 Mario Rotondi1
Mario Rotondi1 Martina Molteni
Martina Molteni Valentina Capelli
Valentina Capelli Michele Marinò
Michele Marinò Luca Chiovato
Luca Chiovato- 1Unit of Internal Medicine and Endocrinology, Laboratory for Endocrine Disruptors, ICS-Maugeri IRCCS, University of Pavia, Pavia, Italy
- 2Department of Internal Medicine and Therapeutics, and Department of Medical and Surgical Sciences, University of Pavia, Pavia, Italy
- 3Endocrinology Unit I, Department of Clinical and Experimental Medicine, University of Pisa, University Hospital of Pisa, Pisa, Italy
Alemtuzumab, a humanized anti-CD52 monoclonal antibody, is approved for the treatment of active relapsing-remitting multiple sclerosis (MS). Alemtuzumab induces a rapid and prolonged depletion of lymphocytes from the circulation, which results in a profound immuno-suppression status followed by an immune reconstitution phase. Secondary to reconstitution autoimmune diseases represent the most common side effect of Alemtuzumab treatment. Among them, Graves’ disease (GD) is the most frequent one with an estimated prevalence ranging from 16.7 to 41.0% of MS patients receiving Alemtuzumab. Thyrotropin (TSH) receptor (R)-reactive B cells are typically observed in GD and eventually present this autoantigen to T-cells, which, in turn, secrete several pro-inflammatory cytokines and chemokines. Given that reconstitution autoimmunity is more frequently characterized by autoantibody-mediated diseases rather than by destructive Th1-mediated disorders, it is not surprising that GD is the most commonly reported side effect of Alemtuzumab treatment in patients with MS. On the other hand, immune reconstitution GD was not observed in a large series of patients with rheumatoid arthritis treated with Alemtuzumab. This negative finding supports the view that patients with MS are intrinsically more at risk for developing Alemtuzumab-related thyroid dysfunctions and in particular of GD. From a clinical point of view, Alemtuzumab-induced GD is characterized by a surprisingly high rate of remission, both spontaneous and after antithyroid drugs, as well as by a spontaneous shift to hypothyroidism, which is supposed to result from a change from stimulating to blocking TSH-receptor antibodies. These immune and clinical peculiarities support the concept that antithyroid drugs should be the first-line treatment in Alemtuzumab-induced Graves’ hyperthyroidism.
Alemtuzumab as an Immunomodulating Drug
Alemtuzumab is a humanized monoclonal antibody that has been approved for the treatment of active relapsing-remitting (RR) multiple sclerosis (MS) (1, 2). As a main pharmacologic action, Alemtuzumab targets the cell-surface antigen CD52. CD52 is a cell-surface glycoprotein with a still poorly understood function. CD52 is expressed on the surface of more than 95% T and B cells, of monocytes and of some dendritic cells, and, although to a lesser extent, even on natural killer cells and other leukocytes (3). The binding of Alemtuzumab to lymphocytes induces cellular lysis leading to their rapid and prolonged depletion from the circulation (4).
The acute immuno-suppressive effect of Alemtuzumab is followed by the homeostatic reconstitution of immune cells. Typically, monocytes and B cells recover first, followed by CD4+ T cells. Changes in lymphocyte subsets result in an increased number of T regulatory (Treg) cells and of memory T and B lymphocytes; an increased production of anti-inflammatory cytokines also occurs (5). These events produce a profound rebalance of the immune system (6, 7).
Circulating lymphocytes disappear within a few minutes after the administration of Alentuzumab. B cells recover within 3 months and a dominance of mature naïve cells (CD19+ CD23+ CD27−) over the memory B cells occurs. CD4+ T cell counts are restored after 35 months, while CD8+ T cell counts are restored after 20 months. The faster recovery of the latter subset of T cells might be related to the development of autoimmune diseases (8). For at least 9 months after the administration of Alemtuzumab, most circulating T cells are represented by effector memory CD4+ and CD8+ cells. Baker et al. recently described the kinetics of lymphocyte subset reconstitution after Alemtuzumab (9). After depletion, B cells repopulated much more rapidly than T cells in general and Treg in particular (9). In this scenario, the reconstitution of B cells without adequate regulatory control by T cells may explain the high prevalence of post-Alentuzumab autoimmunity (9, 10).
Alentuzumab-induced lymphocytopenia is followed by the homeostatic growth of T cells, which is stimulated by the T cell receptor–self peptide complex. The process results in the appearance of an oligoclonal cell population, which tends to autoreactivity. New T cell populations have typical aspects of memory T cells, such as lower dependency to co-stimulation, need for lower antigen doses than naïve cells, and faster secrection of inflammatory cytokines when re-stimulated (6–8). The above described immune derangements lead to a reduced self-tolerance. In most patients, the proliferation of regulatory lymphocytes is unable to prevent autoimmune deseases, possibly because T cells undergo a faster homeostatic growth, which increases their resistance to regulation (8). Patients who developed autoimmunity after Alentuzumab treatment also show high basal levels of IL-21, a cytokine which increases the growth of auto-reactive T cells. In general, the cytokine expression is skewed to the Th2 profile, in agreement with the high B cell counts (11, 12).
