
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 20 April 2018
Sec. Cancer Endocrinology
Volume 9 - 2018 | https://doi.org/10.3389/fendo.2018.00178
This article is part of the Research Topic Personalized Medicine in Cancer Research View all 7 articles
 Caterina Mancarella1*
Caterina Mancarella1* Michela Pasello1
Michela Pasello1 Maria Cristina Manara1
Maria Cristina Manara1 Lisa Toracchio1
Lisa Toracchio1 Evelina Fiorenza Sciandra1
Evelina Fiorenza Sciandra1 Piero Picci1,2
Piero Picci1,2 Katia Scotlandi1*
Katia Scotlandi1*
Insulin-like growth factor 2 (IGF2) mRNA-binding protein 3 (IGF2BP3) is an oncofetal protein that binds RNA, thereby influencing the fate of target transcripts. IGF2BP3 is synthesized de novo in cancer, where it promotes proliferation, drug resistance, and metastasis via both IGF2-dependent and IGF2-independent mechanisms. Ewing sarcoma (ES) is a rare bone and soft tissue tumor in which the IGF system plays a pivotal role. This study aimed to investigate the effect of IGF2BP3 on the regulation of the IGF system in ES. Among the components of the IGF axis, a direct significant correlation was identified between IGF2BP3 and IGF1R at mRNA and protein levels in two independent series of clinical specimens from patients with localized ES. After the formal demonstration of a direct association between IGF2BP3 and IGF1R mRNA using ribo-immunoprecipitation assay, we performed in vitro studies using A673 and TC-71 ES cell lines to demonstrate that IGF2BP3 loss promotes the downregulation of IGF1R and a decreased biological response to IGF1, represented by reduced migration and cell growth. Additionally, the compensatory activation of insulin receptor (IR) and its mitogenic ligand IGF2 is triggered in some but not all cell lines in response to IGF2BP3-mediated IGF1R loss. These findings have therapeutic implications because cells with a decreased expression of IGF2BP3/IGF1R axis but an increased expression of the IR/IGF2 loop display higher sensitivity to the dual inhibitor OSI-906 than do control cells. Therefore, studies on IGF2BP3, which was confirmed as a post-transcriptional regulator of IGF1R, provide a step forward in the identification of new mechanisms regulating the IGF system. In addition, our results demonstrate that the detection of IGF2BP3 expression should be combined with the assessment of the IGF1R/IR ratio to predict cell responses to anti-IGF1R/IR agents.
Insulin-like growth factor 2 (IGF2) mRNA-binding proteins (IGF2BPs) represent a family of oncofetal RNA-binding proteins (RBPs), including the paralogs IGF2BP1, 2, and 3, which control the localization, translation, and stability of mRNAs (1). IGF2BPs are mainly expressed during embryogenesis and absent in adult tissues (2, 3), but in vitro studies have demonstrated that IGF2BP1 and 3 are synthesized de novo in cancer, where they act as oncogenes promoting malignant processes including cell polarization, migration, morphological determination, proliferation, differentiation, and drug sensitivity (4–8). Structurally, the three proteins share 56% amino acid sequence homology and are characterized by two RNA-recognition motifs in the N-terminal region and four hnRNP-K homology domains in the C-terminal region, which are particularly involved in the binding, at 5′- or 3′-untranslated regions, of targets RNAs (9). IGF2BPs are located in the cytoplasm, mainly around the nucleus and in cell protrusions, as ribonucleoprotein granules acting as “safe-houses” for transcript targets (10). The comprehensive identification of IGF2BP targets is still under investigation, but the network of RNAs whose stability is regulated by each IGF2BP may predict the phenotype of IGF2BP-expressing cells and determine the involvement of these proteins in various crucial cellular functions [reviewed in Ref. (6, 11, 12)]. Each IGF2BP exhibits distinct RNA-binding properties and associates with variable targets, but all IGF2BPs are reported to associate with IGF2 mRNA (2). IGF2, together with IGF1 and insulin, represents extracellular ligand belonging to the IGF system that also includes three receptors [insulin receptor (IR), IGF1R, and mannose 6-phosphate receptor (M6P/IGF2R)], and six known types of circulating IGF-binding proteins (13). Upon ligand binding, the kinase domains of the receptors are activated, and phosphorylation cascades are initiated through the PI3K (14) or MAPK (15) pathway with an impact on vital processes under physiological as well as pathological conditions including cancer. In tumor cells, the IGF system drives cell proliferation, differentiation, and survival during anoikis and after conventional and targeted therapies [for reviews, see Ref. (16, 17)]. Thus, IGF signaling, particularly involving IGF1R, has become an attractive target for the development of novel anticancer agents.
Among the IGF2BPs, IGF2BP3 has been particularly described to favor IGF2 translation, thereby activating IGF signaling and promoting cell growth, proliferation, and resistance to ionic irradiation in different tumor types (18–20). Therefore, in this study, we investigated the value of IGF2BP3 as a novel regulator of the IGF system in Ewing sarcoma (ES), the second most frequent primary tumor of the bone affecting children and adolescents. The clinical history of ES remains disappointing because, despite an overall cure of approximately 70% in cases with localized disease, survival is lower than 30% in patients with metastatic disease. In addition, the reduction of severe treatment-related side effects still represents an urgent clinical need (21, 22). However, the discovery of novel treatment opportunities may not be imminent because ES is characterized by a recurrent EWS/ETS chromosomal translocation (EWS-FLI1 in most of the cases) but otherwise an overall stable genome (23), leading to a general paucity of druggable mutations. The IGF system is a key participant in ES development and progression (24–28), and the functions of this system are mainly sustained by the autocrine production of IGF1, which induces constitutive IGF1R activation. The clinical use of agents blocking this axis has demonstrated remarkable efficacy in approximately 15% of ES patients (29–31) and has highlighted the need of more profound knowledge of this system to identify biomarkers of response. In the present study, we show that IGF2BP3 directly favors IGF1R, but not IGF2, translation in ES, supporting cell growth and migration in response to IGF1 and that different levels of IGF2BP3 expression can impact ES response to anti-IGF1R/IR agents.
