 Illana Gozes1*
Illana Gozes1* Marc C. Patterson2
Marc C. Patterson2 Anke Van Dijck3
Anke Van Dijck3 R. Frank Kooy3Joseph N. Peeden4Jacob A. Eichenberger5Angela Zawacki-Downing6
R. Frank Kooy3Joseph N. Peeden4Jacob A. Eichenberger5Angela Zawacki-Downing6 Sandra Bedrosian-Sermone6
Sandra Bedrosian-Sermone6
- 1The Lily and Avraham Gildor Chair for the Investigation of Growth Factors, Elton Laboratory for Neuroendocrinology, Department of Human Molecular Genetics and Biochemistry, Sackler Faculty of Medicine, Adams Super Center for Brain Studies and Sagol School for Neuroscience, Tel Aviv University, Tel Aviv, Israel
- 2Division of Child and Adolescent Neurology, Pediatrics and Medical Genetics, Mayo Clinic Children’s Center Rochester, Rochester, MN, USA
- 3Cognitive Genetics Group, Department of Medical Genetics, University of Antwerp, Antwerp, Belgium
- 4Diagnostic Clinic, East Tennessee Children’s Hospital and Clinical Assistant Professor of Medicine at the University of Tennessee, Knoxville, TN, USA
- 5Physician Informaticist, Children’s Hospital of Georgia at Augusta University, Augusta, GA, USA
- 6ADNP Kids Research Foundation, Brush Prairie, WA, USA
Background: Activity-dependent neuroprotective protein (ADNP) is one of the most prevalent de novo mutated genes in syndromic autism spectrum disorders, driving a general interest in the gene and the syndrome.
Aim: The aim of this study was to provide a detailed developmental case study of ADNP p.Tyr719* mutation toward improvements in (1) diagnostic procedures, (2) phenotypic scope, and (3) interventions.
Methods: Longitudinal clinical and parental reports.
Results: AD (currently 11-year-old) had several rare congenital anomalies including imperforate anus that was surgically repaired at 2 days of age. Her findings were craniofacial asymmetries, global developmental delay, autistic behaviors (loss of smile and inability to make eye contact at the age of 15 months), and slow thriving as she gradually matures. Comprehensive diagnostic procedures at 3 years resulted in no definitive diagnosis. With parental persistence, AD began walking at 3.5 years (skipping crawling). At the age of 8.5 years, AD was subjected to whole exome sequencing, compared to the parents and diagnosed as carrying an ADNP p.Tyr719* mutation, a causal recurring mutation in ADNP (currently ~17/80 worldwide). Brain magnetic resonance imaging demonstrated mild generalized cerebral volume loss with reduced posterior white matter. AD is non-verbal, communicating with signs and word approximations. She continues to make slow but forward developmental progress, and her case teaches newly diagnosed children within the ADNP Kids Research Foundation.
Conclusion: This case study emphasizes the importance of diagnosis and describes, for the first time, early motor intervention therapies. Detailed developmental profile of selected cases leads to better treatments.
Introduction
Looking at neuroglial interactions, we (Gozes group) discovered activity-dependent neuroprotective protein (ADNP), as a protein secreted from glial cells in the presence of vasoactive intestinal peptide (VIP), which mediates VIP’s neuroprotective activity (1). Further cloning (2) revealed high conservation and specificity to vertebrates (3). The large human ADNP (hADNP) gene structure (~40 kb) includes five exons and four introns with alternative splicing of an untranslated second exon (chromosome 20q12-13.2, a region associated with aggressive tumor growth). As we described (2, 3), hADNP is also mutated in cancer.
Knocking out ADNP in mice resulted in embryonic death at the time of neural tube closure, revealing that ADNP is crucial for brain formation (4). ADNP haploinsufficient mice survive but show learning and memory deficits (5), in a sex-dependent manner (6). At the protein level, we have shown multiple crucial interactions for ADNP including direct binding to the chromatin remodeling complex SWI/SNF and interaction with heterochromatin protein 1 alpha (7, 8). This study was extended to show that ADNP interacts with all HP1 proteins toward histone posttranslational modification (9). At the transcriptional level, ADNP binds to the locus control region of the beta globin gene to regulate globin transcription (10) as well as the promoter regions of apolipoprotein E, cathepsin C, cathepsin Z, metallothionein 1, neurogenin 1, and myosin regulatory light chain 2 (8). At the RNA splicing level (11), ADNP interacts with Brahma, a component of the SWI/SNF complex regulating alternative splicing that shows a similar developmental expression pattern to ADNP. Immunoprecipitations further suggested binding between ADNP and polypyrimidine tract-binding protein-associated splicing factor (PSF), with PSF being a direct regulator of the microtubule-associated tau transcript splicing. Further interaction with the protein translation machinery was shown with the eukaryotic translation initiation factor 4E (eIF4E) (6) through direct binding as well as sex and age-dependent regulation. Notably, eIF4E is linked to autism (12). In the neuronal cell cytoplasm, ADNP is critical for neurite outgrowth and maintenance (13) through direct interaction by its SIP motif with microtubule end binding proteins (14, 15). The microtubule system is tightly associated with the autophagy system (16), and ADNP binds the microtubule-associated protein 1 light chain 3B (17). With these key regulatory functions ADNP controls the expression of >400 genes during embryonic development (8) and of thousands of hippocampal genes postnatally, impacting pathways associated with ion channels-synaptic transmission in a sex- and age-dependent manner (18).
