrogenase release into culture supernatants of the H9c2 myocardial cells was detected by colorimetric enzyme-linked immunosorbent assay, using the cytotoxicity detection kit (LDH) from Roche (Roche Diagnostics) as previously described (22).
The siRNAs for ERα were designed and synthesized by GenePharma Corporation (Shanghai, China). The target sequences for ERα was: 5′-CAGGTCCAATTCTGACAAT-3′. The negative control siRNA sequence was scrambled without any specific target: 5′-TTCTCCGAACGTGTCACGT-3′. The transfection of siRNA was performed by using a siPORT NeoFx transfection agent (Ambion, Austin, TX, USA) according to the manufacturer’s instructions.
The wild-type 3′UTR and the miR-494 “seed” mutant 3′UTR of NKRF were synthesized in vitro and cloned into the psi-CHECK2 luciferase reporter plasmid (Promega). The H9c2 cells were co-transfected with psiCHECK-2 plasmid containing wild-type or mutant derivatives, along with the miRNA control or miR-494 mimic. Lysates were collected 24 h after transfection and the luciferase activity was measured by a dual-luciferase reporter system (Promega).
The proteins of the H9c2 cardiomyocytes were lysed with cold RIPA lysis buffer (Beyotime). The protein load was 30 µg/lane in 10% SDS-PAGE, and it was subsequently transferred to nitrocellulose membranes. The bolts were blocked with 5% skim milk powder in 0.1%tris-buffered saline/Tween20 for 2 h and incubated with antibodies (Santa Cruz, Cat. No. sc-365568) against NKRF overnight at 4°C at a dilution of 1:1,000. Then, the membrane was incubated with a secondary horseradish peroxidase-conjugated antibody for 1 h at room temperature. The immunoreactive proteins were visualized using the enhanced chemiluminescence western blotting detection system (Santa Cruz). The chemiluminescent signal from the membranes was quantified by a GeneGnome HR scanner using GeneTools software (SynGene). To control sampling errors, the ratio of band intensities to β-actin was obtained to quantify the relative protein expression level.
All data were expressed as the mean ± SEM. For illustrative purposes, some results are presented as the mean percent control ± SEM. When comparing multiple groups, one-way ANOVA was performed. When ANOVA showed significant statistical differences (P < 0.05), comparisons between each groups were conducted using the Student–Newman–Keuls test. SPSS 13.0 statistical software was used for the data analysis. P < 0.05 was considered statistically significant.
After comparing the miRNA expression profile between the control and E2-treated primary cultured rat cardiomyocytes using an array-based miRNA profiling (Figure 1A), six differentially expressed miRNAs were identified by qRT-PCR. Three were downregulated (miR-10b, miR-188, and miR-200c) and three were upregulated (miR-494, miR-297, and miR-181c) (Figures 1B,C). Because miR-494 is known to exert cardioprotective effects (18), we then further investigated the effect of E2 on miR-494 expression.
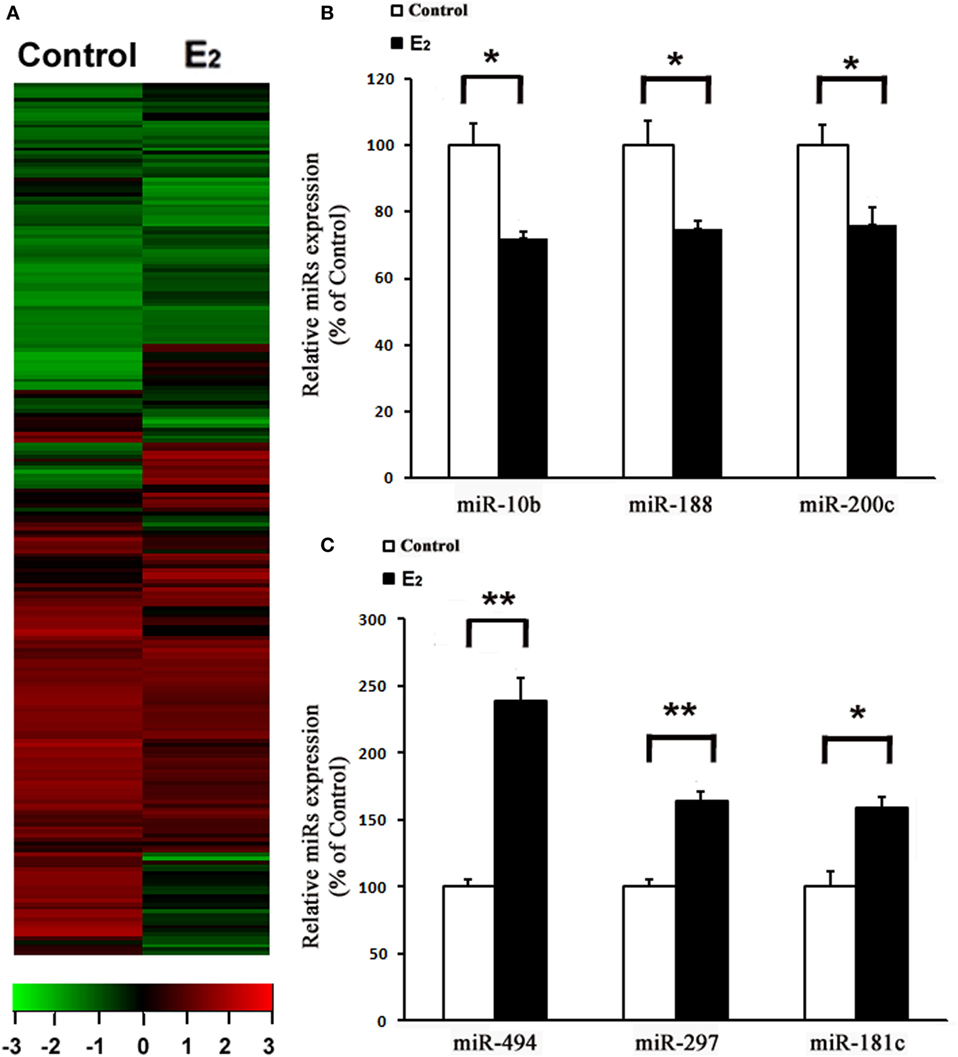
Figure 1. E2 regulates microRNAs (miRs) expression in cardiomyocytes. (A), Heatmap image of microarray data, illustrating differentially expressed miRs between the control and E2-treated cardiomyocytes (n = 3). The scale is −3 to +3 in log10. Red indicates increased expression, and green indicates decreased expression. (B,C) Expressions of downregulated (B) and upregulated (C) miRs were determined by qRT-PCR by performing three independent experiments. All bar graphs represent the means ± SEM. *P < 0.05, **P < 0.01.