OSI-906 (linsitinib) was purchased from Selleckchem (Houston, TX, USA). IGF1 was purchased from Upstate (Waltham, MA, USA). Working dilutions of all drugs were prepared immediately before use.
Human ES cells TC-71 were kindly provided by T.J. Triche (Children’s Hospital, Los Angeles, CA, USA), A673 was provided by American Type Culture Collection (Rockville, MD, USA). Cell lines were authenticated by DNA fingerprinting (last control December 2017) and found mycoplasma-free by the MycoAlert mycoplasma detection kit (Lonza, Basel, Switzerland) (verified every 3 months). A673 and TC-71 cells were stably silenced for IGF2BP3 after transfection with the pLKO.1 vector containing IGF2BP3-specific short hairpin RNA (TRCN0000074673) and selection with puromycin 2 µg/ml (Sigma, St. Louis, MO, USA). Transfections were performed using TransIT-X2 (Mirus, Madison, WI, USA) according to the manufacturer’s protocol. The ES cell lines were grown as previously described (32).
Patients with localized ES who were enrolled in prospective studies and treated at the Rizzoli Institute were included in the present analysis (33). All patients had a diagnosis of ES made on representative specimens from open or needle biopsies based on histological, cytological, immunohistochemical features as well as the molecular presence of the chimeric product derived from ES-specific chromosomal translocations (34). Local treatment, performed after induction chemotherapy, consisted of radiation therapy, surgery or surgery followed by radiation therapy. In patients locally treated by surgery, histologic response to chemotherapy was evaluated according to the method proposed by Picci et al. (35). The patients’ clinical characteristics are summarized in Table 1. Clinical and follow-up data were updated to June 2014. The ethical committee of the Rizzoli Institute approved the study (0041040/2015), and informed consent was obtained.
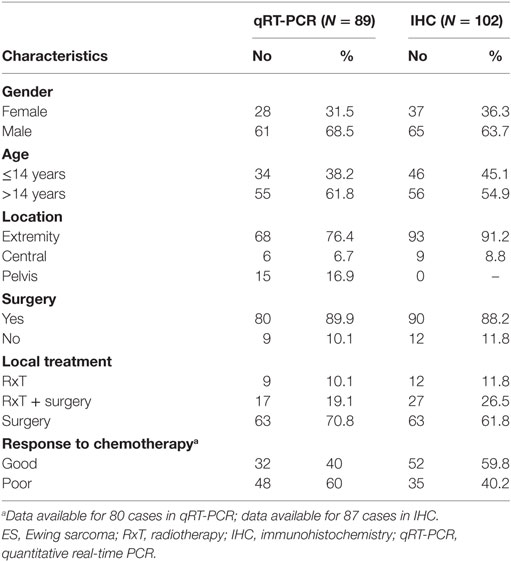
Table 1. Clinico-pathological features of ES patients included in the study.
Total RNA from snap-frozen tissue samples and/or cell lines was isolated using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). RNA quality and quantity were assessed by NanoDrop analysis (NanoDrop ND1000, ThermoFisher Scientific, Waltham, MA, USA) and/or by electrophoresis. To determine whether the extracted RNA was representative of ES, matching tissues were morphologically analyzed after hematoxylin–eosin staining. Oligo dT primers (Applied Biosystems, Foster City, CA, USA) were used to reverse transcribe RNA. Predesigned TaqMan probes (Applied Biosystem) for target genes were used to determine the expression level of the corresponding genes by quantitative real-time PCR (qRT-PCR) using a ViiA™ seven Real-Time PCR System (Applied Biosystems) according to the manufacturer’s instructions. The probes were as follows: IGF2BP3 (Hs00559907_g1), IGF1R (Hs00181385_m1), and ABCG2 (Hs00184979_m1). Primer sets for IGF1 and IGF2 were designed for SYBR Green-mediated quantification as previously reported (36). The Primer Express software (Applied Biosystems) was used to design appropriate primer pairs for the reference gene [glyceraldehyde-3-phosphate dehydrogenase (GAPDH)] as previously reported (36). Total RNA from primary cultures of human mesenchymal stem cells (hMSCs) obtained during surgery was also extracted and used as a calibrator. Two biological replicates were performed. In each replicate, samples were run in triplicate. Relative quantification analysis was performed using the 2−ΔΔCt method (37).
Representative samples of paraffin-embedded ES tumors, at least three cores per patient, were included in a tissue microarray (TMA) and processed for IHC as previously described (38). The TMA was then incubated overnight at 4°C with the following primary antibodies: anti-IGF1Rβ antibody (Santa Cruz Biotechnology, Dallas, TX, USA) diluted 1:50, as previously reported (38), or anti-IGF2BP3 antibody (Santa Cruz Biotechnology) diluted 1:50. The samples were classified based on positivity scores as follows: “low expression” (categorized as 0 for statistical analysis), when no staining or low positivity was observed (intensity of the staining was scored as +/– or + ––), or “high expression” (categorized as 1 for statistical analysis), when a diffused immunostaining was present (intensity of the staining was scored as + + − or + + +).
Cell lysates were prepared and processed as previously described (32). Membranes were incubated overnight with the following primary antibodies: anti-IRβ, anti-IGF2BP3, anti-GAPDH, anti-IGF1Rβ, anti-Lamin B (Santa Cruz Biotechnology), anti-phospho-ERK 1/2, anti-ERK 1/2, anti-phospho-Akt (Ser473), and anti-Akt (Cell Signaling Technology, Beverly, MA, USA) antibodies. Anti-rabbit, anti-mouse (GE Healthcare, Little Chalfont, UK), and anti-goat (Santa Cruz Biotechnology) antibodies conjugated to horseradish peroxidase were used as secondary antibodies.