From a clinical point of view, an ADNP gene deletion was first implicated in delayed cognitive development in a case study in 2007 (19). Further findings included the first de novo p.Lys408Valfs*31 mutation in the ADNP gene in a large cohort of autistic patients (20). Large-scale sequencing study (21), analyzing 2,446 probands, identified an additional p.Tyr719* de novo ADNP mutation and this is the subject of our current publication. Helsmoortel et al. (22) grouped 10 patients with mutations in ADNP. As all patients suffered from autism with intellectual deficiencies and shared characteristic facial features, it was concluded that mutations in ADNP cause a syndromic form of autism. Two additional patients were further described, sharing the reported characteristics (23, 24). Interestingly, Pescosolido et al. described another case of p.Tyr719* de novo ADNP mutation, suggesting that this is a recurrent mutation. Coe et al. (25) also reported five patients with a truncating ADNP mutation in a screening of 4,716 patients with autism/ID. De Rubeis et al. (26) identified three more patients of a total of 3,871 screened and the DDD project reported four novel cases out of 1,133 screened (27). We have summarized the above in an introductory remark to the story of Tony Sermone, presenting an additional ADNP mutation L349Rfs*49 (28). A short synopsis of the clinical phenotype was recently published (29), additional mutations are discovered and information extended (27, 30–33) and an extensive description is in preparation (Van Dijck et al., in preparation). Interestingly, a recent publication suggested that ADNP is one of three major genes associated with autism spectrum disorder (ASD) (34), complementing the original estimation of the ADNP-related syndrome constituting 0.17% of ASD cases (22). These findings mark ADNP as one of the most frequent ASD-associated genes known to date. Therefore, further understanding of the ADNP-related syndrome is of general interest both from a case study point of view as well as from a population perspective. Here, we chose to present one case study and concentrate, for the first time, on delays in motor development.
Methods
Longitudinal clinical and parental reports were collected focusing on diagnosis, phenotypic scope, and interventions. Time points of emphasis: birth, 1–3.5 years and 8.5 to date. All materials were given with parental informed consent.
Results
Initial Clinical Data
Father’s and mother’s age at birth was 32 and 31, respectively. No consanguinity. No affected siblings—parents were carriers of CF. AD was born at 37–38 weeks vaginally (October 1, 2005). She was 50 cm in length and weighed 3.475 kg. Her head circumference was 34 cm. She had Apgar scores of 7 and 8 at 1 and 5 min of life. She was noticed to have low anal atresia and had surgery to repair this at 2 days of age [Table 1 (A), physical anomalies]. She had significant GERD as an infant and young child. At 2 years of age, AD was diagnosed with autism.
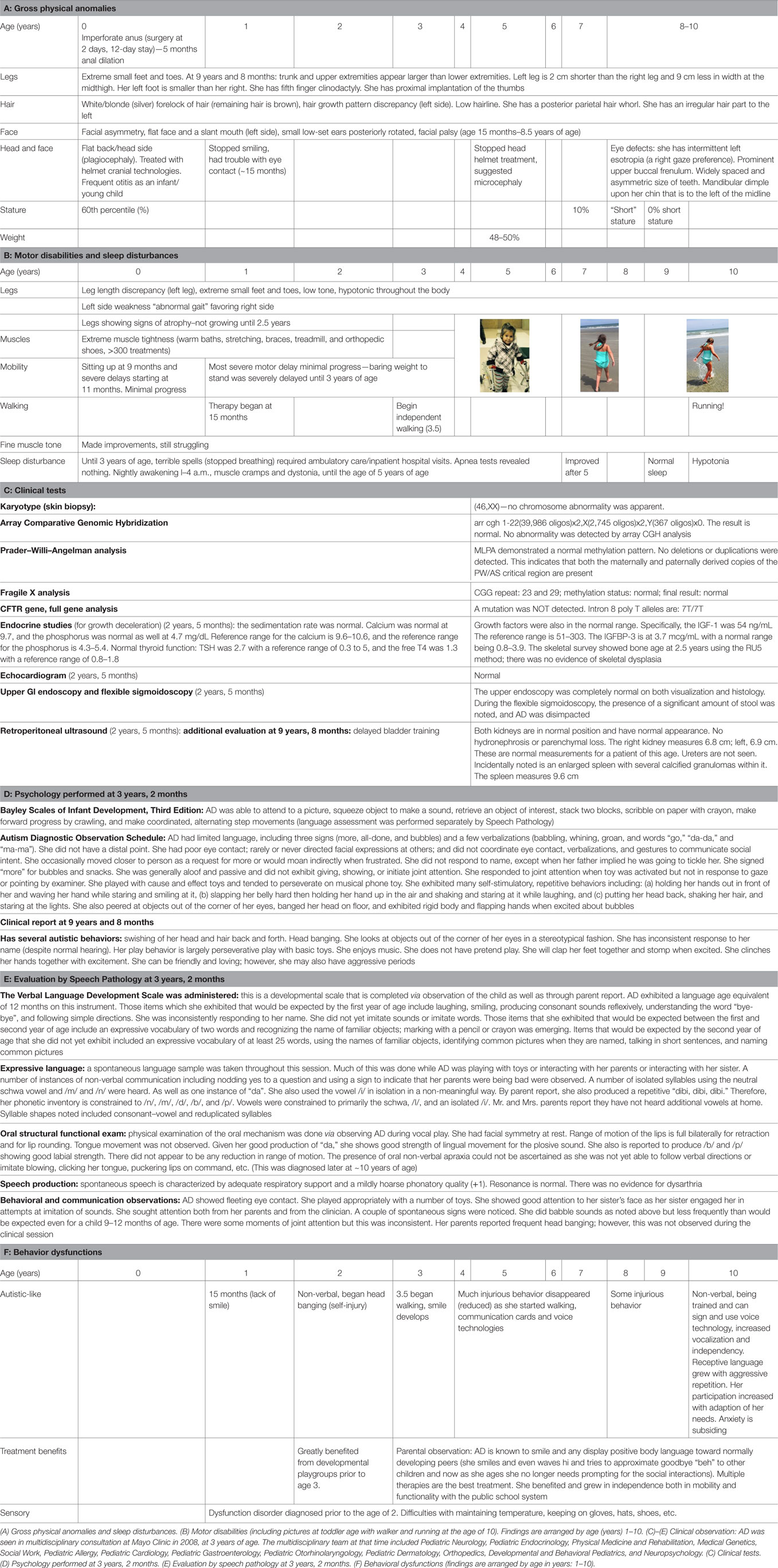
Table 1. Parental/caregiver observations/clinician observations.
Developmental Delay: Focus on Motor Function
AD started sitting at 7–9 months. While her younger brother walked at 1 year of age, she continued to lag severely behind, she could not crawl, walk, or even bare weight to stand on her legs. It seemed that at about 2 years of age, AD’s legs began to show some signs of atrophy, not increasing in shoe size (unlike her siblings), until she began to walk. Currently, at the age of 11, one can still see the disproportion of AD’s small tapered legs from the knees to the feet, compared to the rest of her body. This finding attests to the importance of ADNP in muscle and bone development and to the regulation of key gene/proteins beyond embryonic development (8, 18).