Innate immunity is not affected, and no clinically relevant infection appears after Alentuzumab treatment. This can be due to the maintenance/growth of memory T cells.
The above described immunomodulating actions of Alemtuzumab are responsible for its favorable effects in patients with RRMS (13), but also explain the high prevalence of Alemtuzumab-induced autoimmunity. The latter event received great concern and clinical trials aimed at evaluating potential preventive measures were designed (CAM-THY) (8, 14, 15).
MS and Thyroid Diseases
Multiple sclerosis is a human chronic inflammatory disease of the central nervous system supposed to be a Th1/Th17 type cell-mediated autoimmune disorder (16, 17). Studies aimed at evaluating whether there is an increased prevalence of autoimmune thyroid diseases (AITDs) in patients with MS as compared with healthy controls reported conflicting results. While early studies found an increased prevalence of AITD in patients with MS, more recent surveys reported rates which are consistent with the AITD prevalence in the general population (18–21). According to more recent views, an increased prevalence of AITD would be observed in family members of patients with MS (22). This is a rather intriguing and yet to be a fully elucidated observation (23, 24).
In addition to studies aimed at evaluating the prevalence of AITD in naive patients with MS, the occurrence of AITD, as a side effect of immunomodulatory treatments for MS, was extensively reported (25–27). At present, we know that treatment with interferon-β (IFN-β) increases the risk for worsening and/or de novo appearance of both thyroid autoimmunity and dysfunction. On the other hand, thyroid side effects were not observed following glatiramer acetate (GA) therapy, even in large series of patients with MS who were longitudinally followed for more than 10 years (11, 28, 29).
Specifically designed head-to-head clinical studies demonstrated a similar efficacy of IFN-β and GA, as assessed by their ability to prevent clinical relapses and disease progression in patients with MS (12). However, more effective pharmacologic agents are now available. Among them, Alemtuzumab is currently regarded as an effective second-line treatment in patients with highly active RRMS (1, 2, 13).
TSH-Receptor (TSH-R) as a Major Autoantigen in Graves’ Disease
Graves’ disease (GD), also referred to as toxic diffuse goiter, is commonly regarded as an autoimmune organ-specific disease (30–32). The presence of extra-thyroid manifestations, such as Graves’ orbitopathy and pretibial myxedema, apparently contradicts this classification, which is, however, justified by the TSH-R being a common antigen shared by the thyroid gland and by extra-thyroid tissues (31). The TSH-R, a G-protein coupled receptor with seven transmembrane-spanning domains and a large extracellular portion, is expressed primarily on the surface of thyroid follicular cells, but it is also present in adipocytes, fibroblast, bone cells, and other sites including the heart (33). TSH-R Antiboides (TRAb) encompass stimulating, blocking, and neutral antibodies. In patiens with GD, TRAb mainly have thyroid-stimulating activity (TSAb), which results in hyperthyroidism and goiter formation, TSAb bind only the naturally conformed TSH-R and induce cyclic AMP generation, thyroid cell proliferation, and thyroid hormone synthesis and secretion (34, 35). More rarely, and less functionally dominant, TRAb with thyroid blocking activity have been described in patients with GD (36).
Immune system abnormalities in GD are represented by TSH-R-reactive B cells, which escape deletion and eventually present this thyroid autoantigen to T cells. When activated, T cells secrete several pro-inflammatory cytokines and chemokines (37, 38). Hence both B cells and T cells play a central role in perpetuating the autoimmune cascade in GD (11, 37).
AITDs and Alemtuzumab
Among several autoimmune conditions, which have been reported to occur following Alemtuzumab treatment, the present review will focus on GD, the most frequently observed one. The occurrence of GD in Alemtuzumab-treated patients with secondary progressive MS was first described in 1999 (39). In this early report, a third of MS patients (9/27) receiving the anti-CD52 monoclonal Ab developed GD, with circulating TRAb and hyperthyroidism (39). Besides being the first description, the study by Coles et al. is of great interest, as it clearly shows that GD (and its humoral marker, TRAb) is the most prevalent form of AITD occurring in MS patients treated with Alemtuzumab (39). By contrast, in MS patients, treated with other immunomodulatory therapies (i.e., IFN-β), the most prevalent side effect is represented by euthyroid or hypothyroid autoimmune thyroiditis (26, 27, 40). Thus, a first crucial difference between IFN-β and Alemtuzumab was already evident: they elicited different autoimmune reactions driving the onset of Hashimoto’s thyroiditis and GD, respectively.