Sensitivity to OSI-906 was assessed after 72 h of treatment with the TACS® MTT Cell Proliferation Assay kit (Trevigen, Inc., Gaithersburg, MD, USA) according to the manufacturer’s instructions. Cells were seeded into 96-well plates (2,500 cells/well) in IMDM plus 10% FBS. After 24 h, various concentrations of OSI-906 (0.3–10 µM) were added. Motility assay was performed using Transwell chambers (CoStar, Cambridge, MA, USA). 105 cells in IMDM plus 1% FBS were seeded in the upper compartment, and IMDM plus 1% FBS and IGF1 (50 ng/ml) was added to the lower compartment of the chambers. To assess the response to IGF1, 5 × 104 cells were seeded in 24-well plates, serum starved for 24 h and treated with IGF1 (50 ng/ml) for 24 h. Trypan Blue cell count was employed to assess the response to IGF1. For each independent experiment, samples were in duplicate (Trypan Blue) or triplicate (MTT assay; Motility assay).
For RIP assays, cells plated in 100-mm Ø dishes, to generate 4–6 mg of total protein, were washed twice with PBS, harvested, and extracted for 30 min on ice in 250 µl of ice-cold lysis buffer [100 mM KCl, 5 mM MgCl2, 10 mM HEPES (pH 7.0), 0.5% Nonidet P-40, and 1 mM dithiothreitol (DTT)] supplemented with RNase and protease inhibitors. Extracts were cleared by centrifugation at 4°C for 15 min at 12,000 rpm. Supernatants were collected and diluted in NT-2 buffer [150 mM NaCl, 1 mM MgCl2, 50 mM Tris–HCl (pH 7.4), and 0.05% Nonidet P-40] supplemented with protease and RNase inhibitors and 1 mM DTT. To preclear the cytoplasmic extracts, 50 µl of a suspension of Protein G plus/Protein A agarose beads (Merck Millipore) was added to the supernatant for 1 h at 4°C. For immunoprecipitation, precleared extracts were incubated with 150 µl of Protein G plus/Protein A agarose beads precoated with the same amount of either nonimmune mouse IgG (Santa Cruz Biotechnology) or anti-IGF2BP3 antibody (10 µg) in 800 µl of NT-2 buffer [150 mM NaCl, 1 mM MgCl2, 50 mM Tris–HCl (pH 7.4), and 0.05% Nonidet P-40] containing 1 mM DTT, RNase inhibitor, and protease inhibitors for 4 h at 4°C with rotation. The beads were washed 10 times with ice-cold NT-2 buffer. RNA was extracted by adding TRIzol directly to the beads, and qRT-PCR was performed on equivalent amounts of samples to quantify protein-bound mRNAs.
Correlations between gene expression levels were assessed using Spearman’s rank test while association between protein expression levels was assessed using Fisher’s exact test. All p-values were two-sided. p-Value <0.05 was considered statistically significant. Differences among means were analyzed using Student’s t-test or one-way ANOVA when experimental data included more than two groups.
Data from the literature indicate that IGF2BP3 directly regulates IGF2, thereby enhancing IGF1R activity. Therefore, the expression of IGF1R and IGF2 were evaluated using qRT-PCR in a retrospective series of 89 primary ES specimens and compared with the expression of IGF2BP3 (Table 1). IGF2BP3 expression was not correlated with IGF2 expression (Figure 1A), but a direct correlation was found between IGF1R and IGF2BP3 expression (Figure 1B). This association was confirmed at protein level. We analyzed the protein expression of IGF2BP3 and IGF1R in an independent series of 102 ES patients (Table 1). As observed at the mRNA level, IGF1R protein expression was found to be significantly associated with IGF2BP3 expression [IGF1R: low expression 29/102, high expression 73/102; IGF2BP3: low expression 62/102, high expression 40/102; IGF1R and IGF2BP3 protein levels in agreement (either low expression and high expression) in 61/102 cases; p = 0.001, Fisher’s exact test], further verifying the association between the two molecules.
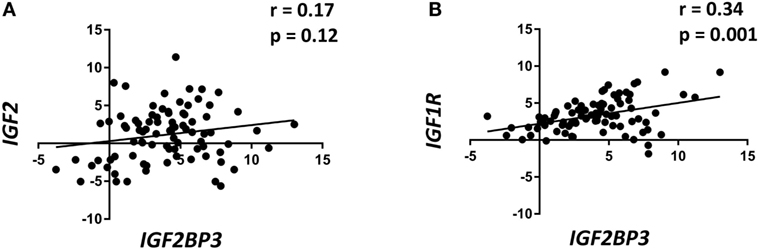
Figure 1. Insulin-like growth factor 2 (IGF2) mRNA-binding protein 3 (IGF2BP3) expression is directly correlated with IGF1R expression in Ewing sarcoma (ES) cases. Scatter plot displaying the correlation between IGF2BP3 and IGF2 (A) or IGF1R (B) mRNA levels in 89 ES cases. Correlation coefficient (r) and p-value were calculated using Spearman’s correlation test. Human mesenchymal stem cells were used as calibrators (2−ΔΔCt = 1).
The interaction between IGF2BP3 and IGF1R mRNA was validated in vitro by RIP assay using anti-IGF2BP3 and control IgG antibodies; after extracting RNA from immunoprecipitated samples, qRT-PCR analysis was used to measure IGF1R levels in each sample. IGF1R was highly enriched in IGF2BP3 immunoprecipitated samples relative to that in IgG immunoprecipitated samples, revealing that IGF2BP3 selectively associates with IGF1R (Figure 2A). To investigate the biological significance of IGF2BP3 binding to IGF1R, we first examined whether IGF2BP3 regulates IGF1R stability. Depletion of IGF2BP3 in ES cells significantly reduced IGF1R mRNA (Figure 2B) and protein levels (Figure 2C). Compared with control cells, IGF2BP3-depleted cells displayed reduced migration (Figure 3A) and growth (Figure 3B) in response to IGF1, confirming that IGF2BP3 levels substantially impact IGF1R-mediated functions in ES cells.
