 Matthew P. Wahl
Matthew P. Wahl Rebecca L. Scalzo
Rebecca L. Scalzo Judith G. Regensteiner
Judith G. Regensteiner Jane E. B. Reusch
Jane E. B. Reusch- 1Division of Endocrinology, University of Colorado School of Medicine, Aurora, CO, United States
- 2Veterans Administration Eastern Colorado Health Care System, Denver, CO, United States
- 3Center for Women’s Health Research, Department of Medicine, University of Colorado School of Medicine, Aurora, CO, United States
- 4Division of General Internal Medicine, University of Colorado School of Medicine, Aurora, CO, United States
The prevalence of diabetes in the United States and globally has been rapidly increasing over the last several decades. There are now estimated to be 30.3 million people in the United States and 422 million people worldwide with diabetes. Diabetes is associated with a greatly increased risk of cardiovascular mortality, which is the leading cause of death in adults with diabetes. While exercise training is a cornerstone of diabetes treatment, people with diabetes have well-described aerobic exercise impairments that may create an additional diabetes-specific barrier to adding regular exercise to their lifestyle. Physiologic mechanisms linked to exercise impairment in diabetes include insulin resistance, cardiac abnormalities, mitochondrial function, and the ability of the body to supply oxygen. In this paper, we highlight the abnormalities of exercise in type 2 diabetes as well as potential therapeutic approaches.
Scope
The prevalence of diabetes is rising rapidly in the United States, with a 75% increase from 1988 to 2010 (1). There are now 30.3 million people in the United States with diabetes (2). Diabetes is associated with a greatly increased risk of cardiovascular mortality, which is the leading cause of death in adults with diabetes (2–4). In 2015, diabetes was the seventh leading cause of death in the United States (2). Total direct and indirect costs associated with diabetes globally may have been as high as 1.3 trillion USD and are expected to rise (5, 6).
Regular exercise is a cornerstone of treatment for diabetes. In the 2018 Diabetes Standards of Care publication, the American Diabetes Association recommends most adults with type 1 and type 2 diabetes (T2D) should engage in 150 min or more of moderate to vigorous intensity physical activity plus two bouts of resistance exercise per week (7). Despite the well-established health benefits of exercise, paradoxically only 51% of adults in the USA meet the recommended aerobic exercise guidelines (8). Exercise training is associated with lowering blood pressure, improving insulin sensitivity, and glucose control, improving lipoprotein profile and playing an important role in weight management among other benefits (9). More recent evidence shows that moderate to high volumes of aerobic activity are associated with significantly lower cardiovascular and overall mortality risks in both type 1 diabetes and T2D (10). In contrast, physical inactivity or sedentarism, is known to have deleterious health effects in people with diabetes (11, 12).
The focus of this review is to outline recent advances in understanding the interaction between diabetes and impairments in aerobic exercise function. There are numerous other chronic conditions that are known to impact exercise capacity including smoking (13), obesity (14, 15), and hypertension (16–18). The mechanisms underlying these impairments may overlap with or differ from those leading to impairments in people with diabetes. Past studies from our group have addressed this potential overlap by controlling for BMI, weight, activity level, and excluding potential participants with hypertension, chronic kidney disease, or smokers (19).
Clinical Impact
People with diabetes have physiologic exercise limitations and decreased cardiorespiratory fitness (CRF). More specifically, people with diabetes have approximately 20% lower maximal oxygen uptake (VO2 max) when compared with those without diabetes (20–25). This is important because reduced VO2 max is linked to increased cardiovascular mortality (11, 20, 22, 26–28). A second exercise limitation is that individuals with T2D have slower oxygen uptake kinetics with constant-load exercise (21, 29), indicating decreased ability to adapt to an acute change in the demand for oxygenation at the beginning of exercise. Our group has reported that, perhaps related to the aforementioned physiologic limitations, people with diabetes report greater perceived exertion compared with their non-diabetic counterparts; that is, exercise seems more challenging to them, even at very low-work rates (30).
We and others have characterized many individual impairments in key cardiac and vascular measures associated with CRF impairment in T2D including insulin resistance, endothelial dysfunction, decreased myocardial perfusion with exercise, abnormally increased pulmonary capillary wedge pressure (PCWP), decreased limb blood flow, and skeletal muscle mitochondrial dysfunction (20–24, 29, 31–36). CRF impairment in T2D is present in youth as well as adults, even those with recent onset diabetes and no clinically apparent cardiovascular disease (23, 24). The CRF impairment appears to be independent from effects of obesity or decreased habitual physical activity (19–24, 37–41). Due to methodologic limitations to date, we lack critical knowledge about how each of the associated abnormalities of CRF mechanistically contributes to the CRF impairments. In addition, we lack information about which of the abnormalities are potentially reversible vs non-reversible. The goal of this review is to discuss underlying mechanisms of decreased CRF in T2D and to highlight newer data suggesting a microvascular contribution to the impaired exercise capacity associated with T2D.
Exercise as Intervention
Given the overwhelming importance CRF plays in health and the importance of exercise training in diabetes management, it is critical to characterize diabetes-related physiologic exercise limitations in order to identify therapeutic strategies to improve CRF. Physical activity has been seen to prevent T2D even in the absence of weight loss (42). Multiple studies have documented that exercise training generally improves exercise function in T2D (20–22, 29, 43–49). It is less clear if a clinical intervention with exercise training reduces mortality in established T2D. For example, the Look Ahead trial did not find a survival advantage associated with lifestyle/exercise intervention (50). In this study, there was a very low-event rate (due in part to the overall decline in CV mortality in people with DM likely related to the use of improved CV risk factor modification over the time period of the study). There were, however, other benefits resulting from lifestyle modification in the Look Ahead Study including better quality of life, lower medical costs, and reduced need of diabetes medications in the intensive lifestyle intervention group (51). We have previously shown that, despite improvement in CRF after formal exercise training, exercise impairments persist in people with T2D relative to similarly trained people without T2D, possibly suggesting a mixture of modifiable and fixed defects (Table 1) (21).
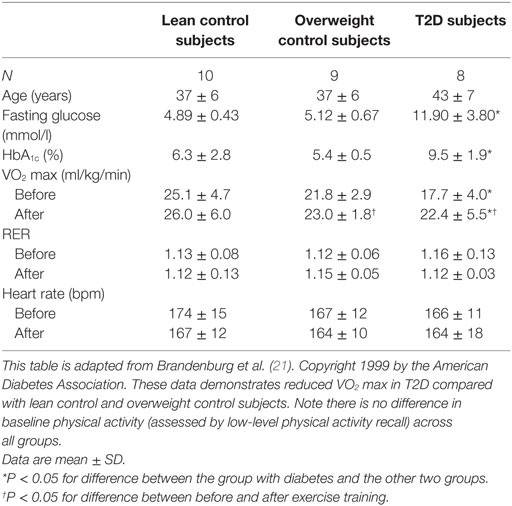
Table 1. Decreased exercise capacity in type 2 diabetes (T2D)—exercise training intervention.
Assessment of Potential Mechanistic Causes of Exercise Impairment in T2D
Previous research from our laboratory has demonstrated a significant positive correlation between insulin sensitivity and VO2 max (20, 31). As proof of concept, we elected to examine whether an insulin sensitizer has the capacity to improve exercise function. Our group investigated the effects of rosiglitazone, a thiazolidinedione (TZD), on exercise capacity in individuals with diabetes (31). TZDs augment insulin responsiveness of the skeletal muscle and adipose tissue. This study demonstrated significantly improved VO2 max (7% increase) in participants with diabetes that were treated for 4 months with rosiglitazone 4 mg/day (31). Insulin sensitivity, measured by HOMA, and endothelial function, measured by ultrasound of the brachial artery, were also significantly improved in the rosiglitazone-treated group. Since multiple physiological factors correlated with improvement in CRF, the relative contributions to improved exercise capacity from changes in insulin action and improvements in vascular function with TZD remain unclear.