Subsequent studies, investigating the efficacy and safety of Alemtuzumab therapy in patients with RRMS, confirmed GD as the main autoimmune sequela of this immunomodulatory treatment. In 2008, Coles et al. published a phase 2 clinical trial (CAMMS223) in which 334 patients with RRMS were randomized to receive either IFN-β-1a three times/week or annual cycles of Alemtuzumab (either 12 or 24 mg/day) for 3 years (41). Among other (rare) autoimmune side effects, such as trombocytopenic purpura and glomerulonephritis due to autoantibodies binding the glomerular basal membrane, they reported a significantly higher rate of thyroid autoimmunity in patients treated with Alemtuzumab as opposed to those receiving IFN-β-1a (22.7 versus 2.8%, respectively) (41). In 2012, two phase-3 trials also reported the occurrence of mild to moderate thyroid dysfunction in nearly 18% of RRMS patients treated with Alemtuzumab (1, 2).
The early series of patients included in the CAMMS223 study (2) was further investigated with the specific aim of evaluating thyroid side effects of Alemtuzumab therapy (42). Daniels et al. prolonged the surveillance period of these patients up to a median time of 57.3 months and a maximum of 80.6 months (42). They confirmed that thyroid dysfunctions more frequently occurred in patients treated with Alemtuzumab as compared to those receiving IFN-β-1a. In particular, 34% of patients treated with Alemtuzumab developed thyroid dysfunctions (39% receiving 12 mg and 29% receiving 24 mg) as compared with a 6.5% rate in those treated with IFN-β-1a. As shown in Table 1, in the Alemtuzumab treatment group, GD was the most prevalent condition, being experienced by nearly 23% of patients (42). Hypothyroidism was observed in 7.4% patients and destructive thyroiditis with thyrotoxicosis in 4.2% of patients (42).
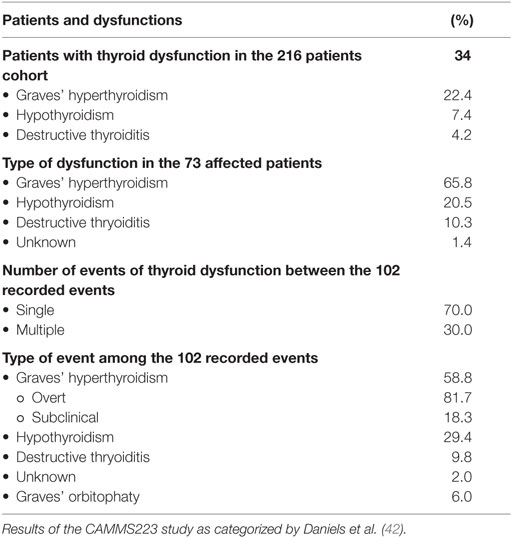
Table 1. Thyroid dysfunction during Alemtuzumab treatment in patients with multiple sclerosis (MS).
Some clinical peculiarities of these patients are worth noting. The first episode of thyroid dysfunction was observed starting from the first year after Alemtuzumab administration. Afterward, the episodes’ prevalence progressively increased each year for the first 3 years (from 4.6 to 16.1%) with a subsequent decrease in the following 4 years (from 11.3 to 5.9%). There was a higher than expected prevalence (52.7%) of patients in whom Graves’ hyperthyroidism (either overt or subclinical) was spontaneously reverted to hypothyroidism (either overt or subclinical) (Table 2). An unusual frequency of patients converting from hyperthyroidism to hypothyroidism and vice versa was also observed. Importantly, the conversion from hyperthyroidism to hypothyroidism was accompanied by the occurrence of TRAb in nearly 77% of patients (Table 3). This observation strongly suggests that the conversion from hyperthyroidism to hypothyroidism was likely due to a shift in the ratio between stimulating and blocking TRAbs. In the routine endocrine practice of sporadic GD, the transition from hyperthyroidism to hypothyroidism is a unusual event, which is mainly observed several years after a successful course of antithyroid drugs and is rarely accompanied by TRAb positivity (43). Taken together, these data indicate that the immune reconstitution occurring after Alemtuzumab treatment is mainly humoral, being directed to the TSH-R as a major autoantigen.
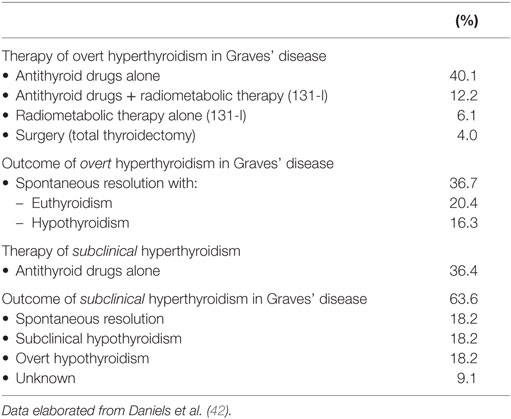
Table 2. Treatment and outcome of Graves’ disease developing after Alemtuzumab therapy.

Table 3. TSH-receptor antibodies (TRAb) and thyroid dysfunction during Alemtuzumab treatment.
