 Farhad Dehkhoda
Farhad Dehkhoda Christine M. M. Lee
Christine M. M. Lee Johan Medina
Johan Medina Andrew J. Brooks
Andrew J. Brooks
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 13 February 2018
Sec. Molecular and Structural Endocrinology
Volume 9 - 2018 | https://doi.org/10.3389/fendo.2018.00035
This article is part of the Research Topic Manipulating and Understanding the Function of Cytokine Receptors, their Partners JAKs and Negative Regulators SOCS View all 8 articles
The growth hormone receptor (GHR), although most well known for regulating growth, has many other important biological functions including regulating metabolism and controlling physiological processes related to the hepatobiliary, cardiovascular, renal, gastrointestinal, and reproductive systems. In addition, growth hormone signaling is an important regulator of aging and plays a significant role in cancer development. Growth hormone activates the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signaling pathway, and recent studies have provided a new understanding of the mechanism of JAK2 activation by growth hormone binding to its receptor. JAK2 activation is required for growth hormone-mediated activation of STAT1, STAT3, and STAT5, and the negative regulation of JAK–STAT signaling comprises an important step in the control of this signaling pathway. The GHR also activates the Src family kinase signaling pathway independent of JAK2. This review covers the molecular mechanisms of GHR activation and signal transduction as well as the physiological consequences of growth hormone signaling.
The growth hormone receptor (GHR) is a member of the class I cytokine receptor family, which includes more than 30 receptors such as the prolactin receptor (PRLR), erythropoietin receptor (EPOR), thrombopoietin receptor (TPOR), granulocyte-macrophage colony-stimulating factor receptor, interleukin-3 receptor, interleukin-6 receptor, and interleukin-7 receptor (1, 2). GHR has been considered the archetypal class I cytokine receptor as it was the first cytokine receptor to be cloned and have its extracellular domain (ECD) crystal structure solved (3). GHR is a 638 amino acid long homodimeric receptor with one cytokine receptor homology domain (CRH), a single-pass transmembrane domain, and cytoplasmic intracellular domain (ICD) (Figure 1). With the exception of GHR, all members of the class I cytokine receptor family contain a WSXWS motif in the ECD. The WSXWS motif is important for expression and stability of the receptor and comprises a consensus sequence for C-mannosylation. For the IL-21R, the WSXWS has been shown to be mannosylated at the first tryptophan where the sugar chain appears to form structurally important interactions that bridge the two fibronectin domains (4, 5). GHR has in place of the WSXWS a similar sequence of YGeFS that has an analogous function in expression and stability of the receptor (1). Cytokine receptors lack an intrinsic protein tyrosine kinase (PTK) activity and therefore rely on binding non-receptor PTKs for their signal transduction. Within the ICD of all class I cytokine receptors is a proline-rich Box1 motif that is located a short distance from the cell membrane. A less conserved Box2 sequence consisting of acidic and aromatic residues is located a short distance C-terminal of the Box1 (1). The Box1 motif acts as a binding site for a cognate Janus kinase (JAK) of which there are four family members, JAK1, JAK2, JAK3, and TYK2 that can bind to specific receptors (Table 1). For GHR, the only JAK family member that binds the receptor is JAK2. GH binding to GHR results in activation of JAK2, which subsequently phosphorylates multiple tyrosine residues on the ICD of the receptor (Figure 2) (6, 7). This provides a scaffold for binding of STAT5a and STAT5b, which are subsequently phosphorylated by JAK2 upon receptor docking (Figure 3) (8). GHR also activates STAT1 and STAT3 via JAK2; however, these STATs do not appear to require binding to the phosphorylated receptor. Other signaling pathways such as the Ras/extracellular signal-regulated kinase (ERK) and PI 3-kinase/Akt are also activated by GHR (7, 9). The consequence of these GH-mediated cellular signaling pathways in a diverse range of cell types is responsible for the large range of physiological processes regulated by GH.
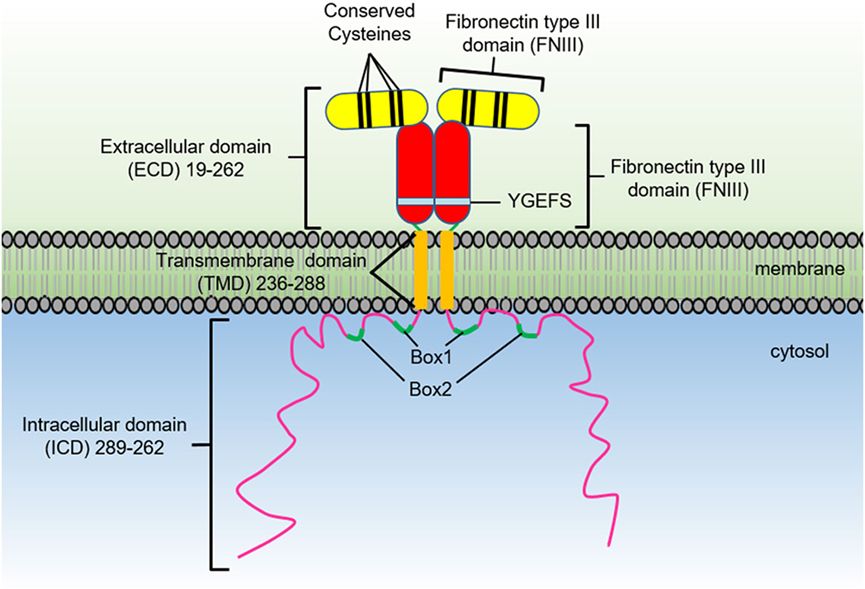
Figure 1. The growth hormone receptor domain organization.
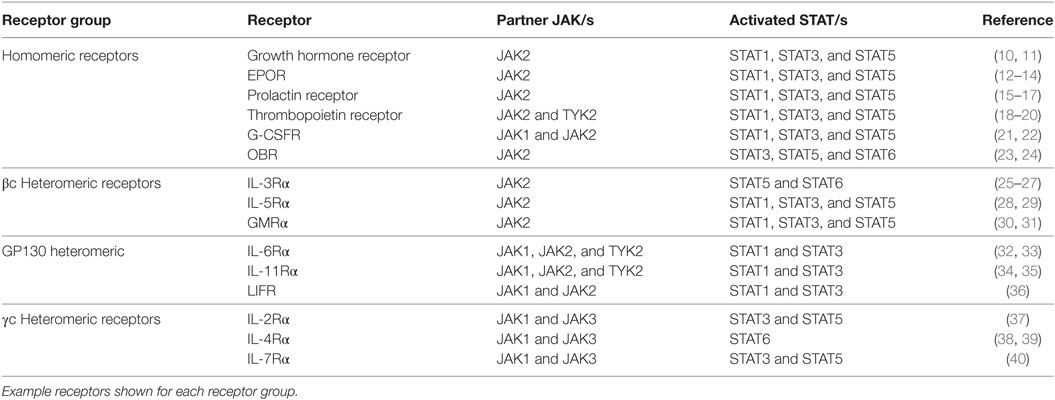
Table 1. Class I cytokine receptors and their Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signaling partners.
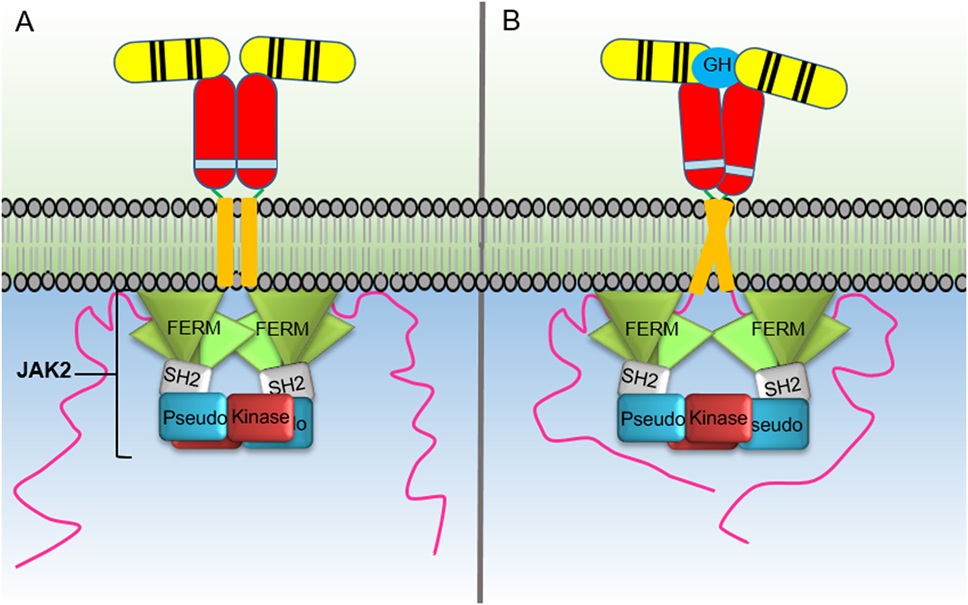
Figure 2. Activation of the growth hormone receptor (GHR) by growth hormone. In the inactive homodimeric GHR, (A) the Janus kinase (JAK) 2 kinase domain (KD) is inhibited in trans by interaction with the pseudokinase domain from the JAK2 bound to the opposing receptor within the homodimer. GH binding to the GHR extracellular domain (B) results in conformational changes that cause the transmembrane domains to transition from a parallel interaction to a left-handed crossover interaction. These structural changes cause a separation of the intracellular domains to the Box1 and Box2 motifs and the associated JAK2 molecules. The movement of the associated JAK2s dissociates the inhibitory interaction of the pseudokinase from the KD and brings the two JAK2 KDs in close proximity resulting in trans phosphorylation and activation.
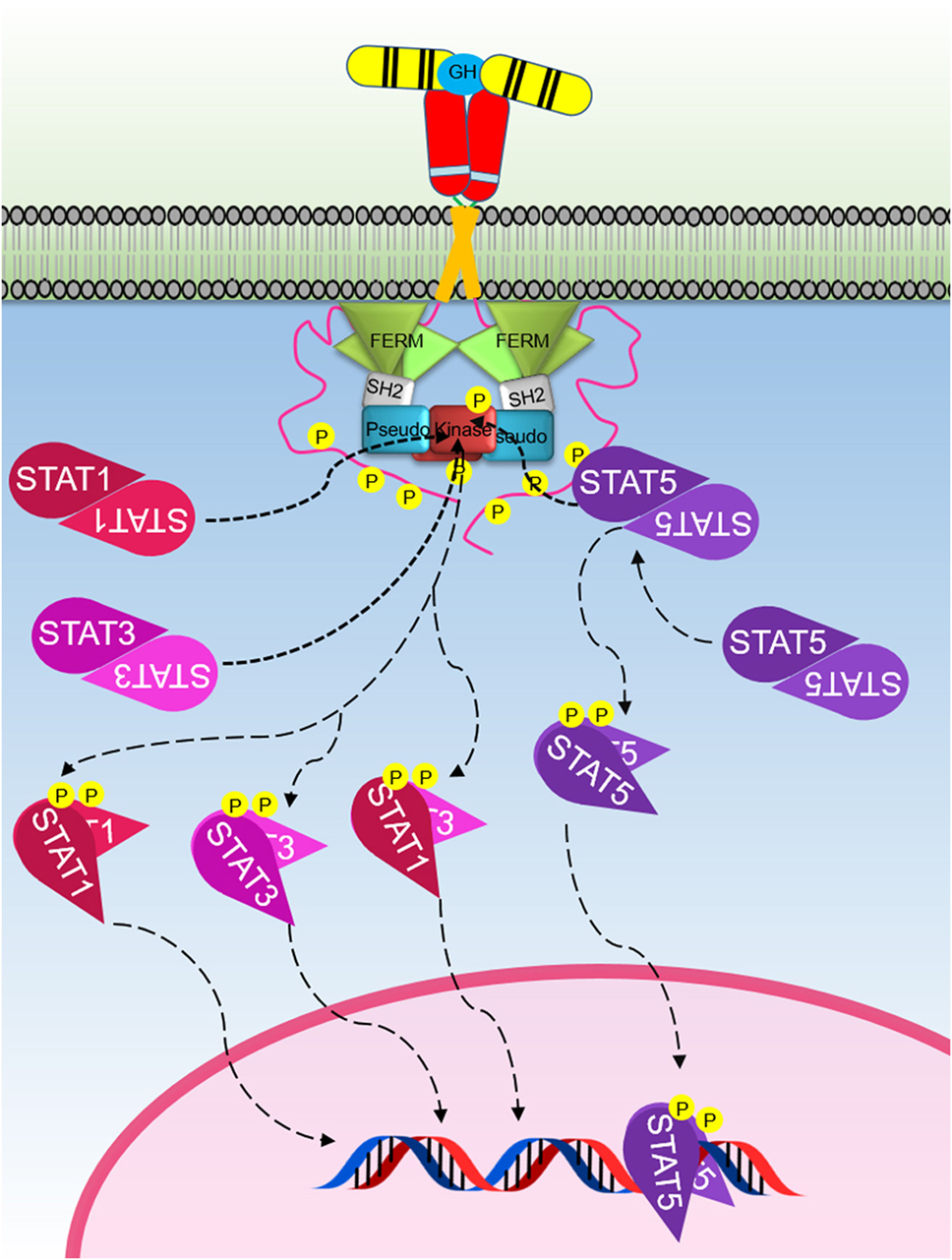
Figure 3. The Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signaling pathway initiated by the activated growth hormone receptor. Activated JAK2 phosphorylates tyrosines on the intracellular domain of the receptor. Inactive-STAT5 dimers bind these phosphorylated tyrosine residues on the receptor, and the STAT5 is subsequently phosphorylated by JAK2 forming different active-STAT5 dimers that are translocated to the nucleus, bind DNA, and act as transcription factors. STAT1 and STAT3 are phosphorylated and activated by JAK2. Active STAT1 and STAT3 form homodimers or heterodimers, are translocated to the nucleus, bind DNA, and act as transcription factors.
The crystal structure of GH bound to the GHR ECD showed that one GH molecule bound two GHR molecules (3). Biophysical studies demonstrated that GH binds initially to a single receptor through its “site 1” motif and then subsequently binds to the second receptor via its “site 2” interactions (41, 42). The major energy contributor residues of the hormone–receptor complex were identified as residues in the receptor hydrophobic patch, where tryptophan104 and tryptophan169 contributed significantly to site 1 interactions. In addition, tryptophan104 plays a key role in the weaker site 2 interaction (43, 44). These studies suggested GH binding causes receptor dimerization, which induces intracellular signal transduction. This dimerization-induced activation model was supported by a study using a hybrid receptor of the GHR ECD fused to the base of the CRH domain of granulocyte colony-stimulating factor receptor (G-CSFR) with the remaining fibronectin type III homology domains (FNIII), transmembrane, and ICD. Monoclonal antibodies to the GHR ECD activated the hybrid receptor whereas monovalent fragments did not; however, the additional FNIII domains may have influenced these data (45). In addition, of eight antibodies that were effective agonists for the GHR/G-CSFR hybrid, only one antibody acted as an agonist with weak activity on full-length GHR, suggesting that receptor dimerization alone is not sufficient for activation (46). Later studies using site 2-modified GH antagonists, which would be predicted to bind only GHR monomer, appeared to bind GHR dimers expressed on cell membranes, thus demonstrating the existence of GHR dimers before GH binding (47). Subsequent studies confirmed that GHR dimers exist on the cell surface, and that dimers form in the endoplasmic reticulum. It was also shown that the extracellular region is not required for maintaining receptor dimers, although the lower FNIII domain may define receptor-dimer specificity (48–52). Data supporting the GHR preformed dimer conclusion were performed independently by more than one group using multiple techniques including co-immunoprecipitation, FRET, BRET, and fluorescence anisotropy (48–51). By analyzing the FRET efficiency with FRET reporters placed at the C-termini of full-length GHR and C-terminal truncations, it was found, in general, that the dimeric-GHR ICDs are positioned at a greater distance apart, the further they are from the transmembrane domain (50). This has been illustrated clearly for the closely related receptor PRLR where a recent full-length structure has been produced, which includes a molecular model of the ICD (53).
Crystallographic studies comparing the unliganded ECD of GHR to the ligand-bound form showed no major change in overall structure; however, a 7–9° of rotation between upper and lower FNIII-type domains of the CRH module was observed, which was small compared with the ligand-induced conformation change shown for the ECD of RTKs (50). It was also evident that ligand binding did not change FRET efficiency of GH receptors with N-terminal reporters confirming lack of major conformational change in the ECDs upon GH binding (50).
Most members of the JAK family are widely expressed in many cell types, except JAK3 whose expression is restricted to cells of the hematopoietic lineage (54). While JAK1 and JAK2 are involved in diverse physiological actions such as hematopoiesis, immunity, development, and growth (1, 55, 56), JAK3 and Tyk2 are predominantly involved in homeostasis of immune system (57, 58).
Janus kinases are relatively large proteins ranging from 120 to 140 kD (~1,150 amino acids) (59). The N-terminal half of these proteins comprises N-terminal band 4.1, ezrin, radixin, moesin (FERM) domain followed by Src homology 2 (SH2) domain, which were primarily defined as Janus homology (JH) domains 3–7 (60). These domains play the major role in binding of JAKs to the cytoplasmic tails of their cognate cytokine receptors (61–63). The first evidence demonstrating the role of the N-terminus of JAK2 in receptor interactions was shown using JAK2 mutants, lacking the 239 N-terminal residues that rendered the JAK2 mutant incapable of binding GHR or the membrane proximal region of the common β chain (64, 65). This was further supported by studies showing that the FERM and SH2 domains of JAK1 are essential for association to oncostatin M receptor (OSMR), and that this binding is also required for upregulating surface expression of the OSMR (66, 67). Truncation studies showed that the membrane proximal 61 amino acids of GP130 are sufficient for signal transduction (68), and later it was shown for GHR that the membrane proximal 20 residues (which includes the Box1 motif) of the ICD is sufficient for JAK2 binding, but not sufficient for GH-induced activation of JAK2 (69). Until recently, little was known about the mechanism of receptor–JAK association or how JAKs gain specificity to their cognate receptors. Previous studies have shown that the Box1 motif, although required for JAK binding, is not sufficient to determine the specificity for a particular JAK to associate with its cytokine receptor, and that additional sequences in the receptor play an important role in specificity since JAK1 and JAK2 were shown to have different preferences for GHR, EPOR, and GP130 chimeras (70). The first crystal structure of a JAK bound to residues of a receptor ICD was shown for the FERM–SH2 of TYK2 in complex with the Box2 region of the interferon-α receptor 1 (IFNAR1) (71). This study revealed that the TYK2 FERM–SH2 domains form a clover shaped structure with the SH2 being the forth leaf. The structure showed the FERM domain has a canonical three-lobe structure (F1–F3), and the SH2 has a preserved SH2-like structure despite lacking the highly conserved arginine residue in the phosphotyrosine binding site (71, 72). Interestingly, the SH2 domain coordinates the interactions with IFNAR1 to form the initial connections with the receptor peptide in the C-terminus. Further insights regarding cytokine receptor recognition by JAKs emerged when the crystal structure of JAK1 bound to peptides derived from the IL-10 receptor (IL10RA) and interferon lambda receptor (IFNLR1) was solved (73). These studies showed the Box1 peptide bound to the F2 domain of the FERM while the Box2 peptide was not resolved in the structure. In addition, the binding kinetics of the IFNLR1 Box1 and Box2 showed that Box1 alone has a strong affinity while Box2 alone had no detectable interaction; however, the full Box1–Box2 peptide had a significantly enhanced affinity over Box1 alone, which was proposed to be due to the increased complex stability from the hydrophobic JAK1–Box2 interaction (73). Although neither of these recent structures showed simultaneous interactions between the JAK molecule and both Box1 and Box2 sequences, geometrically both sequences are capable of binding to single JAK protein (73, 74). The crystal structure of the JAK1 FERM–SH2 bound to peptides from IL10RA and IFNLR1 gave a provocative suggestion that the Box1 peptide bound to the FERM domain extends away from the FERM domain to interact with the SH2 domain of the neighboring JAK1 molecule in the crystal lattice. Further studies would be needed to determine if Box1–Box2 sequences can have relevant interactions with two JAKs simultaneously, although it was suggested that this may have been due to a crystal artifact (73). More recently, a crystal structure of the JAK1 FERM–SH2 has been solved with a continuous IFNLR1 Box1–Box2 peptide suggesting that lack full Box1–Box2 peptide in previous structures may have been a crystal artifact (75). Recently, a JAK2 FERM–SH2 structure has been solved; however, this structure is not in complex with any receptor sequence (76).