Our findings with rosiglitazone and CRF were reproduced by Kadoglou et al. (52). In addition, they demonstrated an added benefit of exercise training plus the TZD (52), a finding which was recently substantiated in an animal model (53). The impact of TZD on exercise capacity appears to differ depending upon disease status, as people with T2D and cardiovascular disease did not improve VO2 max (54). Taken together, these publications support the proof of concept that targeting insulin action in diabetes is associated with improvement in VO2 max in uncomplicated T2D. Rosiglitazone is no longer commonly used in diabetes treatment, due in part to concerns that it increased risk of cardiac events [a claim the FDA later disputed (55)], and thus further research with other insulin sensitizers may be warranted.
Metformin, a known stimulator of AMP-activated protein kinase, has also been evaluated for its effects on CRF. A study published in 2008 by Braun et al. investigated the effects of metformin on exercise capacity in healthy individuals and noted metformin decreased VO2 max by 2.7% when compared with controls (56). A similar study in people with insulin resistance by HOMA IR also demonstrated a significant decrease in VO2 peak with metformin for 3 months (57). However, evaluation of the impact of metformin on VO2 peak has not been determined in people with diabetes. Limited investigations have addressed the response to metformin in people with metabolic disease and the data are mixed regarding CRF and metformin. A recent study showed a significant decrease in VO2 max in people with metabolic syndrome following 6 weeks of metformin administration (58). However, previous investigations in people with prediabetes demonstrated no effect of metformin on CRF when administered without a concomitant exercise intervention and no metformin-associated impairment of the response to an exercise training intervention (59, 60). Metformin is reported to inhibit complex 1 of the mitochondrial respiratory chain (61, 62). It has been postulated that this action of metformin may be responsible for the observed reduced CRF in people with diabetes. As metformin is first-line therapy for the treatment of diabetes, further studies are necessary to determine the impact of this widely used pharmacological agent on CRF in the primary population where it is prescribed.
Epigenetic modifications have been noted in T2D (63) and research also shows that acute exercise results in DNA hypomethylation in promoter regions in human skeletal muscle (64). There are suggestions in the literature that a physiological challenge, such as diabetes, may change these acute epigenetic responses (65). This is an area of intensive investigation under the NIH Common Fund Molecular Transducers of Physical Activity Program. Once the changes in the healthy population are understood, we will be in a better place to understand whether there are differences in the adaptive response in the context of diabetes.
Mitochondrial Dysfunction
Diabetes is associated with mitochondrial dysfunction in cardiac tissue and skeletal muscle (66, 67). In addition to other muscle tissue, we reported that DM affects mitochondrial function in the vasculature (68). This finding is pertinent because mitochondria, when dysfunctional, can decrease normal vasomotion and generate excess vascular reactive oxygen species (ROS) (69, 70). Vascular ROS are related to vascular inflammation and vascular stiffness—precursors of clinical cardiovascular disease (71). Prior studies in healthy subjects demonstrate that exercise improves skeletal muscle and vascular mitochondrial function and decreases mitochondrial damage (72–76). However, our group found that rats with hypertension or metabolic syndrome and mild diabetes did not have the mitochondrial improvement in the aorta that was demonstrated in control rats after exercise training (68). This failure of mitochondrial adaptation in the diabetic rats was unexpected and led us to consider vascular mitochondrial function and turnover as new therapeutic targets in diabetes.
To target mitochondrial dysfunction, we have explored connections between known mitochondrial regulatory pathways and established vascular consequences of diabetes. Endothelial dysfunction is an established vascular abnormality in diabetes and it is known to be regulated, in part, by endothelial nitric oxide synthase (eNOS) (77). Work by Nisoli’s group established nitric oxide (NO) to be an upstream regulator of mitochondrial biogenesis in a variety of tissues (78). We therefore examined the effects that NOS inhibition might have specifically in vascular mitochondrial biogenesis. In a 2013 publication, we noted that inhibition of eNOS blocked mitochondrial adaptation to an exercise intervention in the aorta of Sprague Dawley rats (79)—affirming the important role NO plays in vascular mitochondrial biogenesis and the adaptive response to exercise.
Pancreatic Dysfunction
Onset of exercise in individuals without diabetes is associated with decreased insulin secretion and increased glucagon secretion (80–82). Previous reports suggest that exercise is trophic for the pancreatic beta cell (83, 84). We are unaware of studies conducted to evaluate islet dysfunction and its interaction with exercise capacity. Pancreatic β cell dysfunction and failure are common culprits in the pathogenesis of diabetes. The process of aging can result in impaired carbohydrate metabolism, via both increased insulin resistance and impaired insulin secretion (85–89). Several factors have been shown to contribute to decreased insulin secretion in aging including reduced expression of β(2)-adrenergic receptors (90), decreased calcium signaling (91), and chronic oxidative stress (92). Consequently, it is plausible that aging in people with diabetes could be associated with abnormal ratios of glucagon to insulin with a subsequent impact on exercise capacity (93). It is also important to note the important role that pancreatic β cell mitochondria play in facilitating insulin secretion and that mitochondrial dysfunction in β cells is associated with beta cell failure and thus dysregulation of glucagon (94–96). This principle has been further substantiated recently in a study illustrating the deleterious effect of tacrolimus on β cell mitochondria and subsequent β cell failure (97). It is interesting to speculate that the mitochondrial impairment noted in aging and diabetes could deleteriously impact the metabolic flexibility needed for the mitochondria to adapt to an exercise challenge and further interfere with the acute adaptive response of the islet to an exercise bout. Evaluation of the islet response to exercise in people with uncomplicated diabetes is an area for future investigation.
Targeting NOS Dysfunction
To determine if it was possible to counteract NOS inhibition and improve adaptive response to exercise training, we looked to the incretin class of insulin secretagogues: glucagon-like peptide 1 receptor agonists (GLP-1 RA) and dipeptidyl peptidase 4 inhibitors (DPP4). GLP-1 signals via G-protein-coupled receptors that are highly expressed in the vasculature and have been shown to stimulate eNOS and increase cyclic AMP (cAMP)—leading to enhanced endothelial function and tissue perfusion, plus improved muscle glucose utilization (98–101). To explore the effects of incretins on exercise, vascular function, and mitochondrial adaptation in diabetes, we first examined the effects of saxagliptin (a DPP4 inhibitor) in Goto-Kakazaki rats (a lean rat model of insulin resistant diabetes). Saxagliptin combined with exercise training stimulated eNOS and restored vascular mitochondrial expression (70).
In a more recent study, we treated human subjects with uncomplicated T2D with the GLP-1 RA exenatide (102). Treatment with exenatide, without a concomitant exercise intervention, led to improvement of both diastolic cardiac function and aortic stiffness, but did not improve VO2 max or endothelial dysfunction as measured by flow mediated dilatation (102). These data suggest that an effective stand-alone intervention (without concomitant exercise intervention) would need to impact cardiac, vascular, and muscle function (the latter was not evaluated in this trial). In our clinical study, we did not measure the impact of exenatide on eNOS or NO. It is possible that eNOS dysfunction, including eNOS uncoupling in diabetes, rendered our subjects resistant to cAMP regulation of NO, or that the combination of GLP-1 RA and exercise training is needed to achieve an adaptive response and change VO2 max. These observations suggest that NO may be crucial for exercise adaptation in the vasculature. We are currently exploring other methods to target NOS dysfunction in the context of diabetes.