As shown in Figure 2A, the treatment of H9c2 with increasing concentrations of E2 (0.1–10 nM) caused significant increases in the miR-494 levels in a dose-dependent manner over a 24-h incubation period. The effect of E2 on miR-494 expression was completely eliminated by the ERα-selective antagonist MPP, but not by the ERβ-selective antagonist THC (Figure 2B). In addition, E2-induced miR-494 expression in cardiomyocytes was eliminated by the ERα siRNA treatment (Figure 2C). Transfection of ERα siRNA resulted in an approximately 80% decrease in ERα expression in the H9c2 cells (Figure 2D).
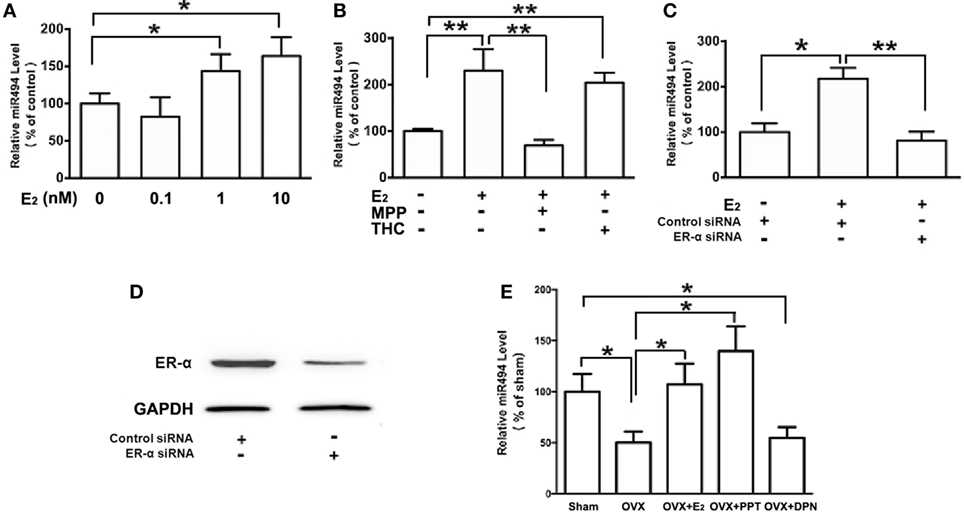
Figure 2. ERα mediates the regulation of miR-494 expression by E2 in cardiomyocytes and myocardium. (A) H9c2 cells were stimulated with E2 at indicated doses for 24 h. Quantitative real-time RT-PCR was used to determine miR-494 expression in cardiomyocytes by performing four independent experiments. (B) Cells were treated with E2 (10 nmol/L) in the absence or presence of ERα-specific antagonist MPP (1 µmol/L), or ERβ-specific antagonist THC (1 µmol/L) for 24 h. Quantitative real-time RT-PCR was used to determine miR-494 expression in cardiomyocytes by performing four independent experiments. (C) Cells were transfected with Control siRNA or ERα siRNA for 24 h and then treated with E2 (10 nmol/L) for another 24 h. Quantitative real-time RT-PCR was used to determine miR-494 expression in cardiomyocytes by performing four independent experiments. (D) Cells were transfected with Control siRNA or ERα siRNA for 24 h. Western blot analysis were used to determine ERα protein expression in cardiomyocytes by performing four independent experiments. (E) Bilateral ovariectomy was performed on female mice to deplete endogenous estrogens. Exogenous E2, ERα selective agonists (PPT) and ERβ selective agonists (DPN) were subcutaneously administered for four weeks. Quantitative real-time RT-PCR was used to determine miR-494 expression in the myocardium (n = 8 in each group). All bar graphs represent the mean ± SEM. *P < 0.05, **P < 0.01.
We then examined the roles of ERα and ERβ in the estrogen upregulation of miR-494 expression in vivo. To verify that the ovariectomy was successful, the weight of the uterus and level of E2 in the serum were determined. The level of serum E2 was found to be significantly lower in OVX mice than that in sham mice (data not shown). The weight of the uterus was also significantly decreased in the OVX group compared with the sham group (data not shown). As shown in Figure 2E, the ovariectomy resulted in decreased miR-494 expression in the myocardium of female mice. The E2 and ERα-specific agonist PPT treatment for 4 weeks significantly increased miR-494 expression in the myocardium whereas the ERβ-specific agonist DPN treatment did not affect miR-494 expression.
As shown in Figure 3, H2O2 (200 µmol/L) treatment of cardiomyocytes caused cell damage by showing an increase in the release of LDH as well as a decrease in cell viability. E2 at 10 nmol/L protected cardiomyocytes against the insult of H2O2 as evidenced by the increased cell survival and reduction of LDH release (Figures 3A,B). Moreover, the cardioprotective effects of E2 were significantly reduced in the presence of the inhibitor of miR-494 (Figures 3A,B). By contrast, the miR-494 mimic protected cardiomyocytes against the insult of H2O2 as evidenced by the increased cell survival and reduction of LDH release (Figures 3C,D).
