 Roberta Malaguarnera
Roberta Malaguarnera Veronica Vella
Veronica Vella Maria Luisa Nicolosi
Maria Luisa Nicolosi Antonino Belfiore
Antonino Belfiore- 1Endocrinology, Department of Health Sciences, University Magna Graecia of Catanzaro, Catanzaro, Italy
- 2School of Human and Social Sciences, “Kore” University of Enna, Enna, Italy
In the past few decades, the incidence of thyroid cancer (TC), namely of its papillary hystotype (PTC), has shown a steady increase worldwide, which has been attributed at least in part to the increasing diagnosis of early stage tumors. However, some evidence suggests that environmental and lifestyle factors can also play a role. Among the potential risk factors involved in the changing epidemiology of TC, particular attention has been drawn to insulin-resistance and related metabolic disorders, such as obesity, type 2 diabetes, and metabolic syndrome, which have been also rapidly increasing worldwide due to widespread dietary and lifestyle changes. In accordance with this possibility, various epidemiological studies have indeed gathered substantial evidence that insulin resistance-related metabolic disorders might be associated with an increased TC risk either through hyperinsulinemia or by affecting other TC risk factors including iodine deficiency, elevated thyroid stimulating hormone, estrogen-dependent signaling, chronic autoimmune thyroiditis, and others. This review summarizes the current literature evaluating the relationship between metabolic disorders characterized by insulin resistance and the risk for TC as well as the possible underlying mechanisms. The potential implications of such association in TC prevention and therapy are discussed.
Introduction
Thyroid cancer (TC) is a relatively rare cancer but represents one of the most common malignancies originating from the endocrine organs (1). It is more frequent in women than in menand is now the third most common cancer in women under the age of 45 in highly developed countries (2). Among various histotypes, differentiated thyroid carcinomas (DTCs) are the most frequent, accounting for approximately 85% of all TCs (3). Increasing incidence of DTCs has been observed worldwide (4–6) in both men and women, although the cancer-specific mortality remains stable (7, 8). TC incidence has increased about twofold in some European countries (7, 9) and up to threefold in North America in the past decades (7, 10). Some studies put emphasis on the supposed “overdiagnosis” of TC consequent to the widespread use of ultrasonography and fine needle biopsy, and point out to the increasing diagnosis of papillary thyroid microcarcinomas (tumors with a diameter of 1 cm or less) (4, 11, 12). However, other studies (5, 6) have reported an increased incidence of TC of all sizes, suggesting that “overdiagnosis” cannot explain all the findings and that TC incidence is truly increasing. A promising hypothesis is that some rising risk factors might favor the molecular alterations typical of papillary TCs (PTCs), thus increasing its incidence.
The known non-modifiable risk factors for TC are age, sex, ethnicity, and genetic predisposition for TC (13–15). However, epidemiological studies suggest that TC incidence is largely dependent on modifiable risk factors, such as environmental carcinogens, diet habits, and lifestyle (16). Environmental pollutants, such as heavy metals, compounds used by industries, non-anthropogenic carcinogens of volcanic origin (17–19), as well as dietary factors (20), and obesity (21) are some of the putative risk factors suspected to play a role in the changing epidemiology of TC. Interestingly, this increasing incidence involves virtually only the papillary histotype, suggesting that some carcinogens may favor specific molecular abnormalities related to this histotype (5, 22).
Insulin Resistance, Hyperinsulinemia, and Epidemiologic and Clinical Aspects of TC: A Possible Link?
Evidence of a Positive Association between TC and Insulin Resistance
Obesity is the most common metabolic disorder associated with insulin resistance and compensative hyperinsulinemia. Obesity has more than doubled its prevalence in the past 30 years reaching a prevalence of 40% in the United States and 30% in Europe. The association of obesity with several cancer histotypes is now well established and has become an area of raising concern in oncology (23). In particular, cancers associated with obesity also pose a therapeutical challenge because they tend to be resistant to conventional as well as to target treatments, to metastasize earlier and to have a worse prognosis (24–26). Approximately 14% of cancer-related deaths in men and 20% in women are partially attributed to obesity.
During the past two decades, several epidemiological studies, although not specifically designed for TC, have consistently suggested that a positive association exists between obesity and TC risk (Table 1). A pooled analysis of 12 case–control studies provided early evidence that body mass index (BMI) and weight at diagnosis were directly related to a higher risk for TC in women [odds ratio (OR) = 1.2 for the highest tertile], but not in men. This association was observed for both PTCs and follicular TCs and in all age groups, although there was a significant heterogeneity between the studies analyzed (27). From 2001 to 2010 several single cohort, case–control, prospective cohort, and cross-sectional studies have confirmed the association between overweigh/obesity and TC risk, although the results are rather inconsistent in men, likely for the smaller number of cancer cases in men and the suboptimal adjustment for potential concomitant risk factors (28–36). However, a meta-analysis based on prospective observational studies, found a positive role of obesity as risk factor for TC in both sexes [relative risk (RR) of 1.33 and 1.14, respectively, for women and men, for each 5-unit increase in BMI] (32). In a prospective study based on self-reported medical history, anthropometric and behavioral factors in 90,713 US radiologic technologists followed for 23 years, an elevated risk for TC was observed for women with a RR of 1.74 (95% CI: 1.03–2.94, P-trend: 0.04) for BMI ≥ 35.0 vs. 18.5–24.9 kg/m2. A similar association was found for men (37).
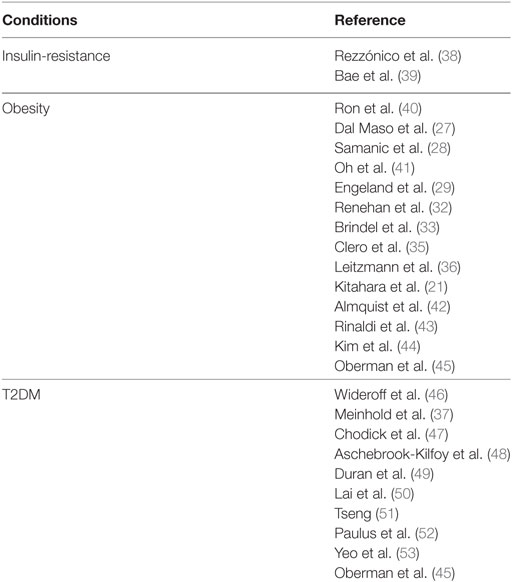
Table 1. Studies regarding a possible association between TC risk and insulin-resistance and related disorders.
In 2011, a pooled analysis of five prospective studies including a large number of incident TC in men, and taking into account several potential risk factors, found that the risk of TC was greater with increased BMI [per 5 kg/m2: hazard risk (HR) in women 1.16 and 1.21 in men]. When considering women and men together, the HR was 1.2 and 1.53, respectively, in overweight and obese subjects. No differences were found among the TC histotypes. This pooled analysis provided the first strong support to the concept that obesity is an independent risk factor for TC in both women and men (21). However, these studies have limitations, as they lack data on fat distribution, amount of lean versus fat mass, fat mass and/or insulin resistance-related biomarkers, and thyroid function parameters. These technical issues and the low HR values often reported impose caution in interpreting the biological significance of these results.
Three studies conducted in 2012 attempted to provide additional clues regarding TC association with insulin-resistance parameters (43, 54, 55). However, the results were conflicting. Two of these studies (43, 55) found an increased risk of TC in subjects with high waist circumference (>102 cm in men and >88 cm in women), a parameter that correlates with visceral adiposity and is a solid readout of insulin resistance. The HR was 1.79 in men (56) and ranged from 1.42 to 1.54 in women (43), suggesting that central adiposity may impact on TC risk. In contrast, the third study (54), conducted in a cohort of postmenopausal women, failed to find an association between TC risk and various adiposity parameters such as waist circumference, waist-hip-ratio, hip circumference, and BMI (54). In a meta-analysis of seven cohort studies, the combined RR of TC was 1.18 (95% CI: 1.11–1.25) for overweight and obesity combined (57).
Another recent pooled analysis (56) included 22 prospective studies investigating the association between anthropometric factors, such as waist circumference, baseline BMI, and BMI gain and the risk of TC. Data showed that all anthropometric factors analyzed were associated with an increased risk of all histotypes of TC originating from follicular cells: HR for height (per 5 cm) = 1.07; BMI (per 5 kg/m2) = 1.06; waist circumference (per 5 cm) = 1.03; young-adult BMI (per 5 kg/m2) = 1.13; and adulthood BMI gain (per 5 kg/m2) = 1.07. Associations for baseline BMI and waist circumference were mitigated after mutual adjustment (HR for waist = 1.02 and for BMI = 1.01). Furthermore, baseline BMI and BMI gain were strongly associated with anaplastic TC (ATC) and TC mortality.
A strong association between BMI and TC clinical–pathological features has been also confirmed by other studies, which found that, in patients affected by papillary TCs (PTCs), overweight and obesity were positively associated with recurrent or residual post-operative locoregional events (58), extrathyroidal invasion and advanced TNM (TNM Classification of Malignant Tumors) stage (44, 59). Taken together, these results suggest that excess adiposity is associated with increased incidence and mortality for TC of follicular origin. However, at least one study has reported an inverse relation of BMI with stage, tumor invasion and recurrence (60), suggesting that more studies are needed to better evaluate the link between TC prognosis and adiposity.
Dysregulation of Adipocytokines As a Possible Contributor to Cancer Risk in Obese Patients
Obesity is strictly associated not only with insulin resistance and hyperinsulinemia but also with a profound dysregulation of adipocytokines secretion (Figure 1). Indeed, adipose tissue has strongly been established as an endocrine organ for its ability to secrete several polypeptides, known as adipokines, which contribute to the pathogenesis of insulin-resistance and related metabolic alterations in obese patients. Two most known adipokines are leptin and adiponectin. Both of them have been studied as potential contributors to the pathophysiology of cancer associated with insulin resistance, beyond their well-known role in energy balance (61). Leptin is generally up-regulated with increasing fat mass and acts as an antiappetite regulator, mainly through specific membrane receptors, the obesity receptors (Ob-Rs). Aberrant expression of leptin and/or its receptor have been found in a variety of malignancies including TC (62, 63). In vitro studies have shown that leptin modulates growth, proliferation and invasion of TC cell lines via activation of various prosurvival signaling pathways such as Janus kinase/signal transducers of activated transcription (JAK/STAT), phosphoinositide-3-kinase (PI3K)/protein kinase B/Akt (PKB/Akt), and/or mitogen-activated protein kinase (MAPK) (62, 63). However, the results have been sometimes contradictory, likely because of dependence on the cell type and cell context.
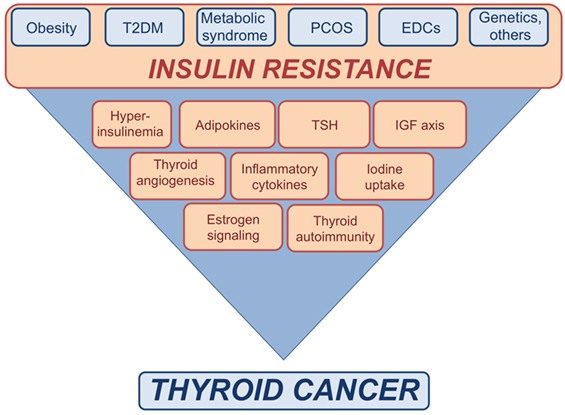
Figure 1. Schematic representation of the possible links between insulin resistance and thyroid cancer (TC). Insulin resistance consequent to metabolic disorders, as well as exposure to endocrine disrupting chemicals (EDCs), genetic factors, and other conditions may affect the risk of TC by inducing or increasing various risk factors.
Adiponectin is the most abundant adipokine negatively correlated with body fat, BMI, insulin-resistance, and inflammation states (61). Adiponectin binds two receptors isoforms (AdipoR1, AdipoR2) and acts as an insulin-sensitizer, anti-inflammatory and anti-tumor agent, the latter by inhibiting cell proliferation and angiogenesis and increasing apoptosis via the involvement of mammalian target of rapamycin (mTOR)/5′ adenosine monophosphate-activated protein kinase (AMPK), MAPK, JAK/STAT, and PI3K/PKB/Akt pathways (61, 64). So far, few studies have investigated the association between adiponectin and TC. One of these studies has shown that TC specimens and cell lines express both AdipoR1 and AdipoR2. However, in the TC cell lines evaluated, recombinant adiponectin did not exert significant biological effects (65).
For both adiponectin and leptin only a limited number of in vivo studies have been performed. Serum leptin levels in papillary thyroid tumor patients were found to be significantly higher than in control subjects, and Ob-Rs expression in TC tissues was significantly associated with more aggressive tumor phenotype (66, 67). However, these studies did not find significant differences in BMI between cancer patients and control subjects (66–72).
Adiponectin levels have been found to be lower in TC patients than in controls (17.00 ± 6.32 vs. 19.26 ± 6.28 µg/ml) (65). Besides, individuals in the highest tertile of adiponectin levels showed a lower risk for TC (OR = 0.29; 95% CI: 0.14−0.55) (65). Conversely, in a prospective cohort study of patients with end-stage renal disease, low adiponectin levels were an independent predictor of developing cancer, with the TC being the second more common malignancy (71). Yet, in a single-cohort study of patients affected by PTC, tumor expression of adiponectin receptors (both AdipoR1 and AdipoR2) was positively correlated with the tumor aggressiveness.
In summary, several studies suggest a significant association between obesity, visceral adiposity and altered adipocytokine profile with TC risk and aggressiveness. However, at least some of these studies have significant limitations with regard to study design, including lack of adjustment for potential confounders, and/or limited statistic power. Therefore, more studies are needed to confirm these conclusions. As a practical implication of these data, one study found that in obese patients with established risk factors [family history, radiation exposure, Hashimoto’s thyroiditis (HT), elevated thyroid stimulating hormone (TSH)], ultrasound screening for TC is cost-effective (73).
The Interplay of Insulin Resistance with Other Putative Risk Factors for TC
Insulin Resistance and TSH
In follicular well-differentiated thyroid cells, signaling mediated by pituitary TSH represents the major pathway, which primes thyroid cells to undergo cell cycle progression, DNA synthesis, and cell proliferation (74). The key role of TSH signaling in thyroid carcinogenesis is supported by large epidemiological studies showing a strong association between serum TSH levels and TC development and progression (75–77) (Figure 1). However, even in differentiated hystotypes, suppression of TSH is not enough to avoid or block local invasion and distant metastases. This observation suggests that the mitogenic effect of TSH on human thyrocytes is modulated by other factors including insulin, insulin growth factor-1 (IGF-1), insulin growth factor-2 (IGF-2), and epidermal growth factor (EGF) (74, 78–83).
Classically, TSH induced growth in thyrocytes occurs mainly through the TSHR-dependent increase in cyclic adenosine monophosphate (cAMP), which in turn activates protein kinase A (PKA)-dependent and -independent pathways including: cAMP/PKA/cAMP response element-binding protein (CREB), cAMP/PKA/exchange factor directly activated by cAMP 1/Ras-related protein 1(Rap1)/extracellular signal-regulated kinases (ERKs)/ETS transcription factor, protein kinase C (PKC)/nuclear factor kappa-light-chain-enhancer of activated B cells, nuclear factor κB, PKC/proto-oncogene c-Raf (c-Raf)/ERK/MAPK-activated protein kinase-1 (p90rsk), and rat sarcoma virus protein (Ras)/c-Raf/ERK cascades (84–87).
Moreover, full activation of mitogenesis results from the crosstalk between TSH downstream pathways with other signaling networks, such as PI3K/Akt/mTOR, serine/threonine-protein kinase B-Raf, (B-Raf)/MAPK, proto-oncogene protein Wnt-1, (Wnt)/β-catenin, activated by several tyrosine kinase receptors (RTKs) (85, 88). Studies carried out in normal and tumor thyrocytes have especially highlighted the importance of the functional crosstalk between TSH-cAMP and insulin/IGF axis, which occurs at multiple levels (74, 89).
Notably, the IGF axis plays an important role in regulating normal growth and development in the thyroid (90, 91), partially by modulating the expression of thyroid transcription factor 2, which mediates the transcription of thyroid specific genes such as thyroglobulin (Tg) and thyroperoxidase (TPO) (74, 92–95). As mentioned above, the crosstalk between TSH and insulin/IGF axis appears also to play a role in thyroid tumorigenesis. Indeed, in TC cells and tissue specimens, both IGF-1 receptor (IGF-1R) and insulin receptor (IR) are often overexpressed, representing an early event in thyroid carcinogenesis (96, 97). IR expression is also stimulated by TSH, via cAMP (98). IR, exists in two isoforms (IR-A and IR-B), and in cancer is predominantly expressed as the “promitogenic” isoform A (IR-A), which binds with high affinity not only insulin but also IGF-2 (97, 99–101). In TC, the activation of the autocrine IGF-2/IR-A loop was found to correlate with cellular dedifferentiation and tumor progression and aggressiveness. Indeed, the relative abundance of IR-A is approximately 40% in normal thyrocytes and increases to over 70% in TC cells with undifferentiated or stem-like phenotype (90, 97) that also produce IGF-2 (97). Interestingly, IGF-1R expression is also high in differentiated cancers but decreases somehow with cancer dedifferentiation (102–105). In agreement with these data, phosphorylated IGF-1Rs are highly expressed in the majority of TCs but tend to be low in aggressive tumors (106). Interestingly, IGF-1R expression in PTC appears to be higher in patients with type 2 diabetes mellitus (T2DM) than in non-diabetic patients (107). Taken together, these data suggest that both IGF-1R and IR-A play a role in TC. However, the IGF-2/IR-A loop appears to be more important than the IGF-1/IGF-1R loop in thyroid cells with dedifferentiated and stem-like phenotype (108) involved in tumor progression and metastasis (90).