Other agents with “insulin sensitizing” mechanisms have been examined as exercise mimetics in non-diabetic preclinical models and healthy subjects, including AMPK stimulation and PPAR delta ligands. In a 2008 study, administration of an orally active AMPK agonist, AICAR, for 4 weeks resulted in a 44% enhancement of running endurance in sedentary mice (103). In a 2017 study, sedentary mice administered a PPAR delta ligand were able to run ~100 min longer than control mice before exhaustion, a 70% increase (104). Studies in diabetic models employing these agents are either underway or not yet conducted.
Sodium glucose transporter type 2 (SGLT2) inhibitors are a relatively new class of diabetes medications with demonstrated cardiovascular morbidity and mortality benefit in humans (105, 106). SGLT2 inhibitors have recently been found to improve insulin sensitivity in the skeletal muscle of diabetic rats (107) and in ex vivo human epicardial adipose tissue (108). SGLT inhibition with phlorizin was shown by Li et al. to improve endothelial dysfunction by way of increasing NO levels in human umbilical vein endothelial cells (109). We are not aware of any published studies directly addressing the effect of SGLT2 inhibition on CRF, except a beneficial impact in the context of heart failure (105, 106, 110). In light of the recent reports demonstrating improvements in cardiac function, aortic stiffness, and renal perfusion, it is plausible that SGLT2 inhibition will counteract the pathophysiological factors contributing to exercise impairment in diabetes (111–116). Further studies evaluating the impact of SGLT2 inhibition on insulin sensitivity, NO, and functional exercise capacity in diabetes are warranted.
Muscle Perfusion-Microvasculature
The role of NOS in vascular adaptation to sheer stress and exercise is accepted. Still, it is unclear what the consequences of impaired NOS function are on skeletal muscle and cardiac function. Importantly, it is controversial whether the skeletal muscle and heart NOS isoforms (eNOS and neuronal NOS, nNOS) contribute to exercise training adaptation or whether it is NOS mediated blood flow that contributes to the NOS-dependent adaptive response. McConnell has reported that skeletal muscle mitochondrial adaptation to exercise training is unaffected by deletion of either eNOS or nNOS (117). Another recent study indicates that alterations in muscle microvascular blood flow after an exercise bout are needed to garner the bout effect of exercise on insulin action (118). In a parallel line of investigation, Liu and Barrett report that insulin-mediated microvascular recruitment is essential for skeletal muscle insulin action and that this response requires NOS stimulation (119, 120). Taken together, these reports support a model wherein skeletal muscle insulin action involves coordination of blood flow-dependent insulin delivery to the skeletal muscle and intact skeletal muscle insulin signaling.
Similar to the relationship between muscle microvascular perfusion and insulin action, cardiac and skeletal muscle perfusion are critical for physiological function during a bout of exercise. Our previous work demonstrated decreased myocardial perfusion by sestamibi during exercise in people with T2D—consistent with abnormal tissue perfusion associated with cardiac dysfunction in T2D (19). Further, we have also reported slowed skeletal muscle blood flow at the onset of exercise in people with T2D (22). With regard to skeletal muscle mitochondrial dysfunction, a few key questions are currently unresolved: 1. What is the relationship between in vivo skeletal muscle oxidative capacity and VO2 max? 2. What is the contribution of muscle perfusion to decreased skeletal muscle oxidative capacity? 3. Is the adaptive response to either an exercise bout or exercise training in the skeletal muscle impacted by diabetes?
We postulated that skeletal muscle perfusion limitations due to either capillary rarefication or perfusion heterogeneity may contribute to the limitations observed in muscle oxidative phosphorylation in people with diabetes, and ultimately the associated CRF impairment. In support of this model, we recently published that people with diabetes have similar capillary recruitment with the onset of exercise as controls, yet have decreased oxyhemoglobin depletion during exercise compared with people without diabetes (121). We postulate that decreased skeletal muscle oxidative capacity is due to a combination of skeletal muscle mitochondrial dysfunction and muscle perfusion abnormalities including irregular blood flow distribution. Ongoing work in our laboratory is investigating the relationship between muscle O2 delivery and muscle mitochondrial function.
To add to our understanding of lower effective muscle oxygen delivery in diabetes, we conducted a recent analysis of capillary density, muscle perfusion, and muscle fatigue in collaboration with Jefferson Frisbee’s group (122). This analysis combines a theoretical modeling simulation of blood flow distribution and predicted oxygen extraction across a muscle bed with experimental data from the lean Zucker rat and the metabolic syndrome obese Zucker rat (OZR) to test the contributors to decreased muscle VO2 in metabolic syndrome. Muscle blood flow was lower in the OZR and yet venous oxygen was higher. Simply stated, despite less blood flow delivery to the muscle there was less oxygen extracted to do the same amount of work (Figure 1). Possible explanations for this could be decreased capillary density or decreased effectively perfused capillaries. Based on our simulation, the observed diabetes-related decrease in capillary density only accounted for 20% of the mismatch. The remaining defect in muscle oxygen extraction was due to uneven blood flow (red blood cell) distribution. We were able to confirm that our modeling was correct by treating with an antioxidant (TEMPOL) and observing that we were able to restore ~80% of the mismatch (123). An acute response like this would not be possible if the defect was due to a fixed lesion such as decreased capillary density. Taken together, published findings indicate that the discrepancy in muscle oxygen extraction (muscle VO2) is primarily explained by muscle perfusion heterogeneity (122). In light of the significant improvements in oxygen extraction with the TEMPOL intervention, microvascular perfusion heterogeneity represents an unaddressed therapeutic target for improving tissue oxygenation and muscle function.
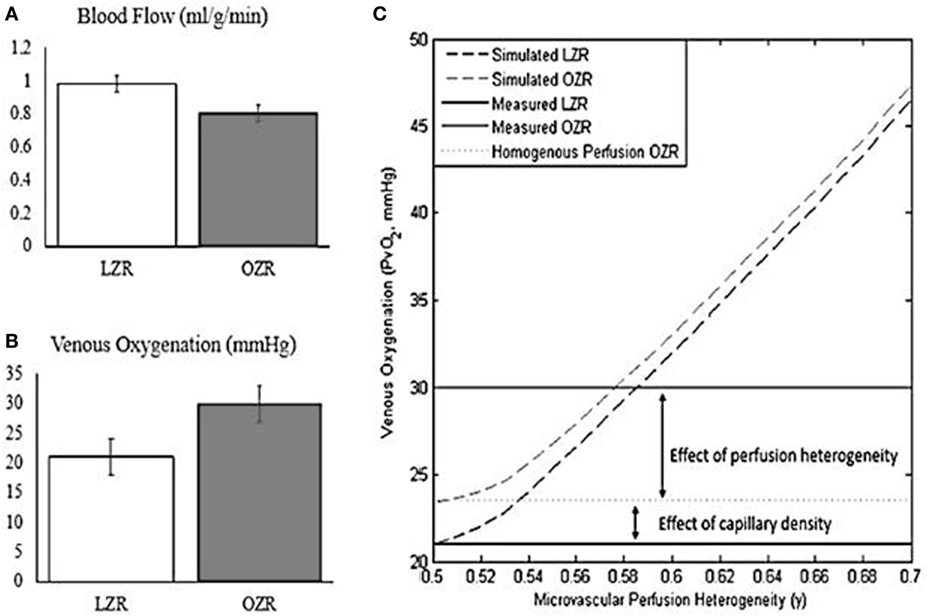
Figure 1. Oxygen extraction fails to compensate for reduced blood flow under conditions of microvascular perfusion heterogeneity. Adapted from Mason McClatchey et al. (123). Copyright 2017 Springer US. (A) Blood flow during exercise is reduced by ~20% in the obese Zucker rat (OZR) relative to the Lean Zucker Rat. (B) Venous oxygenation during exercise is increased in the OZR relative to the Lean Zucker Rat, reflecting impaired oxygen extraction. (C) Computational modeling of oxygen transport reveals that the failure to compensate for reduced blood flow in the OZR can be explained by the combined effects of reduced capillary density and microvascular perfusion heterogeneity.