The C-terminal half of JAK proteins comprises a pseudokinase domain (PKD) followed by a kinase domain (KD), also known as JH2 and JH1, respectively (77). The KD is a typical tyrosine KD and is catalytically active, whereas the adjacent PKD is catalytically inactive and lacks critical residues despite containing a canonical kinase fold (78). The PKD is believed to regulate kinase activity, and its deletion in JAK2 and JAK3 was shown to increase basal activity of the kinase (64, 79–81). Biochemical studies have reported co-immunoprecipitation of JH1 and JH2 of JAK2, and that the pseudokinase acts to inhibit the kinase activity of JAKs (60, 79, 82). Furthermore, recent studies have shown that the PKD of JAK2, unlike the other members of the JAK family, exhibits some catalytic activity via autophosphorylation of two critical residues in the JH2–SH2 linker region, Ser523 and Tyr570, which play an important role in regulating the activity of the KD (82, 83).
Recent studies have made major strides in elucidating the mechanism of JAK2 activation by GHR (84). Activation of the receptor by GH binding was found to result in an increase in distance between the Box2 motifs of the intracellular receptor domains. This revelation required a new model for JAK2 activation by cytokine binding whereby in the cytokine free receptor, the JAK2 KD is inhibited by a JAK2 PKD in trans (Figure 2). In this model, the KD from one JAK2 molecule bound to a Box1–Box2 motif from one receptor is inhibited by interacting with the PKD from a different JAK2 molecule bound to the opposing receptor within the receptor homodimer. Upon GH binding, there is a structural change in the ECD that changes the receptor transmembrane domain interaction from a parallel form to a left-handed crossover interaction. This structural transition leads to a separation of the ICD, at least to the Box1–Box2 motif, and dissociates the JAK2 kinase-pseudokinase trans interaction and brings the KDs in proximity allowing trans phosphorylation and activation (1, 51).
Currently, there is no crystal structure available for the JAK2 pseudokinase-kinase to allow observation of the interaction interface between each domain; however, crystal structures for the individual pseudokinase and KDs have been solved (83, 85). The pseudokinase-kinase crystal structure has presently been solved for only one JAK family member, Tyk2 (86). However, the functional Tyk2 interface was only inferred from the crystal structure as there were three significant potential interaction faces identified in the crystal lattice. It could not be determined directly in this structure as to how the pseudokinase and KDs were joined via the interdomain linker as this linker sequence was not resolved in the crystal structure. The potential dimer interface with the largest buried surface area was excluded from detailed analysis as the distance between the termini of each domain was too great to allow each domain to be linked by the interdomain linker peptide (86). However, for a trans-inhibition interaction model, there would be no requirement for each domain to be linked (87), therefore perhaps this interface with the largest buried surface area may have some biological relevance. The interface with the second largest buried surface area in the Tyk2 crystal lattice did place the C- and N-termini in close enough proximity to be linked by the interdomain linker, and this interface was investigated in detail and mutational data did support the biological significance of this interaction surface. There are a number of activating mutations identified for the JAK2 PKD; however, the location of these mutations suggests there may be two interaction interfaces that are involved in regulating the KD (88). For the Tyk2 pseudokinase-kinase crystal structure, a number of JAK-activating mutations were found to lie directly in the proposed domain interface while a second surface of activating mutations was found peripheral to the proposed dimer interface, which includes the position for the JAK2 V617F mutation commonly identified in myeloproliferative neoplasms (89). Interestingly, some JAK2-activating mutations including V617F have been shown to require interaction with a cytokine receptor for their activity, suggesting that these mutations only disrupt inhibition of the pseudokinase when the JAK2 is bound to a receptor (90–93). Intriguingly, this supports previous suggestions that the JAK2 pseudokinase interaction and inhibition of the KD is different between the receptor bound JAK2 and the receptor free JAK2 (90). Molecular modeling of the proposed JAK2 trans-inhibition interaction of the PKD with the KD placed the V617F in proximity to the activation loop of the KD; however, further direct functional and structural data supporting this interaction are currently not available. Although functional data supporting a trans-inhibition model were obtained when constitutive activation of JAK2 was shown with a JAK2 construct where the pseudokinase and KDs were swapped and this construct showed significant cytokine-independent activation only in the presence of wild-type JAK2 (51).
As the GHR ICD is intrinsically disordered with a number of transient secondary structures (94), the C-termini are likely a long distance apart in the inactivated receptor homodimer in a similar manner as illustrated in a recent full structural model of the PRLR (53). Studies using FRET reporters placed at the C-terminus of GHR have shown a transient increase in FRET shortly after GH addition (95). These data likely represent the flexible receptor ICD interacting with the activated JAK2 KDs and being phosphorylated upon multiple tyrosine residues, which subsequently interact with downstream signaling molecules and negative regulators of signaling.
The STATs are important components of the signaling cascades triggered by various cytokines (Table 1). STATs were initially identified as cytokine-induced transcription factors in interferon treated cells (96–98). To date, seven different members of the STAT family have been identified which includes STAT1–4, 5a, 5b, and 6. STAT5a and 5b are encoded by two different genes with high-sequence homology, except for regions in the N- and C-termini (99). In addition, posttranslational modification and proteolytic cleavage have shown to produce additional forms of STAT1 and STAT3 (100). Although the STAT family members are activated via various cytokines, STAT2 and STAT6 are predominantly activated by IFN-α and IL-4, respectively (101). STAT proteins contain six domains, including an N-terminal domain, coiled coil domain, DNA-binding domain, linker domain, SH2 domain, and transactivation domain (98). Each STAT member can be activated by a JAK via phosphorylation of a conserved tyrosine. Activated STATs translocate to the nucleus where they act as transcriptional regulators. Although STAT members including STAT1, 3, and 5 are activated by JAK2 following GH induction (Figure 3), STAT5 is the predominant transcription factor that delivers GH-induced cell proliferation and actions. Activated STAT5 forms homodimers, whereas active STAT1 and STAT3 form both homodimers and STAT1–STAT3 heterodimers (Figure 3) (7, 102, 103).
Src family kinases are non-receptor tyrosine kinases, which comprise nine members, namely, Src, Lck, Hck, Fyn, Blk, Lyn, Fgr, Yes, and Yrk, all of which share a common multidomain topology consisting of consecutive SH3, SH2, and SH1 KDs and a “unique” region located close to the N-terminus, which differentiates each member of the family. The regulation of SFKs is accomplished through mechanisms involving phosphorylation, dephosphorylation, and complex intramolecular interactions that lead to SH3 and SH2 domain displacement (104, 105). Members of the SFKs are essential for the regulation of immune system function and cellular homeostasis (106). Lyn is a unique member of this family because it acts both as a signal amplifier and also as a signal inhibitor while others are known to only play a single role (107). The major functions of Lyn have been attributed to signaling of hematopoietic growth factor receptors and blood cell development as it is expressed in all blood cells except T lymphocytes (108).
Studies have shown that GH can activate SFKs independent of any JAK2 activation (Figure 4) (109). The SFK member Lyn was found to be associated with the membrane proximal 150 residues of the cytoplasmic domain of human GHR, and Lyn was still shown to be activated by GH when using JAK2-deficient γ2A cells. Furthermore, it was reported that a mutant of the GHR Box1, resulting in loss of GHR-mediated JAK2–STAT signaling, still responded to GH stimulation through the activation of SFKs and ERK1/2 (109, 110). These studies suggested that this novel SFK-dependent pathway involves the activation of PLCγ, and a subsequent release of inositol 1,4,5-triphosphate (IP3) and diacylglycerol which leads to an increase in the cytoplasmic calcium ion concentration-activating Ras guanine nucleotide exchange factor RasGRP1. This process results in RAS activation and initiation of the ERK1/2 signaling pathway (109). Related studies using growth hormone as a ligand confirmed that this JAK2-independent pathway can activate ERK1/2 through the activation of Ras-like small GTPases, RalA, and RalB in NIH-3T3 cells and through the activation of Rap1 and Rap2 (111, 112). Mutations introduced in a loop of the GHR ECD that alters GH-induced conformational changes resulted in impaired ERK1/2 activation but not JAK2 and STAT5 activation. It was shown that GHR interacts with Lyn (an SFK), and that a deficiency in Lyn activation was responsible for the impaired ERK1/2 activation (109). These studies revealed that the extent of JAK2 or SFK signaling is regulated by conformational changes in the ECD. Interestingly, exon 3 deleted GHR, which results in the deletion of 22 amino acids in the ECD exhibits altered ERK signaling, but normal STAT5 and AKT signaling. This study highlighted the physiological importance of altering the signaling strength of these pathways as exon 3-deleted GHR individuals were found to be of higher stature with extended lifespan (113). Studies on closely related cytokine receptors have demonstrated SFK activation for TPOR, EPOR, and PRLR upon ligand binding (114–117). In addition, in a similar manner as shown for GHR, studies illustrate that structural changes in the ECD of EPOR and TPOR alter the extent of JAK2 and SFK signaling independently (118–120). The strength of JAK2 or SFK may also be significantly affected by different expression levels of each kinase in different cell types (109). Evidence also suggests JAK2 may not be the only PTK responsible for the tyrosine phosphorylation of STAT5 following GH stimulation, and that SRC may be taking this role in certain cell types and cytokine receptor systems (115, 121, 122).
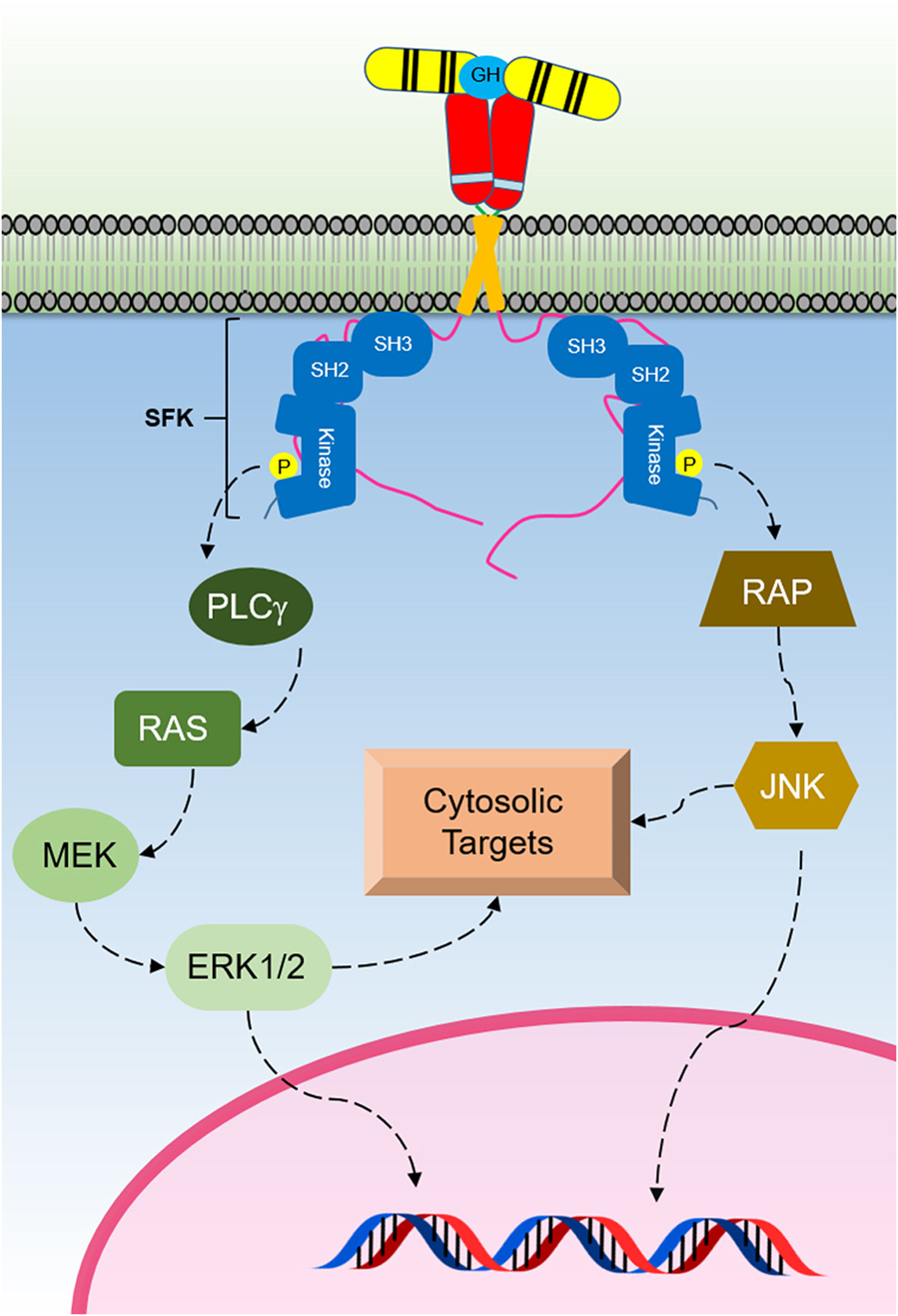
Figure 4. The Src family kinase (SFK) signaling initiated by the activated growth hormone receptor (GHR). SFKs are activated by GH binding to GHR. This signaling pathway activates ERK1/2 that regulates cytosolic targets and gene transcription.
Negative regulation of cytokine receptor signaling is critical to the correct function of cytokine receptors as dysregulated negative regulation of signaling can lead to disease states. Negative regulators of JAK–STAT signaling can be grouped into three main families, and this includes the suppressor of cytokine signaling (SOCS) protein family, protein tyrosine phosphatase (PTP), and protein inhibitor of activated STAT (PIAS). Apart from these protein families, the SH2-domain containing LNK (SH2B3) adaptor protein has been shown to be an important negative regulator in the JAK–STAT pathway. These negative regulators act in different ways to suppress JAK–STAT signaling. They can target JAKs, STATs, and/or receptors and may also involve other proteins of the proteasomal degradation pathway (123). The various mechanisms that are used by these signal regulators in modulating JAK–STAT signaling will be focused on below.
The SOCS protein family consists of SOCS1–7 and CIS. SOCS genes are translated as a result of cytokine-induced activation as part of the JAK–STAT negative feedback loop. SOCS proteins bind JAKs or their receptors and act by (i) abolishing the kinase ability of JAK, (ii) abolishing the ability of JAK to bind to the receptor, (iii) blocking STATs from being recruited to the receptor, and (iv) mediating the ubiquitination of JAKs and STATs for degradation (124).
SOCS1–3 and CIS have been shown to negatively regulate GH-mediated signaling (Figure 5). SOCS2 appears to be the major SOCS-negative regulator of GH signaling as socs2−/− mice are 30–40% larger than WT littermates while other SOCS knockouts (KOs) do not significantly affect size (124, 125). The SOCS proteins contain a conserved SOCS box (126, 127) that is located at the C-terminus which facilitates ubiquitin-mediated proteasomal degradation of their associated target proteins (128). As the SOCS box comprises (i) BC box that interacts with elongin B and C and the (ii) Cul box that recruits Cullin 5, these protein complexes can associate with the RING-box protein 2 and other components to form the E3 ubiquitin ligase complex (128–130). Interestingly, the crystal structure of the SOCS2–Elongin B/C complex revealed that the C-terminus of the SOCS box and the N-terminus of the extended SH2 subdomain (ESS) can interact and maintain the stability of the SH2–SOCS box domain interaction (131). This finding shows that all three domains of SOCS2 (N-terminal ESS, SH2, and SOCS box) can undergo conformational changes and work together to channel their substrates to ubiquitin-mediated degradation. Many of the functional studies on SOCS proteins have focused on SOCS1 and SOCS3. These are the only SOCS that contain the kinase inhibitory region (KIR) at their N-terminal domain. The KIR is able to bind and block the catalytic cleft of JAK thereby inhibiting the ability of JAK kinase to phosphorylate its substrate and consequently inhibiting signaling (132–134). The importance of SOCS2-mediated degradation of GHR has recently been underscored by a study showing that this was the key mechanism that causes an increased risk of developing lung cancer in individuals that carry an SNP in GHR resulting the in the amino acid change P495T in the GHR ICD. This amino acid change was shown to cause a structural change that impaired binding of SOCS2 resulting in reduced degradation of GHR and extended signaling in response to GH (135).