In insulin resistant subjects, the crosstalk between TSH and IGFs axis appears to be enhanced (Figure 1). In fact, obese subjects often show TSH levels at the upper limit of the normal range or slightly increased (109) that seem in relation with the degree of obesity and to the levels of cytokines and other inflammatory markers produced by adipose tissue, including leptin (109–117). Although the actual cause for the hyperthyrotropinemia in obese individuals is still unknown, several mechanisms have been proposed, including increased production of pro-TRH by leptin (118), impaired feedback due to decreased T3 receptors in the hypothalamus (119), changes in peripheral deiodination process of thyroid hormones (119–121), the adaptive response to increased energy expenditure, and chronic low-grade inflammation associated with insulin resistance (122).
A relationship between TSH and insulin-resistance has been also reported in women with polycystic ovary syndrome (PCOS), where mild TSH elevation may be positively related to their metabolic phenotype (123–127). Yet, TSH was also positively correlated with HOMA-IR and BMI in type 2 diabetic patients and in patients with metabolic syndrome (128–134). Although these results have been sometimes controversial, overall they support the positive correlation between insulin resistance and increased TSH serum levels (109) (Table 2).
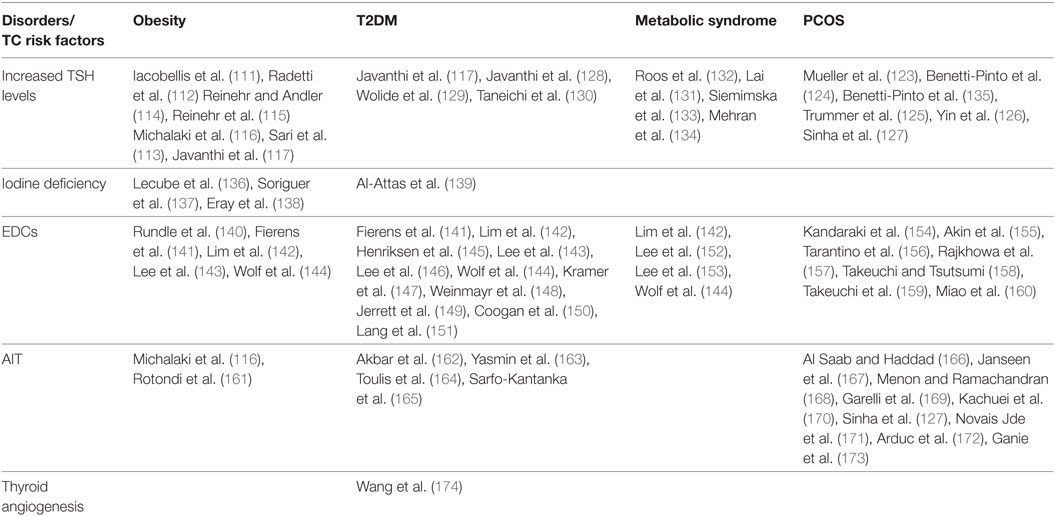
Table 2. Studies showing a possible interplay between insulin resistance related disorders and some TC risk factors.
Hyperinsulinemia itself, a major characteristic of insulin-resistant patients, is considered a determinant of cancer initiation/progression in diabetic/obese patients (175, 176). In the animal model, several studies carried out in hyperinsulinemic male mice overexpressing a dominant-negative, kinase-dead IGF-1R in muscle (MKR mice), have supported the important role of chronic hyperinsulinemia in cancer progression (177, 178). Notably, hyperinsulinemia may increase the growth of orthotopic mammary tumors through direct stimulation of the IR and without the involvement of the IGF-1R (179). However, no such studies have specifically addressed the role of hyperinsulinemia in TC.
Hyperinsulinemia may increase the bioavailability of IGF-1 and IGF-2 by inhibiting the synthesis of IGF-binding protein 1 and 2 and by intensifying IGF-1 hepatic production. The increased bioavailability of IGFs may contribute to tumor progression through the stimulation of IGF-1R, IR/IGF-1R hybrids, and IR-A itself (101). Yet, hyperinsulinemia, by directly activating IR-A, may favors its “non metabolic” functions and the induction of the pro-mitogenic MAPK/mTOR branch. Non-classical molecular partners, such as discoidin domain receptor 1 and G protein-coupled estrogen receptor, can be further recruited by the IGF system activated receptors, thereby favoring cancer cell proliferation and migration (180–183).
Taken together, these studies suggest that, in insulin-resistant patients, the concomitance of increased TSH levels, deregulation of the IGF axis, and hyperinsulinemia, may represent significant risk factors for TC.
Insulin Resistance and Thyroid Angiogenesis
Recently, a study has suggested that insulin-resistance may affect the growth and progression of thyroid nodules by increasing angiogenesis and intranodular vascularization (Figure 1) (174). Indeed, it was found that insulin-resistance and high HbA1c are positively associated with a predominant intranodular flow, and with velocity, pressure and density of intranodular blood vessels, especially in nodules of large size (174) (Table 2). The molecular mechanisms responsible for these findings warrant further investigation. However, it is possible to speculate that insulin may stimulate vascular endothelial growth factor (VEGF) expression and promote proliferation of vascular endothelial cells in thyroid nodules and tumors, as shown in other contexts (184). In line with these findings, in breast cancer patients, both hyperinsulinemia and hyperglycemia may stimulate the secretion of pro-inflammatory factors, such as tumor necrosis factor-α, tumor growth factor-α, tumor growth factor-β, interleukin-8, fibroblast growth factor-2, and VEGF-α, thus contributing to tumor neoangiogenesis (185).
Insulin Resistance and Iodine Deficiency
Iodine is essential for the synthesis and regulation of thyroid hormones. The relationship between iodine intake and TC is complex, as both iodine deficiency and iodine excess have been related to TC development (186, 187). Long-term iodine deficiency has especially been associated with follicular and anaplastic histotypes but also with the papillary histotype (188–190). In animal models, iodine deficiency acts as a weak initiator but a strong promoter of TC, mainly of the follicular type (187). The mechanisms linking the association between iodine deficiency and TC are multiple. Severe iodine deficiency may cause increase of TSH levels (191). However, iodine deficiency could per se favor angiogenesis in TC tissues by increasing VEGF mRNA expression (192) through the activation of the transcription factor hypoxia inducible factor 1a (192). In TC, iodine deficiency may also activate additional signals such as the mTOR/p70S6K pathway (193). Low iodine levels may also promote TC development favoring H2O2-mediated radical reactive oxygen species (ROS) generation, which could result in DNA damage and somatic mutations (191).
Iodine deficiency is also linked to insulin resistance. Several lines of evidences have shown that urinary iodine, which is roughly equal to iodine intake, is markedly decreased in T2DM and obese patients as compared to control subjects, and is negatively correlated with glucose, insulin concentrations and HOMA-IR index (136–139, 194, 195) (Table 2). The physiological pathways connecting insulin resistance with iodine status and the molecular mechanisms by which obese individuals show a reduction in urinary iodine levels are still unclear. It has been proposed that inflammatory cytokines secreted by adipose tissue of insulin resistant patients as well as hyperinsulinemia itself may negatively modulate the expression of sodium/iodine symporter (NIS) on the apical surface of enterocytes, thus inducing a decrease in iodine absorption (136). Taken together, these findings suggest a functional association between iodine deficiency and insulin resistance (Figure 1). However, further studies are needed to better clarify the mechanisms underlying this relationship.
Insulin Resistance and Endocrine Disrupting Chemicals (EDCs)
Various environmental compounds either natural or synthetic, act as EDCs. These substances may affect hormone signaling through different mechanisms. In the thyroid, they may act at different levels: they may interfere with the hypothalamic–pituitary–thyroid axis, induce direct thyroid cell damage, alter peripheral metabolism of thyroid hormones, and/or affect thyrocytes proliferation, increasing the susceptibility to develop DTCs (196). Recently, it has been found that in the volcanic area of Sicily, DTC incidence is abnormally increased possibly through chronic exposure to EDCs of volcanic origin (197), supporting data reported in other volcanic areas (198).
Beyond their intrinsic carcinogenic potential, some EDCs at the concentration found in human plasma, may lead to disturbances in glucose and fat metabolism. Indeed, they alter pancreatic β-cell function in cellular and animal models (199) and inappropriately regulate intracellular lipid homeostasis as well as proliferation and differentiation of adipocytes (200). These observations suggest that some environmental EDCs may represent a risk factor in the etiology of T2DM and other metabolic disorders, particularly in pre-diabetic individuals (199). In support of these evidences, several biological and epidemiological studies have correlated EDCs exposure with obesity, metabolic syndrome, T2DM and other diseases related to insulin-resistance, including cancer (Figure 1) (201, 202) (Table 2). Recently, it has been found that long-term exposure to air pollution is associated with an increase in HOMA index and insulin levels (144, 202). Some EDCs, including certain metals, by disrupting estrogen homeostasis or by mimicking estrogen actions, may lead to a pregnancy-like metabolic state characterized by insulin-resistance and hyperinsulinemia (199). Furthermore, estrogens potentiate insulin proliferative effects (203). Therefore, EDCs may contribute to DTC initiation and progression (204). The observation that DTC is 3-fold more frequent in women than in men (205) support the pivotal role of estrogens in DTC etiopathogenesis. Indeed, 17-β estradiol (E2) is a potent stimulator of benign and malignant thyrocytes, and both estrogen receptor α (ERα) and ERβ are expressed in DTCs (206). Moreover, as it has been seen in other cellular contexts (207–210), it is likely that also in thyrocytes EDCs may induce membrane-initiated rapid signals involving androgen receptors (ARs) and ERs, both of which crosstalk with the IGF axis (211).
In summary, long-term exposure to EDCs is linked to TC development and progression by multiple mechanisms that include direct toxic effects, estrogen-like effects, and worsening of insulin-resistance (Figure 1).
Insulin Resistance and Chronic Autoimmune Thyroiditis (AIT)
The association between AIT and DTC has long been a topic of controversy. Data available so far are conflicting. The coexistence of these two diseases has been reported by numerous studies ranging from 0.5 to 30% (212). A meta-analysis conducted by Singh et al. (213) demonstrated that the incidence rate of HT, the most common AIT, is 2.8 times higher in patients with PTC than in patients affected by benign thyroid diseases, and that patients with HT were affected by PTC twice as often as expected. However, many of the published studies are retrospective, had used variable histological methodologies and definitions and have been subjected to several selection biases. Furthermore, it should be underlined that population-based fine needle aspiration biopsy studies have not confirmed this relationship between HT and DTC (214).
The presence of chronic inflammation in HT acting as an initiating factor in carcinogenesis could represent a potential mechanism responsible for the link between HT and PTC. Moreover, the increase in TSH levels or in TSH receptor stimulating antibodies (TSAb), the imbalance in the amount of chemokines and cytokines favoring a switch from Th2 to Th1 immune response or the presence of insulin-resistance, may provide additional explanations for this association (215, 216).
A link between insulin-resistance and AIT has been reported by several studies (Table 2; Figure 1). For instance, it has been seen that the prevalence of AIT in insulin-resistant individuals is higher compared to normal control subjects. For instance, in PCOS patients, anti-TPO antibodies are present in 19.6–26.9% when compared with 3.3–8.3% of control patients (127, 166–173), whereas the prevalence of AIT ranges from 10 to 43% in T2DM patients (162–165) and from 12.4% (in children) to 10–16% (in adults) in obese patients (116, 161). Recently, obesity has been proposed to be a risk factor for thyroid autoimmunity (217). Data showed a positive correlation between leptin and AIT (r = 0.26; P < 0.001), independent of BMI and fat mass, suggesting the hypothesis that high leptin levels may enhance autoimmune thyroid reaction in a context susceptible to Th-1 immune response (217). Despite these intriguing results, some controversy does remain concerning whether and how insulin resistance prompts the development of AIT. So far, available studies have several limitations, such as restricted number of subjects, biases in the selection of patients and controls, differences in study design, and variability in the use of commercially available assays for anti-TPO antibodies.
Insulin Resistance in the Context of T2DM
Type 2 diabetes mellitus is characterized not only by insulin resistance but also by hyperglycemia with oxidative stress and advanced glycation end products on proteins and macromolecules, as well as by dyslipidemia and chronic low-grade inflammation (218). In some studies T2DM has been associated with increased risk for TC, although the association ratio values were low (75, 76, 219). Indeed, a recent pooled analysis, including five prospective studies from the USA, showed that the hazard ratio for TC was 1.19 (95% CI: 0.84–1.69) in women and 0.96 (95% CI: 0.65–1.42) in men (55) (Table 1).
Many studies have shown an association between glucose metabolism disorders and thyroid morphologic changes in terms of gland echogenicity, goiter and nodules prevalence, and TSH levels (220–223). In a prospective study, T2DM patients showed higher TSH levels than controls (224). T2DM patients had also larger thyroid volumes and an increased prevalence of nodules. Conversely, in a retrospective survey of 1,559 patients with a new diagnosis of TC (from the continuous National Examination Survey, NHANES) an increased prevalence of T2DM was found among patients who were ≤44 years old as compared to control patients (RR 2.32, CI: 1.37–3.66) (52).
Two longitudinal studies showed that a history of T2DM, ascertained by a self-administered questionnaire, is a risk factor for TC (37, 48). In the total cohort, the increase in TC risk was irrelevant, but it was significantly increased in women (HR. 1.46, 95% CI: 1.01–2.10). Case–control and cohort studies conducted in Unites States (37, 45, 48, 225), Canada (226), Europe (46, 227–230), and Asia (231, 232), confirmed an increased TC risk of approximately 20% in diabetic patients, independently of geographic region, study design, and quality analysis. Despite of a high heterogeneity among studies, the observation that the risk is increased among diabetic women, but not among men, has been always confirmed (37, 46, 233). However, the TC risk associated with DM is more evident in the geographic areas of the world with high rates of TC.
A recent study based on a large prospective cohort, the Women’s Health Initiative, reported data at variance with previous results. In this study, 147,934 cancer free women at baseline were followed up for a median time of 15.9 years. No significant association was found between occurrence of TC with diabetes or diabetes treatment (234). Possible explanations for these negative findings include a weak association between TC and diabetes in postmenopausal women, and the lack of information for insulin resistance and hyperinsulinemia. TC risk is also increased in metabolic syndrome characterized by long-standing insulin resistance, confirming the fundamental role of elevated insulin and glucose levels in the pathogenesis of this association (235). Nevertheless, studies on TC risk in T2DM have some limitations because data regarding the metabolic control, the duration of DM, or the presence of chronic complications, have not always been evaluated. Moreover, confounding elements such as treatment, age of patients, comorbidities like obesity, have not always been appropriately taken into account in some studies. For these reasons, results are somehow controversial and should be interpreted with caution.
Several potential mechanisms can be taken into account to explain the association between T2DM and TC including the higher prevalence of abnormal serum TSH levels (236), the effects of elevated insulin and/or glucose levels in affecting cellular energy metabolism [by increasing the intracellular adenosine triphosphate (ATP)/adenosine monophosphate ratio and inactivating AMPK] (237) and immune system (by increasing ROS production and especially nitric oxide) (237, 238) (Figure 1). Moreover, it has been suggested that chronic treatment with some antidiabetic drugs, may favor the association between T2DM and cancer (239).
Clearly, insulin therapy causes chronic peripheral hyperinsulinemia, and several studies have attempted to clarify whether long-term treatment with insulin or insulin analogs may increase the risk of overall cancer mortality and incidence in patients with T2DM (240, 241). Although some studies have suggested that, unlike native insulin, the long-acting insulin analog glargine could be associated with a higher risk for cancer, especially breast cancer (241–246), re-analysis of these data, as well as further studies have found no differences in cancer risk for insulin glargine as compared with native insulin or other insulin analogs (240). Therefore, there is no clear recommendation regarding the use of insulin or insulin analog in relation to cancer risk.
The possible role of insulin secretagogues (sulfonylureas, glinides) has also been studied. Sulfonylureas (SUs) (glibenclamide, glipizide, and glimepiride) are widely used in diabetic patients. Binding to sulfonylurea receptor 1 on pancreatic beta cells, they stimulates insulin release from the intracellular vesicles. Being potent stimulators of insulin secretion, in principle, sulfonylureas might increase cancer risk. However, epidemiological studies have given controversial results sometimes showing increased cancer risk (243, 247–249). Less potent insulin secretagogues, such as glinides do not appear to be associated with cancer risk (250–253). In any case, none of the above-mentioned studies has focused on TC.
Incretin-based therapies include glucagon-like peptide-1 receptor (GLP-1R) agonists and dipeptidyl-peptidase-4 inhibitors, both of which amplify the insulin response to glucose besides having pleiotropic effects. Therapy with GLP-1R agonists has been recently linked to the C-cell hyperplasia and increased medullary, but not follicular, TC in rodents (245, 254). However, this effect has been observed after lifetime exposure to supratherapeutic doses (255). Moreover, as human thyroid tissues express very low levels of GLP-1R this risk seems to be irrelevant. Data from human observational studies and clinical trials have yielded inconclusive results, thus, continuous monitoring of this issue is still required. Patients with T2DM often follow combination therapies with multiple drugs, making these epidemiological studies very difficult. Moreover, many studies have not taken into account the length of treatment, thus introducing time-related bias.
Two main classes of antidiabetic drugs, thiazolidinediones (TZDs) and biguanides act by reducing insulin resistance (insulin sensitizers). Their possible role in TC is discussed below.
Other antidiabetic drugs, such as alpha-glucosidase inhibitors or SGLT-2 inhibitors, do not directly affect insulin levels or insulin resistance. In any case, no data regarding the use of these drugs and the risk of TC are available.