To summarize, muscle perfusion abnormalities are a plausible contributor to decreased VO2 and CRF in diabetes. Recent data in humans and in rodents using TEMPOL and other agents demonstrate that abnormalities in muscle perfusion can be corrected (123). Targeting muscle perfusion is a novel therapeutic direction for improving CRF and the adaptation to exercise intervention in diabetes. Future studies should focus on the impact of interventions targeting cardiac and skeletal muscle blood flow and examining the interaction between perfusion and the adaptive exercise training response.
Cardiac Function
Studies to evaluate cardiac abnormalities using invasive and non-invasive methods revealed that cardiac abnormalities are present in newly diagnosed adults and youth with T2D upon exercise challenge (23, 24). We employed right heart catheterization to investigate cardiac function during exercise in sedentary, overweight premenopausal women with and without uncomplicated T2D. We found no differences in cardiac output at rest or during exercise (19). However, we did observe significantly increased PCWP during exercise in the T2D group suggesting the possibility of subclinical decreased ventricular contractility. This increase in PCWP correlated with decreased myocardial perfusion as assessed using sestamibi during stress testing (19). Similar differences were observed in adolescents with and without diabetes using non-invasive methods (23, 24). A clearer understanding of the long-term consequences of these subclinical CV changes is needed. In addition, it is unknown whether the agents that improve skeletal muscle perfusion will have the same impact on myocardial perfusion. Finally, the relationship between diastolic dysfunction, increased PCWP, and perceived rate of exertion may reveal an impact of cardiac dysfunction on CRF that is distinct from cardiac output. New MRI techniques may help elucidate changes and aid in evaluating the cardiac functional changes responsive to pharmacological or exercise interventions.
Summary
People with even recently diagnosed diabetes have well-described aerobic exercise impairment as evidenced by decreased CRF with both maximal and submaximal workloads when compared with similarly overweight, sedentary non-diabetic subjects. Despite improvement in CRF after exercise training, exercise impairments persist in people with T2D compared with people without T2D for reasons that are not completely clear.
Diabetes is associated with exercise-induced abnormalities in the heart, skeletal muscle, and vasculature (Figure 2). Thus, the abnormalities are multifactorial which makes it unlikely that a single therapeutic approach will alleviate the entire problem. Therefore, approaches to resolve the exercise impairment in T2D should be multifactorial and target the contributors that have been identified across the cardiac, vascular, and skeletal muscle parameters in an integrative approach.
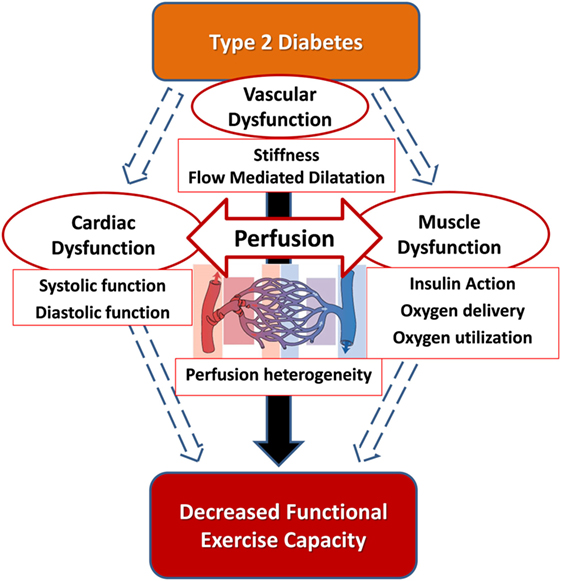
Figure 2. The hypothesis that the integration of cardiac function, macrovascular function, and microvascular function is impaired in type 2 diabetes and correlates with cardiorespiratory fitness impairment.
Author Contributions
MW drafted the manuscript and revised it. RS edited and revised the manuscript. MW and RS developed the figures/tables. JGR and JEBR outlined and edited the manuscript and contributed to generation of data in the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful for P. Mason McClatchey’s contribution to the repurposing of figures for this review. The authors’ sources of funding include: NIH-5T32HL007171 (RS), VA Merit Denver Research Institute (JEBR), CCTSI- UL1RR025780 (JEBR), and the Center for Women’s Health Research (RS, JEBR, and JGR).
References
1. Cheng YJ, Imperatore G, Geiss LS, Wang J, Saydah SH, Cowie CC, et al. Secular changes in the age-specific prevalence of diabetes among U.S. adults: 1988–2010. Diabetes Care (2013) 36(9):2690–6. doi:10.2337/dc12-2074
2. Centers for Disease Control and Prevention. National Diabetes Statistics Report, 2017. Atlanta, GA: Centers for Disease Control and Prevention, US Department of Health and Human Services (2017).
3. Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med (1998) 339(4):229–34. doi:10.1056/NEJM199807233390404
4. Emerging Risk Factors CollaborationSarwar N, Gao P, Seshasai SR, Gobin R, Kaptoge S, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet (2010) 375(9733):2215–22. doi:10.1016/S0140-6736(10)60484-9
5. Bommer C, Heesemann E, Sagalova V, Manne-Goehler J, Atun R, Barnighausen T, et al. The global economic burden of diabetes in adults aged 20–79 years: a cost-of-illness study. Lancet Diabetes Endocrinol (2017) 5(6):423–30. doi:10.1016/S2213-8587(17)30097-9
6. Bommer C, Sagalova V, Heesemann E, Manne-Goehler J, Atun R, Barnighausen T, et al. Global economic burden of diabetes in adults: projections from 2015 to 2030. Diabetes Care (2018) 41(4):dc171962. doi:10.2337/dc17-1962
7. American Diabetes Association. Standards of medical care in diabetes-2018. Diabetes Care (2018) 41(Suppl 1):S152–3. doi:10.2337/dc18-S015
8. Katzmarzyk PT, Lee IM, Martin CK, Blair SN. Epidemiology of physical activity and exercise training in the United States. Prog Cardiovasc Dis (2017) 60(1):3–10. doi:10.1016/j.pcad.2017.01.004
9. Garber CE, Blissmer B, Deschenes MR, Franklin BA, Lamonte MJ, Lee IM, et al. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc (2011) 43(7):1334–59. doi:10.1249/MSS.0b013e318213fefb
10. Sluik D, Buijsse B, Muckelbauer R, Kaaks R, Teucher B, Johnsen NF, et al. Physical activity and mortality in individuals with diabetes mellitus: a prospective study and meta-analysis. Arch Intern Med (2012) 172(17):1285–95. doi:10.1001/archinternmed.2012.3130
11. Wei M, Gibbons LW, Kampert JB, Nichaman MZ, Blair SN. Low cardiorespiratory fitness and physical inactivity as predictors of mortality in men with type 2 diabetes. Ann Intern Med (2000) 132(8):605–11. doi:10.7326/0003-4819-132-8-200004180-00035
12. Cooper AJ, Brage S, Ekelund U, Wareham NJ, Griffin SJ, Simmons RK. Association between objectively assessed sedentary time and physical activity with metabolic risk factors among people with recently diagnosed type 2 diabetes. Diabetologia (2014) 57(1):73–82. doi:10.1007/s00125-013-3069-8
13. de Borba AT, Jost RT, Gass R, Nedel FB, Cardoso DM, Pohl HH, et al. The influence of active and passive smoking on the cardiorespiratory fitness of adults. Multidiscip Respir Med (2014) 9(1):34. doi:10.1186/2049-6958-9-34
14. Mondal H, Mishra SP. Effect of BMI, body fat percentage and fat free mass on maximal oxygen consumption in healthy young adults. J Clin Diagn Res (2017) 11(6):CC17–20. doi:10.7860/JCDR/2017/25465.10039
15. Dumuid D, Lewis LK, Olds TS, Maher C, Bondarenko C, Norton L. Relationships between older adults’ use of time and cardio-respiratory fitness, obesity and cardio-metabolic risk: a compositional isotemporal substitution analysis. Maturitas (2018) 110:104–10. doi:10.1016/j.maturitas.2018.02.003
16. Yoon JH, So WY. Associations of hypertension status with physical fitness variables in Korean women. Iran J Public Health (2013) 42(7):673–80.
