
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 17 January 2018
Sec. Clinical Diabetes
Volume 9 - 2018 | https://doi.org/10.3389/fendo.2018.00002
This article is part of the Research TopicCardiovascular Disease and DiabetesView all 11 articles
 Salvatore De Rosa1†
Salvatore De Rosa1† Biagio Arcidiacono2†
Biagio Arcidiacono2† Eusebio Chiefari2
Eusebio Chiefari2 Antonio Brunetti2*
Antonio Brunetti2* Ciro Indolfi1*
Ciro Indolfi1* Daniela P. Foti2*
Daniela P. Foti2*
Type 2 diabetes mellitus (DM) is a common metabolic disorder predisposing to diabetic cardiomyopathy and atherosclerotic cardiovascular disease (CVD), which could lead to heart failure through a variety of mechanisms, including myocardial infarction and chronic pressure overload. Pathogenetic mechanisms, mainly linked to hyperglycemia and chronic sustained hyperinsulinemia, include changes in metabolic profiles, intracellular signaling pathways, energy production, redox status, increased susceptibility to ischemia, and extracellular matrix remodeling. The close relationship between type 2 DM and CVD has led to the common soil hypothesis, postulating that both conditions share common genetic and environmental factors influencing this association. However, although the common risk factors of both CVD and type 2 DM, such as obesity, insulin resistance, dyslipidemia, inflammation, and thrombophilia, can be identified in the majority of affected patients, less is known about how these factors influence both conditions, so that efforts are still needed for a more comprehensive understanding of this relationship. The genetic, epigenetic, and environmental backgrounds of both type 2 DM and CVD have been more recently studied and updated. However, the underlying pathogenetic mechanisms have seldom been investigated within the broader shared background, but rather studied in the specific context of type 2 DM or CVD, separately. As the precise pathophysiological links between type 2 DM and CVD are not entirely understood and many aspects still require elucidation, an integrated description of the genetic, epigenetic, and environmental influences involved in the concomitant development of both diseases is of paramount importance to shed new light on the interlinks between type 2 DM and CVD. This review addresses the current knowledge of overlapping genetic and epigenetic aspects in type 2 DM and CVD, including microRNAs and long non-coding RNAs, whose abnormal regulation has been implicated in both disease conditions, either etiologically or as cause for their progression. Understanding the links between these disorders may help to drive future research toward an integrated pathophysiological approach and to provide future directions in the field.
Type 2 diabetes mellitus (DM) is a complex metabolic disease in which concomitant insulin resistance and beta-cell impairment lead to hyperglycemia, which is the hallmark of the disease (1). Its prevalence is in rapid and progressive rise, due to the increase in average life expectancy, growing prevalence of obesity, and westernization of lifestyles in developing countries (2, 3), while its long-term complications are the major causes of morbidity, mortality, and exceptional healthcare costs (4, 5).
Cardiovascular disease (CVD) represents a leading health problem worldwide (6). Prospective studies have demonstrated that diabetic patients have a two- to fourfold propensity to develop coronary artery disease (CAD) and myocardial infarction (MI) (7), establishing that type 2 DM is an independent risk factor for stroke and heart disease (8). Indeed, about 70% of type 2 DM at an age ≥65 years die from CVD (7), while type 2 DM patients with no history of CAD have an equal cardiovascular risk as patients with previous MI (9). CVD and type 2 DM share several common pathophysiological features that are summarized in Table 1. Classical cardiovascular risk factors, such as dyslipidemia, hypertension and obesity can also raise the risk of type 2 DM. In particular, insulin resistance and hyperglycemia are associated with a low-grade inflammation, as well as with chronic enhancement of oxidative stress, triggering endothelial dysfunction and promoting atherogenesis (10–12). Among the different soluble mediators associated with the above-mentioned aspects, IL-1β, IL-6, tumor necrosis factor (TNF)-α, and CRP are worth mentioning (13). In addition, it is well documented that type 2 DM is associated with enhancement of platelet and hemostatic activities (14).

Table 1. Common pathophysiology of type 2 diabetes mellitus (DM) and cardiovascular disease (CVD).
Currently, a number of evidences exists, demonstrating that the interaction of type 2 DM and related cardiovascular risk underpin the progressive nature of the vascular damage, leading to atherosclerosis (23), while it is also proved that lifestyle modifications, such as physical activity and weight loss, counteract CVD risk factors in prediabetic individuals (23, 24). As diabetes shares many risk factors with CVD, while some other ones may be independent, this reinforces the postulate proposed by Stern, according to which both diseases come independently from a “common soil” (20). In this scenario, as type 2 DM and CVD are both complex diseases, common risk factors predisposing to these disorders may include shared genetic factors, a setting that has been only partly elucidated.
Many common single-nucleotide polymorphisms (SNPs) have been already associated with an increased risk of CVD and type 2 DM (25), while their search is still ongoing. In addition, novel links between these disorders come from epigenetic studies. In this review, we will try to address the current knowledge about the genetic links between type 2 DM and CVD, and to evidence their potential pathophysiological role in the context of these diseases. We will dedicate a special focus to the high-mobility group A1 (HMGA1) common variant rs139876191, previously identified by us as a susceptibility locus for type 2 DM (26), and recently also associated with MI (27). In addition, we intend to provide an overview about the epigenetic links between type 2 DM and CVD to widen our understanding about the biological mechanisms that join these disorders. More recently, non-coding RNAs have emerged as key regulators of the pathophysiology underlying both type 2 DM and CVD (28–30), adding up to the fast-growing list of common background in the epigenetic regulation between type 2 DM and CVD. However, these mechanisms are often addressed within a specific pathological context, whereas an integrated approach should be preferred in order to capture all potential interlinks between type 2 DM and CVD.
Although the most common forms of type 2 DM and the vast majority of CVD are polygenic, Mendelian forms have also been described for both conditions, in which a single gene mutation can trigger the disease (31, 32). In this regard, heterozygous mutations in candidate genes can be at the basis of familial forms of cardiovascular risk factors, including hypertension, hypercholesterolemia and type 2 DM (32). However, such genes do not automatically predispose to both type 2 DM and CVD. For example, recent studies have described a protective role against type 2 DM of LDL receptor or Apo B gene mutations, the most commonly studied genes for familial hypercholesterolemia. Being this condition characterized by impaired intracellular transport of cholesterol, this suggests a mechanistic role of cholesterol metabolism in type 2 DM (33).
Many research reports have addressed genetic variants associated with CVD or type 2 DM (34, 35), and the list of loci joint to each specific disease is progressively increasing, mostly due to the power of genome-wide association studies (GWAS), combined with the analysis of large cohorts of patients. Up to now, at least 83 loci have been associated with type 2 DM (36), and more than 30 with CVD (37). As type 2 DM and CVD are linked by common pathophysiological mechanisms, share many risk factors, and display highly correlated phenotypes, different approaches—including candidate gene studies, linkage analyses, and GWAS—have been employed to search for genes predisposing to both diseases. Current findings are summarized in Table 2.
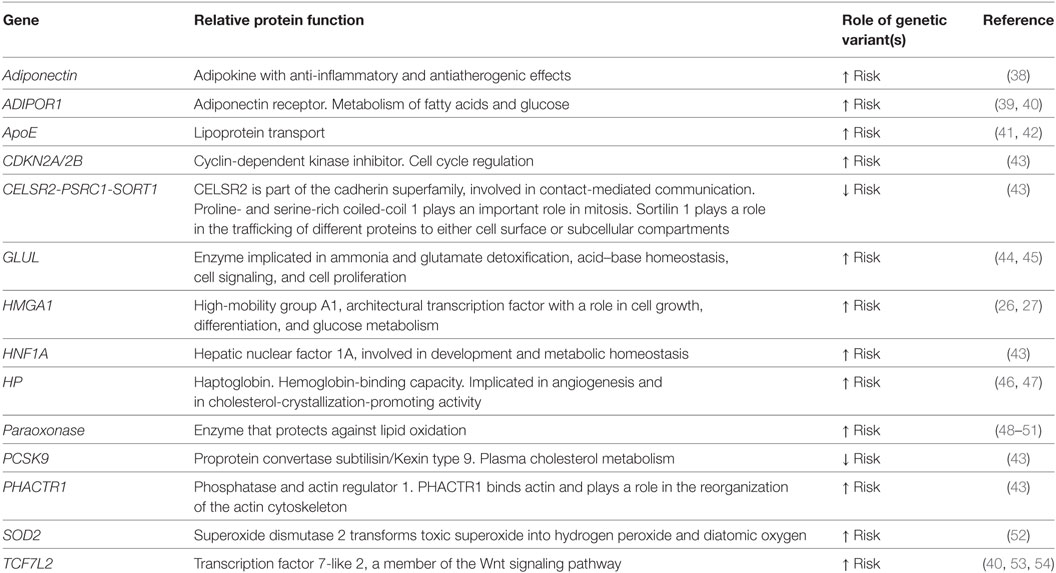
Table 2. Genes whose variants are commonly associated with both type 2 diabetes mellitus and cardiovascular disease.
Among candidate genes, several ones involved in pathways pathophysiologically related to both diseases, have been extensively investigated. One of them, paraoxonase, synthesizes an enzyme bound to high-density lipoprotein (HDL) particles, with a role in protecting LDL from proatherogenic, oxidative modifications. Paraoxonase variants have been described, which lead to reduced enzymatic activity or reduced levels of circulating enzyme, such as the paraoxonase polymorphism Gln-Arg 192, or Met-Leu 54, which are independently associated with both type 2 DM and CVD (48–51). As oxidative stress is a major contributor to atherogenesis in diabetic complications (55), further studies have examined other genes involved in the redox balance. The superoxide dismutase (SOD) 2 is one of the key antioxidant defense systems against free radicals. Ala16Val (rs4880) is the SOD 2 most commonly described gene variant and resulted in a higher risk to develop CVD in diabetic women (52). Other interesting candidate genes for diabetes and CVD are represented by adiponectin and its pathway. Adiponectin is an adipokine with anti-inflammatory and antiatherogenic effects. Reduced levels of this biomolecule, as in obesity, correlate with increased risk for type 2 DM and CVD, whereas higher levels of adiponectin protect from the risk of CVD in diabetes (56, 57). In patients with type 2 DM, the +276 G/T SNP of the adiponectin gene has been reported to be associated with CAD (38). The adiponectin receptor 1 (ADIPOR1) gene has been found to be another interesting candidate gene for CVD in diabetic subjects. In particular, common haplotypes tagging three SNPs (rs7539542, rs10920531, and rs4950894) and causing reduced ADIPOR1 gene expression were found significantly associated with CAD in type 2 DM (39). Furthermore, in type 2 DM, an ADIPOR1 gene promoter variant (rs266729) has been linked with oxidative stress and cardiovascular risk (40).