Possible Implications for TC Prevention and Therapy
Insulin resistance is multifactorial, and genetic factors account for a significant proportion of insulin resistant subjects (256–262). However, physical inactivity and visceral obesity are the most frequent preventable causes of insulin resistance (263–265).
While the underlying biological mechanisms remain to be investigated, insulin resistance seems to be worsened by iodine deficiency in obese and diabetic patients (136–139, 194, 195). Moreover, evidences showing that TSH and estrogens potentiate the growth effects of insulin, lend support to the hypothesis that insulin resistance may significantly affect the risk of TC, especially by interacting with subclinical hypothyroidism, iodine deficiency, and endocrine disruptors with either estrogen-like or antithyroid activity (Figure 1). At least two of these factors, insulin resistance and environmental contamination with endocrine disruptors have been steadily rising in the past decades (204, 266) and it is reasonable to hypothesize that the interplay among these factors may contribute to the worldwide increase of PTCs incidence (16, 17, 20, 21).
Prevention and therapy of visceral obesity and of related disorders, such as T2DM and metabolic syndrome, are the mainstay to limit the spread of insulin resistance in the population. To this aim, and to reduce associated disorders including cancer, several international organizations and scientific societies have issued guidelines that recommend a healthy lifestyle consisting of constant physical activity and a correct diet (267–270). It is worth noting that these lifestyle changes are difficult to attain and maintain for most people and that only a small proportion of obese subjects is able to achieve significant weight loss by these measures. However, relatively small weight losses may bring about significant amelioration of insulin resistance (271, 272). Whether reduction of insulin resistance achieved by lifestyle changes is associated with reduced TC risk is currently unknown.
Exposure to environmental risk factors, such as iodine deficiency and contamination with endocrine disruptors, should be considered as well. Although iodine prophylaxis with iodized salt is now established in most countries, borderline low iodine intake can be still observed, especially in countries where iodine prophylaxis is facultative (187, 273).
Similarly, environmental monitoring for EDCs may reveal geographic areas and places with high EDC contamination (204). Clinical, ultrasound and biochemical screening can now easily diagnose autoimmune thyroid disorders that may also be associated with TC.
The identification of these additional risk factors may allow the recommendation of individualized strategies for TC prevention.
Insulin Sensitizers: A Possible Role in TC Prevention and Therapy
As stated above, only a minority of patients succeeds to change lifestyle and achieve long-lasting weight loss. Therefore, the use of insulin sensitizers has been proposed to reduce insulin resistance and its complications.
Metformin
Metformin is by far the insulin sensitizer most studied in cancer prevention and therapy. A primary effect of metformin is the suppression of the hepatic gluconeogenesis and glucose output, and to increase the peripheral glucose uptake, with consequent reduction in insulin resistance and circulating insulin levels (274).
Interestingly, the use of metformin in diabetic patients has been associated with a lower risk for cancer. A recent meta-analysis of 11 independent studies found an overall 30% statistically significant decrease in cancer risk in patients treated with metformin compared with other diabetic treatments with a promising trend for reduction in overall cancer mortality (275). Similar reductions in cancer risk and mortality was also observed in a second meta-analysis that included 32 articles (276). These and other studies have made a good case for metformin repurposing in cancer chemoprevention. However, a note of caution comes from the fact that these studies regard only diabetic patients, are all retrospective, and results need to be adjusted for multiple variables (277).
Moreover, no specific data on TC were available in these studies, although a recent study performed in Taiwan has shown that the risk of TC is also reduced in diabetic patients treated with metformin (278). However, a second case–control study (279) was unable to find a reduced risk for TC in diabetic patients taking metformin.
In some studies, the use of metformin seems to inhibit the growth of thyroid nodules, which is among risk factors for TC (280–282). In one study, the association of metformin to l-thyroxine was shown to inhibit the growth of thyroid nodules more effectively that l-thyroxine alone (280). In another randomized placebo-controlled clinical trial the use of metformin was also associated with the reduction of small solid thyroid nodules (281). Notably, a recent study showed that metformin therapy in subjects with insulin resistance was effective in decreasing thyroid volume and nodule size (282).
Multiple mechanisms may account for the chemopreventive and anticancer effects of metformin in several cancer histotypes and in TC in particular (283, 284). Relevant to thyroid, additional in vivo effects of metformin that may be linked with chemoprevention of TC may include lowering of TSH serum levels in diabetic patients (285). In fact, metformin potentiated the effect of l-thyroxine in reducing thyroid nodule volume in patients with multinodular goiter (280). However, there is evidence that the TSH lowering effect of metformin is seen only in patients with treated hypothyroidism, but not in euthyroid patients (286). Further studies are needed to fully clarify the potential of metformin as chemopreventive drug in non-diabetic, insulin resistant, euthyroid patients.
As an additional mechanism, sex steroids and sex steroid-mimicking EDCs may induce membrane-initiated signals involving AR and ERs and activation of the IGF system (211, 287). These effects have been demonstrated in prostate cancer cells, but may also operate in other cells sensitive to sex hormones. Interestingly, these membrane-initiated signals may be inhibited by metformin (288), thus supporting the potential role of metformin in cancer chemoprevention.
Apart for its possible role in cancer prevention metformin may also play a role in cancer treatment.
Anticancer actions of metformin are partially ascribed to its ability to activate the liver kinase B1/AMPK pathway and to suppress ATP production through the inhibition of mitochondrial complex I (289–292). Both actions of metformin contribute to the inhibition of the mTOR pathway, a major regulator of cell growth and proliferation (277). Metformin may also inhibit ERK signaling (293) and Ca(2+)-dependent PKC-alpha/ERK and JNK/activator protein 1 pathways (294, 295). It may also reduce Akt activity through serine phosphorylation of IRS-1 (296).
Other effects include the inhibition of transcriptional activity of CREB transcriptional factor (297) through the induction of the AMPK-dependent phosphorylation of CREB cofactor CRTC2 at Ser171, which causes CRTC2 sequestration in the cytoplasm by binding with 14–3–3 proteins (298, 299). In fact, dephosphorylated CRTC2 translocates into the nucleus, where it contributes to the CREB-dependent transcription by stimulating the formation of the complex CREB—CREB-binding protein—CRTC2 (297).
Notably, in metformin-treated patients, intraparenchimal metformin concentrations are generally significantly higher than metformin concentration in the bloodstream. For example, metformin concentration at the level of the portal vein is much higher than in the peripheral circulation, thus exposing liver to very high metformin levels (300). Many other organs, including salivary glands, stomach, small intestine, kidney as well as other organs/tissues are also able to concentrate metformin in dependence of the expression of organic cation transporters (OCTs), such as OCT1–2–3 and organ-specific metformin metabolism (301). Metformin also concentrates in the mitochondrial matrix by approximately 1,000-folds (302). These high concentrations are believed to play an important anticancer role (302, 303).
In TC cell lines, metformin was able to inhibit proliferation, through the downregulation of cyclin D1 expression and activation of AMPK, which in turn inhibits the p70S6K/pS6 signaling pathway. Moreover, in undifferentiated TC cells cultured as thyrospheres and enriched in stem-like cells, metformin inhibited the effects of insulin on growth and sphere formation, and potentiated the inhibitory effects of doxorubicin and cisplatin (304). The ability of metformin to potentiate the cytotoxic effects of chemotherapeutics via AMPK and p53 signaling was confirmed in other studies (305–307). In addition, metformin may inhibit the growth, migration and mesenchymal transition of TC cell lines by inhibiting mTOR (308). Han et al. showed that metformin elicited a dual antiproliferative effect on primary thyroid cultures and TC cells both by reducing circulating insulin and by directly inhibiting cell cycle progression and survival (304). Accordingly, DTCs occurring in metformin-treated diabetic patients were found to be significantly smaller and with increased progression-free survival as compared with the non-metformin groups (309). A higher remission in patients with TCs with cervical lymph node metastasis has also been observed (310).
Taken together, these data suggest that metformin might play a role in prevention and treatment of thyroid nodules and cancer in insulin resistant patients. Several clinical trials are currently under way with the aim to evaluate the efficacy of metformin as an add-on therapy for patients with various cancer histotypes, but none of these is focused on TC (Clinicaltrials.gov).
PPAR-γ Agonists
Thiazolidinediones, also known as glitazones, bind and activate the nuclear receptors PPAR-γ acting as agonists. They are potent insulin sensitizers used in the treatment of patients with T2DM (311). Although both metformin and TZDs decrease hepatic glucose production (312, 313), only TZDs reduce liver fat content (312, 314) and diminish fasting free fatty acid concentrations (315) thus improving skeletal muscle insulin sensitivity and reducing liver steatosis. However, side effects of TZDs, such as weight gain and fluid retention that can precipitate cardiac failure and bone fractures, have limited their use in clinical practice. Troglitazone and rosiglitazone (RGZ) were withdrawn because of hepatotoxicity (316) and suspected to increase cardiovascular risk (317), respectively. In addition, the benefit–risk ratio of pioglitazone (PIO) has been reassessed recently in light of a putatively increased risk of bladder cancer.
In a population-based study (318), it has been found that RGZ was associated with a 30–50% reduced risk of TC. In dose response analysis, the adjusted hazard ratios (95% confidence intervals) were significant for the third tertile of duration of therapy (≥14 months) and cumulative dose ≥1,800 mg (0.53, CI 0.31–0.89) and for age ≥50 years (0.50, CI 0.29–0.87) (318). However, a successive study using PIO did not show the same protective effect on TC risk, even if some limitations related to the patients classification or the presence of confounding factors cannot be excluded. However, the different results obtained with the two glitazones (RGZ and PIO) suggested that, apart from restoring insulin-sensitivity, the two drugs might have differential mechanisms on cancer (319) and thyroid cells (320).
Inhibitors of Insulin/IR-A Signaling
Several studies have highlighted the importance of the insulin/IGF-2/IR-A pathway as a potential target in tumors addicted to this signaling (101, 105). However, for several reasons, targeting this pathway in cancer treatment is not simple. In particular, it is now well accepted that IR and its homolog IGF-1R are functionally interconnected by forming hybrid receptors with an important role in cancer (321), and that targeting either IR or IGF-1R alone results in increased activity of the homolog receptor (322) and resistance to treatment. However, various strategies have been developed to target the insulin/IGF-2/IR-A pathway. Whether these approaches may have specific benefits in insulin resistant patients with cancer is unknown.
In order to avoid or minimize the severe derangement of the glucose metabolism associated with inhibition of total IR, future therapies should possibly aim at specific targeting of IR-A. However, specific antibodies or other drugs able to inhibit the IR-A and not the IR-B are not available and difficult to obtain because of the small differences between the two IR isoforms (323).
The identification of mutations in splicing factors in several malignancies (324, 325) has led to the development of drugs able to counteract the effects of these mutated splicing factors (326, 327). However, whether such drugs may inhibit IR-A formation and favor the IR-B isoform in TC is unknown. Another possible approach is to take advantage of the differential regulation of IR isoform protein maturation. Indeed, furin and paired basic amino acid-cleaving enzyme 4 enzymes, seems to be differentially required for IR-A and IR-B maturation (328, 329), and furin can be inhibited by a number of polyphenols (330).
Finally, various miRNAs have been found to be dysregulated in obesity and insulin resistance (331). Studies are needed to assess whether some of these miRNAs may play a role in the altered IR-A expression in cancer and whether they could be useful tools to normalize the IR-A:IR-B ratio.
Currently available small molecule TK inhibitors lack specificity for IR-A, but are able to coinhibit the IR and IGF-1R. The most studied drugs in this category are Linsitinib (OSI-906) and BMS-754807. Preclinical data, showing a significant efficacy of both drugs either alone or in combination therapies (332), have prompted several phase I–III studies—https://clinicaltrials.gov/ct2/results?term=linsitinib&pg=1 and https://clinicaltrials.gov/ct2/results?term=BMS-754807+&Search=Search. However, no definite evidence of efficacy in a clinical setting has been demonstrated so far.
A different approach for malignancies driven by the IGF-2/IR-A loop is to block IGF-2 using specific antibodies or specific ligand traps. A specific trap for IGF-2 can be obtained using a soluble preparation of the high-affinity domain 11 of M6P/IGF-2R (333, 334), while the soluble form of the IGF-1R combined with the Fc portion of IgG1 can provide a trap for both circulating IGF-1 and IGF-2 (335). These therapies have the advantage to block IR-A stimulation by IGF-2 without impairing the metabolic effects of insulin. However, they do not inhibit the effects of high circulating insulin levels in insulin resistant patients. Preclinical studies are encouraging but clinical data are lacking (335). So far, no studies have addressed the question whether inhibition of the insulin/IGF-2/IR-A signaling by these approaches may provide benefits to patients with TC in the context of insulin resistance.
Summary and Perspectives
Several lines of evidence now support the concept that the activation of the insulin/IR axis plays a role in TC carcinogenesis. In particular, various dysmetabolic conditions characterized by insulin resistance are significantly associated with an increased risk and worse prognosis of TC. Whether and to what extent insulin resistance plays a role in the worldwide, steady increase in PTCs has not been clarified yet. Indeed, several clinical studies performed until now, have reported only a positive association rather than a causative role. Moreover, some of these studies show significant limitations, including lack of adjustment for potential confounders, and/or limited statistic power and/or low OR values. These limitations should be taken into account when considering the physiological/biological significance of these studies. Similarly, more studies are required to elucidate the possible interactions between insulin resistance/hyperinsulinemia and more established TC risk factors, such as radiations, iodine deficiency, endocrine disruptors, and inflammation. Finally, how obesity derived cytokines, and overactivation of the insulin/IGF axis may affect the molecular pathways involved in the pathogenesis of TC should be explored in depth.
However, as for other malignancies associated with insulin resistance, it is to be expected that a correct lifestyle, which includes a healthy diet and physical activity, aimed at preventing obesity and T2DM would exert a beneficial effect also in TC occurrence. For all people living in iodine deficient areas, iodine prophylaxis is mandatory in order to avoid the growth promoting effect of reduced iodine intake on the thyroid gland. It could be hypothesized that additional attention should be lent to people with concomitant insulin resistance in order to avoid the combined effects of hyperinsulinemia and iodine deficiency. In T2DM patients, administration of insulin sensitizers or inhibitors of SGLT2 (336, 337) should be preferred to insulin stimulating drugs and insulin itself. Hopefully, these assumptions will be validated by future studies, although, because of the indolent natural history of most DTCs, such studies may prove to be difficult to perform.
As far as therapy is concerned, surgery, radioactive iodine treatment and TSH suppression by l-thyroxine administration are the cornerstones of DTC treatment. However, no specific therapy exists for poorly differentiated or undifferentiated TCs that have lost the ability to uptake radioiodine. Drugs with multikinase inhibiting activity are increasingly used in these cancers, but so far their effect on cancer mortality is at best uncertain (338). Clearly, new combined therapies are urgently required for these aggressive cancers. Evidences showing that the insulin/IGF axis is frequently activated in these tumors owing to overexpressed IR and IGF-1R and increased local production of IGFs lend support to the possibility that therapies targeting this axis could have a role in these new approaches. The increased awareness that overexpression of IR-A possibly plays a more important role than IGF-1R and may compensate for IGF-1R inhibition, strongly suggests that dual IR and IGF-1R inhibitors should be more efficacious than specific inhibitors of IGF-1R. Moreover, future approaches may explore the efficacy of drugs specifically targeting IR-A or pathways preferentially activated by the IGF-2/IR-A loop. Finally, insulin sensitizers able to reduce peripheral insulin levels could have a role on both prevention and treatment of TC. Certainly, more studies are needed to address the role of insulin and insulin resistance in better and individualized programs of TC prevention and adjuvant therapies. More in general, in spite of several lines of evidence indicating that obesity/insulin resistance-driven mechanisms are associated with cancer development and progression, specific guidelines for cancer prevention and treatment in these patients are lacking and should be considered a desirable aim of precision medicine.
Author Contributions
VV, RM, MN, and AB: substantial contributions to the conception and design of the article; drafting the work; final approval of the version to be published; and agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. VV, RM, and AB: revising it critically for important intellectual content.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported in part by grants from AIRC IG 19242 to AB; Ministero della Salute, Italy (grant 67/GR-2010-2319511) to RM.
Abbreviations
AIT, autoimmune thyroiditis; cAMP, cyclic adenosine monophosphate; AMPK, 5′ adenosine monophosphate-activated protein kinase; ATC, anaplastic thyroid cancer; ATP, adenosine triphosphate; BMI, body mass index; B-Raf, serine/threonine-protein kinase B-Raf; CBP, CREB-binding protein; CRCT2, CREB regulated transcription coactivator 2; CREB, cAMP response element-binding protein; DTC, differentiated thyroid cancer; E2, 17-beta estradiol; EDCs, endocrine disrupting chemicals; EGF, epidermal growth factor; ERKs, extracellular signal-regulated kinases; FGF-2, fibroblast growth factor-2; GPER, G protein-coupled estrogen receptor; HR, hazard risk; HT, Hashimoto’s thyroiditis; IGF-1, insulin growth factor 1; IGF-2, insulin growth factor 2; IL-8, interleukin-8; IR, insulin receptor; JAK/STAT, janus kinase/signal transducers of activated transcription;MAPK, mitogen-activated protein kinase; OR, odds ratio; p90rsk, MAPK-activated protein kinase-1; OCT, organic cation transporter; PCOS, polycystic ovary syndrome; PIO, pioglitazone; PI3K, phosphoinositide-3-kinase; PKC, protein kinase C; PTC, papillary thyroid cancer; PKA, protein kinase A; PKB/Akt, protein kinase B/Akt; c-Raf, proto-oncogene; Rap1, Ras-related protein 1; Ras, rat sarcoma virus protein; RGZ, rosiglitazone; ROS, reactive oxygen species; RR, relative risk; RTKs, tyrosine kinase receptors; TC, thyroid cancer; T2DM, type 2 diabetes mellitus; Tg, thyroglobulin; TPO, thyroperoxidase; mTOR, mammalian target of rapamycin; TSH, thyroid stimulating hormone; TZDs, tiazolidinediones; VEGF, vascular endothelial growth factor; Wnt, proto-oncogene protein Wnt.
