
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 04 May 2018
Sec. Molecular and Structural Endocrinology
Volume 9 - 2018 | https://doi.org/10.3389/fendo.2018.00207
This article is part of the Research Topic Endocrine and Paracrine Role of FGF23 and Klotho in Health and Disease View all 13 articles
 Florian Lang1*
Florian Lang1* Christina Leibrock1,2Lisann Pelzl1Meinrad Gawaz3Burkert Pieske4,5,6Ioana Alesutan4,5,6
Christina Leibrock1,2Lisann Pelzl1Meinrad Gawaz3Burkert Pieske4,5,6Ioana Alesutan4,5,6 Jakob Voelkl4,6
Jakob Voelkl4,6
Medial vascular calcification, a major pathophysiological process associated with cardiovascular disease and mortality, involves osteo-/chondrogenic transdifferentiation of vascular smooth muscle cells (VSMCs). In chronic kidney disease (CKD), osteo-/chondrogenic transdifferentiation of VSMCs and, thus, vascular calcification is mainly driven by hyperphosphatemia, resulting from impaired elimination of phosphate by the diseased kidneys. Hyperphosphatemia with subsequent vascular calcification is a hallmark of klotho-hypomorphic mice, which are characterized by rapid development of multiple age-related disorders and early death. In those animals, hyperphosphatemia results from unrestrained formation of 1,25(OH)2D3 with subsequent retention of calcium and phosphate. Analysis of klotho-hypomorphic mice and mice with vitamin D3 overload uncovered several pathophysiological mechanisms participating in the orchestration of vascular calcification and several therapeutic opportunities to delay or even halt vascular calcification. The present brief review addresses the beneficial effects of bicarbonate, carbonic anhydrase inhibition, magnesium supplementation, mineralocorticoid receptor (MR) blockage, and ammonium salts. The case is made that bicarbonate is mainly effective by decreasing intestinal phosphate absorption, and that carbonic anhydrase inhibition leads to metabolic acidosis, which counteracts calcium-phosphate precipitation and VSMC transdifferentiation. Magnesium supplementation, MR blockage and ammonium salts are mainly effective by interference with osteo-/chondrogenic signaling in VSMCs. It should be pointed out that the, by far, most efficient substances are ammonium salts, which may virtually prevent vascular calcification. Future research will probably uncover further therapeutic options and, most importantly, reveal whether these observations in mice can be translated into treatment of patients suffering from vascular calcification, such as patients with CKD.
Medial vascular calcification is a key pathophysiological process associated with the risk of cardiovascular events in a variety of clinical conditions such as aging, diabetes, and chronic kidney disease (CKD) (1, 2). Accordingly, vascular calcification is a powerful predictor of cardiovascular and all-cause mortality (3–5). Vascular calcification in CKD results mainly from impaired renal phosphate elimination with subsequent hyperphosphatemia and precipitation of calcium-phosphate (6). Accordingly, plasma phosphate concentrations are correlated with the incidence of cardiovascular events, heart failure, and death (7, 8).
Vascular calcification results, at least in part, from an active process in vascular smooth muscle cells (VSMCs) (6). Exposure of VSMCs to enhanced extracellular phosphate concentrations is followed by osteo-/chondrogenic transdifferentiation via complex intracellular signaling pathways (9). Phosphate complexes with calcium to form pro-inflammatory calcium-phosphate nanoparticles (10, 11). Calcium-phosphate crystals are further involved in the formation of protein–mineral complexes, the calciprotein particles (CPPs) (12). These can transform into more toxic secondary CPPs containing crystalline calcium-phosphate (13). Osteo-/chondrogenic signaling cascades in VSMCs can be triggered by calcium-phosphate nanoparticles and/or secondary CPPs (14–20).
Osteo-/chondrogenic signaling involves upregulation of the type III sodium-dependent phosphate transporter PIT1 (also known as SLC20A1) (21, 22). The transdifferentiated VSMCs express osteogenic transcription factors, such as MSH homeobox 2 (MSX2) and core-binding factor alpha 1 (CBFA1, also known as runt-related transcription factor 2, RUNX2) as well as chondrogenic transcription factors such as SRY-Box 9 (SOX9) (23–25) to facilitate, via various complex mechanisms, vascular tissue mineralization (1). Vascular calcification can be prevented by inhibition of CBFA1 (26). The transcription factor NFAT5 (nuclear factor of activated T-cells 5) upregulates CBFA1 expression, an effect mediated by the transcription factor SOX9 (27). Osteo-/chondrogenic reprogramming ultimately upregulates the expression and activity of tissue non-specific alkaline phosphatase (ALPL), an enzyme hydrolyzing the calcification inhibitor pyrophosphate (28). Transdifferentiated VSMCs are also able to secrete matrix vesicles to actively promote tissue mineralization (29). Vascular osteo-/chondrogenic transdifferentiation precedes vascular calcification (30) and has been observed in vasculature of CKD patients (31). Accordingly, osteo-/chondrogenic transdifferentiation predisposes vascular tissue in CKD patients to vascular calcification (32). The orchestration of vascular calcification is, however, still incompletely understood (33).
Valuable insight into mechanisms of vascular calcification was gained by analysis of the klotho-hypomorphic mice (34). Klotho is a transmembrane protein with highest expression in kidney, but also found in parathyroid glands and choroid plexus (34). The extracellular domain of klotho is cleaved off and released into blood (35). Soluble klotho confers protection of kidneys (36) and cardiovascular system (37). Klotho counteracts tissue fibrosis (38, 39), progression of CKD (38), cardiomyopathy (38), vascular calcification (38), and tumor growth (39). Klotho is in part effective by interference with TGFβ1 signaling (39).
Klotho is required for the negative regulation of 25-hydroxyvitamin D3 1-α-hydroxylase (1-α-hydroxylase) by FGF23 and thus for inhibition of 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) production (35, 40). Contrary to CKD patients, production of 1,25(OH)2D3 is excessive in klotho-hypomorphic mice, resulting in elevated phosphate levels (35). Therefore, the mice suffer from severe tissue calcification, mimicking the findings in mice with renal failure (41). These mice further display a wide variety of age-related disorders and early death (34, 35). Conversely, overexpression of klotho increases the life span of mice (42). Apparently, klotho may similarly influence the life span of humans (43). Although 1,25(OH)2D3 may exhibit protective effects during calcification (44), its excessive formation in klotho-hypomorphic mice increases intestinal calcium and phosphate uptake and renal phosphate retention, thus driving the phenotype and tissue calcification (35, 45). The life span of klotho-hypomorphic mice is substantially increased by vitamin D3-deficient diet (45). Moreover, klotho stimulates Na+/K+-ATPase activity (46) and lack of klotho leads to extracellular volume depletion with secondary increase of ADH and aldosterone release (40). Dehydration, in turn, downregulates klotho expression (47). Although the origin of hyperphosphatemia differs between CKD and klotho-hypomorphic mice, both lead to comparable sequelae of vascular calcification (Figure 1).
