 Paras K. Mishra1,2*
Paras K. Mishra1,2* Wei Ying3
Wei Ying3 Shyam Sundar Nandi1Gautam K. Bandyopadhyay3
Shyam Sundar Nandi1Gautam K. Bandyopadhyay3 Kaushik K. Patel1
Kaushik K. Patel1 Sushil K. Mahata3,4*
Sushil K. Mahata3,4*
- 1Department of Cellular and Integrative Physiology, University of Nebraska Medical Center, Omaha, NE, USA
- 2Department of Anesthesiology, University of Nebraska Medical Center, Omaha, NE, USA
- 3Department of Medicine, Metabolic Physiology and Ultrastructural Biology Laboratory, University of California San Diego, La Jolla, CA, USA
- 4Department of Medicine, Metabolic Physiology and Ultrastructural Biology Laboratory, VA San Diego Healthcare System, San Diego, CA, USA
The heart possesses a remarkable inherent capability to adapt itself to a wide array of genetic and extrinsic factors to maintain contractile function. Failure to sustain its compensatory responses results in cardiac dysfunction, leading to cardiomyopathy. Diabetic cardiomyopathy (DCM) is characterized by left ventricular hypertrophy and reduced diastolic function, with or without concurrent systolic dysfunction in the absence of hypertension and coronary artery disease. Changes in substrate metabolism, oxidative stress, endoplasmic reticulum stress, formation of extracellular matrix proteins, and advanced glycation end products constitute the early stage in DCM. These early events are followed by steatosis (accumulation of lipid droplets) in cardiomyocytes, which is followed by apoptosis, changes in immune responses with a consequent increase in fibrosis, remodeling of cardiomyocytes, and the resultant decrease in cardiac function. The heart is an omnivore, metabolically flexible, and consumes the highest amount of ATP in the body. Altered myocardial substrate and energy metabolism initiate the development of DCM. Diabetic hearts shift away from the utilization of glucose, rely almost completely on fatty acids (FAs) as the energy source, and become metabolically inflexible. Oxidation of FAs is metabolically inefficient as it consumes more energy. In addition to metabolic inflexibility and energy inefficiency, the diabetic heart suffers from impaired calcium handling with consequent alteration of relaxation–contraction dynamics leading to diastolic and systolic dysfunction. Sarcoplasmic reticulum (SR) plays a key role in excitation–contraction coupling as Ca2+ is transported into the SR by the SERCA2a (sarcoplasmic/endoplasmic reticulum calcium-ATPase 2a) during cardiac relaxation. Diabetic cardiomyocytes display decreased SERCA2a activity and leaky Ca2+ release channel resulting in reduced SR calcium load. The diabetic heart also suffers from marked downregulation of novel cardioprotective microRNAs (miRNAs) discovered recently. Since immune responses and substrate energy metabolism are critically altered in diabetes, the present review will focus on immunometabolism and miRNAs.
Introduction
Insulin deficiency and/or resistance and elevated plasma glucose level characterize diabetes, a chronic and progressive metabolic disorder. While type 1 diabetes mellitus (T1DM) accounts for 5–10% of all cases of diabetes (1), type 2 diabetes mellitus (T2DM) accounts for the remaining ~90% of all cases of diabetes (2). As of 2015, 415 million people across the globe have diabetes mellitus (DM) (www.diabetesatlas.org), which will cost 12% of all global health expenditures (accounting for $320 billion in the USA alone) (3). The International Diabetic Federation predicts that 552 million people will suffer from diabetes by 2030. T2DM is recognized as an independent risk factor for heart failure (HF). Patients with T2DM have a greater probability of death in established HF; suffer from worse prognosis after myocardial infarction (MI) (4–8); and accounts for 5.2% of all deaths globally (9, 10). T2DM is strongly associated with obesity and sedentary lifestyle coupled with increasingly westernized diet (2, 11). Diabetic patients are also highly susceptible to diastolic dysfunction, ventricular hypertrophy, and decreased myocardial strain (12).
Rubler and colleagues initially reported diabetic cardiomyopathy (DCM) from their observation of cardiac hypertrophy on post-mortem hearts from four diabetic patients who died of HF without cardiovascular disease, which was subsequently followed by various other studies (13–16). The Strong Heart Study, the Cardiovascular Health Study, and the Framingham Study revealed cardiac hypertrophy with compromised systolic and diastolic function in DCM patients (4, 17–19). Of note, diastolic dysfunction has been reported in diabetic hearts without hypertrophy (20–22). In fact, DCM starts with diastolic dysfunction in patients with T1DM or T2DM followed by systolic dysfunction (23–27). Rodent models of T1DM including streptozotocin (STZ)-treated (28) or alloxan-treated animals (29) and T2DM models such as Goto-Kakizaki rat (30), Zucker fatty rats, Zucker diabetic fatty rats, leptin-deficient ob/ob mice, and leptin receptor-deficient db/db mice consistently show the human DCM phenotypes (31–33). Of note, STZ- and alloxan-induced diabetes is characterized by myocardial atrophy including loss of contractile proteins as opposed to cardiac hypertrophy in T2DM models (34–36). In addition, in T1DM animals, the progress of systolic dysfunction is positively correlated with the progress of the magnitude and duration of hyperglycemia (hypoinsulinemic/hyperglycemia → systolic dysfunction) (31, 35–38). By contrast, mouse models of T2DM are characterized by hyperinsulinemia, hyperglycemia (later stages), and hyperlipidemia (hyperinsulinemic/hyperglycemic → hypertrophy and diastolic dysfunction) (31, 39–41).