AD started walking with a walker at 2.5 years of age, skipping crawling [Table 1 (B); Figure 1A]. Walking short distances resulted in skin redness and muscle pain. She began walking independently at the age of 3.5 years. The following year presented daily challenges in her “functional” walking. She often could not stand for more than a few seconds. Climbing stairs was a third milestone, using the strong leg and pushing the weaker leg after. Until about the age of 5, AD had severe muscle tightness that mimicked symptoms of a neuromuscular disorder. The muscle tightness was associated with severe sleep disturbances [Table 1 (B), last raw]. At 9 years and 3 months, the parents noticed that she had more of a delay with gross motor skills than with fine (albeit both delayed). She walked independently, but with a broad based and antalgic gait [Table 1 (B); Figure 1A]. At 10.5 years of age, she was ambulatory and quite flexible with increased range of motion noted particularly in the small joints. Physically, hands were broad, with short and broad fingers and fifth finger clinodactyly. Distal phalanges appeared short and fingernails were spatulate. Feet showed short fourth and fifth toes and deep setting. Toe tip and fingertip pads were prominent [see also Table 1 (A)]. Through parental persistence and AD’s willpower (see treatments below), now at age 11, she is running and riding a bicycle. She still struggles with reciprocating down stairs and is mostly non-verbal (see below).
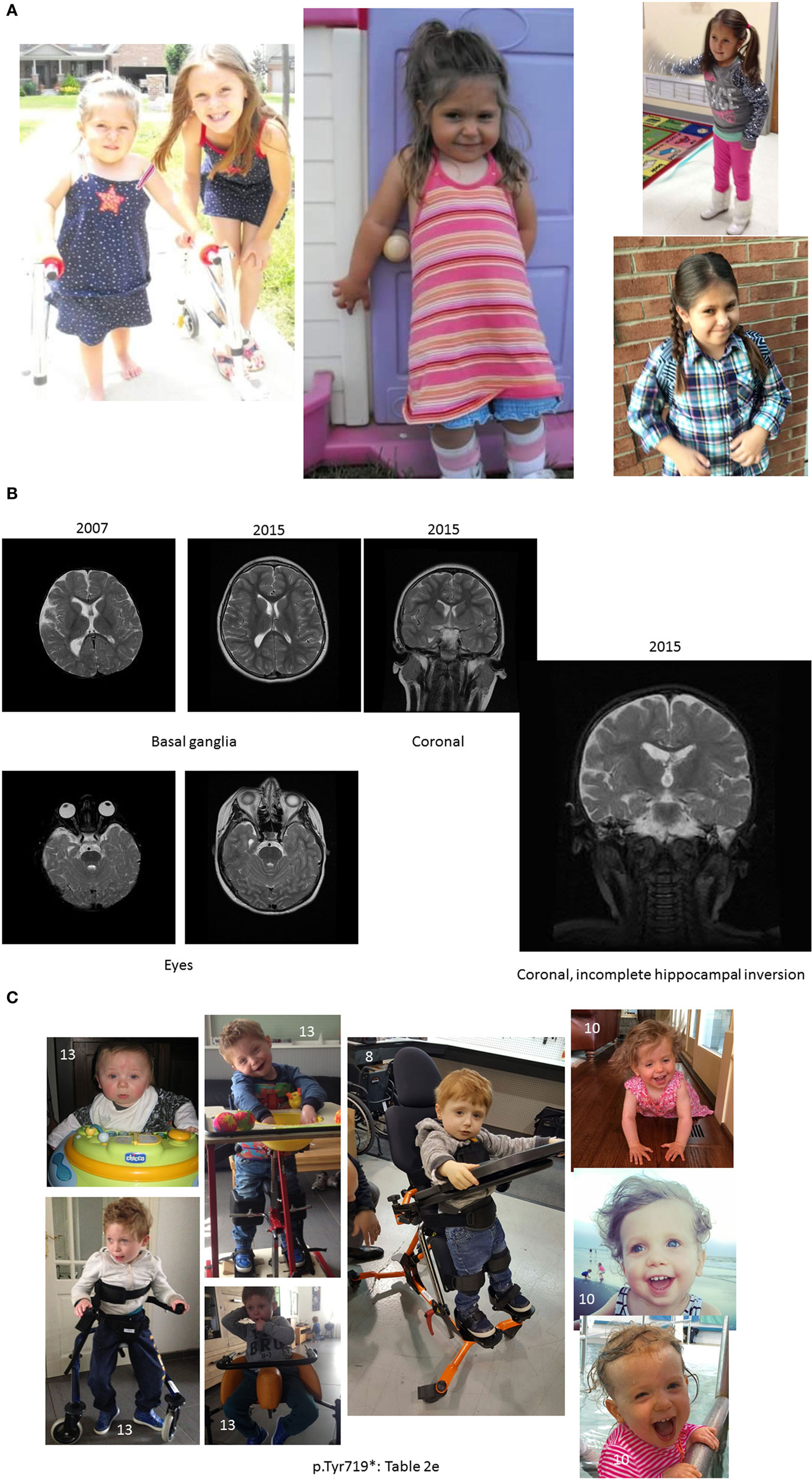
Figure 1. Pictures depicting AD’s development, brain development, and comparison to other children with the same mutation. (A) AD’s development in pictures: starting top left: young AD with her sister’s “push” to walk. AD is barefoot and her legs turned red from a very short walk. Her muscles in her ankles got tight and she exhibited low muscle tone and needed to be stretched several times a day, or she would suffer excruciating pain at night. The painful day walk and nights were full of crying periods. At the same time, a picture was taken, when she finally could lean against things with her leg braces and at a similar time point. Third picture on the top, AD at her first beauty pageant, after months of her sister and mother teaching her how to spin and dance with a baton ribbon. Fourth picture on the bottom, AD at her eighth birthday, just before her diagnosis and submission to brain scans. AD is successful at attending regular primary schools. Her smile keeps the family and caregivers going. (B) Magnetic resonance imaging results (volumetric T2 performed at 2007 and 2015). (C) Pictures of additional children with ADNP p.Tyr719* mutation (featured also in Table 2 (E), all materials given with parental informed consent).