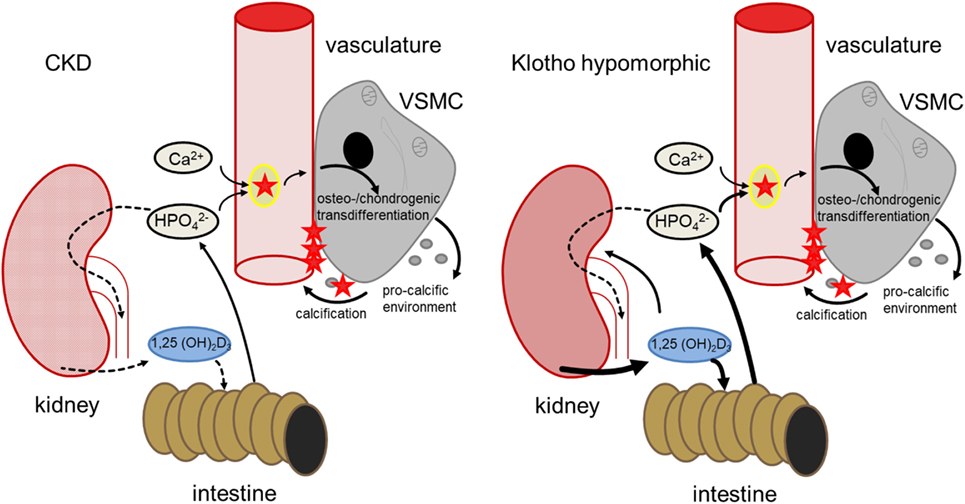
Figure 1. Comparison of hyperphosphatemia due to chronic kidney disease (CKD) (left) and klotho deficiency (right). In CKD, renal failure leads to impaired phosphate elimination and reduced formation of calcitriol [1,25(OH)2D3]. Phosphate overload and formation of secondary calciprotein particles (CPPs) (48) induce an osteogenic remodeling of vascular smooth muscle cells (VSMCs), causing matrix-vesicle release and a pro-calcific environment and subsequent vascular mineralization. In klotho-hypomorphic mice, klotho deficiency causes an unrestrained calcitriol formation. This causes increased intestinal and renal phosphate reabsorption, increasing phosphate levels. Therefore, despite the potential anti-calcific effects of calcitriol (49), this phosphate overload exceeds the capabilities of anti-calcific mechanisms in the body and may induce a comparable osteo-/chondrogenic transdifferentiation of VSMCs. Although the origin of hyperphosphatemia differs between CKD and kl/kl mice, both presumably suffer from excessive phosphate concentrations and increased formation of secondary CPPs to induce a similar VSMC-mediated calcification.
The present brief review addresses attempts to interfere with vascular calcification, premature aging and early death of klotho-hypomorphic mice and similar models. We anticipate that insights from maneuvers successful in klotho-hypomorphic mice may improve our understanding of the mechanisms underlying calcifications in patients with CKD.
Most CKD patients and klotho-hypomorphic mice suffer from acidosis (50, 51), which may further enhance plasma phosphate concentrations (52) and aggravate CKD (53–57). Conversely, alkali administration may slow the progression of CKD (53–56). In contrast to rats, in which metabolic acidosis has been shown to slow the progression of renal disease (58–60), in CKD patients, the deterioration of renal function is accelerated by acidosis and slowed by bicarbonate treatment (56, 61, 62).
Bicarbonate treatment of klotho-hypomorphic mice decreased tissue calcification and increased the average life span of those mice (63). Bicarbonate treatment did not significantly modify plasma concentrations of 1,25(OH)2D3 and calcium, but significantly decreased plasma phosphate concentrations and plasma aldosterone concentrations (63). Bicarbonate treatment was presumably primarily effective by decreasing intestinal phosphate absorption and renal phosphate reabsorption (63). Alkalinization of the intestinal lumen is expected to compromise phosphate solubility and absorption.
Extracellular pH can be modified by treatment with carbonic anhydrase inhibitors, such as acetazolamide (64). The diuretic interferes with proximal tubular bicarbonate reabsorption and, thus, leads to renal bicarbonate loss and acidosis (64). Extracellular pH has a profound effect on calcium and phosphate solubility, which is enhanced by acidification and decreased by alkalinization (65). Moreover, acidosis counteracts vascular calcification by downregulation of PIT1 expression (58, 65, 66) and inhibition of renal tubular phosphate reabsorption with increase of renal phosphate elimination (67).
Acetazolamide treatment of klotho-hypomorphic mice blunted the calcifications in trachea, lung, kidney, stomach, intestine, and vascular tissues, reversed the excessive aortic Alpl transcript levels as a marker of aortic osteo-/chondrogenic signaling, increased the plasma concentrations of the calcification counteracting proteins osteoprotegerin, osteopontin as well as fetuin-A (68–70) and, thus, tripled the life span despite unaltered plasma concentrations of FGF23, 1,25(OH)2D3, calcium and phosphate (64). In vitro, acidic medium prevented the phosphate-induced upregulation of ALPL mRNA expression in primary human aortic smooth muscle cells, indicating that extracellular acidosis interferes with osteo-/chondrogenic transdifferentiation of VSMCs (64). Acidic conditions may impair the formation of small calcium-phosphate complexes during hyperphosphatemia and, thus, hinder VSMC osteo-/chondrogenic transdifferentiation.
It should be kept in mind that the bicarbonaturia and, thus, systemic acidosis following carboanhydrase inhibitor treatment depends on renal function and may, thus, be lacking in CKD patients.
In CKD patients, lower serum magnesium levels are associated with vascular calcification (71) and are predictive for increased arterial stiffness and mortality (72). Previous in vitro studies have shown that magnesium treatment is able inhibit phosphate-induced VSMCs calcification (73–75). Magnesium is able to interfere with hydroxyapatite formation (76). Also, magnesium interferes with osteo-/chondrogenic reprogramming of VSMCs.
Experiments in mice treated with excessive levels of vitamin D3, mimicking excessive vitamin D receptor activation during klotho deficiency, revealed magnesium supplementation as a further potential treatment to reduce the progression of vascular calcification (77). Vitamin D3 overload was followed by extensive vascular calcification and upregulation of aortic osteoinduction as shown by expression of the osteogenic markers Msx2, Cbfa1, and Alpl (77). Those effects were blunted by additional treatment with MgCl2. Vitamin D3 overload upregulated the aortic expression of calcium-sensing receptor (CASR), an effect augmented by additional MgCl2 supplementation (77). Magnesium can activate CASR (78) and CASR activation in VSMCs inhibits osteo-/chondrogenic remodeling and calcification (79).
Those in vivo observations were supported by in vitro experiments using primary human aortic VSMCs. Addition of MgCl2 to the VSMCs cell culture medium reversed the phosphate-induced calcification and osteo-/chondrogenic signaling, effects paralleled by upregulation of CASR expression. The protective effects of MgCl2 were virtually abrogated by the CASR antagonist NPS-2143 or by silencing of the CASR gene (77). Thus, magnesium supplementation may reduce the progression of vascular calcification at least in part by activating CASR. Magnesium supplementation may thus be beneficial in CKD patients (80). Recently, a first pilot trial indicated that magnesium supplementation is safe in CKD patients and is able to reduce serum calcification propensity (81).