Autophagy is reduced in the mouse hearts of OVE26 (a transgenic model of insulinopenic diabetes) and STZ-induced diabetic mouse hearts (42–44). Metformin has been shown to prevent DCM by stimulating AMP-activated protein kinase (AMPK) activity and enhancing autophagic capacity (43).
Recently, DM is identified as a microRNA (miRNA)-related disease (45), and several diabetic complications are associated with differential expressions of various miRNAs (46). Further, miRNAs play a vital role in the regulation of metabolism (47) and since DM is a metabolic disease it is logical to examine the role of miRNAs in DM. Thus the present review will focus on altered metabolism of glucose and fatty acids (FAs) as well as immune responses in diabetes.
Decreased Glucose Uptake and Metabolism
The heart consumes about 6 kg of ATP, or ~20 times its own weight, per day (48) that comes from the breakdown of fat, carbohydrate, protein, ketone bodies, or lactate. Of note, the amount of ATP in the heart is small (~10 mM, enough for only a few beats) compared with the demand (~10,000 times greater) (49). About 95% of total energy is generated from oxidative phosphorylation of FAs and glucose (50–52). A dramatic metabolic shift takes place in diabetic heart, as they rely almost completely on FAs for their energy source. As for example, 46 atoms of oxygen are required to generate 105 molecules of ATP from oxidation of 1 molecule of palmitate. By contrast, oxidation of 1 molecule of glucose utilizes 12 atoms of oxygen to generate 31 molecules of ATP. Therefore, oxidation of FAs consumes ~0.3 oxygen molecules more than glucose to generate each molecule of ATP. Thus, the diabetic heart suffers from metabolic inflexibility due to its reliance on FAs. The lack of insulin production in T1DM patients causes a dramatic decrease in cardiac glucose uptake (53, 54) where hyperglycemia increases glucose oxidation and mitochondrial generation of superoxide (55–57). Increased production of superoxide damages DNA and activates poly (ADP ribose) polymerase 1 (PARP-1) (58), which mediates inflammation and fibrosis in liver (59). PARP-1 inhibition improves cardiac function (60) and prevents hyperglycemia-induced pathological processes (61). While decreased glucose transporter type 4 (Glut4) expression in T1DM animals causes decreased glucose uptake in cardiac and skeletal muscle (62, 63), glucose uptake is impaired in T2DM hearts by decreased expression and translocation of Glut4/Glut1 (64, 65). Diabetic db/db mice show decreased glucose oxidation and increased reliance on FAs, indicating that insulin resistance is not responsible for metabolic switch (66–69). The high rate of FA oxidation in T2DM patients and rodents increases production of acetyl CoA and NADH, resulting in activation of pyruvate dehydrogenase kinase 4 (PDK4). PDK4 is also activated by peroxisome proliferator-activated receptor alpha (PPARα), which is overexpressed in diabetic rodents (70–73). Activated PDK4 inhibits pyruvate dehydrogenase complex, thereby preventing oxidation of pyruvate (74, 75) (Figure 1). In addition, increased accumulation of FAs and their derivatives fatty acyl CoA, diacylglycerol, and ceramide activate protein kinase C, c-Jun N-terminal kinases, mammalian target of rapamycin, and inhibitor of κB kinase β with consequent decrease in insulin signaling (76–79).