References
1. Hundahl SA, Fleming ID, Fremgen AM, Menck HR. A national cancer data base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985-1995 [see comments]. Cancer (1998) 83(12):2638–48. doi:10.1002/(SICI)1097-0142(19981215)83:12<2638::AID-CNCR31>3.0.CO;2-1
2. Thyroid Cancer Facts and Figures. National Cancer Institute Surveillance, Epidemiology, and End Results Program. (2015). Available from: http://seer.cancer.gov/statfacts/html/thyro.html
3. Pathak KA, Leslie WD, Klonisch TC, Nason RW. The changing face of thyroid cancer in a population-based cohort. Cancer Med (2013) 2(4):537–44. doi:10.1002/cam4.103
4. Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973-2002. JAMA (2006) 295(18):2164–7. doi:10.1001/jama.295.18.2164
5. Chen AY, Jemal A, Ward EM. Increasing incidence of differentiated thyroid cancer in the United States, 1988-2005. Cancer (2009) 115(16):3801–7. doi:10.1002/cncr.24416
6. Blomberg M, Feldt-Rasmussen U, Andersen KK, Kjaer SK. Thyroid cancer in Denmark 1943-2008, before and after iodine supplementation. Int J Cancer (2012) 131(10):2360–6. doi:10.1002/ijc.27497
7. Kilfoy BA, Zheng T, Holford TR, Han X, Ward MH, Sjodin A, et al. International patterns and trends in thyroid cancer incidence, 1973-2002. Cancer Causes Control (2009) 20(5):525–31. doi:10.1007/s10552-008-9260-4
8. Brito JP, Davies L. Is there really an increased incidence of thyroid cancer? Curr Opin Endocrinol Diabetes Obes (2014) 21(5):405–8. doi:10.1097/MED.0000000000000094
9. McNally RJ, Blakey K, James PW, Gomez Pozo B, Basta NO, Hale J. Increasing incidence of thyroid cancer in Great Britain, 1976-2005: age-period-cohort analysis. Eur J Epidemiol (2012) 27(8):615–22. doi:10.1007/s10654-012-9710-x
10. Morris LG, Myssiorek D. Improved detection does not fully explain the rising incidence of well-differentiated thyroid cancer: a population-based analysis. Am J Surg (2010) 200(4):454–61. doi:10.1016/j.amjsurg.2009.11.008
11. Gschwandtner E, Klatte T, Swietek N, Bures C, Kober F, Ott J, et al. Increase of papillary thyroid microcarcinoma and a plea for restrictive treatment: a retrospective study of 1,391 prospective documented patients. Surgery (2016) 159(2):503–11. doi:10.1016/j.surg.2015.06.015
12. Hay ID, Hutchinson ME, Gonzalez-Losada T, McIver B, Reinalda ME, Grant CS, et al. Papillary thyroid microcarcinoma: a study of 900 cases observed in a 60-year period. Surgery (2008) 144(6):980–7; discussion 987–8. doi:10.1016/j.surg.2008.08.035
13. Imaizumi M, Usa T, Tominaga T, Neriishi K, Akahoshi M, Nakashima E, et al. Radiation dose-response relationships for thyroid nodules and autoimmune thyroid diseases in Hiroshima and Nagasaki atomic bomb survivors 55-58 years after radiation exposure. JAMA (2006) 295(9):1011–22. doi:10.1001/jama.295.9.1011
14. Preston-Martin S, Franceschi S, Ron E, Negri E. Thyroid cancer pooled analysis from 14 case-control studies: what have we learned? Cancer Causes Control (2003) 14(8):787–9. doi:10.1023/A:1026312203045
15. Iribarren C, Haselkorn T, Tekawa IS, Friedman GD. Cohort study of thyroid cancer in a San Francisco Bay area population. Int J Cancer (2001) 93(5):745–50. doi:10.1002/ijc.1377
16. Leenhardt L, Grosclaude P. [Epidemiology of thyroid carcinoma over the world]. Ann Endocrinol (Paris) (2011) 72(2):136–48. doi:10.1016/j.ando.2011.03.025
17. Kung TM, Ng WL, Gibson JB. Volcanoes and carcinoma of the thyroid: a possible association. Arch Environ Health (1981) 36(5):265–7. doi:10.1080/00039896.1981.10667635
18. Duntas LH, Doumas C. The ‘rings of fire’ and thyroid cancer. Hormones (Athens) (2009) 8(4):249–53. doi:10.14310/horm.2002.1242
19. Malandrino P, Scollo C, Marturano I, Russo M, Tavarelli M, Attard M, et al. Descriptive epidemiology of human thyroid cancer: experience from a regional registry and the “volcanic factor”. Front Endocrinol (2013) 4:65. doi:10.3389/fendo.2013.00065
20. Markaki I, Linos D, Linos A. The influence of dietary patterns on the development of thyroid cancer. Eur J Cancer (2003) 39(13):1912–9. doi:10.1016/S0959-8049(03)00432-5
21. Kitahara CM, Platz EA, Freeman LE, Hsing AW, Linet MS, Park Y, et al. Obesity and thyroid cancer risk among U.S. men and women: a pooled analysis of five prospective studies. Cancer Epidemiol Biomarkers Prev (2011) 20(3):464–72. doi:10.1158/1055-9965.EPI-10-1220
22. Rego-Iraeta A, Perez-Mendez LF, Mantinan B, Garcia-Mayor RV. Time trends for thyroid cancer in northwestern Spain: true rise in the incidence of micro and larger forms of papillary thyroid carcinoma. Thyroid (2009) 19(4):333–40. doi:10.1089/thy.2008.0210
23. Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer (2004) 4(8):579–91. doi:10.1038/nrc1408
24. Duggan C, Irwin ML, Xiao L, Henderson KD, Smith AW, Baumgartner RN, et al. Associations of insulin resistance and adiponectin with mortality in women with breast cancer. J Clin Oncol (2011) 29(1):32–9. doi:10.1200/JCO.2009.26.4473
25. Pathak S, Pandanaboyana S, Daniels I, Smart N, Prasad KR. Obesity and colorectal liver metastases: mechanisms and management. Surg Oncol (2016) 25(3):246–51. doi:10.1016/j.suronc.2016.05.021
26. Iyengar NM, Gucalp A, Dannenberg AJ, Hudis CA. Obesity and cancer mechanisms: tumor microenvironment and inflammation. J Clin Oncol (2016) 34(35):4270–6. doi:10.1200/JCO.2016.67.4283
27. Dal Maso L, La Vecchia C, Franceschi S, Preston-Martin S, Ron E, Levi F, et al. A pooled analysis of thyroid cancer studies. V. Anthropometric factors. Cancer Causes Control (2000) 11(2):137–44. doi:10.1023/A:1008938520101
28. Samanic C, Gridley G, Chow WH, Lubin J, Hoover RN, Fraumeni JF Jr. Obesity and cancer risk among white and black United States veterans. Cancer Causes Control (2004) 15(1):35–43. doi:10.1023/B:CACO.0000016573.79453.ba
29. Engeland A, Tretli S, Akslen LA, Bjorge T. Body size and thyroid cancer in two million Norwegian men and women. Br J Cancer (2006) 95(3):366–70. doi:10.1038/sj.bjc.6603249
30. Rapp K, Schroeder J, Klenk J, Stoehr S, Ulmer H, Concin H, et al. Obesity and incidence of cancer: a large cohort study of over 145,000 adults in Austria. Br J Cancer (2005) 93(9):1062–7. doi:10.1038/sj.bjc.6602819
31. Samanic C, Chow WH, Gridley G, Jarvholm B, Fraumeni JF Jr. Relation of body mass index to cancer risk in 362,552 Swedish men. Cancer Causes Control (2006) 17(7):901–9. doi:10.1007/s10552-006-0023-9
32. Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet (2008) 371(9612):569–78. doi:10.1016/S0140-6736(08)60269-X
33. Brindel P, Doyon F, Rachedi F, Boissin JL, Sebbag J, Shan L, et al. Anthropometric factors in differentiated thyroid cancer in French Polynesia: a case-control study. Cancer Causes Control (2009) 20(5):581–90. doi:10.1007/s10552-008-9266-y
34. Mijovic T, How J, Pakdaman M, Rochon L, Gologan O, Hier MP, et al. Body mass index in the evaluation of thyroid cancer risk. Thyroid (2009) 19(5):467–72. doi:10.1089/thy.2008.0386
35. Clero E, Leux C, Brindel P, Truong T, Anger A, Teinturier C, et al. Pooled analysis of two case-control studies in New Caledonia and French Polynesia of body mass index and differentiated thyroid cancer: the importance of body surface area. Thyroid (2010) 20(11):1285–93. doi:10.1089/thy.2009.0456
36. Leitzmann MF, Brenner A, Moore SC, Koebnick C, Park Y, Hollenbeck A, et al. Prospective study of body mass index, physical activity and thyroid cancer. Int J Cancer (2010) 126(12):2947–56. doi:10.1002/ijc.24913
37. Meinhold CL, Ron E, Schonfeld SJ, Alexander BH, Freedman DM, Linet MS, et al. Nonradiation risk factors for thyroid cancer in the US Radiologic Technologists Study. Am J Epidemiol (2010) 171(2):242–52. doi:10.1093/aje/kwp354
38. Rezzonico JN, Rezzonico M, Pusiol E, Pitoia F, Niepomniszcze H. Increased prevalence of insulin resistance in patients with differentiated thyroid carcinoma. Metab Syndr Relat Disord (2009) 7(4):375–80. doi:10.1089/met.2008.0062
39. Bae MJ, Kim SS, Kim WJ, Yi YS, Jeon YK, Kim BH, et al. High prevalence of papillary thyroid cancer in Korean women with insulin resistance. Head Neck (2016) 38(1):66–71. doi:10.1002/hed.23848
40. Ron E, Kleinerman RA, Boice JD Jr, LiVolsi VA, Flannery JT, Fraumeni JF Jr. A population-based case-control study of thyroid cancer. J Natl Cancer Inst (1987) 79(1):1–12.
41. Oh SW, Yoon YS, Shin SA. Effects of excess weight on cancer incidences depending on cancer sites and histologic findings among men: Korea National Health Insurance Corporation Study. J Clin Oncol (2005) 23(21):4742–54. doi:10.1200/JCO.2005.11.726
42. Almquist M, Johansen D, Bjorge T, Ulmer H, Lindkvist B, Stocks T, et al. Metabolic factors and risk of thyroid cancer in the metabolic syndrome and cancer project (Me-Can). Cancer Causes Control (2011) 22(5):743–51. doi:10.1007/s10552-011-9747-2
43. Rinaldi S, Lise M, Clavel-Chapelon F, Boutron-Ruault MC, Guillas G, Overvad K, et al. Body size and risk of differentiated thyroid carcinomas: findings from the EPIC study. Int J Cancer (2012) 131(6):E1004–14. doi:10.1002/ijc.27601
44. Kim HJ, Kim NK, Choi JH, Sohn SY, Kim SW, Jin SM, et al. Associations between body mass index and clinico-pathological characteristics of papillary thyroid cancer. Clin Endocrinol (Oxf) (2013) 78(1):134–40. doi:10.1111/j.1365-2265.2012.04506.x
45. Oberman B, Khaku A, Camacho F, Goldenberg D. Relationship between obesity, diabetes and the risk of thyroid cancer. Am J Otolaryngol (2015) 36(4):535–41. doi:10.1016/j.amjoto.2015.02.015
46. Wideroff L, Gridley G, Mellemkjaer L, Chow WH, Linet M, Keehn S, et al. Cancer incidence in a population-based cohort of patients hospitalized with diabetes mellitus in Denmark. J Natl Cancer Inst (1997) 89(18):1360–5. doi:10.1093/jnci/89.18.1360
47. Chodick G, Heymann AD, Rosenmann L, Green MS, Flash S, Porath A, et al. Diabetes and risk of incident cancer: a large population-based cohort study in Israel. Cancer Causes Control (2010) 21(6):879–87. doi:10.1007/s10552-010-9515-8
48. Aschebrook-Kilfoy B, Sabra MM, Brenner A, Moore SC, Ron E, Schatzkin A, et al. Diabetes and thyroid cancer risk in the National Institutes of Health-AARP diet and health study. Thyroid (2011) 21(9):957–63. doi:10.1089/thy.2010.0396
49. Duran AO, Anil C, Gursoy A, Nar A, Altundag O, Tutuncu NB. The relationship between glucose metabolism disorders and malignant thyroid disease. Int J Clin Oncol (2013) 18(4):585–9. doi:10.1007/s10147-012-0435-3
50. Lai GY, Park Y, Hartge P, Hollenbeck AR, Freedman ND. The association between self-reported diabetes and cancer incidence in the NIH-AARP diet and health study. J Clin Endocrinol Metab (2013) 98(3):E497–502. doi:10.1210/jc.2012-3335
51. Tseng CH. Diabetes and thyroid cancer mortality: a 12-year prospective follow-up of Taiwanese. Eur J Clin Invest (2013) 43(6):595–601. doi:10.1111/eci.12086
52. Paulus YM, Riedel ER, Sabra MM, Tuttle RM, Kalin MF. Prevalence of diabetes mellitus in patients with newly evaluated papillary thyroid cancer. Thyroid Res (2014) 7:7. doi:10.1186/1756-6614-7-7
53. Yeo Y, Ma SH, Hwang Y, Horn-Ross PL, Hsing A, Lee KE, et al. Diabetes mellitus and risk of thyroid cancer: a meta-analysis. PLoS One (2014) 9(6):e98135. doi:10.1371/journal.pone.0098135
54. Kabat GC, Kim MY, Thomson CA, Luo J, Wactawski-Wende J, Rohan TE. Anthropometric factors and physical activity and risk of thyroid cancer in postmenopausal women. Cancer Causes Control (2012) 23(3):421–30. doi:10.1007/s10552-011-9890-9
55. Kitahara CM, Platz EA, Park Y, Hollenbeck AR, Schatzkin A, Berrington de Gonzalez A. Body fat distribution, weight change during adulthood, and thyroid cancer risk in the NIH-AARP diet and health study. Int J Cancer (2012) 130(6):1411–9. doi:10.1002/ijc.26161
56. Kitahara CM, McCullough ML, Franceschi S, Rinaldi S, Wolk A, Neta G, et al. Anthropometric factors and thyroid cancer risk by histological subtype: pooled analysis of 22 prospective studies. Thyroid (2016) 26(2):306–18. doi:10.1089/thy.2015.0319
57. Zhao ZG, Guo XG, Ba CX, Wang W, Yang YY, Wang J, et al. Overweight, obesity and thyroid cancer risk: a meta-analysis of cohort studies. J Int Med Res (2012) 40(6):2041–50. doi:10.1177/030006051204000601
58. Tresallet C, Seman M, Tissier F, Buffet C, Lupinacci RM, Vuarnesson H, et al. The incidence of papillary thyroid carcinoma and outcomes in operative patients according to their body mass indices. Surgery (2014) 156(5):1145–52. doi:10.1016/j.surg.2014.04.020
59. Harari A, Endo B, Nishimoto S, Ituarte PH, Yeh MW. Risk of advanced papillary thyroid cancer in obese patients. Arch Surg (2012) 147(9):805–11. doi:10.1001/archsurg.2012.713
60. Paes JE, Hua K, Nagy R, Kloos RT, Jarjoura D, Ringel MD. The relationship between body mass index and thyroid cancer pathology features and outcomes: a clinicopathological cohort study. J Clin Endocrinol Metab (2010) 95(9):4244–50. doi:10.1210/jc.2010-0440
61. Paz-Filho G, Lim EL, Wong ML, Licinio J. Associations between adipokines and obesity-related cancer. Front Biosci (2011) 16:1634–50. doi:10.2741/3810
62. Cheng SP, Yin PH, Chang YC, Lee CH, Huang SY, Chi CW. Differential roles of leptin in regulating cell migration in thyroid cancer cells. Oncol Rep (2010) 23(6):1721–7. doi:10.3892/or_00000817
63. Uddin S, Hussain AR, Siraj AK, Khan OS, Bavi PP, Al-Kuraya KS. Role of leptin and its receptors in the pathogenesis of thyroid cancer. Int J Clin Exp Pathol (2011) 4(7):637–43.