Vascular smooth muscle cells express the MR (82) and MR stimulation by aldosterone triggers the osteo-/chondrogenic signaling (82–87) by upregulation of PIT1 expression (87, 88), leading to expression of osteogenic transcription factors and enzymes and subsequent mineralization (87). Klotho-hypomorphic mice develop renal sodium loss and hyperaldosteronism (89). Hyperaldosteronism presumably contributes to the stimulation of vascular calcification in klotho-hypomorphic mice (40, 87, 90) and CKD patients (91). Accordingly, treatment with the MR antagonist spironolactone reduces the extent of vascular calcification in klotho-hypomorphic mice and rats with adenine-induced renal failure (88) and reduces cardio-/cerebrovascular mortality in dialysis patients (92). Spironolactone treatment of klotho-hypomorphic mice reduced aortic PIT1-dependent osteoinductive signaling, but increased cystatin-C levels (87). MR blockade with spironolactone may particularly suppress the progression of vascular calcification in patients with hyperaldosteronism.
Spironolactone may be effective even at normal levels of circulating aldosterone (93, 94). Aldosterone is produced not only in adrenal glands, but in diverse tissues (95–98) including the vasculature (99). Aldosterone synthase (also known as CYP11B2) is expressed during calcifying conditions and, thus, aldosterone may be produced in VSMCs (99, 100). Vascular aldosterone production is particularly important under pathological conditions (97). Vascular aldosterone may foster development of hypertension (101). CYP11B2 is upregulated in atheroma-plaques (102) and contributes to oxidative stress (103). In accordance, high-phosphate treatment increased aldosterone synthase expression in VSMCs (90) and silencing of aldosterone synthase attenuated the phosphate-induced osteo-/chondrogenic transdifferentiation and calcification in vitro. Similarly, aldosterone synthase expression is higher in coronary arteries from patients with impaired renal function and correlated with CBFA1 expression. Aldosterone synthase expression in VSMCs is upregulated by disruption of APEX1-dependent gene suppression (90). Accordingly, APEX1 is protective against VSMC calcification (90, 104).
Aldosterone synthase expression is similarly enhanced in klotho-hypomorphic mice (90). In those mice, aortic osteo-/chondrogenic signaling is decreased by spironolactone, but not by adrenalectomy and in adrenalectomized klotho-hypomorphic mice, spironolactone treatment still significantly blunts aortic osteoinductive reprogramming (90).
Mineralocorticoid receptor antagonism may, thus, be a therapeutic option for hyperphosphatemic patients even in the absence of hyperaldosteronism (86). Spironolactone may further protect VSMCs in diabetes (105), which may lead to upregulation of vascular aldosterone synthase (100).
The effects of spironolactone in CKD patients are under study (106, 107). Clinical trials indicate that spironolactone treatment reduces morbidity and mortality in hemodialysis patients (92). MR inhibition may cause a transient reduction of renal function and promote hyperkalemia, but has been shown to be relatively safe in CKD patients (92, 108).
Besides its acidifying effect on extracellular pH (109, 110), may dissociate to H+ and NH3 which easily crosses membranes, thus entering cells and cellular compartments (111). In acidic intracellular compartments NH3 binds H+ and is trapped as (112). The binding of H+ alkalinizes acidic cellular compartments (113) and the intracellular/intra-compartimental accumulation of swells cells and acidic intracellular compartments (114–116). Cell swelling may downregulate the cell volume sensitive transcription factor NFAT5 (117, 118). Moreover, alkalinization of acidic cellular compartments may interfere with the maturation of several proteins including TGFβ1 (119), a key factor in the regulation of osteo-/chondrogenic signaling of VSMCs (120–122).
Treatment of klotho-hypomorphic mice with NH4Cl containing drinking water prevented soft tissue and vascular calcifications and increased their life span more than 12- (♂) or 4-fold (♀) without significantly affecting extracellular pH or plasma concentrations of 1,25(OH)2D3, calcium, and phosphate (123). Tissue calcification and aging were further delayed in klotho-hypomorphic mice by NH4NO3 (124).
NH4Cl prevents vascular calcification apparently not by inducing acidosis. Untreated klotho-hypomorphic mice suffer from respiratory acidosis resulting from severe lung emphysema (123). NH4Cl treatment prevents the development of lung emphysema and, thus, respiratory acidosis (123). Instead, NH4Cl induces a metabolic acidosis of similar extracellular pH as in untreated mice (123).
NH4Cl treatment prevents development of extracellular volume depletion, thus normalizing ADH release and plasma aldosterone levels (40). The decrease of plasma aldosterone concentrations following NH4Cl treatment presumably contributes to the decrease of vascular calcification. However, the effect of NH4Cl on survival and calcification (123) is, by far, larger than that of aldosterone receptor blockade (87).
NH4Cl treatment is presumably mainly effective by interference with osteo-/chondrogenic transdifferentiation of VSMCs (123). In aortic tissue of klotho-hypomorphic mice and in phosphate treated VSMCs in vitro, NH4Cl disrupted the increased expression of osteogenic and chondrogenic markers CBFA1 and SOX9 and of ALPL (123). Osteo-/chondrogenic reprogramming is paralleled by VSMCs senescence (125), and thus vascular aging (126). NH4Cl treatment reversed the upregulation of PAI-1, p21, and GLB1, key elements in the orchestration of senescence (127). TGFβ1 expression was upregulated in aortic tissue of klotho-hypomorphic mice and in phosphate treated VSMCs, which in turn triggers cellular senescence, osteo-/chondrogenic reprogramming and aging (128) and is decreased by NH4Cl treatment (123). NH4Cl further impairs maturation of TGFβ1 (119). TGFβ1 is a stimulator of NFAT5 expression (129). NH4Cl treatment reverses the enhanced expression of NFAT5 in klotho-hypomorphic mice and in phosphate treated VSMCs (123). Addition of exogenous TGFβ1 protein or NFAT5 overexpression triggers osteo-/chondrogenic reprogramming of VSMCs in vitro, which cannot be reversed by NH4Cl, indicating that NH4Cl is effective upstream of mature TGFβ1 (123). NH4Cl treatment is, at least in part, effective by disrupting TGFβ1-dependent osteo-/chondrogenic signaling (123) and, thus, vascular calcification (130).
Further in vitro experiments revealed that inhibition of the vacuolar H+ ATPase with bafilomycin A1 or following dissipation of the pH gradient across the membranes of acidic cellular compartments with methylamine similarly disrupted phosphate-induced TGFβ1-dependent osteo-/chondrogenic signaling in VSMCs (131), supporting the hypothesis that vascular acidic cellular compartments are necessary for promoting vascular calcification.
A concern of NH4Cl treatment is the cerebral ammonia toxicity (114–116). However, the employed NH4Cl dosage does apparently not lead to toxic ammonia concentrations as NH4Cl treated male klotho-hypomorphic mice reached a life span close to that of untreated wild-type mice, and behavioral studies did not reveal any defect in NH4Cl treated wild-type mice (123). Needless to say, NH4Cl treatment may be hazardous in patients with hepatic failure.
Klotho-hypomorphic mice and mice with vitamin D3 overload suffer from severe vascular calcification. Experiments in those animals shed novel light on the mechanisms orchestrating vascular calcification and led to the discovery of several powerful therapeutic opportunities (Figure 2). The most effective treatment turned out to be NH4Cl, but also acetazolamide, spironolactone, bicarbonate, or magnesium supplementation were able to reduce the progression of vascular calcification in vivo. Further studies are required to fully define advantages and disadvantages of those treatments, to possibly uncover additional therapeutic options and—most importantly—to clarify whether the successful treatments in mice can be translated into avoidance of vascular calcification in human disease, such as CKD, diabetes, and (premature) aging.
