
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 06 November 2017
Sec. Systems Endocrinology
Volume 8 - 2017 | https://doi.org/10.3389/fendo.2017.00294
 Jakob Triebel1*
Jakob Triebel1* Christin J. Friedrich1
Christin J. Friedrich1 Andreas Leuchs1
Andreas Leuchs1 Gonzalo Martínez de la Escalera2
Gonzalo Martínez de la Escalera2 Carmen Clapp2
Carmen Clapp2 Thomas Bertsch1
Thomas Bertsch1
Background: A dysregulation of the generation of vasoinhibin hormones by proteolytic cleavage of prolactin (PRL) has been brought into context with diabetic retinopathy, retinopathy of prematurity, preeclampsia, pregnancy-induced hypertension, and peripartum cardiomyopathy. Factors governing vasoinhibin generation are incompletely characterized, and the composition of vasoinhibin isoforms in human tissues or compartments, such as the circulation, is unknown. The aim of this study was to determine the possible contribution of PRL point mutations to the generation of vasoinhibins as well as to project their role in vasoinhibin-related diseases.
Methods: Prolactin sequences, point mutations, and substrate specificity information about the PRL cleaving enzymes cathepsin D, matrix metalloproteinases 8 and 13, and bone-morphogenetic protein 1 were retrieved from public databases. The consequences of point mutations in regard to their possible effect on vasoinhibin levels were projected on the basis of a score indicating the suitability of a particular sequence for enzymatic cleavage that result in vasoinhibin generation. The relative abundance and type of vasoinhibin isoforms were estimated by comparing the relative cleavage efficiency of vasoinhibin-generating enzymes.
Results: Six point mutations leading to amino acid substitutions in vasoinhibin-generating cleavage sites were found and projected to either facilitate or inhibit vasoinhibin generation. Four mutations affecting vasoinhibin generation in cancer tissues were found. The most likely composition of the relative abundance of vasoinhibin isoforms is projected to be 15 > 17.2 > 16.8 > 17.7 > 18 kDa vasoinhibin.
Conclusion: Prolactin point mutations are likely to influence vasoinhibin levels by affecting the proteolysis efficiency of vasoinhibin-generating enzymes and should be monitored in patients with vasoinhibin-related diseases. Attempts to characterize vasoinhibin-related diseases should include the 15, 17.2, 16.8, 17.7, and 18 kDa vasoinhibin isoforms.
The regulation of blood vessel growth, pressure, permeability, and dilation is essential for all vertebrate species and is in part executed by hormones such as vasoinhibins (1–3). Apart from their vascular effects, vasoinhibins exert non-vascular actions, which include the stimulation of vasopressin release (4), proinflammatory actions (5) thrombolytic effects (6), and the stimulation of anxiety- and depression-related behaviors (7). Vasoinhibins are generated by the proteolytic cleavage of their precursor molecule, the pituitary hormone prolactin (PRL). This cleavage takes place in various anatomical compartments and tissues, such as the pituitary gland (8), the retina (9), the heart (10), and the cartilage (11). As the cleaving enzymes, matrix metalloproteinases (MMP), cathepsin D, and bone-morphogenetic protein 1 (BMP-1) utilize multiple cleavage sites along the PRL amino acid sequence, the molecular mass of the vasoinhibin isoforms varies from 11 to 18 kDa (11–13). The regulation of vasoinhibins occurs at the hypothalamic, pituitary, and peripheral level demonstrating typical features of an endocrine axis. Consequently, the organizational principle of the regulation of vasoinhibins has been described as the PRL/vasoinhibin axis (14).
Dysregulation of the PRL/vasoinhibin axis has been brought into context with diabetic retinopathy, retinopathy of prematurity (9, 15, 16), preeclampsia, and pregnancy-induced hypertension (10, 17–21). In regard to the PRL/vasoinhibin axis, the shared hallmark of these diseases is the dysregulation of vasoinhibin levels, that means they may either be too low to execute a physiological function—such as the suppression of angiogenesis in the eye—or too high, which may lead to detrimental effects on the microvasculature of the heart, as proposed for the etiopathology of peripartum cardiomyopathy (PPCM). The interplay between two factors appears to be decisive for such dysregulation: the availability of the substrate, that is the level of pituitary PRL secretion, and the activity of the PRL-cleaving enzyme(s), cathepsin D, MMP, and BMP-1. Here, it is hypothesized that a third factor may be of relevance for the generation of vasoinhibins: the PRL amino acid sequence, or, more precisely, point mutations in the PRL gene which translate into amino acid substitutions within the PRL protein sequence, with the effect of enhancing or inhibiting enzymatic cleavage and, thereby, altering the levels and type of the generated vasoinhibins.
Vasoinhibins have been detected in the circulation and in other biological fluids and tissue extracts, but the techniques used for their detection are not quantitative, nor specific enough to provide unambiguous identification of their respective isoform(s). In consequence, the concentration and type of vasoinhibin isoforms are unknown (22). This is a major obstacle limiting the characterization of vasoinhibins’ emerging roles in the abovementioned diseases (23). Besides microfluidics- and mass-spectrometry-based assays (15, 24), the only established technique to measure vasoinhibins is immunoprecipitation with anti-PRL antibodies followed by SDS-PAGE and Western blotting. Quantitative immunoassays usually employ monoclonal antibodies, but the development of anti-vasoinhibin monoclonal antibodies able to discriminate between PRL and vasoinhibins has been delayed due to uncertainties about which isoform should be targeted—a problem aggravated by the observation that the type of vasoinhibin isoform appears to vary in physiological and pathological conditions. Here, we used a bioinformatic approach to project how the enzymatic cleavage-efficiency generating vasoinhibins is affected by known PRL point mutations. Furthermore, we have estimated the relative vasoinhibin isoform composition expected on the basis of the efficacy of their generation process.