Clinical Test Results and Growth Curve
Age of 3 Years and 2 Months: (Mayo Clinic)
1. Static encephalopathy of presumed antenatal onset and genetic etiology
2. Severe global developmental delay (the diagnosis of intellectual disability is typically not made at this age)
3. ASD
4. Multiple congenital anomalies (imperforate anus, dental anomalies, facial asymmetry, and poliosis)
5. Microcephaly [head circumference, 45.5 cm (<2%)]
6. Generalized hypotonia, axial greater than appendicular
7. Normal stature and mass [weight 14.7 kg (61%); length, 90 cm (10%)]
8. Hyperopic astigmatism
Extensive investigations showed normal or negative results [Table 1 (C)], those also included tests for Rett syndrome and a skeletal survey (bone, age 2.5 years). Psychological and speech evaluations were also included showing global delays as indicated below.
At 9 Years and 3 Months
AD’s weight was 27.1 kg. Her height 114.7 cm, occipitofrontal circumference 50 cm. She has continued problems with constipation/obstipation.
At 10.5 Years of Age
She was short statured (height 128 cm)1 with normal weight and head circumference (34.5 kg and 52 cm, respectively). She was still mostly non-verbal. She communicated with her family with signs and word approximations. She demonstrated autistic stereotypies as well as sensory-seeking behaviors including head banging and skin picking.
Currently, AD continues to make slow but forward developmental progress, demonstrating the importance of early and extensive therapies (see below).
Psychological Test Results (3 Years and 2 Months, Mayo Clinic)
Bayley Scales of Infant Development, Third Edition: (SS = 55, first percentile) 11-month-old level [Table 1 (D)]. Motor skills (SS = 49, <0.1st percentile) 15-month-old level for fine motor skills to 9-month-old level for gross motor skills.
Child Development Inventory (MCDI): general development 16-month-old level, social maturity, 23-month-old level; self-help skills, 15-month-old level; fine motor and language skills, 16-month-old level; and gross motor skills 11–12 months.
Behavior Assessment System for Children, Second Edition–Parent and Teacher Rating Scales: “at risk” for attention problems and sad/irritable mood.
Conners Teachers Rating Scale, Revised: limited adaptive skills, including socialization and functional communication.
School Situation Questionnaire: AD’s behavior was not reported to be problematic in most school situations.
Autism Diagnostic Observation Schedule: communication (RS = 4, cutoffs = 2/4), reciprocal social interaction (RS = 14, cutoffs = 4/7), combined algorithm (RS = 18, cutoffs = 7/12); limited play skills (RS = 4) and stereotyped behavior and restricted interests (RS = 5). Consistent with ASD [Table 1 (D)].
Evaluation by Speech Pathology (3 Years and 2 Months)
The Verbal Language Development Scale: age equivalent of 12 months.
Expressive language: a number of instances of non-verbal communication observed and a number of isolated syllables; no recognizable words.
Oral structural functional exam: facial symmetry at rest; range of motion of the lips was full bilaterally for retraction and for lip rounding; reported adequate tongue movement and labial strength. No reduction in range of motion.
Speech production: adequate respiratory support and a mildly hoarse phonatory quality (+1). Resonance is normal. No evidence for dysarthria.
Behavioral and communication observations: fleeting eye contact; played appropriately with a number of toys; good attention to her sister; sought attention both from her parents and from examiner. Babbled sounds less frequently than would be expected even for a child of 9–12 months. Inconsistent joint attention. At ~10 years of age, verbal apraxia was diagnosed [Table 1 (E)].
Brain Magnetic Resonance Imaging (MRI) Results: Mild Generalized Cerebral Volume Loss with Reduced Posterior White Matter at 15.5 Months and at 9 Years
Scans demonstrated mild generalized cerebral volume loss with reduced posterior white matter. The 2007 study showed periatrial signal hyperintensity that had resolved by 2015. The corpus callosum appeared relatively proportional to the overall volume loss but slightly foreshortened, consistent with brachycephaly. The right lateral ventricle was more asymmetric than normal and much of that can be attributed to the incompletely inverted hippocampus on that side with a low-lying fornix. The frontal and temporal extra-axial spaces were enlarged in 2007, but had resolved by 2015. Craniofacial dysmorphism (prominent forehead, broad nose, and thin upper lip) was apparent on the 2015 scan [Table 1 (A); Figure 1B]. Similarly, at 10.5 years clinical observations, craniofacies showed tall forehead with high anterior hairline, heavy brows giving eyes a deep-set appearance, right exotropia, and long eyelashes. Philtrum was short and deep, and the upper lip was thin. There were creases on the earlobes.
Molecular Test Results: Part of a Growing Syndrome
Primary diagnosis (Dr. Peeden) was obtained at 8.5 years showing ADNP de novo gene mutation: g.49509094G>C; cDNA: c.2157C>G; protein: p.Tyr719*. Diagnosis was obtained by whole exome sequencing compared to the parents. With the ADNP kids belonging to a family of affected children suffering many similar abnormalities, further solutions are on the horizon. To provide information, advocacy and emotional support to families worldwide [e.g., Ref. (28)], an ADNP Syndrome Parents Group was established on the social networking site Facebook™. This is facing the time, like other support groups, for versatile diseases (35). Currently ~80 families are assembled, connected through the original set up of ADNP ASD mutations2. The parents formed a table of traits3, which are shown for AD [Table 2 (A–D)]. Several other ADNP-associated traits are visible when looking at Table 2 including auditory impediments, suggesting an involvement of ADNP in the development of all senses. AD was spared heart abnormalities and seizures; however, as these affect other ADNP children, she is closely observed (28).
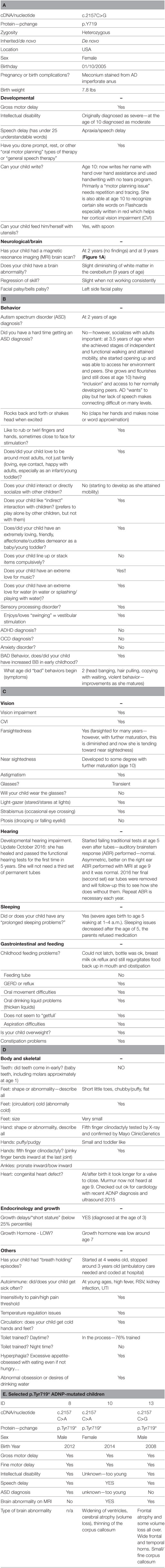
Table 2. Activity-dependent neuroprotective protein (ADNP) phenotype data questionnaire (Facebook™, A–D); (E) selected children with ADNP p.Tyr719* mutation (featured also in Figure 1C); Facebook data.