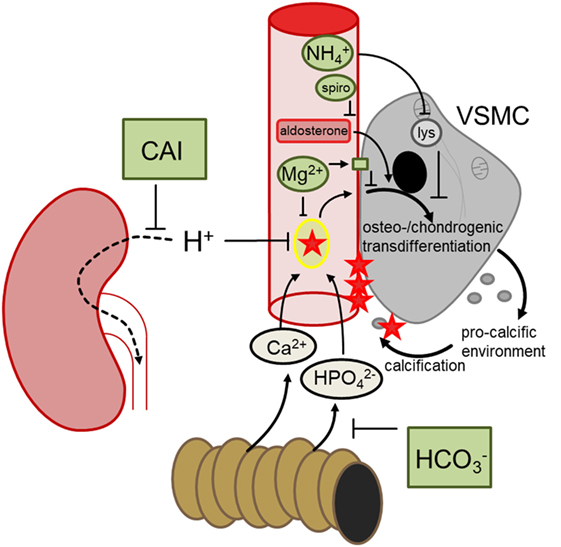
Figure 2. Hypothetical mechanisms involved in the discussed treatments preventing vascular calcification. During hyperphosphatemia, phosphate complexes with calcium and forms secondary calciprotein particles, inducing osteo-/chondrogenic transdifferentiation of vascular smooth muscle cells (VSMCs), which in turn generates a pro-calcific environment and subsequent active tissue mineralization. Oral bicarbonate treatment may impair intestinal phosphate reabsorption, ameliorating hyperhosphatemia. Renal carboanhydrase inhibition causes proton retention, which may reduce the formation of calcium-phosphate nanoparticles. Magnesium may similarly directly prevent calcium-phosphate complexation and stimulates the calcium sensing receptor in VSMCs, blunting osteo-chondrogenic transdifferentiation. This transdifferentiation is also directly stimulated by aldosterone, which may be blunted by spironolactone (spiro). Ammoniumchloride leads to lysosomal (lys) alkalinization which appears to dissipate the osteo-chondrogenic transdifferentiation of VSMCs. These mechanisms may provide the basis to develop a therapeutic approach to reduce the burden of vascular calcification.
All authors listed have made substantial, direct and intellectual contribution to the work, and approved it for publication.
CL is employed by the company Fresenius Kabi Deutschland GmbH, Bad Homburg, Germany. All other authors declare no competing interests.
The authors acknowledge the meticulous preparation of the manuscript by L. Subasic.
Experiments in the author’s laboratories were supported by the European Union Seventh Framework Programme (FP7/2007-2013-603288-SysVasc), the Deutsche Forschungsgemeinschaft (La315-15, AL2054/1-1, VO2259/2-1), Else Kröner-Fresenius-Stiftung, and the Berlin Institute of Health (BIH) Translational Postdoc Grant. The authors acknowledge support by Deutsche Forschungsgemeinschaft and Open Access Publishing Fund of University of Tübingen.
1. Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res (2011) 109(6):697–711. doi:10.1161/CIRCRESAHA.110.234914
2. Stabley JN, Towler DA. Arterial calcification in diabetes mellitus: preclinical models and translational implications. Arterioscler Thromb Vasc Biol (2017) 37(2):205–17. doi:10.1161/ATVBAHA.116.306258
3. Blacher J, Guerin AP, Pannier B, Marchais SJ, London GM. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension (2001) 38(4):938–42. doi:10.1161/hy1001.096358
4. London GM, Guérin AP, Marchais SJ, Métivier F, Pannier B, Adda H. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant (2003) 18(9):1731–40. doi:10.1093/ndt/gfg414
5. Foley RN, Parfrey PS, Sarnak MJ. Epidemiology of cardiovascular disease in chronic renal disease. J Am Soc Nephrol (1998) 9(12 Suppl):S16–23.
6. Giachelli CM. Vascular calcification: in vitro evidence for the role of inorganic phosphate. J Am Soc Nephrol (2003) 14(9 Suppl 4):S300–4. doi:10.1097/01.ASN.0000081663.52165.66
7. Tonelli M, Sacks F, Pfeffer M, Gao Z, Curhan G; Cholesterol and Recurrent Events Trial Investigators. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation (2005) 112(17):2627–33. doi:10.1161/CIRCULATIONAHA.105.553198
8. Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis (1998) 31(4):607–17. doi:10.1053/ajkd.1998.v31.pm9531176
9. Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res (2001) 89(12):1147–54. doi:10.1161/hh2401.101070
10. Dautova Y, Kozlova D, Skepper JN, Epple M, Bootman MD, Proudfoot D. Fetuin-A and albumin alter cytotoxic effects of calcium phosphate nanoparticles on human vascular smooth muscle cells. PLoS One (2014) 9(5):e97565. doi:10.1371/journal.pone.0097565
11. Dautova Y, Kapustin AN, Pappert K, Epple M, Okkenhaug H, Cook SJ, et al. Calcium phosphate particles stimulate interleukin-1beta release from human vascular smooth muscle cells: a role for spleen tyrosine kinase and exosome release. J Mol Cell Cardiol (2018) 115:82–93. doi:10.1016/j.yjmcc.2017.12.007
12. Paloian NJ, Giachelli CM. A current understanding of vascular calcification in CKD. Am J Physiol Renal Physiol (2014) 307(8):F891–900. doi:10.1152/ajprenal.00163.2014
13. Hocher B, Pasch A. Hope for CKD-MBD patients: new diagnostic approaches for better treatment of CKD-MBD. Kidney Dis (Basel) (2017) 3(1):8–14. doi:10.1159/000477244
14. Pasch A. Novel assessments of systemic calcification propensity. Curr Opin Nephrol Hypertens (2016) 25(4):278–84. doi:10.1097/MNH.0000000000000237
15. Holt SG, Smith ER. Fetuin-A-containing calciprotein particles in mineral trafficking and vascular disease. Nephrol Dial Transplant (2016) 31(10):1583–7. doi:10.1093/ndt/gfw048
16. Kuro OM. A phosphate-centric paradigm for pathophysiology and therapy of chronic kidney disease. Kidney Int Suppl (2011) (2013) 3(5):420–6. doi:10.1038/kisup.2013.88
17. Brylka L, Jahnen-Dechent W. The role of fetuin-A in physiological and pathological mineralization. Calcif Tissue Int (2013) 93(4):355–64. doi:10.1007/s00223-012-9690-6
18. Herrmann M, Kinkeldey A, Jahnen-Dechent W. Fetuin-A function in systemic mineral metabolism. Trends Cardiovasc Med (2012) 22(8):197–201. doi:10.1016/j.tcm.2012.07.020
19. Jahnen-Dechent W, Heiss A, Schäfer C, Ketteler M. Fetuin-A regulation of calcified matrix metabolism. Circ Res (2011) 108(12):1494–509. doi:10.1161/CIRCRESAHA.110.234260
20. Aghagolzadeh M, Hochberg LR, Cash SS, Truccolo W. Predicting seizures from local field potentials recorded via intracortical microelectrode arrays. Conf Proc IEEE Eng Med Biol Soc (2016) 2016:6353–6. doi:10.1109/EMBC.2016.7592181
21. Li X, Yang HY, Giachelli CM. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ Res (2006) 98(7):905–12. doi:10.1161/01.RES.0000216409.20863.e7
22. Masuda M, Miyazaki-Anzai S, Keenan AL, Shiozaki Y, Okamura K, Chick WS, et al. Activating transcription factor-4 promotes mineralization in vascular smooth muscle cells. JCI Insight (2016) 1(18):e88646. doi:10.1172/jci.insight.88646