Data concerning the suitability of cleavage site amino acid sequence for proteolytic cleavage by cathepsin D, MMP, and BMP-1 with subsequent generation of vasoinhibins were retrieved from databases, and a projection for single amino acid substitutions was performed. For this purpose, human PRL gene transcripts (PRL-001, transcript ID: ENST00000306482.1, PRL 202, ID: ENST00000617911.4, PRL 201, ID: ENST00000615510.4) and amino acid sequences were retrieved from ENSEMBL1 (25), as were missense variants, with detected point mutations leading to amino acid substitutions within the PRL sequence. The amino acid sequences of the three transcripts were aligned using MEGA, version 7 (clustal W alignment) (26). Mutations of the PRL gene in cancer were identified in ENSEMBL and details were retrieved from COSMIC (Catalogue of Somatic Mutations in Cancer2) (27). Furthermore, the MEROPS database of proteolytic enzymes (28), their substrates, and inhibitors3 was interrogated to obtain information about the cleavage site specificity of cathepsin D (MEROPS ID: A01.009), MMP-8 (M10.002), MMP-13 (M10.013) and BMP-1 (M12.005). The following five vasoinhibin isoforms, for which specific cleavage site information are available, were selected for this analysis: the 16.8, 17.2, 17.7, 18, and 15 kDa vasoinhibin isoform. Each cleavage site generating vasoinhibins was defined by the 8 amino acids neighboring the cleavage site (P1–P4, and P1’–P4’) meaning four residues toward the N-terminus and four toward the C-terminus of the uncleaved PRL sequence. The specificity matrix for each enzyme was retrieved and entries of numbers of cleavages for each amino acid at a particular position of the cleavage site were added up to build an “8P”-score—a surrogate parameter for the “suitability” of a given sequence for cleavage by the respective enzyme. Missense variants were matched with the cleavage sites of five vasoinhibin isoforms and the effect of the amino acid substitutions on the 8P-score was projected according to the MEROPS specificity matrix of the respective enzyme. An example of how the 8P-score was generated for the ancestral cleavage sites, including the effect of an amino acid substitution, is presented in Figure 1.
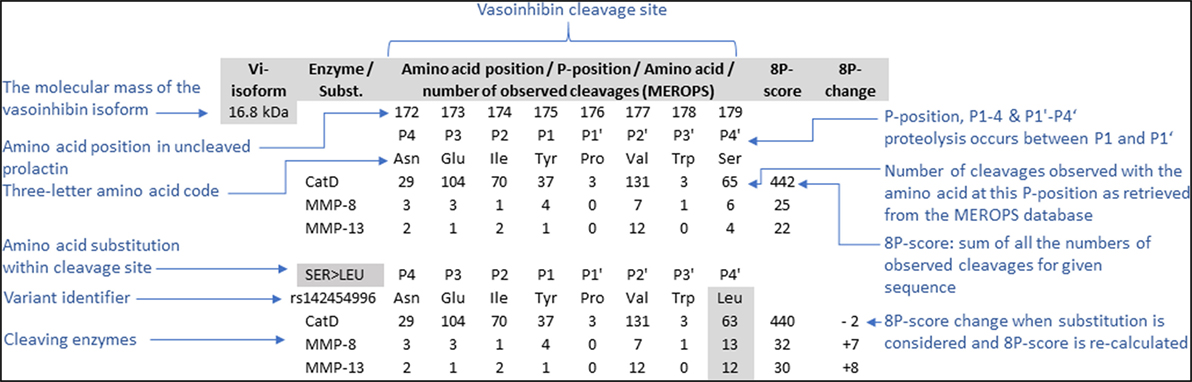
Figure 1. To project the consequences of single amino acid substitutions in vasoinhibin-generating cleavage sites of the prolactin amino acid sequence, an 8P-score was used. The score consists of the sum of the numbers of observed cleavages by the enzyme indicated with a particular amino acid on each of the eight positions (P1’–P4’, P1–P4), retrieved from the MEROPS database (28). The cleavage occurs between positions P1’ and P1. The effect of amino acid substitutions was projected by recalculation of the score after replacement of the amino acid and adjusting the corresponding number of observed cleavages.
Six point mutations leading to a single amino acid substitution in cleavage sites affecting four vasoinhibin isoforms were found (Table 1). A SER/LEU substitution at P4’ of the cleavage site of the 16.8 kDa vasoinhibin isoform results in a slightly higher 8P-score for MMP-8 (+7 units) and MMP-13 (+8), whereas cleavage by cathepsin D is slightly reduced (−2). The TRP/CYS substitution at P3’ leads to a higher score for cathepsin D (+8), whereas the score for MMP-8 and MMP-13 demonstrates only minor and no change, respectively (+1; 0). Greater changes are observed due to the same substitution (TRP/CYS) and the SER/LEU substitution in the cleavage site of the 17.2 kDa vasoinhibin isoform. The TRP/CYS substitution leads to a reduction of the 8P-score by 21 units and the SER/LEU substitution to an increase of +69 units, both for cleavage by cathepsin D. A SER/ALA substitution in the cleavage site of the 17.7 kDa vasoinhibin isoform leads to slightly higher scores for MMP-8 (+2) and MMP-13 (+2). The MET/VAL substitution at P3 in the cleavage site for the 15 kDa vasoinhibin isoform leads to a higher score (+68) for cathepsin D. Four COSMIC point mutations causing amino acid substitutions in cleavage sites affecting the generation of three vasoinhibin isoforms were found and analyzed in the same manner (Table 2).