One of the most associated spot for MI and CAD, identified by GWA strategies in cohorts of different ethnicities (58, 59), is a 58 Kb non-coding region on chromosome 9p21, localized close to the CDKN2A and CDKN2B genes, in the context of a known non-coding RNA locus (ANRIL). This same region has turned out to be associated with type 2 DM and several cancers in some studies (60–63). Intriguingly, while the proximity to CDKN2A and CDKN2B, two genes with a role in cell cycle inhibition and tumor suppression, may explain a causal association with cancer, the 9p21 locus does not contain described genes for CAD, and is not linked with major cardiovascular risk factors, such as plasma lipoproteins, and hypertension. As mentioned before, several studies, but not all, have found the association of this locus with type 2 DM (60–62, 64, 65). In this regard, it has been reported that susceptibility to CAD and diabetes is encoded by distinct, tightly linked SNPs on chromosome 9p21, thereby sustaining an independent association, with the ANRIL locus, of CAD and type 2 DM susceptibility (66). On the other hand, the putative molecular role of this locus in human CVD and type 2 DM has not been yet definitively identified. In fact, while mice lacking the orthologous region on chromosome 4 showed a reduction in cdkn2a and cdkn2b expression in several tissues, as well as increased incidence of cancers and increased proliferation of vascular smooth muscle cells (VSMCs), this condition was not associated with accelerated atherosclerosis (67). Moreover, studies aimed at evaluating CDKN2A/2B and lncANRIL levels in patients have provided conflicting data (68–70), underlying our current limit to interpret results from the non-coding genome. Recently, it has been hypothesized that the regulation of CDKN2B gene expression by lncANRIL could be involved in glucose homeostasis (71), while in diabetic patients, high glucose could alter ANRIL expression, favoring cell adhesion and cell proliferation, thereby leading to atherosclerosis (72). Other molecular mechanisms through which lncANRIL are associated with diabetes and its cardiovascular complications, however, remain unclear.
In another important study, 12 loci, previously identified by GWAS as predictors of coronary heart disease (CHD) in the general population, were investigated in three CHD case–control studies of diabetic patients. Among them, five variants, rs4977574 (CDKN2A/2B), rs12526453 (PHACTR1), rs646776 (CELSR2-PSRC1-SORT1), rs2259816 (HNF1A), and rs11206510 (PCSK9), showed a significant association with the risk for CHD also in type 2 DM (43). Among the type 2 DM susceptibility genes investigated by GWAS, the transcription factor 7-like 2 gene (TCF7L2) has been identified as one of the most significant (73). TCF7L2 variants have been found to be associated with CVD in some (40, 53), but not in all (74) reports, although the association between TCF7L2 risk alleles and CAD was not higher in diabetic individuals. Subsequent studies analyzed the association of three TCF7L2 variants (rs7903146, rs12255372, and rs11196205) with CAD in 1,650 patients that underwent coronary angiography, and found that these variants were more strongly associated with CAD in diabetic patients than in non-diabetics (54).
Other genetic variants may confer more CHD risk in patients with type 2 DM than in non-diabetic subjects. An example is a polymorphism in the promoter region (−308) of the TNF-α gene, whose association with type 2 DM is even stronger in diabetic women (75). Also, as the apolipoprotein E (apo E) polymorphisms are known to modulate the risk for CVD in type 2 DM, many studies, but not all, have shown that the ApoE4 allele is related to a greater susceptibility for CVD in the presence of type 2 DM (41, 42). Another important challenge refers to the identification of diabetes-specific susceptibility genes for CVD. In this regard, interesting studies have addressed the haptoglobin (HP) gene polymorphisms. HP is a serum protein that binds free hemoglobin, and prevents hemoglobin-induced oxidation. It is synthesized by two alleles, HP1 and HP2, the former encoded by 5 exons, and the latter by 7 exons, obtained by the intragenic duplications of exons 3 and 4. No significant association was shown between HP phenotype and CVD risk, whereas the HP2 allele is strongly related to CVD in type 2 DM patients (46). The molecular explanations that may justify this specific association include the reduced ability of HP2, with respect to HP1, to prevent the oxidative stress driven by glycated hemoglobin (46, 76). Further studies have demonstrated that, in a large, type 2 DM-enriched cohort of Americans of European ancestry, the HP2-2 phenotype significantly associates with CVD mortality, triglyceride levels, and subclinical atherosclerosis, in the form of increased carotid-media thickness, but not of calcified arterial plaques (47). Also, a recent GWAS investigated the link between glutamate-ammonia ligase (GLUL) gene polymorphism and CHD, demonstrating that the association was specific for type 2 DM patients (44). Further studies confirmed the association of the rs10911021 GLUL variant with type 2 DM, and demonstrated that this polymorphism does not affect amino acid metabolism. However, although apparently counterintuitive, it is associated with lower HDL cholesterol levels, and large HDL particles (45).
These and other examples of type 2 DM-specific associated variants, while enriching our knowledge about CVD risk factors, contribute to the debate about the “common soil” hypothesis for type 2 DM and CVD (20, 77). In this context, only few significant loci for type 2 DM and CVD, identified by large-scale GWAS, had shown to be shared between both diseases. Starting from this provocative observation, new strategies have been used to identify novel and ethnic-specific genetic links between CVD and type 2 DM. For example, studies have been carried out using an integrative pathway and network analysis combined with GWAS in more than 15,000 women from three different ethnicities, leading to the identification of eight major pathways shared by type 2 DM and CVD in all ethnic groups (78). In these studies, key driver genes, influencing the extra-cellular matrix composition, such as COL1A1, COL3A1, and ELN, that had been cross-validated in mouse models for type 2 DM and CVD, have also emerged. Interestingly, few peculiar pathways related to specific ethnic groups were identified (78). In addition, in the past years, attempts have been made to assess a more reliable disease susceptibility for CVD in type 2 DM by analyzing cumulative genetic risk from multiple loci rather than from single SNPs (79, 80). As an example, two genetic risk scores have been successfully used to predict CVD and CVD fatal outcomes using patients from the Diabetes Heart Study (81).
High-mobility group A1 is a small, non-histonic nuclear protein, with pleiotropic effects involved in the regulation of embryogenesis, oncogenesis and tumor progression, cell differentiation, as well as inflammation (82–84). As an architectural transcription factor, it binds to the minor groove of AT-rich regions of DNA, and alters the chromatin conformation, facilitating the assembly and stability of stereospecific DNA–protein complexes called “enhanceosomes,” which drive gene transcription (85–87). Many studies from our group have demonstrated the role of HMGA1 in the transcriptional control of glucose metabolism, being a key regulator of the insulin receptor (INSR), insulin-like growth factor binding protein 1 (IGFBP1), retinol binding-protein 4 (RBP4), visfatin, and insulin (INS) genes (88–93), as well as an important mediator of insulin action (94). Defects in HMGA1 protein, or the association with functional HMGA1 variants, among which the most common rs139876191 variant (previously named rs146052672), cause a decrease in INSR expression and a trans-ethnic increased susceptibility to either type 2 DM (26, 95–98) or metabolic syndrome (99). Besides its effects on glucose homeostasis, HMGA1 plays a role in adipogenesis and lipid metabolism (100–102), while the HMGA1 rs139876191 variant correlates with body mass index, and reduced HDL levels in patients with metabolic syndrome and type 2 DM (97, 99).
Also, HMGA1 plays a critical role in the development and progression of the atherosclerotic plaque by promoting the proliferation and the migration of VSMCs to the neointima, and by inducing the expression of several inflammatory cytokines, adhesion molecules, including CD44, and chemokines (103, 104). On the other hand, by activating the matrix metalloproteinase 9 (MMP-9), and the vascular endothelial growth factor (VEGF), HMGA1 is essential for vascular repair and neoangiogenesis, whereas its lack causes impairment of both vascular protection from injuries and of neovascularization (92, 105, 106). Recently, the functional HMGA1 rs139876191 variant has been found to be associated with acute MI, independently of type 2 DM or other cardiovascular risk factors, such as hypertension, obesity, and gender, suggesting that HMGA1 may represent a new candidate gene for acute MI and a marker for cardiovascular risk (27). Although further studies in other populations are needed to confirm this association, due to its pathophysiological role in insulin resistance, glucose homeostasis, lipid metabolism, inflammation and vascular repair, HMGA1 may represent a convincing molecular link between type 2 DM and MI.
Epigenetic processes are defined as heritable modifications in gene expression that occur in the absence of changes in the DNA sequence, and include DNA methylation, histone acetylation, and RNA-based mechanisms. These processes are cell-specific, susceptible to modifications, and responsive to the environment, and should be taken into account to better understand otherwise hidden causes of diseases.
New research investigations have addressed the link between epigenetic factors, type 2 DM and CVD. Hyperglycemia, for example, can induce epigenetic changes that lead to the overexpression of genes implicated in vascular inflammation. In particular, hyperglycemia has been shown to activate the NF-kB signaling pathway in cultured THP-1 monocytes, leading to the production of MCP-1 and other inflammatory factors, and to the expression of adhesion molecules in endothelial cells, providing a plausible molecular mechanism for endothelial dysfunction and atherosclerosis (107). On the other hand, clinical studies have demonstrated that early intensive control of glycemia in diabetic patients is crucial to prevent chronic micro- and macrovascular complications, reinforcing the notion that glycemia may have a longstanding influence on clinical outcomes, a phenomenon called “metabolic memory” (108).
In support of an epigenetic role of hyperglycemia, it has been demonstrated, in aortic endothelial cells, that exposure to high glucose correlates with the inverse acetylation of the histone H3K9/K14 and modified DNA methylation patterns (109). Several histone lysine modifications have also been described following transient high glucose levels that may account for a persistent transcriptional induction of the RELA gene, encoding for the p65 subunit of NF-kB, even after subsequent incubation of endothelial cells with normal glucose concentrations (110). Altogether, the net result of this activity leads to the transcriptional activation of some target genes implicated in the endothelial dysfunction, and the repression of other ones (111). Acetylation or hyperacetylation may also occur, being responsible for the increased expression of HMOX1, MMP10, SLC7A11, MMP1, MCP-1, and ICAM genes (109). Hyperglycemia is, however, not the only inducer of epigenetic modifications. Many other pathophysiologic mechanisms that may be operative in diabetes, independently from glucotoxicity, like ROS, PKC activation, and AGEs have been described to induce also epigenetic changes (112). In particular, ROS production is able to significantly induce the CpG hypomethylation of the p66Shc promoter and, at the same time, an increment in the H3 histone acetylation. Thus, ROS-induced epigenetic modifications are associated with higher levels of p66Shc, a mitochondrial adaptor that modulates the intracellular redox state, and with significant activation of PKC, therefore sustaining endothelial dysfunction and vascular damages (111, 112).