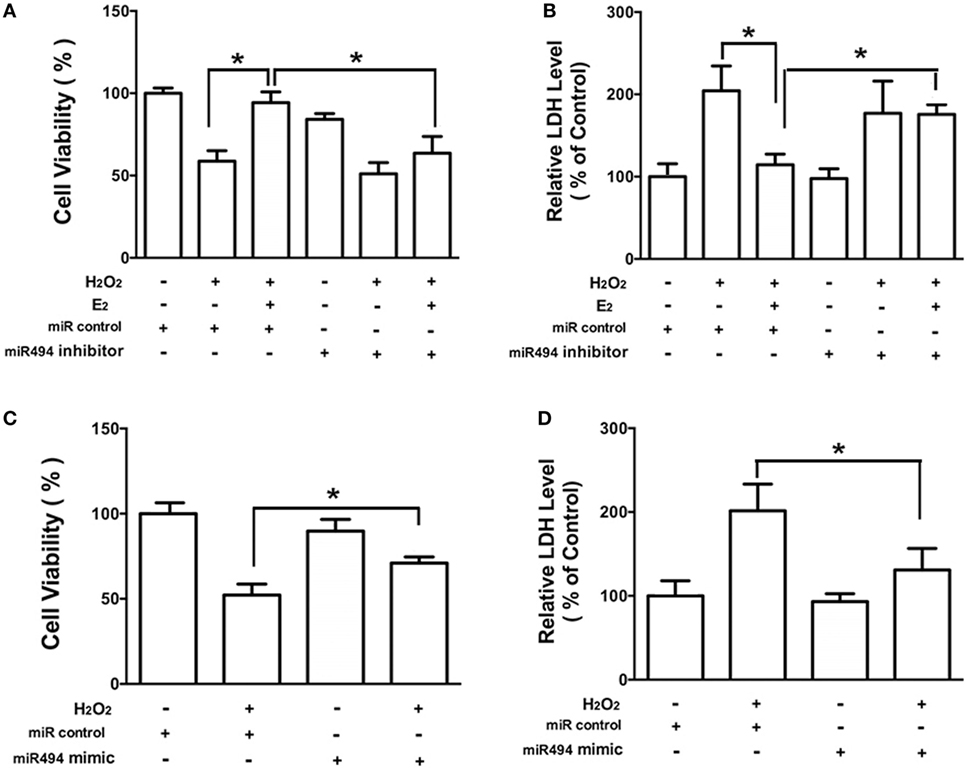
Figure 3. miR-494 contributes to the protective effects of E2 against oxidative stress in cardiomyocytes. (A,B) H9c2 cells were transfected with control microRNA (miR) or miR-494 inhibitor for 24 h and then treated with E2 (10 nmol/L) for another 24 h. Cells were then exposed to H2O2 (200 µmol/L) for 4 h. Overall cell viability was assessed by MTT (A) and supernatant lactate dehydrogenase (LDH) concentration (B). (C,D) H9c2 cells were transfected with control miR or miR-494 mimics for 24 h. Cells were then exposed to H2O2 (200 µmol/L) for 4 h. Overall cell viability was assessed by MTT (C) and supernatant LDH concentration (D). All bar graphs represent the means ± SEM from four independent experiments. *P < 0.05.
MicroRNAs are known to negatively regulate the stability and translation of target protein-coding mRNAs at the 3′UTR (14). To identify potential targets for miR-494, we used a consensus approach with three widely used types of software (miRanda, TargetScan, and PicTar) to perform the target prediction. After overlapping prediction results, NKRF, a suppression factor for NF-κB, was selected to be a putative miR-494 target gene. The mRNA of NKRF contains a putative binding site for miR-494 in its 3′-UTR, and each site is broadly conservative among mammals (Figure 4A). As shown in Figure 4B, transfection with miR-494 significantly decreased both the mRNA and protein expression of NKRF in cardiomyocytes. To address whether NKRF was directly regulated by miR-494, we transfected NKRF 3′UTR luciferase reporter constructs together with a miR-494 mimic into cardiomyocytes and observed a significant reduction in luciferase activity in the co-transfection with miR-494 compared with that in the cells co-transfected with the nontargeting miR control (Figure 4C). By contrast, no reduction in luciferase activity was detected upon co-transfection of miR-494 when the putative miR-494 binding sequence in the NKRF 3′UTR luciferase reporter construct was mutated (Figure 4C). These findings suggest that NKRF expression is suppressed in cardiomyocytes by miR-494 binding to response elements in its 3′UTR.
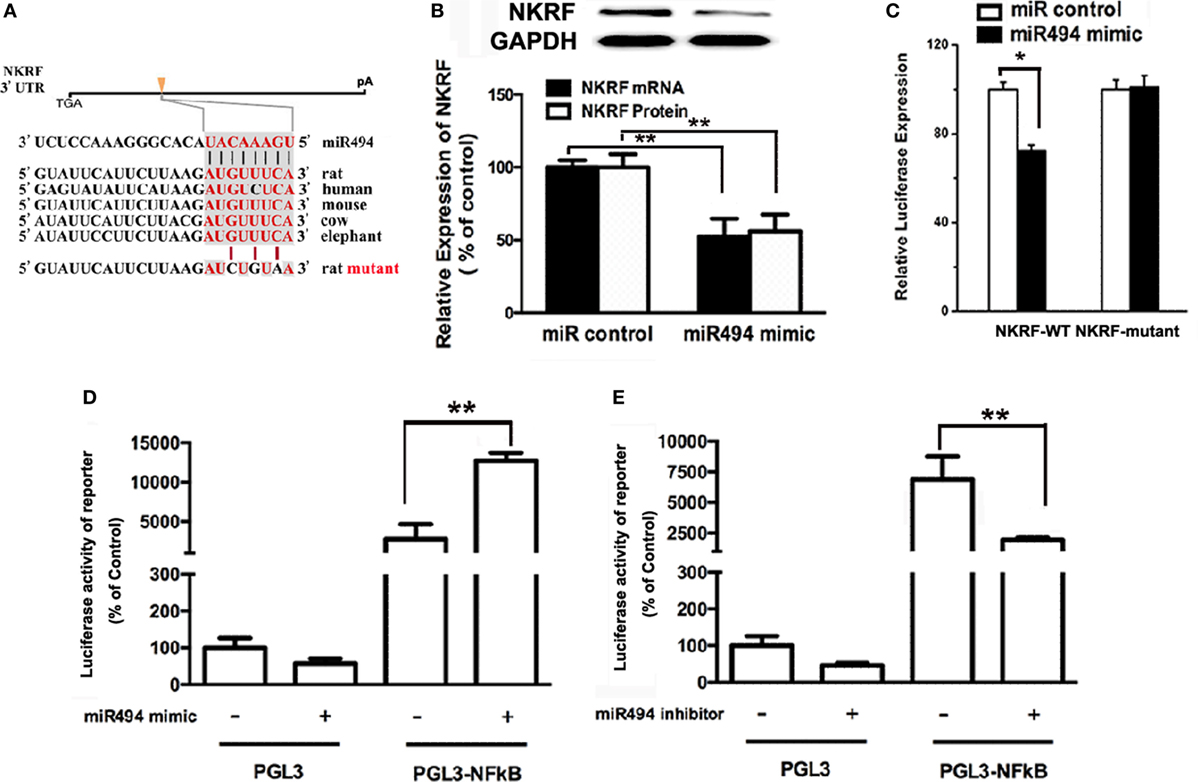
Figure 4. miR-494 targets NF-κB repressing factor (NKRF), thus activating NF-κB. (A) NKRF contained highly conserved miR-494-mRNA interaction motifs within their 3′ untranslated regions (UTRs), and the sequences of the wild-type (WT) and mutant 3′UTR of NKRF for the luciferase reporter assay are shown. (B) H9c2 were transfected with microRNA (miR) control or miR-494 mimics (200 nM) for 24 h. Quantitative real-time RT-PCR and western blot analysis were used to determine the NKRF mRNA and protein expression in cardiomyocytes, respectively. (C) H9c2 co-transfected with 0.4 µg of WT or mutant NKRF 3′UTR reporter plasmids in the presence of miR control or miR-494 mimics (200 nM). 24 h later, luciferase activity was measured using dual-luciferase assay. (D) H9c2 co-transfected with 0.4 µg of pGL3 or pGL3-NFκB reporter plasmids in the presence of miR control or miR-494 mimic (200 nM). 24 h later, luciferase activity was measured using dual-luciferase assay. (E) H9c2 co-transfected with 0.4ug pGL3 or pGL3-NFκB reporter plasmids in the presence of miR control or miR-494 inhibitor (200 nM). 24 h later, luciferase activity was measured using dual-luciferase assay. All bar graphs represent the mean ± SEM from four independent experiments. *P < 0.05, **P < 0.01.