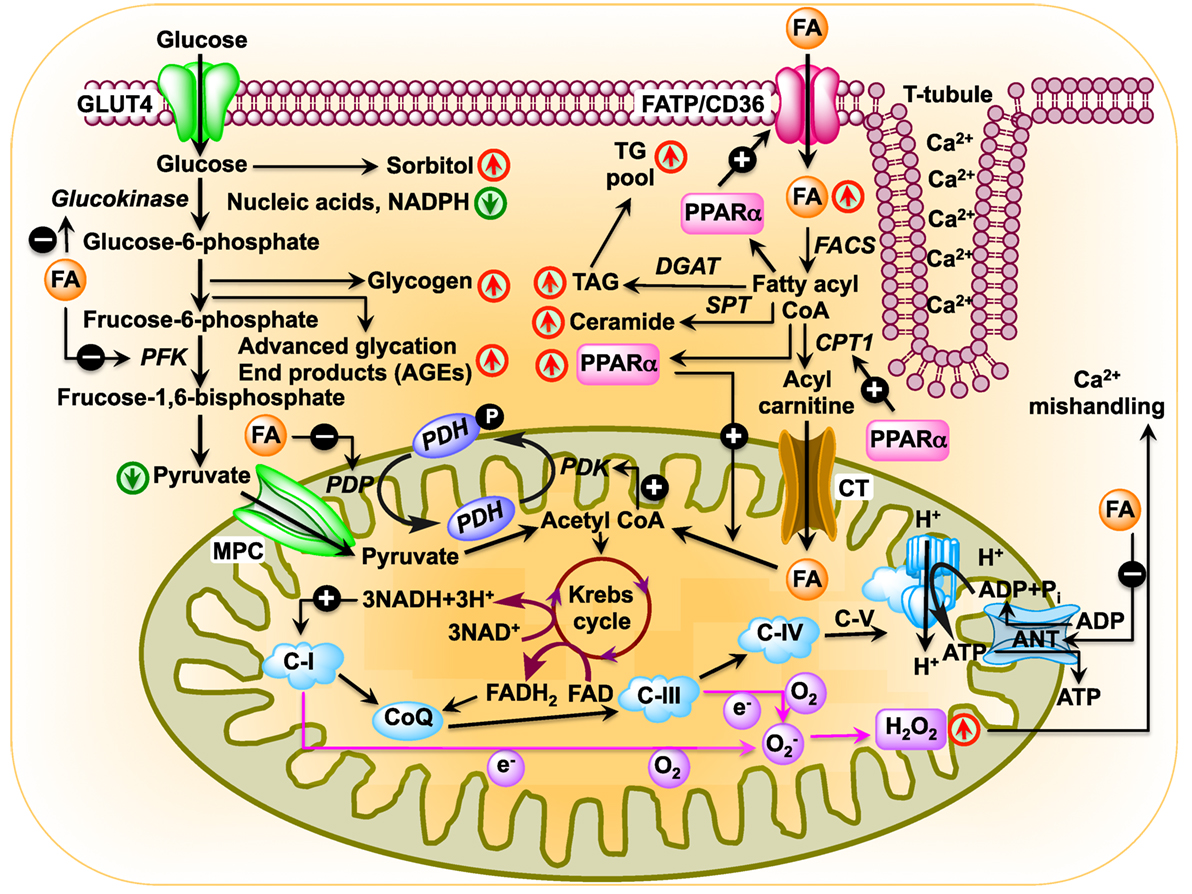
Figure 1. Schematic diagram showing changes in cardiac metabolism in diabetic cardiomyopathy. In the diabetic heart, glucose oxidation is inhibited at multiple steps: (i) uptake of glucose is inhibited by reduced expression of glucose transporter Glut4 as well as by blunted translocation of Glut4 in response to insulin (64, 65); (ii) inhibition of hexokinase activity by fatty acids (FAs) resulting in reduced conversion of glucose to glucose-6-phosphate (80); (iii) inhibition of phosphofructokinase activity by FA, leading to reduced formation of fructose-1,6-bisphosphate by fructose-6-phosphate (69); (iv) inhibition of pyruvate dehydrogenase phosphatase activity by FA resulting in reduced pyruvate dehydrogenase (PDH) activity, which leads to reduced conversion of pyruvate to acetyl CoA. In the diabetic heart, PPARα expression is activated by increased FA uptake (81, 82). Activated PPARα upregulates PDH kinase 4 enzymes, which inhibits PDH resulting in reduced production of acetyl CoA (83–85). FA transporters CD36 and FA transport protein import FAs into the cell. After import, FAs can be stored as triacylglycerol (TAG) or converted to fatty acyl CoA by fatty acyl-CoA synthetase (FACS). Carnitine palmitoyltransferase 1 (CPT1) transfers the acyl group of fatty acyl CoA to carnitine, which then shuttles into the mitochondria by carnitine translocase (CT). PPARα activates transcription of CPT1 (86). In the matrix, CPT2 reconverts the acylcarnitine back into free carnitine and fatty acyl CoA (87), which is then converted to acetyl CoA that can be used in the tricarboxylic acid to produce adenosine triphosphate by β-oxidation. Diabetes upregulates mitochondrial generation of reactive oxygen species (57, 88, 89), which affects Ca2+ signaling (90).