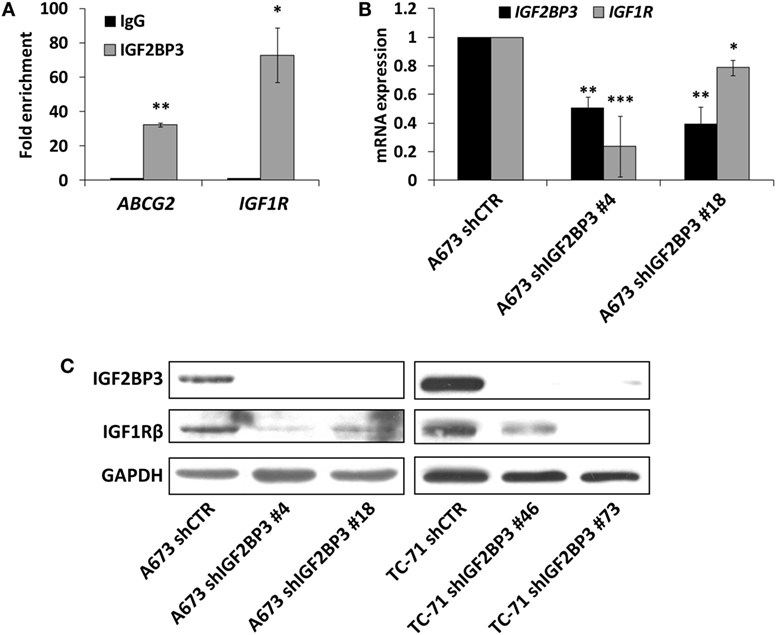
Figure 2. Assessment of IGF1R as an insulin-like growth factor 2 mRNA-binding protein 3 (IGF2BP3) mRNA target in Ewing sarcoma (ES) cells. (A) Quantitative real-time PCR analysis of IGF2BP3-associated mRNAs isolated from the cytoplasmic extracts of A673 cells by immunoprecipitation using an anti-IGF2BP3 antibody. Non-immune goat IgG was used as a negative control. ABCG2 was used as positive control. Columns represent the mean values of two independent experiments, in which samples were run in triplicates, and the bars represent the SE. *p < 0.05, **p < 0.01, Student’s t-test. (B) mRNA expression levels of IGF2BP3 and IGF1R in IGF2BP3-depleted or empty vector-transfected (shCTR) A673 ES cells. GAPDH was used as a housekeeping gene. Columns represent the mean values of at least two independent experiments, in which samples were run in triplicates, and the bars represent the SE. *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA with respect to shCTR. (C) Western blotting showing IGF2BP3 and IGF1R expression in IGF2BP3-depleted or empty vector-transfected (shCTR) A673 (left) or TC-71 (right) ES cells. GAPDH was used for normalization. Data from one experiment representative of three.
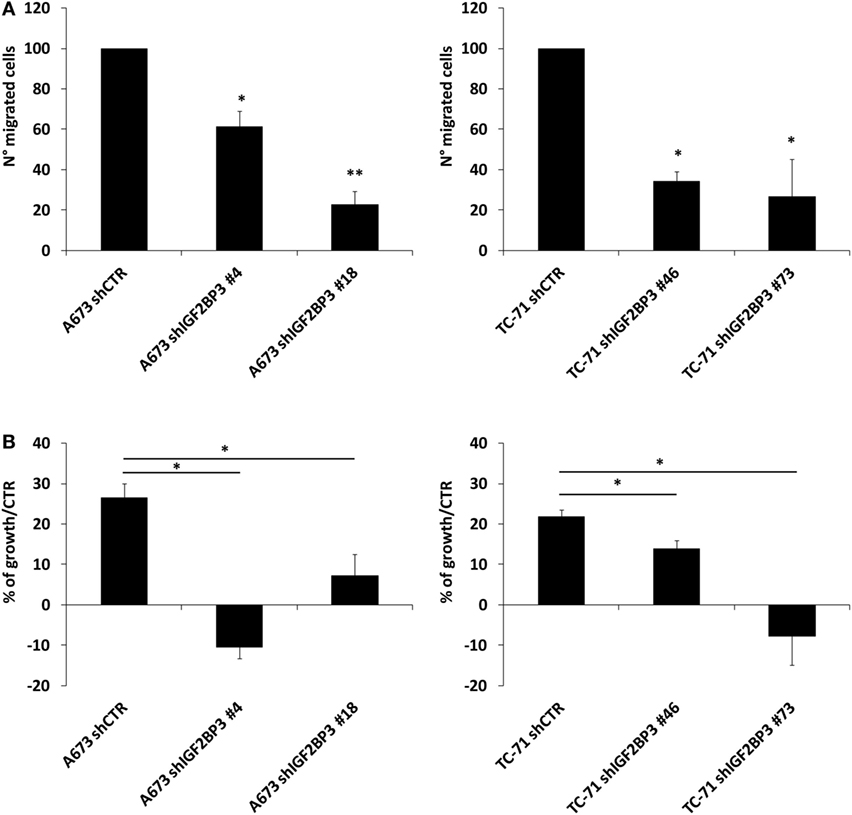
Figure 3. Evaluation of the oncogenic potential of insulin-like growth factor 2 mRNA-binding protein 3 (IGF2BP3)/IGF1R in Ewing sarcoma (ES) cell lines. (A) Migration of IGF2BP3-depleted or empty vector-transfected (shCTR) A673 or TC-71 ES cells in response to IGF1 (50 ng/ml) stimulation. Columns represent the mean values of two independent experiments, in which samples were run in triplicate, and the bars represent the SE. (B) Growth capacity of IGF2BP3-depleted or empty vector-transfected (shCTR) A673 or TC-71 ES cells after 24 h of IGF1 (50 ng/ml) stimulation. Trypan blue staining was used for cell counting. Columns represent the mean values of two independent experiments, in which samples were run in duplicates, and the bars represent the SE. (A,B) *p < 0.05, **p < 0.01, one-way ANOVA.
It has been demonstrated that ES cells can rapidly counteract IGF1R blockade induced by selective anti-IGF1R antibodies by switching from the canonical IGF1/IGF1R pathway to the alternative IGF2/IR signaling pathway (36, 39). Thus, we postulated that a similar mechanism of resistance is triggered by IGF2BP3. Indeed, compared with control cells, in addition to exhibiting decreased expression of IGF1R, IGF2BP3-silenced A673 cells displayed increased levels of IR (Figure 4A) and IGF2 (Figure 4C). Additionally, as a consequence of the activation of this compensatory loop, phosphorylation of downstream signaling components Akt and ERK 1/2 was maintained in IGF2BP3-silenced cells (Figure 4A). However, this was not a general phenomenon, because we were unable to observe the same alterations in the TC-71 experimental models in which IR was downregulated similarly to IGF1R (Figure 4B), and marked decreased level of Akt phosphorylation was observed (Figure 4B), but no modulation in the expression of the ligands was detected (Figure 4D) when the cells were depleted of IGF2BP3.