AD shares the same mutation with several other children [e.g., Ref. (24)]. A parent questionnaire of 17 individuals with ADNP p.Tyr719* or a close mutation indicated cognitive (including speech) and motor delays, as well as autistic features and >50% brain abnormalities, which are further investigated. Figure 1C shows representative pictures of three additional ADNP p.Tyr719* children exhibiting potential facial similarities and using standing and walking devices [Table 2 (E)]. The ADNP p.Tyr719* is an integral part of the ADNP-related syndrome encompassing other ADNP mutations (Van Dijck et al., in preparation).
Treatment Modalities and Future Perspectives
In general, a helmet treatment was implemented for cranial asymmetry [Table 1 (A)].
Treatments for Motor Disabilities
Proved to be successful and should be implemented in future cases. In this respect, all children with ADNP mutations show motor delays (see text footnote 3).
The painful doubt whether AD would be wheelchair bound drove the parents to seek help. Physical therapy began at the age of 15 months. She had multitude therapies at home, collaborative clinic, early intervention, developmental playgroups, and private clinic/water therapy. For example, she had physical and occupational therapy, hippotherapy (involving an occupational therapist, a physiotherapist, or a speech and language therapist working with AD and a horse to present challenges and to promote different postural responses) and aquatic therapy. At one point, she was given up to 10 therapies a week. At the age of 2.5, her therapist and the orthotics4 tailor made shoes with an inside lift to accommodate her left leg shortness. She then began to wear ankle foot orthosis brace to her knee, covering her entire foot. The physical therapist devised fabric braces with Velcro with metal bars that did not allow her to bend her knee, forcing her to bare weight. Her father has further built a “standing device” made of wood and PVC pipes to aid in her “timed” standing to ensure that she would not incur atrophy in her lower limbs/legs. Following this, parents were advised of gait trainers and other newly engineered products that help children like AD. AD further used a surfboard therapy machine that helped her core balance and many mirrors and began making a connection. Basic standing on her own was her first milestone, followed by the use of a pediatric walker [Table 1 (B), picture].
Walking short distances resulted in skin redness and muscle pain, which were relieved with warm baths and pool therapy. The parents further used a treadmill program (e.g., http://www.taap-project.com/) in physical therapy to strengthen AD and built new pathways and signals to help her motor planning and focus on the repetition of steps. Metronome was a program5 that they have also used that proved to be successful. Notably, the physical therapist persisted, despite AD’s severe unknown motor movement disorder and continued to increase the “frequency” of land and water therapies toward success.
She was treated with over 300 h combined physical and aquatic therapies between the ages of 1–5 years. Without a medical billing code for doctors to use, the term “global delay” did not guarantee insurance coverage nor did it describe the medical complexity of AD. This resulted in expenses of over $2,000 per summer during the walk therapies. Currently, AD gets biweekly physical therapies to maintain and further improve her mobility and has begun growth hormone therapy (age 10), aiming to bring her back to the growth chart.
Treatment of Speech Disabilities
AD was recently diagnosed with apraxia of speech [Table 1 (E)] and traditional speech therapy may not benefit her as much as focus on oral motor “planning.” She is now receiving this specialized therapy as her evaluation showed she desires to speak, tries and makes approximations, but similar to walking cannot put it together.
The Z-VIBES6 oral motor tool has been most successful for the children with ADNP mutation who speak (closed ADNP Syndrome PARENTS GROUP, Facebook). AD has used it as a small child, with no obvious results, but now it seems to be having a significant effect.
Additionally, the Kaufmann kit,7 which uses cards to teach children to combine consonants and vowels to form words while controlling for oral-motor difficulty, is also helping AD. As a small child she was not able to understand, but now these cards are used in combination with PROMPT therapy (Prompts for Restructuring Oral Muscular Phonetic Targets8). With physical cueing and repetition of the cards, AD now has some approximations for sounds such as “meh,” “na,” and “beh,” for “me,” “no,” and “bye,” respectively.
Over the years, AD’s educational team has indicated that the autism symptoms have become less prevalent. With access to a speaking device (a card system) and multimodal communication system, AD is now flourishing. Her improved communication shows an amazing personality, which is humorous and lively.
Face to the Future: Treatment Using Novel Biologically Active ADNP Peptides
From a scientific point of view, we strive to have tailored drugs to treat the ADNP children, early enough to avoid confounding developmental delays. In this respect, we (Gozes) have been developing NAP (NAPVSIPQ) the shortest active snippet of ADNP (1). NAP increases ADNP’s activity at the cellular level in terms of cellular protection (14) as well as synaptic plasticity (14). On the whole animal level, it provides cognitive protection in mice with ADNP deficiencies (5). NAP has also shown protective activity in over 20 cell culture systems and in over 25 animal models. Notable examples include schizophrenia (36–38), Alzheimer’s disease (39–41), frontotemporal dementia (42, 43), amyotrophic lateral sclerosis (44), and most importantly cognitive deficiencies associated with ASD (5) as well as developmental delays linked to prenatal and postnatal toxicities (45–47). NAP, also called davunetide, has shown activity in clinical trials. It increased cognitive scores in a Phase IIa clinical trial in patients suffering from amnestic mild cognitive impairment (48, 49) and protected functional daily activities (50) as well as brain cellular activity (measured by MRI) (51), in schizophrenia patients. NAP (davunetide) is safe and bioavailable (52, 53). Clinical studies in the ADNP-related syndrome are currently planned with NAP—now called CP201 (Coronis Neurosciences9).
Conclusion
The described complex syndrome of motor dysfunction, autism, speech delay, and facial dysmorphism should trigger gene sequencing and early diagnosis of children with the ADNP syndrome (also known as the Helsmoortel–Van der Aa syndrome10).