It has been well-recognized that NKRF negatively regulates the activity of NF-κB through a direct protein–protein interaction (23). To determine the effect of miR-494 on NF-κB activity, H9c2 cells were transfected with an NF-κB report plasmid. As shown in Figures 4D,E, we found that the miR-494 mimic significantly stimulated, while the miR-494 inhibitor decreased the luciferase activity of the NF-κB report gene. Taken together, these findings suggest that miR-494 targets NKRF, thus activating NF-κB.
As shown in Figure 5A, the treatment of H9c2 with increasing concentration of E2 (0.1–10 nM) caused significant decreases in the NKRF levels in a dose-dependent manner over a 24-h incubation period. The treatment of the cells with E2 significantly increased the luciferase activity of the NF-κB report gene (Figure 5C). Notably, both the inhibitory effect of E2 on NKRF expression and the stimulatory effect of E2 on NF-κB activity were significantly reduced in the presence of the miR-494 inhibitor (Figures 5B,C).
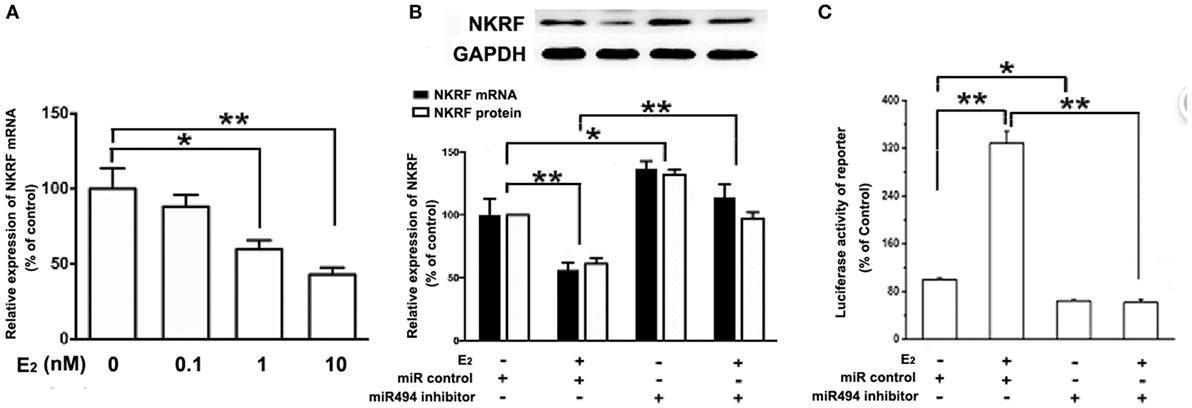
Figure 5. E2 inhibits (NF-κB) repressing factor (NKRF), thus activating NF-κB through a miR-494-dependent mechanism. (A) H9c2 cells were stimulated with E2 at indicated doses for 24 h. Quantitative real-time RT-PCR was used to determine NKRF mRNA expression in cardiomyocytes. (B) H9c2 cells were transfected with control microRNA (miR) or miR-494 inhibitor for 24 h and then treated with E2 (10 nmol/L) for another 24 h. Quantitative real-time RT-PCR and western blot analysis were used to determine NKRF mRNA and protein expression in cardiomyocytes, respectively. (C) H9c2 co-transfected with 0.4 µg of pGL3 or pGL3-NFκB reporter plasmids in the presence of miR control or miR-494 inhibitor (200 nM) for 24 h, and then treated with E2 (10 nmol/L) for another 24 h. Luciferase activity was measured using dual-luciferase assay. All bar graphs represent mean ± SEM from four independent experiments. *P < 0.05, **P < 0.01.
As shown in Figure 6, the H2O2 (200 µmol/L) treatment of cardiomyocytes caused cell damage by showing an increase in the release of LDH and a decrease in cell viability. Both E2 (Figures 6A,B) and the miR-494 mimic (Figures 6C,D) protected cardiomyocytes against the insult of H2O2 as evidenced by increased cell survival and the reduction in LDH release. These cardioprotective effects of E2 and the miR-494 mimic were significantly reduced in the presence of PDTC (1 µmol/L), an NF-κB inhibitor.