64. Pazaitou-Panayiotou K, Polyzos SA, Mantzoros CS. Obesity and thyroid cancer: epidemiologic associations and underlying mechanisms. Obes Rev (2013) 14(12):1006–22. doi:10.1111/obr.12070
65. Mitsiades N, Pazaitou-Panayiotou K, Aronis KN, Moon HS, Chamberland JP, Liu X, et al. Circulating adiponectin is inversely associated with risk of thyroid cancer: in vivo and in vitro studies. J Clin Endocrinol Metab (2011) 96(12):E2023–8. doi:10.1210/jc.2010-1908
66. Hsieh CJ, Wang PW, Wang ST, Liu RT, Tung SC, Chien WY, et al. Serum leptin concentrations of patients with sequential thyroid function changes. Clin Endocrinol (Oxf) (2002) 57(1):29–34. doi:10.1046/j.1365-2265.2002.01543.x
67. Akinci M, Kosova F, Cetin B, Aslan S, Ari Z, Cetin A. Leptin levels in thyroid cancer. Asian J Surg (2009) 32(4):216–23. doi:10.1016/S1015-9584(09)60397-3
68. Hedayati M, Yaghmaei P, Pooyamanesh Z, Zarif Yeganeh M, Hoghooghi Rad L. Leptin: a correlated peptide to papillary thyroid carcinoma? J Thyroid Res (2011) 2011:832163. doi:10.4061/2011/832163
69. Cheng SP, Chi CW, Tzen CY, Yang TL, Lee JJ, Liu TP, et al. Clinicopathologic significance of leptin and leptin receptor expressions in papillary thyroid carcinoma. Surgery (2010) 147(6):847–53. doi:10.1016/j.surg.2009.11.004
70. Lin SY, Huang SC, Sheu WH. Circulating adiponectin concentrations were related to free thyroxine levels in thyroid cancer patients after thyroid hormone withdrawal. Metabolism (2010) 59(2):195–9. doi:10.1016/j.metabol.2009.06.032
71. Park JT, Yoo TH, Chang TI, Lee DH, Lee JH, Lee JE, et al. Insulin resistance and lower plasma adiponectin increase malignancy risk in nondiabetic continuous ambulatory peritoneal dialysis patients. Metabolism (2011) 60(1):121–6. doi:10.1016/j.metabol.2010.02.006
72. Cheng SP, Liu CL, Hsu YC, Chang YC, Huang SY, Lee JJ. Expression and biologic significance of adiponectin receptors in papillary thyroid carcinoma. Cell Biochem Biophys (2013) 65(2):203–10. doi:10.1007/s12013-012-9419-1
73. Cham S, Zanocco K, Sturgeon C, Yeh MW, Harari A. Risk-based ultrasound screening for thyroid cancer in obese patients is cost-effective. Thyroid (2014) 24(6):975–86. doi:10.1089/thy.2013.0470
74. Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocr Rev (2001) 22(5):631–56. doi:10.1210/edrv.22.5.0444
75. Boelaert K, Horacek J, Holder RL, Watkinson JC, Sheppard MC, Franklyn JA. Serum thyrotropin concentration as a novel predictor of malignancy in thyroid nodules investigated by fine-needle aspiration. J Clin Endocrinol Metab (2006) 91(11):4295–301. doi:10.1210/jc.2006-0527
76. Haymart MR, Repplinger DJ, Leverson GE, Elson DF, Sippel RS, Jaume JC, et al. Higher serum thyroid stimulating hormone level in thyroid nodule patients is associated with greater risks of differentiated thyroid cancer and advanced tumor stage. J Clin Endocrinol Metab (2008) 93(3):809–14. doi:10.1210/jc.2007-2215
77. Fiore E, Rago T, Provenzale MA, Scutari M, Ugolini C, Basolo F, et al. Lower levels of TSH are associated with a lower risk of papillary thyroid cancer in patients with thyroid nodular disease: thyroid autonomy may play a protective role. Endocr Relat Cancer (2009) 16(4):1251–60. doi:10.1677/ERC-09-0036
78. Tramontano D, Cushing GW, Moses AC, Ingbar SH. Insulin-like growth factor-I stimulates the growth of rat thyroid cells in culture and synergizes the stimulation of DNA synthesis induced by TSH and Graves’-IgG. Endocrinology (1986) 119(2):940–2. doi:10.1210/endo-119-2-940
79. Kanamori A, Abe Y, Yajima Y, Manabe Y, Ito K. Epidermal growth factor receptors in plasma membranes of normal and diseased human thyroid glands. J Clin Endocrinol Metab (1989) 68(5):899–903. doi:10.1210/jcem-68-5-899
80. Haugen DR, Akslen LA, Varhaug JE, Lillehaug JR. Expression of c-erbB-2 protein in papillary thyroid carcinomas. Br J Cancer (1992) 65(6):832–7. doi:10.1038/bjc.1992.177
81. Belfiore A, Gangemi P, Costantino A, Russo G, Santonocito GM, Ippolito O, et al. Negative/low expression of the Met/hepatocyte growth factor receptor identifies papillary thyroid carcinomas with high risk of distant metastases. J Clin Endocrinol Metab (1997) 82(7):2322–8. doi:10.1210/jcem.82.7.4104
82. Eggo MC, Hopkins JM, Franklyn JA, Johnson GD, Sanders DS, Sheppard MC. Expression of fibroblast growth factors in thyroid cancer. J Clin Endocrinol Metab (1995) 80(3):1006–11. doi:10.1210/jcem.80.3.7533768
83. Milazzo G, La Rosa GL, Catalfamo R, Vigneri R, Belfiore A. Effect of TSH in human thyroid cells: evidence for both mitogenic and antimitogenic effects. J Cell Biochem (1992) 49(3):231–8. doi:10.1002/jcb.240490305
84. Marians RC, Ng L, Blair HC, Unger P, Graves PN, Davies TF. Defining thyrotropin-dependent and -independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proc Natl Acad Sci U S A (2002) 99(24):15776–81. doi:10.1073/pnas.242322099
85. Garcia-Jimenez C, Santisteban P. TSH signalling and cancer. Arq Bras Endocrinol Metabol (2007) 51(5):654–71. doi:10.1590/S0004-27302007000500003
86. De Felice M, Postiglione MP, Di Lauro R. Minireview: thyrotropin receptor signaling in development and differentiation of the thyroid gland: insights from mouse models and human diseases. Endocrinology (2004) 145(9):4062–7. doi:10.1210/en.2004-0501
87. Richards JS. New signaling pathways for hormones and cyclic adenosine 3’,5’-monophosphate action in endocrine cells. Mol Endocrinol (2001) 15(2):209–18. doi:10.1210/mend.15.2.0606
88. Frasca F, Vella V, Nicolosi ML, Messina RL, Giani F, Lotta S, et al. Thyroid cancer cell resistance to gefitinib depends on the constitutive oncogenic activation of the ERK pathway. J Clin Endocrinol Metab (2013) 98(6):2502–12. doi:10.1210/jc.2012-3623
89. Medina DL, Suzuki K, Pietrarelli M, Okajima F, Kohn LD, Santisteban P. Role of insulin and serum on thyrotropin regulation of thyroid transcription factor-1 and pax-8 genes expression in FRTL-5 thyroid cells. Thyroid (2000) 10(4):295–303. doi:10.1089/thy.2000.10.295
90. Malaguarnera R, Frasca F, Garozzo A, Giani F, Pandini G, Vella V, et al. Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J Clin Endocrinol Metab (2011) 96(3):766–74. doi:10.1210/jc.2010-1255
91. Dumont JE, Lamy F, Roger P, Maenhaut C. Physiological and pathological regulation of thyroid cell proliferation and differentiation by thyrotropin and other factors. Physiol Rev (1992) 72(3):667–97.
92. Riedemann J, Macaulay VM. IGF1R signalling and its inhibition. Endocr Relat Cancer (2006) 13(Suppl 1):S33–43. doi:10.1677/erc.1.01280
93. Mohan S, Baylink DJ, Pettis JL. Insulin-like growth factor (IGF)-binding proteins in serum – do they have additional roles besides modulating the endocrine IGF actions? J Clin Endocrinol Metab (1996) 81(11):3817–20. doi:10.1210/jcem.81.11.8923818
94. Santisteban P, Acebron A, Polycarpou-Schwarz M, Di Lauro R. Insulin and insulin-like growth factor I regulate a thyroid-specific nuclear protein that binds to the thyroglobulin promoter. Mol Endocrinol (1992) 6(8):1310–7. doi:10.1210/mend.6.8.1406708
95. Ortiz L, Zannini M, Di Lauro R, Santisteban P. Transcriptional control of the forkhead thyroid transcription factor TTF-2 by thyrotropin, insulin, and insulin-like growth factor I. J Biol Chem (1997) 272(37):23334–9. doi:10.1074/jbc.272.37.23334
96. Vella V, Sciacca L, Pandini G, Mineo R, Squatrito S, Vigneri R, et al. The IGF system in thyroid cancer: new concepts. Mol Pathol (2001) 54(3):121–4. doi:10.1136/mp.54.3.121
97. Vella V, Pandini G, Sciacca L, Mineo R, Vigneri R, Pezzino V, et al. A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J Clin Endocrinol Metab (2002) 87(1):245–54. doi:10.1210/jcem.87.1.8142
98. Condorelli G, Formisano P, Miele C, Beguinot F. Thyrotropin regulates autophosphorylation and kinase activity in both the insulin and the insulin-like growth factor-I receptors in FRTL5 cells. Endocrinology (1992) 130(3):1615–25. doi:10.1210/endo.130.3.1311244
99. Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol (1999) 19(5):3278–88. doi:10.1128/MCB.19.5.3278
100. Sciacca L, Costantino A, Pandini G, Mineo R, Frasca F, Scalia P, et al. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene (1999) 18(15):2471–9. doi:10.1038/sj.onc.1202600
101. Belfiore A, Malaguarnera R. Insulin receptor and cancer. Endocr Relat Cancer (2011) 18(4):R125–47. doi:10.1530/ERC-11-0074
102. Pandini G, Vigneri R, Costantino A, Frasca F, Ippolito A, Fujita-Yamaguchi Y, et al. Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: evidence for a second mechanism of IGF-I signaling. Clin Cancer Res (1999) 5(7):1935–44.
103. Papa V, Gliozzo B, Clark GM, McGuire WL, Moore D, Fujita-Yamaguchi Y, et al. Insulin-like growth factor-I receptors are overexpressed and predict a low risk in human breast cancer. Cancer Res (1993) 53(16):3736–40.
104. Koda M, Sulkowski S, Garofalo C, Kanczuga-Koda L, Sulkowska M, Surmacz E. Expression of the insulin-like growth factor-I receptor in primary breast cancer and lymph node metastases: correlations with estrogen receptors alpha and beta. Horm Metab Res (2003) 35(11–12):794–801. doi:10.1055/s-2004-814152
105. Malaguarnera R, Belfiore A. The insulin receptor: a new target for cancer therapy. Front Endocrinol (2011) 2:93. doi:10.3389/fendo.2011.00093
106. Chakravarty G, Santillan AA, Galer C, Adams HP, El-Naggar AK, Jasser SA, et al. Phosphorylated insulin like growth factor-I receptor expression and its clinico-pathological significance in histologic subtypes of human thyroid cancer. Exp Biol Med (Maywood) (2009) 234(4):372–86. doi:10.3181/0809-RM-284
107. Yan Y, Hu F, Wu W, Ma R, Huang H. Expression characteristics of proteins of IGF-1R, p-Akt, and survivin in papillary thyroid carcinoma patients with type 2 diabetes mellitus. Medicine (Baltimore) (2017) 96(12):e6393. doi:10.1097/MD.0000000000006393
108. Giani F, Vella V, Nicolosi ML, Fierabracci A, Lotta S, Malaguarnera R, et al. Thyrospheres from normal or malignant thyroid tissue have different biological, functional, and genetic features. J Clin Endocrinol Metab (2015) 100(9):E1168–78. doi:10.1210/JC.2014-4163
109. Biondi B. Thyroid and obesity: an intriguing relationship. J Clin Endocrinol Metab (2010) 95(8):3614–7. doi:10.1210/jc.2010-1245
110. Neves JS, Castro Oliveira S, Souteiro P, Pedro J, Magalhaes D, Guerreiro V, et al. Effect of weight loss after bariatric surgery on thyroid-stimulating hormone levels in patients with morbid obesity and normal thyroid function. Obes Surg (2017). doi:10.1007/s11695-017-2792-5
111. Iacobellis G, Ribaudo MC, Zappaterreno A, Iannucci CV, Leonetti F. Relationship of thyroid function with body mass index, leptin, insulin sensitivity and adiponectin in euthyroid obese women. Clin Endocrinol (Oxf) (2005) 62(4):487–91. doi:10.1111/j.1365-2265.2005.02247.x
112. Radetti G, Kleon W, Buzi F, Crivellaro C, Pappalardo L, di Iorgi N, et al. Thyroid function and structure are affected in childhood obesity. J Clin Endocrinol Metab (2008) 93(12):4749–54. doi:10.1210/jc.2008-0823
113. Sari R, Balci MK, Altunbas H, Karayalcin U. The effect of body weight and weight loss on thyroid volume and function in obese women. Clin Endocrinol (Oxf) (2003) 59(2):258–62. doi:10.1046/j.1365-2265.2003.01836.x
114. Reinehr T, Andler W. Thyroid hormones before and after weight loss in obesity. Arch Dis Child (2002) 87(4):320–3. doi:10.1136/adc.87.4.320
115. Reinehr T, Isa A, de Sousa G, Dieffenbach R, Andler W. Thyroid hormones and their relation to weight status. Horm Res (2008) 70(1):51–7. doi:10.1159/000129678
116. Michalaki MA, Vagenakis AG, Leonardou AS, Argentou MN, Habeos IG, Makri MG, et al. Thyroid function in humans with morbid obesity. Thyroid (2006) 16(1):73–8. doi:10.1089/thy.2006.16.73
117. Jayanthi R, Srinivasan AR, Hanifah M, Maran AL. Associations among insulin resistance, triacylglycerol/high density lipoprotein (TAG/HDL ratio) and thyroid hormone levels – a study on type 2 diabetes mellitus in obese and overweight subjects. Diabetes Metab Syndr (2016). doi:10.1016/j.dsx.2016.12.020
118. Nillni EA, Vaslet C, Harris M, Hollenberg A, Bjorbak C, Flier JS. Leptin regulates prothyrotropin-releasing hormone biosynthesis. Evidence for direct and indirect pathways. J Biol Chem (2000) 275(46):36124–33. doi:10.1074/jbc.M003549200
119. Burman KD, Smallridge RC, Osburne R, Dimond RC, Whorton NE, Kesler P, et al. Nature of suppressed TSH secretion during undernutrition: effect of fasting and refeeding on TSH responses to prolonged TRH infusions. Metabolism (1980) 29(1):46–52. doi:10.1016/0026-0495(80)90097-9
120. Harris M, Aschkenasi C, Elias CF, Chandrankunnel A, Nillni EA, Bjoorbaek C, et al. Transcriptional regulation of the thyrotropin-releasing hormone gene by leptin and melanocortin signaling. J Clin Invest (2001) 107(1):111–20. doi:10.1172/JCI10741
121. Ciavardelli D, Bellomo M, Crescimanno C, Vella V. Type 3 deiodinase: role in cancer growth, stemness, and metabolism. Front Endocrinol (2014) 5:215. doi:10.3389/fendo.2014.00215
122. Fontenelle LC, Feitosa MM, Severo JS, Freitas TE, Morais JB, Torres-Leal FL, et al. Thyroid function in human obesity: underlying mechanisms. Horm Metab Res (2016) 48(12):787–94. doi:10.1055/s-0042-121421
123. Mueller A, Schofl C, Dittrich R, Cupisti S, Oppelt PG, Schild RL, et al. Thyroid-stimulating hormone is associated with insulin resistance independently of body mass index and age in women with polycystic ovary syndrome. Hum Reprod (2009) 24(11):2924–30. doi:10.1093/humrep/dep285
124. Benetti-Pinto CL, Piccolo VB, Yela DA, Garmes H. Thyroid-stimulating hormone and insulin resistance: their association with polycystic ovary syndrome without overt hypothyroidism. Rev Bras Ginecol Obstet (2017) 39(5):224–8. doi:10.1055/s-0037-1601436
125. Trummer C, Schwetz V, Giuliani A, Obermayer-Pietsch B, Lerchbaum E. Impact of elevated thyroid-stimulating hormone levels in polycystic ovary syndrome. Gynecol Endocrinol (2015) 31(10):819–23. doi:10.3109/09513590.2015.1062864
126. Yin D, Ruan X, Tian X, Du J, Zhao Y, Cui Y, et al. The relationship between thyroid function and metabolic changes in Chinese women with polycystic ovary syndrome. Gynecol Endocrinol (2017) 33(4):332–5. doi:10.1080/09513590.2016.1273895
127. Sinha U, Sinharay K, Saha S, Longkumer TA, Baul SN, Pal SK. Thyroid disorders in polycystic ovarian syndrome subjects: a tertiary hospital based cross-sectional study from Eastern India. Indian J Endocrinol Metab (2013) 17(2):304–9. doi:10.4103/2230-8210.109714
128. Jayanthi R, Srinivasan AR, Gopal N, Ramaswamy R. Association of divalent cations and insulin resistance with thyroid hormones in patients with type 2 diabetes mellitus. Diabetes Metab Syndr (2017). doi:10.1016/j.dsx.2017.07.010
129. Wolide AD, Zawdie B, Alemayehu T, Tadesse S. Association between thyroid hormone parameters and dyslipidemia among type 2 diabetes mellitus patients: comparative cross-sectional study. Diabetes Metab Syndr (2016). doi:10.1016/j.dsx.2016.12.041
130. Taneichi H, Sasai T, Ohara M, Honma H, Nagasawa K, Takahashi T, et al. Higher serum free triiodothyronine levels within the normal range are associated with metabolic syndrome components in type 2 diabetic subjects with euthyroidism. Tohoku J Exp Med (2011) 224(3):173–8. doi:10.1620/tjem.224.173