23. Shroff RC, McNair R, Figg N, Skepper JN, Schurgers L, Gupta A, et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation (2008) 118(17):1748–57. doi:10.1161/CIRCULATIONAHA.108.783738
24. Shao JS, Cheng SL, Pingsterhaus JM, Charlton-Kachigian N, Loewy AP, Towler DA. Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J Clin Invest (2005) 115(5):1210–20. doi:10.1172/JCI24140
25. Xu Z, Ji G, Shen J, Wang X, Zhou J, Li L. SOX9 and myocardin counteract each other in regulating vascular smooth muscle cell differentiation. Biochem Biophys Res Commun (2012) 422(2):285–90. doi:10.1016/j.bbrc.2012.04.149
26. Lin ME, Chen TM, Wallingford MC, Nguyen NB, Yamada S, Sawangmake C, et al. Runx2 deletion in smooth muscle cells inhibits vascular osteochondrogenesis and calcification but not atherosclerotic lesion formation. Cardiovasc Res (2016) 112(2):606–16. doi:10.1093/cvr/cvw205
27. Caron MM, van der Windt AE, Emans PJ, van Rhijn LW, Jahr H, Welting TJ. Osmolarity determines the in vitro chondrogenic differentiation capacity of progenitor cells via nuclear factor of activated T-cells 5. Bone (2013) 53(1):94–102. doi:10.1016/j.bone.2012.11.032
28. Sheen CR, Kuss P, Narisawa S, Yadav MC, Nigro J, Wang W, et al. Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification. J Bone Miner Res (2015) 30(5):824–36. doi:10.1002/jbmr.2420
29. Chen NX, Moe SM. Pathophysiology of vascular calcification. Curr Osteoporos Rep (2015) 13(6):372–80. doi:10.1007/s11914-015-0293-9
30. Pai A, Leaf EM, El-Abbadi M, Giachelli CM. Elastin degradation and vascular smooth muscle cell phenotype change precede cell loss and arterial medial calcification in a uremic mouse model of chronic kidney disease. Am J Pathol (2011) 178(2):764–73. doi:10.1016/j.ajpath.2010.10.006
31. Koleganova N, Piecha G, Ritz E, Schirmacher P, Müller A, Meyer HP, et al. Arterial calcification in patients with chronic kidney disease. Nephrol Dial Transplant (2009) 24(8):2488–96. doi:10.1093/ndt/gfp137
32. Shroff RC, McNair R, Skepper JN, Figg N, Schurgers LJ, Deanfield J, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol (2010) 21(1):103–12. doi:10.1681/ASN.2009060640
33. Mizobuchi M, Towler D, Slatopolsky E. Vascular calcification: the killer of patients with chronic kidney disease. J Am Soc Nephrol (2009) 20(7):1453–64. doi:10.1681/ASN.2008070692
34. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature (1997) 390(6655):45–51. doi:10.1038/36285
35. Kuro-o M. Klotho, phosphate and FGF-23 in ageing and disturbed mineral metabolism. Nat Rev Nephrol (2013) 9(11):650–60. doi:10.1038/nrneph.2013.111
36. Neyra JA, Hu MC. Potential application of klotho in human chronic kidney disease. Bone (2017) 100:41–9. doi:10.1016/j.bone.2017.01.017
37. Xie J, Yoon J, An SW, Kuro-o M, Huang CL. Soluble klotho protects against uremic cardiomyopathy independently of fibroblast growth factor 23 and phosphate. J Am Soc Nephrol (2015) 26(5):1150–60. doi:10.1681/ASN.2014040325
38. Lu X, Hu MC. Klotho/FGF23 axis in chronic kidney disease and cardiovascular disease. Kidney Dis (Basel) (2017) 3(1):15–23. doi:10.1159/000452880
39. Mencke R, Olauson H, Hillebrands JL. Effects of klotho on fibrosis and cancer: a renal focus on mechanisms and therapeutic strategies. Adv Drug Deliv Rev (2017) 121:85–100. doi:10.1016/j.addr.2017.07.009
40. Fischer SS, Kempe DS, Leibrock CB, Rexhepaj R, Siraskar B, Boini KM, et al. Hyperaldosteronism in klotho-deficient mice. Am J Physiol Renal Physiol (2010) 299(5):F1171–7. doi:10.1152/ajprenal.00233.2010
41. Alesutan I, Feger M, Tuffaha R, Castor T, Musculus K, Buehling SS, et al. Augmentation of phosphate-induced osteo-/chondrogenic transformation of vascular smooth muscle cells by homoarginine. Cardiovasc Res (2016) 110(3):408–18. doi:10.1093/cvr/cvw062
42. Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, et al. Suppression of aging in mice by the hormone klotho. Science (2005) 309(5742):1829–33. doi:10.1126/science.1112766
43. Invidia L, Salvioli S, Altilia S, Pierini M, Panourgia MP, Monti D, et al. The frequency of klotho KL-VS polymorphism in a large Italian population, from young subjects to centenarians, suggests the presence of specific time windows for its effect. Biogerontology (2010) 11(1):67–73. doi:10.1007/s10522-009-9229-z
44. Lau WL, Leaf EM, Hu MC, Takeno MM, Kuro-o M, Moe OW, et al. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int (2012) 82(12):1261–70. doi:10.1038/ki.2012.322
45. Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol (2003) 17(12):2393–403. doi:10.1210/me.2003-0048
46. Sopjani M, Alesutan I, Dërmaku-Sopjani M, Gu S, Zelenak C, Munoz C, et al. Regulation of the Na+/K+ ATPase by klotho. FEBS Lett (2011) 585(12):1759–64. doi:10.1016/j.febslet.2011.05.021
47. Tang C, Pathare G, Michael D, Fajol A, Eichenmüller M, Lang F. Downregulation of klotho expression by dehydration. Am J Physiol Renal Physiol (2011) 301(4):F745–50. doi:10.1152/ajprenal.00037.2011
48. Smith ER, Hewitson TD, Hanssen E, Holt SG. Biochemical transformation of calciprotein particles in uraemia. Bone (2018) 110:355–67. doi:10.1016/j.bone.2018.02.023
49. Mathew S, Lund RJ, Chaudhary LR, Geurs T, Hruska KA. Vitamin D receptor activators can protect against vascular calcification. J Am Soc Nephrol (2008) 19(8):1509–19. doi:10.1681/ASN.2007080902
50. Alexander RT, Woudenberg-Vrenken TE, Buurman J, Dijkman H, van der Eerden BC, van Leeuwen JP, et al. Klotho prevents renal calcium loss. J Am Soc Nephrol (2009) 20(11):2371–9. doi:10.1681/ASN.2008121273
51. Kovesdy CP. Metabolic acidosis and kidney disease: does bicarbonate therapy slow the progression of CKD? Nephrol Dial Transplant (2012) 27(8):3056–62. doi:10.1093/ndt/gfs291
52. Barsotti G, Lazzeri M, Cristofano C, Cerri M, Lupetti S, Giovannetti S. The role of metabolic acidosis in causing uremic hyperphosphatemia. Miner Electrolyte Metab (1986) 12(2):103–6.