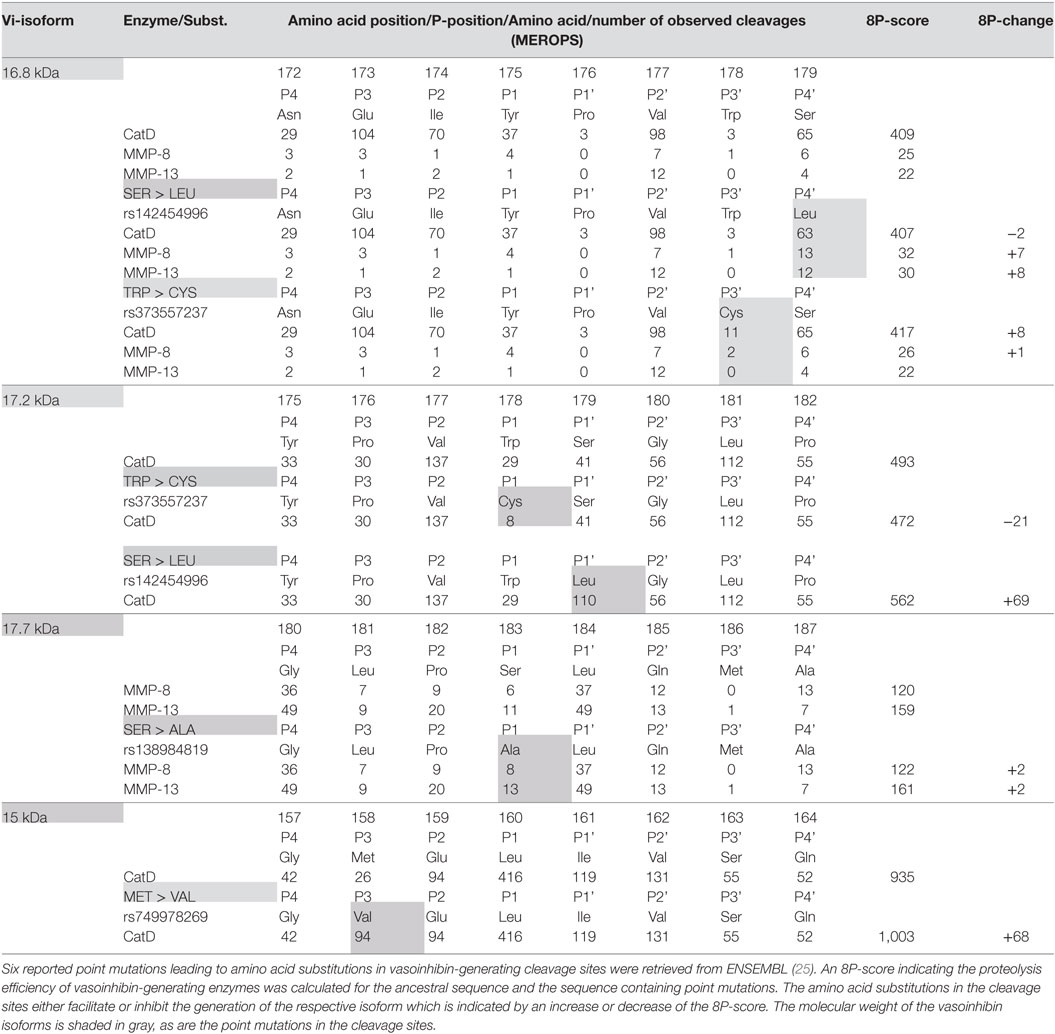
Table 1. Effect of single amino acid substitutions on vasoinhibin isoforms generation.
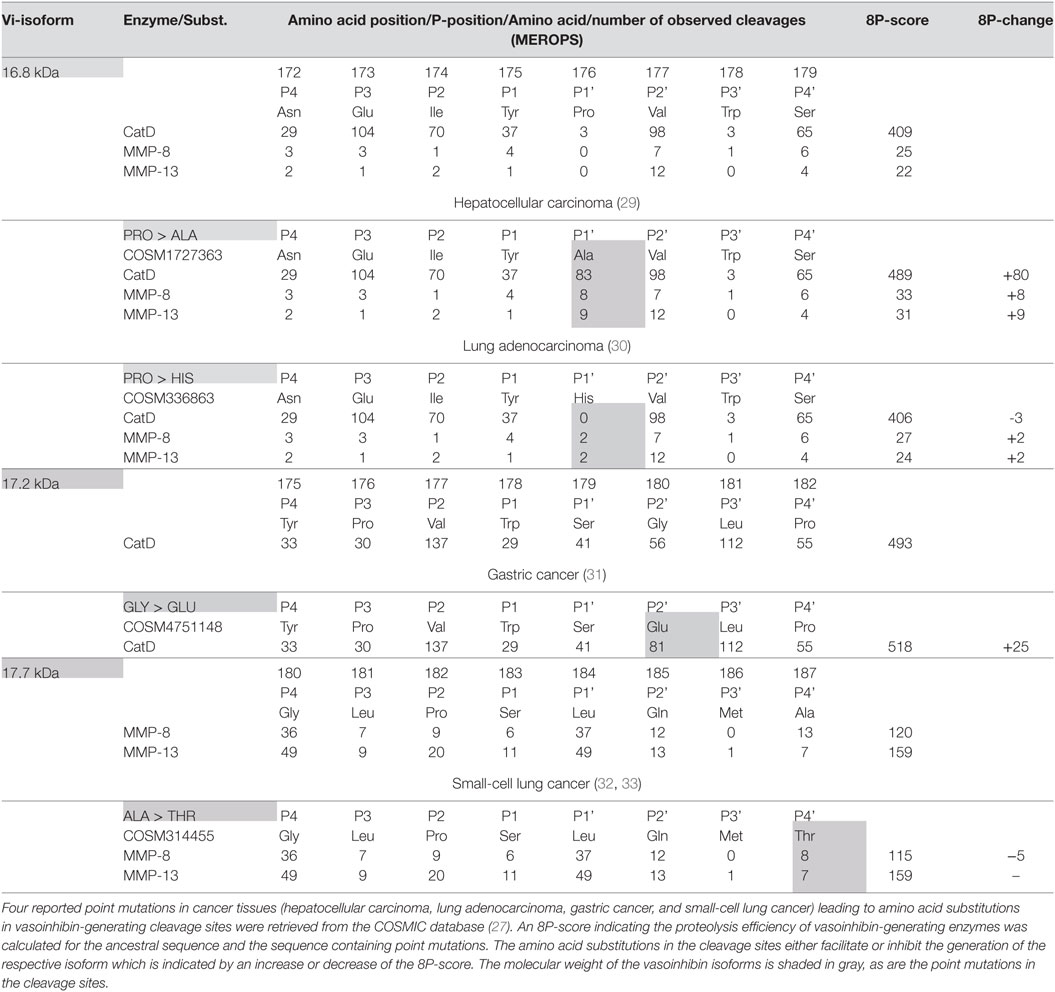
Table 2. Effect of single amino acid substitutions observed in cancer on vasoinhibin isoforms generation.