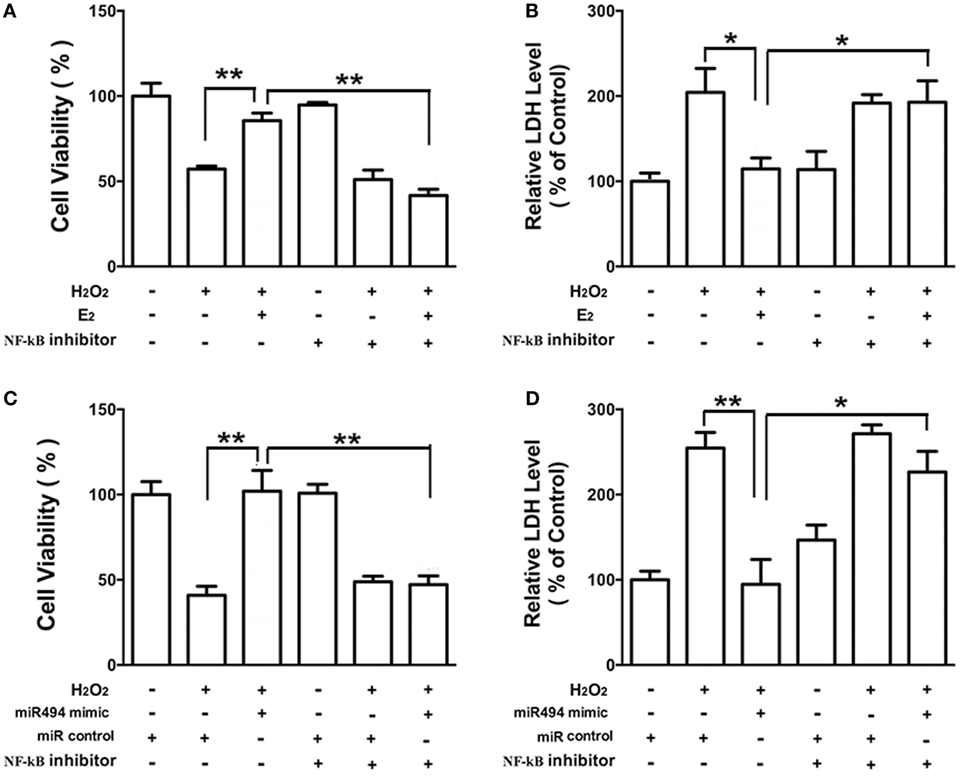
Figure 6. The protective effects of E2 and miR-494 against oxidative stress in cardiomyocytes are eliminated by NF-κB inhibitor. (A,B) H9c2 cells were incubated with E2 (10 nmol/L) in the absence or presence of NF-κB inhibitor (PDTC, 1 µmol/L) for 24 h. Cells were then exposed to H2O2 (200 µmol/L) for 4 h. Overall cell viability was assessed by MTT (A) and supernatant lactate dehydrogenase (LDH) concentration (B). (C,D) H9c2 cells were transfected with control microRNA (miR) or mimics of miR-494 in the absence or presence of NF-κB inhibitor (PDTC, 1 µmol/L) for 24 h. Cells were then exposed to H2O2 (200 µmol/L) for 4 h. Overall cell viability was assessed by MTT (C) and supernatant LDH concentration (D). All bar graphs represent the mean ± SEM from four independent experiments. *P < 0.05, **P < 0.01.
In this study, we tested the hypothesis that estrogenic regulation of miRNA expression is involved in estrogen-mediated cardioprotection against oxidative stress. miRNAs are powerful endogenous inhibitors of target genes at the posttranscriptional levels. Using array-based miRNA profiling, we found that six miRs were significantly regulated by estrogen treatment, with three upregulated and three downregulated in cardiomyocytes. Of these six miRs, miR-494 had the highest expression and the most significant difference. The cardioprotective effect of miR-494 against I/R-induced injury has been reported. However, the potential role of miR-494 in estrogen-mediated cardioprotection has not been explored.
Previous studies have described a link between miR-494 and cellular injuries in a cell-specific manner. For example, miR-494 contributes to inflammatory or adhesion molecule-induced kidney injury after I/R by inhibiting the expression of activating transcription factor 3 (24). miR-494 inhibition protects nucleus pulposus cells from TNF-α-induced apoptosis by targeting JunD (10, 11). By contrast, Sun et al. reported that miR-494 upregulates HIF-1α expression through activating the PI3K/Akt pathway, thus protecting against hypoxia-induced apoptosis in human liver cell line L02 cells (25). MicroRNA-494 has also been found to improve functional recovery and inhibits apoptosis in rats after spinal cord injury (26). In the myocardium, Wang et al. demonstrated that miR-494 has cardioprotective effects against ischemia/reperfusion-induced cardiac injury via targeting apoptotic factors (18). Consistent with the findings of Wang et al., this study indicated that miR-494 per se exerted protective effects against oxidative stress-induced injuries in cardiomyocytes. In addition, the protective effects of estrogen against oxidative stress were blocked by the miR-494 inhibitor, suggesting that miR-494 contributed to estrogen-mediated cardioprotection.
The estrogenic modulation of miRNA expression has been widely described, especially in the pathogenesis of tumor development and metastasis (14). As for miR-494, previous studies show that estrogen treatment results in the upregulation of miR-494 expression in the human mammary adenocarcinoma cell MCF-7 (27) and the human hepatocarcinoma cell line HuH-7 (28), thus indicating a possible role of miR-494 in the estrogen-mediated downregulation of tissue factor pathway inhibitor α and protein S, respectively. In the present study, a dose-dependent induction of miR-494 expression levels was detected in cardiomyocytes treated with estrogen. In addition, the inhibitory effect of estrogen on NKRF, a target gene of miR-494, was blocked by the miR-494 inhibitor. These findings suggest a possible role for miR-494 in the estrogenic regulation of NKRF at the posttranscriptional level.
Estrogens exert their biological effects via numerous mechanisms, one of which is through the two classic nuclear estrogen receptors, ERα and ERβ. Although both are involved in estrogen cardioprotection, the relative contribution and prevalence of each receptor may be dependent on the type of cardiac injury. ERα has been found to exert the cardioprotective effect of estrogens against acute I/R injury through upregulating some endogenous cardioprotective molecules such as stromal cell-derived factor-1 (29). ERα even supports the survival of cardiomyocytes indirectly through post-infarct cardiac c-kit+ cells accumulating in the peri-infarct myocardium (30). Our previous study also demonstrated that ERα mediates estrogen upregulation of CSE in cardiomyocytes, which contributes to the protective effects of estrogen against oxidative stress (10, 11). For the role of ERβ, a number of studies have demonstrated that ERβ exhibits myocardial protection following ischemia (31–33). Recently, the role for ERβ in reducing hypertrophy in females was also reported (34). However, the present study showed that ERα but not ERβ mediated E2 maintenance of miR-494 expression in cardiomyocytes and the myocardium, again corroborating the distinct functions of ERα and ERβ in the heart.