17. Mazic S, Suzic Lazic J, Dekleva M, Antic M, Soldatovic I, Djelic M, et al. The impact of elevated blood pressure on exercise capacity in elite athletes. Int J Cardiol (2015) 180:171–7. doi:10.1016/j.ijcard.2014.11.125
18. Jae SY, Kurl S, Franklin BA, Laukkanen JA. Changes in cardiorespiratory fitness predict incident hypertension: a population-based long-term study. Am J Hum Biol (2017) 29(3):e22932. doi:10.1002/ajhb.22932
19. Regensteiner JG, Bauer TA, Reusch JE, Quaife RA, Chen MY, Smith SC, et al. Cardiac dysfunction during exercise in uncomplicated type 2 diabetes. Med Sci Sports Exerc (2009) 41(5):977–84. doi:10.1249/MSS.0b013e3181942051
20. Regensteiner JG, Sippel J, McFarling ET, Wolfel EE, Hiatt WR. Effects of non-insulin-dependent diabetes on oxygen consumption during treadmill exercise. Med Sci Sports Exerc (1995) 27(6):875–81. doi:10.1249/00005768-199505000-00007
21. Brandenburg SL, Reusch JE, Bauer TA, Jeffers BW, Hiatt WR, Regensteiner JG. Effects of exercise training on oxygen uptake kinetic responses in women with type 2 diabetes. Diabetes Care (1999) 22(10):1640–6. doi:10.2337/diacare.22.10.1640
22. Bauer TA, Reusch JE, Levi M, Regensteiner JG. Skeletal muscle deoxygenation following the onset of moderate exercise suggests slowed microvascular blood flow kinetics in type 2 diabetes. Diabetes Care (2007) 30(11):2880–5. doi:10.2337/dc07-0843
23. Nadeau KJ, Zeitler PS, Bauer TA, Brown MS, Dorosz JL, Draznin B, et al. Insulin resistance in adolescents with type 2 diabetes is associated with impaired exercise capacity. J Clin Endocrinol Metab (2009) 94(10):3687–95. doi:10.1210/jc.2008-2844
24. Nadeau KJ, Regensteiner JG, Bauer TA, Brown MS, Dorosz JL, Hull A, et al. Insulin resistance in adolescents with type 1 diabetes and its relationship to cardiovascular function. J Clin Endocrinol Metab (2010) 95(2):513–21. doi:10.1210/jc.2009-1756
25. Ross R, Blair SN, Arena R, Church TS, Despres JP, Franklin BA, et al. Importance of assessing cardiorespiratory fitness in clinical practice: a case for fitness as a clinical vital sign: a scientific statement from the American Heart Association. Circulation (2016) 134(24):e653–99. doi:10.1161/CIR.0000000000000461
26. Lee CD, Blair SN, Jackson AS. Cardiorespiratory fitness, body composition, and all-cause and cardiovascular disease mortality in men. Am J Clin Nutr (1999) 69(3):373–80. doi:10.1093/ajcn/69.3.373
27. Wei M, Kampert JB, Barlow CE, Nichaman MZ, Gibbons LW, Paffenbarger RS Jr, et al. Relationship between low cardiorespiratory fitness and mortality in normal-weight, overweight, and obese men. JAMA (1999) 282(16):1547–53. doi:10.1001/jama.282.16.1547
28. Weintraub WS, Daniels SR, Burke LE, Franklin BA, Goff DC Jr, Hayman LL, et al. Value of primordial and primary prevention for cardiovascular disease: a policy statement from the American Heart Association. Circulation (2011) 124(8):967–90. doi:10.1161/CIR.0b013e3182285a81
29. Regensteiner JG, Bauer TA, Reusch JE, Brandenburg SL, Sippel JM, Vogelsong AM, et al. Abnormal oxygen uptake kinetic responses in women with type II diabetes mellitus. J Appl Physiol (1985) (1998) 85(1):310–7. doi:10.1152/jappl.1998.85.1.310
30. Huebschmann AG, Reis EN, Emsermann C, Dickinson LM, Reusch JE, Bauer TA, et al. Women with type 2 diabetes perceive harder effort during exercise than nondiabetic women. Appl Physiol Nutr Metab (2009) 34(5):851–7. doi:10.1139/H09-074
31. Regensteiner JG, Bauer TA, Reusch JE. Rosiglitazone improves exercise capacity in individuals with type 2 diabetes. Diabetes Care (2005) 28(12):2877–83. doi:10.2337/diacare.28.12.2877
32. Subudhi AW, Dimmen AC, Roach RC. Effects of acute hypoxia on cerebral and muscle oxygenation during incremental exercise. J Appl Physiol (1985) (2007) 103(1):177–83. doi:10.1152/japplphysiol.01460.2006
33. MacAnaney O, Reilly H, O’Shea D, Egana M, Green S. Effect of type 2 diabetes on the dynamic response characteristics of leg vascular conductance during exercise. Diab Vasc Dis Res (2011) 8(1):12–21. doi:10.1177/1479164110389625
34. Jones WS, Duscha BD, Robbins JL, Duggan NN, Regensteiner JG, Kraus WE, et al. Alteration in angiogenic and anti-angiogenic forms of vascular endothelial growth factor-A in skeletal muscle of patients with intermittent claudication following exercise training. Vasc Med (2012) 17(2):94–100. doi:10.1177/1358863X11436334
35. Macananey O, O’Shea D, Warmington SA, Green S, Egana M. Gymnasium-based unsupervised exercise maintains benefits in oxygen uptake kinetics obtained following supervised training in type 2 diabetes. Appl Physiol Nutr Metab (2012) 37(4):599–609. doi:10.1139/h2012-012
36. Huebschmann AG, Kohrt WM, Herlache L, Wolfe P, Daugherty S, Reusch JE, et al. Type 2 diabetes exaggerates exercise effort and impairs exercise performance in older women. BMJ Open Diabetes Res Care (2015) 3(1):e000124. doi:10.1136/bmjdrc-2015-000124
37. Schneider SH, Amorosa LF, Khachadurian AK, Ruderman NB. Studies on the mechanism of improved glucose control during regular exercise in type 2 (non-insulin-dependent) diabetes. Diabetologia (1984) 26(5):355–60. doi:10.1007/BF00266036
38. Kjaer M, Hollenbeck CB, Frey-Hewitt B, Galbo H, Haskell W, Reaven GM. Glucoregulation and hormonal responses to maximal exercise in non-insulin-dependent diabetes. J Appl Physiol (1985) (1990) 68(5):2067–74. doi:10.1152/jappl.1990.68.5.2067
39. Mac Ananey O, Malone J, Warmington S, O’Shea D, Green S, Egana M. Cardiac output is not related to the slowed O2 uptake kinetics in type 2 diabetes. Med Sci Sports Exerc (2011) 43(6):935–42. doi:10.1249/MSS.0b013e3182061cdb
40. Gusso S, Hofman P, Lalande S, Cutfield W, Robinson E, Baldi JC. Impaired stroke volume and aerobic capacity in female adolescents with type 1 and type 2 diabetes mellitus. Diabetologia (2008) 51(7):1317–20. doi:10.1007/s00125-008-1012-1
41. Wilkerson DP, Poole DC, Jones AM, Fulford J, Mawson DM, Ball CI, et al. Older type 2 diabetic males do not exhibit abnormal pulmonary oxygen uptake and muscle oxygen utilization dynamics during submaximal cycling exercise. Am J Physiol Regul Integr Comp Physiol (2011) 300(3):R685–92. doi:10.1152/ajpregu.00479.2010