A projection of the relative composition of vasoinhibin isoforms was performed, showing the relative proteolysis efficiency of four PRL-cleaving enzymes and the corresponding vasoinhibin isoform generated, respectively (Figure 2). From this projection, it is most likely that the most abundant isoform is the 15 kDa vasoinhibin, followed by the 17.2 kDa, and the 16.8 kDa isoform, all of which are generated by cathepsin D, and MMP-8 and MMP-13 in case of the 16.8 kDa isoform. The 17.7 and 18 kDa vasoinhibin isoforms demonstrate much lower scores and are, thus, likely to be less abundant (Figure 1). Of note, the general order of composition 15 > 17.2 > 16.8 > 17.7 > 18 kDa isoform is maintained in all scenarios considering all mutations tested, although with slight variation.
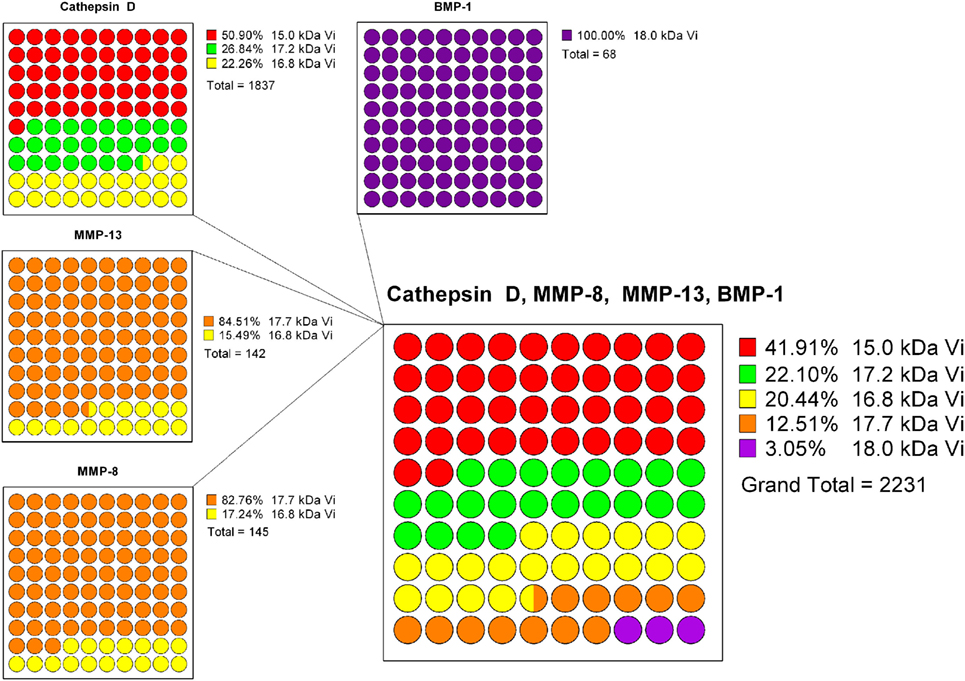
Figure 2. To estimate the composition of vasoinhibin isoforms, a projection based on the known prolactin-cleavage sites of vasoinhibin-generating enzymes (Cathepsin D, MMP-8/13, and BMP-1) and their calculated cleavage efficiency (8P-score) was performed. Cathepsin D generates the 15, the 17.2, and the 16.8 kDa vasoinhibin isoform, with the highest cleavage efficiency at the site generating the 15 kDa isoform. The 8P-scores of each Cathepsin D cleavage site were added and represented in parts of a whole graph. The same analysis was performed for MMP-13, MMP-8, and BMP-1. Finally, the percent representation of the cleavage efficiency of all enzymes and vasoinhibin isoforms was combined. It appears that, on the basis of the cleavage efficiency of vasoinhibin-generating enzymes, the 15 kDa vasoinhibin isoform would be most abundant (41.91%), followed by the 17.2 kDa (22.10%), the 16.8 kDa (20.44%), the 17.7 kDa (12.51%), and the 18 kDa (3.05%) isoform.
The present analysis demonstrates that besides substrate (PRL) availability and enzyme activity, point mutations in the human PRL gene leading to amino acid substitutions in cleavage sites may determine vasoinhibin levels. The amino acid substitutions appear to influence enzymatic cleavage of PRL, as some amino acids facilitate the cleavage, whereas others reduce the cleavage efficiency. This is a relevant finding, as factors and controlling mechanisms governing the generation of vasoinhibins are incompletely understood.
The present analysis is based on the enzyme-substrate specificity data from the MEROPS database of proteolytic enzymes, in which data from experimental observations are listed. Hence, the projection of the 8P-score, as performed in the present analysis, is based on experimental evidence. However, it remains a projection and is as such theoretical, especially since the 8P-score is a compound score summarizing the effects observed on 8 amino acid positions, which may not necessarily be representative and complex interactions arising from the complete cleavage site sequence, which are independent from the calculated score, may be missed. Also, since the 8P-score is based on the linear sequence of amino acids, effects arising from the three-dimensional structure of the protein in solution, such as effects of the organizational degree of the amino acids at the cleavage site or their various degrees of solvent exposure, are not being considered. Hence, these projections should be tested under experimental conditions evaluating the proteolysis of PRL with and without point mutations at cleavage site regions. Piwnica et al. (12) reported some substitutions and their effects on vasoinhibin generation. They showed that substitution of Leucine with Lysine at position 160 abolishes the cathepsin D cleavage generating the 15 kDa vasoinhibin isoform. This experimental observation corresponds with the 8P-score used in this study, as the score demonstrates a decline from 935 in the ancestral sequence to 525 (−410) in the sequence with p.L160K. Another observation by Piwnica et al. was that the substitution of proline by leucine at position 176 dramatically increases cleavage efficiency. Again, this is confirmed by corresponding changes in the 8P-score, which increases by +107 (16.8 kDa vasoinhibin, cathepsin D), +37 (MMP-8) and +49 (MMP-13), and +69 (17.2 kDa vasoinhibin, cathepsin D), respectively. Finally, the observation by Piwnica et al. of an increased cleavage by substitution of Proline at position 176 by alanine corresponds with respective changes in the 8P-score (16.8 kDa vasoinhibin: +80 for cathepsin D, +8 for MMP-8, +9 for MMP-13; 17.2 kDa vasoinhibin: +5 for cathepsin D).