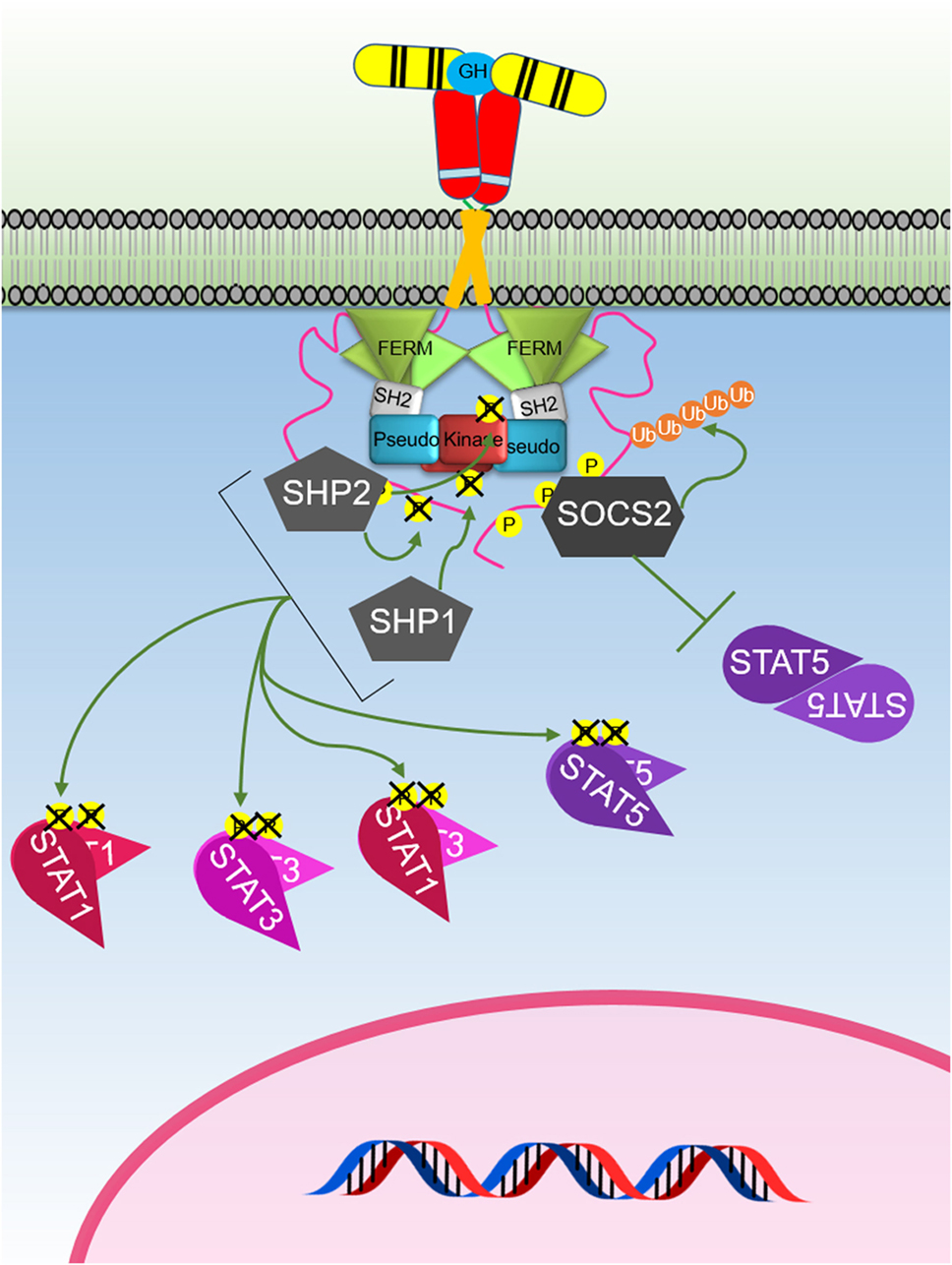
Figure 5. The major negative regulation of growth hormone receptor (GHR) signaling. GH-activated GHR results in suppressor of cytokine signaling (SOCS) 2 binding to a phosphorylated tyrosine on the GHR intracellular domain. This blocks signal transducer and activator of transcription (STAT) 5 binding and induces ubiquitination of the GHR leading to targeted degradation of the receptor. SHP1 and SHP2 act to dephosphorylate and inactivate Janus kinase 2 and STATs, while SHP2 is known to also dephosphorylate the receptor.
Tyrosine phosphatases, as their name suggests, act to inhibit signaling by dephosphorylating their substrate. There are four groups of PTPs (based on their substrate recognition): (i) non-receptor PTPs such as SHP1 and SHP2, (ii) receptor tyrosine phosphatases such as CD45, (iii) dual-specificity phosphatases, and (iv) low molecular weight phosphatases (136). According to their subcellular localization, PTPs can affect either JAKs or STATs. Those that are located on the membrane or in the cytoplasm will act on JAKs whereas those that are localized to the nucleus act on STATs. Both the non-TM PTPs and receptor protein tyrosine phosphatases are cysteine based and recognize only tyrosine phosphorylated residues and not serine or threonine residues (137, 138).
Several PTPs have been implicated in regulating GH signaling including SHP1, SHP2, PTP-1B, and PTP-H1 (139–144). The well-studied SHP1 (PTPN6; also known as HCP, PTP1C, SH-PTP1, and Hcph) is generally expressed in hematopoietic cell lineages whereas SHP2 (PTPN11; also known as PTP1D, SYP, SH-PTP2, and SH-PTP3) has ubiquitous expression. Both SHP1 and SHP2 have high sequence and domain homology whereby they contain two SH2 domains at their N-terminal region, denoted as N- and C-SH2 domains, and a PTP catalytic domain. They also possess a C-terminal tail with tyrosine residues that can be phosphorylated to regulate PTP activity (145, 146). Interestingly, SOCS1 can aid SHP1 in downregulating JAK–STAT signaling via the erythropoietin and PRLR. It has been proposed that the activation of the receptor by ligand binding recruits SHP1 to the JAK2–receptor complex and results in the phosphorylation of the C-terminus of SHP1 where the phosphorylated residues act as docking sites for the Grab2/SOCS1 complex to bind (147). SOCS1 is subsequently released and then binds to JAK2 to inhibit its kinase activity (147). This proposed mechanism suggests that negative regulators can associate with each other to regulate signaling.
Mouse studies with deficiency of SHP1 or SHP2 highlight the importance of these phosphatases. The motheaten phenotype is due to a splicing mutation in SHP1 causing an absence of detectable protein and mice that have severe disorders of hematopoietic cells resulting in death within 3 weeks after birth (148). Mice with a targeted deletion within the N-terminal SH2 domain result in embryonic lethality (149). Mutation of SHP2 accounts for more than half of the cases of Noonan syndrome, characterized by proportional short stature, facial dysmorphia, and heart disease (150). These mutations in SHP2 result in hyperactive phosphatase activity, notably with elevated basal activity and result in inhibition of GH-induced insulin-like growth factor-1 (IGF-1) production (143, 151). SHP2 has been shown to associate with a specific phosphorylated tyrosine residue in GHR with high affinity and mutation of this tyrosine to phenylalanine results in prolonged GH activation of STAT5 (140). Studies have shown an intriguing regulation of GHR signaling by IGF-1R, which suggested that IGF-1R positively regulates GH-mediated signaling by preventing PTP-1B suppression of GH activation of STAT5 (144).
Protein inhibitor of activated STATs are constitutively expressed, and, similar to SOCS, are involved in the negative feedback loop of cytokine-induced JAK–STAT signaling. Different PIAS proteins affect different STAT transcription factors. PIAS proteins may affect signaling by (i) inhibiting the DNA-binding activity of STATs, (ii) substrate SUMOylation, or (iii) recruit other transcriptional co-repressors. While PIASs are able to associate with STATs, they are also able to regulate other transcription factors including SMADs, androgen receptor, c-JUN, and p53. The PIAS family of transcriptional co-regulators consist of PIAS1, PIASxα, PIASxβ, PIAS3, PIAS3L (PIAS3β), PIASy, and PIASyE6. Although PIAS proteins are known as negative regulators of JAK–STAT signaling, their specific role in GH-mediated signaling is not well studied.
The LNK adaptor protein is also involved in inhibiting downstream JAK–STAT signaling. LNK is encoded by the SH2B3 gene and is part of the family of SH2-containing adaptor protein that includes SH2B (or SH2B1) and APS (or SH2B2). However, these adaptor proteins have different functions as the binding of SH2B or APS to JAK2 enhances its kinase activity whereas LNK binding to JAK2 inhibits its kinase activity (152). LNK is mainly expressed in hematopoietic cells and is known to be important for regulating a number of signaling pathways mediated by the TPOR (MPL), erythropoietin receptor (EPOR), stem cell factor receptor (KIT and SCFR), macrophage colony-stimulating factor receptor (CSF1R and C-FMS), and platelet-derived growth factor receptor (PDGFRB). Although LNK has been shown to be expressed in a range of tissue including muscle, brain, testis, and hematopoietic cells (153), its ability to regulate GHR signaling is currently not clear.
The effects of cellular signaling from GHR activation are responsible for a vast array of important physiological roles. Growth hormone is secreted by the anterior pituitary gland and not only has a role in increasing bone length, bone density, and muscle mass during childhood and adolescence but also importantly in the regulation of metabolism of lipids, carbohydrates, and body water throughout life (Figure 6) (154). The effects of GH are exerted by binding to the GH receptors on target cells, which in turn stimulates the production and secretion of IGF-1 from many tissues, mainly the liver (155). Since 1957 when IGF-1 and IGF-2 were identified and first designated as “sulfation factors” (156), the interest in the study of these molecules that structurally resembled proinsulin increased, especially when IGF-1 was found to be the mediator of the anabolic and mitogenic activity of GH (157). IGFs were first named as somatomedins due to their concentration dependence by GH regulation. A subsequent isolation and amino acid sequence determination of two homogeneous polypeptides from purified non-suppressible insulin-like activity factors (158–160) established the current designation of these molecules as insulin-like growth factors (IGFs) 1 and 2 (161). IGF-1 is a 70 amino acid peptide with a molecular weight of 7,649 Da. It has the ability to bind to the insulin receptor, although with low affinity. Most IGF-1 is secreted by the liver and acts as an endocrine hormone, although it can be secreted by many other tissues (162). One of the main roles for which IGF-1 has promoted subsequent research is its involvement in growth and its relation to growth hormone. Exogenous IGF-1 was shown to stimulate growth when administered to hypophysectomized rats (163, 164). Furthermore, children with IGF-1 deficiency-primary GH insensitivity or children with Laron syndrome who were treated with biosynthetic IGF-1 (165) showed increases in their serum alkaline phosphatase and serum procollagen and IGF-binding protein-3 (IGFBP-3) (165–167). This treatment was subsequently widely used in other parts of the world (168, 169). In terms of efficacy, GH and IGF-1 both stimulated linear growth but some variables including the greater growth deficit in infants with Laron syndrome than those with isolated growth hormone deficiency, insufficient IGF-1 dose, or the IGF-1 dependency on the GH-linked stem cell population of prechondrocytes made GH more efficient in terms of linear growth stimulation (170, 171). However, IGF-1 was shown to be an important growth-related hormone that has a GH-independent growth stimulating effects that in some cases acts synergistically with GH (172). On the other hand, as described later, GHR signaling has been shown to have growth promoting effects independent of IGF-1.
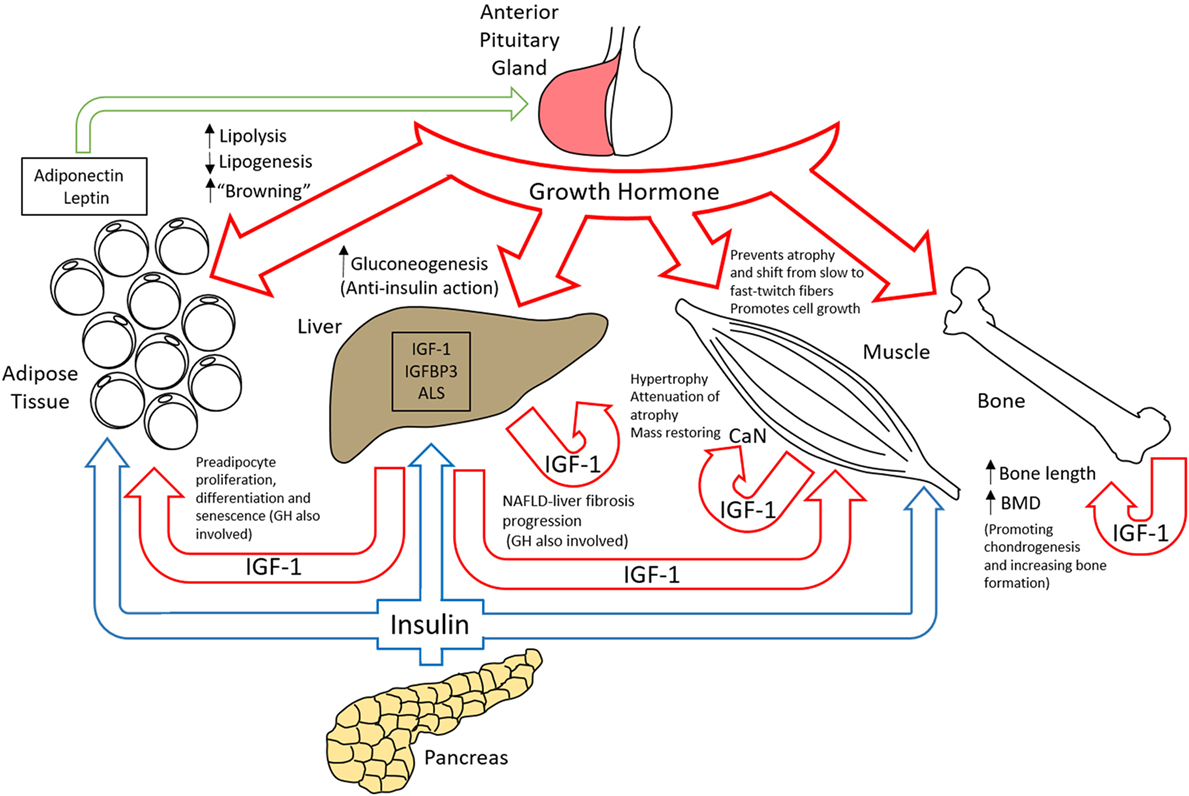
Figure 6. Organ-specific roles of the GH/insulin-like growth factor-1 (IGF-1) axis.
The anti-insulin action of GH is very well established (173) as liver, skeletal muscle, and adipose tissue develop insulin resistance induced by GH administration (174–177). Liver IGF-1-deficient mice show a reduction of around 75% in circulating IGF-1 levels and as a consequence due to the lack of negative feedback, these mice have about a fourfold increase in GH production. These liver IGF-1-deficient mice were crossed with mice that express a mutant GH that acts as an antagonist, and the resulting mice showed improved insulin sensitivity and decreased blood glucose and insulin levels (175). An important function of GH besides the control of growth is to provide a mechanism to cope with periods of food deprivation. In this way, GH stimulates lipolysis and provides free fatty acids and glycerol as substrates for energy metabolism and also inhibits insulin-induced suppression of hepatic gluconeogenesis counteracting insulin action (178). IGF-1 binds mainly to IGF-1 receptors located in the skeletal muscle and/or hybrid insulin/IGF-1 receptors, and it has been postulated as the mediator of enhanced insulin action as it binds insulin receptors with very low affinity (179). The interaction between IGF-1 and GH effects has made it difficult to determine whether IGF-1’s effect is due to the enhancement of insulin action or the suppression of GH secretion (180). Some studies suggested phosphatidylinositol 3-kinase as the key enzyme involved in the regulation of glucose uptake into cells by GH (175, 181, 182) while others found this mechanism to be of low significance as these experiments have not been replicated in humans (176, 177).
GH receptors and IGF-1 receptors are abundant on not only adipocytes but also preadipocytes, fibroblasts, various immune cells, and endothelial cells (183–188). The connection between GH and energy metabolism is not just restricted to the promotion of lipolysis or the prevention of the lipogenesis (189, 190) but GH together with IGF-1 plays a crucial role in preadipocyte proliferation, differentiation, and senescence (191–194). Moreover, GH action alters the level of adiponectin and leptin in the circulation, and these adipokines can at the same time, control the GH/IGF axis by modulating GH output from the pituitary gland (195). Furthermore, GH was found to induce the so-called “browning” of adipocytes by increasing the amount of beige adipocytes in the white adipose tissue and as a consequence, decreasing the store of excess lipid as white adipocytes, which may function as a protective mechanism against obesity (196). In obesity, the spontaneous (197, 198) and stimulated (199) pulsatile patterns of GH secretion governed by the hypothalamus are blunted, and the accumulation of abdominal adiposity, particularly visceral adipose tissue mass, was found to be a stronger negative determinant of GH secretion than other factors including age, sex, or generalized obesity (200–202). In relation to GHR expression, its level was found to be influenced by nutritional status (203). For instance, white adipose tissue from obese women showed to have a lower expression of total GHR mRNA compared with those from lean women, and the expression of GHR mRNA was also found to be higher in visceral white adipose tissue than in subcutaneous white adipose tissue in the lean women group (204).
The significance of the GH/IGF-1 axis in the muscular system was highlighted when it was found that calcineurin (CaN), a calcium-calmodulin-dependent protein phosphatase that activates slow isoform of troponin I and myoglobin genes, was regulated by IGF-1 and that inhibiting CaN blocked IGF-mediated hypertrophy of cultured skeletal muscle cells (205). In addition, as shown using an animal model of congestive heart failure (CHF), GH prevented the atrophy and the shift from slow-twitch fibers to fast-twitch and more fatigable glycolytic fibers which works as an adaptation mechanism of the skeletal muscle to a low cardiac output characterizing the muscle atrophy as a common sign of patients with advanced CHF (206). GH has been shown to promote the cell growth of the skeletal muscle by facilitating the fusion of myoblasts with nascent myotubes, and that IGF-1 expression in myotubes is not regulated by GH (207). On the other hand, IGF-1 has been implicated in skeletal muscle hypertrophy, attenuation of age-related skeletal muscle atrophy, and restoring and improvement of muscle mass when virally delivered (208). The diversity of actions of both components of this axis in promoting the growth of skeletal muscle increases the bases to explain their synergistic effect in the development of cardiac diseases characterized by hyperplasic and hypertrophic muscle fibers. GH was also found to promote an increase in mitochondrial oxidative capacity and abundance of several mitochondrial genes when acutely administered (209) while exogenous administration of IGF-1 was associated with a reduced susceptibility of dystrophic muscles to contraction-induced injury (210). In addition to the roles of the GH/IGF axis in pathological conditions of the muscular system, studies have reported an important role of the GH/IGF axis in physiological conditions of the tendon and skeletal muscle. Both circulating GH and IGF increase in response to exercise (211, 212) and although the IGF-1 response to exercise is not as pronounced as the GH one, it is not necessarily a consequence of GH release (213, 214) as it was shown by the fact that IGF-1 can rise in response to exercise in patients with pituitary insufficiency (212). Circulating GH consistently increases after high-intensity exercise in adult males (215), and its level was related to the central effort in the brain to perform exercise (216). In the same way, local levels of IGF-1 were also induced in the muscular tissue subjected to both long- and short-term loading (217–219).
In relation to the bone, IGF-1 is an important mediator of the effects of GH and modulates bone growth through its paracrine action in the growth plate (220) as it was demonstrated that the selective deletion of the igf1 gene in the liver and a consequent decrease in systemic IGF-1 did not affect the animal’s body and femoral growth (221). However, a targeted disruption of igf1 expression in mouse chondrocytes reduced bone length (222). GH may act directly on skeletal cells having IGF-1-independent effects on bone growth (223, 224). In this regard, IGF-1 effects are produced by the enhancement of the function of mature osteoblasts (225) and promotion of osteoblast differentiation (226) and bone formation (227–229). Furthermore, higher levels of IGF-1 are associated with higher bone mineral density in adult obese individuals (230).
The liver has important roles related to the function of the GH/IGF-1 axis besides the production and secretion of IGF-1 induced by GH. It also has a role in the production of IGFBP-3 by Kupffer cells (231, 232). IGF-binding proteins (IGFBPs) antagonize the activity of IGFs due to their high affinity to the IGF-1 receptor (227). Of the six IGFBPs, IGFBP-3 is the major binding protein for IGF-1 that modulates its biological effects (233). More than 80% of the circulating IGF-1 forms a complex with IGFBP-3 and the acid-labile subunit (ALS), which prolongs the half-life of serum IGF-1 (227, 234). Furthermore, IGFBP-3 levels are directly and indirectly modulated by GH and IGF-1 (232, 235) and are altered by nutritional deficiencies and liver function (236, 237).