42. Hamman RF, Wing RR, Edelstein SL, Lachin JM, Bray GA, Delahanty L, et al. Effect of weight loss with lifestyle intervention on risk of diabetes. Diabetes Care (2006) 29(9):2102–7. doi:10.2337/dc06-0560
43. Bouchard C, Leon AS, Rao DC, Skinner JS, Wilmore JH, Gagnon J. The HERITAGE family study. Aims, design, and measurement protocol. Med Sci Sports Exerc (1995) 27(5):721–9. doi:10.1249/00005768-199505000-00015
44. Morss GM, Jordan AN, Skinner JS, Dunn AL, Church TS, Earnest CP, et al. Dose response to exercise in women aged 45–75 yr (DREW): design and rationale. Med Sci Sports Exerc (2004) 36(2):336–44. doi:10.1249/01.MSS.0000113738.06267.E5
45. Slentz CA, Aiken LB, Houmard JA, Bales CW, Johnson JL, Tanner CJ, et al. Inactivity, exercise, and visceral fat. STRRIDE: a randomized, controlled study of exercise intensity and amount. J Appl Physiol (1985) (2005) 99(4):1613–8. doi:10.1152/japplphysiol.00124.2005
46. Thompson AM, Mikus CR, Rodarte RQ, Distefano B, Priest EL, Sinclair E, et al. Inflammation and exercise (INFLAME): study rationale, design, and methods. Contemp Clin Trials (2008) 29(3):418–27. doi:10.1016/j.cct.2007.09.009
47. Sisson SB, Katzmarzyk PT, Earnest CP, Bouchard C, Blair SN, Church TS. Volume of exercise and fitness nonresponse in sedentary, postmenopausal women. Med Sci Sports Exerc (2009) 41(3):539–45. doi:10.1249/MSS.0b013e3181896c4e
48. Regensteiner JG, Bauer TA, Huebschmann AG, Herlache L, Weinberger HD, Wolfel EE, et al. Sex differences in the effects of type 2 diabetes on exercise performance. Med Sci Sports Exerc (2015) 47(1):58–65. doi:10.1249/MSS.0000000000000371
49. Ring M, Eriksson MJ, Fritz T, Nyberg G, Ostenson CG, Krook A, et al. Influence of physical activity and gender on arterial function in type 2 diabetes, normal and impaired glucose tolerance. Diab Vasc Dis Res (2015) 12(5):315–24. doi:10.1177/1479164115588548
50. Look ARG, Wing RR, Bolin P, Brancati FL, Bray GA, Clark JM, et al. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med (2013) 369(2):145–54. doi:10.1056/NEJMoa1212914
51. Pi-Sunyer X. The look AHEAD trial: a review and discussion of its outcomes. Curr Nutr Rep (2014) 3(4):387–91. doi:10.1007/s13668-014-0099-x
52. Kadoglou NP, Iliadis F, Sailer N, Athanasiadou Z, Vitta I, Kapelouzou A, et al. Exercise training ameliorates the effects of rosiglitazone on traditional and novel cardiovascular risk factors in patients with type 2 diabetes mellitus. Metabolism (2010) 59(4):599–607. doi:10.1016/j.metabol.2009.09.002
53. Kim JC. The effect of exercise training combined with PPARgamma agonist on skeletal muscle glucose uptake and insulin sensitivity in induced diabetic obese Zucker rats. J Exerc Nutrition Biochem (2016) 20(2):42–50. doi:10.20463/jenb.2016.06.20.2.6
54. McGuire DK, Abdullah SM, See R, Snell PG, McGavock J, Szczepaniak LS, et al. Randomized comparison of the effects of rosiglitazone vs. placebo on peak integrated cardiovascular performance, cardiac structure, and function. Eur Heart J (2010) 31(18):2262–70. doi:10.1093/eurheartj/ehq228
55. Hiatt WR, Kaul S, Smith RJ. The cardiovascular safety of diabetes drugs—insights from the rosiglitazone experience. N Engl J Med (2013) 369(14):1285–7. doi:10.1056/NEJMp1309610
56. Braun B, Eze P, Stephens BR, Hagobian TA, Sharoff CG, Chipkin SR, et al. Impact of metformin on peak aerobic capacity. Appl Physiol Nutr Metab (2008) 33(1):61–7. doi:10.1139/H07-144
57. Cadeddu C, Nocco S, Deidda M, Cadeddu F, Bina A, Demuru P, et al. Cardiopulmonary and endothelial effects of metformin treatment in an insulin resistant population. Int J Cardiol (2012) 158(2):302–4. doi:10.1016/j.ijcard.2012.04.113
58. Paul AA, Dkhar SA, Kamalanathan S, Thabah MM, George M, Chandrasekaran I, et al. Effect of metformin on exercise capacity in metabolic syndrome. Diabetes Metab Syndr (2017) 11(Suppl 1):S403–6. doi:10.1016/j.dsx.2017.03.025
59. Malin SK, Gerber R, Chipkin SR, Braun B. Independent and combined effects of exercise training and metformin on insulin sensitivity in individuals with prediabetes. Diabetes Care (2012) 35(1):131–6. doi:10.2337/dc11-0925
60. Cadeddu C, Nocco S, Lucia C, Deidda M, Bina A, Fabio O, et al. Effects of metformin and exercise training, alone or in association, on cardio-pulmonary performance and quality of life in insulin resistance patients. Cardiovasc Diabetol (2014) 13:93. doi:10.1186/1475-2840-13-93
61. Leverve XM, Guigas B, Detaille D, Batandier C, Koceir EA, Chauvin C, et al. Mitochondrial metabolism and type-2 diabetes: a specific target of metformin. Diabetes Metab (2003) 29(4 Pt 2):S88–94. doi:10.1016/S1262-3636(03)72792-X
62. Batandier C, Guigas B, Detaille D, El-Mir MY, Fontaine E, Rigoulet M, et al. The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J Bioenerg Biomembr (2006) 38(1):33–42. doi:10.1007/s10863-006-9003-8
63. Barres R, Osler ME, Yan J, Rune A, Fritz T, Caidahl K, et al. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab (2009) 10(3):189–98. doi:10.1016/j.cmet.2009.07.011
64. Barres R, Yan J, Egan B, Treebak JT, Rasmussen M, Fritz T, et al. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab (2012) 15(3):405–11. doi:10.1016/j.cmet.2012.01.001
65. Sparks LM. Exercise training response heterogeneity: physiological and molecular insights. Diabetologia (2017) 60(12):2329–36. doi:10.1007/s00125-017-4461-6
66. Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes (2002) 51(10):2944–50. doi:10.2337/diabetes.51.10.2944
67. Bugger H, Abel ED. Mitochondria in the diabetic heart. Cardiovasc Res (2010) 88(2):229–40. doi:10.1093/cvr/cvq239
68. Knaub LA, McCune S, Chicco AJ, Miller M, Moore RL, Birdsey N, et al. Impaired response to exercise intervention in the vasculature in metabolic syndrome. Diab Vasc Dis Res (2013) 10(3):222–38. doi:10.1177/1479164112459664
69. Davidson SM. Endothelial mitochondria and heart disease. Cardiovasc Res (2010) 88(1):58–66. doi:10.1093/cvr/cvq195