53. Loniewski I, Wesson DE. Bicarbonate therapy for prevention of chronic kidney disease progression. Kidney Int (2014) 85(3):529–35. doi:10.1038/ki.2013.401
54. Nath KA, Hostetter MK, Hostetter TH. Pathophysiology of chronic tubulo-interstitial disease in rats. Interactions of dietary acid load, ammonia, and complement component C3. J Clin Invest (1985) 76(2):667–75. doi:10.1172/JCI112020
55. Phisitkul S, Hacker C, Simoni J, Tran RM, Wesson DE. Dietary protein causes a decline in the glomerular filtration rate of the remnant kidney mediated by metabolic acidosis and endothelin receptors. Kidney Int (2008) 73(2):192–9. doi:10.1038/sj.ki.5002647
56. Wesson DE, Jo CH, Simoni J. Angiotensin II receptors mediate increased distal nephron acidification caused by acid retention. Kidney Int (2012) 82(11):1184–94. doi:10.1038/ki.2012.267
57. Torres VE, Mujwid DK, Wilson DM, Holley KH. Renal cystic disease and ammoniagenesis in Han:SPRD rats. J Am Soc Nephrol (1994) 5(5):1193–200.
58. Mendoza FJ, Lopez I, Montes de Oca A, Perez J, Rodriguez M, Aguilera-Tejero E. Metabolic acidosis inhibits soft tissue calcification in uremic rats. Kidney Int (2008) 73(4):407–14. doi:10.1038/sj.ki.5002646
59. Jara A, Felsenfeld AJ, Bover J, Kleeman CR. Chronic metabolic acidosis in azotemic rats on a high-phosphate diet halts the progression of renal disease. Kidney Int (2000) 58(3):1023–32. doi:10.1046/j.1523-1755.2000.00260.x
60. Jara A, Chacón C, Ibaceta M, Valdivieso A, Felsenfeld AJ. Effect of ammonium chloride and dietary phosphorus in the azotaemic rat. I. Renal function and biochemical changes. Nephrol Dial Transplant (2004) 19(8):1986–92. doi:10.1093/ndt/gfh311
61. Dobre M, Rahman M, Hostetter TH. Current status of bicarbonate in CKD. J Am Soc Nephrol (2015) 26(3):515–23. doi:10.1681/ASN.2014020205
62. de Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol (2009) 20(9):2075–84. doi:10.1681/ASN.2008111205
63. Leibrock CB, Voelkl J, Kohlhofer U, Quintanilla-Martinez L, Kuro-O M, Lang F. Bicarbonate-sensitive calcification and lifespan of klotho-deficient mice. Am J Physiol Renal Physiol (2016) 310(1):F102–8. doi:10.1152/ajprenal.00037.2015
64. Leibrock CB, Alesutan I, Voelkl J, Michael D, Castor T, Kohlhofer U, et al. Acetazolamide sensitive tissue calcification and aging of klotho-hypomorphic mice. J Mol Med (Berl) (2016) 94(1):95–106. doi:10.1007/s00109-015-1331-x
65. Yonova D. Vascular calcification and metabolic acidosis in end stage renal disease. Hippokratia (2009) 13(3):139–40.
66. Villa-Bellosta R, Sorribas V. Compensatory regulation of the sodium/phosphate cotransporters NaPi-IIc (SCL34A3) and Pit-2 (SLC20A2) during Pi deprivation and acidosis. Pflugers Arch (2010) 459(3):499–508. doi:10.1007/s00424-009-0746-z
67. Biber J, Hernando N, Forster I, Murer H. Regulation of phosphate transport in proximal tubules. Pflugers Arch (2009) 458(1):39–52. doi:10.1007/s00424-008-0580-8
68. Voelkl J, Pakladok T, Lin Y, Viereck R, Lebedeva A, Kukuk D, et al. Up-regulation of hepatic alpha-2-HS-glycoprotein transcription by testosterone via androgen receptor activation. Cell Physiol Biochem (2014) 33(6):1911–20. doi:10.1159/000362968
69. Zhou S, Fang X, Xin H, Li W, Qiu H, Guan S. Osteoprotegerin inhibits calcification of vascular smooth muscle cell via down regulation of the notch1-RBP-Jkappa/Msx2 signaling pathway. PLoS One (2013) 8(7):e68987. doi:10.1371/journal.pone.0068987
70. Paloian NJ, Leaf EM, Giachelli CM. Osteopontin protects against high phosphate-induced nephrocalcinosis and vascular calcification. Kidney Int (2016) 89(5):1027–36. doi:10.1016/j.kint.2015.12.046
71. Meema HE, Oreopoulos DG, Rapoport A. Serum magnesium level and arterial calcification in end-stage renal disease. Kidney Int (1987) 32(3):388–94. doi:10.1038/ki.1987.222
72. João Matias P, Azevedo A, Laranjinha I, Navarro D, Mendes M, Ferreira C, et al. Lower serum magnesium is associated with cardiovascular risk factors and mortality in haemodialysis patients. Blood Purif (2014) 38(3–4):244–52. doi:10.1159/000366124
73. Louvet L, Metzinger L, Büchel J, Steppan S, Massy ZA. Magnesium attenuates phosphate-induced deregulation of a microRNA signature and prevents modulation of Smad1 and osterix during the course of vascular calcification. Biomed Res Int (2016) 2016:7419524. doi:10.1155/2016/7419524
74. Louvet L, Bazin D, Büchel J, Steppan S, Passlick-Deetjen J, Massy ZA. Characterisation of calcium phosphate crystals on calcified human aortic vascular smooth muscle cells and potential role of magnesium. PLoS One (2015) 10(1):e0115342. doi:10.1371/journal.pone.0115342
75. Montezano AC, Zimmerman D, Yusuf H, Burger D, Chignalia AZ, Wadhera V, et al. Vascular smooth muscle cell differentiation to an osteogenic phenotype involves TRPM7 modulation by magnesium. Hypertension (2010) 56(3):453–62. doi:10.1161/HYPERTENSIONAHA.110.152058
76. Ter Braake AD, Tinnemans PT, Shanahan CM, Hoenderop JGJ, de Baaij JHF. Magnesium prevents vascular calcification in vitro by inhibition of hydroxyapatite crystal formation. Sci Rep (2018) 8(1):2069. doi:10.1038/s41598-018-20241-3