Risk factors for the development of Alemtuzumab-induced GD were a family history of thyroid diseases, female sex, younger age, smoking habit, lower administered dose of the monoclonal Ab, and pretretment postivity for thyroid peroxidase (TPO) antibody (Ab). However, TPOAb had a minor relevance as a risk factor, due to the low frequency of pre-teratment positive results for this autoimmune marker (Table 4). Daniels et al. found that only 16/206 (8%) patients were positive for TPOAb at baseline. Among them, the prevalence of thyroid dysfunction after Alentuzumab treatment was 69%, a much higher rate than the 31% one observed in patients who were TPOAb negative at baseline (44). However, the majority (85%) of patients developing a thyroid disorder were negative for TPOAb before Alentuzumab treatment. Therefore, regardless of the pretreatment TPOAb status, patients may develop a thyroid disfunction and should have thyroid function tests performed periodically (41).

Table 4. TPOAb and thyroid dysfunction during Alemtuzumab treatment.
The remission rate of Alemtuzumab-induced Graves’ hyperthyroidism, either spontaneous or after antithyroid drug treatment (Table 2), was also higher (78%) than what commonly observed in the sporadic form of the disease (32).
In a futher observational cohort study, Tuohy et al. (45) re-evaluated 87 patients with RRMS who had been treated with Alemtuzumab in investigator-led studies in Cambridge from 1999 to 2012. This series included 67 patients of the CAMMS224 trial (18) and 20 of the SM3 trial (46). Among the 86 patients who completed the study, 35 (41%) developed a thyroid dysfunction, which was diagnosed as Graves’ hyperthyroidism in 22 (63%) and as hypothyroidism with positive tests for TPOAb in 12 (34%) of them. The main limitation of this study is that TRAb were not measured. At the present, large series studies aimed at evaluating the occurrence of thyroid dysfunctions in RRMS patients treated with Alemtuzumab reported a prevalence ranging from 16.7 to 41% (Table 5). The remaining published studies on the occurrence of GD in Alemtuzumab-treated patients mainly consist of single case or small series reports, which are summarized in Table 6. In the majority of these reports, patients developing AITD were taking Alemtuzumab for a RRMS (47–50). However, reconstitution GD was also described in patients receiving Alemtuzumab for other clinical conditions. The development of Graves’ hyperthyroidism was described by Walsh et al. in 11% of patients treated with Alemtuzumab for vasculitis (51). Other reports include (i) a young kidney transplant recipient who developed GD 4 years after Alemtuzumab treatment (52) and (ii) three pediatric cases of thyroid autoimmune diseases in patients receiving Alemtuzumab after hematopoietic cell transplantation (53).