70. Keller AC, Knaub LA, Miller MW, Birdsey N, Klemm DJ, Reusch JE. Saxagliptin restores vascular mitochondrial exercise response in the Goto-Kakizaki rat. J Cardiovasc Pharmacol (2015) 65(2):137–47. doi:10.1097/FJC.0000000000000170
71. Keller AC, Knaub LA, McClatchey PM, Connon CA, Bouchard R, Miller MW, et al. Differential mitochondrial adaptation in primary vascular smooth muscle cells from a diabetic rat model. Oxid Med Cell Longev (2016) 2016:8524267. doi:10.1155/2016/8524267
72. Brierley EJ, Johnson MA, James OF, Turnbull DM. Effects of physical activity and age on mitochondrial function. QJM (1996) 89(4):251–8. doi:10.1093/qjmed/89.4.251
73. Spina RJ, Chi MM, Hopkins MG, Nemeth PM, Lowry OH, Holloszy JO. Mitochondrial enzymes increase in muscle in response to 7–10 days of cycle exercise. J Appl Physiol (1985) (1996) 80(6):2250–4. doi:10.1152/jappl.1996.80.6.2250
74. Starritt EC, Angus D, Hargreaves M. Effect of short-term training on mitochondrial ATP production rate in human skeletal muscle. J Appl Physiol (1985) (1999) 86(2):450–4. doi:10.1152/jappl.1999.86.2.450
75. Ji LL. Exercise-induced modulation of antioxidant defense. Ann N Y Acad Sci (2002) 959:82–92. doi:10.1111/j.1749-6632.2002.tb02085.x
76. Young CG, Knight CA, Vickers KC, Westbrook D, Madamanchi NR, Runge MS, et al. Differential effects of exercise on aortic mitochondria. Am J Physiol Heart Circ Physiol (2005) 288(4):H1683–9. doi:10.1152/ajpheart.00136.2004
77. Montagnani M, Chen H, Barr VA, Quon MJ. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser(1179). J Biol Chem (2001) 276(32):30392–8. doi:10.1074/jbc.M103702200
78. Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science (2003) 299(5608):896–9. doi:10.1126/science.1079368
79. Miller MW, Knaub LA, Olivera-Fragoso LF, Keller AC, Balasubramaniam V, Watson PA, et al. Nitric oxide regulates vascular adaptive mitochondrial dynamics. Am J Physiol Heart Circ Physiol (2013) 304(12):H1624–33. doi:10.1152/ajpheart.00987.2012
80. Krotkiewski M, Lonnroth P, Mandroukas K, Wroblewski Z, Rebuffe-Scrive M, Holm G, et al. The effects of physical training on insulin secretion and effectiveness and on glucose metabolism in obesity and type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia (1985) 28(12):881–90. doi:10.1007/BF00703130
81. Kirwan JP, Kohrt WM, Wojta DM, Bourey RE, Holloszy JO. Endurance exercise training reduces glucose-stimulated insulin levels in 60- to 70-year-old men and women. J Gerontol (1993) 48(3):M84–90. doi:10.1093/geronj/48.3.M84
82. Marliss EB, Vranic M. Intense exercise has unique effects on both insulin release and its roles in glucoregulation: implications for diabetes. Diabetes (2002) 51(Suppl 1):S271–83. doi:10.2337/diabetes.51.2007.S271
83. Kiraly MA, Bates HE, Kaniuk NA, Yue JT, Brumell JH, Matthews SG, et al. Swim training prevents hyperglycemia in ZDF rats: mechanisms involved in the partial maintenance of beta-cell function. Am J Physiol Endocrinol Metab (2008) 294(2):E271–83. doi:10.1152/ajpendo.00476.2007
84. Park S, Hong SM, Sung SR. Exendin-4 and exercise promotes beta-cell function and mass through IRS2 induction in islets of diabetic rats. Life Sci (2008) 82(9–10):503–11. doi:10.1016/j.lfs.2007.12.018
85. DeFronzo RA. Glucose intolerance and aging. Diabetes Care (1981) 4(4):493–501. doi:10.2337/diacare.4.4.493
86. Fink RI, Kolterman OG, Griffin J, Olefsky JM. Mechanisms of insulin resistance in aging. J Clin Invest (1983) 71(6):1523–35. doi:10.1172/JCI110908
87. Iozzo P, Beck-Nielsen H, Laakso M, Smith U, Yki-Jarvinen H, Ferrannini E. Independent influence of age on basal insulin secretion in nondiabetic humans. European Group for the Study of Insulin Resistance. J Clin Endocrinol Metab (1999) 84(3):863–8. doi:10.1210/jcem.84.3.5542
88. Szoke E, Shrayyef MZ, Messing S, Woerle HJ, van Haeften TW, Meyer C, et al. Effect of aging on glucose homeostasis: accelerated deterioration of beta-cell function in individuals with impaired glucose tolerance. Diabetes Care (2008) 31(3):539–43. doi:10.2337/dc07-1443
89. De Tata V. Age-related impairment of pancreatic beta-cell function: pathophysiological and cellular mechanisms. Front Endocrinol (2014) 5:138. doi:10.3389/fendo.2014.00138
90. Santulli G, Lombardi A, Sorriento D, Anastasio A, Del Giudice C, Formisano P, et al. Age-related impairment in insulin release: the essential role of beta(2)-adrenergic receptor. Diabetes (2012) 61(3):692–701. doi:10.2337/db11-1027
91. Lin Y, Sun Z. Antiaging gene Klotho enhances glucose-induced insulin secretion by up-regulating plasma membrane levels of TRPV2 in MIN6 beta-cells. Endocrinology (2012) 153(7):3029–39. doi:10.1210/en.2012-1091
92. Bonomini F, Rodella LF, Rezzani R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis (2015) 6(2):109–20. doi:10.14336/AD.2014.0305
93. Rogers MA, Hagberg JM, Martin WH III, Ehsani AA, Holloszy JO. Decline in VO2max with aging in master athletes and sedentary men. J Appl Physiol (1985) (1990) 68(5):2195–9. doi:10.1152/jappl.1990.68.5.2195
94. Ishihara H, Maechler P, Gjinovci A, Herrera PL, Wollheim CB. Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells. Nat Cell Biol (2003) 5(4):330–5. doi:10.1038/ncb951
95. Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science (2005) 307(5708):384–7. doi:10.1126/science.1104343
96. Lu H, Koshkin V, Allister EM, Gyulkhandanyan AV, Wheeler MB. Molecular and metabolic evidence for mitochondrial defects associated with beta-cell dysfunction in a mouse model of type 2 diabetes. Diabetes (2010) 59(2):448–59. doi:10.2337/db09-0129
97. Lombardi A, Trimarco B, Iaccarino G, Santulli G. Impaired mitochondrial calcium uptake caused by tacrolimus underlies beta-cell failure. Cell Commun Signal (2017) 15(1):47. doi:10.1186/s12964-017-0203-0
98. Gros R, You X, Baggio LL, Kabir MG, Sadi AM, Mungrue IN, et al. Cardiac function in mice lacking the glucagon-like peptide-1 receptor. Endocrinology (2003) 144(6):2242–52. doi:10.1210/en.2003-0007
99. Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation (2008) 117(18):2340–50. doi:10.1161/CIRCULATIONAHA.107.739938