Increased Formation of Advanced Glycation end Products (AGEs)
In the diabetic heart, glucose forms covalent adducts with the plasma proteins through a non-enzymatic reaction between the free amino groups of proteins and carbonyl groups of reducing sugars, resulting in the formation of stable glycosylation products by Amadori rearrangement, which is called glycation (91–94). Glycated proteins undergo a series of oxidation, dehydration, and cyclization reactions to form long-lived AGEs (95, 96). Both AGE and its receptor RAGE are overexpressed in diabetes (97) leading to the generation of reactive oxygen species (ROS) and subsequent activation of RAS–MAP kinase pathway (98). Activation of RAS–MAPK pathway in turn activates NF-κB pathway resulting in decreases in contractile proteins such as α-actin and myosin ATPase activity (35, 36) and shifts in myosin heavy chain isoforms from α to β with consequent development of decreased systolic tension (36–38, 97, 99). In diabetes, increased serum levels of AGEs show positive correlation with ventricular isovolumetric relaxation time, arterial stiffness, and carotid intimal thickness (100–102). Treatment of diabetic animals with aminoguanidine (an inhibitor of AGE formation) (103, 104) or with alagebrium (ALT-711; disrupts AGE cross-link) (105) restored LV function and reduced myocardial collagen, highlighting the importance of AGE in cardiac dysfunction. AGEs also impair collagen degradation by matrix metalloproteinases (MMPs), such as MMP2, resulting in increased fibrosis (106, 107). Fibrosis increases myocardial stiffness and impairs diastolic function (104). In T1DM heart, AGEs also induce cross-linking of SERCA2a pump, thereby attenuating sarcoplasmic reticulum (SR) Ca2+ reuptake (108, 109) with consequent attenuation of the maximum and minimum rate of pressure change in the ventricle and LV developed pressure (108). Of note, the type 2 ryanodine receptor-dependent Ca2+ release not only plays critical roles for excitation–contraction coupling in cardiomyocytes but plays crucial roles in the regulation of insulin secretion and glucose homeostasis (110, 111). Genetic ablation of the RAGE gene improves hemodynamic dysfunction, thereby providing AGE/RAGE pathway as a potential therapeutic target to alleviate cardiac dysfunction in diabetes.
Increased FA Uptake and Metabolism
The heart has a limited capacity for de novo synthesis of FAs. Therefore, it relies heavily on the circulating FAs (112). FAs translocate from blood to cardiomyocytes using three FA transporters: cluster of differentiation 36 (CD36), FA transport protein 1, and the plasma membrane form of FA-binding protein (113–116). Increased PPARα expression in diabetic hearts (70–73, 117) augments transcription of FA transporters. About 75% of the translocated FAs are transferred to mitochondria for the generation of ATP and the rest are converted to triacylglycerol (TAG) for future use (118). Translocated FAs are activated by esterification to fatty acyl CoA by the action of cytosolic fatty acyl-CoA synthetase (FACS). Carnitine palmitoyltransferase 1 (CPT1) exchanges the CoA moiety of fatty acyl CoA for carnitine resulting in the formation of acylcarnitine. Acylcarnitine is transported across the inner mitochondrial membrane into the matrix by carnitine–acylcarnitine translocase. PPARα augments transcription of CPT1 (119, 120). In the matrix, CPT2 reconverts the acylcarnitine back into free carnitine and fatty acyl CoA. PPARα increases transcription of CPT2 (120). Fatty acyl CoA is then converted to acetyl CoA for β-oxidation and generation of ATP. PPARα increases conversion of fatty acyl CoA in the mitochondrial matrix to acetyl CoA. Thus, PPARα plays critical roles in metabolic reprograming in diabetic hearts.
Since the diabetic heart relies on FAs for ATP generation, it consumes ~30% more oxygen compared with non-diabetic heart to generate similar levels of ATP (87, 121) and generate the same or the reduced amounts of contractile force (41). This disproportionate use of FAs also alters cellular ATP shuttling as long-chain acyl CoA derivatives inhibit the adenine nucleotide translocator for the transport of ATP from mitochondria to the cytosol (122–124), eventuating in inefficient delivery of ATP to myofibrils that affects cardiac contractility.