131. Lai Y, Wang J, Jiang F, Wang B, Chen Y, Li M, et al. The relationship between serum thyrotropin and components of metabolic syndrome. Endocr J (2011) 58(1):23–30. doi:10.1507/endocrj.K10E-272
132. Roos A, Bakker SJ, Links TP, Gans RO, Wolffenbuttel BH. Thyroid function is associated with components of the metabolic syndrome in euthyroid subjects. J Clin Endocrinol Metab (2007) 92(2):491–6. doi:10.1210/jc.2006-1718
133. Sieminska L, Wojciechowska C, Walczak K, Borowski A, Marek B, Nowak M, et al. Associations between metabolic syndrome, serum thyrotropin, and thyroid antibodies status in postmenopausal women, and the role of interleukin-6. Endokrynol Pol (2015) 66(5):394–403. doi:10.5603/EP.2015.0049
134. Mehran L, Amouzegar A, Tohidi M, Moayedi M, Azizi F. Serum free thyroxine concentration is associated with metabolic syndrome in euthyroid subjects. Thyroid (2014) 24(11):1566–74. doi:10.1089/thy.2014.0103
135. Benetti-Pinto CL, Berini Piccolo VR, Garmes HM, Teatin Juliato CR. Subclinical hypothyroidism in young women with polycystic ovary syndrome: an analysis of clinical, hormonal, and metabolic parameters. Fertil Steril (2013) 99(2):588–92. doi:10.1016/j.fertnstert.2012.10.006
136. Lecube A, Zafon C, Gromaz A, Fort JM, Caubet E, Baena JA, et al. Iodine deficiency is higher in morbid obesity in comparison with late after bariatric surgery and non-obese women. Obes Surg (2015) 25(1):85–9. doi:10.1007/s11695-014-1313-z
137. Soriguer F, Valdes S, Morcillo S, Esteva I, Almaraz MC, de Adana MS, et al. Thyroid hormone levels predict the change in body weight: a prospective study. Eur J Clin Invest (2011) 41(11):1202–9. doi:10.1111/j.1365-2362.2011.02526.x
138. Eray E, Sari F, Ozdem S, Sari R. Relationship between thyroid volume and iodine, leptin, and adiponectin in obese women before and after weight loss. Med Princ Pract (2011) 20(1):43–6. doi:10.1159/000322075
139. Al-Attas OS, Al-Daghri NM, Alkharfy KM, Alokail MS, Al-Johani NJ, Abd-Alrahman SH, et al. Urinary iodine is associated with insulin resistance in subjects with diabetes mellitus type 2. Exp Clin Endocrinol Diabetes (2012) 120(10):618–22. doi:10.1055/s-0032-1323816
140. Rundle A, Hoepner L, Hassoun A, Oberfield S, Freyer G, Holmes D, et al. Association of childhood obesity with maternal exposure to ambient air polycyclic aromatic hydrocarbons during pregnancy. Am J Epidemiol (2012) 175(11):1163–72. doi:10.1093/aje/kwr455
141. Fierens S, Mairesse H, Heilier JF, De Burbure C, Focant JF, Eppe G, et al. Dioxin/polychlorinated biphenyl body burden, diabetes and endometriosis: findings in a population-based study in Belgium. Biomarkers (2003) 8(6):529–34. doi:10.1080/1354750032000158420
142. Lim JS, Lee DH, Jacobs DR Jr. Association of brominated flame retardants with diabetes and metabolic syndrome in the U.S. population, 2003-2004. Diabetes Care (2008) 31(9):1802–7. doi:10.2337/dc08-0850
143. Lee DH, Lee IK, Song K, Steffes M, Toscano W, Baker BA, et al. A strong dose-response relation between serum concentrations of persistent organic pollutants and diabetes: results from the National Health and Examination Survey 1999-2002. Diabetes Care (2006) 29(7):1638–44. doi:10.2337/dc06-0543
144. Wolf K, Popp A, Schneider A, Breitner S, Hampel R, Rathmann W, et al. Association between long-term exposure to air pollution and biomarkers related to insulin resistance, subclinical inflammation, and adipokines. Diabetes (2016) 65(11):3314–26. doi:10.2337/db15-1567
145. Henriksen GL, Ketchum NS, Michalek JE, Swaby JA. Serum dioxin and diabetes mellitus in veterans of Operation Ranch Hand. Epidemiology (1997) 8(3):252–8. doi:10.1097/00001648-199705000-00005
146. Lee DH, Lind PM, Jacobs DR Jr, Salihovic S, van Bavel B, Lind L. Polychlorinated biphenyls and organochlorine pesticides in plasma predict development of type 2 diabetes in the elderly: the prospective investigation of the vasculature in uppsala seniors (PIVUS) study. Diabetes Care (2011) 34(8):1778–84. doi:10.2337/dc10-2116
147. Kramer U, Herder C, Sugiri D, Strassburger K, Schikowski T, Ranft U, et al. Traffic-related air pollution and incident type 2 diabetes: results from the SALIA cohort study. Environ Health Perspect (2010) 118(9):1273–9. doi:10.1289/ehp.0901689
148. Weinmayr G, Hennig F, Fuks K, Nonnemacher M, Jakobs H, Mohlenkamp S, et al. Long-term exposure to fine particulate matter and incidence of type 2 diabetes mellitus in a cohort study: effects of total and traffic-specific air pollution. Environ Health (2015) 14:53. doi:10.1186/s12940-015-0031-x
149. Jerrett M, Brook R, White LF, Burnett RT, Yu J, Su J, et al. Ambient ozone and incident diabetes: a prospective analysis in a large cohort of African American women. Environ Int (2017) 102:42–7. doi:10.1016/j.envint.2016.12.011
150. Coogan PF, White LF, Jerrett M, Brook RD, Su JG, Seto E, et al. Air pollution and incidence of hypertension and diabetes mellitus in black women living in Los Angeles. Circulation (2012) 125(6):767–72. doi:10.1161/CIRCULATIONAHA.111.052753
151. Lang IA, Galloway TS, Scarlett A, Henley WE, Depledge M, Wallace RB, et al. Association of urinary bisphenol A concentration with medical disorders and laboratory abnormalities in adults. JAMA (2008) 300(11):1303–10. doi:10.1001/jama.300.11.1303
152. Lee DH, Lee IK, Jin SH, Steffes M, Jacobs DR Jr. Association between serum concentrations of persistent organic pollutants and insulin resistance among nondiabetic adults: results from the National Health and Nutrition Examination Survey 1999-2002. Diabetes Care (2007) 30(3):622–8. doi:10.2337/dc06-2190
153. Lee DH, Lee IK, Porta M, Steffes M, Jacobs DR Jr. Relationship between serum concentrations of persistent organic pollutants and the prevalence of metabolic syndrome among non-diabetic adults: results from the National Health and Nutrition Examination Survey 1999-2002. Diabetologia (2007) 50(9):1841–51. doi:10.1007/s00125-007-0755-4
154. Kandaraki E, Chatzigeorgiou A, Livadas S, Palioura E, Economou F, Koutsilieris M, et al. Endocrine disruptors and polycystic ovary syndrome (PCOS): elevated serum levels of bisphenol A in women with PCOS. J Clin Endocrinol Metab (2011) 96(3):E480–4. doi:10.1210/jc.2010-1658
155. Akin L, Kendirci M, Narin F, Kurtoglu S, Saraymen R, Kondolot M, et al. The endocrine disruptor bisphenol A may play a role in the aetiopathogenesis of polycystic ovary syndrome in adolescent girls. Acta Paediatr (2015) 104(4):e171–7. doi:10.1111/apa.12885
156. Tarantino G, Valentino R, Di Somma C, D’Esposito V, Passaretti F, Pizza G, et al. Bisphenol A in polycystic ovary syndrome and its association with liver-spleen axis. Clin Endocrinol (Oxf) (2013) 78(3):447–53. doi:10.1111/j.1365-2265.2012.04500.x
157. Rajkhowa M, Bicknell J, Jones M, Clayton RN. Insulin sensitivity in women with polycystic ovary syndrome: relationship to hyperandrogenemia. Fertil Steril (1994) 61(4):605–12. doi:10.1016/S0015-0282(16)56633-3
158. Takeuchi T, Tsutsumi O. Serum bisphenol A concentrations showed gender differences, possibly linked to androgen levels. Biochem Biophys Res Commun (2002) 291(1):76–8. doi:10.1006/bbrc.2002.6407
159. Takeuchi T, Tsutsumi O, Ikezuki Y, Takai Y, Taketani Y. Positive relationship between androgen and the endocrine disruptor, bisphenol A, in normal women and women with ovarian dysfunction. Endocr J (2004) 51(2):165–9. doi:10.1507/endocrj.51.165
160. Miao M, Yuan W, Yang F, Liang H, Zhou Z, Li R, et al. Associations between bisphenol A exposure and reproductive hormones among female workers. Int J Environ Res Public Health (2015) 12(10):13240–50. doi:10.3390/ijerph121013240
161. Rotondi M, Leporati P, La Manna A, Pirali B, Mondello T, Fonte R, et al. Raised serum TSH levels in patients with morbid obesity: is it enough to diagnose subclinical hypothyroidism? Eur J Endocrinol (2009) 160(3):403–8. doi:10.1530/EJE-08-0734
162. Akbar DH, Ahmed MM, Al-Mughales J. Thyroid dysfunction and thyroid autoimmunity in Saudi type 2 diabetics. Acta Diabetol (2006) 43(1):14–8. doi:10.1007/s00592-006-0204-8
163. Yasmin T, Ghafoor F, Malik T, Ruhy Naz S, Khan AU. Pattern of thyroid autoimmunity in type 1 and type 2 diabetics. J Coll Physicians Surg Pak (2006) 16(12):751–4. doi:12.2006/JCPSP.751754
164. Toulis K, Tsekmekidou X, Potolidis E, Didangelos T, Gotzamani-Psarrakou A, Zebekakis P, et al. Thyroid autoimmunity in the context of type 2 diabetes mellitus: implications for vitamin D. Int J Endocrinol (2015) 2015:710363. doi:10.1155/2015/710363
165. Sarfo-Kantanka O, Sarfo FS, Ansah EO, Yorke E, Akpalu J, Nkum BC, et al. Frequency and determinants of thyroid autoimmunity in Ghanaian type 2 diabetes patients: a case-control study. BMC Endocr Disord (2017) 17(1):2. doi:10.1186/s12902-016-0152-4
166. Al-Saab R, Haddad S. Detection of thyroid autoimmunity markers in euthyroid women with polycystic ovary syndrome: a case-control study from Syria. Int J Endocrinol Metab (2014) 12(3):e17954. doi:10.5812/ijem.17954
167. Janssen OE, Mehlmauer N, Hahn S, Offner AH, Gartner R. High prevalence of autoimmune thyroiditis in patients with polycystic ovary syndrome. Eur J Endocrinol (2004) 150(3):363–9. doi:10.1530/eje.0.1500363
168. Menon M, Ramachandran V. Antithyroid peroxidase antibodies in women with polycystic ovary syndrome. J Obstet Gynaecol India (2017) 67(1):61–5. doi:10.1007/s13224-016-0914-y
169. Garelli S, Masiero S, Plebani M, Chen S, Furmaniak J, Armanini D, et al. High prevalence of chronic thyroiditis in patients with polycystic ovary syndrome. Eur J Obstet Gynecol Reprod Biol (2013) 169(2):248–51. doi:10.1016/j.ejogrb.2013.03.003
170. Kachuei M, Jafari F, Kachuei A, Keshteli AH. Prevalence of autoimmune thyroiditis in patients with polycystic ovary syndrome. Arch Gynecol Obstet (2012) 285(3):853–6. doi:10.1007/s00404-011-2040-5
171. Novais Jde S, Benetti-Pinto CL, Garmes HM, Jales RM, Juliato CR. Polycystic ovary syndrome and chronic autoimmune thyroiditis. Gynecol Endocrinol (2015) 31(1):48–51. doi:10.3109/09513590.2014.958990
172. Arduc A, Aycicek Dogan B, Bilmez S, Imga Nasiroglu N, Tuna MM, Isik S, et al. High prevalence of Hashimoto’s thyroiditis in patients with polycystic ovary syndrome: does the imbalance between estradiol and progesterone play a role? Endocr Res (2015) 40(4):204–10. doi:10.3109/07435800.2015.1015730
173. Ganie MA, Marwaha RK, Aggarwal R, Singh S. High prevalence of polycystic ovary syndrome characteristics in girls with euthyroid chronic lymphocytic thyroiditis: a case-control study. Eur J Endocrinol (2010) 162(6):1117–22. doi:10.1530/EJE-09-1012
174. Wang K, Yang Y, Wu Y, Chen J, Zhang D, Mao X, et al. The association between insulin resistance and vascularization of thyroid nodules. J Clin Endocrinol Metab (2015) 100(1):184–92. doi:10.1210/jc.2014-2723
175. Gallagher EJ, LeRoith D. Obesity and diabetes: the increased risk of cancer and cancer-related mortality. Physiol Rev (2015) 95(3):727–48. doi:10.1152/physrev.00030.2014
176. Ben-Shmuel S, Rostoker R, Scheinman EJ, LeRoith D. Metabolic syndrome, type 2 diabetes, and cancer: epidemiology and potential mechanisms. Handb Exp Pharmacol (2016) 233:355–72. doi:10.1007/164_2015_12
177. Rostoker R, Abelson S, Bitton-Worms K, Genkin I, Ben-Shmuel S, Dakwar M, et al. Highly specific role of the insulin receptor in breast cancer progression. Endocr Relat Cancer (2015) 22(2):145–57. doi:10.1530/ERC-14-0490
178. Zelenko Z, Gallagher EJ, Antoniou IM, Sachdev D, Nayak A, Yee D, et al. EMT reversal in human cancer cells after IR knockdown in hyperinsulinemic mice. Endocr Relat Cancer (2016) 23(9):747–58. doi:10.1530/ERC-16-0142
179. Gallagher EJ, Alikhani N, Tobin-Hess A, Blank J, Buffin NJ, Zelenko Z, et al. Insulin receptor phosphorylation by endogenous insulin or the insulin analog AspB10 promotes mammary tumor growth independent of the IGF-I receptor. Diabetes (2013) 62(10):3553–60. doi:10.2337/db13-0249
180. Malaguarnera R, Nicolosi ML, Sacco A, Morcavallo A, Vella V, Voci C, et al. Novel cross talk between IGF-IR and DDR1 regulates IGF-IR trafficking, signaling and biological responses. Oncotarget (2015) 6(18):16084–105. doi:10.18632/oncotarget.3177
181. Vella V, Malaguarnera R, Nicolosi ML, Palladino C, Spoleti C, Massimino M, et al. Discoidin domain receptor 1 modulates insulin receptor signaling and biological responses in breast cancer cells. Oncotarget (2017) 8(26):43248–70. doi:10.18632/oncotarget.18020
182. De Marco P, Romeo E, Vivacqua A, Malaguarnera R, Abonante S, Romeo F, et al. GPER1 is regulated by insulin in cancer cells and cancer-associated fibroblasts. Endocr Relat Cancer (2014) 21(5):739–53. doi:10.1530/ERC-14-0245
183. Mata R, Palladino C, Nicolosi ML, Lo Presti AR, Malaguarnera R, Ragusa M, et al. IGF-I induces upregulation of DDR1 collagen receptor in breast cancer cells by suppressing MIR-199a-5p through the PI3K/AKT pathway. Oncotarget (2016) 7(7):7683–700. doi:10.18632/oncotarget.6524
184. Miele C, Rochford JJ, Filippa N, Giorgetti-Peraldi S, Van Obberghen E. Insulin and insulin-like growth factor-I induce vascular endothelial growth factor mRNA expression via different signaling pathways. J Biol Chem (2000) 275(28):21695–702. doi:10.1074/jbc.M000805200
185. Rose DP, Vona-Davis L. The cellular and molecular mechanisms by which insulin influences breast cancer risk and progression. Endocr Relat Cancer (2012) 19(6):R225–41. doi:10.1530/ERC-12-0203
186. Zimmermann MB, Galetti V. Iodine intake as a risk factor for thyroid cancer: a comprehensive review of animal and human studies. Thyroid Res (2015) 8:8. doi:10.1186/s13044-015-0020-8
187. Zimmermann MB, Boelaert K. Iodine deficiency and thyroid disorders. Lancet Diabetes Endocrinol (2015) 3(4):286–95. doi:10.1016/S2213-8587(14)70225-6
188. Lind P, Langsteger W, Molnar M, Gallowitsch HJ, Mikosch P, Gomez I. Epidemiology of thyroid diseases in iodine sufficiency. Thyroid (1998) 8(12):1179–83. doi:10.1089/thy.1998.8.1179
189. Besic N, Hocevar M, Zgajnar J. Lower incidence of anaplastic carcinoma after higher iodination of salt in Slovenia. Thyroid (2010) 20(6):623–6. doi:10.1089/thy.2009.0404
190. Pellegriti G, Frasca F, Regalbuto C, Squatrito S, Vigneri R. Worldwide increasing incidence of thyroid cancer: update on epidemiology and risk factors. J Cancer Epidemiol (2013) 2013:965212. doi:10.1155/2013/965212
191. Krohn K, Maier J, Paschke R. Mechanisms of disease: hydrogen peroxide, DNA damage and mutagenesis in the development of thyroid tumors. Nat Clin Pract Endocrinol Metab (2007) 3(10):713–20. doi:10.1038/ncpendmet0621
192. Gerard AC, Humblet K, Wilvers C, Poncin S, Derradji H, de Ville de Goyet C, et al. Iodine-deficiency-induced long lasting angiogenic reaction in thyroid cancers occurs via a vascular endothelial growth factor-hypoxia inducible factor-1-dependent, but not a reactive oxygen species-dependent, pathway. Thyroid (2012) 22(7):699–708. doi:10.1089/thy.2011.0387
193. Craps J, Joris V, De Jongh B, Sonveaux P, Horman S, Lengele B, et al. Involvement of mTOR and regulation by AMPK in early iodine deficiency-induced thyroid microvascular activation. Endocrinology (2016) 157(6):2545–59. doi:10.1210/en.2015-1911
194. Skrypnyk NV. [Correlation between adipocytokines with iodine deficiency criteria in patients with metabolic syndrome and type 2 diabetes mellitus in Carpathians region]. Lik Sprava (2010) (1–2):70–7.