In silico analysis of the NKRF 3′-UTR sequence identified one putative miR-494 binding site. In our study, we performed in vitro validation of miR-494 to negatively regulate NKRF expression in cardiomyocytes. The direct interaction of miR-494 with NKRF mRNA was assessed by NKRF 3′-UTR-dependent luciferase activity and co-transfection with a miR-494 mimic that led to significantly suppressed luciferase activity. Mutation of the predicted miR-494 binding site abrogated the effect of miR-494 on the NKRF 3′-UTR luciferase activity. Overexpression of the miR-494 mimic also led to significantly reduced NKRF mRNA and protein levels that was consist with the luciferase data. NKRF is well-recognized as a suppression factor for NF-κB (23). We found that the miR-494 mimic significantly stimulated, while the miR-494 inhibitor decreased the luciferase activity of the NF-κB report gene, suggesting that miR-494 targets NKRF, thus activating NF-κB. To our knowledge, this is the first report indicating that miR-494 directly regulates NKRF expression in cardiomyocytes and supports the potential involvement of miR-494 in the regulation of the NKRF/NFκB signaling pathway.
The transcription factor NF-κB has been implicated in the regulation of immune cell maturation, cell survival, and inflammation in many cell types, including cardiac myocytes. Prolonged activation of NF-κB appears to be detrimental, promoting heart failure by eliciting signals that trigger chronic inflammation through enhanced elaboration of cytokines, leading to endoplasmic reticulum stress responses and cell death (35). However, accumulating studies have documented a potent role for NF-κB in the regulation of cardiac myocyte survival through the repression of apoptotic cell death triggered by hypoxia, chemotherapeutic agent, or ischemic/reperfusion myocardial injury (36–38).
Oxidative stress is considered one of the main causative factors in various cardiovascular disorders (39). Given that NF-κB targets a number of important cellular antioxidants including manganese superoxide dismutase (MnSOD, or SOD2), Copper-Zinc superoxide dismutase (Cu,Zn-SOD, or SOD1), ferritin heavy chain, metallothionein-3 (MT3), and NAD(P)H dehydrogenase [quinone] 1 (NQO1), the protective effects of NF-κB against oxidative stress-induced injuries have been widely reported in a variety of cell types including cardiomyocytes (39, 40). For example, the NF-κB signaling pathway attenuates oxidative stress and plays a protective role in cardiomyocytes through the regulation of MnSOD in a setting of acute pressure overload (41). In addition, some agents, such as hesperetin (38), SIRT3 (42), and metformin (43), have shown beneficial effects against oxidative stress-induced cardiotoxicity, which is, at least partly, mediated by the activation of NF-κB. In the myocardium, estrogens have been shown to exert protective effects against oxidative stress. Estrogen is also found to activate NF-κB in adult cardiomyocytes (44). Consistent with these studies, we found that estrogen activated NF-κB through a miR-494-dependent mechanism. The protective effects of both miR-494 and estrogen against oxidative stress in cardiomyocytes were eliminated by the NF-κB inhibitor. Taken together, these results suggest that miR-494 may link estrogenic cardioprotection with NF-κB activation in cardiomyocytes.
Although numerous experimental in vitro and in vivo studies have provided reliable evidence supporting estrogen’s cardiovascular benefits (5, 8, 9, 12, 30), clinical trials report contradictory results regarding cardiovascular effects of menopausal hormone use (45). For example, the Women’s Health Initiative (WHI) clinical trial enrolls participants whose average age at initiation of treatment was 63 years with the age range of 50–79 years (46). Women with a uterus were given oral conjugated equine estrogen (CEE) with continuous combined medroxyprogesterone acetate (MPA). Women who had undergone hysterectomy alone or with either unilateral or bilateral oophorectomy were treated with CEE alone. The CEE + MPA study of the WHI demonstrated increased adverse cardiovascular events including cardiac events, strokes, and pulmonary embolisms. However, in the CEE alone cohort, the combined endpoints of coronary heart disease events and coronary revascularization were lower in women randomized to treatment who were between the ages of 50–59 compared to women in age strata > 60 years (47). In an 11-year cumulative follow-up study of the CEE alone cohort, myocardial infarction was lower in the 50–59 age strata than in women who had been randomized to treatment at older ages (48). Additionally, a large Danish open-label study (Danish Osteoporosis Prevention Study) enrolled women between the ages of 45–58 years (49). The treatments were either triphasic estradiol and norethisterone acetate or estradiol a day if they had a hysterectomy. At 11 years follow-up, there were significantly fewer myocardial infarctions and lower incidence of heart failure in the treated women compared to the control group. These clinical trials suggest that the timing of initiation of hormone therapy in relation to menopause onset or age might influence cardiovascular risk. The limitation of the present study is that it seems difficult to extrapolate the data obtained from experimental studies to humans to indicate estrogen’s cardiovascular benefits.
In summary, this study demonstrates for the first time that E2 stimulates miR-494 expression via ERα in both cardiomyocytes and the myocardium of female mice. ERα-mediated upregulation of miR-494 may contribute to estrogen protection of cardiomyocytes against oxidative stress. Furthermore, we identify NKRF as a miR-494 target in cardiomyocytes. Estrogen inhibits NKRF expression through ERα-mediated upregulation of miR-494 in cardiomyocytes, leading to the activation of NF-κB, which in turn results in an increase in antioxidative defense.
All animal protocols were approved by the Ethical Committee of Experimental Animals of Second Military Medical University.
X-YZ and XN drafted the manuscript; Z-PT, WZ, and J-kD prepared figures; X-YZ and J-QL edited and revised the manuscript critically for important intellectual content; Z-PT, WZ, J-kD, XN, X-YZ, and J-QL approved the final version of the manuscript submitted.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by Natural Science Foundation of China (No. 31571227, No. 31671213, and No. 31371197), and Key Laboratory of Exercise and Health Sciences of Ministry of Education of Shanghai University of Sport.
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fendo.2018.00215/full#supplementary-material.