Inflammation, Innate, and Adaptive Immune Responses
Metabolic disturbances induce subcellular low-grade inflammation in the heart (125). Inflammation is a key pathogenic feature of lipid excess and diabetes. The innate immune system comprising of neutrophils, dendritic cells, macrophages, mast cells, and eosinophils also induces chronic metabolic inflammation (126, 127). Myocardial inflammation is implicated in the development of DCM (128–131). Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), a primary regulator of inflammatory responses, is activated in the heart upon exposure to FAs or glucose (132, 133). NF-κB induces not only the expression of pro-inflammatory cytokines, such as tumor necrosis factor alpha (TNFα), interleukin 6 (IL6), pro-IL1β, and pro-IL18, but it also induces the expression of NLR family pyrin domain-containing 3 (NLRP3) inflammasome (134). Activated RAGE also triggers an inflammatory response by heterodimerizing with TLR-4 leading to the production of pro-IL1β, Pro-IL18, and NLRP3 (135). Activated NLRP3 inflammasome activates caspase-1 and mediates the processing and release of pro-inflammatory cytokines IL1β and IL18 resulting in inflammatory cell infiltration and amplification of the inflammatory response (125, 136–138). Likewise, depletion of NLRP3 attenuates inflammation and cardiomyopathy in T2DM rats (137). Of note, activated inflammasomes play critical roles in the pathogenesis of HF (139). Resident immune cells in the resting heart include the following: macrophages, residing near endothelial cells or within the interstitial space (140–143); mast cells that are responsible for early triggers of immune responses (144); a small number of adaptive immune cells: B cells and regulatory T (TReg) cell subsets (142, 145, 146); and dendritic cells that test sample antigens (142, 147) (Figure 2A). The differential expression of major histocompatibility complex (MHC) class II and CC chemokine receptor 2 (CCR2) distinguishes three different subsets of cardiac macrophages: MHC class IIhigh (CCR2−), MHC class IIlow (CCR2−), and CCR2+ macrophages. The first two are the preponderant macrophages in the heart, derived from embryogenic progenitors and renewed through in situ proliferation, rather than through monocyte input. By contrast, CCR2+ macrophages derive from and replenished by circulating blood monocytes, which comprise of Ly6Chigh and Ly6Clow (148–150). Studies in Ccr2 knockout mice (lacking circulating monocytes) reveal increased cardiac pathology (151, 152). The loss of Ly6Chigh monocytes prevents hypertension-induced cardiac fibrosis and improves cardiac function after MI (141, 153, 154). Monnerat et al. suggest that diabetes enhances IL1β production from cardiac MHC IIhigh pro-inflammatory macrophages through activation of TLR2–NLRP3 inflammasome axis (155). The increased level of IL1β leads to a reduction in potassium current and an increase in calcium sparks in cardiomyocytes, which cause cardiac arrhythmias (156). By contrast, M2-like macrophages (CD206+F4/80+CD11b+) exert profound functions on tissue repair in heart depending on IL4 secretion (156). Recent studies implicate TNFβ producing B cells as a major contributor to myocardial fibrosis (153, 157). Antigen and cytokine stimulation are known to differentiate naive T cells into distinct T cell subpopulations that include T helper cells and CD4+CD25+FOXP3+ TReg cells (158). TReg cells comprise a subset of CD4+ lymphocytes that suppress activation, proliferation, and effector responses of both innate and adaptive immune cells (159–161). As opposed to B cells, depletion of TReg cells aggravates myocardial fibrosis and adoptive transfer of exogenous TReg cells into these mice attenuates the extent of myocardial fibrosis (158). The following pro-inflammatory changes with oxidative stress and decreased cardiac function were detected in STZ-induced rat model of T1DM (162): (i) significant increases in myocardial intercellular adhesion molecule 1 and vascular cell adhesion molecule 1, (ii) increased expression of beta2-leukotrienes-integrins+ (CD18+, CD11a+, CD11b+), (iii) increased expression of TNFα, and (iv) IL1β (Figure 2B). Treatment of STZ-induced DCM rats with irbesartan (AT-1 receptor antagonist) has been reported to improve cardiac functions by attenuating cardiac inflammation (IL1β, TNFα, and TGFβ) and restoration of MMP activity with consequent decrease in fibrosis (107). Similar results were reported after neutralization of TNFα (163) or genetic deletion of neurokinin receptor B (164) in rodent models of T1DM. Subsequently, several studies confirmed the pro-inflammatory phenotypes in diabetic rodent heart (165–167). As opposed to metabolic responses, immune responses in T1DM and T2DM are comparable as both of them show consistent activation of pro-inflammatory transcription factor NF-κB. Cytokines (i) increase formation of peroxynitrite, which play critical roles in cardiac dysfunction (168), (ii) exert direct effects on the function of SR as well as on the regulation of SR calcium ATPase expression (168, 169), and (iii) increase fibrosis (170). Treatment of rats and humans with statins (171), renin angiotensin aldosterone system (RAAS) inhibitors (107), metformin (172), and thiazolidinediones (173) reduces inflammation in the heart and improve cardiac function.