Further studies have investigated the associations between epigenetic modifications and cardio-metabolic phenotypes, such as obesity, dyslipidemia, insulin resistance, inflammation, and hypertension, in relation to CVD risk (113). In a recent study, peripheral blood mononuclear cells were used to measure histone deacetylases (HDACs) activity and expression in relation to glycemia, inflammation and insulin resistance in patients with type 2 DM. Low-grade chronic inflammation and insulin resistance induced HDAC3 activity and expression, and correlated positively with circulating levels of TNF-α, IL-6, and other proinflammatory markers, and negatively with Sirt1 expression (114).
Several reports have demonstrated a correlation between DNA methylation and cardiovascular risk. The susceptibility haplotype rs8050136 of the FTO gene, a prominent gene associated with increased risk for obesity and CVD, displayed increased levels of methylation (115); a similar mechanism has been hypothesized for the rs9939609 polymorphism (116). In another candidate gene study, an association between IGF2 methylation and lipid profile alterations was found in obese children. In particular, IGF2 hypermethylation was associated with higher triglyceride/HDL-cholesterol ratio, representing an epigenetic marker of metabolic risk (117). Another study that combined genome-wide transcriptome and CpG methylation profiling by array, reported many differentially methylated predicted sites in adipose tissue from insulin-resistant patients compared to controls, which included genes involved in insulin signaling and in the interaction with integrins (118). Altered methylation were also found in IL18, CD44, CD48, CD38, Cd37, CX3CL1, CXCR1, CXCR2, CXCL1, IGF1R, APOB48R, LEF1, GIPR, GRB10, SIRT2, HDAC4, DNMT3A, LEPR, and LEP genes that were already found to be strongly and independently associated with insulin resistance (118–121). In addition, polarization of adipose tissue macrophages from an anti-inflammatory (M2) to a proinflammatory phenotype (M1) in obese mice was shown to involve the methylation of the PPARγ promoter (122). Finally, there are evidences that MI susceptibility risk may be influenced by epigenetic changes occurring in the prenatal environment (123).
MicroRNAs are small single-strand RNA molecules that influence their target genes at a posttranscriptional level, thereby regulating many biological processes. Since their discovery about two decades ago, numerous miRNAs have been described to be associated with a multitude of diseases, including type 2 DM and CVD (28, 124, 125). In particular, with reference to type 2 DM, miRNAs have shown to be involved in regulating beta cell function, insulin response, glucose homeostasis, as well as the pathogenesis of diabetic vascular complications (126, 127). Research in this field has highlighted new mechanistic links between diabetes and CVD (128), with many evidences proving the involvement of distinct miRNAs in the pathological steps that lead to atherosclerosis (Figure 1).
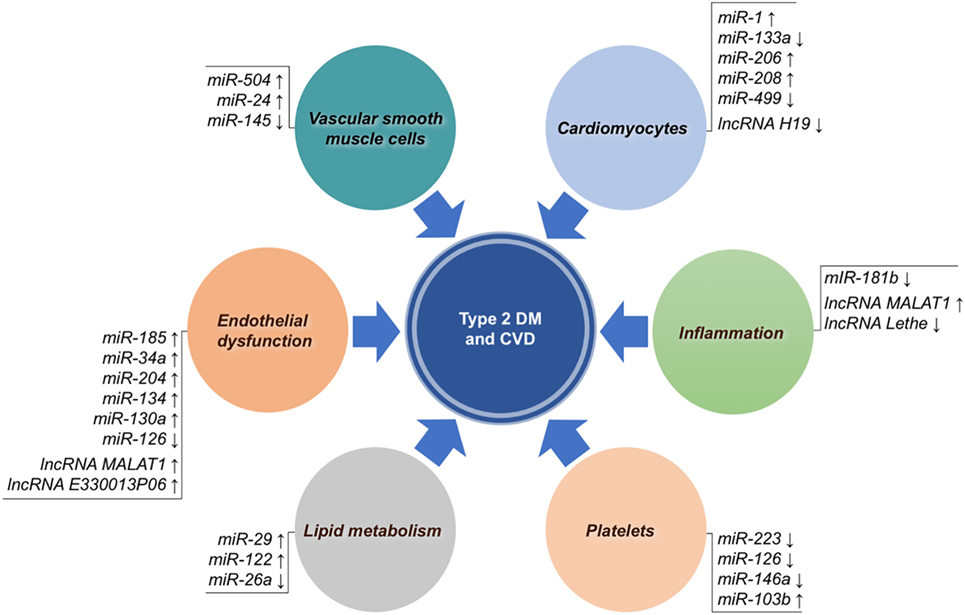
Figure 1. Non-coding RNAs associated with both type 2 diabetes mellitus (DM) and cardiovascular disease (CVD). MicroRNAs (miRNAs) and long non-coding RNAs (lncRNAs) are grouped according to their main biological mechanism involved in atherosclerotic CVD. Arrows indicate overexpression (↑) or underexpression (↓).
In vitro and in vivo studies concerning the mechanisms that are responsible for the endothelial dysfunction in diabetes demonstrated that, in the presence of high glucose concentrations, upregulation of miR-185 reduced the expression of the glutathione peroxidase-1 (GPx-1) gene, which encodes an enzyme that is important in the prevention of oxidative stress (129); instead upregulation of miR-34a and miR-204 contributed to endothelial cell senescence by impairing SIRT-1 expression and function (130, 131). In the endothelium, miR-126 exerts proangiogenic, and anti-inflammatory activities. At a functional level, it enhances VEGF and fibroblast growth factor activities, contributing to vascular integrity and angiogenesis (132, 133), recruits progenitor cells through the chemokine CXCL12 (134), while it suppresses inflammation by inhibiting TNF-α, ROS, and NADPH oxidase via HMGB1 (135). Consistently, miR-126 levels are down-regulated in both myocardial tissue and plasma from type 2 diabetic patients without any known anamnestic data for CVD (136, 137), and in patients with CAD (138), suggesting that it could represent a new diagnostic marker for diabetes and CVD. Other studies in endothelial colony-forming cells, as well as in progenitor endothelial cells (EPCs) exposed to high glucose, demonstrated that miR-134 and miR-130a affected cell motility and apoptosis, respectively (139, 140).
In diabetes, VSMCs loose their contractility and acquire proliferative and migratory properties, facilitating the onset of pathological processes relevant to CVD (141). miR-145 has proved to reduce its level in the presence of high glucose, to impair myocardin gene expression via Klf4, and to facilitate VSMC proliferation (29, 142). In this context, a role of miR-504 and miR-24 in promoting VSMC proliferation and migration, has also been reported (143, 144).
An important issue is the link between lipid metabolism and miRNAs in diabetic CVD. Several important genes implicated in lipid synthesis or processing, like FoxA2, Ppargcla, Hmgcs2, and Abdhd5 have been shown to be dysregulated by miR-29 in Zucker diabetic fatty rats (145), while HNF-4 alpha was found to be raised by increased levels of miR-122 in diabetic mice and insulin-resistant HepG2 cells (146). Both miR-122 and HNF-4 alpha were able to upregulate the expression of SREBP-1 and FAAS genes, causing abnormal cholesterol homeostasis and high levels of fatty acid and triglyceride synthesis (146). Finally, decreased levels of miR-26a have been reported in obese mice, in which they contribute to increased fatty acid synthesis, and to obesity-related metabolic complications (147).
Platelets are key partaker in CVD and their involvement in the development of cardiovascular complications is strengthened in diabetes (148). Platelets play an important role in the pathophysiology of thrombosis and represent an important source of different RNA species, including pseudogenes, intronic transcripts, non-coding RNAs, and antisense transcripts (149, 150). These molecules can be released by platelets through microvescicles, contributing to the horizontal transfer of molecular signals delivered through the bloodstream to specific sites of action (151). The downregulation of miR-223, miR-126, or 146a observed in diabetic and hyperglycemic patients (137, 152) has been associated with increased platelet reactivity and aggregation (153, 154). In line with these findings, silencing of miR-223 in mice caused a hyperreactive and hyperadhesive platelet phenotype, and was associated with calpain activation through the increased expression of beta1 integrin, kindlin-3, and factor XIII (153, 155). Moreover, the modulation of the expression levels of platelet miRNAs can also be measured in plasma. In fact, plasma levels of miR-223 and miR-126 are decreased in diabetics (137, 156). This leads to the upregulation of the P2Y12 receptor, as well as P-selectin, further contributing to platelet dysfunction (156). As a result of this interaction, activation level of platelets in type 2 DM is increased (149, 156, 157). Consistently with this, circulating miR-223 levels are independent predictors of high on-treatment platelet reactivity (158). Another interesting mechanism linking platelets and diabetes involves miR-103b, a platelet-derived biomarker proposed for the early diagnosis of type 2 DM, and the secreted frizzled-related protein-4 (SFRP4), a potential biomarker of early β cell dysfunction and diabetes. In fact, platelet-derived miR-103b is able to downregulate SFRP4, whose expression levels are significantly increased in pancreatic islets and in the blood of patients with prediabetes or overt diabetes (159). These interesting results identify miR-103b as a novel potential marker of prediabetes and diabetes, and disclose a novel potential therapeutic target in type 2 DM.
Macrophages also play a key role in atherosclerotic plaques. Unbalanced production of proinflammatory molecules from adipose tissue contributes to the polarization of macrophages toward the M1 phenotype and their accumulation within the vessel wall (160, 161). It has been demonstrated in vitro and in vivo that in the presence of high glucose or in insulin-resistant states, endothelial cells decreased miR-181b expression, while the production of this miR, through the inhibition of AKT Ser 473 phosphorylation, was associated with a M2 anti-inflammatory response, but not with antiproliferative effects (162). These results are compatible with an inhibitory role of miR-181b in atherosclerosis.