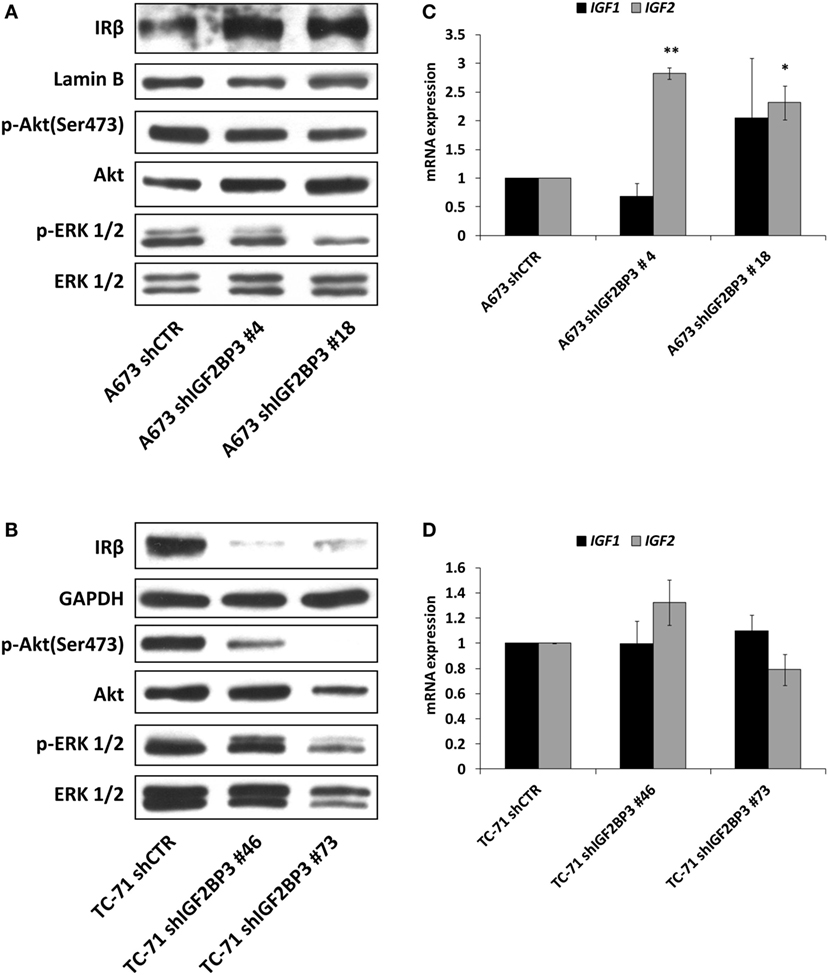
Figure 4. Insulin receptor (IR) signaling in response to insulin-like growth factor 2 (IGF2) mRNA-binding protein 3 (IGF2BP3)-mediated IGF1R regulation. (A,B) Western blotting depicting the expression of IR and phosphorylation of Akt and ERK 1/2 in IGF2BP3-depleted or empty vector-transfected (shCTR) (A) A673 or (B) TC-71 Ewing sarcoma (ES) cells. GAPDH, Lamin B, and total proteins were used for normalization purposes. Data from one experiment representative of three. (C,D) mRNA level of IGF1 and IGF2, expressed as 2−ΔΔCt, in IGF2BP3-depleted or empty vector-transfected (shCTR) (C) A673 or (D) TC-71 cells. Columns represent the mean values of at least two independent experiments, in which samples were run in triplicates, and the bars represent the SE. *p < 0.05, **p < 0.01, one-way ANOVA compared with shCTR.
The data presented above indicate that IGF2BP3 directly and indirectly affects the expression of IGF system components. Therefore, we tested the sensitivity of ES cells with differential expression of IGF2BP3 to anti-IGF agents. Based on the reported role of IGF1R as a major driver of cancer development and progression, several pharmacological inhibitors were developed including the small-molecule inhibitor OSI-906, which can block both IGF1R and IR. Accordingly, compared with control cells, IGF2BP3-depleted A673 cells, which showed decreased IGF1R but increased IR/IGF2 expression and active PI3K and MAPK signaling, displayed an increased sensitivity to the dual inhibitor OSI-906 (Figure 5A). However, this was not the case for IGF2BP3-depleted TC-71 cells, where no compensatory mechanism mediated by IR signaling was observed (Figure 5B).
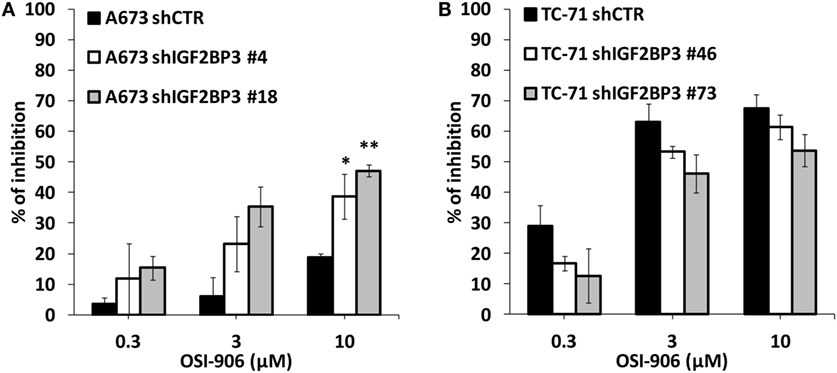
Figure 5. Efficacy of agents blocking the IGF system in insulin-like growth factor 2 mRNA-binding protein 3 (IGF2BP3)-depleted models. Cell growth was assessed using the MTT assay after 72 h of exposure to OSI-906 at indicated doses in IGF2BP3-depleted or empty vector-transfected (shCTR) A673 (A) and TC-71 (B) Ewing sarcoma cells. Results are displayed as the percentage of growth inhibition relative to that in untreated control cells. Histograms represent the mean of three independent experiments, in which each sample was in triplicate, and bars represent the SE. *p < 0.05, **p < 0.01, one-way ANOVA compared with shCTR.