There are additional vasoinhibin isoforms smaller than 15 kDa that were not considered in the present study since no reliable information about their precise PRL-cleavage sites, either by sequencing or by mass spectrometry, could be retrieved. Also, other MMP (MMP-3, 1, 2, and 9) known to generate vasoinhibins (11) were not considered and the consequences of the mutations in PRL sequence might be different from those projected for MMP-8 and 13. The latter were selected since they cleave PRL with the highest relative rate.
The present analysis proposes that point mutations in the PRL gene, which result in single amino acid substitutions in the PRL sequence, impact the levels and isoform composition of vasoinhibins by altering the efficacy of PRL-cleaving enzymes. These point mutations may, therefore, constitute risk or protective factors affecting diseases associated with a dysregulation of the PRL/vasoinhibin axis in terms of deficient or excessive vasoinhibin generation. One such disease is PPCM, defined by the Heart Failure Association of the European Society of Cardiology Working Group on Peripartum Cardiomyopathy (PPCM) as: “PPCM is an idiopathic cardiomyopathy presenting with heart failure secondary to left ventricular systolic dysfunction toward the end of pregnancy or in the month following delivery, where no other cause of heart failure is found. It is a diagnosis of exclusion. The left ventricle may not be dilated but the ejection fraction (EF) is nearly always reduced below 45%” (34). PPCM is a rare disease, which occurred with a frequency of 1 case/3189 live births and an estimated mortality of 1.36–2.05% (confidence interval 0.29–10.8%) from 1990 to 2002 in the United States (35). Age, race, multiple gestations, and geographical region are associated factors recently reviewed and not discussed here (36–39). As mentioned in the above definition of PPCM, the disease is idiopathic, meaning that no specific causal pathological mechanism could be identified. However, mechanistic research revealed that the 16 kDa vasoinhibin isoform, also referred to as 16 kDa PRL, could be causally related to the development of PPCM (10). PRL, colloquially referred to as the “nursing hormone,” physiologically rises during pregnancy in preparation for lactation and remains elevated in the postpartum period (40). Because of the known, potentially detrimental effects of vasoinhibins on blood vessels (41–43), it was suggested that a higher than normal cleavage of PRL, resulting in an excessive generation of the 16 kDa vasoinhibin isoform, impairs myocardial microvascularization and is a causal trigger of PPCM (10). Consistent with this notion, a higher activity of the vasoinhibin-generating enzyme cathepsin D was demonstrated in the plasma of patients with PPCM (10), and a new therapy for PPCM using the dopamine D2 receptor agonists, cabergoline, and bromocriptine is currently being evaluated in a multicenter clinical trial (NCT00998556) (17). The concept underlying this putative therapy is the inhibition of the generation of vasoinhibins by substrate depletion, i.e., the inhibition of pituitary PRL secretion by activation of dopamine D2 receptors in lactotrophs. However, even if the vasoinhibin-related concept of PPCM etiopathology is substantiated, unanswered questions remain. Despite consideration of known associated factors, the risk of PPCM cannot be reliably predicted and the clinical course is variable, ranging from relatively mild cases to more severe, prolonged cases and also lethal outcomes. It will remain an open question why patients with similar risk factors—high PRL levels and high PRL-cleaving enzyme activity—could experience either no, mild, or severe PPCM. A missense PRL variant affecting the sequence at which proteases act to generate vasoinhibins, could increase cleavage to vasoinhibins, and may constitute a “second hit” in the pathogenesis of PPCM; whereas a variant that results in decreased cleavage may constitute a protective factor, as, despite high activity of cleaving enzymes, the levels of vasoinhibins generated would be lower and less detrimental effects would be expected.
A similar concept can be applied to diabetic retinopathy, in which vasoinhibins play a protective role in terms of their inhibitory effect on retinal neovascularizations, a hallmark of proliferative diabetic retinopathy. Furthermore, vasoinhibins participate in maintaining the normal retinal vasculature (44) and corneal avascularity (45). Raising circulating PRL levels leads to vasoinhibin accumulation in the retina (9), and the elevation of intraocular vasoinhibins prevents and reverses diabetes-induced excessive retinal vasopermeability in rats (46, 47) and angiogenesis in a mouse model of retinopathy of prematurity (48). Also, immunosequestering ocular vasoinhibins stimulates adult rat retinal angiogenesis (44) and reduces the apoptosis of the ocular hyaloid vasculature in neonatal rats (49). As in the etiopathology of PPCM, PRL point mutations affecting the amino acid sequence at vasoinhibin-generating cleavage sites, may, by modifying their levels and composition, enhance or diminish vasoinhibin effects. In consequence, these mutations may account for inter-individual differences in scenarios in which PRL levels as well as enzyme activities are similar, but observed vasoinhibin-related effects vary.
Four mutations in cancer tissues likely affecting the generation of vasoinhibins were found using the COSMIC database. Three of these mutations facilitated PRL-cleavage, whereas a fourth demonstrated a minor negative effect (Table 2). This is of relevance as vasoinhibins (the 16 kDa vasoinhibin isoform) reduced angiogenesis in a breast cancer model (50) and adenovirus-mediated vasoinhibin expression in prostate cancer cells lowered their ability to form tumors in a xenograft animal model (51). Another study reported that the 16 kDa vasoinhibin isoform reduced the growth and microvascular density of tumors derived from implanted human colon cancer cells in mice (52). Furthermore, adenovirus-mediated gene transfer of the 16 kDa vasoinhibin inhibited tumor growth in a subcutaneous mouse melanoma model and reduced the size and number of lung metastases (53). Considering these studies, the generation of vasoinhibins in cancer tissues may be a factor debilitating tumor fitness because two classical hallmarks of cancer (54)—the induction of angiogenesis and the activation of invasion and metastasis—are compromised. On the other hand, vasoinhibins can act as potent proinflammatory cytokines stimulating iNOS expression and NO production by pulmonary fibroblasts (5, 55) and these effects may contribute to inflammation-mediated restrain of tumor growth.