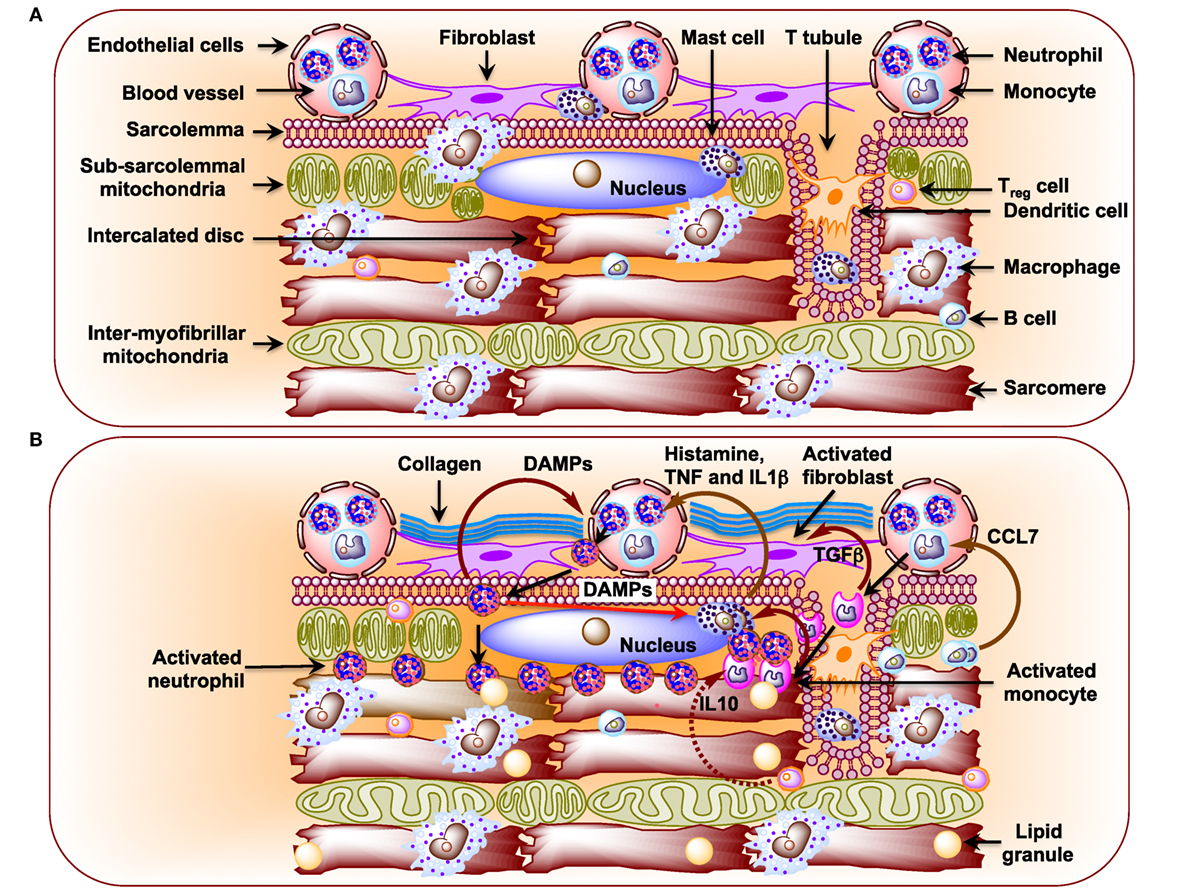
Figure 2. (A) Schematic diagram showing immune cells in the healthy heart. Macrophages are the preponderant immune cells in the resting heart and reside primarily surrounding endothelial cells and also in the interstitium among cardiomyocytes (141–143). The less preponderant immune cells include the following: mast cells, dendritic cells, B cells, and regulatory T (TReg) cells (142, 144, 147). Neutrophils and monocytes, in general, are not detected in the resting heart. (B) Schematic diagram showing infiltration of neutrophils and monocytes from the circulation and their effects on resident immune cells in the diabetic cardiomyopathy (DCM) heart. In DCM, activated B cells release CCL7 that activates blood monocytes (146). Upon infiltration activated monocytes stimulate mast cells to release histamine, TNF, and interleukin 1β (IL1β), which activate neutrophils in circulation (144, 174). Activated neutrophils infiltrate heart and activate mast cells through damage-associated molecular patterns as well as blood neutrophils. Activated monocytes secrete TGFβ, which activates fibroblasts to induce formation of collagen.
miRNA in Diabetic Hearts
MicroRNAs are highly conserved endogenous small non-coding RNAs, ~22 nucleotides in length, that regulate gene expression by binding to partially complementary sequences of mRNA (175). The failing hearts consistently show chronic immune activation and aberrant miRNA expression (176). Thus, miR-155 plays an important role in the mammalian immune systems as well as during HF and is abundantly expressed in T-cells, B-cells, and monocytes (177–180). miRNAs are also differentially expressed during HF (181). STZ-induced diabetic heart expresses higher levels of miR-195 and silencing of miR-195 reduces DCM (182). Likewise, miR-141 is increased in diabetic heart and affects mitochondrial function and ATP generation (183). Palmitate-stimulated neonatal rat cardiomyocytes (NRCs) and diet-induced obese (DIO) mouse heart also showed increased expression of miR-451, which decreases LKB1/AMPK signaling (184). Expression of miR-133a reduces Glut4 expression with consequent decrease in insulin-mediated glucose uptake in NRCs (185). While overexpression of miR-223 in NRCs significantly increased glucose uptake by increasing total Glut4 level and its translocation, inhibition of miR-223 in the heart resulted in a significant decrease in Glut4 expression (186). In contrast to the findings in NRCs, expression of miR-133a is decreased in the hearts of diabetic mice and is associated with increased fibrosis. Of note, overexpression of miR-133a in the heart attenuates cardiac fibrosis (187). Murine miR-322 has recently been shown to provide cardioprotection against consequences of hyperinsulinemia and hyperlipidemia (188). In Ins+/– Akita mice, a model for T1DM, the majority of miRNAs are downregulated in the heart (189), including miR-133a which regulates contractility of the diabetic heart (190). Even after treatment with insulin, which normalizes blood glucose levels, there are several miRNAs that remain differentially regulated in the diabetic heart, and they can potentially contribute to pathological remodeling of the diabetic heart (191). These miRNAs could be a potential target for developing a novel therapeutic strategy for the treatment of diabetic HF.