The rationale for the development of agents against IGF1R was provided by experimental evidence showing that the receptor is necessary for malignant transformation, while low IGF activity was shown to protect against cancer (40). Despite promising preclinical results, the negative results obtained by phase 2/3 trials using selective anti-IGF1R antibodies have prompted the cessation of several pharma programs and blocked the research on the IGF system. However, even the failed studies included patients who received some benefit, especially ES patients. On one hand, this evidence confirmed the therapeutic importance of the IGF1R pathway, but on the other, it supported the need for deeper insights into the IGF system to help the identification of biomarkers of response [reviewed in Ref. (41)]. In this study, we demonstrate that IGF2BP3 is a novel regulator of IGF1R expression, with implications for both cellular malignancy and therapy. Direct activating mutations in IGF1R have not been described, and regulation of IGF1R gene expression is generally achieved at the level of transcription (42). In ES cells, EWS-FLI1, the oncogenic driver of this tumor (43), was found to directly bind to the IGF1R promoter and sustain IGF1R expression (44, 45). In this study, we provide evidence of an RBP-mediated post-transcriptional regulation of IGF1R, proposing the possibility of an additional regulatory mechanism that operates in ES cells. In contrast to other tumors in which the effect of IGF2BP3 on the IGF system is primarily mediated by IGF2 mRNA regulation (4, 20), in ES, IGF2BP3 mainly affects IGF1R. The expression of IGF1R and IGF2BP3 are correlated in clinical ES samples, while in experimental models, although the small number of independent experiments suggests caution in the interpretation of the data, we demonstrated that IGF2BP3 can bind to and stabilize IGF1R mRNA, favoring the expression of the receptor and its functions. In fact, when IGF2BP3 is silenced, ES cells express low levels of IGF1R and respond less to the IGF1 stimulus in terms of cell growth and migration. No direct relationship was observed between IGF2BP3 and the ligands IGF1 and IGF2. However, IGF2, together with IR, may be induced at least in some cell lines to activate an IR-mediated mitogenic pathway that can compensate for the IGF2BP3-mediated repression of IGF1R. Therefore, IGF2BP3 can directly or indirectly module the sensitivity of cells to anti-IGF1R/IR agents, and this molecule should thus be added to the list of proteins that are analyzed for personalized treatment with agents targeting the IGF system. This is highly required especially because clinical responses to anti-IGF1R is observed in approximately 15% in ES, the tumor type with the best response to this targeted approach, while clinical response was registered in only a small (1–5%) but distinct proportion of patients with solid tumors (46, 47). In thyroid tumors (20), the high expression of IGF2BP3 sustains IGF2 levels and leads to an increased sensitivity to OSI-906. In contrast, in ES, the low expression of IGF2BP3 inhibits IGF1R expression and indirectly induces IR/IGF2 activation, leading to an increased sensitivity to the dual inhibitor OSI-906. Considering that the upregulation of IR and IGF2 after IGF2BP3 depletion did not appear to be a general phenomenon, reflective of heterogeneity in tumor response, we strongly advocate for the need to evaluate the IGF1R/IR ratio together with the expression of IGF2BP3 to predict therapeutic response to agents targeting IGF1R and/or IR before and during any treatment.
Overall, our data indicate that IGF2BP3 represents a novel regulator of IGF1R that has a wide impact on the IGF system in ES cells. This interaction strongly affects response to IGF1, one of the major growth factors influencing ES malignancy. The results of this study provide early, preclinical evidence that the assessment of IGF2BP3 expression combined with the IGF1R/IR ratio may be used to predict therapeutic responses to the dual IGF1R/IR inhibitor OSI-906, providing new insights for a more precise and rational administration of anti-IGF system agents in ES.
This study was carried out in accordance with the recommendations of the ethical committee of the Rizzoli Institute that approved the study (0041040/2015). All subjects gave written informed consent.
CM and KS conception and design of the study. CM, MP, MCM, EFS, and LT acquisition of data. CM, MP, PP, and KS analysis and interpretation of data. CM, MP, and KS drafting or revising the work. All the authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Associazione “Un Sorriso con Luca” for encouraging our research.
This work was supported by the Italian Association for Cancer Research (IG2016_18451 to KS) and Italian Ministry of Health (RF Bando 2016 to KS). Caterina Mancarella was awarded the “Guglielmina Lucatello e Gino Mazzega” fellowship granted by Fondazione Italiana per la Ricerca sul Cancro- FIRC (FIRC project code: 17984).