195. Mansourian AR. A review on the metabolic disorders of iodine deficiency. Pak J Biol Sci (2011) 14(7):412–24. doi:10.3923/pjbs.2011.412.424
196. Boelaert K, Syed AA, Manji N, Sheppard MC, Holder RL, Gough SC, et al. Prediction of cure and risk of hypothyroidism in patients receiving 131I for hyperthyroidism. Clin Endocrinol (Oxf) (2009) 70(1):129–38. doi:10.1111/j.1365-2265.2008.03291.x
197. Pellegriti G, De Vathaire F, Scollo C, Attard M, Giordano C, Arena S, et al. Papillary thyroid cancer incidence in the volcanic area of Sicily. J Natl Cancer Inst (2009) 101(22):1575–83. doi:10.1093/jnci/djp354
198. Arnbjornsson E, Arnbjornsson A, Olafsson A. Thyroid cancer incidence in relation to volcanic activity. Arch Environ Health (1986) 41(1):36–40. doi:10.1080/00039896.1986.9935763
199. Alonso-Magdalena P, Quesada I, Nadal A. Endocrine disruptors in the etiology of type 2 diabetes mellitus. Nat Rev Endocrinol (2011) 7(6):346–53. doi:10.1038/nrendo.2011.56
200. Swedenborg E, Ruegg J, Makela S, Pongratz I. Endocrine disruptive chemicals: mechanisms of action and involvement in metabolic disorders. J Mol Endocrinol (2009) 43(1):1–10. doi:10.1677/JME-08-0132
201. Casals-Casas C, Desvergne B. Endocrine disruptors: from endocrine to metabolic disruption. Annu Rev Physiol (2011) 73:135–62. doi:10.1146/annurev-physiol-012110-142200
202. Holmes D. Endocrine disruptors: air pollution linked to insulin resistance. Nat Rev Endocrinol (2016) 12(12):688. doi:10.1038/nrendo.2016.168
203. Lanzino M, Morelli C, Garofalo C, Panno ML, Mauro L, Ando S, et al. Interaction between estrogen receptor alpha and insulin/IGF signaling in breast cancer. Curr Cancer Drug Targets (2008) 8(7):597–610. doi:10.2174/156800908786241104
204. Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, et al. Endocrine-disrupting chemicals: an endocrine society scientific statement. Endocr Rev (2009) 30(4):293–342. doi:10.1210/er.2009-0002
205. Henderson BE, Ross RK, Pike MC, Casagrande JT. Endogenous hormones as a major factor in human cancer. Cancer Res (1982) 42(8):3232–9.
206. Derwahl M, Nicula D. Estrogen and its role in thyroid cancer. Endocr Relat Cancer (2014) 21(5):T273–83. doi:10.1530/ERC-14-0053
207. Huff MO, Todd SL, Smith AL, Elpers JT, Smith AP, Murphy RD, et al. Arsenite and cadmium activate MAPK/ERK via membrane estrogen receptors and G-protein coupled estrogen receptor signaling in human lung adenocarcinoma cells. Toxicol Sci (2016) 152(1):62–71. doi:10.1093/toxsci/kfw064
208. Liu Z, Yu X, Shaikh ZA. Rapid activation of ERK1/2 and AKT in human breast cancer cells by cadmium. Toxicol Appl Pharmacol (2008) 228(3):286–94. doi:10.1016/j.taap.2007.12.017
209. Di Donato M, Cernera G, Giovannelli P, Galasso G, Bilancio A, Migliaccio A, et al. Recent advances on bisphenol-A and endocrine disruptor effects on human prostate cancer. Mol Cell Endocrinol (2017) 457:35–42. doi:10.1016/j.mce.2017.02.045
210. Vinas R, Watson CS. Bisphenol S disrupts estradiol-induced nongenomic signaling in a rat pituitary cell line: effects on cell functions. Environ Health Perspect (2013) 121(3):352–8. doi:10.1289/ehp.1205826
211. Genua M, Pandini G, Sisci D, Castoria G, Maggiolini M, Vigneri R, et al. Role of cyclic AMP response element-binding protein in insulin-like growth factor-I receptor up-regulation by sex steroids in prostate cancer cells. Cancer Res (2009) 69(18):7270–7. doi:10.1158/0008-5472.CAN-09-0088
212. Feldt-Rasmussen U, Rasmussen AK. Autoimmunity in differentiated thyroid cancer: significance and related clinical problems. Hormones (Athens) (2010) 9(2):109–17. doi:10.14310/horm.2002.1261
213. Singh B, Shaha AR, Trivedi H, Carew JF, Poluri A, Shah JP. Coexistent Hashimoto’s thyroiditis with papillary thyroid carcinoma: impact on presentation, management, and outcome. Surgery (1999) 126(6):1070–6; discussion 1076–7. doi:10.1067/msy.2099.101431
214. Jankovic B, Le KT, Hershman JM. Clinical review: Hashimoto’s thyroiditis and papillary thyroid carcinoma: is there a correlation? J Clin Endocrinol Metab (2013) 98(2):474–82. doi:10.1210/jc.2012-2978
215. Anand A, Singh KR, Kushwaha JK, Hussain N, Sonkar AA. Papillary thyroid cancer and Hashimoto’s thyroiditis: an association less understood. Indian J Surg Oncol (2014) 5(3):199–204. doi:10.1007/s13193-014-0325-4
216. Ehlers M, Schott M. Hashimoto’s thyroiditis and papillary thyroid cancer: are they immunologically linked? Trends Endocrinol Metab (2014) 25(12):656–64. doi:10.1016/j.tem.2014.09.001
217. Marzullo P, Minocci A, Tagliaferri MA, Guzzaloni G, Di Blasio A, De Medici C, et al. Investigations of thyroid hormones and antibodies in obesity: leptin levels are associated with thyroid autoimmunity independent of bioanthropometric, hormonal, and weight-related determinants. J Clin Endocrinol Metab (2010) 95(8):3965–72. doi:10.1210/jc.2009-2798
218. Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R. Diabetes and cancer. Endocr Relat Cancer (2009) 16(4):1103–23. doi:10.1677/ERC-09-0087
219. Polyzos SA, Kita M, Efstathiadou Z, Poulakos P, Slavakis A, Sofianou D, et al. Serum thyrotropin concentration as a biochemical predictor of thyroid malignancy in patients presenting with thyroid nodules. J Cancer Res Clin Oncol (2008) 134(9):953–60. doi:10.1007/s00432-008-0373-7
220. Junik R, Kozinski M, Debska-Kozinska K. Thyroid ultrasound in diabetic patients without overt thyroid disease. Acta Radiol (2006) 47(7):687–91. doi:10.1080/02841850600806308
221. Pimenta WP, Mazeto GM, Callegaro CF, Shibata SA, Marins LV, Yamashita S, et al. [Thyroid disorders in diabetic patients]. Arq Bras Endocrinol Metabol (2005) 49(2):234–40. doi:10.1590/S0004-27302005000200009
222. Duran AO, Anil C, Gursoy A, Nar A, Inanc M, Bozkurt O, et al. Thyroid volume in patients with glucose metabolism disorders. Arq Bras Endocrinol Metabol (2014) 58(8):824–7. doi:10.1590/0004-2730000003418
223. Blanc E, Ponce C, Brodschi D, Nepote A, Barreto A, Schnitman M, et al. Association between worse metabolic control and increased thyroid volume and nodular disease in elderly adults with metabolic syndrome. Metab Syndr Relat Disord (2015) 13(5):221–6. doi:10.1089/met.2014.0158
224. Anil C, Akkurt A, Ayturk S, Kut A, Gursoy A. Impaired glucose metabolism is a risk factor for increased thyroid volume and nodule prevalence in a mild-to-moderate iodine deficient area. Metabolism (2013) 62(7):970–5. doi:10.1016/j.metabol.2013.01.009
225. Ahluwalia IB, Mack KA, Murphy W, Mokdad AH, Bales VS. State-specific prevalence of selected chronic disease-related characteristics – behavioral risk factor surveillance system, 2001. MMWR Surveill Summ (2003) 52(8):1–80.
226. Johnson JA, Bowker SL, Richardson K, Marra CA. Time-varying incidence of cancer after the onset of type 2 diabetes: evidence of potential detection bias. Diabetologia (2011) 54(9):2263–71. doi:10.1007/s00125-011-2242-1
227. Adami HO, McLaughlin J, Ekbom A, Berne C, Silverman D, Hacker D, et al. Cancer risk in patients with diabetes mellitus. Cancer Causes Control (1991) 2(5):307–14. doi:10.1007/BF00051670
228. Hemminki K, Li X, Sundquist J, Sundquist K. Risk of cancer following hospitalization for type 2 diabetes. Oncologist (2010) 15(6):548–55. doi:10.1634/theoncologist.2009-0300
229. Stocks T, Borena W, Strohmaier S, Bjorge T, Manjer J, Engeland A, et al. Cohort profile: the metabolic syndrome and cancer project (Me-Can). Int J Epidemiol (2010) 39(3):660–7. doi:10.1093/ije/dyp186
230. La Vecchia C, Negri E, Franceschi S, D’Avanzo B, Boyle P. A case-control study of diabetes mellitus and cancer risk. Br J Cancer (1994) 70(5):950–3. doi:10.1038/bjc.1994.427
231. Zhan YS, Feng L, Tang SH, Li WG, Xu M, Liu TF, et al. Glucose metabolism disorders in cancer patients in a Chinese population. Med Oncol (2010) 27(2):177–84. doi:10.1007/s12032-009-9189-9
232. Kuriki K, Hirose K, Tajima K. Diabetes and cancer risk for all and specific sites among Japanese men and women. Eur J Cancer Prev (2007) 16(1):83–9. doi:10.1097/01.cej.0000228404.37858.40
233. Inoue M, Iwasaki M, Otani T, Sasazuki S, Noda M, Tsugane S. Diabetes mellitus and the risk of cancer: results from a large-scale population-based cohort study in Japan. Arch Intern Med (2006) 166(17):1871–7. doi:10.1001/archinte.166.17.1871
234. Luo J, Phillips L, Liu S, Wactawski-Wende J, Margolis KL. Diabetes, diabetes treatment, and risk of thyroid cancer. J Clin Endocrinol Metab (2016) 101(3):1243–8. doi:10.1210/jc.2015-3901
235. Hsu IR, Kim SP, Kabir M, Bergman RN. Metabolic syndrome, hyperinsulinemia, and cancer. Am J Clin Nutr (2007) 86(3):s867–71.
236. Celani MF, Bonati ME, Stucci N. Prevalence of abnormal thyrotropin concentrations measured by a sensitive assay in patients with type 2 diabetes mellitus. Diabetes Res (1994) 27(1):15–25.
237. Dang CV, Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci (1999) 24(2):68–72. doi:10.1016/S0968-0004(98)01344-9
238. Cowey S, Hardy RW. The metabolic syndrome: a high-risk state for cancer? Am J Pathol (2006) 169(5):1505–22. doi:10.2353/ajpath.2006.051090
239. Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, et al. Diabetes and cancer: a consensus report. Diabetes Care (2010) 33(7):1674–85. doi:10.2337/dc10-0666
240. But A, De Bruin ML, Bazelier MT, Hjellvik V, Andersen M, Auvinen A, et al. Cancer risk among insulin users: comparing analogues with human insulin in the CARING five-country cohort study. Diabetologia (2017). doi:10.1007/s00125-017-4312-5
241. Tseng CH. Treatment with human insulin does not increase thyroid cancer risk in patients with type 2 diabetes. Eur J Clin Invest (2014) 44(8):736–42. doi:10.1111/eci.12290
242. Suissa S, Azoulay L, Dell’Aniello S, Evans M, Vora J, Pollak M. Long-term effects of insulin glargine on the risk of breast cancer. Diabetologia (2011) 54(9):2254–62. doi:10.1007/s00125-011-2190-9
243. Ruiter R, Visser LE, van Herk-Sukel MP, Coebergh JW, Haak HR, Geelhoed-Duijvestijn PH, et al. Risk of cancer in patients on insulin glargine and other insulin analogues in comparison with those on human insulin: results from a large population-based follow-up study. Diabetologia (2012) 55(1):51–62. doi:10.1007/s00125-011-2312-4
244. Peeters PJ, Bazelier MT, Leufkens HG, Auvinen A, van Staa TP, de Vries F, et al. Insulin glargine use and breast cancer risk: associations with cumulative exposure. Acta Oncol (2016) 55(7):851–8. doi:10.3109/0284186X.2016.1155736
245. Smith U, Gale EA. Does diabetes therapy influence the risk of cancer? Diabetologia (2009) 52(9):1699–708. doi:10.1007/s00125-009-1441-5
246. Mannucci E, Monami M, Balzi D, Cresci B, Pala L, Melani C, et al. Doses of insulin and its analogues and cancer occurrence in insulin-treated type 2 diabetic patients. Diabetes Care (2010) 33(9):1997–2003. doi:10.2337/dc10-0476
247. Currie CJ. The longest ever randomised controlled trial of insulin glargine: study design and HbA(1c) findings. Diabetologia (2009) 52(10):2234–5; author reply 2236–9. doi:10.1007/s00125-009-1477-6
248. Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care (2006) 29(2):254–8. doi:10.2337/dc06-0997
249. Yang X, Ko GT, So WY, Ma RC, Yu LW, Kong AP, et al. Associations of hyperglycemia and insulin usage with the risk of cancer in type 2 diabetes: the Hong Kong diabetes registry. Diabetes (2010) 59(5):1254–60. doi:10.2337/db09-1371
250. Chang CH, Lin JW, Wu LC, Lai MS, Chuang LM. Oral insulin secretagogues, insulin, and cancer risk in type 2 diabetes mellitus. J Clin Endocrinol Metab (2012) 97(7):E1170–5. doi:10.1210/jc.2012-1162
251. Simo R, Plana-Ripoll O, Puente D, Morros R, Mundet X, Vilca LM, et al. Impact of glucose-lowering agents on the risk of cancer in type 2 diabetic patients. The Barcelona case-control study. PLoS One (2013) 8(11):e79968. doi:10.1371/journal.pone.0079968
252. Lin CM, Huang HL, Chu FY, Fan HC, Chen HA, Chu DM, et al. Association between gastroenterological malignancy and diabetes mellitus and anti-diabetic therapy: a nationwide, population-based cohort study. PLoS One (2015) 10(5):e0125421. doi:10.1371/journal.pone.0125421
253. Chiu CC, Huang CC, Chen YC, Chen TJ, Liang Y, Lin SJ, et al. Increased risk of gastrointestinal malignancy in patients with diabetes mellitus and correlations with anti-diabetes drugs: a nationwide population-based study in Taiwan. Intern Med (2013) 52(9):939–46. doi:10.2169/internalmedicine.52.8276
254. Filippatos TD, Liberopoulos EN, Elisaf MS. Dapagliflozin in patients with type 2 diabetes mellitus. Ther Adv Endocrinol Metab (2015) 6(1):29–41. doi:10.1177/2042018814558243
255. Knudsen LB. Liraglutide: the therapeutic promise from animal models. Int J Clin Pract Suppl (2010) 167:4–11. doi:10.1111/j.1742-1241.2010.02499.x
256. Sakul H, Pratley R, Cardon L, Ravussin E, Mott D, Bogardus C. Familiality of physical and metabolic characteristics that predict the development of non-insulin-dependent diabetes mellitus in Pima Indians. Am J Hum Genet (1997) 60(3):651–6.
257. Elbein SC, Hasstedt SJ, Wegner K, Kahn SE. Heritability of pancreatic beta-cell function among nondiabetic members of Caucasian familial type 2 diabetic kindreds. J Clin Endocrinol Metab (1999) 84(4):1398–403. doi:10.1210/jcem.84.4.5604
258. Watanabe RM, Valle T, Hauser ER, Ghosh S, Eriksson J, Kohtamaki K, et al. Familiality of quantitative metabolic traits in Finnish families with non-insulin-dependent diabetes mellitus. Finland-United States Investigation of NIDDM Genetics (FUSION) Study investigators. Hum Hered (1999) 49(3):159–68. doi:10.1159/000022865
259. Lehtovirta M, Kaprio J, Forsblom C, Eriksson J, Tuomilehto J, Groop L. Insulin sensitivity and insulin secretion in monozygotic and dizygotic twins. Diabetologia (2000) 43(3):285–93. doi:10.1007/s001250050046
260. Lin HF, Boden-Albala B, Juo SH, Park N, Rundek T, Sacco RL. Heritabilities of the metabolic syndrome and its components in the Northern Manhattan Family Study. Diabetologia (2005) 48(10):2006–12. doi:10.1007/s00125-005-1892-2
261. Bellia A, Giardina E, Lauro D, Tesauro M, Di Fede G, Cusumano G, et al. “The Linosa Study”: epidemiological and heritability data of the metabolic syndrome in a Caucasian genetic isolate. Nutr Metab Cardiovasc Dis (2009) 19(7):455–61. doi:10.1016/j.numecd.2008.11.002
262. Carmelli D, Cardon LR, Fabsitz R. Clustering of hypertension, diabetes, and obesity in adult male twins: same genes or same environments? Am J Hum Genet (1994) 55(3):566–73.