Chronic liver disease (CLD) has been shown to correlate with high IGFBP-1 and low IGFBP-3, with IGFBP-3 being a major limiting factor for the reported low circulating IGF-1 levels (237). A similar observation was reported showing a significant relation between cirrhosis and circulating and venous ALS levels, which were decreased in relation to liver dysfunction (238). In non-alcoholic fatty liver disease (NAFLD) and advanced fibrosis, it was shown that the levels of GH, IGF-1, and IGFBP-3 varied according to the severity of the steatosis, which differed from the variation in samples taken from patients with hepatitis C virus-related CLD (239). The same study reported that patients with NAFLD and advanced fibrosis had high levels of GH and low levels of IGF-1 and IGFBP-3 (239). However, patients with NAFLD have significantly lower levels of serum GH in comparison with controls (240). This is not the case in cirrhotic patients where GH levels in serum were reported to be elevated (237) as the GH secretion rate was found to be increased (241). On the other hand, as IGF-1 and IGFBP-3 are regulated by GH secretion, the lack of negative feedback in cirrhotic patients with low levels of IGF-1 may be the cause of the exaggerated secretion of GH (239).
Circulating GH/IGF-1 levels peak in the second decade of life (242, 243) with this axis being the main regulator of postnatal growth while modulating other important physiological processes as well (244). Subsequently, secretion of GH and IGF-1 rapidly declines to very low levels in people aged 60 years or more, an event that was named “somatopause” (242, 245). This fact supported related studies that led to the belief that recombinant human GH was a potent antiaging agent. However, it was later found that while the effects on body composition from rhGH treatment were minor, adverse effects were significant and include the development of diabetes mellitus (246). On the other hand, decreasing GH/IGF signaling in various species including mice extended their lifespan (247), which was also related to a reduced risk of several age-related diseases including cancer (248). In a study of people with GHR deficiency, individuals showed IGF-1 deficiency and appeared to be protected against age-related diseases (249). It is interesting to note that although KO mice for GH receptor/GH binding protein and for GH-releasing hormone showed an extended longevity (250, 251), this effect was not observed when using targeted deletion of the GH receptor in the liver, muscle, or adipose tissue (Table 3) (252). As low levels of GH/IGF were also reported to delay puberty and reproduction in KO models, it seems feasible that this axis may be working by shifting a “biological timer” and delaying aging as a consequence, which may defer as well the arrival of age-related diseases (253). In contrast to the animal models of the absence of GHR signaling, humans with GHR deficiency do not live longer; however, the interpretation of this is complicated by a high rate of deaths caused by alcohol toxicity, liver cirrhosis, convulsive disorders, and other non-age-related deaths such as accidents (249). A recent study investigating a common polymorphism resulting in exon 3-deleted GHR and resulting in increased GH sensitivity found that this polymorphism also correlated with a striking increase in life span in males of around 10 years (113).
As GH/IGF signaling induces growth mechanisms, inhibiting this signaling should suppress cell proliferation and a reduction in the cancer risk; however, other important mechanisms that may be affected by this process include a decrease in immune responses and reduced favorable metabolic adjustments (266). Moreover, being the process of aging characterized by a low-grade inflammation that is linked to several age-related diseases (267), the inhibition of the mechanisms that lead to it may consequently affect the development of these pathologies. According to these data, using GH antagonists or somatostatin analogs should slow aging (268). Currently, these drugs are being used for the treatment of acromegaly. Although these drugs show beneficial effects, such as somatostatin analogs, which normalize IGF-1 levels in around 45% of treated patients as is the case for lanreotide (269), they have also been reported to inhibit insulin secretion triggering the development of diabetes mellitus and aggravating the insulin resistance found in acromegalic patients (268, 270). Moreover, they have shown to produce other major adverse effects on the gastrointestinal tract (271). Therefore, it becomes necessary to understand the process of aging as a whole to detect the best target for treating age-related diseases.
GH and its specific receptor, GHR, are involved in multiple biological and physiological actions contributing to cell proliferation and differentiation. Dysregulation of GH–IGF-1 axis can amplify the synergistic effect of GH and IGF-1 to promote uncontrolled cell proliferation, cell movement, angiogenesis, and suppress apoptosis to increase risk of neoplasia (272). The excessive production of GH and associated complications has been studied long before its broader role in human health was discovered. Acromegaly is an endocrine condition, which is manifested with excess secretion of pituitary GH accompanied by an elevated level of circulatory IGF-1 (273). This is usually associated with high mortality and morbidity, mostly due to cardiovascular and/or respiratory diseases, renal failure, and elevated risk of multiple cancers especially colorectal cancer (274–277). A review of over 300 case–control and cohort studied used final height as an indication of GH–IGF-1 action and found that individuals over 175 cm have a 20% higher chance of developing prostate cancer, a 22 and 20–60% higher chance of breast and colorectal cancers, respectively, when compared with individuals below 160 cm (278, 279). Also, another study of 14-year-old females found that girls in the top fifth of height have an adjusted relative risk of over 1.5 for breast cancer, exceeding BMI and age of menarche as examined risk factors in the study (280). In addition, meta-analysis of published research investigating the association of IGF-1–IGF-2 and their binding proteins IGFBP-1–6 with prostate cancer confirmed that elevated circulating level of IGF-1 positively correlates with higher prostate cancer risk (281). On the other hand, this study found no or little evidence for the association of IGF-2, IGFBP-1, -2, and -3 with increased prostate cancer risk. In stark contrast, GHR-deficient individuals show a lack of deaths from cancer (249). A recent meta-analysis showed that increased height associated with an increased risk of lung cancer (282). The link between GHR signaling and lung cancer has been shown in two independent genetic studies that identified an SNP in GHR that correlated with an increased risk of lung cancer (283, 284). Recently, a study showed that functional consequence of this GHR SNP was an impaired interaction with SOCS2 resulting in an extended signaling following GH stimulation (135).
The link between acromegaly and increased risk of certain diseases especially colorectal cancers is well established, and there is a consensus on this issue even though there are some studies reporting contradictory results (285). However, the extent of this association is unclear since some studies have reported a 7.6-fold increase in the rate of colorectal cancers in acromegalic patients compared with control groups (275), whereas another study has reported as low as twofold increased risk of colorectal cancer in acromegalic patients (286, 287). Furthermore, a recent study investigating the prevalence of malignancies among 160 acromegalic patients over the period of 32 years using mammography, colonoscopy, thyroid, and prostate ultrasonography found thyroid cancer as the most frequent cancer (n = 17, 10.6%) followed by breast and colorectal cancers (288).
GH has been prescribed for children with GH deficiency since late 1950s using hormone extracted from the pituitary gland (289), and it was successfully produced in 1985 using recombinant DNA technology and approved to be administered for treatment of various disorders (290). However, in the past couple of decades, there has been a growing debate on GH treatment and its diabetogenic effects, impaired skeletal growth, and increased risk of de novo or recurrent cancers (272). Long-term survival of childhood cancer patients who have gone under total body or cranial irradiation in preparation for bone marrow transplantation or as a part of treatment for brain tumors or acute lymphoblastic leukemia are at higher risk of developing GH deficiency (285, 291). Although some initial studies reported recurrent brain tumors as a frequent cause of death in children treated with GH (292), a study that followed 180 children treated with GH who had brain tumors concluded that there was not a substantial trend in relative risk of recurrence with cumulative time for which GH treatment had been administrated and GH does not result in elevated risk of recurrent brain tumors (293). However, the study also suggested continued surveillance based on the rising trend in mortality relative risks with longer follow-ups. Initial studies had reported leukemia incidences in children treated with GH replacement in Japan (294, 295); however, later follow-up studies were unsuccessful in establishing a link between GH therapy and leukemia when patients with existing risk factors were excluded (296). Another comprehensive study from Genentech’s National Cooperative Growth Study examined 40,000 children undergoing GH therapy with prolonged follow-ups and compared them to age-matched general public without known leukemia risk factors and showed a comparable incidence of leukemia for GH recipients (297, 298).
Growth hormone receptor has in some settings been observed to have a high degree of localization to the nucleus (272), and this nuclear localization has been shown to be induced by GH (299, 300). This nuclear localization has particularly been observed in proliferating cells including a range of different cancers such as colorectal carcinoma, melanoma, uterine cervical neoplasms, breast cancer, and hepatocellular carcinoma. Nuclear localized GHR has been observed in a number of species such as pig (300), rat (299), and fish (301). By targeting the GHR to the nucleus by fusing a nuclear localization signal to the receptor, it was found in the absence of GH that several genes known to be involved in oncogenesis were upregulated, and further studies provided evidence that the GHR can bind to a transcriptional regulator (299, 302). Studies also suggest that autocrine GH production may play an important role in cancer (272).
Gene KO (Table 2) and targeted mutant GHR mouse models (Table 3) have enabled us to better understand the role of GH signaling in postnatal growth and development. Hepatocytes generate the majority of circulating IGF-1 that is found in the blood. As IGF-1 expression is induced as a result of GH binding to GHR, hepatic IGF-1 levels are predominantly regulated by GH that is produced by the pituitary. The Somatomedin hypothesis states that postnatal growth that is induced by GH is mediated by hepatic IGF-1 (previously known as Somatomedin C or sulfation factor) in an endocrine manner (156). However, studies have shown that postnatal growth is actually mediated by IGF-1 in an autocrine/paracrine manner instead (303). To elucidate this, mice that were deficient of hepatic igf1 using the Cre–loxP system were generated (303). These mice have markedly reduced circulating IGF-1 levels (approximately 80% reduction), a slight reduction in body length while body mass was maintained (221, 304). By contrast, igf1 KO in the whole mouse led to mice with dramatically reduced body mass and length (305–307). However, several organs were relatively larger, e.g., liver, brain, kidney, and heart (307, 308). The increase in liver size (liver:body weight ratio) could be explained by the increase in circulating GH as a result of the lack of the IGF-1-mediated negative feedback mechanism, where circulating IGF-1 produced by the liver can regulate secretion of GH and consequently the size of the liver (164, 309–311). In summary, this study showed that although most circulating IGF-1 is derived from hepatocytes, it is not the main IGF-1 that is required for postnatal growth and development (303).

Table 2. Growth hormone receptor (GHR) transgenic mice.

Table 3. Growth hormone receptor (GHR) conditional knockout (KO) mice.
GH deficiency leads to reduced bone mineral density, but administration of GH is able to subvert this effect (312, 313). Previously, growth has been thought to be directly mediated by IGF-1, instead of GH (156). Studies using ghr KO mice showed significantly decreased body mass as well as smaller liver and brain mass/size than control mice (250, 310, 314–319) and growth delay (250, 254). Complete igf1 KO mice also demonstrated a similar phenotype (305–307). However, this resulted in perinatal lethality of most mice whereas those that survived were only approximately half the size of the control mice (320). Consistent with this observation, patients with IGF1 gene deficiency are growth retarded (321).
In mice that have combined KO of ghr and igf1, the growth deficiency is significantly more severe than in each individual KO (322), indicating that GH has growth mediating effects that are independent of IGF-1. Studies have shown that GH is capable to directly induce growth. When GH is infused locally into one leg of a hypophysectomized (GH-deficient) rat, that leg grows significantly longer than the other (323). Interestingly, in socs2 KO mice, GH was shown to promote linear growth by local mechanisms independent of IGF-1 (324).
Postnatal growth of mice has been shown to rely on signaling mediated by JAK2 and STAT5, downstream from the GHR (110, 255, 256). Mice with ghr Box1 deletion by substitution of each proline in the Box1 to alanine (which eliminates JAK2 binding and signaling, while still allowing signaling to occur via the SFK–ERK1/2 pathway) have obstructed growth with a similar phenotype observed in mice with ghr KO (109, 110). A similar growth phenotype is also observed in mice with a truncated ghr (at amino acid position 391), which eliminates all GH-mediated STAT5 signaling while still allowing for JAK2, STAT1, and STAT3 activation (110). These mouse models demonstrate the crucial involvement of JAK2 and STAT5 in regulating postnatal growth by GH. However, they also show that GH-mediated SFK signaling does not play a significant role in regulating growth. Nevertheless, it is possible that the relative levels of JAK2 or SFKs in certain cell types may be a determinant as to which kinase would be dominant for signaling (110).
Although we have learnt a vast amount on the molecular mechanism of GHR activation, signaling, physiological aspects, and roles of GH signaling in disease states, there is still much to learn. In particular, this is due to the wide range of physiological roles that GH has, making it an important player in many biological conditions and diseases. At the molecular and cellular level, the activation and role of SFK signaling is still currently not well understood. As our knowledge of the molecular signaling nature of GHR increases, our ability to specifically target the receptor and its signaling pathways for a diverse range of therapeutic purposes should also increase.
All the authors contributed equally. All the authors contributed to drafting and editing the manuscript. All the authors contributed to figures and tables.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the project grant APP1084797 from the National Health and Medical Research Council (NHMRC) of Australia. The authors would like to acknowledge the support from The University of Queensland Diamantina Institute. CMML was supported by Hardy Brothers Scholarship for Research in Blood Cancer at the UQ Diamantina Institute.
1. Waters MJ, Brooks AJ. JAK2 activation by growth hormone and other cytokines. Biochem J (2015) 466(1):1–11. doi:10.1042/BJ20141293
2. Brooks AJ, Dehkhoda F, Kragelund BB. Cytokine receptors. In: Belfiore A, LeRoith D, editors. Principles of Endocrinology and Hormone Action. Springer International Publishing (2016). p. 1–29.
3. de Vos AM, Ultsch M, Kossiakoff AA. Human growth hormone and extracellular domain of its receptor: crystal structure of the complex. Science (1992) 255(5042):306–12. doi:10.1126/science.1549776
4. Baumgartner JW, Wells CA, Chen CM, Waters MJ. The role of the WSXWS equivalent motif in growth hormone receptor function. J Biol Chem (1994) 269(46):29094–101.
5. Hamming OJ, Kang L, Svensson A, Karlsen JL, Rahbek-Nielsen H, Paludan SR, et al. Crystal structure of interleukin-21 receptor (IL-21R) bound to IL-21 reveals that sugar chain interacting with WSXWS motif is integral part of IL-21R. J Biol Chem (2012) 287(12):9454–60. doi:10.1074/jbc.M111.311084
6. Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol (2003) 3(11):900–11. doi:10.1038/nri1226
7. Brooks AJ, Waters MJ. The growth hormone receptor: mechanism of activation and clinical implications. Nat Rev Endocrinol (2010) 6(9):515–25. doi:10.1038/nrendo.2010.123
8. Brooks AJ, Wooh JW, Tunny KA, Waters MJ. Growth hormone receptor; mechanism of action. Int J Biochem Cell Biol (2008) 40(10):1984–9. doi:10.1016/j.biocel.2007.07.008
9. Waters MJ, Brooks AJ. Growth hormone receptor: structure function relationships. Horm Res Paediatr (2011) 76(Suppl 1):12–6. doi:10.1159/000329138
10. Argetsinger LS, Campbell GS, Yang X, Witthuhn BA, Silvennoinen O, Ihle JN, et al. Identification of JAK2 as a growth hormone receptor-associated tyrosine kinase. Cell (1993) 74(2):237–44. doi:10.1016/0092-8674(93)90415-M
11. Smit LS, Meyer DJ, Billestrup N, Norstedt G, Schwartz J, Carter-Su C. The role of the growth hormone (GH) receptor and JAK1 and JAK2 kinases in the activation of Stats 1, 3, and 5 by GH. Mol Endocrinol (1996) 10(5):519–33. doi:10.1210/mend.10.5.8732683
12. Witthuhn BA, Quelle FW, Silvennoinen O, Yi T, Tang B, Miura O, et al. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell (1993) 74(2):227–36. doi:10.1016/0092-8674(93)90414-L
13. Chu J, Gui CY, Fan J, Tang XD, Qiao RL. STAT1 is involved in signal transduction in the EPO induced HEL cells. Cell Res (1998) 8(2):105–17. doi:10.1038/cr.1998.11
14. Kirito K, Nakajima K, Watanabe T, Uchida M, Tanaka M, Ozawa K, et al. Identification of the human erythropoietin receptor region required for Stat1 and Stat3 activation. Blood (2002) 99(1):102–10. doi:10.1182/blood.V99.1.102
15. Lebrun JJ, Ali S, Sofer L, Ullrich A, Kelly PA. Prolactin-induced proliferation of Nb2 cells involves tyrosine phosphorylation of the prolactin receptor and its associated tyrosine kinase JAK2. J Biol Chem (1994) 269(19):14021–6.
16. DaSilva L, Rui H, Erwin RA, Howard OM, Kirken RA, Malabarba MG, et al. Prolactin recruits STAT1, STAT3 and STAT5 independent of conserved receptor tyrosines TYR402, TYR479, TYR515 and TYR580. Mol Cell Endocrinol (1996) 117(2):131–40. doi:10.1016/0303-7207(95)03738-1
17. Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev (1997) 11(2):179–86. doi:10.1101/gad.11.2.179
18. Ezumi Y, Takayama H, Okuma M. Thrombopoietin, c-Mpl ligand, induces tyrosine phosphorylation of Tyk2, JAK2, and STAT3, and enhances agonists-induced aggregation in platelets in vitro. FEBS Lett (1995) 374(1):48–52. doi:10.1016/0014-5793(95)01072-M
19. Drachman JG, Kaushansky K. Dissecting the thrombopoietin receptor: functional elements of the Mpl cytoplasmic domain. Proc Natl Acad Sci U S A (1997) 94(6):2350–5. doi:10.1073/pnas.94.6.2350
20. Schulze H, Ballmaier M, Welte K, Germeshausen M. Thrombopoietin induces the generation of distinct Stat1, Stat3, Stat5a and Stat5b homo- and heterodimeric complexes with different kinetics in human platelets. Exp Hematol (2000) 28(3):294–304. doi:10.1016/S0301-472X(99)00154-X
21. Tian SS, Tapley P, Sincich C, Stein RB, Rosen J, Lamb P. Multiple signaling pathways induced by granulocyte colony-stimulating factor involving activation of JAKs, STAT5, and/or STAT3 are required for regulation of three distinct classes of immediate early genes. Blood (1996) 88(12):4435–44.
22. Shimoda K, Feng J, Murakami H, Nagata S, Watling D, Rogers NC, et al. Jak1 plays an essential role for receptor phosphorylation and Stat activation in response to granulocyte colony-stimulating factor. Blood (1997) 90(2):597–604.