1. Christiansen J, Kolte AM, Hansen T, Nielsen FC. IGF2 mRNA-binding protein 2: biological function and putative role in type 2 diabetes. J Mol Endocrinol (2009) 43:187–95. doi:10.1677/JME-09-0016
2. Nielsen J, Christiansen J, Lykke-Andersen J, Johnsen AH, Wewer UM, Nielsen FC. A family of insulin-like growth factor II mRNA-binding proteins represses translation in late development. Mol Cell Biol (1999) 19:1262–70. doi:10.1128/MCB.19.2.1262
3. Yisraeli JK. VICKZ proteins: a multi-talented family of regulatory RNA-binding proteins. Biol Cell (2005) 97:87–96. doi:10.1042/BC20040151
4. Liao B, Hu Y, Herrick DJ, Brewer G. The RNA-binding protein IMP-3 is a translational activator of insulin-like growth factor II leader-3 mRNA during proliferation of human K562 leukemia cells. J Biol Chem (2005) 280:18517–24. doi:10.1074/jbc.M500270200
5. Ueki A, Shimizu T, Masuda K, Yamaguchi SI, Ishikawa T, Sugihara E, et al. Up-regulation of Imp3 confers in vivo tumorigenicity on murine osteosarcoma cells. PLoS One (2012) 7:e50621. doi:10.1371/journal.pone.0050621
6. Bell JL, Wachter K, Muhleck B, Pazaitis N, Kohn M, Lederer M, et al. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression? Cell Mol Life Sci (2013) 70:2657–75. doi:10.1007/s00018-012-1186-z
7. Samanta S, Pursell B, Mercurio AM. IMP3 protein promotes chemoresistance in breast cancer cells by regulating breast cancer resistance protein (ABCG2) expression. J Biol Chem (2013) 288:12569–73. doi:10.1074/jbc.C112.442319
8. Hsu KF, Shen MR, Huang YF, Cheng YM, Lin SH, Chow NH, et al. Overexpression of the RNA-binding proteins Lin28B and IGF2BP3 (IMP3) is associated with chemoresistance and poor disease outcome in ovarian cancer. Br J Cancer (2015) 113:414–24. doi:10.1038/bjc.2015.254
9. Chao JA, Patskovsky Y, Patel V, Levy M, Almo SC, Singer RH. ZBP1 recognition of beta-actin zipcode induces RNA looping. Genes Dev (2010) 24:148–58. doi:10.1101/gad.1862910
10. Nielsen J, Kristensen MA, Willemoes M, Nielsen FC, Christiansen J. Sequential dimerization of human zipcode-binding protein IMP1 on RNA: a cooperative mechanism providing RNP stability. Nucleic Acids Res (2004) 32:4368–76. doi:10.1093/nar/gkh754
11. Lederer M, Bley N, Schleifer C, Huttelmaier S. The role of the oncofetal IGF2 mRNA-binding protein 3 (IGF2BP3) in cancer. Semin Cancer Biol (2014) 29:3–12. doi:10.1016/j.semcancer.2014.07.006
12. Degrauwe N, Suva ML, Janiszewska M, Riggi N, Stamenkovic I. IMPs: an RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev (2016) 30:2459–74.
13. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer (2008) 8:915–28. doi:10.1038/nrc2536
14. Giorgetti S, Ballotti R, Kowalski-Chauvel A, Tartare S, Van Obberghen E. The insulin and insulin-like growth factor-I receptor substrate IRS-1 associates with and activates phosphatidylinositol 3-kinase in vitro. J Biol Chem (1993) 268:7358–64.
15. Grey A, Chen Q, Xu X, Callon K, Cornish J. Parallel phosphatidylinositol-3 kinase and p42/44 mitogen-activated protein kinase signaling pathways subserve the mitogenic and antiapoptotic actions of insulin-like growth factor I in osteoblastic cells. Endocrinology (2003) 144:4886–93. doi:10.1210/en.2003-0350
16. Casa AJ, Dearth RK, Litzenburger BC, Lee AV, Cui X. The type I insulin-like growth factor receptor pathway: a key player in cancer therapeutic resistance. Front Biosci (2008) 13:3273–87. doi:10.2741/2925
17. Chitnis MM, Yuen JS, Protheroe AS, Pollak M, Macaulay VM. The type 1 insulin-like growth factor receptor pathway. Clin Cancer Res (2008) 14:6364–70. doi:10.1158/1078-0432.CCR-07-4879
18. Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell (2010) 141:129–41. doi:10.1016/j.cell.2010.03.009
19. Fawzy IO, Hamza MT, Hosny KA, Esmat G, Abdelaziz AI. Abrogating the interplay between IGF2BP1, 2 and 3 and IGF1R by let-7i arrests hepatocellular carcinoma growth. Growth Factors (2016) 34:42–50. doi:10.3109/08977194.2016.1169532
20. Panebianco F, Kelly LM, Liu P, Zhong S, Dacic S, Wang X, et al. THADA fusion is a mechanism of IGF2BP3 activation and IGF1R signaling in thyroid cancer. Proc Natl Acad Sci U S A (2017) 114:2307–12. doi:10.1073/pnas.1614265114
21. Paioli A, Luksch R, Fagioli F, Tamburini A, Cesari M, Palmerini E, et al. Chemotherapy-related toxicity in patients with non-metastatic Ewing sarcoma: influence of sex and age. J Chemother (2014) 26:49–56. doi:10.1179/1973947813Y.0000000103
22. Gaspar N, Hawkins DS, Dirksen U, Lewis IJ, Ferrari S, Le Deley MC, et al. Ewing sarcoma: current management and future approaches through collaboration. J Clin Oncol (2015) 33:3036–46. doi:10.1200/JCO.2014.59.5256
23. Crompton BD, Stewart C, Taylor-Weiner A, Alexe G, Kurek KC, Calicchio ML, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov (2014) 4:1326–41. doi:10.1158/2159-8290.CD-13-1037
24. Scotlandi K, Benini S, Sarti M, Serra M, Lollini PL, Maurici D, et al. Insulin-like growth factor I receptor-mediated circuit in Ewing’s sarcoma/peripheral neuroectodermal tumor: a possible therapeutic target. Cancer Res (1996) 56:4570–4.
25. Scotlandi K, Benini S, Nanni P, Lollini PL, Nicoletti G, Landuzzi L, et al. Blockage of insulin-like growth factor-I receptor inhibits the growth of Ewing’s sarcoma in athymic mice. Cancer Res (1998) 58:4127–31.