1. Van Dyke M, Greer S, Odom E, Schieb L, Vaughan A, Kramer M, et al. Heart disease death rates among blacks and whites aged ≥35 years – United States, 1968–2015. MMWR Surveill Summ (2018) 67:1–11. doi:10.15585/mmwr.ss6705a1
2. Yang PC, Clancy CE. Gender-based differences in cardiac diseases. J Biomed Res (2011) 25:81–9. doi:10.1016/S1674-8301(11)60010-9
3. Mendelsohn M, Karas R. Molecular and cellular basis of cardiovascular gender differences. Science (2005) 308:1583–7. doi:10.1126/science.1112062
4. Rossouw JE, Prentice RL, Manson JE, Wu L, Barad D, Barnabei VM, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA (2007) 297:1465–77. doi:10.1001/jama.297.13.1465
5. Babiker FA, DeWindt LJ, Van Eickels M, Grohe C, Meyer R, Doevendans PA. Estrogenic hormone action in the heart: regulatory network and function. Cardiovasc Res (2002) 53:709–19. doi:10.1016/S0008-6363(01)00526-0
6. Miller VM, Duckles SP. Vascular actions of estrogens: functional implications. Pharmacol Rev (2008) 60:210–41. doi:10.1124/pr.107.08002
7. Zegura B, Keber I, Sebestjen M, Koenig W. Double blind, randomized study of estradiol replacement therapy on markers of inflammation, coagulation and fibrinolysis. Atherosclerosis (2003) 168:123–9. doi:10.1016/S0021-9150(03)00088-1
8. Arias-Loza PA, Muehlfelder M, Pelzer T. Estrogen and estrogen receptors in cardiovascular oxidative stress. Pflugers Arch (2013) 465:739–46. doi:10.1007/s00424-013-1247-7
9. Kim JK, Pedram A, Razandi M, Levin ER. Estrogen prevents cardiomyocyte apoptosis through inhibition of reactive oxygen species and differential regulation of p38 kinase isoforms. J Biol Chem (2006) 281:6760–7. doi:10.1074/jbc.M511024200
10. Wang L, Tang ZP, Zhao W, Cong BH, Lu JQ, Tang XL, et al. miR-22/Sp-1 links estrogens with the up-regulation of cystathionine γ-lyase in myocardium, which contributes to estrogenic cardioprotection against oxidative stress. Endocrinology (2015) 156:2124–37. doi:10.1210/en.2014-1362
11. Wang T, Li P, Ma X, Tian P, Han C, Zang J, et al. MicroRNA-494 inhibition protects nucleus pulposus cells from TNF-α-induced apoptosis by targeting JunD. Biochimie (2015) 115:1–7. doi:10.1016/j.biochi.2015.04.011
12. Gardner JD, Murray DB, Voloshenyuk TG, Brower GL, Bradley JM, Janicki JS. Estrogen attenuates chronic volume overload induced structural and functional remodeling in male rat hearts. Am J Physiol Heart Circ Physiol (2010) 298:H497–504. doi:10.1152/ajpheart.00336.2009
13. Kararigas G, Fliegner D, Gustafsson JÅ, Regitz-Zagrosek V. Role of the estrogen/estrogen-receptor-beta axis in the genomic response to pressure overload-induced hypertrophy. Physiol Genomics (2011) 43:438–46. doi:10.1152/physiolgenomics.00199.2010
14. Klinge CM. miRNAs and estrogen action. Trends Endocrinol Metab (2012) 23:223–33. doi:10.1016/j.tem.2012.03.002
15. Deng J, Zhong Q. Advanced research on the microRNA mechanism in heart failure. Int J Cardiol (2016) 220:61–4. doi:10.1016/j.ijcard.2016.06.185
16. Pinti MV, Hathaway QA, Hollander JM. Role of microRNA in metabolic shift during heart failure. Am J Physiol Heart Circ Physiol (2017) 312:H33–45. doi:10.1152/ajpheart.00341.2016
17. Queirós AM, Eschen C, Fliegner D, Kararigas G, Dworatzek E, Westphal C, et al. Sex- and estrogen-dependent regulation of a miRNA network in the healthy and hypertrophied heart. Int J Cardiol (2013) 169:331–8. doi:10.1016/j.ijcard.2013.09.002
18. Wang X, Zhang X, Ren XP, Chen J, Liu H, Yang J, et al. MicroRNA-494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion-induced cardiac injury. Circulation (2010) 122:1308–18. doi:10.1161/CIRCULATIONAHA.110.964684
19. Chu C, Zheng G, Hu S, Zhang J, Xie S, Ma W, et al. Epididymal region-specific miRNA expression and DNA methylation and their roles in controlling gene expression in rats. PLoS One (2015) 10:e0124450. doi:10.1371/journal.pone.0124450
20. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods (2001) 25:402–8. doi:10.1006/meth.2001.1262
21. Zhu X, Liu S, Liu Y, Wang S, Ni X. Glucocorticoids suppress cystathionine gamma-lyase expression and H2S production in lipopolysaccharide-treated macrophages. Cell Mol Life Sci (2010) 67:1119–32. doi:10.1007/s00018-009-0250-9
22. Tjoa ML, Cindrova-Davies T, Spasic-Boskovic O, Bianchi DW, Burton GJ. Trophoblastic oxidative stress and the release of cell-free feto-placental DNA. Am J Pathol (2006) 169:400–4. doi:10.2353/ajpath.2006.060161
23. Niedick I, Froese N, Oumard A, Mueller PP, Nourbakhsh M, Hauser H, et al. Nucleolar localization and mobility analysis of the NF-kappaB repressing factor NRF. J Cell Sci (2004) 117:3447–58. doi:10.1242/jcs.01129
24. Lan YF, Chen HH, Lai PF, Cheng CF, Huang YT, Lee YC, et al. MicroRNA-494 reduces ATF3 expression and promotes AKI. J Am Soc Nephrol (2012) 23:2012–23. doi:10.1681/ASN.2012050438
25. Sun G, Zhou Y, Li H, Guo Y, Shan J, Xia M, et al. Over-expression of microRNA-494 up-regulates hypoxia-inducible factor-1 alpha expression via PI3K/Akt pathway and protects against hypoxia-induced apoptosis. J Biomed Sci (2013) 20:100. doi:10.1186/1423-0127-20-100
26. Zhu H, Xie R, Liu X, Shou J, Gu W, Gu S, et al. MicroRNA-494 improves functional recovery and inhibits apoptosis by modulating PTEN/AKT/mTOR pathway in rats after spinal cord injury. Biomed Pharmacother (2017) 92:879–87. doi:10.1016/j.biopha.2017.05.143