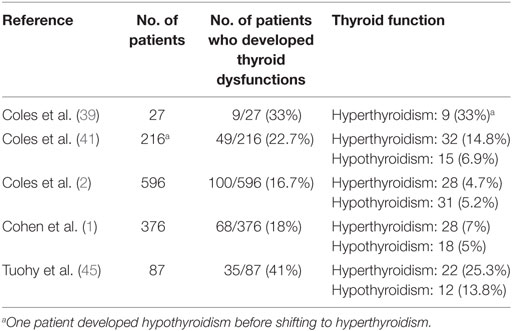
Table 5. Studies reporting Alemtuzumab-related thyroid dysfunction.
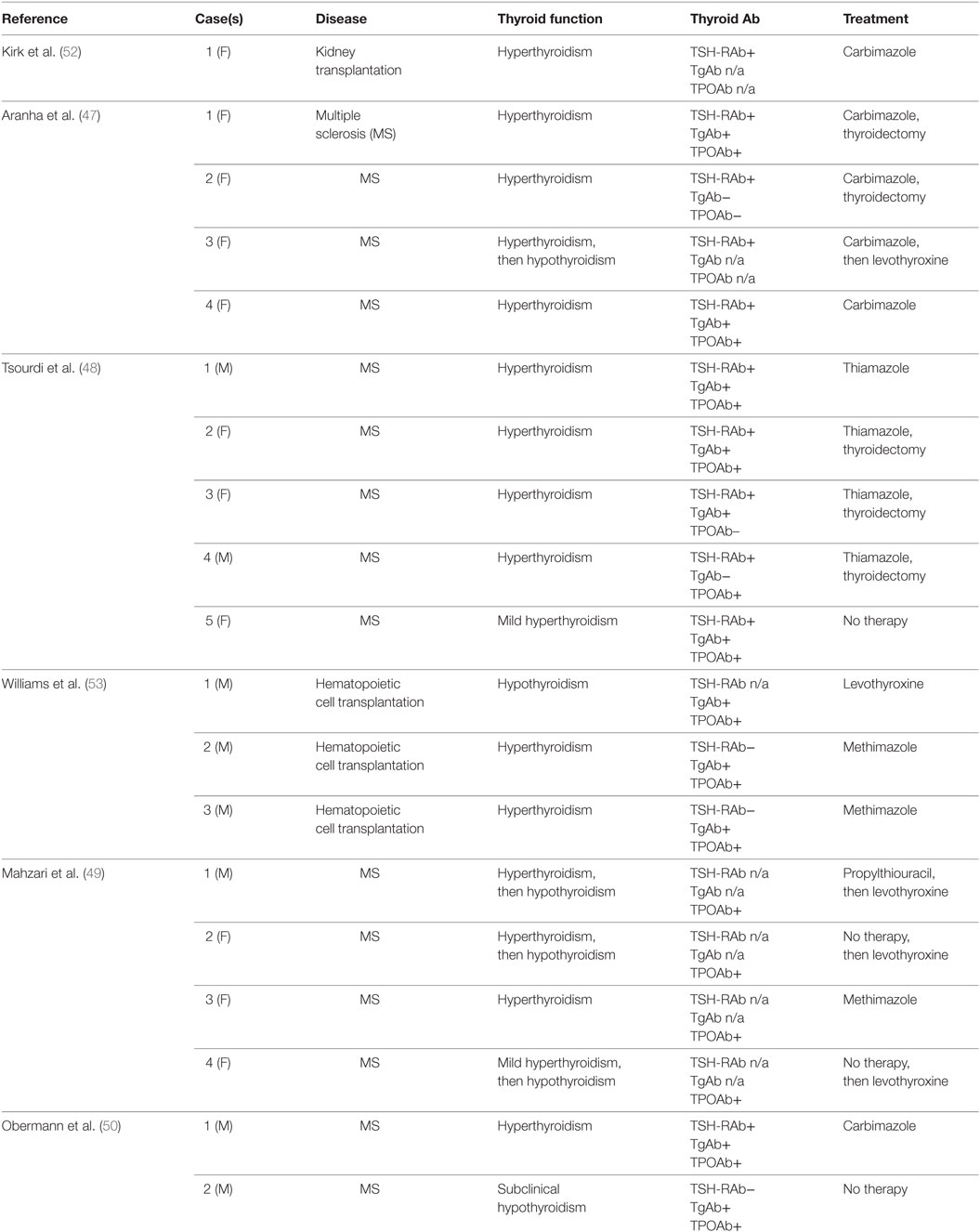
Table 6. Case reports of patients developing autoimmune thyroid diseases following Alemtuzumab administration.
At variance with all the above described studies, the development of immune reconstition GD was not observed in a large series of patients with rheumatoid arthritis treated with Alemtuzumab (54). These negative findings support the view that patients with MS bear a higher intrinsic risk for the development of Alemtuzumab-related thyroid dysfunctions. Further support to the above statement stems from the notion that while Alemtuzumab is increasingly prescribed in chronic lymphocytic leukemia, no case of Graves’ hyperthyroidism has been reported in these patients.
Final Remarks
Reconstitution GD may occur during the recovery phase of Alemtuzumab-induced CD52 cells depletion. Because reconstitution autoimmunity is more frequently related to autoantibody-mediated diseases rather than to destructive, Th1-mediated disorders (i.e., Hashimoto’s thyroiditis), it is not surprising that GD is the most commonly reported side effects of Alemtuzumab treatment.
The reason why, as compared with patients bearing other clinical conditions, those with MS carry a higher risk for the development of GD after Alemtuzumab treatment remains unknown. Genetic factors and/or specific clinical aspects of MS, such as the cytokine/chemokine milieu and/or the RR clinical course, might play a role, but there is still no definite proof at this regard.
From a clinical point of view, there are peculiar aspects of Alemtuzumab-induced GD. First, hyperthyroid patients have an unusualy high rate of spontaneous shift to hypothyroidism. This shift is supposed to result from a change from stimulating to blocking TRAb. Second, the remission rate of Graves’ hyperthyroidism, both spontaneous and after antithyroid drugs, is unexpectedly high, suggesting a less aggressive disease (14). This observation implies that antithyroid drugs should be the first-line treatment in patients with Alemtuzumab-induced Graves’ hyperthyroidism.