Studies reported the detection of one, two, or several vasoinhibin isoforms in biological fluids such as plasma and urine from patients, but also in tissue lysates (15, 18, 20, 56). This alone indicates that the composition of vasoinhibins varies during physiological states and disease conditions. Such variation is to be expected because the type of PRL cleavage depends on the cellular and extracellular location and activity of vasoinhibin-generating enzymes, and on additional microenvironmental factors affecting enzyme kinetics, such as pH and temperature (57). We did not consider these additional factors, which is why the projected vasoinhibin composition of 15 > 17.2 > 16.8 > 17.7 > 18 kDa isoform is only a rough approximation of what can be expected to occur in vivo. However, the projection is useful as it points to the need to consider more than one vasoinhibin isoform when detecting vasoinhibins in biological fluids. Furthermore, the projected composition question investigations in which isoforms were assigned the molecular mass of 16.8 kDa (3, 10), because the 15 and 17.2 kDa vasoinhibin isoforms are projected to be more abundant. Moreover, if only one cathepsin D cleaved isoform is detected, mechanisms must be present suppressing the generation of the other cathepsin D-generated isoforms (58). The problem extends to studies of vasoinhibin-related diseases and points out that differentiation between vasoinhibin isoforms and complete characterization of their generation process should be a focus in future investigations (22, 23). This is a prerequisite to resolve uncertainties about which isoforms should be targeted when manufacturing anti-vasoinhibin monoclonal antibodies for the development of quantitative immunoassays and for further characterization of vasoinhibin physiology and related diseases. The general order of vasoinhibin composition 15 > 17.2 > 16.8 > 17.7 > 18 kDa isoform did not change in consequence of a mutation. Hence, the mutations appear more relevant for the levels of single vasoinhibin isoforms and not for the general order of vasoinhibin composition. However, the large change (+69 points) seen in the 8P-score in case of the 17.2 kDa isoform, when SER is substituted by LEU in position 1 (Table 1), for example, demonstrates that other mutations not yet detected may indeed change the general order of vasoinhibin composition. Whether such change would be relevant or not cannot be ascertained momentarily, as comparative analyses of mixtures of vasoinhibin isoforms have not yet been performed.
Our work proposes that—besides substrate availability and enzyme activity—point mutations in the PRL gene may impact vasoinhibin generation and vasoinhibin-related disease outcomes. This hypothesis is testable as point mutations affecting cleavage site regions can be evaluated in patients with vasoinhibin-related diseases and the proteolytic cleavage of PRL carrying these mutations can be performed in in vitro assays. The study of only one vasoinhibin isoform when investigating vasoinhibin levels in patients appears insufficient. This underscores the need to heighten the focus on the neglected issue of addressing the various vasoinhibin isoforms and to differentiate between them. This will facilitate the ongoing process of understanding the functional role of vasoinhibins under health and disease.
JT designed research and wrote the manuscript. AL, CF, and JT performed data analysis. GE, CC, and TB edited and revised the manuscript. All authors approved the final version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the National Council of Science and Technology of Mexico (CONACYT), grant 220574 to CC.
1. Clapp C, Thebault S, Macotela Y, Moreno-Carranza B, Triebel J, Martinez de la Escalera G. Regulation of blood vessels by prolactin and vasoinhibins. Adv Exp Med Biol (2015) 846:83–95. doi:10.1007/978-3-319-12114-7_4
2. Clapp C, Thebault S, Jeziorski MC, Martinez De La Escalera G. Peptide hormone regulation of angiogenesis. Physiol Rev (2009) 89(4):1177–215. doi:10.1152/physrev.00024.2009
3. Chang AS, Grant R, Tomita H, Kim HS, Smithies O, Kakoki M. Prolactin alters blood pressure by modulating the activity of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A (2016) 113(44):12538–43. doi:10.1073/pnas.1615051113
4. Mejia S, Torner LM, Jeziorski MC, Gonzalez C, Morales MA, de la Escalera GM, et al. Prolactin and 16K prolactin stimulate release of vasopressin by a direct effect on hypothalamo-neurohypophyseal system. Endocrine (2003) 20(1–2):155–62. doi:10.1385/ENDO:20:1-2:155
5. Corbacho AM, Nava G, Eiserich JP, Noris G, Macotela Y, Struman I, et al. Proteolytic cleavage confers nitric oxide synthase inducing activity upon prolactin. J Biol Chem (2000) 275(18):13183–6. doi:10.1074/jbc.275.18.13183
6. Bajou K, Herkenne S, Thijssen VL, D’Amico S, Nguyen NQ, Bouche A, et al. PAI-1 mediates the antiangiogenic and profibrinolytic effects of 16K prolactin. Nat Med (2014) 20(7):741–7. doi:10.1038/nm.3552
7. Zamorano M, Ledesma-Colunga MG, Adan N, Vera-Massieu C, Lemini M, Mendez I, et al. Prolactin-derived vasoinhibins increase anxiety- and depression-related behaviors. Psychoneuroendocrinology (2014) 44:123–32. doi:10.1016/j.psyneuen.2014.03.006
8. Cruz-Soto ME, Cosio G, Jeziorski MC, Vargas-Barroso V, Aguilar MB, Carabez A, et al. Cathepsin D is the primary protease for the generation of adenohypophyseal vasoinhibins: cleavage occurs within the prolactin secretory granules. Endocrinology (2009) 150(12):5446–54. doi:10.1210/en.2009-0390
9. Arnold E, Rivera JC, Thebault S, Moreno-Paramo D, Quiroz-Mercado H, Quintanar-Stephano A, et al. High levels of serum prolactin protect against diabetic retinopathy by increasing ocular vasoinhibins. Diabetes (2010) 59(12):3192–7. doi:10.2337/db10-0873
10. Hilfiker-Kleiner D, Kaminski K, Podewski E, Bonda T, Schaefer A, Sliwa K, et al. A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell (2007) 128(3):589–600. doi:10.1016/j.cell.2006.12.036
11. Macotela Y, Aguilar MB, Guzman-Morales J, Rivera JC, Zermeno C, Lopez-Barrera F, et al. Matrix metalloproteases from chondrocytes generate an antiangiogenic 16 kDa prolactin. J Cell Sci (2006) 119(Pt 9):1790–800. doi:10.1242/jcs.02887
12. Piwnica D, Touraine P, Struman I, Tabruyn S, Bolbach G, Clapp C, et al. Cathepsin D processes human prolactin into multiple 16K-like N-terminal fragments: study of their antiangiogenic properties and physiological relevance. Mol Endocrinol (2004) 18(10):2522–42. doi:10.1210/me.2004-0200