The case of AD highlights global developmental delays, with emphasis on motor functions, which was not described before. While AD had not always been referred to the appropriate specialized therapies, her family persistence and ultimate success in motor development should help guide newly diagnosed children. This report teaches modes of interventions and paves the path to basic discoveries on ADNP’s involvement in muscle development.
AD’s major impediment nowadays is her inability to talk, a trait that has been lagging behind. In this respect, better understanding of ADNP’s association with muscle development may shed light on speech development as well.
The more we understand about the ADNP protein and gene, the better we can help the families and patients with syndromic developmental dysfunctions gain enhanced functionality and integration into a healthy society coupled with ADNP replacement/enhancement therapies, such as CP201 (Coronis Neurosciences; see text footnote 9).
Ethics Statement
This study was supported by Tel Aviv University, the study is descriptive the parent is an author. Informed consents were given for all data by the respective parents.
Author Notes
Recently (at about age 11) AD was diagnosed with a suspected form of dysarthria—https://www.ncbi.nlm.nih.gov/pubmedhealth/PMHT0027115/. The number of ADNP syndrome children has now increased to 107 within the ADNP network (http://www.adnpfoundation.org/), including 23 children sharing a similar mutation with AD (Table 2).
Author Contributions
IG compiled the data and wrote/edited the paper; MP provided the in-depth clinical analysis at 3 years of age and the longitudinal MRI data; AVD and FK read the manuscript and provided clinical input; JP provided the diagnosis and longitudinal clinical data, JE provided critical editorial inputs; AZ-D initiated the study and provided the most extensive parental story, and SB-S compiled the data in Table 2, read, and commented on the paper.
Conflict of Interest Statement
Coronis Neurosciences is developing NAP (davunetide, CP201) for autism. Professor Gozes is the Chief Scientific Officer (Consultant).
Acknowledgments
We thank Dr. Sara S. Cathey, Clinical Geneticist, Greenwood Genetic Center, Charleston Office, North Charleston, SC, USA, AD’s current geneticist. We also thank for his input Dr. Gonzalez-Lamuño Leguina, Domingo, Hospital U. “Marqués de Valdecilla,” Santander, Spain. We further thank Gal Hacohen Kleiman and Shlomo Sragovich for critical reading of the manuscript. This study was supported by ERA-NET Neuron, Grant acronym AUTISYN, AMN Foundation and the Spanish, French (Drs. Ronith and Armand Stemmer) and Canadian Friends of Tel Aviv University Additional support was obtained from the ADNP Kids Research Foundation http://www.adnpfoundation.org/. CP201 is planned for clinical trials (Coronis Neuroscience, Coronis Neurosciences, http://www.coronisns.com/, IG, Chief Scientific Officer). From the family point of view, AD’s story is told expressively and with great sensitivity on the parent WEB page—http://www.adnpkids.com/. We thank the reviewers of this paper for excellent input. A comprehensive description of the ADNP-related syndrome is currently in preparation (Van Dijck et al., 2017).
Footnotes
- ^https://www.cdc.gov/growthcharts/data/set1clinical/cj41c022.pdf.
- ^http://www.omim.org/entry/611386.
- ^http://www.adnpkids.com/.
- ^https://en.wikipedia.org/wiki/Orthotics.
- ^https://www.youtube.com/watch?v=WR1lC6UYh9I.
- ^http://www.arktherapeutic.com/z-vibes-all/.
- ^http://www.northernspeech.com/early-intervention-language-and-speech/kaufman-k-slp-treatment-kit-1-ndash-basic-level/.
- ^http://www.promptinstitute.com/?page=FamiliesWIP.
- ^http://www.coronisns.com/.
- ^https://rarediseases.info.nih.gov/diseases/12931/adnp-syndrome.
References
1. Bassan M, Zamostiano R, Davidson A, Pinhasov A, Giladi E, Perl O, et al. Complete sequence of a novel protein containing a femtomolar-activity-dependent neuroprotective peptide. J Neurochem (1999) 72:1283–93. doi: 10.1046/j.1471-4159.1999.0721283.x
2. Zamostiano R, Pinhasov A, Gelber E, Steingart RA, Seroussi E, Giladi E, et al. Cloning and characterization of the human activity-dependent neuroprotective protein. J Biol Chem (2001) 276:708–14. doi:10.1074/jbc.M007416200
3. Gozes I, Yeheskel A, Pasmanik-Chor M. Activity-dependent neuroprotective protein (ADNP): a case study for highly conserved chordata-specific genes shaping the brain and mutated in cancer. J Alzheimers Dis (2015) 45:57–73. doi:10.3233/JAD-142490
4. Pinhasov A, Mandel S, Torchinsky A, Giladi E, Pittel Z, Goldsweig AM, et al. Activity-dependent neuroprotective protein: a novel gene essential for brain formation. Brain Res Dev Brain Res (2003) 144:83–90. doi:10.1016/S0165-3806(03)00162-7
5. Vulih-Shultzman I, Pinhasov A, Mandel S, Grigoriadis N, Touloumi O, Pittel Z, et al. Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J Pharmacol Exp Ther (2007) 323:438–49. doi:10.1124/jpet.107.129551
6. Malishkevich A, Amram N, Hacohen-Kleiman G, Magen I, Giladi E, Gozes I. Activity-dependent neuroprotective protein (ADNP) exhibits striking sexual dichotomy impacting on autistic and Alzheimer’s pathologies. Transl Psychiatry (2015) 5:e501. doi:10.1038/tp.2014.138
7. Mandel S, Gozes I. Activity-dependent neuroprotective protein constitutes a novel element in the SWI/SNF chromatin remodeling complex. J Biol Chem (2007) 282:34448–56. doi:10.1074/jbc.M704756200
8. Mandel S, Rechavi G, Gozes I. Activity-dependent neuroprotective protein (ADNP) differentially interacts with chromatin to regulate genes essential for embryogenesis. Dev Biol (2007) 303:814–24. doi:10.1016/j.ydbio.2006.11.039
9. Mosch K, Franz H, Soeroes S, Singh PB, Fischle W. HP1 recruits activity-dependent neuroprotective protein to H3K9me3 marked pericentromeric heterochromatin for silencing of major satellite repeats. PLoS One (2011) 6:e15894. doi:10.1371/journal.pone.0015894
10. Dresner E, Malishkevich A, Arviv C, Leibman Barak S, Alon S, Ofir R, et al. Novel evolutionary-conserved role for the activity-dependent neuroprotective protein (ADNP) family that is important for erythropoiesis. J Biol Chem (2012) 287:40173–85. doi:10.1074/jbc.M112.387027