Diabetes mellitus is a metabolic disease, and miRNAs play a crucial role in the regulation of metabolism (47). Increased levels of plasma cholesterol and triglyceride are common in diabetes, and liver specific ablation of miR-122, the most abundant miRNA in the liver, reduces plasma cholesterol and triglyceride levels (192, 193). The intracellular cholesterol and FA homeostasis are controlled by miR-33a and miR-33b, which target genes involved in cholesterol export including adenosine triphosphate-binding cassette transporters (194–196). Endogenous inhibition or knockout of miR-33 leads to increased plasma high-density lipid levels (194–197). MiR-223 controls the expression of Glut4 gene in cardiomyocytes, and miR-223 is upregulated while Glut4 is downregulated in human diabetic hearts (186). The switch of glycolysis to FA oxidation is regulated by PPARδ, which is regulated by the miR-199/miR-214 cluster. The miR-199/miR-214 cluster downregulates PPARδ and impairs FA oxidation (198). ROS stimulates apoptosis by mitochondrial cytochrome c release and ceramide generation (199). In rat cardiomyocytes, high glucose upregulates miR-34a and miR-1 that reduces the levels of B-cell lymphoma 2 (Bcl-2) and insulin-like growth factor 1 (Igf-1) genes, respectively, and induces apoptosis (200, 201). Recently, Kuwabara et al. has elegantly shown that miR-451 plays a key role in exacerbating lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway (184). MiR-133a, the most abundant miRNA in the heart, is downregulated in the diabetic mice heart with consequent induction of cardiac hypertrophy (202) and fibrosis (187). Lack of miR-133a also causes contractile dysfunction in the diabetic mice heart (190). These changes cause diastolic dysfunction, which if untreated leads to potential systolic dysfunction (28).
Conclusion and Future Perspectives
Cardiovascular disease has remained the leading cause of mortality and morbidity in individuals with diabetes. DCM is emerging as an increasing health concern with the epidemic rise in DM worldwide. Animal studies have clearly shown that glycemic control at an early stage prevents the development of DCM, and that certain anti-diabetic drugs exert anti-remodeling effects. While a large body of epidemiological evidence (50,000 T2DM patients) indicate a positive correlation between blood glucose level and/or HbA1c and the risk of HF (203–205), a meta-analysis of randomized controlled trials (37,229 patients) showed no effect of intensive glycemic control on the risk of HF in T2DM patients (206).
Therapeutic approach for DCM depends mainly on (i) glycemic control, (ii) glucose-lowering drug administration, (iii) improvement of autophagy, and (iv) an active life style. Earliest detection, helped by current research on miRNAs, will enhance therapeutic efficacy. The current burst of scientific evidence for the potential use of circulating miRNAs as biomarkers for cardiomyopathy is generating hopes that someday soon detection of specific miRNAs in biofluids of patients will help early treatment of both diabetes and cardiomyopathy.
Author Contributions
SM conceived the idea, wrote the immunometabolism part of the manuscript, and made the schematic diagrams. PM wrote the microRNA part of the manuscript and contributed in correcting the final draft of the manuscript. SN contributed to drafting and correcting of the final version of the manuscript. WY, KP, and GB participated in discussion and reviewed/edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors gratefully acknowledge financial support from the National Institutes of Health grants HL-116205 and HL113281 to PM, R56 HL124104 and P01 HL62222 to KP, and American Heart Association Postdoctoral fellowship award 16 POST 30180003 to SN. SM lab is supported by home-equity loan.
References
1. Raskin P, Mohan A. Emerging treatments for the prevention of type 1 diabetes. Expert Opin Emerg Drugs (2010) 15(2):225–36. doi: 10.1517/14728211003694631
2. Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature (2001) 414(6865):782–7. doi:10.1038/414782a
3. Farag YM, Gaballa MR. Diabesity: an overview of a rising epidemic. Nephrol Dial Transplant (2011) 26(1):28–35. doi:10.1093/ndt/gfq576
4. Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA (1979) 241(19):2035–8.