13. Ge G, Fernandez CA, Moses MA, Greenspan DS. Bone morphogenetic protein 1 processes prolactin to a 17-kDa antiangiogenic factor. Proc Natl Acad Sci U S A (2007) 104(24):10010–5. doi:10.1073/pnas.0704179104
14. Triebel J, Bertsch T, Bollheimer C, Rios-Barrera D, Pearce CF, Hufner M, et al. Principles of the prolactin/vasoinhibin axis. Am J Physiol Regul Integr Comp Physiol (2015) 309(10):R1193–203. doi:10.1152/ajpregu.00256.2015
15. Triebel J, Huefner M, Ramadori G. Investigation of prolactin-related vasoinhibin in sera from patients with diabetic retinopathy. Eur J Endocrinol (2009) 161(2):345–53. doi:10.1530/EJE-09-0130
16. Triebel J, Macotela Y, de la Escalera GM, Clapp C. Prolactin and vasoinhibins: endogenous players in diabetic retinopathy. IUBMB Life (2011) 63(10):806–10. doi:10.1002/iub.518
17. Haghikia A, Podewski E, Berliner D, Sonnenschein K, Fischer D, Angermann CE, et al. Rationale and design of a randomized, controlled multicentre clinical trial to evaluate the effect of bromocriptine on left ventricular function in women with peripartum cardiomyopathy. Clin Res Cardiol (2015) 104(11):911–7. doi:10.1007/s00392-015-0869-5
18. Nakajima R, Ishida M, Kamiya CA, Yoshimatsu J, Suzuki M, Hirota A, et al. Elevated vasoinhibin derived from prolactin and cathepsin D activities in sera of patients with preeclampsia. Hypertens Res (2015) 38(12):899–901. doi:10.1038/hr.2015.99
19. Gonzalez C, Parra A, Ramirez-Peredo J, Garcia C, Rivera JC, Macotela Y, et al. Elevated vasoinhibins may contribute to endothelial cell dysfunction and low birth weight in preeclampsia. Lab Invest (2007) 87(10):1009–17. doi:10.1038/labinvest.3700662
20. Leanos-Miranda A, Marquez-Acosta J, Cardenas-Mondragon GM, Chinolla-Arellano ZL, Rivera-Leanos R, Bermejo-Huerta S, et al. Urinary prolactin as a reliable marker for preeclampsia, its severity, and the occurrence of adverse pregnancy outcomes. J Clin Endocrinol Metab (2008) 93(7):2492–9. doi:10.1210/jc.2008-0305
21. Masumoto A, Masuyama H, Takamoto N, Akahori Y, Hiramatsu Y. Expression of antiangiogenic prolactin fragments in the placentas of women with pregnancy induced hypertension. Acta Med Okayama (2010) 64(4):249–55. doi:10.18926/AMO/40133
22. Triebel J, Bertsch T, Martinez de la Escalera G, Clapp C. On the path toward classifying hormones of the vasoinhibin-family. Front Endocrinol (2015) 6:16. doi:10.3389/fendo.2015.00016
23. Triebel J, Clapp C, Martinez de la Escalera G, Bertsch T. Remarks on the prolactin hypothesis of peripartum cardiomyopathy. Front Endocrinol (2017) 8:77. doi:10.3389/fendo.2017.00077
24. Waybright TJ, Xu X, Faupel-Badger JM, Xiao Z. Preparation of human serum for prolactin measurement by multiple reaction monitoring mass spectrometry. Methods Mol Biol (2013) 1002:195–203. doi:10.1007/978-1-62703-360-2_16
25. Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, et al. Ensembl 2015. Nucleic Acids Res (2015) 43(Database issue):D662–9. doi:10.1093/nar/gku1010
26. Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol (2016) 33(7):1870–4. doi:10.1093/molbev/msw054
27. Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res (2017) 45(D1):D777–83. doi:10.1093/nar/gkw1121
28. Rawlings ND. Using the MEROPS database for investigation of lysosomal peptidases, their inhibitors, and substrates. Methods Mol Biol (2017) 1594:213–26. doi:10.1007/978-1-4939-6934-0_14
29. Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res (2013) 23(9):1422–33. doi:10.1101/gr.154492.113
30. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell (2012) 150(6):1107–20. doi:10.1016/j.cell.2012.08.029
31. Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet (2014) 46(6):573–82. doi:10.1038/ng.2983
32. George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature (2015) 524(7563):47–53. doi:10.1038/nature14664
33. Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet (2012) 44(10):1104–10. doi:10.1038/ng.2396
34. Sliwa K, Hilfiker-Kleiner D, Petrie MC, Mebazaa A, Pieske B, Buchmann E, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of peripartum cardiomyopathy: a position statement from the Heart Failure Association of the European Society of Cardiology Working Group on peripartum cardiomyopathy. Eur J Heart Fail (2010) 12(8):767–78. doi:10.1093/eurjhf/hfq120
35. Mielniczuk LM, Williams K, Davis DR, Anthony T, Lemery R, Green MS, et al. Frequency of Peripartum Cardiomyopathy. Am J Cardio (2006) 97(12):1765–8. doi:10.1016/j.amjcard.2006.01.039
36. Arany Z, Elkayam U. Peripartum cardiomyopathy. Circulation (2016) 133(14):1397–409. doi:10.1161/CIRCULATIONAHA.115.020491
37. Bello NA, Arany Z. Molecular mechanisms of peripartum cardiomyopathy: a vascular/hormonal hypothesis. Trends Cardiovasc Med (2015) 25(6):499–504. doi:10.1016/j.tcm.2015.01.004
38. Brar SS, Khan SS, Sandhu GK, Jorgensen MB, Parikh N, Hsu JW, et al. Incidence, mortality, and racial differences in peripartum cardiomyopathy. Am J Cardiol (2007) 100(2):302–4. doi:10.1016/j.amjcard.2007.02.092
39. Ersboll AS, Damm P, Gustafsson F, Vejlstrup NG, Johansen M. Peripartum cardiomyopathy: a systematic literature review. Acta Obstet Gynecol Scand (2016) 95(11):1205–19. doi:10.1111/aogs.13005
40. Tyson JE, Hwang P, Guyda H, Friesen HG. Studies of prolactin secretion in human pregnancy. Am J Obstet Gynecol (1972) 113(1):14–20. doi:10.1016/0002-9378(72)90446-2
41. Clapp C, Aranda J, Gonzalez C, Jeziorski MC, Martinez de la Escalera G. Vasoinhibins: endogenous regulators of angiogenesis and vascular function. Trends Endocrinol Metab (2006) 17(8):301–7. doi:10.1016/j.tem.2006.08.002
42. Clapp C, Gonzalez C, Macotela Y, Aranda J, Rivera JC, Garcia C, et al. Vasoinhibins: a family of N-terminal prolactin fragments that inhibit angiogenesis and vascular function. Front Horm Res (2006) 35:64–73. doi:10.1159/000094309
43. Clapp C, Martial JA, Guzman RC, Rentier-Delure F, Weiner RI. The 16-kilodalton N-terminal fragment of human prolactin is a potent inhibitor of angiogenesis. Endocrinology (1993) 133(3):1292–9. doi:10.1210/endo.133.3.7689950
44. Aranda J, Rivera JC, Jeziorski MC, Riesgo-Escovar J, Nava G, Lopez-Barrera F, et al. Prolactins are natural inhibitors of angiogenesis in the retina. Invest Ophthalmol Vis Sci (2005) 46(8):2947–53. doi:10.1167/iovs.05-0173
45. Duenas Z, Torner L, Corbacho AM, Ochoa A, Gutierrez-Ospina G, Lopez-Barrera F, et al. Inhibition of rat corneal angiogenesis by 16-kDa prolactin and by endogenous prolactin-like molecules. Invest Ophthalmol Vis Sci (1999) 40(11):2498–505.