11. Schirer Y, Malishkevich A, Ophir Y, Lewis J, Giladi E, Gozes I. Novel marker for the onset of frontotemporal dementia: early increase in activity-dependent neuroprotective protein (ADNP) in the face of Tau mutation. PLoS One (2014) 9:e87383. doi:10.1371/journal.pone.0087383
12. Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature (2013) 493:371–7. doi:10.1038/nature11628
13. Mandel S, Spivak-Pohis I, Gozes I. ADNP differential nucleus/cytoplasm localization in neurons suggests multiple roles in neuronal differentiation and maintenance. J Mol Neurosci (2008) 35:127–41. doi:10.1007/s12031-007-9013-y
14. Oz S, Kapitansky O, Ivashco-Pachima Y, Malishkevich A, Giladi E, Skalka N, et al. The NAP motif of activity-dependent neuroprotective protein (ADNP) regulates dendritic spines through microtubule end binding proteins. Mol Psychiatry (2014) 19:1115–24. doi:10.1038/mp.2014.97
15. Gozes I, Sragovich S, Schirer Y, Idan-Feldman A. D-SAL and NAP: two peptides sharing a SIP domain. J Mol Neurosci (2016) 59(2):220–31. doi:10.1007/s12031-015-0701-8
16. Esteves AR, Gozes I, Cardoso SM. The rescue of microtubule-dependent traffic recovers mitochondrial function in Parkinson’s disease. Biochim Biophys Acta (2014) 1842:7–21. doi:10.1016/j.bbadis.2013.10.003
17. Merenlender-Wagner A, Malishkevich A, Shemer Z, Udawela M, Gibbons A, Scarr E, et al. Autophagy has a key role in the pathophysiology of schizophrenia. Mol Psychiatry (2015) 20:126–32. doi:10.1038/mp.2013.174
18. Ponting CP, Goodstadt L. Separating derived from ancestral features of mouse and human genomes. Biochem Soc Trans (2009) 37:734–9. doi:10.1042/BST0370734
19. Borozdin W, Graham JM Jr, Böhm D, Bamshad MJ, Spranger S, Burke L, et al. Multigene deletions on chromosome 20q13.13-q13.2 including SALL4 result in an expanded phenotype of Okihiro syndrome plus developmental delay. Hum Mutat (2007) 28:830. doi:10.1002/humu.9502
20. O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature (2012) 485:246–50. doi:10.1038/nature10989
21. O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science (2012) 338:1619–22. doi:10.1126/science.1227764
22. Helsmoortel C, Vulto-van Silfhout AT, Coe BP, Vandeweyer G, Rooms L, van den Ende J, et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat Genet (2014) 46:380–4. doi:10.1038/ng.2899
23. Vandeweyer G, Helsmoortel C, Van Dijck A, Vulto-van Silfhout AT, Coe BP, Bernier R, et al. The transcriptional regulator ADNP links the BAF (SWI/SNF) complexes with autism. Am J Med Genet C Semin Med Genet (2014) 166C:315–26. doi:10.1002/ajmg.c.31413
24. Pescosolido MF, Schwede M, Johnson Harrison A, Schmidt M, Gamsiz ED, Chen WS, et al. Expansion of the clinical phenotype associated with mutations in activity-dependent neuroprotective protein. J Med Genet (2014) 51:587–9. doi:10.1136/jmedgenet-2014-102444
25. Coe BP, Witherspoon K, Rosenfeld JA, van Bon BW, Vulto-van Silfhout AT, Bosco P, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet (2014) 46:1063–71. doi:10.1038/ng.3092
26. De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature (2014) 515:209–15. doi:10.1038/nature13772
27. Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature (2015) 519:223–8. doi:10.1038/nature14135
28. Gozes I, Helsmoortel C, Vandeweyer G, Van der Aa N, Kooy F, Sermone SB. The compassionate side of neuroscience: Tony Sermone’s undiagnosed genetic journey-ADNP mutation. J Mol Neurosci (2015) 56:751–7. doi:10.1007/s12031-015-0586-6
29. Van Dijck A, Helsmoortel C, Vandeweyer G, Kooy F. ADNP-Related Intellectual Disability and Autism Spectrum Disorder. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington (2016).
30. Krajewska-Walasek M, Jurkiewicz D, Piekutowska-Abramczuk D, Kucharczyk M, Chrzanowska KH, Jezela-Stanek A, et al. Additional data on the clinical phenotype of Helsmoortel-Van der Aa syndrome associated with a novel truncating mutation in ADNP gene. Am J Med Genet A (2016) 170:1647–50. doi:10.1002/ajmg.a.37641
31. Alvarez-Mora MI, Calvo Escalona R, Puig Navarro O, Madrigal I, Quintela I, Amigo J, et al. Comprehensive molecular testing in patients with high functioning autism spectrum disorder. Mutat Res (2016) 78(4–785):46–52. doi:10.1016/j.mrfmmm.2015.12.006
32. D’Gama AM, Pochareddy S, Li M, Jamuar SS, Reiff RE, Lam AT, et al. Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron (2015) 88:910–7. doi:10.1016/j.neuron.2015.11.009
33. Iossifov I, Levy D, Allen J, Ye K, Ronemus M, Lee YH, et al. Low load for disruptive mutations in autism genes and their biased transmission. Proc Natl Acad Sci U S A (2015) 112:E5600–7. doi:10.1073/pnas.1516376112
34. Larsen E, Menashe I, Ziats MN, Pereanu W, Packer A, Banerjee-Basu S. A systematic variant annotation approach for ranking genes associated with autism spectrum disorders. Mol Autism (2016) 7:44. doi:10.1186/s13229-016-0103-y
35. Craig D, Strivens E. Facing the times: a young onset dementia support group: facebook style. Australas J Ageing (2016) 35:48–53. doi:10.1111/ajag.12264
36. Merenlender-Wagner A, Pikman R, Giladi E, Andrieux A, Gozes I. NAP (davunetide) enhances cognitive behavior in the STOP heterozygous mouse – a microtubule-deficient model of schizophrenia. Peptides (2010) 31:1368–73. doi:10.1016/j.peptides.2010.04.011
37. Vaisburd S, Shemer Z, Yeheskel A, Giladi E, Gozes I. Risperidone and NAP protect cognition and normalize gene expression in a schizophrenia mouse model. Sci Rep (2015) 5:16300. doi:10.1038/srep16300
38. Merenlender-Wagner A, Shemer Z, Touloumi O, Lagoudaki R, Giladi E, Andrieux A, et al. New horizons in schizophrenia treatment: autophagy protection is coupled with behavioral improvements in a mouse model of schizophrenia. Autophagy (2014) 10:2324–32. doi:10.4161/15548627.2014.984274
39. Matsuoka Y, Gray AJ, Hirata-Fukae C, Minami SS, Waterhouse EG, Mattson MP, et al. Intranasal NAP administration reduces accumulation of amyloid peptide and tau hyperphosphorylation in a transgenic mouse model of Alzheimer’s disease at early pathological stage. J Mol Neurosci (2007) 31:165–70.