27. Ali HO, Arroyo AB, González-Conejero R, Stavik B, Iversen N, Sandset PM, et al. The role of microRNA-27a/b and microRNA-494 in estrogen-mediated downregulation of tissue factor pathway inhibitor α. J Thromb Haemost (2016) 14:1226–37. doi:10.1111/jth.13321
28. Tay JW, Romeo G, Hughes QW, Baker RI. Micro-ribonucleic acid 494 regulation of protein S expression. J Thromb Haemost (2013) 11:1547–55. doi:10.1111/jth.12331
29. Huang C, Gu H, Wang Y, Wang M. Estrogen-induced SDF-1 production is mediated by estrogen receptor-α in female hearts after acute ischemia and reperfusion. Surgery (2011) 150:197–203. doi:10.1016/j.surg.2011.05.010
30. Brinckmann M, Kaschina E, Altarche-Xifró W, Curato C, Timm M, Grzesiak A, et al. Estrogen receptor α supports cardiomyocytes indirectly through post-infarct cardiac c-kit cells. J Mol Cell Cardiol (2009) 47:66–75. doi:10.1016/j.yjmcc.2009.03.014
31. Wang M, Crisostomo PR, Markel T, Wang Y, Lillemoe KD, Meldrum DR. Estrogen receptor β mediates acute myocardial protection following ischemia. Surgery (2008) 144:233–8. doi:10.1016/j.surg.2008.03.009
32. Wang M, Wang Y, Weil B, Abarbanell A, Herrmann J, Tan J, et al. Estrogen receptor β mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am J Physiol Regul Integr Comp Physiol (2009) 296:R972–8. doi:10.1152/ajpregu.00045.2009
33. Yu HP, Shimizu T, Choudhry MA, Hsieh YC, Suzuki T, Bland KI, et al. Mechanism of cardioprotection following trauma-hemorrhagic shock by a selective estrogen receptor-β agonist: up-regulation of cardiac heat shock factor-1 and heat shock proteins. J Mol Cell Cardiol (2006) 40:185–94. doi:10.1016/j.yjmcc.2005.10.001
34. Gürgen D, Hegner B, Kusch A, Catar R, Chaykovska L, Hoff U, et al. Estrogen receptor-β signals left ventricular hypertrophy sex differences in normotensive deoxycorticosterone acetate-salt mice. Hypertension (2011) 57:648–54. doi:10.1161/HYPERTENSIONAHA.110.166157
35. Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-κB in the heart: to be or not to NF-κB. Circ Res (2011) 108:1122–32. doi:10.1161/CIRCRESAHA.110.226928
36. Maekawa N, Wada H, Kanda T, Niwa T, Yamada Y, Saito K, et al. Improved myocardial ischemia/reperfusion injury in mice lacking tumor necrosis factor-alpha. J Am Coll Cardiol (2002) 39:1229–35. doi:10.1016/S0735-1097(02)01738-2
37. Mustapha S, Kirshner A, De Moissac D, Kirshenbaum LA. A direct requirement of nuclear factor-kappa B for suppression of apoptosis in ventricular myocytes. Am J Physiol Heart Circ Physiol (2000) 279:H939–45. doi:10.1152/ajpheart.2000.279.3.H939
38. Trivedi PP, Kushwaha S, Tripathi DN, Jena GB. Cardioprotective effects of hesperetin against doxorubicin-induced oxidative stress and DNA damage in rat. Cardiovasc Toxicol (2011) 11:215–25. doi:10.1007/s12012-011-9114-2
39. Fearon IM, Faux SP. Oxidative stress and cardiovascular disease: novel tools give (free) radical insight. J Mol Cell Cardiol (2009) 47:372–81. doi:10.1016/j.yjmcc.2009.05.013
40. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res (2011) 21:103–15. doi:10.1038/cr.2010.178
41. Rui T, Kvietys PR. NFkappaB and AP-1 differentially contribute to the induction of Mn-SOD and eNOS during the development of oxidant tolerance. FASEB J (2005) 19:1908–10. doi:10.1096/fj.05-4028fje
42. Chen CJ, Fu YC, Yu W, Wang W. SIRT3 protects cardiomyocytes from oxidative stress-mediated cell death by activating NF-κB. Biochem Biophys Res Commun (2013) 430:798–803. doi:10.1016/j.bbrc.2012.11.066
43. Asensio-López MC, Sánchez-Más J, Pascual-Figal DA, Abenza S, Pérez-Martínez MT, Valdés M, et al. Involvement of ferritin heavy chain in the preventive effect of metformin against doxorubicin-induced cardiotoxicity. Free Radic Biol Med (2013) 57:188–200. doi:10.1016/j.freeradbiomed.2012.09.009
44. Stice JP, Chen L, Kim SC, Jung JS, Tran AL, Liu TT, et al. 17β-Estradiol, aging, inflammation, and the stress response in the female heart. Endocrinology (2011) 152:1589–98. doi:10.1210/en.2010-0627
45. Miller VM, Harman SM. An update on hormone therapy in postmenopausal women: mini-review for the basic scientist. Am J Physiol Heart Circ Physiol (2017) 313(5):H1013–21. doi:10.1152/ajpheart.00383.2017
46. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA (2002) 288:321–33. doi:10.1001/jama.288.3.321
47. Hsia J, Langer RD, Manson JE, Kuller L, Johnson KC, Hendrix SL, et al. Conjugated equine estrogens and coronary heart disease: the Women’s Health Initiative. Arch Intern Med (2006) 166:357–65. doi:10.1001/archinte.166.3.357
48. Manson JE, Allison MA, Rossouw JE, Carr JJ, Langer RD, Hsia J, et al. Estrogen therapy and coronary-artery calcification. N Engl J Med (2007) 356:2591–602. doi:10.1056/NEJMoa071513
Keywords: miR-494, estrogen, cardiomyocytes, oxidative stress, (NF-κB) repressing factor
Citation: Tang Z-P, Zhao W, Du J-k, Ni X, Zhu X-Y and Lu J-Q (2018) miR-494 Contributes to Estrogen Protection of Cardiomyocytes Against Oxidative Stress via Targeting (NF-κB) Repressing Factor. Front. Endocrinol. 9:215. doi: 10.3389/fendo.2018.00215
Received: 02 November 2017; Accepted: 16 April 2018;
Published: 14 May 2018
Edited by:
Yan-ling Wang, Institute of Zoology (CAS), ChinaReviewed by:
Shuangbo Kong, Xiamen University, ChinaCopyright: © 2018 Tang, Zhao, Du, Ni, Zhu and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Yan Zhu, eGlhb3lhbnpodUBzbW11LmVkdS5jbg==;
Jian-Qiang Lu, bHVqaWFucWlhbmdAc3VzLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.