46. Garcia C, Aranda J, Arnold E, Thebault S, Macotela Y, Lopez-Casillas F, et al. Vasoinhibins prevent retinal vasopermeability associated with diabetic retinopathy in rats via protein phosphatase 2A-dependent eNOS inactivation. J Clin Invest (2008) 118(6):2291–300. doi:10.1172/JCI34508
47. Ramirez M, Wu Z, Moreno-Carranza B, Jeziorski MC, Arnold E, Diaz-Lezama N, et al. Vasoinhibin gene transfer by adenoassociated virus type 2 protects against VEGF- and diabetes-induced retinal vasopermeability. Invest Ophthalmol Vis Sci (2011) 52(12):8944–50. doi:10.1167/iovs.11-8190
48. Pan H, Nguyen NQ, Yoshida H, Bentzien F, Shaw LC, Rentier-Delrue F, et al. Molecular targeting of antiangiogenic factor 16K hPRL inhibits oxygen-induced retinopathy in mice. Invest Ophthalmol Vis Sci (2004) 45(7):2413–9. doi:10.1167/iovs.03-1001
49. Duenas Z, Rivera JC, Quiroz-Mercado H, Aranda J, Macotela Y, Montes de Oca P, et al. Prolactin in eyes of patients with retinopathy of prematurity: implications for vascular regression. Invest Ophthalmol Vis Sci (2004) 45(7):2049–55. doi:10.1167/iovs.03-1346
50. Faupel-Badger JM, Ginsburg E, Fleming JM, Susser L, Doucet T, Vonderhaar BK. 16 kDa prolactin reduces angiogenesis, but not growth of human breast cancer tumors in vivo. Horm Cancer (2010) 1(2):71–9. doi:10.1007/s12672-010-0012-z
51. Kim J, Luo W, Chen DT, Earley K, Tunstead J, Yu-Lee LY, et al. Antitumor activity of the 16-kDa prolactin fragment in prostate cancer. Cancer Res (2003) 63(2):386–93.
52. Bentzien F, Struman I, Martini JF, Martial J, Weiner R. Expression of the antiangiogenic factor 16K hPRL in human HCT116 colon cancer cells inhibits tumor growth in Rag1(-/-) mice. Cancer Res (2001) 61(19):7356–62.
53. Nguyen NQ, Cornet A, Blacher S, Tabruyn SP, Foidart JM, Noel A, et al. Inhibition of tumor growth and metastasis establishment by adenovirus-mediated gene transfer delivery of the antiangiogenic factor 16K hPRL. Mol Ther (2007) 15(12):2094–100. doi:10.1038/sj.mt.6300294
54. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
55. Macotela Y, Mendoza C, Corbacho AM, Cosio G, Eiserich JP, Zentella A, et al. 16K prolactin induces NF-kappaB activation in pulmonary fibroblasts. J Endocrinol (2002) 175(3):R13–8. doi:10.1677/joe.0.175R013
56. Zepeda-Romero LC, Vazquez-Membrillo M, Adan-Castro E, Gomez-Aguayo F, Gutierrez-Padilla JA, Angulo-Castellanos E, et al. Higher prolactin and vasoinhibin serum levels associated with incidence and progression of retinopathy of prematurity. Pediatr Res (2017) 81(3):473–9. doi:10.1038/pr.2016.241
57. Clapp C, Thebault S, Martinez de la Escalera G. Role of prolactin and vasoinhibins in the regulation of vascular function in mammary gland. J Mammary Gland Biol Neoplasia (2008) 13(1):55–67. doi:10.1007/s10911-008-9067-7
Keywords: prolactin mutations, vasoinhibins, prolactin/vasoinhibin axis, peripartum cardiomyopathy, 16 kDa prolactin
Citation: Triebel J, Friedrich CJ, Leuchs A, Martínez de la Escalera G, Clapp C and Bertsch T (2017) Human Prolactin Point Mutations and Their Projected Effect on Vasoinhibin Generation and Vasoinhibin-Related Diseases. Front. Endocrinol. 8:294. doi: 10.3389/fendo.2017.00294
Received: 09 June 2017; Accepted: 13 October 2017;
Published: 06 November 2017
Edited by:
Nils P. Krone, University of Sheffield, United KingdomReviewed by:
Xavier Bonnefont, UMR5203 Institut de génomique fonctionnelle (IGF), FranceCopyright: © 2017 Triebel, Friedrich, Leuchs, Martínez de la Escalera, Clapp and Bertsch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jakob Triebel, amFrb2IudHJpZWJlbEBnbXguZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.