Other miRNAs, abundantly expressed in cardiomyocytes, such as miR-1 and miR-133a, seem to be crucial in preventing myocardial dysfunction. Both these miRNAs have been shown to be reduced in ischemic myocardial tissue, in left ventricular hypertrophy, and in diabetic cardiomyopathy (163, 164). Among the molecular mechanisms proposed for miR-133a, the repression of serum response factor, which plays a role in myoblast proliferation, of RhoA (a protein involved in GDP-GTP cycling), Cdc42 (a kinase implicated in hypertrophy), and Nelf-A/WHSC2 nuclear factor (165).
Many cardiac-enriched miRNAs have been reported to be responsive to hyperglycemia, including miR133a, miR-1, and miR-206, with the last two favoring the apoptosis of cardiomyocytes through the negative regulation of the heath shock protein 60 (166). Recent evidences demonstrated that miR-208 and miR-499, together with miR-1 and miR-133, could play a role into the molecular mechanisms leading to the differentiation of stem cells into cardiomyocytes (167). In fact, the involvement of miR-133a in the modulation of contractility was recently demonstrated in streptozotocin-induced diabetic rats (168), in which miR-133a overexpression was able to improve contractility through the upregulation of tyrosine aminotransferase, a known regulator of norepinephrine production and β-adrenergic receptors (168). These latter findings are particularly interesting, as we could recently demonstrate that miR-133a transcoronary concentration has an interesting prognostic potential in patients with CVD (169). Less data is currently available on the involvement of miR-208 in diabetic heart disease. A proposed mechanism for this miRNA implicated a role in the regulation of myosin heavy chain gene expression (170). On the other hand, functional studies showed that miR-499 protects cardiomyocytes from ischemic damage and apoptosis via the suppression of calcineurin-mediated dephosphorylation of dynamin-related protein-1 (171).
Specific miRNAs, such as miR-15, -16, -26a, -196a2, and Let-7a (172) are able to modulate HMGA1, whose association with acute MI, type 2 DM, and cardiovascular risk has already been discussed (26, 27, 99). Also, HMGA1 can specifically induce the expression of miR-10b, -21, -125b, -221, -222, or inhibit the production of miR-34a and -603, all of which are involved in several aspects of cardiovascular pathophysiology (173), thereby further supporting the notion that a complex relationship indeed exists between HMGA1 and miRNAs in this context (29, 174).
Long non-coding RNAs include non-protein coding transcripts longer than 200 nucleotides (175, 176). They have both nuclear and cytoplasmic location and work as signal amplifiers for biological activity, regulating gene expression through a variety of partly explored molecular mechanisms, including the interaction or competition with other RNAs, DNA binding proteins, and specific regulatory DNA sequences (176, 177). New increasing evidences show the involvement of lncRNAs in human diseases (178), such as cardiometabolic diseases (179–182). For example, in the context of atherosclerosis (Figure 1), experimental studies have shown altered expression of lncRNAs in several processes implicated in SMC proliferation, endothelial function, inflammatory cells, lipid metabolism and obesity, as well as with insulin resistance (183), while clinical studies have demonstrated that circulating lncRNAs could be potentially used to predict type 2 DM (182) or the outcome of heart failure (184). However, data from this kind of studies are still initial and in progress. The first lncRNA robustly associated with CVD and type 2 DM has been lncANRIL, a locus identified by GWA studies, already widely discussed in this review in the Section “Genetic Polymorphisms.” Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is an lncRNA particularly expressed in the nucleus and physiologically implicated in the regulation of endothelial cell function. It has been demonstrated that hyperglicemia alters MALAT1 expression, leading to micro- and macrovascular damages (185–187). In particular, at a molecular level, MALAT1, by targeting serum amyloid antigen 3, a proinflammatory ligand, has been shown to induce the expression of IL-6 and TNF-α, as well as ROS production, thereby promoting endothelial dysfunction (187). Recently, the lncRNA H19, which has a role in limiting body weight and cell proliferation, was found to be markedly reduced in a mouse model of diabetic cardiomyopathy as a consequence of hyperglycemia (188). In an elegant study, it was demonstrated that lncRNA H19, via mIR-675, targets VDAC1, a mitochondrial porin that plays a role in ATP transport, regulating cardiomyocyte apoptosis (188). In other cases, lnc RNAs have been implicated in diabetic vascular complications through mechanisms linked to macrophage-mediated inflammation. By transcriptome profiling of bone marrow-derived macrophages from db/db and diet-induced insulin-resistant type 2 diabetic mice, an increase in lncRNA E330013P06 has been observed, demonstrating that this lncRNA promoted foam cell formation and endothelial dysfunction through the expression of inflammatory genes like Nos2, IL6 and ptgs2 (189). Also, a recent study using RAW264.7, as well as bone-derived macrophages, showed that lncRNA Lethe exerted an anti-inflammatory role by inhibiting the translocation of NF-kB transcription factor to the nucleus, and that in the presence of high glucose concentrations, lncRNA Lethe expression was reduced, with a consequent increment in NOX2 gene expression and ROS production (190).
In this review, we provide an overlook about the main genetic and epigenetic factors linking type 2 DM and CVD, with a particular emphasis on the pathophysiological mechanisms involved. We addressed known genetic variants shared by both conditions, and the most relevant epigenetic mechanisms involved in their interplay. However, as a lower amount of solid evidence is available to date about epigenetics in this pathophysiological context, further research will be necessary to validate, in patients with type 2 DM, the results obtained so far in vitro and in vivo, in animal models. A deeper understanding of gene networks, intracellular pathways, and cell-to-cell communication mechanisms will allow the identification of novel biomarkers, as well as new therapeutic targets to exploit in the management of CVD in patients with type 2 DM.
SR and BA prepared the first draft of the manuscript; EC was involved in the literature search; AB, CI, and DPF critically revised the manuscript and wrote the final version of the article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet (2005) 9467:1333–46. doi:10.1016/S0140-6736(05)61032-X
2. Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care (2004) 27:1047–53. doi:10.2337/diacare.27.10.2569-a
3. Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world – a growing challenge. N Engl J Med (2007) 356:213–5. doi:10.1056/NEJMp068177
4. Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract (2014) 103:137–49. doi:10.1016/j.diabres.2013.11.002
5. Krolewski AS, Warram JH, Freire MB. Epidemiology of late diabetic complications. A basis for the development and evaluation of preventive programs. Endocrinol Metab Clin North Am (1996) 2:217–42. doi:10.1016/S0889-8529(05)70322-4
6. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation (2017) 135:e146–603. doi:10.1161/CIR.0000000000000485
7. Kannel WB, McGee DL. Diabetes and cardiovascular disease. JAMA (1979) 241:2035–8. doi:10.1001/jama.1979.03290450033020
8. Grundy SM, Benjamin IJ, Burke GL, Chait A, Eckel RH, Howard BV, et al. Diabetes and cardiovascular disease: a statement for health professionals from the American Heart Association. Circulation (1999) 100:1134–46. doi:10.1161/01.CIR.100.10.1134
9. Kim JA, Koh KK, Quon MJ. The union of vascular and metabolic actions of insulin in sickness and in health. Arterioscler Thromb Vasc Biol (2005) 25(5):889–91. doi:10.1161/01.ATV.0000164044.42910.6b
10. Woods M, Mitchell JA, Wood EG, Barker S, Walcot NR, Rees GM, et al. Endothelin-1 is induced by cytokines in human vascular smooth muscle cells: evidence for intracellular endothelin-converting enzyme. Mol Pharmacol (1999) 55:902–9.
11. Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med (2004) 350:664–71. doi:10.1056/NEJMoa031314
12. Low Wang CC, Hess CN, Goldfine AB. Clinical update: cardiovascular disease in diabetes mellitus: atherosclerotic cardiovascular disease and heart failure in type 2 diabetes mellitus – mechanisms, management, and clinical considerations. Circulation (2016) 133:24. doi:10.1161/CIRCULATIONAHA.116.022194
13. Carr ME. Diabetes mellitus: a hypercoagulable state. J Diabetes Complications (2001) 15:44–54. doi:10.1016/S1056-8727(00)00132-X
14. Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med (1999) 340:115–26. doi:10.1056/NEJM199901143400207
15. Haffner SM, Stern MP, Hazuda HP, Mitchell BD, Patterson JK. Cardiovascular risk factors in confirmed prediabetic individuals. Does the clock for coronary heart disease start ticking before the onset of clinical diabetes? JAMA (1990) 263:2893–8. doi:10.1001/jama.263.21.2893
16. Festa A, D’Agostino R, Howard G, Mykkänen L, Tracy RP, Haffner SM. Chronic subclinical inflammation as part of the insulin resistance syndrome: the insulin resistance atherosclerosis study (IRAS). Circulation (2000) 102:42–7. doi:10.1161/01.CIR.102.1.42
17. Otani H. Oxidative stress as pathogenesis of cardiovascular risk associated with metabolic syndrome. Antioxid Redox Signal (2011) 15:1911–26. doi:10.1089/ars.2010.3739
18. Andreotti F, Becker RC. Atherothrombotic disorders: new insights from hematology. Circulation (2005) 111:1855–63. doi:10.1161/01.CIR.0000160361.73423.23
19. Sowers JR, Epstein M, Frohlich ED. Diabetes, hypertension, and cardiovascular disease: an update. Hypertension (2001) 37:1053–9. doi:10.1161/01.HYP.37.4.1053
20. Stern MP. Diabetes and cardiovascular disease. The “common soil” hypothesis. Diabetes (1995) 44:369–74. doi:10.2337/diabetes.44.4.369
21. Wu L, Parhofer KG. Diabetic dyslipidemia. Metabolism (2014) 63:1469–79. doi:10.1016/j.metabol.2014.08.010
22. Wilson PW, Kannel WB. Obesity, diabetes, and risk of cardiovascular disease in the elderly. Am J Geriatr Cardiol (2002) 11:119–23. doi:10.1111/j.1076-7460.2002.00998.x
23. Grundy SM. Pre-diabetes, metabolic syndrome, and cardiovascular risk. J Am Coll Cardiol (2012) 59:7. doi:10.1016/j.jacc.2011.08.080
24. Greco M, Chiefari E, Montalcini T, Accattato F, Costanzo FS, Pujia A, et al. Early effects of a hypocaloric, Mediterranean diet on laboratory parameters in obese individuals. Mediators Inflamm (2014) 2014:750860. doi:10.1155/2014/750860
25. Ma RCW. Genetics of cardiovascular and renal complications in diabetes. J Diabetes Investig (2016) 7:139–54. doi:10.1111/jdi.12391
26. Chiefari E, Tanyolaç S, Paonessa F, Pullinger CR, Capula C, Iiritano S, et al. Functional variants of the HMGA1 gene and type 2 diabetes mellitus. JAMA (2011) 305:9. doi:10.1001/jama.2011.207
27. De Rosa S, Chiefari E, Salerno N, Ventura V, D’Ascoli GL, Arcidiacono B, et al. HMGA1 is a novel candidate gene for myocardial infarction susceptibility. Int J Cardiol (2017) 227:331–4. doi:10.1016/j.ijcard.2016.11.088
28. De Rosa S, Curcio A, Indolfi C. Emerging role of microRNAs in cardiovascular diseases. Circ J (2014) 78:567–75. doi:10.1253/circj.CJ-14-0086
29. Gareri C, De Rosa S, Indolfi C. MicroRNAs for restenosis and thrombosis after vascular injury. Circ Res (2016) 118:1170–84. doi:10.1161/CIRCRESAHA.115.308237
30. Iaconetti C, Gareri C, Polimeni A, Indolfi C. Non-coding RNAs: the "dark matter" of cardiovascular pathophysiology. Int J Mol Sci (2013) 14:19987–20018. doi:10.3390/ijms141019987
31. American Diabetes Association. Standards of medical care in diabetes-2017, classification and diagnosis of diabetes. Diabetes Care (2017) 40:S11–24. doi:10.2337/dc17-S005
32. O’Donnell CJ, Nabel EG. Genomics of cardiovascular disease. N Engl J Med (2011) 365:2098–109. doi:10.1056/NEJMra1105239
33. Besseling J, Kastelein JJ, Defesche JC, Hutten BA, Hovingh GK. Association between familial hypercholesterolemia and prevalence of type 2 diabetes mellitus. JAMA (2015) 313:1029–36. doi:10.1001/jama.2015.1206
34. Mohlke KL, Boehnke M, Abecasis GR. Metabolic and cardiovascular traits: an abundance of recently identified common genetic variants. Hum Mol Genet (2008) 17(R2):R102–8. doi:10.1093/hmg/ddn275
35. Kathiresan S, Srivastava D. Genetics of human cardiovascular disease. Cell (2012) 148:1242–57. doi:10.1016/j.cell.2012.03.001
36. Wang X, Strizivh G, Hu Y, Wang T, Kaplan RC, Qi Q. Genetic markers of type 2 diabetes: progress in genome-wide association studies and clinical application for risk prediction. J Diabetes (2016) 8:24–35. doi:10.1111/1753-0407.12323
37. Ashar FN, Arking DE. Genomics of complex cardiovascular disease. 2nd ed. In: Kumar D, Eng C, editors. Genomic Medicine: Principles and Practice. New York, NY: Oxford University Press (2014). p. 316–36.