26. Scotlandi K, Avnet S, Benini S, Manara MC, Serra M, Cerisano V, et al. Expression of an IGF-I receptor dominant negative mutant induces apoptosis, inhibits tumorigenesis and enhances chemosensitivity in Ewing’s sarcoma cells. Int J Cancer (2002) 101:11–6. doi:10.1002/ijc.10537
27. Scotlandi K, Manara MC, Nicoletti G, Lollini PL, Lukas S, Benini S, et al. Antitumor activity of the insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 in musculoskeletal tumors. Cancer Res (2005) 65:3868–76. doi:10.1158/0008-5472.CAN-04-3192
28. Scotlandi K, Picci P. Targeting insulin-like growth factor 1 receptor in sarcomas. Curr Opin Oncol (2008) 20:419–27. doi:10.1097/CCO.0b013e328302edab
29. Buck E, Gokhale PC, Koujak S, Brown E, Eyzaguirre A, Tao N, et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol Cancer Ther (2010) 9:2652–64. doi:10.1158/1535-7163.MCT-10-0318
30. Olmos D, Postel-Vinay S, Molife LR, Okuno SH, Schuetze SM, Paccagnella ML, et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohort study. Lancet Oncol (2010) 11:129–35. doi:10.1016/S1470-2045(09)70354-7
31. Pappo AS, Patel SR, Crowley J, Reinke DK, Kuenkele KP, Chawla SP, et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol (2011) 29:4541–7. doi:10.1200/JCO.2010.34.0000
32. Rocchi A, Manara MC, Sciandra M, Zambelli D, Nardi F, Nicoletti G, et al. CD99 inhibits neural differentiation of human Ewing sarcoma cells and thereby contributes to oncogenesis. J Clin Invest (2010) 120:668–80. doi:10.1172/JCI36667
33. Ferrari S, Sundby Hall K, Luksch R, Tienghi A, Wiebe T, Fagioli F, et al. Nonmetastatic Ewing family tumors: high-dose chemotherapy with stem cell rescue in poor responder patients. Results of the Italian Sarcoma Group/Scandinavian Sarcoma Group III protocol. Ann Oncol (2011) 22:1221–7. doi:10.1093/annonc/mdq573
34. Toomey EC, Schiffman JD, Lessnick SL. Recent advances in the molecular pathogenesis of Ewing’s sarcoma. Oncogene (2010) 29:4504–16. doi:10.1038/onc.2010.205
35. Picci P, Bohling T, Bacci G, Ferrari S, Sangiorgi L, Mercuri M, et al. Chemotherapy-induced tumor necrosis as a prognostic factor in localized Ewing’s sarcoma of the extremities. J Clin Oncol (1997) 15:1553–9. doi:10.1200/JCO.1997.15.4.1553
36. Garofalo C, Mancarella C, Grilli A, Manara MC, Astolfi A, Marino MT, et al. Identification of common and distinctive mechanisms of resistance to different anti-IGF-IR agents in Ewing’s sarcoma. Mol Endocrinol (2012) 26:1603–16. doi:10.1210/me.2012-1142
37. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (2001) 25:402–8. doi:10.1006/meth.2001.1262
38. Scotlandi K, Manara MC, Serra M, Marino MT, Ventura S, Garofalo C, et al. Expression of insulin-like growth factor system components in Ewing’s sarcoma and their association with survival. Eur J Cancer (2011) 47:1258–66. doi:10.1016/j.ejca.2011.01.007
39. Garofalo C, Manara MC, Nicoletti G, Marino MT, Lollini PL, Astolfi A, et al. Efficacy of and resistance to anti-IGF-1R therapies in Ewing’s sarcoma is dependent on insulin receptor signaling. Oncogene (2011) 30:2730–40. doi:10.1038/onc.2010.640
40. Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, Wei M, Madia F, Cheng CW, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med (2011) 3:70ra13. doi:10.1126/scitranslmed.3001845
41. Mancarella C, Scotlandi K. IGF system in sarcomas: a crucial pathway with many unknowns to exploit for therapy. J Mol Endocrinol (2017). doi:10.1530/JME-17-0250
42. Werner H, Sarfstein R. Transcriptional and epigenetic control of IGF1R gene expression: implications in metabolism and cancer. Growth Horm IGF Res (2014) 24:112–8. doi:10.1016/j.ghir.2014.03.006
43. Lessnick SL, Ladanyi M. Molecular pathogenesis of Ewing sarcoma: new therapeutic and transcriptional targets. Annu Rev Pathol (2012) 7:145–59. doi:10.1146/annurev-pathol-011110-130237
44. de Alava E, Panizo A, Antonescu CR, Huvos AG, Pardo-Mindan FJ, Barr FG, et al. Association of EWS-FLI1 type 1 fusion with lower proliferative rate in Ewing’s sarcoma. Am J Pathol (2000) 156:849–55. doi:10.1016/S0002-9440(10)64953-X
45. Amaral AT, Garofalo C, Frapolli R, Manara MC, Mancarella C, Uboldi S, et al. Trabectedin efficacy in Ewing sarcoma is greatly increased by combination with anti-IGF signaling agents. Clin Cancer Res (2015) 21:1373–82. doi:10.1158/1078-0432.CCR-14-1688
46. Jones RL, Kim ES, Nava-Parada P, Alam S, Johnson FM, Stephens AW, et al. Phase I study of intermittent oral dosing of the insulin-like growth factor-1 and insulin receptors inhibitor OSI-906 in patients with advanced solid tumors. Clin Cancer Res (2015) 21:693–700. doi:10.1158/1078-0432.CCR-14-0265
47. Puzanov I, Lindsay CR, Goff L, Sosman J, Gilbert J, Berlin J, et al. A phase I study of continuous oral dosing of OSI-906, a dual inhibitor of insulin-like growth factor-1 and insulin receptors, in patients with advanced solid tumors. Clin Cancer Res (2015) 21:701–11. doi:10.1158/1078-0432.CCR-14-0303
Keywords: insulin-like growth factor 2 mRNA-binding protein 3, IGF1R, anti-IGF agents, Ewing sarcoma, personalized treatment
Citation: Mancarella C, Pasello M, Manara MC, Toracchio L, Sciandra EF, Picci P and Scotlandi K (2018) Insulin-Like Growth Factor 2 mRNA-Binding Protein 3 Influences Sensitivity to Anti-IGF System Agents Through the Translational Regulation of IGF1R. Front. Endocrinol. 9:178. doi: 10.3389/fendo.2018.00178
Received: 14 February 2018; Accepted: 03 April 2018;
Published: 20 April 2018
Edited by:
Haim Werner, Tel Aviv University, IsraelReviewed by:
Leon Bach, Monash University, AustraliaCopyright: © 2018 Mancarella, Pasello, Manara, Toracchio, Sciandra, Picci and Scotlandi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caterina Mancarella, Y2F0ZXJpbmEubWFuY2FyZWxsYUBpb3IuaXQ=;
Katia Scotlandi, a2F0aWEuc2NvdGxhbmRpQGlvci5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.