Author Contributions
LC, MR, MaM, PL, VC, and MiM designed the study and reviewed the literature on thyroid side effects of Alemtuzumab. LC and MR wrote the manuscript. All the authors revised the paper and approved the final edition.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung HP, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet (2012) 380(9856):1819–28. doi:10.1016/S0140-6736(12)61769-3
2. Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet (2012) 380(9856):1829–39. doi:10.1016/S0140-6736(12)61768-1
3. Rao SP, Sancho J, Campos-Rivera J, Boutin PM, Severy PB, Weeden T, et al. Human peripheral blood mononuclear cells exhibit heterogeneous CD52 expression levels and show differential sensitivity to alemtuzumab mediated cytolysis. PLoS One (2012) 7(6):e39416. doi:10.1371/journal.pone.0039416
4. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol (2004) 14(2):164–74. doi:10.1111/j.1750-3639.2004.tb00049.x
5. Wiendl H, Kieseier B. Multiple sclerosis: reprogramming the immune repertoire with alemtuzumab in MS. Nat Rev Neurol (2013) 9(3):125–6. doi:10.1038/nrneurol.2013.2
6. Milo R. Therapeutic strategies targeting B-cells in multiple sclerosis. Autoimmun Rev (2016) 15(7):714–8. doi:10.1016/j.autrev.2016.03.006
7. Jones JL, Coles AJ. Mode of action and clinical studies with alemtuzumab. Exp Neurol (2014) 262(Pt A):37–43. doi:10.1016/j.expneurol.2014.04.018
8. Weetman A. Immune reconstitution syndrome and the thyroid. Best Pract Res Clin Endocrinol Metab (2009) 23(6):693–702. doi:10.1016/j.beem.2009.07.003
9. Baker D, Herrod SS, Alvarez-Gonzalez C, Giovannoni G, Schmierer K. Interpreting lymphocyte reconstitution data from the pivotal phase 3 trials of alemtuzumab. JAMA Neurol (2017) 74(8):961–9. doi:10.1001/jamaneurol.2017.0676
10. Steinman L. Induction of new autoimmune diseases after alemtuzumab therapy for multiple sclerosis learning from adversity. JAMA Neurol (2017) 74(8):907–8. doi:10.1001/jamaneurol.2017.0325
11. Rotondi M, Stufano F, Lagonigro MS, La Manna L, Zerbini F, Ghilotti S, et al. Interferon-β but not glatiramer acetate stimulates CXCL10 secretion in primary cultures of thyrocytes: a clue for understanding the different risks of thyroid dysfunctions in patients with multiple sclerosis treated with either of the two drugs. J Neuroimmunol (2011) 234(1–2):161–4. doi:10.1016/j.jneuroim.2011.01.013
12. Mikol DD, Barkhof F, Chang P, Coyle PK, Jeffery DR, Schwid SR, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol (2008) 7(10):903–14. doi:10.1016/S1474-4422(08)70200-X
13. Coles AJ, Fox E, Vladic A, Gazda SK, Brinar V, Selmaj KW, et al. Alemtuzumab more effective than interferon β-1a at 5-year follow-up of CAMMS223 clinical trial. Neurology (2012) 78(14):1069–78. doi:10.1212/WNL.0b013e31824e8ee7
14. Weetman AP. Graves’ disease following immune reconstitution or immunomodulatory treatment: should we manage it any differently? Clin Endocrinol (Oxf) (2014) 80(5):629–32. doi:10.1111/cen.12427
15. ClinicalTrials.gov. Keratinocyte Growth Factor to Prevent Autoimmunity After Alemtuzumab Treatment of Multiple Sclerosis. (2017). NCT01712945. Available from: https://clinicaltrials.gov/ct2/show/NCT01712945
16. Batocchi AP, Rotondi M, Caggiula M, Frisullo G, Odoardi F, Nociti V, et al. Leptin as a marker of multiple sclerosis activity in patients treated with interferon-beta. J Neuroimmunol (2003) 139(1–2):150–4. doi:10.1016/S0165-5728(03)00154-1
17. Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol (2010) 162(1):1–11. doi:10.1111/j.1365-2249.2010.04143.x
18. Durelli L, Oggero A, Verdun E, Isoardo GL, Barbero P, Bergamasco B, et al. Thyroid function and anti-thyroid antibodies in MS patients screened for interferon treatment. A multicenter study. J Neurol Sci (2001) 193(1):17–22. doi:10.1016/S0022-510X(01)00637-2
19. Marrie RA, Yu BN, Leung S, Elliott L, Warren S, Wolfson C, et al. The incidence and prevalence of fibromyalgia are higher in multiple sclerosis than the general population: a population-based study. Mult Scler Relat Disord (2012) 1(4):162–7. doi:10.1016/j.msard.2012.06.001
20. Niederwieser G, Buchinger W, Bonelli RM, Berghold A, Reisecker F, Költringer P, et al. Prevalence of autoimmune thyroiditis and non-immune thyroid disease in multiple sclerosis. J Neurol (2003) 250(6):672–5. doi:10.1007/s00415-003-1053-9