38. Bacci S, Menzaghi C, Ercolino T, Ma X, Rauseo A, Salvemini L, et al. The +276 G/T single nucleotide polymorphism of the adiponectin gene is associated with coronary artery disease in type 2 diabetic patients. Diabetes Care (2004) 27:2015–20. doi:10.2337/diacare.27.8.2015
39. Soccio T, Zhang YY, Bacci S, Mlynarski W, Placha G, Raggio G, et al. Common haplotypes at the adiponectin receptor 1 (ADIPOR1) locus are associated with increased risk of coronary artery disease in type 2 diabetes. Diabetes (2006) 55(2763):2770. doi:10.2337/db06-0613
40. Sousa AG, Selvatici L, Krieger JE, Pereira AC. Association between genetics of diabetes, coronary artery disease, and macrovascular complications: exploring a common ground hypothesis. Rev Diabet Stud (2011) 8:230–44. doi:10.1900/RDS.2011.8.230
41. Ukkola O, Kervinen K, Salmela PI, von Dickhoff K, Laakso M, Kesaniemi YA. Apolipoprotein E phenotype is related to macro- and microangiopathy in patients with non-insuli dependent diabetes mellitus. Atherosclerosis (1993) 101:9–15. doi:10.1016/0021-9150(93)90096-D
42. El-Lebedy D, Raslan HM, Mohammed AM. Apolipoprotein E gene polymorphism and risk of type 2 diabetes and cardiovascular disease. Cardiovasc Diabetol (2016) 15:12. doi:10.1186/s12933-016-0329-1
43. Qi L, Parast L, Cai T, Powers C, Gervino EV, Hauser TH, et al. Genetic susceptibility to coronary heart disease in type 2 diabetes: 3 independent studies. J Am Coll Cardiol (2011) 58:2675–82. doi:10.1016/j.jacc.2011.08.054
44. Qi L, Qi Q, Prudente S, Mendonca C, Andreozzi F, di Pietro N, et al. Association between a genetic variant related to glutamic acid metabolism and coronary heart disease in individuals with type 2 diabetes. JAMA (2013) 310:821–8. doi:10.1001/jama.2013.276305
45. Beaney KE, Cooper JA, McLachlan S, Wannamethee SG, Jefferis BJ, Whincup P, et al. Variant rs10911021 that associates with coronary heart disease in type 2 diabetes, is associated with lower concentrations of circulating HDL cholesterol and large HDL particle but not with amino acids. Cardiovasc Diabetol (2016) 15:115. doi:10.1186/s12933-016-0435-0
46. Levy AP. Haptoglobin: a major susceptibility gene for diabetic cardiovascular disease. Isr Med Assoc J (2004) 6:308–10.
47. Adams JN, Cox AJ, Freedman BI, Langefeld C, Carr JJ, Bowden DW. Genetic analysis of haptoglobin polymorphisms with cardiovascular disease and type 2 diabetes in the diabetes heart study. Cardiovasc Diabetol (2013) 12:31. doi:10.1186/1475-2840-12-31
48. Ruiz J, Blanché H, James RW, Garin MC, Vaisse C, Charpentier G, et al. Gln-Arg192 polymorphism of paraoxonase and coronary heart disease in type 2 diabetes. Lancet (1995) 346:869–72. doi:10.1016/S0140-6736(95)92709-3
49. Serrato M, Marian AJ. A variant of human paraoxonase/arylesterase (HUMPONA) gene is a risk factor for coronary artery disease. J Clin Invest (1995) 96:30005–8. doi:10.1172/JCI118373
50. Blatter Garin MC, James P, Blanché H, Passa P, Froguel P, Ruiz J. Paraoxonase polymorphism Met-Leu54 is associated with modified serum concentration of the enzyme. A possible link between the paraoxonase gene and increased risk of cardiovascular disease. J Clin Invest (1997) 99:62–6. doi:10.1172/JCI119134
51. Pfohl M, Koch M, Enderle MD, Kuehn R, Fuellhase J, Karsch KR, et al. Paraoxonase 192 Gln/Arg gene polymorphism, coronary artery disease, and myocardial infarction in type 2 diabetes. Diabetes (1999) 48:623–7. doi:10.2337/diabetes.48.3.623
52. Jones DA, Prior SL, Tang TS, Bain SC, Hurel SJ, Humphries SE, et al. Association between the rs4880 superoxide dismutase 2(C>T) gene variant and coronary heart disease in diabetes mellitus. Diabetes Res Clin Pract (2010) 90:196–201. doi:10.1016/j.diabres.2010.07.009
53. Sousa AG, Marquezine GF, Lemos PA, Martinez E, Lopes N, Hueb WA, et al. TCF7L2 polymorphism rs7903146 is associated with coronary artery disease severity and mortality. PLoS One (2009) 11:e7697. doi:10.1371/journal.pone.0007697
54. Muendlein A, Saely CH, Geller-Rhomberg S, Sonde-Regger G, Rein P, Winder T, et al. Single nucleotide polymorphisms of TCF7L2 are linked to diabetic coronary atherosclerosis. PLoS One (2011) 3:e17978. doi:10.1371/journal.pone.0017978
55. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res (2010) 107:1058–70. doi:10.1161/CIRCRESAHA.110223545
56. Shibata R, Ouchi N, Murohara T. Adiponectin and cardiovascular disease. Circ J (2009) 74:608–14. doi:10.1253/circj.CJ-09-0057
57. Schulze MB, Shai I, Rimm EB, Li T, Rifai N, Hu FB. Adiponectin and future coronary heart disease events among men with type 2 diabetes. Diabetes (2005) 54:534–9. doi:10.2337/diabetes.54.2.534
58. Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature (2007) 447:661–78. doi:10.1038/nature05911
59. Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science (2007) 316:1491–3. doi:10.1126/science.1142842
60. Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research, Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science (2007) 316:1331–6. doi:10.1126/science.1142358
61. Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science (2007) 316:1341–5. doi:10.1126/science.1142382
62. Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliot KS, Lango H, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science (2007) 316:1336–41. doi:10.1126/science.1142364
63. Li Z, Yu X, Shen J. ANRIL: a pivotal tumor suppressor long non-coding RNA in human cancers. Tumour Biol (2016) 37:5657–61. doi:10.1007/s13277-016-4808-5
64. Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature (2007) 445:881–5. doi:10.1038/nature05616
65. Salonen JT, Uimari P, Aalto JM, Pirskanen M, Kaikkonen J, Tdorova B, et al. Type 2 diabetes whole-genome association study in four populations: the DiaGen consortium. Am J Hum Genet (2007) 81:338–45. doi:10.1086/520599
66. Broadbent HM, Peden JF, Lorkowski S, Goel A, Ongen H, Green F, et al. Susceptibility to coronary heart disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum Mol Genet (2008) 17:806–14. doi:10.1093/hmg/ddm352
67. Visel A, Zhu Y, May D, Afzal V, Gong E, Attanasio C, et al. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature (2010) 464:409–12. doi:10.1038/nature08801
68. Cunnington MS, Santibanez Koref M, Mayosi BM, Burn J, Keavney B. Chromosome 9p21 SNPs associated with multiple disease phenotypes correlate with ANRIL expression. PLoS Genet (2010) 6:e1000899. doi:10.1371/journal.pgen.1000899
69. Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Hentzman N, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-γ signalling response. Nature (2011) 470:264–8. doi:10.1038/nature09753
70. Jarinova O, Stewart AF, Roberts R, Wells G, Lau P, Naing T, et al. Functional analysis of the chromosome 9p21.3 coronary artery diseaes risk locus. Arterioscler Thromb Vasc Biol (2009) 29:1671–7. doi:10.1161/ATVBAHA.109.189522
71. Bantubungi K, Hannou SA, Caron-Houde S, Vallez E, Baron M, Lucas A, et al. Cdkn2a/p16Ink4a regulates fasting-induced hepatic gluconeogenesis through the PKA-CREB-PGC1α pathway. Diabetes (2014) 63:3199–209. doi:10.2337/db13-1921
72. Holdt LM, Hoffmann S, Sass K, Langenberger D, Scholz M, Krohn K, et al. Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet (2013) 9:e1003588. doi:10.1371/journal.pgen.1003588
73. Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet (2006) 38:320–3. doi:10.1038/ng1732