263. Hardy OT, Czech MP, Corvera S. What causes the insulin resistance underlying obesity? Curr Opin Endocrinol Diabetes Obes (2012) 19(2):81–7. doi:10.1097/MED.0b013e3283514e13
264. Booth FW, Roberts CK, Laye MJ. Lack of exercise is a major cause of chronic diseases. Compr Physiol (2012) 2(2):1143–211. doi:10.1002/cphy.c110025
265. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature (2006) 444(7121):840–6. doi:10.1038/nature05482
266. Califf R. Insulin resistance: a global epidemic in need of effective therapies. Eur Heart J Suppl (2003) 5(Suppl C):C13–8. doi:10.1016/S1520-765X(03)90045-9
267. Darnton-Hill I, Nishida C, James WP. A life course approach to diet, nutrition and the prevention of chronic diseases. Public Health Nutr (2004) 7(1A):101–21. doi:10.1079/PHN2003584
268. Stockley L. Toward public health nutrition strategies in the European Union to implement food based dietary guidelines and to enhance healthier lifestyles. Public Health Nutr (2001) 4(2A):307–24.
269. Trubswasser U, Branca F. Nutrition policy is taking shape in Europe. Public Health Nutr (2009) 12(3):295–306. doi:10.1017/S1368980009004753
270. Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med (2002) 346(6):393–403. doi:10.1056/NEJMoa012512
271. Wadden TA, Webb VL, Moran CH, Bailer BA. Lifestyle modification for obesity: new developments in diet, physical activity, and behavior therapy. Circulation (2012) 125(9):1157–70. doi:10.1161/CIRCULATIONAHA.111.039453
272. McLaughlin T, Abbasi F, Kim HS, Lamendola C, Schaaf P, Reaven G. Relationship between insulin resistance, weight loss, and coronary heart disease risk in healthy, obese women. Metabolism (2001) 50(7):795–800. doi:10.1053/meta.2001.24210
273. Andersson M, Karumbunathan V, Zimmermann MB. Global iodine status in 2011 and trends over the past decade. J Nutr (2012) 142(4):744–50. doi:10.3945/jn.111.149393
274. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia (2017) 60(9):1577–85. doi:10.1007/s00125-017-4342-z
275. Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila) (2010) 3(11):1451–61. doi:10.1158/1940-6207.CAPR-10-0157
276. Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS One (2012) 7(3):e33411. doi:10.1371/journal.pone.0033411
277. Heckman-Stoddard BM, Gandini S, Puntoni M, Dunn BK, DeCensi A, Szabo E. Repurposing old drugs to chemoprevention: the case of metformin. Semin Oncol (2016) 43(1):123–33. doi:10.1053/j.seminoncol.2015.09.009
278. Tseng CH. Metformin reduces thyroid cancer risk in Taiwanese patients with type 2 diabetes. PLoS One (2014) 9(10):e109852. doi:10.1371/journal.pone.0109852
279. Becker C, Jick SS, Meier CR, Bodmer M. No evidence for a decreased risk of thyroid cancer in association with use of metformin or other antidiabetic drugs: a case-control study. BMC Cancer (2015) 15:719. doi:10.1186/s12885-015-1719-6
280. Rezzonico J, Rezzonico M, Pusiol E, Pitoia F, Niepomniszcze H. Metformin treatment for small benign thyroid nodules in patients with insulin resistance. Metab Syndr Relat Disord (2011) 9(1):69–75. doi:10.1089/met.2010.0026
281. Karimifar M, Aminorroaya A, Amini M, Mirfendereski T, Iraj B, Feizi A, et al. Effect of metformin on thyroid stimulating hormone and thyroid volume in patients with prediabetes: a randomized placebo-controlled clinical trial. J Res Med Sci (2014) 19(11):1019–26.
282. Anil C, Kut A, Atesagaoglu B, Nar A, Bascil Tutuncu N, Gursoy A. Metformin decreases thyroid volume and nodule size in subjects with insulin resistance: a preliminary study. Med Princ Pract (2016) 25(3):233–6. doi:10.1159/000442821
283. Meng X, Xu S, Chen G, Derwahl M, Liu C. Metformin and thyroid disease. J Endocrinol (2017) 233(1):R43–51. doi:10.1530/JOE-16-0450
284. Ben Sahra I, Le Marchand-Brustel Y, Tanti JF, Bost F. Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Mol Cancer Ther (2010) 9(5):1092–9. doi:10.1158/1535-7163.MCT-09-1186
285. Vigersky RA, Filmore-Nassar A, Glass AR. Thyrotropin suppression by metformin. J Clin Endocrinol Metab (2006) 91(1):225–7. doi:10.1210/jc.2005-1210
286. Fournier JP, Yin H, Yu OH, Azoulay L. Metformin and low levels of thyroid-stimulating hormone in patients with type 2 diabetes mellitus. CMAJ (2014) 186(15):1138–45. doi:10.1503/cmaj.140688
287. Pandini G, Mineo R, Frasca F, Roberts CT Jr, Marcelli M, Vigneri R, et al. Androgens up-regulate the insulin-like growth factor-I receptor in prostate cancer cells. Cancer Res (2005) 65(5):1849–57. doi:10.1158/0008-5472.CAN-04-1837
288. Malaguarnera R, Sacco A, Morcavallo A, Squatrito S, Migliaccio A, Morrione A, et al. Metformin inhibits androgen-induced IGF-IR up-regulation in prostate cancer cells by disrupting membrane-initiated androgen signaling. Endocrinology (2014) 155(4):1207–21. doi:10.1210/en.2013-1925
289. Lei Y, Yi Y, Liu Y, Liu X, Keller ET, Qian CN, et al. Metformin targets multiple signaling pathways in cancer. Chin J Cancer (2017) 36(1):17. doi:10.1186/s40880-017-0184-9
290. Sosnicki S, Kapral M, Weglarz L. Molecular targets of metformin antitumor action. Pharmacol Rep (2016) 68(5):918–25. doi:10.1016/j.pharep.2016.04.021
291. Dowling RJ, Goodwin PJ, Stambolic V. Understanding the benefit of metformin use in cancer treatment. BMC Med (2011) 9:33. doi:10.1186/1741-7015-9-33
292. El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem (2000) 275(1):223–8. doi:10.1074/jbc.275.1.223
293. Malki A, Youssef A. Antidiabetic drug metformin induces apoptosis in human MCF breast cancer via targeting ERK signaling. Oncol Res (2011) 19(6):275–85. doi:10.3727/096504011X13021877989838
294. Kim HG, Hien TT, Han EH, Hwang YP, Choi JH, Kang KW, et al. Metformin inhibits P-glycoprotein expression via the NF-kappaB pathway and CRE transcriptional activity through AMPK activation. Br J Pharmacol (2011) 162(5):1096–108. doi:10.1111/j.1476-5381.2010.01101.x
295. Hwang YP, Jeong HG. Metformin blocks migration and invasion of tumour cells by inhibition of matrix metalloproteinase-9 activation through a calcium and protein kinase Calpha-dependent pathway: phorbol-12-myristate-13-acetate-induced/extracellular signal-regulated kinase/activator protein-1. Br J Pharmacol (2010) 160(5):1195–211. doi:10.1111/j.1476-5381.2010.00762.x
296. Kumar N, Dey CS. Metformin enhances insulin signalling in insulin-dependent and -independent pathways in insulin resistant muscle cells. Br J Pharmacol (2002) 137(3):329–36. doi:10.1038/sj.bjp.0704878
297. He L, Sabet A, Djedjos S, Miller R, Sun X, Hussain MA, et al. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell (2009) 137(4):635–46. doi:10.1016/j.cell.2009.03.016
298. Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, et al. TORCs: transducers of regulated CREB activity. Mol Cell (2003) 12(2):413–23. doi:10.1016/j.molcel.2003.08.013
299. Screaton RA, Conkright MD, Katoh Y, Best JL, Canettieri G, Jeffries S, et al. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell (2004) 119(1):61–74. doi:10.1016/j.cell.2004.09.015
300. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J (2000) 348(Pt 3):607–14. doi:10.1042/0264-6021:3480607
301. Wilcock C, Bailey CJ. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica (1994) 24(1):49–57. doi:10.3109/00498259409043220
302. Carvalho C, Correia S, Santos MS, Seica R, Oliveira CR, Moreira PI. Metformin promotes isolated rat liver mitochondria impairment. Mol Cell Biochem (2008) 308(1–2):75–83. doi:10.1007/s11010-007-9614-3
303. Andrzejewski S, Gravel SP, Pollak M, St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab (2014) 2:12. doi:10.1186/2049-3002-2-12
304. Chen G, Xu S, Renko K, Derwahl M. Metformin inhibits growth of thyroid carcinoma cells, suppresses self-renewal of derived cancer stem cells, and potentiates the effect of chemotherapeutic agents. J Clin Endocrinol Metab (2012) 97(4):E510–20. doi:10.1210/jc.2011-1754
305. Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res (2011) 71(9):3196–201. doi:10.1158/0008-5472.CAN-10-3471
306. Rocha GZ, Dias MM, Ropelle ER, Osorio-Costa F, Rossato FA, Vercesi AE, et al. Metformin amplifies chemotherapy-induced AMPK activation and antitumoral growth. Clin Cancer Res (2011) 17(12):3993–4005. doi:10.1158/1078-0432.CCR-10-2243
307. Petrushev B, Tomuleasa C, Soritau O, Aldea M, Pop T, Susman S, et al. Metformin plus PIAF combination chemotherapy for hepatocellular carcinoma. Exp Oncol (2012) 34(1):17–24.
308. Han B, Cui H, Kang L, Zhang X, Jin Z, Lu L, et al. Metformin inhibits thyroid cancer cell growth, migration, and EMT through the mTOR pathway. Tumour Biol (2015) 36(8):6295–304. doi:10.1007/s13277-015-3315-4
309. Klubo-Gwiezdzinska J, Costello J Jr, Patel A, Bauer A, Jensen K, Mete M, et al. Treatment with metformin is associated with higher remission rate in diabetic patients with thyroid cancer. J Clin Endocrinol Metab (2013) 98(8):3269–79. doi:10.1210/jc.2012-3799
310. Jang EK, Kim WG, Kwon H, Choi YM, Jeon MJ, Kim TY, et al. Metformin is associated with a favorable outcome in diabetic patients with cervical lymph node metastasis of differentiated thyroid cancer. Eur Thyroid J (2015) 4(3):181–8. doi:10.1159/000437365
311. Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med (2006) 355(23):2427–43. doi:10.1056/NEJMoa066224
312. Juurinen L, Kotronen A, Graner M, Yki-Jarvinen H. Rosiglitazone reduces liver fat and insulin requirements and improves hepatic insulin sensitivity and glycemic control in patients with type 2 diabetes requiring high insulin doses. J Clin Endocrinol Metab (2008) 93(1):118–24. doi:10.1210/jc.2007-1825
313. Phielix E, Szendroedi J, Roden M. The role of metformin and thiazolidinediones in the regulation of hepatic glucose metabolism and its clinical impact. Trends Pharmacol Sci (2011) 32(10):607–16. doi:10.1016/j.tips.2011.06.006
314. Tiikkainen M, Hakkinen AM, Korsheninnikova E, Nyman T, Makimattila S, Yki-Jarvinen H. Effects of rosiglitazone and metformin on liver fat content, hepatic insulin resistance, insulin clearance, and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes (2004) 53(8):2169–76. doi:10.2337/diabetes.53.8.2169
315. Hevener AL, Reichart D, Janez A, Olefsky J. Thiazolidinedione treatment prevents free fatty acid-induced insulin resistance in male wistar rats. Diabetes (2001) 50(10):2316–22. doi:10.2337/diabetes.50.10.2316
316. Shibuya A, Watanabe M, Fujita Y, Saigenji K, Kuwao S, Takahashi H, et al. An autopsy case of troglitazone-induced fulminant hepatitis. Diabetes Care (1998) 21(12):2140–3. doi:10.2337/diacare.21.12.2140
317. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med (2007) 356(24):2457–71. doi:10.1056/NEJMoa072761
318. Tseng CH. Rosiglitazone may reduce thyroid cancer risk in patients with type 2 diabetes. Ann Med (2013) 45(8):539–44. doi:10.3109/07853890.2013.851865
319. Vella V, Nicolosi ML, Giuliano S, Bellomo M, Belfiore A, Malaguarnera R. PPAR-gamma agonists as antineoplastic agents in cancers with dysregulated IGF axis. Front Endocrinol (2017) 8:31. doi:10.3389/fendo.2017.00031
320. Tseng CH. Pioglitazone and thyroid cancer risk in Taiwanese patients with type 2 diabetes 2. J Diabetes (2014) 6(5):448–50. doi:10.1111/1753-0407.12149
321. Frasca F, Pandini G, Vigneri R, Goldfine ID. Insulin and hybrid insulin/IGF receptors are major regulators of breast cancer cells. Breast Dis (2003) 17:73–89. doi:10.3233/BD-2003-17108
322. Buck E, Gokhale PC, Koujak S, Brown E, Eyzaguirre A, Tao N, et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol Cancer Ther (2010) 9(10):2652–64. doi:10.1158/1535-7163.MCT-10-0318
323. Belfiore A, Malaguarnera R, Vella V, Lawrence MC, Sciacca L, Frasca F, et al. Insulin receptor isoforms in physiology and disease: an updated view. Endocr Rev (2017) 38(5):379–431. doi:10.1210/er.2017-00073
324. Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature (2012) 486(7403):405–9. doi:10.1038/nature11154
325. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell (2012) 150(6):1107–20. doi:10.1016/j.cell.2012.08.029
326. Scott LM, Rebel VI. Acquired mutations that affect pre-mRNA splicing in hematologic malignancies and solid tumors. J Natl Cancer Inst (2013) 105(20):1540–9. doi:10.1093/jnci/djt257
327. Bonnal S, Vigevani L, Valcarcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov (2012) 11(11):847–59. doi:10.1038/nrd3823
328. Kara I, Poggi M, Bonardo B, Govers R, Landrier JF, Tian S, et al. The paired basic amino acid-cleaving enzyme 4 (PACE4) is involved in the maturation of insulin receptor isoform B: an opportunity to reduce the specific insulin receptor-dependent effects of insulin-like growth factor 2 (IGF2). J Biol Chem (2015) 290(5):2812–21. doi:10.1074/jbc.M114.592543
329. Roebroek AJ, Taylor NA, Louagie E, Pauli I, Smeijers L, Snellinx A, et al. Limited redundancy of the proprotein convertase furin in mouse liver. J Biol Chem (2004) 279(51):53442–50. doi:10.1074/jbc.M407152200
330. Zhu J, Van de Ven WJ, Verbiest T, Koeckelberghs G, Chen C, Cui Y, et al. Polyphenols can inhibit furin in vitro as a result of the reactivity of their auto-oxidation products to proteins. Curr Med Chem (2013) 20(6):840–50. doi:10.2174/092986713805076702
331. Vivacqua A, De Marco P, Belfiore A, Maggiolini M. Recent advances on the role of microRNAs in both insulin resistance and cancer. Curr Pharm Des (2017) 23(25):3658–66. doi:10.2174/1381612823666170622105123
332. Scagliotti GV, Novello S. The role of the insulin-like growth factor signaling pathway in non-small cell lung cancer and other solid tumors. Cancer Treat Rev (2012) 38(4):292–302. doi:10.1016/j.ctrv.2011.07.008
333. Prince SN, Foulstone EJ, Zaccheo OJ, Williams C, Hassan AB. Functional evaluation of novel soluble insulin-like growth factor (IGF)-II-specific ligand traps based on modified domain 11 of the human IGF2 receptor. Mol Cancer Ther (2007) 6(2):607–17. doi:10.1158/1535-7163.MCT-06-0509
334. Frago S, Nicholls RD, Strickland M, Hughes J, Williams C, Garner L, et al. Functional evolution of IGF2:IGF2R domain 11 binding generates novel structural interactions and a specific IGF2 antagonist. Proc Natl Acad Sci U S A (2016) 113(20):E2766–75. doi:10.1073/pnas.1513023113
335. Wang N, Rayes RF, Elahi SM, Lu Y, Hancock MA, Massie B, et al. The IGF-Trap: novel inhibitor of carcinoma growth and metastasis. Mol Cancer Ther (2015) 14(4):982–93. doi:10.1158/1535-7163.MCT-14-0751
336. Scheen AJ. SGLT2 inhibitors: benefit/risk balance. Curr Diab Rep (2016) 16(10):92. doi:10.1007/s11892-016-0789-4
337. Avogaro A, Giaccari A, Fioretto P, Genovese S, Purrello F, Giorgino F, et al. A consensus statement for the clinical use of the renal sodium-glucose co-transporter-2 inhibitor dapagliflozin in patients with type 2 diabetes mellitus. Expert Rev Clin Pharmacol (2017) 10(7):763–72. doi:10.1080/17512433.2017.1322507
338. Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, et al. 2015 American Thyroid Association Management Guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: the American Thyroid Association Guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid (2016) 26(1):1–133. doi:10.1089/thy.2015.0020
Keywords: insulin resistance, insulin, thyroid cancer, obesity, type 2 diabetes, insulin growth factor, metformin, insulin sensitizers
Citation: Malaguarnera R, Vella V, Nicolosi ML and Belfiore A (2017) Insulin Resistance: Any Role in the Changing Epidemiology of Thyroid Cancer? Front. Endocrinol. 8:314. doi: 10.3389/fendo.2017.00314
Received: 04 September 2017; Accepted: 30 October 2017;
Published: 14 November 2017
Edited by:
Haim Werner, Tel Aviv University, IsraelReviewed by:
Pierre De Meyts, de Duve Institute, BelgiumEddy Karnieli, Technion Israel Institute of Technology, Israel
Copyright: © 2017 Malaguarnera, Vella, Nicolosi and Belfiore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Veronica Vella, dmVyb25pY2EudmVsbGFAdW5pa29yZS5pdA==;
Antonino Belfiore, YmVsZmlvcmVAdW5pY3ouaXQ=
†These authors have equally contributed to this work.