77. Alesutan I, Tuffaha R, Auer T, Feger M, Pieske B, Lang F, et al. Inhibition of osteo/chondrogenic transformation of vascular smooth muscle cells by MgCl2 via calcium-sensing receptor. J Hypertens (2017) 35(3):523–32. doi:10.1097/HJH.0000000000001202
78. Saidak Z, Brazier M, Kamel S, Mentaverri R. Agonists and allosteric modulators of the calcium-sensing receptor and their therapeutic applications. Mol Pharmacol (2009) 76(6):1131–44. doi:10.1124/mol.109.058784
79. Molostvov G, Hiemstra TF, Fletcher S, Bland R, Zehnder D. Arterial expression of the calcium-sensing receptor is maintained by physiological pulsation and protects against calcification. PLoS One (2015) 10(10):e0138833. doi:10.1371/journal.pone.0138833
80. Apetrii M, Covic A, Massy ZA. Magnesium supplementation: a consideration in dialysis patients. Semin Dial (2018) 31(1):11–4. doi:10.1111/sdi.12653
81. Bressendorff I, Hansen D, Schou M, Silver B, Pasch A, Bouchelouche P, et al. Oral magnesium supplementation in chronic kidney disease stages 3 and 4: efficacy, safety, and effect on serum calcification propensity-A prospective randomized double-blinded placebo-controlled clinical trial. Kidney Int Rep (2017) 2(3):380–9. doi:10.1016/j.ekir.2016.12.008
82. Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ Res (2005) 96(6):643–50. doi:10.1161/01.RES.0000159937.05502.d1
83. Jaffe IZ, Tintut Y, Newfell BG, Demer LL, Mendelsohn ME. Mineralocorticoid receptor activation promotes vascular cell calcification. Arterioscler Thromb Vasc Biol (2007) 27(4):799–805. doi:10.1161/01.ATV.0000258414.59393.89
84. Wu SY, Yu YR, Cai Y, Jia LX, Wang X, Xiao CS, et al. Endogenous aldosterone is involved in vascular calcification in rat. Exp Biol Med (Maywood) (2012) 237(1):31–7. doi:10.1258/ebm.2011.011175
85. Lang F, Ritz E, Alesutan I, Voelkl J. Impact of aldosterone on osteoinductive signaling and vascular calcification. Nephron Physiol (2014) 128(1–2):40–5. doi:10.1159/000368268
86. Lang F, Ritz E, Voelkl J, Alesutan I. Vascular calcification – is aldosterone a culprit? Nephrol Dial Transplant (2013) 28(5):1080–4. doi:10.1093/ndt/gft041
87. Voelkl J, Alesutan I, Leibrock CB, Quintanilla-Martinez L, Kuhn V, Feger M, et al. Spironolactone ameliorates PIT1-dependent vascular osteoinduction in klotho-hypomorphic mice. J Clin Invest (2013) 123(2):812–22. doi:10.1172/JCI64093
88. Tatsumoto N, Yamada S, Tokumoto M, Eriguchi M, Noguchi H, Torisu K, et al. Spironolactone ameliorates arterial medial calcification in uremic rats: the role of mineralocorticoid receptor signaling in vascular calcification. Am J Physiol Renal Physiol (2015) 309(11):F967–79. doi:10.1152/ajprenal.00669.2014
89. Andrukhova O, Slavic S, Smorodchenko A, Zeitz U, Shalhoub V, Lanske B, et al. FGF23 regulates renal sodium handling and blood pressure. EMBO Mol Med (2014) 6(6):744–59. doi:10.1002/emmm.201303716
90. Alesutan I, Voelkl J, Feger M, Kratschmar DV, Castor T, Mia S, et al. Involvement of vascular aldosterone synthase in phosphate-induced osteogenic transformation of vascular smooth muscle cells. Sci Rep (2017) 7(1):2059. doi:10.1038/s41598-017-01882-2
91. Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int (1982) 21(1):98–101. doi:10.1038/ki.1982.14
92. Matsumoto Y, Mori Y, Kageyama S, Arihara K, Sugiyama T, Ohmura H, et al. Spironolactone reduces cardiovascular and cerebrovascular morbidity and mortality in hemodialysis patients. J Am Coll Cardiol (2014) 63(6):528–36. doi:10.1016/j.jacc.2013.09.056
93. Ando K, Ohtsu H, Uchida S, Kaname S, Arakawa Y, Fujita T, et al. Anti-albuminuric effect of the aldosterone blocker eplerenone in non-diabetic hypertensive patients with albuminuria: a double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol (2014) 2(12):944–53. doi:10.1016/S2213-8587(14)70194-9
94. Toyonaga J, Tsuruya K, Ikeda H, Noguchi H, Yotsueda H, Fujisaki K, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant (2011) 26(8):2475–84. doi:10.1093/ndt/gfq750
95. Slight SH, Joseph J, Ganjam VK, Weber KT. Extra-adrenal mineralocorticoids and cardiovascular tissue. J Mol Cell Cardiol (1999) 31(6):1175–84. doi:10.1006/jmcc.1999.0963
96. Le Goascogne C, Robel P, Gouézou M, Sananès N, Baulieu EE, Waterman M. Neurosteroids: cytochrome P-450scc in rat brain. Science (1987) 237(4819):1212–5. doi:10.1126/science.3306919
97. Gomez-Sanchez CE, Gomez-Sanchez EP. Editorial: cardiac steroidogenesis – new sites of synthesis, or much ado about nothing? J Clin Endocrinol Metab (2001) 86(11):5118–20. doi:10.1210/jc.86.11.5118
98. Xue C, Siragy HM. Local renal aldosterone system and its regulation by salt, diabetes, and angiotensin II type 1 receptor. Hypertension (2005) 46(3):584–90. doi:10.1161/01.HYP.0000175814.18550.c0
99. Hatakeyama H, Miyamori I, Fujita T, Takeda Y, Takeda R, Yamamoto H. Vascular aldosterone. Biosynthesis and a link to angiotensin II-induced hypertrophy of vascular smooth muscle cells. J Biol Chem (1994) 269(39):24316–20.