23. Ghilardi N, Ziegler S, Wiestner A, Stoffel R, Heim MH, Skoda RC. Defective STAT signaling by the leptin receptor in diabetic mice. Proc Natl Acad Sci U S A (1996) 93(13):6231–5. doi:10.1073/pnas.93.13.6231
24. Ghilardi N, Skoda RC. The leptin receptor activates Janus kinase 2 and signals for proliferation in a factor-dependent cell line. Mol Endocrinol (1997) 11(4):393–9. doi:10.1210/mend.11.4.9907
25. Silvennoinen O, Witthuhn BA, Quelle FW, Cleveland JL, Yi T, Ihle JN. Structure of the murine Jak2 protein-tyrosine kinase and its role in interleukin 3 signal transduction. Proc Natl Acad Sci U S A (1993) 90(18):8429–33. doi:10.1073/pnas.90.18.8429
26. Quelle FW, Shimoda K, Thierfelder W, Fischer C, Kim A, Ruben SM, et al. Cloning of murine Stat6 and human Stat6, Stat proteins that are tyrosine phosphorylated in responses to IL-4 and IL-3 but are not required for mitogenesis. Mol Cell Biol (1995) 15(6):3336–43. doi:10.1128/MCB.15.6.3336
27. Callus BA, Mathey-Prevot B. Interleukin-3-induced activation of the JAK/STAT pathway is prolonged by proteasome inhibitors. Blood (1998) 91(9):3182–92.
28. Caldenhoven E, van Dijk T, Raaijmakers JA, Lammers JW, Koenderman L, De Groot RP. Activation of the STAT3/acute phase response factor transcription factor by interleukin-5. J Biol Chem (1995) 270(43):25778–84. doi:10.1074/jbc.270.43.25778
29. Mui AL, Wakao H, O’Farrell AM, Harada N, Miyajima A. Interleukin-3, granulocyte-macrophage colony stimulating factor and interleukin-5 transduce signals through two STAT5 homologs. EMBO J (1995) 14(6):1166–75.
30. Brizzi MF, Aronica MG, Rosso A, Bagnara GP, Yarden Y, Pegoraro L. Granulocyte-macrophage colony-stimulating factor stimulates JAK2 signaling pathway and rapidly activates p93fes, STAT1 p91, and STAT3 p92 in polymorphonuclear leukocytes. J Biol Chem (1996) 271(7):3562–7. doi:10.1074/jbc.271.7.3562
31. Rosen RL, Winestock KD, Chen G, Liu X, Hennighausen L, Finbloom DS. Granulocyte-macrophage colony-stimulating factor preferentially activates the 94-kD STAT5A and an 80-kD STAT5A isoform in human peripheral blood monocytes. Blood (1996) 88(4):1206–14.
32. Stahl N, Boulton TG, Farruggella T, Ip NY, Davis S, Witthuhn BA, et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science (1994) 263(5143):92–5. doi:10.1126/science.8272873
33. Guschin D, Rogers N, Briscoe J, Witthuhn B, Watling D, Horn F, et al. A major role for the protein tyrosine kinase JAK1 in the JAK/STAT signal transduction pathway in response to interleukin-6. EMBO J (1995) 14(7):1421–9.
34. Ernst M, Najdovska M, Grail D, Lundgren-May T, Buchert M, Tye H, et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J Clin Invest (2008) 118(5):1727–38. doi:10.1172/JCI34944
35. Buchert M, Burns CJ, Ernst M. Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene (2016) 35(8):939–51. doi:10.1038/onc.2015.150
36. Kodama H, Fukuda K, Pan J, Makino S, Baba A, Hori S, et al. Leukemia inhibitory factor, a potent cardiac hypertrophic cytokine, activates the JAK/STAT pathway in rat cardiomyocytes. Circ Res (1997) 81(5):656–63. doi:10.1161/01.RES.81.5.656
37. Johnston JA, Bacon CM, Finbloom DS, Rees RC, Kaplan D, Shibuya K, et al. Tyrosine phosphorylation and activation of STAT5, STAT3, and Janus kinases by interleukins 2 and 15. Proc Natl Acad Sci U S A (1995) 92(19):8705–9. doi:10.1073/pnas.92.19.8705
38. Brunn GJ, Falls EL, Nilson AE, Abraham RT. Protein-tyrosine kinase-dependent activation of STAT transcription factors in interleukin-2- or interleukin-4-stimulated T lymphocytes. J Biol Chem (1995) 270(19):11628–35. doi:10.1074/jbc.270.19.11628
39. Haque SJ, Wu Q, Kammer W, Friedrich K, Smith JM, Kerr IM, et al. Receptor-associated constitutive protein tyrosine phosphatase activity controls the kinase function of JAK1. Proc Natl Acad Sci U S A (1997) 94(16):8563–8. doi:10.1073/pnas.94.16.8563
40. Lin JX, Migone TS, Tsang M, Friedmann M, Weatherbee JA, Zhou L, et al. The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity (1995) 2(4):331–9. doi:10.1016/1074-7613(95)90141-8
41. Cunningham BC, Ultsch M, De Vos AM, Mulkerrin MG, Clauser KR, Wells JA. Dimerization of the extracellular domain of the human growth hormone receptor by a single hormone molecule. Science (1991) 254(5033):821–5. doi:10.1126/science.1948064
42. Wells JA. Binding in the growth hormone receptor complex. Proc Natl Acad Sci U S A (1996) 93(1):1–6. doi:10.1073/pnas.93.1.1
43. Clackson T, Ultsch MH, Wells JA, de Vos AM. Structural and functional analysis of the 1:1 growth hormone:receptor complex reveals the molecular basis for receptor affinity. J Mol Biol (1998) 277(5):1111–28. doi:10.1006/jmbi.1998.1669
44. Walsh ST, Jevitts LM, Sylvester JE, Kossiakoff AA. Site2 binding energetics of the regulatory step of growth hormone-induced receptor homodimerization. Protein Sci (2003) 12(9):1960–70. doi:10.1110/ps.03133903
45. Fuh G, Cunningham BC, Fukunaga R, Nagata S, Goeddel DV, Wells JA. Rational design of potent antagonists to the human growth hormone receptor. Science (1992) 256(5064):1677–80. doi:10.1126/science.256.5064.1677
46. Rowlinson SW, Behncken SN, Rowland JE, Clarkson RW, Strasburger CJ, Wu Z, et al. Activation of chimeric and full-length growth hormone receptors by growth hormone receptor monoclonal antibodies. A specific conformational change may be required for full-length receptor signaling. J Biol Chem (1998) 273(9):5307–14. doi:10.1074/jbc.273.9.5307
47. Ross RJ, Leung KC, Maamra M, Bennett W, Doyle N, Waters MJ, et al. Binding and functional studies with the growth hormone receptor antagonist, B2036-PEG (pegvisomant), reveal effects of pegylation and evidence that it binds to a receptor dimer. J Clin Endocrinol Metab (2001) 86(4):1716–23. doi:10.1210/jcem.86.4.7403
48. Gent J, van Kerkhof P, Roza M, Bu G, Strous GJ. Ligand-independent growth hormone receptor dimerization occurs in the endoplasmic reticulum and is required for ubiquitin system-dependent endocytosis. Proc Natl Acad Sci U S A (2002) 99(15):9858–63. doi:10.1073/pnas.152294299
49. Gent J, Van Den Eijnden M, Van Kerkhof P, Strous GJ. Dimerization and signal transduction of the growth hormone receptor. Mol Endocrinol (2003) 17(5):967–75. doi:10.1210/me.2002-0261
50. Brown RJ, Adams JJ, Pelekanos RA, Wan Y, McKinstry WJ, Palethorpe K, et al. Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nat Struct Mol Biol (2005) 12(9):814–21. doi:10.1038/nsmb977
51. Brooks AJ, Dai W, O’Mara ML, Abankwa D, Chhabra Y, Pelekanos RA, et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science (2014) 344(6185):1249783. doi:10.1126/science.1249783
52. Liu Y, Jiang J, Lepik B, Zhang Y, Zinn KR, Frank SJ. Subdomain 2, not the transmembrane domain, determines the dimerization partner of growth hormone receptor and prolactin receptor. Endocrinology (2017) 158(10):3235–48. doi:10.1210/en.2017-00469
53. Bugge K, Papaleo E, Haxholm GW, Hopper JT, Robinson CV, Olsen JG, et al. A combined computational and structural model of the full-length human prolactin receptor. Nat Commun (2016) 7:11578. doi:10.1038/ncomms11578
54. Suzuki K, Nakajima H, Saito Y, Saito T, Leonard WJ, Iwamoto I. Janus kinase 3 (Jak3) is essential for common cytokine receptor gamma chain (gamma(c))-dependent signaling: comparative analysis of gamma(c), Jak3, and gamma(c) and Jak3 double-deficient mice. Int Immunol (2000) 12(2):123–32. doi:10.1093/intimm/12.2.123
55. Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell (1998) 93(3):397–409. doi:10.1016/S0092-8674(00)81168-X
56. Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell (1998) 93(3):373–83. doi:10.1016/S0092-8674(00)81166-6
57. Velazquez L, Fellous M, Stark GR, Pellegrini S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell (1992) 70(2):313–22. doi:10.1016/0092-8674(92)90105-L
58. Nosaka T, van Deursen JM, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, et al. Defective lymphoid development in mice lacking Jak3. Science (1995) 270(5237):800–2. doi:10.1126/science.270.5237.800
59. Pellegrini S, Dusanter-Fourt I. The structure, regulation and function of the Janus kinases (JAKs) and the signal transducers and activators of transcription (STATs). Eur J Biochem (1997) 248(3):615–33. doi:10.1111/j.1432-1033.1997.00615.x
60. Yamaoka K, Saharinen P, Pesu M, Holt VE III, Silvennoinen O, O’Shea JJ. The Janus kinases (Jaks). Genome Biol (2004) 5(12):253. doi:10.1186/gb-2004-5-12-253
61. Chen M, Cheng A, Chen YQ, Hymel A, Hanson EP, Kimmel L, et al. The amino terminus of JAK3 is necessary and sufficient for binding to the common gamma chain and confers the ability to transmit interleukin 2-mediated signals. Proc Natl Acad Sci U S A (1997) 94(13):6910–5. doi:10.1073/pnas.94.13.6910
62. Cacalano NA, Migone TS, Bazan F, Hanson EP, Chen M, Candotti F, et al. Autosomal SCID caused by a point mutation in the N-terminus of Jak3: mapping of the Jak3-receptor interaction domain. EMBO J (1999) 18(6):1549–58. doi:10.1093/emboj/18.6.1549
63. He K, Wang X, Jiang J, Guan R, Bernstein KE, Sayeski PP, et al. Janus kinase 2 determinants for growth hormone receptor association, surface assembly, and signaling. Mol Endocrinol (2003) 17(11):2211–27. doi:10.1210/me.2003-0256
64. Frank SJ, Yi W, Zhao Y, Goldsmith JF, Gilliland G, Jiang J, et al. Regions of the JAK2 tyrosine kinase required for coupling to the growth hormone receptor. J Biol Chem (1995) 270(24):14776–85. doi:10.1074/jbc.270.24.14776
65. Zhao Y, Wagner F, Frank SJ, Kraft AS. The amino-terminal portion of the JAK2 protein kinase is necessary for binding and phosphorylation of the granulocyte-macrophage colony-stimulating factor receptor beta c chain. J Biol Chem (1995) 270(23):13814–8. doi:10.1074/jbc.270.23.13814
66. Radtke S, Hermanns HM, Haan C, Schmitz-Van De Leur H, Gascan H, Heinrich PC, et al. Novel role of Janus kinase 1 in the regulation of oncostatin M receptor surface expression. J Biol Chem (2002) 277(13):11297–305. doi:10.1074/jbc.M100822200
67. Radtke S, Haan S, Jorissen A, Hermanns HM, Diefenbach S, Smyczek T, et al. The Jak1 SH2 domain does not fulfill a classical SH2 function in Jak/STAT signaling but plays a structural role for receptor interaction and up-regulation of receptor surface expression. J Biol Chem (2005) 280(27):25760–8. doi:10.1074/jbc.M500822200
68. Murakami M, Narazaki M, Hibi M, Yawata H, Yasukawa K, Hamaguchi M, et al. Critical cytoplasmic region of the interleukin-6 signal transducer Gp130 is conserved in the cytokine receptor family. Proc Natl Acad Sci U S A (1991) 88(24):11349–53. doi:10.1073/pnas.88.24.11349
69. Frank SJ, Gilliland G, Kraft AS, Arnold CS. Interaction of the growth hormone receptor cytoplasmic domain with the JAK2 tyrosine kinase. Endocrinology (1994) 135(5):2228–39. doi:10.1210/endo.135.5.7956946
70. Tanner JW, Chen W, Young RL, Longmore GD, Shaw AS. The conserved box-1 motif of cytokine receptors is required for association with Jak kinases. J Biol Chem (1995) 270(12):6523–30. doi:10.1074/jbc.270.12.6523
71. Wallweber HJ, Tam C, Franke Y, Starovasnik MA, Lupardus PJ. Structural basis of recognition of interferon-alpha receptor by tyrosine kinase 2. Nat Struct Mol Biol (2014) 21(5):443–8. doi:10.1038/nsmb.2807
72. McNally R, Eck MJ. JAK-cytokine receptor recognition, unboxed. Nat Struct Mol Biol (2014) 21(5):431–3. doi:10.1038/nsmb.2824
73. Ferrao R, Wallweber HJ, Ho H, Tam C, Franke Y, Quinn J, et al. The structural basis for class II cytokine receptor recognition by JAK1. Structure (2016) 24(6):897–905. doi:10.1016/j.str.2016.03.023
74. Babon JJ, Liau NP, Kershaw NJ. JAK1 takes a FERM hold of type II cytokine receptors. Structure (2016) 24(6):840–2. doi:10.1016/j.str.2016.05.007
75. Zhang D, Wlodawer A, Lubkowski J. Crystal structure of a complex of the intracellular domain of interferon lambda receptor 1 (IFNLR1) and the FERM/SH2 domains of human JAK1. J Mol Biol (2016) 428(23):4651–68. doi:10.1016/j.jmb.2016.10.005
76. McNally R, Toms AV, Eck MJ. Crystal structure of the FERM-SH2 module of human Jak2. PLoS One (2016) 11(5):e0156218. doi:10.1371/journal.pone.0156218
77. O’Shea JJ, Gadina M, Chen XM. Structure of a Janus kinase: molecular insights and prospects for optimizing a new class of immunosuppressants. Blood (2005) 106(3):765–6. doi:10.1182/blood-2005-05-1947
78. Silvennoinen O, Ungureanu D, Niranjan Y, Hammaren H, Bandaranayake R, Hubbard SR. New insights into the structure and function of the pseudokinase domain in JAK2. Biochem Soc Trans (2013) 41(4):1002–7. doi:10.1042/BST20130005
79. Saharinen P, Takaluoma K, Silvennoinen O. Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol Cell Biol (2000) 20(10):3387–95. doi:10.1128/MCB.20.10.3387-3395.2000
80. Saharinen P, Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J Biol Chem (2002) 277(49):47954–63. doi:10.1074/jbc.M205156200
81. Saharinen P, Vihinen M, Silvennoinen O. Autoinhibition of Jak2 tyrosine kinase is dependent on specific regions in its pseudokinase domain. Mol Biol Cell (2003) 14(4):1448–59. doi:10.1091/mbc.E02-06-0342
82. Ungureanu D, Wu J, Pekkala T, Niranjan Y, Young C, Jensen ON, et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat Struct Mol Biol (2011) 18(9):971–6. doi:10.1038/nsmb.2099
83. Bandaranayake RM, Ungureanu D, Shan Y, Shaw DE, Silvennoinen O, Hubbard SR. Crystal structures of the JAK2 pseudokinase domain and the pathogenic mutant V617F. Nat Struct Mol Biol (2012) 19(8):754–9. doi:10.1038/nsmb.2348
84. Brooks AJ, Waters MJ. Rewriting the mechanism of JAK2 activation. Cell Cycle (2015) 14(3):285–6. doi:10.1080/15384101.2015.1006533
85. Lucet IS, Fantino E, Styles M, Bamert R, Patel O, Broughton SE, et al. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood (2006) 107(1):176–83. doi:10.1182/blood-2005-06-2413
86. Lupardus PJ, Ultsch M, Wallweber H, Bir Kohli P, Johnson AR, Eigenbrot C. Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc Natl Acad Sci U S A (2014) 111(22):8025–30. doi:10.1073/pnas.1401180111
87. Waters MJ, Brooks AJ, Chhabra Y. A new mechanism for growth hormone receptor activation of JAK2, and implications for related cytokine receptors. JAKSTAT (2014) 3:e29569. doi:10.4161/jkst.29569
88. Babon JJ, Lucet IS, Murphy JM, Nicola NA, Varghese LN. The molecular regulation of Janus kinase (JAK) activation. Biochem J (2014) 462(1):1–13. doi:10.1042/BJ20140712
89. Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene (2013) 32(21):2601–13. doi:10.1038/onc.2012.347
90. Funakoshi-Tago M, Pelletier S, Moritake H, Parganas E, Ihle JN. Jak2 FERM domain interaction with the erythropoietin receptor regulates Jak2 kinase activity. Mol Cell Biol (2008) 28(5):1792–801. doi:10.1128/MCB.01447-07
91. Lu X, Huang LJ, Lodish HF. Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. J Biol Chem (2008) 283(9):5258–66. doi:10.1074/jbc.M707125200
92. Wernig G, Gonneville JR, Crowley BJ, Rodrigues MS, Reddy MM, Hudon HE, et al. The Jak2V617F oncogene associated with myeloproliferative diseases requires a functional FERM domain for transformation and for expression of the Myc and Pim proto-oncogenes. Blood (2008) 111(7):3751–9. doi:10.1182/blood-2007-07-102186
93. Gorantla SP, Dechow TN, Grundler R, Illert AL, Zum Buschenfelde CM, Kremer M, et al. Oncogenic JAK2V617F requires an intact SH2-like domain for constitutive activation and induction of a myeloproliferative disease in mice. Blood (2010) 116(22):4600–11. doi:10.1182/blood-2009-07-236133
94. Haxholm GW, Nikolajsen LF, Olsen JG, Fredsted J, Larsen FH, Goffin V, et al. Intrinsically disordered cytoplasmic domains of two cytokine receptors mediate conserved interactions with membranes. Biochem J (2015) 468(3):495–506. doi:10.1042/BJ20141243
95. Biener-Ramanujan E, Ramanujan VK, Herman B, Gertler A. Spatio-temporal kinetics of growth hormone receptor signaling in single cells using FRET microscopy. Growth Horm IGF Res (2006) 16(4):247–57. doi:10.1016/j.ghir.2006.06.001
96. Schindler C, Shuai K, Prezioso VR, Darnell JE. Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science (1992) 257(5071):809–13. doi:10.1126/science.1496401
97. Darnell JE, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (1994) 264(5164):1415–21. doi:10.1126/science.8197455
98. Darnell JE Jr. STATs and gene regulation. Science (1997) 277(5332):1630–5. doi:10.1126/science.277.5332.1630
99. Leonard WJ. Role of Jak kinases and STATS in cytokine signal transduction. Int J Hematol (2001) 73(3):271–7. doi:10.1007/Bf02981951
100. Schaefer TS, Sanders LK, Nathans D. Cooperative transcriptional activity of Jun and Stat3-beta, a short-form of Stat3. Proc Natl Acad Sci U S A (1995) 92(20):9097–101. doi:10.1073/pnas.92.20.9097
101. Hou JZ, Schindler U, Henzel WJ, Ho TC, Brasseur M, Mcknight SL. An interleukin-4-induced transcription factor – Il-4 Stat. Science (1994) 265(5179):1701–6. doi:10.1126/science.8085155
102. Gouilleux F, Pallard C, Dusanterfourt I, Wakao H, Haldosen LA, Norstedt G, et al. Prolactin, growth-hormone, erythropoietin and granulocyte-macrophage colony-stimulating factor induce Mgf-Stat5 DNA-binding activity. EMBO J (1995) 14(9):2005–13.