40. Matsuoka Y, Jouroukhin Y, Gray AJ, Ma L, Hirata-Fukae C, Li HF, et al. A neuronal microtubule-interacting agent, NAPVSIPQ, reduces tau pathology and enhances cognitive function in a mouse model of Alzheimer’s disease. J Pharmacol Exp Ther (2008) 325:146–53. doi:10.1124/jpet.107.130526
41. Gozes I, Giladi E, Pinhasov A, Bardea A, Brenneman DE. Activity-dependent neurotrophic factor: intranasal administration of femtomolar-acting peptides improve performance in a water maze. J Pharmacol Exp Ther (2000) 293:1091–8.
42. Shiryaev N, Jouroukhin Y, Giladi E, Polyzoidou E, Grigoriadis NC, Rosenmann H, et al. NAP protects memory, increases soluble tau and reduces tau hyperphosphorylation in a tauopathy model. Neurobiol Dis (2009) 34:381–8. doi:10.1016/j.nbd.2009.02.011
43. Quraishe S, Cowan CM, Mudher A. NAP (davunetide) rescues neuronal dysfunction in a Drosophila model of tauopathy. Mol Psychiatry (2013) 18:834–42. doi:10.1038/mp.2013.32
44. Jouroukhin Y, Ostritsky R, Assaf Y, Pelled G, Giladi E, Gozes I. NAP (davunetide) modifies disease progression in a mouse model of severe neurodegeneration: protection against impairments in axonal transport. Neurobiol Dis (2013) 56:79–94. doi:10.1016/j.nbd.2013.04.012
45. Sokolowska P, Passemard S, Mok A, Schwendimann L, Gozes I, Gressens P. Neuroprotective effects of NAP against excitotoxic brain damage in the newborn mice: implications for cerebral palsy. Neuroscience (2011) 173:156–68. doi:10.1016/j.neuroscience.2010.10.074
46. Rotstein M, Bassan H, Kariv N, Speiser Z, Harel S, Gozes I. NAP enhances neurodevelopment of newborn apolipoprotein E-deficient mice subjected to hypoxia. J Pharmacol Exp Ther (2006) 319:332–9. doi:10.1124/jpet.106.106898
47. Spong CY, Abebe DT, Gozes I, Brenneman DE, Hill JM. Prevention of fetal demise and growth restriction in a mouse model of fetal alcohol syndrome. J Pharmacol Exp Ther (2001) 297:774–9.
48. Gozes I, Stewart A, Morimoto B, Fox A, Sutherland K, Schmeche D. Addressing Alzheimer’s disease tangles: from NAP to AL-108. Curr Alzheimer Res (2009) 6:455–60. doi:10.2174/156720509789207895
49. Morimoto BH, Schmechel D, Hirman J, Blackwell A, Keith J, Gold M, et al. A double-blind, placebo-controlled, ascending-dose, randomized study to evaluate the safety, tolerability and effects on cognition of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Dement Geriatr Cogn Disord (2013) 35:325–36. doi:10.1159/000348347
50. Javitt DC, Buchanan RW, Keefe RS, Kern R, McMahon RP, Green MF, et al. Effect of the neuroprotective peptide davunetide (AL-108) on cognition and functional capacity in schizophrenia. Schizophr Res (2012) 136:25–31. doi:10.1016/j.schres.2011.11.001
51. Jarskog LF, Dong Z, Kangarlu A, Colibazzi T, Girgis RR, Kegeles LS, et al. Effects of davunetide on N-acetylaspartate and choline in dorsolateral prefrontal cortex in patients with schizophrenia. Neuropsychopharmacology (2013) 38:1245–52. doi:10.1038/npp.2013.23
52. Gozes I, Morimoto BH, Tiong J, Fox A, Sutherland K, Dangoor D, et al. NAP: research and development of a peptide derived from activity-dependent neuroprotective protein (ADNP). CNS Drug Rev (2005) 11:353–68. doi:10.1111/j.1527-3458.2005.tb00053.x
Keywords: activity-dependent neuroprotective protein, case study, mutation, nonsense, motor delays, autism spectrum disorder
Citation: Gozes I, Patterson MC, Van Dijck A, Kooy RF, Peeden JN, Eichenberger JA, Zawacki-Downing A and Bedrosian-Sermone S (2017) The Eight and a Half Year Journey of Undiagnosed AD: Gene Sequencing and Funding of Advanced Genetic Testing Has Led to Hope and New Beginnings. Front. Endocrinol. 8:107. doi: 10.3389/fendo.2017.00107
Received: 15 September 2016; Accepted: 02 May 2017;
Published: 19 May 2017
Edited by:
Hubert Vaudry, University of Rouen, FranceReviewed by:
Lidia V. Gabis, Sheba at Tel Hashomer, IsraelNils Lambrecht, University of California at Irvine, United States
Copyright: © 2017 Gozes, Patterson, Van Dijck, Kooy, Peeden, Eichenberger, Zawacki-Downing and Bedrosian-Sermone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Illana Gozes, aWdvemVzQHBvc3QudGF1LmFjLmls