100. Matsuzawa Y, Suematsu S, Saito J, Omura M, Nishikawa T. Vascular aldosterone production at the pre-diabetic stage of young Otsuka Long-Evans Tokushima Fatty (OLETF) rats, compared with Long-Evans Tokushima Otsuka (LETO) rats. Molecules (2013) 18(12):15636–47. doi:10.3390/molecules181215636
101. Takeda Y, Miyamori I, Inaba S, Furukawa K, Hatakeyama H, Yoneda T, et al. Vascular aldosterone in genetically hypertensive rats. Hypertension (1997) 29(1 Pt 1):45–8. doi:10.1161/01.HYP.29.1.45
102. Ayari H, Legedz L, Cerutti C, Lantelme P, Feugier P, Gustin MP, et al. Mutual amplification of corticosteroids and angiotensin systems in human vascular smooth muscle cells and carotid atheroma. J Mol Med (Berl) (2014) 92(11):1201–8. doi:10.1007/s00109-014-1193-7
103. Ohmine T, Miwa Y, Takahashi-Yanaga F, Morimoto S, Maehara Y, Sasaguri T. The involvement of aldosterone in cyclic stretch-mediated activation of NADPH oxidase in vascular smooth muscle cells. Hypertens Res (2009) 32(8):690–9. doi:10.1038/hr.2009.76
104. Lee KM, Lee EO, Lee YR, Joo HK, Park MS, Kim CS, et al. APE1/Ref-1 inhibits phosphate-induced calcification and osteoblastic phenotype changes in vascular smooth muscle cells. Int J Mol Sci (2017) 18(10):2053. doi:10.3390/ijms18102053
105. Silva MA, Bruder-Nascimento T, Cau SB, Lopes RA, Mestriner FL, Fais RS, et al. Spironolactone treatment attenuates vascular dysfunction in type 2 diabetic mice by decreasing oxidative stress and restoring NO/GC signaling. Front Physiol (2015) 6:269. doi:10.3389/fphys.2015.00269
106. Hill NR, Lasserson D, Thompson B, Perera-Salazar R, Wolstenholme J, Bower P, et al. Benefits of aldosterone receptor antagonism in chronic kidney disease (BARACK D) trial-a multi-centre, prospective, randomised, open, blinded end-point, 36-month study of 2,616 patients within primary care with stage 3b chronic kidney disease to compare the efficacy of spironolactone 25 mg once daily in addition to routine care on mortality and cardiovascular outcomes versus routine care alone: study protocol for a randomized controlled trial. Trials (2014) 15:160. doi:10.1186/1745-6215-15-160
107. Ng KP, Jain P, Heer G, Redman V, Chagoury OL, Dowswell G, et al. Spironolactone to prevent cardiovascular events in early-stage chronic kidney disease (STOP-CKD): study protocol for a randomized controlled pilot trial. Trials (2014) 15:158. doi:10.1186/1745-6215-15-158
108. Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int (2006) 70(12):2116–23. doi:10.1038/sj.ki.5001854
109. Nowik M, Kampik NB, Mihailova M, Eladari D, Wagner CA. Induction of metabolic acidosis with ammonium chloride (NH4Cl) in mice and rats – species differences and technical considerations. Cell Physiol Biochem (2010) 26(6):1059–72. doi:10.1159/000323984
110. Mohebbi N, Perna A, van der Wijst J, Becker HM, Capasso G, Wagner CA. Regulation of two renal chloride transporters, AE1 and pendrin, by electrolytes and aldosterone. PLoS One (2013) 8(1):e55286. doi:10.1371/journal.pone.0055286
111. Roos A, Boron WF. Intracellular pH. Physiol Rev (1981) 61(2):296–434. doi:10.1152/physrev.1981.61.2.296
112. Ling H, Ardjomand P, Samvakas S, Simm A, Busch GL, Lang F, et al. Mesangial cell hypertrophy induced by NH4Cl: role of depressed activities of cathepsins due to elevated lysosomal pH. Kidney Int (1998) 53(6):1706–12. doi:10.1046/j.1523-1755.1998.00952.x
113. Völkl H, Friedrich F, Häussinger D, Lang F. Effect of cell volume on acridine orange fluorescence in hepatocytes. Biochem J (1993) 295(Pt 1):11–4. doi:10.1042/bj2950011
114. Haussinger D, Gorg B. Interaction of oxidative stress, astrocyte swelling and cerebral ammonia toxicity. Curr Opin Clin Nutr Metab Care (2010) 13(1):87–92. doi:10.1097/MCO.0b013e328333b829
115. Jayakumar AR, Norenberg MD. The Na-K-Cl Co-transporter in astrocyte swelling. Metab Brain Dis (2010) 25(1):31–8. doi:10.1007/s11011-010-9180-3
116. Pasantes-Morales H, Vazquez-Juarez E. Transporters and channels in cytotoxic astrocyte swelling. Neurochem Res (2012) 37(11):2379–87. doi:10.1007/s11064-012-0777-2
117. Burg MB, Ferraris JD, Dmitrieva NI. Cellular response to hyperosmotic stresses. Physiol Rev (2007) 87(4):1441–74. doi:10.1152/physrev.00056.2006
118. Handler JS, Kwon HM. Cell and molecular biology of organic osmolyte accumulation in hypertonic renal cells. Nephron (2001) 87(2):106–10. doi:10.1159/000045897
119. Basque J, Martel M, Leduc R, Cantin AM. Lysosomotropic drugs inhibit maturation of transforming growth factor-beta. Can J Physiol Pharmacol (2008) 86(9):606–12. doi:10.1139/y08-063
120. Liu X, Bai C, Gong D, Yuan Y, Han L, Lu F, et al. Pleiotropic effects of transforming growth factor-beta1 on pericardial interstitial cells. Implications for fibrosis and calcification in idiopathic constrictive pericarditis. J Am Coll Cardiol (2011) 57(15):1634–5. doi:10.1016/j.jacc.2010.10.054
121. Pai AS, Giachelli CM. Matrix remodeling in vascular calcification associated with chronic kidney disease. J Am Soc Nephrol (2010) 21(10):1637–40. doi:10.1681/ASN.2010040349
122. Tachi K, Takami M, Sato H, Mochizuki A, Zhao B, Miyamoto Y, et al. Enhancement of bone morphogenetic protein-2-induced ectopic bone formation by transforming growth factor-beta1. Tissue Eng Part A (2011) 17(5–6):597–606. doi:10.1089/ten.tea.2010.0094
123. Leibrock CB, Alesutan I, Voelkl J, Pakladok T, Michael D, Schleicher E, et al. NH4Cl treatment prevents tissue calcification in klotho deficiency. J Am Soc Nephrol (2015) 26(10):2423–33. doi:10.1681/ASN.2014030230
124. Leibrock CB, Feger M, Voelkl J, Kohlhofer U, Quintanilla-Martinez L, Kuro-o M, et al. Partial reversal of tissue calcification and extension of life span following ammonium nitrate treatment of klotho-deficient mice. Kidney Blood Press Res (2016) 41(1):99–107. doi:10.1159/000443411
125. Mackenzie NC, MacRae VE. The role of cellular senescence during vascular calcification: a key paradigm in aging research. Curr Aging Sci (2011) 4(2):128–36. doi:10.2174/1874609811104020128
126. Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res (2012) 111(2):245–59. doi:10.1161/CIRCRESAHA.111.261388
127. Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell (2006) 5(2):187–95. doi:10.1111/j.1474-9726.2006.00199.x
128. Wang N, Wang X, Xing C, Sun B, Yu X, Hu J, et al. Role of TGF-beta1 in bone matrix production in vascular smooth muscle cells induced by a high-phosphate environment. Nephron Exp Nephrol (2010) 115(3):e60–8. doi:10.1159/000313831
129. Hiyama A, Gogate SS, Gajghate S, Mochida J, Shapiro IM, Risbud MV. BMP-2 and TGF-beta stimulate expression of beta1,3-glucuronosyl transferase 1 (GlcAT-1) in nucleus pulposus cells through AP1, TonEBP, and Sp1: role of MAPKs. J Bone Miner Res (2010) 25(5):1179–90. doi:10.1359/jbmr.091202
130. Kanno Y, Into T, Lowenstein CJ, Matsushita K. Nitric oxide regulates vascular calcification by interfering with TGF-signalling. Cardiovasc Res (2008) 77(1):221–30. doi:10.1093/cvr/cvm049
Keywords: vascular calcification, bicarbonate, carbonic anhydrase inhibitors, magnesium, mineralocorticoid receptor, ammonium salts, osteogenic signaling, phosphate
Citation: Lang F, Leibrock C, Pelzl L, Gawaz M, Pieske B, Alesutan I and Voelkl J (2018) Therapeutic Interference With Vascular Calcification—Lessons From Klotho-Hypomorphic Mice and Beyond. Front. Endocrinol. 9:207. doi: 10.3389/fendo.2018.00207
Received: 12 February 2018; Accepted: 13 April 2018;
Published: 04 May 2018
Edited by:
Reinhold Gottfried Erben, Veterinärmedizinische Universität Wien, AustriaReviewed by:
Daniel Cejka, Krankenhaus der Elisabethinen, AustriaCopyright: © 2018 Lang, Leibrock, Pelzl, Gawaz, Pieske, Alesutan and Voelkl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Florian Lang, Zmxvcmlhbi5sYW5nQHVuaS10dWViaW5nZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.