103. Han YL, Leaman DW, Watling D, Rogers NC, Groner B, Kerr IM, et al. Participation of JAK and STAT proteins in growth hormone-induced signaling. J Biol Chem (1996) 271(10):5947–52. doi:10.1074/jbc.271.10.5947
104. Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene (2004) 23(48):7918–27. doi:10.1038/sj.onc.1208081
105. Engen JR, Wales TE, Hochrein JM, Meyn MA III, Banu Ozkan S, Bahar I, et al. Structure and dynamic regulation of Src-family kinases. Cell Mol Life Sci (2008) 65(19):3058–73. doi:10.1007/s00018-008-8122-2
106. Latour S, Veillette A. Proximal protein tyrosine kinases in immunoreceptor signaling. Curr Opin Immunol (2001) 13(3):299–306. doi:10.1016/S0952-7915(00)00219-3
107. Hibbs ML, Harder KW. The duplicitous nature of the Lyn tyrosine kinase in growth factor signaling. Growth Factors (2006) 24(2):137–49. doi:10.1080/08977190600581327
108. Lowell CA. Src-family kinases: rheostats of immune cell signaling. Mol Immunol (2004) 41(6–7):631–43. doi:10.1016/j.molimm.2004.04.010
109. Rowlinson SW, Yoshizato H, Barclay JL, Brooks AJ, Behncken SN, Kerr LM, et al. An agonist-induced conformational change in the growth hormone receptor determines the choice of signalling pathway. Nat Cell Biol (2008) 10(6):740–7. doi:10.1038/ncb1737
110. Barclay JL, Kerr LM, Arthur L, Rowland JE, Nelson CN, Ishikawa M, et al. In vivo targeting of the growth hormone receptor (GHR) Box1 sequence demonstrates that the GHR does not signal exclusively through JAK2. Mol Endocrinol (2010) 24(1):204–17. doi:10.1210/me.2009-0233
111. Zhu T, Ling L, Lobie PE. Identification of a JAK2-independent pathway regulating growth hormone (GH)-stimulated p44/42 mitogen-activated protein kinase activity. GH activation of Ral and phospholipase D is Src-dependent. J Biol Chem (2002) 277(47):45592–603. doi:10.1074/jbc.M201385200
112. Ling L, Zhu T, Lobie PE. Src-CrkII-C3G-dependent activation of Rap1 switches growth hormone-stimulated p44/42 MAP kinase and JNK/SAPK activities. J Biol Chem (2003) 278(29):27301–11. doi:10.1074/jbc.M302516200
113. Ben-Avraham D, Govindaraju DR, Budagov T, Fradin D, Durda P, Liu B, et al. The GH receptor exon 3 deletion is a marker of male-specific exceptional longevity associated with increased GH sensitivity and taller stature. Sci Adv (2017) 3(6):e1602025. doi:10.1126/sciadv.1602025
114. Tilbrook PA, Ingley E, Williams JH, Hibbs ML, Klinken SP. Lyn tyrosine kinase is essential for erythropoietin-induced differentiation of J2E erythroid cells. EMBO J (1997) 16(7):1610–9. doi:10.1093/emboj/16.7.1610
115. Chin H, Arai A, Wakao H, Kamiyama R, Miyasaka N, Miura O. Lyn physically associates with the erythropoietin receptor and may play a role in activation of the Stat5 pathway. Blood (1998) 91(10):3734–45.
116. Fresno Vara JA, Carretero MV, Geronimo H, Ballmer-Hofer K, Martin-Perez J. Stimulation of c-Src by prolactin is independent of Jak2. Biochem J (2000) 345(Pt 1):17–24. doi:10.1042/0264-6021:3450017
117. Lannutti BJ, Drachman JG. Lyn tyrosine kinase regulates thrombopoietin-induced proliferation of hematopoietic cell lines and primary megakaryocytic progenitors. Blood (2004) 103(10):3736–43. doi:10.1182/blood-2003-10-3566
118. Seubert N, Royer Y, Staerk J, Kubatzky KF, Moucadel V, Krishnakumar S, et al. Active and inactive orientations of the transmembrane and cytosolic domains of the erythropoietin receptor dimer. Mol Cell (2003) 12(5):1239–50. doi:10.1016/S1097-2765(03)00389-7
119. Liu W, Kawahara M, Ueda H, Nagamune T. The influence of domain structures on the signal transduction of chimeric receptors derived from the erythropoietin receptor. J Biochem (2009) 145(5):575–84. doi:10.1093/jb/mvp013
120. Staerk J, Defour JP, Pecquet C, Leroy E, Antoine-Poirel H, Brett I, et al. Orientation-specific signalling by thrombopoietin receptor dimers. EMBO J (2011) 30(21):4398–413. doi:10.1038/emboj.2011.315
121. Manabe N, Kubota Y, Kitanaka A, Ohnishi H, Taminato T, Tanaka T. Src transduces signaling via growth hormone (GH)-activated GH receptor (GHR) tyrosine-phosphorylating GHR and STAT5 in human leukemia cells. Leuk Res (2006) 30(11):1391–8. doi:10.1016/j.leukres.2006.03.024
122. Ingley E. Functions of the Lyn tyrosine kinase in health and disease. Cell Commun Signal (2012) 10(1):21. doi:10.1186/1478-811X-10-21
123. Flores-Morales A, Greenhalgh CJ, Norstedt G, Rico-Bautista E. Negative regulation of growth hormone receptor signaling. Mol Endocrinol (2006) 20(2):241–53. doi:10.1210/me.2005-0170
124. Linossi EM, Babon JJ, Hilton DJ, Nicholson SE. Suppression of cytokine signaling: the SOCS perspective. Cytokine Growth Factor Rev (2013) 24(3):241–8. doi:10.1016/j.cytogfr.2013.03.005
125. Metcalf D, Greenhalgh CJ, Viney E, Willson TA, Starr R, Nicola NA, et al. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature (2000) 405(6790):1069–73. doi:10.1038/35016611
126. Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, et al. A family of cytokine-inducible inhibitors of signalling. Nature (1997) 387(6636):917–21. doi:10.1038/43206
127. Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, et al. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci U S A (1998) 95(1):114–9. doi:10.1073/pnas.95.1.114
128. Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci U S A (1999) 96(5):2071–6. doi:10.1073/pnas.96.5.2071
129. Kamura T, Sato S, Haque D, Liu L, Kaelin WG Jr, Conaway RC, et al. The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev (1998) 12(24):3872–81. doi:10.1101/gad.12.24.3872
130. Kamura T, Maenaka K, Kotoshiba S, Matsumoto M, Kohda D, Conaway RC, et al. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev (2004) 18(24):3055–65. doi:10.1101/gad.1252404
131. Bullock AN, Debreczeni JE, Edwards AM, Sundstrom M, Knapp S. Crystal structure of the SOCS2-elongin C-elongin B complex defines a prototypical SOCS box ubiquitin ligase. Proc Natl Acad Sci U S A (2006) 103(20):7637–42. doi:10.1073/pnas.0601638103
132. Sasaki A, Yasukawa H, Suzuki A, Kamizono S, Syoda T, Kinjyo I, et al. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells (1999) 4(6):339–51. doi:10.1046/j.1365-2443.1999.00263.x
133. Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, et al. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J (1999) 18(5):1309–20. doi:10.1093/emboj/18.5.1309
134. Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, et al. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol (2013) 20(4):469–76. doi:10.1038/nsmb.2519
135. Chhabra Y, Wong HY, Nikolajsen LF, Steinocher H, Papadopulos A, Tunny KA, et al. A growth hormone receptor SNP promotes lung cancer by impairment of SOCS2-mediated degradation. Oncogene (2017) 37(4):489–501. doi:10.1038/onc.2017.352
136. Neel BG, Tonks NK. Protein tyrosine phosphatases in signal transduction. Curr Opin Cell Biol (1997) 9(2):193–204. doi:10.1016/S0955-0674(97)80063-4
137. Xu Y, Fisher GJ. Receptor type protein tyrosine phosphatases (RPTPs) – roles in signal transduction and human disease. J Cell Commun Signal (2012) 6(3):125–38. doi:10.1007/s12079-012-0171-5
138. Bohmer FD, Friedrich K. Protein tyrosine phosphatases as wardens of STAT signaling. JAKSTAT (2014) 3(1):e28087. doi:10.4161/jkst.28087
139. Ram PA, Waxman DJ. Interaction of growth hormone-activated STATs with SH2-containing phosphotyrosine phosphatase SHP-1 and nuclear JAK2 tyrosine kinase. J Biol Chem (1997) 272(28):17694–702. doi:10.1074/jbc.272.28.17694
140. Stofega MR, Herrington J, Billestrup N, Carter-Su C. Mutation of the SHP-2 binding site in growth hormone (GH) receptor prolongs GH-promoted tyrosyl phosphorylation of GH receptor, JAK2, and STAT5B. Mol Endocrinol (2000) 14(9):1338–50. doi:10.1210/mend.14.9.0513
141. Pasquali C, Curchod ML, Walchli S, Espanel X, Guerrier M, Arigoni F, et al. Identification of protein tyrosine phosphatases with specificity for the ligand-activated growth hormone receptor. Mol Endocrinol (2003) 17(11):2228–39. doi:10.1210/me.2003-0011
142. Pilecka I, Patrignani C, Pescini R, Curchod ML, Perrin D, Xue Y, et al. Protein-tyrosine phosphatase H1 controls growth hormone receptor signaling and systemic growth. J Biol Chem (2007) 282(48):35405–15. doi:10.1074/jbc.M705814200
143. De Rocca Serra-Nedelec A, Edouard T, Treguer K, Tajan M, Araki T, Dance M, et al. Noonan syndrome-causing SHP2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced ERK hyperactivation, which contributes to short stature. Proc Natl Acad Sci U S A (2012) 109(11):4257–62. doi:10.1073/pnas.1119803109
144. Gan Y, Zhang Y, Buckels A, Paterson AJ, Jiang J, Clemens TL, et al. IGF-1R modulation of acute GH-induced STAT5 signaling: role of protein tyrosine phosphatase activity. Mol Endocrinol (2013) 27(11):1969–79. doi:10.1210/me.2013-1178
145. Bennett AM, Tang TL, Sugimoto S, Walsh CT, Neel BG. Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor beta to Ras. Proc Natl Acad Sci U S A (1994) 91(15):7335–9. doi:10.1073/pnas.91.15.7335
146. Lorenz U, Ravichandran KS, Pei D, Walsh CT, Burakoff SJ, Neel BG. Lck-dependent tyrosyl phosphorylation of the phosphotyrosine phosphatase SH-PTP1 in murine T cells. Mol Cell Biol (1994) 14(3):1824–34. doi:10.1128/MCB.14.3.1824
147. Minoo P, Zadeh MM, Rottapel R, Lebrun JJ, Ali S. A novel SHP-1/Grb2-dependent mechanism of negative regulation of cytokine-receptor signaling: contribution of SHP-1 C-terminal tyrosines in cytokine signaling. Blood (2004) 103(4):1398–407. doi:10.1182/blood-2003-07-2617
148. Tsui FW, Martin A, Wang J, Tsui HW. Investigations into the regulation and function of the SH2 domain-containing protein-tyrosine phosphatase, SHP-1. Immunol Res (2006) 35(1–2):127–36. doi:10.1385/IR:35:1:127
149. Saxton TM, Henkemeyer M, Gasca S, Shen R, Rossi DJ, Shalaby F, et al. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J (1997) 16(9):2352–64. doi:10.1093/emboj/16.9.2352
150. Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet (2001) 29(4):465–8. doi:10.1038/ng772
151. Keilhack H, David FS, McGregor M, Cantley LC, Neel BG. Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. J Biol Chem (2005) 280(35):30984–93. doi:10.1074/jbc.M504699200
152. Carter-Su C, Schwartz J, Argetsinger LS. Growth hormone signaling pathways. Growth Horm IGF Res (2016) 28:11–5. doi:10.1016/j.ghir.2015.09.002
153. Gery S, Koeffler HP. Role of the adaptor protein LNK in normal and malignant hematopoiesis. Oncogene (2013) 32(26):3111–8. doi:10.1038/onc.2012.435
154. Melmed S, Polonsky KS, Larsen PR, Kronenberg H. Williams Textbook of Endocrinology. Philadelphia: Elseiver (2015).
155. Schwander JC, Hauri C, Zapf J, Froesch ER. Synthesis and secretion of insulin-like growth factor and its binding protein by the perfused rat liver: dependence on growth hormone status. Endocrinology (1983) 113(1):297–305. doi:10.1210/endo-113-1-297
156. Daughaday WH, Hall K, Raben MS, Salmon WD Jr, van den Brande JL, van Wyk JJ. Somatomedin: proposed designation for sulphation factor. Nature (1972) 235(5333):107. doi:10.1038/235107a0
157. Laron Z. Somatomedin-1 (recombinant insulin-like growth factor-1): clinical pharmacology and potential treatment of endocrine and metabolic disorders. BioDrugs (1999) 11(1):55–70. doi:10.2165/00063030-199911010-00006
158. Rinderknecht E, Humbel RE. Polypeptides with nonsuppressible insulin-like and cell-growth promoting activities in human serum: isolation, chemical characterization, and some biological properties of forms I and II. Proc Natl Acad Sci U S A (1976) 73(7):2365–9.
159. Rinderknecht E, Humbel RE. The amino acid sequence of human insulin-like growth factor I and its structural homology with proinsulin. J Biol Chem (1978) 253(8):2769–76.
160. Rinderknecht E, Humbel RE. Primary structure of human insulin-like growth factor II. FEBS Lett (1978) 89(2):283–6. doi:10.1016/0014-5793(78)80237-3
161. Rinderknecht E, Humbel RE. Amino-terminal sequences of two polypeptides from human serum with nonsuppressible insulin-like and cell-growth-promoting activities: evidence for structural homology with insulin B chain. Proc Natl Acad Sci U S A (1976) 73(12):4379–81.
162. D’Ercole AJ, Applewhite GT, Underwood LE. Evidence that somatomedin is synthesized by multiple tissues in the fetus. Dev Biol (1980) 75(2):315–28. doi:10.1016/0012-1606(80)90166-9
163. Schoenle E, Zapf J, Humbel RE, Froesch ER. Insulin-like growth factor I stimulates growth in hypophysectomized rats. Nature (1982) 296(5854):252–3. doi:10.1038/296252a0
164. Guler HP, Zapf J, Scheiwiller E, Froesch ER. Recombinant human insulin-like growth factor I stimulates growth and has distinct effects on organ size in hypophysectomized rats. Proc Natl Acad Sci U S A (1988) 85(13):4889–93. doi:10.1073/pnas.85.13.4889
165. Laron Z, Klinger B, Jensen LT, Erster B. Biochemical and hormonal changes induced by one week of administration of rIGF-I to patients with Laron type dwarfism. Clin Endocrinol (Oxf) (1991) 35(2):145–50. doi:10.1111/j.1365-2265.1991.tb03513.x
166. Kanety H, Karasik A, Klinger B, Silbergeld A, Laron Z. Long-term treatment of Laron type dwarfs with insulin-like growth factor-1 increases serum insulin-like growth factor-binding protein-3 in the absence of growth hormone activity. Acta Endocrinol (Copenh) (1993) 128(2):144–9.