74. Bielinski SJ, Pankow JS, Folsom AR, North KE, Boerwinkle E. TCF7L2 single nucleotide polymorphisms, cardiovascular disease and all-cause mortality: the atherosclerosis risk in communities (ARIC) study. Diabetologia (2008) 6:968–70. doi:10.1007/s00125-008-1004-1
75. Vendrell J, Fernandez-Real JM, Gutierrez C, Zamora A, Simon I, Bardaji A, et al. A polymorphism in the promoter of the tumor necrosis factor-alpha gene (-308) is associated with coronary heart diseaes in type 2 diabetic patients. Atherosclerosis (2003) 167:257–64. doi:10.1016/S0021-9150(02)00429-X
76. Asleh R, Marsch S, Shilkrut M, Binah O, Guetta J, Lejbkowicz F, et al. Genetically determined heterogeneity in hemoglobin scavenging and susceptibility to diabetic cardiovascular disease. Circ Res (2003) 92:1193–200. doi:10.1161/01.RES.0000076889.23082.F1
77. Bowden DW, Cox AJ. Diabetes: unravelling the enigma of type 2 DM and cardiovascular disease. Nat Rev Endocrinol (2013) 9:632–3. doi:10.1038/nrendo.2013.192
78. Chan KHK, Huang YT, Meng Q, Wu C, Reiner A, Sobel EM, et al. Shared molecular pathways and gene networks for cardiovascular disease and type 2 diabetes mellitus in women across diverse ethnicities. Circ Cardiovasc Genet (2014) 7:911–9. doi:10.1161/CIRCGENETICS.114.000676
79. Qi Q, Meigs JB, Rexrode KM, Hu FB, Qi L. Diabetes genetic predisposition score and cardiovascular complications among patients with type 2 diabetes. Diabetes Care (2013) 36:737–9. doi:10.2337/dc12-0852
80. Pfister R, Barnes D, Luben RN, Khaw KT, Wareham NJ, Langenberg C. Individual and cumulative effect of type 2 diabetes genetic susceptibility variants on risk of coronary heart disease. Diabetologia (2011) 54:2283–7. doi:10.1007/s00125-011-2206-5
81. Cox AJ, Hsu FC, Ng MC, Langefeld CD, Freedman BI, Carr JJ, et al. Genetic risk score associations with cardiovascular disease and mortality in the diabetes heart study. Diabetes Care (2014) 37:1157–64. doi:10.2337/dc13-1514
82. Reeves R. Molecular biology of HMGA proteins: hubs of nuclear function. Gene (2001) 277:63–81. doi:10.1016/S0378-1119(01)00689-8
83. Sgarra R, Zammitti S, Lo Sardo A, Maurizio E, Arnoldo L, Pegoraro S, et al. HMGA molecular network: from transcriptional regulation to chromatin remodeling. Biochim Biophys Acta (2010) 1799:37–47. doi:10.1016/j.bbagrm.2009.08.009
84. Schuldenfrei A, Belton A, Kowalski J, Talbot CC Jr, Di Cello F, Poh W, et al. HMGA1 drives stem cell, inflammatory pathway, and cell cycle progression genes during lymphoid tumorigenesis. BMC Genomics (2011) 12:539. doi:10.1186/1471-2164-12-549
85. Yie J, Merika M, Munshi N, Chen G, Thanos D. The role of HMG I(Y) in the assembly and function of the IFN-beta enhanceosome. EMBO J (1999) 18:3074–89. doi:10.1093/emboj/18.11.3074
86. Arnoldo L, Sgarra R, Chiefari E, Iiritano S, Arcidiacono B, Pegoraro S, et al. A novel mechanism of post-translational modulation of HMGA1 functions by the histone chaperone nucleophosmin. Sci Rep (2015) 5:8552. doi:10.1038/srep08552
87. Paonessa F, Foti D, Costa V, Chiefari E, Leone F, Luciano F, et al. Activator protein-2 overexpression accounts for increased insulin receptor expression in human breast cancer. Cancer Res (2006) 10:5085–93. doi:10.1158/0008-5472.CAN-05-3678
88. Foti D, Iuliano R, Chiefari E, Brunetti A. A nucleoprotein complex containing Sp1, C/EBP beta, and HMGI-Y controls human insulin receptor gene expression. Mol Cell Biol (2003) 23:2720–32. doi:10.1128/MCB.23.8.2720-2732.2003
89. Costa V, Foti D, Paonessa F, Chiefari E, Palaia L, Brunetti G, et al. The insulin receptor: a new nticancer target for peroxisome proliferator-activated receptor-gamma (PPARgamma) and thiazolidinedione-PPARgamma agonists. Endocr Relat Cancer (2008) 15:325–35. doi:10.1677/ERC-07-0226
90. Iiritano S, Chiefari E, Ventura V, Arcidiacono B, Possidente K, Nocera A, et al. The HMGA1-IGF-I/IGFBP system: a novel pathway for modulating glucose uptake. Mol Endocrinol (2012) 6:1578–89. doi:10.1210/me.2011-1379
91. Bianconcini A, Lupo A, Capone S, Quadro L, Monti M, Zurlo D, et al. Transcriptional activity of the murine retinol-binding protein gene is regulated by a multiprotein complex containing HMGA1, p54 nrb/NonO, protein-associated splicing factor (PSF) and steroidogenic factor 1 (SF1)/liver receptor homologue 1 (LHR-1). Int J Biochem Cell Biol (2009) 41:2189–203. doi:10.1016/j.biocel.2009.04.011
92. Messineo S, Laria AE, Arcidiacono B, Chiefari E, Luque Huertas RM, Foti DP, et al. Cooperation between HMGA1 and HIF-1 contributes to hypoxia-induced VEGF and visfatin gene expression in 3T3-L1 adipocytes. Front Endocrinol (2016) 7:73. doi:10.3389/fendo2016.00073
93. Arcidiacono B, Iiritano S, Chiefari E, Brunetti FS, Gu G, Foti DP, et al. Cooperation between HMGA1, PDX-1, and MafA is essential for glucose-induced insulin transcription in pancreatic beta cells. Front Endocrinol (2015) 5:237. doi:10.3389/fendo.2014.00237
94. Chiefari E, Nevolo MT, Arcidiacono B, Maurizio E, Nocera A, Iiritano S, et al. HMGA1 is a novel downstream nuclear target of the insulin recptor signaling pathway. Sci Rep (2012) 2:251. doi:10.1038/srep00251
95. Foti D, Chiefari E, Fedele M, Iuliano R, Brunetti L, Paonessa F, et al. Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in human and mice. Nat Med (2005) 7:765–73. doi:10.1038/nm1254
96. Chiefari E, Iiritano S, Paonessa F, Le Pera I, Arcidiacono B, Filocamo M, et al. Pseudogene-mediated posttranscriptional silencing of HMGA1 can result in insulin resistance and type 2 diabetes. Nat Commun (2010) 1:40. doi:10.1038/ncomms1040
97. Pullinger CR, Goldfine ID, Tanyolaç S, Movsesyan I, Faynboym M, Durlach V, et al. Evidence that an HMGA1 gene variant associates with type 2 diabetes, body mass index, and high-density lipoprotein cholesterol in a Hispanic-American population. Metab Syndr Relat Disord (2014) 12:1. doi:10.1089/met.2013.0086
98. Bianco A, Chiefari E, Nobile CG, Foti D, Pavia M, Brunetti A. The association between HMGA1 rs146052672 variant and type 2 diabetes: a transethnic meta-analysis. PLoS One (2015) 10:8. doi:10.1371/journal.pone.0136077
99. Chiefari E, Tanyolaç S, Iiritano S, Sciacqua A, Capula C, Arcidiacono B, et al. A polymorphism of HMGA1 is associated with increased risk of metabolic syndrome and related components. Sci Rep (2013) 3:1491. doi:10.1038/srep01491
100. Melillo RM, Pierantoni GM, Scala S, Battista S, Fedele M, Stella A, et al. Critical role of the HMGI(Y) proteins in adipocytic cell growth and differentiation. Mol Cell Biol (2001) 21:2485–95. doi:10.1128/MCB.21.7.2485-2495.2001
101. Arce-Cerezo A, Garcia M, Rodriguez-Nuevo A, Crosa-Bonell M, Enguix N, Pero A, et al. HMGA1 overexpression in adiose tissue impairs adipogenesis and prevents diet-induced obesity and insulin resistance. Sci Rep (2015) 5:14487. doi:10.1038/srep14487
102. Treff NR, Pouchnik D, Dement GA, Britt RL, Reeves R. High-mobility group A1a protein regulates Ras/ERK signaling in MCF-7 human breast cancer cells. Oncogene (2004) 23:777–85. doi:10.1038/sj.onc.1207167
103. Foster LC, Wiesel P, Huggins GS, Pañares R, Chin MT, Pellacani A, et al. Role of activating protein-1 and high mobility group-I(Y) protein in the induction of CD44 gene expression by interleukin-1beta in vascular smooth muscle cells. FASEB J (2000) 14:368–78.