21. Sloka JS, Phillips PW, Stefanelli M, Joyce C. Co-occurrence of autoimmune thyroid disease in a multiple sclerosis cohort. J Autoimmune Dis (2005) 2:9. doi:10.1186/1740-2557-2-9
22. Broadley SA, Deans J, Sawcer SJ, Clayton D, Compston DA. Autoimmune disease in first-degree relatives of patients with multiple sclerosis. A UK survey. Brain (2000) 123(Pt 6):1102–11. doi:10.1093/brain/123.6.1102
23. Jenkins RC, Weetman AP. Disease associations with autoimmune thyroid disease. Thyroid (2002) 12(11):977–88. doi:10.1089/105072502320908312
24. Weetman AP. Diseases associated with thyroid autoimmunity: explanations for the expanding spectrum. Clin Endocrinol (Oxf) (2011) 74(4):411–8. doi:10.1111/j.1365-2265.2010.03855.x
25. Caraccio N, Dardano A, Manfredonia F, Manca L, Pasquali L, Iudice A, et al. Long-term follow-up of 106 multiple sclerosis patients undergoing interferon-beta 1a or 1b therapy: predictive factors of thyroid disease development and duration. J Clin Endocrinol Metab (2005) 90(7):4133–7. doi:10.1210/jc.2004-2326
26. Monzani F, Caraccio N, Meucci G, Lombardo F, Moscato G, Casolaro A, et al. Effect of 1-year treatment with interferon-beta1b on thyroid function and autoimmunity in patients with multiple sclerosis. Eur J Endocrinol (1999) 141(4):325–31. doi:10.1530/eje.0.1410325
27. Rotondi M, Oliviero A, Profice P, Mone CM, Biondi B, Del Buono A, et al. Occurrence of thyroid autoimmunity and dysfunction throughout a nine-month follow-up in patients undergoing interferon-beta therapy for multiple sclerosis. J Endocrinol Invest (1998) 21(11):748–52. doi:10.1007/BF03348040
28. Rotondi M, Bergamaschi R, Chiovato L. Disease modifying therapies in multiple sclerosis: could a baseline thyroid check-up drive the therapeutic choice between interferon-β and glatiramer acetate? Mult Scler (2014) 20(14):1918–9. doi:10.1177/1352458514533387
29. Frisullo G, Calabrese M, Tortorella C, Paolicelli D, Ragonese P, Annovazzi P, et al. Thyroid autoimmunity and dysfunction in multiple sclerosis patients during long-term treatment with interferon beta or glatiramer acetate: an Italian multicenter study. Mult Scler (2014) 20(9):1265–8. doi:10.1177/1352458514521311
30. Leporati P, Groppelli G, Zerbini F, Rotondi M, Chiovato L. Etiopathogenesis of Basedow’s disease. Trends and current aspects. Nuklearmedizin (2015) 54(5):204–10. doi:10.3413/Nukmed-0739-15-04
31. Marinò M, Latrofa F, Menconi F, Chiovato L, Vitti P. Role of genetic and non-genetic factors in the etiology of Graves’ disease. J Endocrinol Invest (2015) 38(3):283–94. doi:10.1007/s40618-014-0214-2
32. Bartalena L, Chiovato L, Vitti P. Management of hyperthyroidism due to Graves’ disease: frequently asked questions and answers (if any). J Endocrinol Invest (2016) 39(10):1105–14. doi:10.1007/s40618-016-0505-x
33. Bahn RS, Dutton CM, Natt N, Joba W, Spitzweg C, Heufelder AE. Thyrotropin receptor expression in Graves’ orbital adipose/connective tissues: potential autoantigen in Graves’ ophthalmopathy. J Clin Endocrinol Metab (1998) 83(3):998–1002. doi:10.1210/jcem.83.3.4676
34. Morshed SA, Davies TF. Graves’ disease mechanisms: the role of stimulating, blocking, and cleavage region TSH receptor antibodies. Horm Metab Res (2015) 47(10):727–34. doi:10.1055/s-0035-1559633
35. Inaba H, De Groot LJ, Akamizu T. Thyrotropin receptor epitope and human leukocyte antigen in Graves’ disease. Front Endocrinol (2016) 7:120. doi:10.3389/fendo.2016.00120
36. Chiovato L, Fiore E, Vitti P, Rocchi R, Rago T, Dokic D, et al. Outcome of thyroid function in Graves’ patients treated with radioiodine: role of thyroid-stimulating and thyrotropin-blocking antibodies and of radioiodine-induced thyroid damage. J Clin Endocrinol Metab (1998) 83(1):40–6. doi:10.1210/jcem.83.1.4492
37. Rotondi M, Chiovato L, Romagnani S, Serio M, Romagnani P. Role of chemokines in endocrine autoimmune diseases. Endocr Rev (2007) 28(5):492–520. doi:10.1210/er.2006-0044
38. Rotondi M, Chiovato L. The chemokine system as a therapeutic target in autoimmune thyroid diseases: a focus on the interferon-γ inducible chemokines and their receptor. Curr Pharm Des (2011) 17(29):3202–16. doi:10.2174/138161211798157559