167. Klinger B, Jensen LT, Silbergeld A, Laron Z. Insulin-like growth factor-I raises serum procollagen levels in children and adults with Laron syndrome. Clin Endocrinol (Oxf) (1996) 45(4):423–9. doi:10.1046/j.1365-2265.1996.7990809.x
168. Backeljauw PF, Underwood LE. Prolonged treatment with recombinant insulin-like growth factor-I in children with growth hormone insensitivity syndrome – a clinical research center study. GHIS Collaborative Group. J Clin Endocrinol Metab (1996) 81(9):3312–7. doi:10.1210/jcem.81.9.8784089
169. Ranke MB, Savage MO, Chatelain PG, Preece MA, Rosenfeld RG, Wilton P. Long-term treatment of growth hormone insensitivity syndrome with IGF-I. Results of the European Multicentre Study. The Working Group on Growth Hormone Insensitivity Syndromes. Horm Res (1999) 51(3):128–34. doi:10.1159/000023345
170. Green H, Morikawa M, Nixon T. A dual effector theory of growth-hormone action. Differentiation (1985) 29(3):195–8. doi:10.1111/j.1432-0436.1985.tb00316.x
171. Ohlsson C, Bengtsson BA, Isaksson OG, Andreassen TT, Slootweg MC. Growth hormone and bone. Endocr Rev (1998) 19(1):55–79. doi:10.1210/edrv.19.1.0324
172. Laron Z. Insulin-like growth factor 1 (IGF-1): a growth hormone. Mol Pathol (2001) 54(5):311–6. doi:10.1136/mp.54.5.311
173. Clemmons DR. Roles of insulin-like growth factor-I and growth hormone in mediating insulin resistance in acromegaly. Pituitary (2002) 5(3):181–3. doi:10.1023/A:1023321421760
174. Tagliaferri M, Scacchi M, Pincelli AI, Berselli ME, Silvestri P, Montesano A, et al. Metabolic effects of biosynthetic growth hormone treatment in severely energy-restricted obese women. Int J Obes Relat Metab Disord (1998) 22(9):836–41. doi:10.1038/sj.ijo.0800669
175. Yakar S, Setser J, Zhao H, Stannard B, Haluzik M, Glatt V, et al. Inhibition of growth hormone action improves insulin sensitivity in liver IGF-1-deficient mice. J Clin Invest (2004) 113(1):96–105. doi:10.1172/JCI17763
176. Jessen N, Djurhuus CB, Jorgensen JO, Jensen LS, Moller N, Lund S, et al. Evidence against a role for insulin-signaling proteins PI 3-kinase and Akt in insulin resistance in human skeletal muscle induced by short-term GH infusion. Am J Physiol Endocrinol Metab (2005) 288(1):E194–9. doi:10.1152/ajpendo.00149.2004
177. Nielsen C, Gormsen LC, Jessen N, Pedersen SB, Moller N, Lund S, et al. Growth hormone signaling in vivo in human muscle and adipose tissue: impact of insulin, substrate background, and growth hormone receptor blockade. J Clin Endocrinol Metab (2008) 93(7):2842–50. doi:10.1210/jc.2007-2414
178. Rizza RA, Mandarino LJ, Gerich JE. Effects of growth hormone on insulin action in man. Mechanisms of insulin resistance, impaired suppression of glucose production, and impaired stimulation of glucose utilization. Diabetes (1982) 31(8 Pt 1):663–9. doi:10.2337/diabetes.31.8.663
179. Moses AC, Young SC, Morrow LA, O’Brien M, Clemmons DR. Recombinant human insulin-like growth factor I increases insulin sensitivity and improves glycemic control in type II diabetes. Diabetes (1996) 45(1):91–100. doi:10.2337/diab.45.1.91
180. Clemmons DR. The relative roles of growth hormone and IGF-1 in controlling insulin sensitivity. J Clin Invest (2004) 113(1):25–7. doi:10.1172/JCI20660
181. Barbour LA, Mizanoor Rahman S, Gurevich I, Leitner JW, Fischer SJ, Roper MD, et al. Increased P85alpha is a potent negative regulator of skeletal muscle insulin signaling and induces in vivo insulin resistance associated with growth hormone excess. J Biol Chem (2005) 280(45):37489–94. doi:10.1074/jbc.M506967200
182. del Rincon JP, Iida K, Gaylinn BD, McCurdy CE, Leitner JW, Barbour LA, et al. Growth hormone regulation of p85alpha expression and phosphoinositide 3-kinase activity in adipose tissue: mechanism for growth hormone-mediated insulin resistance. Diabetes (2007) 56(6):1638–46. doi:10.2337/db06-0299
183. Fisker S, Kristensen K, Rosenfalck AM, Pedersen SB, Ebdrup L, Richelsen B, et al. Gene expression of a truncated and the full-length growth hormone (GH) receptor in subcutaneous fat and skeletal muscle in GH-deficient adults: impact of GH treatment. J Clin Endocrinol Metab (2001) 86(2):792–6. doi:10.1210/jcem.86.2.7250
184. Fisker S, Hansen B, Fuglsang J, Kristensen K, Ovesen P, Orskov H, et al. Gene expression of the GH receptor in subcutaneous and intraabdominal fat in healthy females: relationship to GH-binding protein. Eur J Endocrinol (2004) 150(6):773–7. doi:10.1530/eje.0.1500773
185. Fleenor D, Arumugam R, Freemark M. Growth hormone and prolactin receptors in adipogenesis: STAT-5 activation, suppressors of cytokine signaling, and regulation of insulin-like growth factor I. Horm Res (2006) 66(3):101–10. doi:10.1159/000093667
186. Lyuh E, Kim HJ, Kim M, Lee JK, Park KS, Yoo KY, et al. Dose-specific or dose-dependent effect of growth hormone treatment on the proliferation and differentiation of cultured neuronal cells. Growth Horm IGF Res (2007) 17(4):315–22. doi:10.1016/j.ghir.2007.03.002
187. Hattori N. Expression, regulation and biological actions of growth hormone (GH) and ghrelin in the immune system. Growth Horm IGF Res (2009) 19(3):187–97. doi:10.1016/j.ghir.2008.12.001
188. Lu C, Kumar PA, Fan Y, Sperling MA, Menon RK. A novel effect of growth hormone on macrophage modulates macrophage-dependent adipocyte differentiation. Endocrinology (2010) 151(5):2189–99. doi:10.1210/en.2009-1194
189. Richelsen B, Pedersen SB, Borglum JD, Moller-Pedersen T, Jorgensen J, Jorgensen JO. Growth hormone treatment of obese women for 5 wk: effect on body composition and adipose tissue LPL activity. Am J Physiol (1994) 266(2 Pt 1):E211–6.
190. Moller N, Jorgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev (2009) 30(2):152–77. doi:10.1210/er.2008-0027
191. Nam SY, Lobie PE. The mechanism of effect of growth hormone on preadipocyte and adipocyte function. Obes Rev (2000) 1(2):73–86. doi:10.1046/j.1467-789x.2000.00015.x
192. Tchkonia T, Lenburg M, Thomou T, Giorgadze N, Frampton G, Pirtskhalava T, et al. Identification of depot-specific human fat cell progenitors through distinct expression profiles and developmental gene patterns. Am J Physiol Endocrinol Metab (2007) 292(1):E298–307. doi:10.1152/ajpendo.00202.2006
193. Cartwright MJ, Schlauch K, Lenburg ME, Tchkonia T, Pirtskhalava T, Cartwright A, et al. Aging, depot origin, and preadipocyte gene expression. J Gerontol A Biol Sci Med Sci (2010) 65(3):242–51. doi:10.1093/gerona/glp213
194. Garten A, Schuster S, Kiess W. The insulin-like growth factors in adipogenesis and obesity. Endocrinol Metab Clin North Am (2012) 41(2):283–295, v–vi. doi:10.1016/j.ecl.2012.04.011
195. Childs GV, Akhter N, Haney A, Syed M, Odle A, Cozart M, et al. The somatotrope as a metabolic sensor: deletion of leptin receptors causes obesity. Endocrinology (2011) 152(1):69–81. doi:10.1210/en.2010-0498
196. Lin L, Saha PK, Ma X, Henshaw IO, Shao L, Chang BH, et al. Ablation of ghrelin receptor reduces adiposity and improves insulin sensitivity during aging by regulating fat metabolism in white and brown adipose tissues. Aging Cell (2011) 10(6):996–1010. doi:10.1111/j.1474-9726.2011.00740.x
197. Iranmanesh A, Lizarralde G, Veldhuis JD. Age and relative adiposity are specific negative determinants of the frequency and amplitude of growth hormone (GH) secretory bursts and the half-life of endogenous GH in healthy men. J Clin Endocrinol Metab (1991) 73(5):1081–8. doi:10.1210/jcem-73-5-1081
198. Veldhuis JD, Iranmanesh A, Ho KK, Waters MJ, Johnson ML, Lizarralde G. Dual defects in pulsatile growth hormone secretion and clearance subserve the hyposomatotropism of obesity in man. J Clin Endocrinol Metab (1991) 72(1):51–9. doi:10.1210/jcem-72-1-51
199. Williams T, Berelowitz M, Joffe SN, Thorner MO, Rivier J, Vale W, et al. Impaired growth hormone responses to growth hormone-releasing factor in obesity. A pituitary defect reversed with weight reduction. N Engl J Med (1984) 311(22):1403–7. doi:10.1056/NEJM198411293112203
200. Vahl N, Jorgensen JO, Jurik AG, Christiansen JS. Abdominal adiposity and physical fitness are major determinants of the age associated decline in stimulated GH secretion in healthy adults. J Clin Endocrinol Metab (1996) 81(6):2209–15. doi:10.1210/jcem.81.6.8964853
201. Vahl N, Jorgensen JO, Skjaerbaek C, Veldhuis JD, Orskov H, Christiansen JS. Abdominal adiposity rather than age and sex predicts mass and regularity of GH secretion in healthy adults. Am J Physiol (1997) 272(6 Pt 1):E1108–16.
202. Clasey JL, Weltman A, Patrie J, Weltman JY, Pezzoli S, Bouchard C, et al. Abdominal visceral fat and fasting insulin are important predictors of 24-hour GH release independent of age, gender, and other physiological factors. J Clin Endocrinol Metab (2001) 86(8):3845–52. doi:10.1210/jcem.86.8.7731
203. Goodyer CG, Zogopoulos G, Schwartzbauer G, Zheng H, Hendy GN, Menon RK. Organization and evolution of the human growth hormone receptor gene 5’-flanking region. Endocrinology (2001) 142(5):1923–34. doi:10.1210/endo.142.5.8170
204. Erman A, Veilleux A, Tchernof A, Goodyer CG. Human growth hormone receptor (GHR) expression in obesity: I. GHR mRNA expression in omental and subcutaneous adipose tissues of obese women. Int J Obes (Lond) (2011) 35(12):1511–9. doi:10.1038/ijo.2011.23
205. Semsarian C, Wu MJ, Ju YK, Marciniec T, Yeoh T, Allen DG, et al. Skeletal muscle hypertrophy is mediated by a Ca2+-dependent calcineurin signalling pathway. Nature (1999) 400(6744):576–81. doi:10.1038/23054
206. Dalla Libera L, Ravara B, Volterrani M, Gobbo V, Della Barbera M, Angelini A, et al. Beneficial effects of GH/IGF-1 on skeletal muscle atrophy and function in experimental heart failure. Am J Physiol Cell Physiol (2004) 286(1):C138–44. doi:10.1152/ajpcell.00114.2003
207. Sotiropoulos A, Ohanna M, Kedzia C, Menon RK, Kopchick JJ, Kelly PA, et al. Growth hormone promotes skeletal muscle cell fusion independent of insulin-like growth factor 1 up-regulation. Proc Natl Acad Sci U S A (2006) 103(19):7315–20. doi:10.1073/pnas.0510033103
208. Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci U S A (1998) 95(26):15603–7. doi:10.1073/pnas.95.26.15603
209. Short KR, Moller N, Bigelow ML, Coenen-Schimke J, Nair KS. Enhancement of muscle mitochondrial function by growth hormone. J Clin Endocrinol Metab (2008) 93(2):597–604. doi:10.1210/jc.2007-1814
210. Schertzer JD, Ryall JG, Lynch GS. Systemic administration of IGF-I enhances oxidative status and reduces contraction-induced injury in skeletal muscles of mdx dystrophic mice. Am J Physiol Endocrinol Metab (2006) 291(3):E499–505. doi:10.1152/ajpendo.00101.2006
211. Roth J, Glick SM, Yalow RS, Berson SA. Secretion of human growth hormone: physiologic and experimental modification. Metabolism (1963) 12:577–9.
212. Bang P, Brandt J, Degerblad M, Enberg G, Kaijser L, Thoren M, et al. Exercise-induced changes in insulin-like growth factors and their low molecular weight binding protein in healthy subjects and patients with growth hormone deficiency. Eur J Clin Invest (1990) 20(3):285–92. doi:10.1111/j.1365-2362.1990.tb01857.x
213. Wallace JD, Cuneo RC, Baxter R, Orskov H, Keay N, Pentecost C, et al. Responses of the growth hormone (GH) and insulin-like growth factor axis to exercise, GH administration, and GH withdrawal in trained adult males: a potential test for GH abuse in sport. J Clin Endocrinol Metab (1999) 84(10):3591–601. doi:10.1210/jcem.84.10.6037
214. Heinemeier KM, Mackey AL, Doessing S, Hansen M, Bayer ML, Nielsen RH, et al. GH/IGF-I axis and matrix adaptation of the musculotendinous tissue to exercise in humans. Scand J Med Sci Sports (2012) 22(4):e1–7. doi:10.1111/j.1600-0838.2012.01459.x
215. Felsing NE, Brasel JA, Cooper DM. Effect of low and high intensity exercise on circulating growth hormone in men. J Clin Endocrinol Metab (1992) 75(1):157–62. doi:10.1210/jcem.75.1.1619005
216. Kjaer M, Secher NH, Bach FW, Galbo H. Role of motor center activity for hormonal changes and substrate mobilization in humans. Am J Physiol (1987) 253(5 Pt 2):R687–95.
217. Bamman MM, Shipp JR, Jiang J, Gower BA, Hunter GR, Goodman A, et al. Mechanical load increases muscle IGF-I and androgen receptor mRNA concentrations in humans. Am J Physiol Endocrinol Metab (2001) 280(3):E383–90. doi:10.1152/ajpendo.2001.280.3.E383
218. Hameed M, Lange KH, Andersen JL, Schjerling P, Kjaer M, Harridge SD, et al. The effect of recombinant human growth hormone and resistance training on IGF-I mRNA expression in the muscles of elderly men. J Physiol (2004) 555(Pt 1):231–40. doi:10.1113/jphysiol.2003.051722
219. Heinemeier KM, Bjerrum SS, Schjerling P, Kjaer M. Expression of extracellular matrix components and related growth factors in human tendon and muscle after acute exercise. Scand J Med Sci Sports (2013) 23(3):e150–61. doi:10.1111/j.1600-0838.2011.01414.x
220. Schlechter NL, Russell SM, Spencer EM, Nicoll CS. Evidence suggesting that the direct growth-promoting effect of growth hormone on cartilage in vivo is mediated by local production of somatomedin. Proc Natl Acad Sci U S A (1986) 83(20):7932–4. doi:10.1073/pnas.83.20.7932
221. Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, et al. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A (1999) 96(13):7324–9. doi:10.1073/pnas.96.13.7324
222. Govoni KE, Lee SK, Chung YS, Behringer RR, Wergedal JE, Baylink DJ, et al. Disruption of insulin-like growth factor-I expression in type IIalphaI collagen-expressing cells reduces bone length and width in mice. Physiol Genomics (2007) 30(3):354–62. doi:10.1152/physiolgenomics.00022.2007
223. Wang J, Zhou J, Cheng CM, Kopchick JJ, Bondy CA. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol (2004) 180(2):247–55. doi:10.1677/joe.0.1800247
224. Wu S, Yang W, De Luca F. Insulin-like growth factor-independent effects of growth hormone on growth plate chondrogenesis and longitudinal bone growth. Endocrinology (2015) 156(7):2541–51. doi:10.1210/en.2014-1983
225. Walsh S, Jefferiss CM, Stewart K, Beresford JN. IGF-I does not affect the proliferation or early osteogenic differentiation of human marrow stromal cells. Bone (2003) 33(1):80–9. doi:10.1016/S8756-3282(03)00165-0
226. Ochiai H, Okada S, Saito A, Hoshi K, Yamashita H, Takato T, et al. Inhibition of insulin-like growth factor-1 (IGF-1) expression by prolonged transforming growth factor-beta1 (TGF-beta1) administration suppresses osteoblast differentiation. J Biol Chem (2012) 287(27):22654–61. doi:10.1074/jbc.M111.279091
227. Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL, et al. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest (2002) 110(6):771–81. doi:10.1172/JCI15463
228. Guntur AR, Rosen CJ. IGF-1 regulation of key signaling pathways in bone. Bonekey Rep (2013) 2:437. doi:10.1038/bonekey.2013.171
229. Yan J, Herzog JW, Tsang K, Brennan CA, Bower MA, Garrett WS, et al. Gut microbiota induce IGF-1 and promote bone formation and growth. Proc Natl Acad Sci U S A (2016) 113(47):E7554–63. doi:10.1073/pnas.1607235113
230. Fornari R, Marocco C, Francomano D, Fittipaldi S, Lubrano C, Bimonte VM, et al. Insulin growth factor-1 correlates with higher bone mineral density and lower inflammation status in obese adult subjects. Eat Weight Disord (2017). doi:10.1007/s40519-017-0362-4
231. Arany E, Afford S, Strain AJ, Winwood PJ, Arthur MJ, Hill DJ. Differential cellular synthesis of insulin-like growth factor binding protein-1 (IGFBP-1) and IGFBP-3 within human liver. J Clin Endocrinol Metab (1994) 79(6):1871–6. doi:10.1210/jcem.79.6.7527416
232. Scharf J, Ramadori G, Braulke T, Hartmann H. Synthesis of insulinlike growth factor binding proteins and of the acid-labile subunit in primary cultures of rat hepatocytes, of Kupffer cells, and in cocultures: regulation by insulin, insulinlike growth factor, and growth hormone. Hepatology (1996) 23(4):818–27. doi:10.1053/jhep.1996.v23.pm0008666337
233. Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev (1995) 16(1):3–34. doi:10.1210/edrv-16-1-3
234. Baxter RC, Martin JL, Beniac VA. High molecular weight insulin-like growth factor binding protein complex. Purification and properties of the acid-labile subunit from human serum. J Biol Chem (1989) 264(20):11843–8.
235. Villafuerte BC, Koop BL, Pao CI, Gu L, Birdsong GG, Phillips LS. Coculture of primary rat hepatocytes and nonparenchymal cells permits expression of insulin-like growth factor binding protein-3 in vitro. Endocrinology (1994) 134(5):2044–50. doi:10.1210/endo.134.5.7512496
236. Fichter MM, Pirke KM, Holsboer F. Weight loss causes neuroendocrine disturbances: experimental study in healthy starving subjects. Psychiatry Res (1986) 17(1):61–72. doi:10.1016/0165-1781(86)90042-9
237. Donaghy A, Ross R, Gimson A, Hughes SC, Holly J, Williams R. Growth hormone, insulinlike growth factor-1, and insulinlike growth factor binding proteins 1 and 3 in chronic liver disease. Hepatology (1995) 21(3):680–8. doi:10.1002/hep.1840210313
238. Moller S, Juul A, Becker U, Henriksen JH. The acid-labile subunit of the ternary insulin-like growth factor complex in cirrhosis: relation to liver dysfunction. J Hepatol (2000) 32(3):441–6. doi:10.1016/S0168-8278(00)80395-5
239. Chishima S, Kogiso T, Matsushita N, Hashimoto E, Tokushige K. The relationship between the growth hormone/insulin-like growth factor system and the histological features of nonalcoholic fatty liver disease. Intern Med (2017) 56(5):473–80. doi:10.2169/internalmedicine.56.7626
240. Xu L, Xu C, Yu C, Miao M, Zhang X, Zhu Z, et al. Association between serum growth hormone levels and nonalcoholic fatty liver disease: a cross-sectional study. PLoS One (2012) 7(8):e44136. doi:10.1371/journal.pone.0044136
241. Cuneo RC, Hickman PE, Wallace JD, Teh BT, Ward G, Veldhuis JD, et al. Altered endogenous growth hormone secretory kinetics and diurnal GH-binding protein profiles in adults with chronic liver disease. Clin Endocrinol (Oxf) (1995) 43(3):265–75. doi:10.1111/j.1365-2265.1995.tb02031.x
242. Zadik Z, Chalew SA, McCarter RJ Jr, Meistas M, Kowarski AA. The influence of age on the 24-hour integrated concentration of growth hormone in normal individuals. J Clin Endocrinol Metab (1985) 60(3):513–6. doi:10.1210/jcem-60-3-513
243. Yamamoto H, Sohmiya M, Oka N, Kato Y. Effects of aging and sex on plasma insulin-like growth factor I (IGF-I) levels in normal adults. Acta Endocrinol (Copenh) (1991) 124(5):497–500.