100. Chai W, Dong Z, Wang N, Wang W, Tao L, Cao W, et al. Glucagon-like peptide 1 recruits microvasculature and increases glucose use in muscle via a nitric oxide-dependent mechanism. Diabetes (2012) 61(4):888–96. doi:10.2337/db11-1073
101. Dong Z, Chai W, Wang W, Zhao L, Fu Z, Cao W, et al. Protein kinase A mediates glucagon-like peptide 1-induced nitric oxide production and muscle microvascular recruitment. Am J Physiol Endocrinol Metab (2013) 304(2):E222–8. doi:10.1152/ajpendo.00473.2012
102. Scalzo RL, Moreau KL, Ozemek C, Herlache L, McMillin S, Gilligan S, et al. Exenatide improves diastolic function and attenuates arterial stiffness but does not alter exercise capacity in individuals with type 2 diabetes. J Diabetes Complications (2017) 31(2):449–55. doi:10.1016/j.jdiacomp.2016.10.003
103. Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell (2008) 134(3):405–15. doi:10.1016/j.cell.2008.06.051
104. Fan W, Waizenegger W, Lin CS, Sorrentino V, He MX, Wall CE, et al. PPARdelta promotes running endurance by preserving glucose. Cell Metab (2017) 25(5):1186–93.e4. doi:10.1016/j.cmet.2017.04.006
105. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med (2015) 373(22):2117–28. doi:10.1056/NEJMoa1504720
106. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med (2017) 377(7):644–57. doi:10.1056/NEJMoa1611925
107. O’Brien TP, Jenkins EC, Estes SK, Castaneda AV, Ueta K, Farmer TD, et al. Correcting postprandial hyperglycemia in Zucker diabetic fatty rats with an SGLT2 inhibitor restores glucose effectiveness in the liver and reduces insulin resistance in skeletal muscle. Diabetes (2017) 66(5):1172–84. doi:10.2337/db16-1410
108. Diaz-Rodriguez E, Agra RM, Fernandez AL, Adrio B, Garcia-Caballero T, Gonzalez-Juanatey JR, et al. Effects of dapagliflozin on human epicardial adipose tissue: modulation of insulin resistance, inflammatory chemokine production, and differentiation ability. Cardiovasc Res (2018) 114(2):336–46. doi:10.1093/cvr/cvx186
109. Li CY, Wang LX, Dong SS, Hong Y, Zhou XH, Zheng WW, et al. Phlorizin exerts direct protective effects on palmitic acid (PA)-induced endothelial dysfunction by activating the PI3K/AKT/eNOS signaling pathway and increasing the levels of nitric oxide (NO). Med Sci Monit Basic Res (2018) 24:1–9. doi:10.12659/MSMBR.907775
110. Radholm K, Figtree G, Perkovic V, Solomon SD, Mahaffey KW, de Zeeuw D, et al. Canagliflozin and heart failure in type 2 diabetes mellitus: results from the CANVAS program (Canagliflozin Cardiovascular Assessment Study). Circulation (2018). doi:10.1161/CIRCULATIONAHA.118.034222
111. Cherney DZ, Perkins BA, Soleymanlou N, Har R, Fagan N, Johansen OE, et al. The effect of empagliflozin on arterial stiffness and heart rate variability in subjects with uncomplicated type 1 diabetes mellitus. Cardiovasc Diabetol (2014) 13:28. doi:10.1186/1475-2840-13-28
112. Chilton R, Tikkanen I, Cannon CP, Crowe S, Woerle HJ, Broedl UC, et al. Effects of empagliflozin on blood pressure and markers of arterial stiffness and vascular resistance in patients with type 2 diabetes. Diabetes Obes Metab (2015) 17(12):1180–93. doi:10.1111/dom.12572
113. Fioretto P, Zambon A, Rossato M, Busetto L, Vettor R. SGLT2 inhibitors and the diabetic kidney. Diabetes Care (2016) 39(Suppl 2):S165–71. doi:10.2337/dcS15-3006
114. Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med (2016) 375(4):323–34. doi:10.1056/NEJMoa1515920
115. Habibi J, Aroor AR, Sowers JR, Jia G, Hayden MR, Garro M, et al. Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc Diabetol (2017) 16(1):9. doi:10.1186/s12933-016-0489-z
116. Packer M, Anker SD, Butler J, Filippatos G, Zannad F. Effects of sodium-glucose cotransporter 2 inhibitors for the treatment of patients with heart failure: proposal of a novel mechanism of action. JAMA Cardiol (2017) 2(9):1025–9. doi:10.1001/jamacardio.2017.2275
117. Wadley GD, Choate J, McConell GK. NOS isoform-specific regulation of basal but not exercise-induced mitochondrial biogenesis in mouse skeletal muscle. J Physiol (2007) 585(Pt 1):253–62. doi:10.1113/jphysiol.2007.141309
118. Sjoberg KA, Frosig C, Kjobsted R, Sylow L, Kleinert M, Betik AC, et al. Exercise increases human skeletal muscle insulin sensitivity via coordinated increases in microvascular perfusion and molecular signaling. Diabetes (2017) 66(6):1501–10. doi:10.2337/db16-1327
119. Barrett EJ, Eggleston EM, Inyard AC, Wang H, Li G, Chai W, et al. The vascular actions of insulin control its delivery to muscle and regulate the rate-limiting step in skeletal muscle insulin action. Diabetologia (2009) 52(5):752–64. doi:10.1007/s00125-009-1313-z
120. Wang H, Wang AX, Aylor K, Barrett EJ. Nitric oxide directly promotes vascular endothelial insulin transport. Diabetes (2013) 62(12):4030–42. doi:10.2337/db13-0627
121. Mason McClatchey P, Bauer TA, Regensteiner JG, Schauer IE, Huebschmann AG, Reusch JEB. Dissociation of local and global skeletal muscle oxygen transport metrics in type 2 diabetes. J Diabetes Complications (2017) 31(8):1311–7. doi:10.1016/j.jdiacomp.2017.05.004
122. McClatchey PM, Frisbee JC, Reusch JEB. A conceptual framework for predicting and addressing the consequences of disease-related microvascular dysfunction. Microcirculation (2017) 24(6):e12359. doi:10.1111/micc.12359
Keywords: insulin resistance, mitochondrial dysfunction, nitric oxide synthase, microvascular, muscle perfusion
Citation: Wahl MP, Scalzo RL, Regensteiner JG and Reusch JEB (2018) Mechanisms of Aerobic Exercise Impairment in Diabetes: A Narrative Review. Front. Endocrinol. 9:181. doi: 10.3389/fendo.2018.00181
Received: 26 January 2018; Accepted: 04 April 2018;
Published: 18 April 2018
Edited by:
Jan Polák, Charles University, CzechiaReviewed by:
Hidetaka Hamasaki, Hamasaki Clinic, JapanAngela Lombardi, Albert Einstein College of Medicine, United States
Copyright: © 2018 Wahl, Scalzo, Regensteiner and Reusch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jane E. B. Reusch, amFuZS5yZXVzY2gmI3gwMDA0MDt1Y2RlbnZlci5lZHU=