39. Coles AJ, Wing M, Smith S, Coraddu F, Greer S, Taylor C, et al. Pulsed monoclonal antibody treatment and autoimmune thyroid disease in multiple sclerosis. Lancet (1999) 354(9191):1691–5. doi:10.1016/S0140-6736(99)02429-0
40. Rotondi M, Mazziotti G, Biondi B, Manganella G, Del Buono AD, Montella P, et al. Long-term treatment with interferon-beta therapy for multiple sclerosis and occurrence of Graves’ disease. J Endocrinol Invest (2000) 23(5):321–4. doi:10.1007/BF03343730
41. Coles AJ, Compston DA, Selmaj KW, Lake SL, Moran S, Margolin DH, et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med (2008) 359(17):1786–801. doi:10.1056/NEJMoa0802670
42. Daniels GH, Vladic A, Brinar V, Zavalishin I, Valente W, Oyuela P, et al. Alemtuzumab-related thyroid dysfunction in a phase 2 trial of patients with relapsing-remitting multiple sclerosis. J Clin Endocrinol Metab (2014) 99(1):80–9. doi:10.1210/jc.2013-2201
43. Wood LC, Ingbar SH. Hypothyroidism as a late sequela in patient with Graves’ disease treated with antithyroid agents. J Clin Invest (1979) 64(5):1429–36. doi:10.1172/JCI109601
44. Topliss DJ. Clinical update in aspects of the management of autoimmune thyroid diseases. Endocrinol Metab (Seoul) (2016) 31(4):493–9. doi:10.3803/EnM.2016.31.4.493
45. Tuohy O, Costelloe L, Hill-Cawthorne G, Bjornson I, Harding K, Robertson N, et al. Alemtuzumab treatment of multiple sclerosis: long-term safety and efficacy. J Neurol Neurosurg Psychiatry (2015) 86(2):208–15. doi:10.1136/jnnp-2014-307721
46. Somerfield J, Hill-Cawthorne GA, Lin A, Zandi MS, McCarthy C, Jones JL, et al. A novel strategy to reduce the immunogenicity of biological therapies. J Immunol (2010) 185(1):763–8. doi:10.4049/jimmunol.1000422
47. Aranha AA, Amer S, Reda ES, Broadley SA, Davoren PM. Autoimmune thyroid disease in the use of alemtuzumab for multiple sclerosis: a review. Endocr Pract (2013) 19(5):821–8. doi:10.4158/EP13020.RA
48. Tsourdi E, Gruber M, Rauner M, Blankenburg J, Ziemssen T, Hofbauer LC. Graves’ disease after treatment with alemtuzumab for multiple sclerosis. Hormones (Athens) (2015) 14(1):148–53. doi:10.14310/horm.2002.1501
49. Mahzari M, Arnaout A, Freedman MS. Alemtuzumab induced thyroid disease in multiple sclerosis: a review and approach to management. Can J Neurol Sci (2015) 42(5):284–91. doi:10.1017/cjn.2015.48
50. Obermann M, Ruck T, Pfeuffer S, Baum J, Wiendl H, Meuth SG. Simultaneous early-onset immune thrombocytopenia and autoimmune thyroid disease following alemtuzumab treatment in relapsing-remitting multiple sclerosis. Mult Scler (2016) 22(9):1235–41. doi:10.1177/1352458516638558
51. Walsh M, Chaudhry A, Jayne D. Long-term follow-up of relapsing/refractory anti-neutrophil cytoplasm antibody associated vasculitis treated with the lymphocyte depleting antibody alemtuzumab (CAMPATH-1H). Ann Rheum Dis (2008) 67(9):1322–7. doi:10.1136/ard.2007.081661
52. Kirk AD, Hale DA, Swanson SJ, Mannon RB. Autoimmune thyroid disease after renal transplantation using depletional induction with alemtuzumab. Am J Transplant (2006) 6(5 Pt 1):1084–5. doi:10.1111/j.1600-6143.2006.01258.x
53. Williams KM, Dietzen D, Hassoun AA, Fennoy I, Bhatia M. Autoimmune thyroid disease following alemtuzumab therapy and hematopoietic cell transplantation in pediatric patients with sickle cell disease. Pediatr Blood Cancer (2014) 61(12):2307–9. doi:10.1002/pbc.25102
Keywords: Graves’ disease, Alemtuzumab, multiple sclerosis, autoimmune thyroid disease, reconstitution syndrome
Citation: Rotondi M, Molteni M, Leporati P, Capelli V, Marinò M and Chiovato L (2017) Autoimmune Thyroid Diseases in Patients Treated with Alemtuzumab for Multiple Sclerosis: An Example of Selective Anti-TSH-Receptor Immune Response. Front. Endocrinol. 8:254. doi: 10.3389/fendo.2017.00254
Received: 26 July 2017; Accepted: 15 September 2017;
Published: 28 September 2017
Edited by:
Cesidio Giuliani, Università degli Studi “G. d’Annunzio” Chieti – Pescara, ItalyReviewed by:
Motoyasu Saji, The Ohio State University Columbus, United StatesAmanda Katherine Huber, University of Michigan, United States
Copyright: © 2017 Rotondi, Molteni, Leporati, Capelli, Marinò and Chiovato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luca Chiovato, bHVjYS5jaGlvdmF0b0BpY3NtYXVnZXJpLml0