104. Schlueter C, Hauke S, Loeschke S, Wenk HH, Bullerdiek J. HMGA1 proteins in human atherosclerotic plaques. Pathol Res Pract (2005) 201:101–7. doi:10.1016/j.prp.2004.11.010
105. Bloch M, Prock A, Paonessa F, Benz V, Baehr IN, Herbst L, et al. High-mobility group A1 protein: a new coregulator of peroxisome proliferator-activated receptor-gamma mediated transrepression in the vasculature. Circ Res (2012) 110:394–405. doi:10.1161/CIRCRESAHA.111.253658
106. Chiefari E, Ventura V, Capula C, Randazzo G, Scorcia V, Fedele M, et al. A polymorphism of HMGA1 protects against proliferative diabetic retinopathy by impairing HMGA1-induced VEGFA expression. Sci Rep (2016) 6:39429. doi:10.1038/srep39429
107. Shanmugam N, Reddy MA, Guha M, Natarajam R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes (2003) 52:1256–64. doi:10.2337/diabetes.52.5.1256
108. Ceriello A. The emerging challenge in diabetes: the “metabolic memory”. Vascul Pharmacol (2012) 57:133–8. doi:10.1016/vph.2012.05.005
109. Pirola L, Balcerczyk A, Tothill RW, Haviv I, Kaspi A, Lunke S, et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res (2011) 21(10):1601–15. doi:10.1101/gr.116095.110
110. Brasacchio D, Okabe J, Tikellis C, Balcerczyk A, George P, Baker EK, et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes (2009) 58:1229–36. doi:10.2337/db08-1666
111. Keating S, Plutzky J, El-Osta A. Epigenetic changes in diabetic and cardiovascular risk. Circ Res (2016) 118:1706–22. doi:10.1161/CIRCRESAHA.116.306819
112. Paneni F, Volpe M, Lüscher TF, Cosentino F. SIRT1, p66(Shc), and Set7/9 in vascular hyperglycemic memory: bringing all the strands together. Diabetes (2013) 62:1800–7. doi:10.2337/db12-1648
113. Aslibekyan S, Class SA, Arnett DK. Clinical applications of epigenetics in cardiovascular disease: the long road ahead. Transl Res (2015) 165:143–53. doi:10.1016/j.trsl.2014.04.004
114. Sathiskumar C, Prabu P, Balakumar M, Lenin R, Prabhu D, Anjana RM, et al. Augmentation of histone deacetylase 3 (HDAC3) epigenetic signature at the interface of proinflammation and insulin resistance in patients with type 2 diabetes. Clin Epigenetics (2016) 8:125. doi:10.1186/s13148-016-0293-3
115. Bell CG, Finer S, Lindgren CM, Wilson GA, Rakyan VK, Teschendorff AE, et al. Integrated genetic and epigenetic analysis identifies haplotype-specific methylation in the FTO type 2 diabetes and obesity susceptibility locus. PLoS One (2010) 5:e14040. doi:10.1371/journal.pone.0014040
116. Liu C, Mou S, Pan C. The FTO gene rs9939609 polymorphism predict risk of cardiovascular disease: a systematic review and metanalysis. PLoS One (2013) 8:e71901. doi:10.1371/journal.pone.0071901
117. Deodati A, Inzaghi E, Liguori A, Puglianiello A, Germani D, Brufani C, et al. IGF2 methylation is associated with lipid profile in obese children. Horm Res Paediatr (2013) 79:361–7. doi:10.1159/000351707
118. Arner P, Sahlqvist AS, Sinha I, Xu H, Yao X, Waterworth D, et al. The epigenetic signature of systemic insulin resistance in obese women. Diabetologia (2016) 59:2393–405. doi:10.1007/s00125-016-4074-5
119. Nilsson E, Jansson PA, Perfilyev A, Volkov P, Pedersen M, Svensson MK, et al. Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes (2014) 63:2962–76. doi:10.2337/db13-1459
120. Rönn T, Volkov P, Gillberg L, Kokosar M, Perfilyev A, Jacobsen AL, et al. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum Mol Genet (2015) 24:3792–813. doi:10.1093/hmg/ddv124
121. Benton MC, Johnstone A, Eccles D, Harmon B, Hayes MT, Lea RA, et al. An analysis of DNAmethylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss. Genome Biol (2015) 16:8. doi:10.1186/s13059-014-0569-x
122. Yang X, Wang X, Liu D, Yu L, Xue B, Shi H. Epigenetic regulation of macrophage polarization by DNAmethyltransferase 3b. Mol Endocrinol (2014) 28:565–74. doi:10.1210/me.2013-1293
123. Talens RP, Jukema JW, Trompet S, Kremer D, Westendorp RG, Lumey LH, et al. Hypermethylation at loci sensitive to the prenatal environment is associated with increased incidence of myocardial infarction. Int J Epidemiol (2012) 41:106–15. doi:10.1093/ije/dyr153
124. Romaine SP, Tomaszewski M, Condorelli G, Samani NJ. MicroRNAs in cardiovascular disease: an introduction for clinicians. Heart (2015) 101:921–8. doi:10.1136/heartjnl-2013-305402
125. Hashimoto N, Tanaka T. Role of miRNAs in the pathogenesis and susceptibility of diabetes mellitus. J Hum Genet (2017) 62:141–50. doi:10.1038/jhg.2016.150
126. Kwak SH, Park KS. Recent progress in genetic and epigenetic research on type 2 diabetes. Exp Mol Med (2016) 48:e220. doi:10.1038/emm.2016.7
127. Ding Y, Sun X, Shan PF. MicroRNAs and cardiovascular disease in diabetes mellitus. Biomed Res Int (2017) 2017:4080364. doi:10.1155/2017/4080364
128. Diao X, Shen E, Wang X, Hu B. Differentially expressed microRNAs and their target genes in the hearts of streptozotocin-induced diabetic mice. Mol Med Rep (2011) 4:633–40. doi:10.3892/mmr.2011.489
129. La Sala L, Cattaneo M, De Nigris V, Pujadas G, Testa R, Bonfigli AR, et al. Oscillating glucose induces microRNA-185 and impairs an efficient antioxidant response in human endothelial cells. Cardiovasc Diabetol (2016) 15:71. doi:10.1186/s12933-016-0390-9
130. Arunachalam G, Lakshmanan AP, Samuel SM, Triggle CR, Ding H. Molecular interplay between microRNA-34a and sirtuin1 in hyperglycemia-mediated impaired angiogenesis in endothelial cells: effects of metformin. J Pharmacol Exp Ther (2016) 356:314–23. doi:10.1124/jpet.115.226894
131. Vikram A, Kim Y, Kumar S, Li Q, Kassan M, Jacobs JS, et al. Vascular microRNA-204 is remotely governed by the microbiome and impairs endothelium-dependent vasorelaxation by downregulating sirtuin1. Nat Commun (2016) 7:12565. doi:10.1038/ncomms12565
132. Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, et al. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell (2008) 15:261–71. doi:10.1016/j.devcel.2008.07.002
133. Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, et al. miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell (2008) 15:272–84. doi:10.1016/j.devcel.2008.07.008
134. Zernecke A, Bidzhekov K, Noels H, Shagdarsuren E, Gan L, Denecke B, et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci Signal (2009) 2:ra81. doi:10.1126/scisignal.2000610
135. Tang ST, Wang F, Shao M, Wang Y, Zhu HQ. MicroRNA-126 suppresses inflammation in endothelial cells under hyperglycemic conditions by targeting HMGB1. Vascul Pharmacol (2017) 88:48–55. doi:10.1016/j.vph.2016.12.002
136. Rawal S, Munasinghe PE, Shindikar A, Paulin J, Cameron V, Manning P, et al. Down-regulation of proangiogenic microRNA-126 and microRNA-132 are early modulators of diabetic cardiac microangiopathy. Cardiovasc Res (2017) 113:90–101. doi:10.1093/cvr/cvw235
137. Zampetaki A, Kiechl S, Drozdov I, Willeit P, Mayr U, Prokopi M, et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ Res (2010) 107:810–7. doi:10.1161/CIRCRESAHA.110.226357
138. Fichtlscherer S, De Rosa S, Fox H, Schwietz T, Fischer A, Liebetrau C, et al. Circulating microRNAs in patients with coronary artery disease. Circ Res (2010) 107:677–84. doi:10.1161/CIRCRESAHA.109.215566
139. Wang HW, Su SH, Wang YL, Chang ST, Liao KH, Lo HH, et al. MicroRNA-134 contributes to glucose-induced endothelial cell dysfunction and this effect can be reversed by far-infrared irradiation. PLoS One (2016) 11:e0147067. doi:10.137/journal.pone.0147067
140. Xu Q, Meng S, Liu B, Li MQ, Li Y, Fang L, et al. MicroRNA-130a regulates autophagy of endothelial progenitor cells through Runx3. Clin Exp Pharmacol Physiol (2014) 41:351–7. doi:10.1111/1440-1681.12227
141. Maegdefessel L, Rayner KJ, Leeper NJ. MicroRNA regulation of vascular smooth muscle function and phenotype: early career committee contribution. Arterioscler Thromb Vasc Biol (2015) 35:2–6. doi:10.1161/ATVBAHA.114.304877
142. Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, et al. MiR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature (2009) 460:705–10. doi:10.1038/nature08195
143. Reddy MA, Das S, Zhuo C, Jin W, Wang M, Lanting L, et al. Regulation of vascular smooth muscle cell dysfunction under diabetic conditions by MIR-504. Arterioscler Thromb Vasc Biol (2016) 36:864–73. doi:10.1161/ATVBAHA.115.306770
144. Yang J, Chen L, Ding J, Fan Z, Li S, Wu H, et al. MicroRNA-24 inhibits high glucose-induced vascular smooth muscle cell proliferation and migration by targeting HMGB1. Gene (2016) 586:268–73. doi:10.1016/j.gene.2016.04.027
145. Kurtz CL, Peck BCE, Fannin EE, Beysen C, Miao J, Landstreet SR, et al. MicroRNA-29 finetunes the expression of key FOXA2-activated lipid metabolism genes and is dysregulated in animal models of insulin resistance and diabetes. Diabetes (2014) 63:3141–8. doi:10.2337/db13-1015
146. Wei S, Zhang M, Yu Y, Xue H, Lan X, Liu S, et al. HNF-4α regulated miR-122 contributes to development of gluconeogenesis and lipid metabolism disorders in type 2 diabetic mice and in palmitate treated HepG2 cells. Eur J Pharmacol (2016) 791:254–63. doi:10.1016/j.ejphar.2016.08.038
147. Fu X, Dong B, Tian Y, Lefebvre P, Meng Z, Wang X, et al. MicroRNA-26a regulates insulin sensitivity and metabolism of glucose and lipids. J Clin Invest (2015) 125:2497–509. doi:10.1172/JCI75438