244. Vitale G, Barbieri M, Kamenetskaya M, Paolisso G. GH/IGF-I/insulin system in centenarians. Mech Ageing Dev (2016) 165(Pt B):107–14. doi:10.1016/j.mad.2016.12.001
245. Toogood AA, O’Neill PA, Shalet SM. Beyond the somatopause: growth hormone deficiency in adults over the age of 60 years. J Clin Endocrinol Metab (1996) 81(2):460–5. doi:10.1210/jcem.81.2.8636250
246. Liu H, Bravata DM, Olkin I, Nayak S, Roberts B, Garber AM, et al. Systematic review: the safety and efficacy of growth hormone in the healthy elderly. Ann Intern Med (2007) 146(2):104–15. doi:10.7326/0003-4819-146-2-200701160-00005
247. Fontana L, Partridge L, Longo VD. Extending healthy life span – from yeast to humans. Science (2010) 328(5976):321–6. doi:10.1126/science.1172539
248. Milman S, Huffman DM, Barzilai N. The somatotropic axis in human aging: framework for the current state of knowledge and future research. Cell Metab (2016) 23(6):980–9. doi:10.1016/j.cmet.2016.05.014
249. Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, Wei M, Madia F, Cheng CW, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med (2011) 3(70):70ra13. doi:10.1126/scitranslmed.3001845
250. Coschigano KT, Clemmons D, Bellush LL, Kopchick JJ. Assessment of growth parameters and life span of GHR/BP gene-disrupted mice. Endocrinology (2000) 141(7):2608–13. doi:10.1210/endo.141.7.7586
251. Coschigano KT, Holland AN, Riders ME, List EO, Flyvbjerg A, Kopchick JJ. Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin, and insulin-like growth factor I levels and increased life span. Endocrinology (2003) 144(9):3799–810. doi:10.1210/en.2003-0374
252. Bartke A, List EO, Kopchick JJ. The somatotropic axis and aging: benefits of endocrine defects. Growth Horm IGF Res (2016) 27:41–5. doi:10.1016/j.ghir.2016.02.002
253. Rincon M, Rudin E, Barzilai N. The insulin/IGF-1 signaling in mammals and its relevance to human longevity. Exp Gerontol (2005) 40(11):873–7. doi:10.1016/j.exger.2005.06.014
254. Zhou Y, Xu BC, Maheshwari HG, He L, Reed M, Lozykowski M, et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc Natl Acad Sci U S A (1997) 94(24):13215–20. doi:10.1073/pnas.94.24.13215
255. Rowland JE, Lichanska AM, Kerr LM, White M, d’Aniello EM, Maher SL, et al. In vivo analysis of growth hormone receptor signaling domains and their associated transcripts. Mol Cell Biol (2005) 25(1):66–77. doi:10.1128/MCB.25.1.66-77.2005
256. Rowland JE, Kerr LM, White M, Noakes PG, Waters MJ. Heterozygote effects in mice with partial truncations in the growth hormone receptor cytoplasmic domain: assessment of growth parameters and phenotype. Endocrinology (2005) 146(12):5278–86. doi:10.1210/en.2005-0939
257. Fan Y, Menon RK, Cohen P, Hwang D, Clemens T, DiGirolamo DJ, et al. Liver-specific deletion of the growth hormone receptor reveals essential role of growth hormone signaling in hepatic lipid metabolism. J Biol Chem (2009) 284(30):19937–44. doi:10.1074/jbc.M109.014308
258. Li X, Bartke A, Berryman DE, Funk K, Kopchick JJ, List EO, et al. Direct and indirect effects of growth hormone receptor ablation on liver expression of xenobiotic metabolizing genes. Am J Physiol Endocrinol Metab (2013) 305(8):E942–50. doi:10.1152/ajpendo.00304.2013
259. List EO, Berryman DE, Funk K, Jara A, Kelder B, Wang F, et al. Liver-specific GH receptor gene-disrupted (LiGHRKO) mice have decreased endocrine IGF-I, increased local IGF-I, and altered body size, body composition, and adipokine profiles. Endocrinology (2014) 155(5):1793–805. doi:10.1210/en.2013-2086
260. Gesing A, Wang F, List EO, Berryman DE, Masternak MM, Lewinski A, et al. Expression of apoptosis-related genes in liver-specific growth hormone receptor gene-disrupted mice is sex dependent. J Gerontol A Biol Sci Med Sci (2015) 70(1):44–52. doi:10.1093/gerona/glu008
261. Wu Y, Liu C, Sun H, Vijayakumar A, Giglou PR, Qiao R, et al. Growth hormone receptor regulates beta cell hyperplasia and glucose-stimulated insulin secretion in obese mice. J Clin Invest (2011) 121(6):2422–6. doi:10.1172/JCI45027
262. Vijayakumar A, Wu Y, Sun H, Li X, Jeddy Z, Liu C, et al. Targeted loss of GHR signaling in mouse skeletal muscle protects against high-fat diet-induced metabolic deterioration. Diabetes (2012) 61(1):94–103. doi:10.2337/db11-0814
263. List EO, Berryman DE, Ikeno Y, Hubbard GB, Funk K, Comisford R, et al. Removal of growth hormone receptor (GHR) in muscle of male mice replicates some of the health benefits seen in global GHR-/- mice. Aging (Albany NY) (2015) 7(7):500–12. doi:10.18632/aging.100766
264. Mavalli MD, DiGirolamo DJ, Fan Y, Riddle RC, Campbell KS, van Groen T, et al. Distinct growth hormone receptor signaling modes regulate skeletal muscle development and insulin sensitivity in mice. J Clin Invest (2010) 120(11):4007–20. doi:10.1172/JCI42447
265. List EO, Berryman DE, Funk K, Gosney ES, Jara A, Kelder B, et al. The role of GH in adipose tissue: lessons from adipose-specific GH receptor gene-disrupted mice. Mol Endocrinol (2013) 27(3):524–35. doi:10.1210/me.2012-1330
266. Bartke A, Sun LY, Longo V. Somatotropic signaling: trade-offs between growth, reproductive development, and longevity. Physiol Rev (2013) 93(2):571–98. doi:10.1152/physrev.00006.2012
267. Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S. Inflammaging and ‘Garb-aging’. Trends Endocrinol Metab (2017) 28(3):199–212. doi:10.1016/j.tem.2016.09.005
268. Junnila RK, List EO, Berryman DE, Murrey JW, Kopchick JJ. The GH/IGF-1 axis in ageing and longevity. Nat Rev Endocrinol (2013) 9(6):366–76. doi:10.1038/nrendo.2013.67
269. Chanson P, Borson-Chazot F, Kuhn JM, Blumberg J, Maisonobe P, Delemer B, et al. Control of IGF-I levels with titrated dosing of lanreotide Autogel over 48 weeks in patients with acromegaly. Clin Endocrinol (Oxf) (2008) 69(2):299–305. doi:10.1111/j.1365-2265.2008.03208.x
270. Davies RR, Turner SJ, Alberti KG, Johnston DG. Somatostatin analogues in diabetes mellitus. Diabet Med (1989) 6(2):103–11. doi:10.1111/j.1464-5491.1989.tb02096.x
271. Parkinson C, Drake WM, Roberts ME, Meeran K, Besser GM, Trainer PJ. A comparison of the effects of pegvisomant and octreotide on glucose, insulin, gastrin, cholecystokinin, and pancreatic polypeptide responses to oral glucose and a standard mixed meal. J Clin Endocrinol Metab (2002) 87(4):1797–804. doi:10.1210/jcem.87.4.8432
272. Chhabra Y, Waters MJ, Brooks AJ. Role of the growth hormone–IGF-1 axis in cancer. Expert Rev Endocrinol Metab (2011) 6:71–84. doi:10.1586/eem.10.73
273. Bustin SA, Jenkins PJ. The growth hormone-insulin-like growth factor-I axis and colorectal cancer. Trends Mol Med (2001) 7(10):447–54. doi:10.1016/S1471-4914(01)02104-9
274. Sacca L, Cittadini A, Fazio S. Growth-hormone and the heart. Endocr Rev (1994) 15(5):555–73. doi:10.1210/er.15.5.555
275. Jenkins PJ, Besser M. Clinical perspective: acromegaly and cancer: a problem. J Clin Endocrinol Metab (2001) 86(7):2935–41. doi:10.1210/jcem.86.7.7634
276. Lichanska AM, Waters MJ. How growth hormone controls growth, obesity and sexual dimorphism. Trends Genet (2008) 24(1):41–7. doi:10.1016/j.tig.2007.10.006
277. Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest (2009) 119(11):3189–202. doi:10.1172/Jci39375
278. Gunnell D, Okasha M, Smith GD, Oliver SE, Sandhu J, Holly JM. Height, leg length, and cancer risk: a systematic review. Epidemiol Rev (2001) 23(2):313–42. doi:10.1093/oxfordjournals.epirev.a000809
279. Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature (2010) 467(7317):832–8. doi:10.1038/nature09410
280. Ahlgren M, Melbye M, Wohlfahrt J, Sorensen TIA. Growth patterns and the risk of breast cancer in women. N Engl J Med (2004) 351(16):1619–26. doi:10.1056/NEJMoa040576
281. Rowlands MA, Gunnell D, Harris R, Vatten LJ, Holly JMP, Martin RM. Circulating insulin-like growth factor peptides and prostate cancer risk: a systematic review and meta-analysis. Int J Cancer (2009) 124(10):2416–29. doi:10.1002/ijc.24202
282. Wang F, Xu X, Yang J, Min L, Liang S, Chen Y. Height and lung cancer risk: a meta-analysis of observational studies. PLoS One (2017) 12(9):e0185316. doi:10.1371/journal.pone.0185316
283. Rudd MF, Webb EL, Matakidou A, Sellick GS, Williams RD, Bridle H, et al. Variants in the GH-IGF axis confer susceptibility to lung cancer. Genome Res (2006) 16(6):693–701. doi:10.1101/gr.5120106
284. Cao G, Lu H, Feng J, Shu J, Zheng D, Hou Y. Lung cancer risk associated with Thr495Pro polymorphism of GHR in Chinese population. Jpn J Clin Oncol (2008) 38(4):308–16. doi:10.1093/jjco/hyn007
285. Jenkins PJ, Mukherjee A, Shalet SM. Does growth hormone cause cancer? Clin Endocrinol (Oxf) (2006) 64(2):115–21. doi:10.1111/j.1365-2265.2005.02404.x
286. Renehan AG, O’Connell J, O’Halloran D, Shanahan F, Potten CS, O’Dwyer ST, et al. Acromegaly and colorectal cancer: a comprehensive review of epidemiology, biological mechanisms, and clinical implications. Horm Metab Res (2003) 35(11–12):712–25. doi:10.1055/s-2004-814150
287. Renehan AG, Brennan BM. Acromegaly, growth hormone and cancer risk. Best Pract Res Clin Endocrinol Metab (2008) 22(4):639–57. doi:10.1016/j.beem.2008.08.011
288. Dagdelen S, Cinar N, Erbas T. Increased thyroid cancer risk in acromegaly. Pituitary (2014) 17(4):299–306. doi:10.1007/s11102-013-0501-5
289. Blizzard RM. History of growth hormone therapy. Indian J Pediatr (2012) 79(1):87–91. doi:10.1007/s12098-011-0609-4
290. Chernausek SD. Growth and development how safe is growth hormone therapy for children? Nat Rev Endocrinol (2010) 6(5):251–3. doi:10.1038/nrendo.2010.43
291. Shalet SM, Clayton PE, Price DA. Growth and pituitary-function in children treated for brain-tumors or acute lymphoblastic-leukemia. Horm Res (1988) 30(2–3):53–61. doi:10.1159/000181029
292. Taback SP, Dean HJ. Mortality in Canadian children with growth hormone (GH) deficiency receiving GH therapy 1967-1992. The Canadian Growth Hormone Advisory Committee. J Clin Endocrinol Metab (1996) 81(5):1693–6. doi:10.1210/jcem.81.5.8626817
293. Swerdlow AJ, Reddingius RE, Higgins CD, Spoudeas HA, Phipps K, Qiao Z, et al. Growth hormone treatment of children with brain tumors and risk of tumor recurrence. J Clin Endocrinol Metab (2000) 85(12):4444–9. doi:10.1210/jcem.85.12.7044
294. Watanabe S, Tsunematsu Y, Fujimoto J, Komiyama A. Leukemia in patients treated with growth-hormone. Lancet (1988) 1(8595):1159–1159. doi:10.1016/S0140-6736(88)91968-X
295. Wada E, Murata M, Watanabe S. Acute lymphoblastic leukemia following treatment with human growth hormone in a boy with possible preanemic Fanconi’s anemia. Jpn J Clin Oncol (1989) 19(1):36–9.
296. Nishi Y, Tanaka T, Takano K, Fujieda K, Igarashi Y, Hanew K, et al. Recent status in the occurrence of leukemia in growth hormone-treated patients in Japan. GH Treatment Study Committee of the Foundation for Growth Science, Japan. J Clin Endocrinol Metab (1999) 84(6):1961–5. doi:10.1210/jcem.84.6.5716
297. Allen DB, Rundle AC, Graves DA, Blethen SL. Risk of leukemia in children treated with human growth hormone: review and reanalysis. J Pediatr (1997) 131(1):S32–6. doi:10.1016/S0022-3476(97)70008-8
298. Chae HW, Kim DH, Kim HS. Growth hormone treatment and risk of malignancy. Korean J Pediatr (2015) 58(2):41–6. doi:10.3345/kjp.2015.58.2.41
299. Conway-Campbell BL, Wooh JW, Brooks AJ, Gordon D, Brown RJ, Lichanska AM, et al. Nuclear targeting of the growth hormone receptor results in dysregulation of cell proliferation and tumorigenesis. Proc Natl Acad Sci U S A (2007) 104(33):13331–6. doi:10.1073/pnas.0600181104
300. Lan HN, Hong P, Li RN, Shan AS, Zheng X. Growth hormone-specific induction of the nuclear localization of porcine growth hormone receptor in porcine hepatocytes. Domest Anim Endocrinol (2017) 61:39–47. doi:10.1016/j.domaniend.2017.05.003
301. Figueiredo MA, Boyle RT, Sandrini JZ, Varela AS Jr, Marins LF. High level of GHR nuclear translocation in skeletal muscle of a hyperplasic transgenic zebrafish. J Mol Endocrinol (2016) 56(1):47–54. doi:10.1530/JME-15-0185
302. Conway-Campbell BL, Brooks AJ, Robinson PJ, Perani M, Waters MJ. The extracellular domain of the growth hormone receptor interacts with coactivator activator to promote cell proliferation. Mol Endocrinol (2008) 22(9):2190–202. doi:10.1210/me.2008-0128
303. Liu JL, Yakar S, LeRoith D. Conditional knockout of mouse insulin-like growth factor-1 gene using the Cre/loxP system. Proc Soc Exp Biol Med (2000) 223(4):344–51. doi:10.1111/j.1525-1373.2000.22349.x
304. Sjogren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, et al. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc Natl Acad Sci U S A (1999) 96(12):7088–92. doi:10.1073/pnas.96.12.7088
305. Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell (1993) 75(1):73–82. doi:10.1016/S0092-8674(05)80085-6
306. Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell (1993) 75(1):59–72. doi:10.1016/S0092-8674(05)80084-4
307. Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S, Dalton D, et al. IGF-I is required for normal embryonic growth in mice. Genes Dev (1993) 7(12B):2609–17. doi:10.1101/gad.7.12b.2609
308. Liu JL, LeRoith D. Insulin-like growth factor I is essential for postnatal growth in response to growth hormone. Endocrinology (1999) 140(11):5178–84. doi:10.1210/endo.140.11.7151
309. Holder AT, Spencer EM, Preece MA. Effect of bovine growth hormone and a partially pure preparation of somatomedin on various growth parameters in hypopituitary dwarf mice. J Endocrinol (1981) 89(2):275–82. doi:10.1677/joe.0.0890275
310. Shea BT, Hammer RE, Brinster RL. Growth allometry of the organs in giant transgenic mice. Endocrinology (1987) 121(6):1924–30. doi:10.1210/endo-121-6-1924
311. Behringer RR, Lewin TM, Quaife CJ, Palmiter RD, Brinster RL, D’Ercole AJ. Expression of insulin-like growth factor I stimulates normal somatic growth in growth hormone-deficient transgenic mice. Endocrinology (1990) 127(3):1033–40. doi:10.1210/endo-127-3-1033
312. Rosen T, Hansson T, Granhed H, Szucs J, Bengtsson BA. Reduced bone mineral content in adult patients with growth hormone deficiency. Acta Endocrinol (Copenh) (1993) 129(3):201–6.
313. Saggese G, Baroncelli GI, Bertelloni S, Cinquanta L, Di Nero G. Effects of long-term treatment with growth hormone on bone and mineral metabolism in children with growth hormone deficiency. J Pediatr (1993) 122(1):37–45. doi:10.1016/S0022-3476(05)83484-5
314. Chandrashekar V, Bartke A, Coschigano KT, Kopchick JJ. Pituitary and testicular function in growth hormone receptor gene knockout mice. Endocrinology (1999) 140(3):1082–8. doi:10.1210/endo.140.3.6557
315. Dominici FP, Arostegui Diaz G, Bartke A, Kopchick JJ, Turyn D. Compensatory alterations of insulin signal transduction in liver of growth hormone receptor knockout mice. J Endocrinol (2000) 166(3):579–90. doi:10.1677/joe.0.1660579
316. Keene DE, Suescun MO, Bostwick MG, Chandrashekar V, Bartke A, Kopchick JJ. Puberty is delayed in male growth hormone receptor gene-disrupted mice. J Androl (2002) 23(5):661–8.
317. Berryman DE, List EO, Coschigano KT, Behar K, Kim JK, Kopchick JJ. Comparing adiposity profiles in three mouse models with altered GH signaling. Growth Horm IGF Res (2004) 14(4):309–18. doi:10.1016/j.ghir.2004.02.005
318. Egecioglu E, Bjursell M, Ljungberg A, Dickson SL, Kopchick JJ, Bergstrom G, et al. Growth hormone receptor deficiency results in blunted ghrelin feeding response, obesity, and hypolipidemia in mice. Am J Physiol Endocrinol Metab (2006) 290(2):E317–25. doi:10.1152/ajpendo.00181.2005
319. Izzard AS, Emerson M, Prehar S, Neyses L, Trainer P, List EO, et al. The cardiovascular phenotype of a mouse model of acromegaly. Growth Horm IGF Res (2009) 19(5):413–9. doi:10.1016/j.ghir.2008.12.006
320. Powell-Braxton L, Hollingshead P, Giltinan D, Pitts-Meek S, Stewart T. Inactivation of the IGF-I gene in mice results in perinatal lethality. Ann N Y Acad Sci (1993) 692:300–1. doi:10.1111/j.1749-6632.1993.tb26240.x
321. Woods KA, Camacho-Hubner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med (1996) 335(18):1363–7. doi:10.1056/NEJM199610313351805
322. Lupu F, Terwilliger JD, Lee K, Segre GV, Efstratiadis A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev Biol (2001) 229(1):141–62. doi:10.1006/dbio.2000.9975
323. Nilsson A, Isgaard J, Lindahl A, Peterson L, Isaksson O. Effects of unilateral arterial infusion of GH and IGF-I on tibial longitudinal bone growth in hypophysectomized rats. Calcif Tissue Int (1987) 40(2):91–6. doi:10.1007/BF02555711
Keywords: growth hormone, growth hormone receptor, Janus kinase 2, Src family kinase, insulin-like growth factor 1, suppressor of cytokine signaling
Citation: Dehkhoda F, Lee CMM, Medina J and Brooks AJ (2018) The Growth Hormone Receptor: Mechanism of Receptor Activation, Cell Signaling, and Physiological Aspects. Front. Endocrinol. 9:35. doi: 10.3389/fendo.2018.00035
Received: 24 July 2017; Accepted: 29 January 2018;
Published: 13 February 2018
Edited by:
Stefan N. Constantinescu, Ludwig Cancer Research, BelgiumReviewed by:
Sandra Pellegrini, Institut Pasteur, FranceCopyright: © 2018 Dehkhoda, Lee, Medina and Brooks. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew J. Brooks, YS5icm9va3NAdXEuZWR1LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.