148. Grove EL, Gregersen S. Antiplatelet therapy in patients with diabetes mellitus. Curr Vasc Pharmacol (2012) 10:494–505. doi:10.2174/157016112800812818
149. Landry P, Plante I, Ouellet DL, Perron MP, Rousseau G, Provost P. Existence of a microRNA pathway in anucleate platelets. Nat Struct Mol Biol (2009) 16:961–6. doi:10.1038/nsmb.1651
150. Bray PF, McKenzie SE, Edelstein LC, Nagalla S, Delgrosso K, Ertel A, et al. The complex transcriptional landscape of the anucleate human platelet. BMC Genomics (2013) 14:1. doi:10.1186/1471-2164-14-1
151. Iaconetti C, Sorrentino S, De Rosa S, Indolfi C. Exosomal miRNAs in heart disease. Physiology (Bethesda) (2016) 31:16–24. doi:10.1152/physiol.00029.2015
152. Duan X, Zhan Q, Song B, Zeng S, Zhou J, Long Y, et al. Detection of platelet microRNA expression in patients with diabetes mellitus with or without ischemic stroke. J Diabetes Complications (2014) 28:705–10. doi:10.1016/j.diacomp.2014
153. Elgheznawy A, Shi L, Hu J, Wittig I, Laban H, Pircher J, et al. Dicer cleavage by calpain determines platelet microRNA levels and function in diabetes. Circ Res (2015) 117:157–65. doi:10.1161/CIRCRESAHA.117.305784
154. Carino A, De Rosa S, Sorrentino S, Polimeni A, Sabatino J, Caiazzo G, et al. Modulation of circulating microRNAs levels during the switch from clopidogrel to ticagrelor. Biomed Res Int (2016) 2016:3968206. doi:10.1155/2016/3968206
155. Lugli G, Larson J, Martone ME, Jones Y, Smalheiser NR. Dicer and eIF2c are enriched at postsynaptic densities in adult mouse brain and are modified by neuronal activity in a calpain-dependent manner. J Neurochem (2005) 94:896–905. doi:10.1111/j.1471-4159.2005.03224.x
156. Fejes Z, Póliska S, Czimmerer Z, Káplár M, Penyige A, Gál Szabó G, et al. Hyperglycemia suppresses microRNA expression in platelets to increase P2RY12 and SELP levels in type 2 diabetes mellitus. Thromb Haemost (2017) 117:529–42. doi:10.1160/TH16-04-0322
157. Shi R, Zhou X, Ji WJ, Zhang YY, Ma YQ, Zhang JQ, et al. The emerging role of miR-223 in platelet reactivity: implications in antiplatelet therapy. Biomed Res Int (2015) 2015:981841. doi:10.1155/2015/98184
158. Zhang YY, Zhou X, Ji WJ, Shi R, Lu RY, Li JL, et al. Decreased circulating microRNA-223 level predicts high on-treatment platelet reactivity in patients with troponin-negative non-ST elevation acute coronary syndrome. J Thromb Thrombolysis (2014) 38:65–72. doi:10.1016/j.thromres.2013.02.015
159. Luo M, Li R, Deng X, Ren M, Chen N, Zeng M, et al. Platelet-derived miR-103b as a novel biomarker for the early diagnosis of type 2 diabetes. Acta Diabetol (2015) 52:943–9. doi:10.1007/s00592-015-0733-0
160. Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol (2012) 32:463–88. doi:10.1615/CritRevImmunol.v32.i6.10
161. Stoger L, Gijbels MJ, van der Velden S, Manca M, van der Loos CM, Biessen EA, et al. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis (2012) 225:461–8. doi:10.1016/j.atherosclerosis.2012.09.013
162. Sun X, Lin J, Zhang Y, Kang S, Belkin N, Waea AK, et al. MicroRNA-181b improves glucose homeostasis and insulin sensitivity by regulating endothelial function in white adipose tissue. Circ Res (2016) 118:810–21. doi:10.1161/CIRCRESAHA.115.308166
163. Kuwabara Y, Ono K, Horie T, Nishi H, Nagao K, Kinoshita M, et al. Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circ Cardiovasc Genet (2011) 4:446–54. doi:10.1161/CIRCGENETICS.110.958975
164. de Golzalo-Calvo D, van der Meer RW, Rijzewijk LJ, Smit JW, Revuelta-Lopez E, Nasarre L, et al. Serum microRNA-1 and microRNA-133a levels reflect myocardial steatosis in uncomplicated type 2 diabetes. Sci Rep (2017) 7:47. doi:10.1038/s41598-017-00070-6
165. Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med (2007) 13:613–8. doi:10.1038/nm1582
166. Shan YX, Liu TJ, Su HF, Samsamshariat A, Mestril R, Wang PH. Hsp10 and Hsp60 modulate Bcl-2 family and mitochondria apoptosis signaling induced by doxorubicin in cardiac muscle cells. J Mol Cell Cardiol (2003) 35:1135–43. doi:10.1016/S0022-2828(03)00229-3
167. Rawal S, Manning P, Katare R. Cardiovascular microRNAs: as modulators and diagnostic biomarkers of diabetic heart disease. Cardiovasc Diabetol (2014) 13:44. doi:10.1186/1475-2840-13-44
168. Nandi SS, Zheng H, Sharma NM, Shahshahan HR, Patel KP, Mishra PK. Lack of miR-113a decreases contractility of diabetic hearts: a role for novel cross talk between tyrosine aminotransferase and tyrosine hydroxylase. Diabetes (2016) 65:3075–90. doi:10.2337/db16-0023
169. De Rosa R, De Rosa S, Leistner D, Boeckel JN, Keller T, Fichtlscherer S, et al. Transcoronary concentration gradient of microRNA-133a and outcome in patients with coronary artery disease. Am J Cardiol (2017) 120:15–24. doi:10.1016/j.amjcard.2017.03.264
170. Babiarz JE, Ravon M, Sridhar S, Ravindran P, Swanson B, Bitter H, et al. Determination of the human cardiomyocyte mRNA and miRNA differentiation network by fine-scale profiling. Stem Cells Dev (2012) 21:1956–65. doi:10.1089/scd.2011.0357
171. Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, et al. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin related protein-1. Nat Med (2011) 17:71–8. doi:10.1038/nm.2282
172. Palmieri D, D’Angelo D, Valentino T, De Martino I, Ferraro A, Wierinckx A, et al. Downregulation of HMGA-targeting microRNAs has a critical role in human pituitary tumorigenesis. Oncogene (2012) 31:3857–65. doi:10.1038/onc.2011.557
173. Mussnich P, D’Angelo D, Leone D, Croce CM, Fusco A. The high mobility group A proteins contribute to thyroid transformation by regulation miR-603 and miR-10b expression. Mol Oncol (2013) 7:531–42. doi:10.1016/j.molonc.2013.01.002
174. Gareri C, Iaconetti C, Sorrentino S, Covello C, De Rosa S, Indolfi C. miR-125a-5p modulates phenotypic switch of vascular smooth muscle cells by targeting ETS-1. J Mol Biol (2017) 429:1817–28. doi:10.1016/j.jmb.2017.05.008
175. Lee JT. Epigenetic regulation by long noncoding RNAs. Science (2012) 338:1435–9. doi:10.1126/science.1231776
176. Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet (2014) 15:7–21. doi:10.1038/nrg3606
177. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell (2011) 43:904–14. doi:10.1016/j.molcel.2011.08.018
178. Esteller M. Non-coding RNAs in human disease. Nat Rev Genet (2011) 12:861–74. doi:10.1038/nrg3074
179. Elia L, Condorelli G. RNA (epi)genetics in cardiovascular diseases. J Mol Cell Cardiol (2015) 89:11–6. doi:10.1016/j.yjmcc.2015.07.012
180. Aryal B, Rotllan N, Fernández-Hernando C. Noncoding RNAs and atherosclerosis. Curr Atheroscler Rep (2014) 16:407. doi:10.1007/s11883-014-014-0407-3
181. Dechamethakun S, Muramatsu M. Long noncoding RNA variations in cardiometabolic diseases. J Hum Genet (2017) 62:97–104. doi:10.1038/jhg.2016.70
182. Carter G, Miladinovic B, Patel AA, Deland L, Mastorides S, Patel NA. Circulating long noncoding RNA GAS5 levels are correlated to prevalence of type 2 diabetes mellitus. BBA Clin (2015) 4:102–7. doi:10.1016/j.bbacli.2015.09.001
183. Li H, Zhu H, Ge J. Long noncoding RNA: recent updates in atherosclerosis. Int J Biol Sci (2017) 12:898–910. doi:10.7150/ijbs.14430
184. Kumarswamy R, Bauters C, Volkmann I, Maury F, Fetisch J, Holzmann A, et al. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ Res (2014) 114:1569–75. doi:10.1161/CIRCRESAHA.114.303915
185. Michalik KM, You X, Manavski Y, Doddaballapur A, Zornig M, Braun T, et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res (2014) 114:1389–97. doi:10.1161/CIRCRESAHA.114.303265
186. Liu JY, Yao J, Li XM, Song YC, Wang XQ, Li YJ, et al. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis (2014) 5:e1506. doi:10.1038/cddis.2014.466
187. Puthanveetil P, Chen S, Feng B, Gautam A, Chakrabarti S. Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J Cell Mol Med (2015) 19:1418–25. doi:10.1111/jcmm.12576
188. Li X, Wang H, Yao B, Xu W, Chen J, Zhou X. lncRNA H19/miR-675 axis regulates cardiomyocyte apoptosis by targeting VDAC1 in diabetic cardiomyopathy. Sci Rep (2016) 6:36340. doi:10.1038/srep36340
189. Reddy MA, Chen Z, Park JT, Wang M, Lanting L, Zhang Q, et al. Regulation of inflammatory phenotype in macrophages by a diabetes-induced long noncoding RNA. Diabetes (2014) 63:4249–61. doi:10.2337/db14-0298
Keywords: type 2 diabetes mellitus, cardiovascular disease, genetic polymorphisms, high-mobility group A1 variant, epigenetics
Citation: De Rosa S, Arcidiacono B, Chiefari E, Brunetti A, Indolfi C and Foti DP (2018) Type 2 Diabetes Mellitus and Cardiovascular Disease: Genetic and Epigenetic Links. Front. Endocrinol. 9:2. doi: 10.3389/fendo.2018.00002
Received: 27 November 2017; Accepted: 03 January 2018;
Published: 17 January 2018
Edited by:
Gaetano Santulli, Columbia University, United StatesReviewed by:
Paras Kumar Mishra, University of Nebraska Medical Center, United StatesCopyright: © 2018 De Rosa, Arcidiacono, Chiefari, Brunetti, Indolfi and Foti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Brunetti, YnJ1bmV0dGlAdW5pY3ouaXQ=;
Ciro Indolfi, aW5kb2xmaUB1bmljei5pdA==;
Daniela P. Foti, Zm90aUB1bmljei5pdA==
†Equal coauthorship.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.