5. MacDonald MR, Petrie MC, Varyani F, Ostergren J, Michelson EL, Young JB, et al. Impact of diabetes on outcomes in patients with low and preserved ejection fraction heart failure: an analysis of the Candesartan in heart failure: assessment of reduction in mortality and morbidity (CHARM) programme. Eur Heart J (2008) 29(11):1377–85. doi:10.1093/eurheartj/ehn153
6. MacDonald MR, Petrie MC, Hawkins NM, Petrie JR, Fisher M, McKelvie R, et al. Diabetes, left ventricular systolic dysfunction, and chronic heart failure. Eur Heart J (2008) 29(10):1224–40. doi:10.1093/eurheartj/ehn156
7. Cubbon RM, Adams B, Rajwani A, Mercer BN, Patel PA, Gherardi G, et al. Diabetes mellitus is associated with adverse prognosis in chronic heart failure of ischaemic and non-ischaemic aetiology. Diab Vasc Dis Res (2013) 10(4):330–6. doi:10.1177/1479164112471064
8. Bell DS. Diabetic cardiomyopathy. Diabetes Care (2003) 26(10):2949–51. doi:10.2337/diacare.26.10.2949
9. Roglic G, Unwin N, Bennett PH, Mathers C, Tuomilehto J, Nag S, et al. The burden of mortality attributable to diabetes: realistic estimates for the year 2000. Diabetes Care (2005) 28(9):2130–5. doi:10.2337/diacare.28.9.2130
10. Nolan CJ, Damm P, Prentki M. Type 2 diabetes across generations: from pathophysiology to prevention and management. Lancet (2011) 378(9786):169–81. doi:10.1016/S0140-6736(11)60614-4
11. Astrup A, Dyerberg J, Selleck M, Stender S. Nutrition transition and its relationship to the development of obesity and related chronic diseases. Obes Rev (2008) 9(Suppl 1):48–52. doi:10.1111/j.1467-789X.2007.00438.x
12. Fuentes-Antras J, Picatoste B, Ramirez E, Egido J, Tunon J, Lorenzo O. Targeting metabolic disturbance in the diabetic heart. Cardiovasc Diabetol (2015) 14:17. doi:10.1186/s12933-015-0173-8
13. Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol (1972) 30(6):595–602. doi:10.1016/0002-9149(72)90595-4
14. de Simone G, Devereux RB, Chinali M, Lee ET, Galloway JM, Barac A, et al. Diabetes and incident heart failure in hypertensive and normotensive participants of the Strong Heart Study. J Hypertens (2010) 28(2):353–60. doi:10.1097/HJH.0b013e3283331169
15. Regan TJ, Lyons MM, Ahmed SS, Levinson GE, Oldewurtel HA, Ahmad MR, et al. Evidence for cardiomyopathy in familial diabetes mellitus. J Clin Invest (1977) 60(4):884–99. doi:10.1172/JCI108843
16. Shehadeh A, Regan TJ. Cardiac consequences of diabetes mellitus. Clin Cardiol (1995) 18(6):301–5. doi:10.1002/clc.4960180604
17. Devereux RB, Roman MJ, Paranicas M, O’Grady MJ, Lee ET, Welty TK, et al. Impact of diabetes on cardiac structure and function: the Strong Heart Study. Circulation (2000) 101(19):2271–6. doi:10.1161/01.CIR.101.19.2271
18. Lee M, Gardin JM, Lynch JC, Smith VE, Tracy RP, Savage PJ, et al. Diabetes mellitus and echocardiographic left ventricular function in free-living elderly men and women: the Cardiovascular Health Study. Am Heart J (1997) 133(1):36–43. doi:10.1016/S0002-8703(97)70245-X
19. Liu JE, Palmieri V, Roman MJ, Bella JN, Fabsitz R, Howard BV, et al. The impact of diabetes on left ventricular filling pattern in normotensive and hypertensive adults: the Strong Heart Study. J Am Coll Cardiol (2001) 37(7):1943–9. doi:10.1016/S0735-1097(01)01230-X
20. Schannwell CM, Schneppenheim M, Perings S, Plehn G, Strauer BE. Left ventricular diastolic dysfunction as an early manifestation of diabetic cardiomyopathy. Cardiology (2002) 98(1–2):33–9. doi:10.1159/000064682
21. Ha JW, Lee HC, Kang ES, Ahn CM, Kim JM, Ahn JA, et al. Abnormal left ventricular longitudinal functional reserve in patients with diabetes mellitus: implication for detecting subclinical myocardial dysfunction using exercise tissue Doppler echocardiography. Heart (2007) 93(12):1571–6. doi:10.1136/hrt.2006.101667
22. Acar G, Akcay A, Sokmen A, Ozkaya M, Guler E, Sokmen G, et al. Assessment of atrial electromechanical delay, diastolic functions, and left atrial mechanical functions in patients with type 1 diabetes mellitus. J Am Soc Echocardiogr (2009) 22(6):732–8. doi:10.1016/j.echo.2009.03.028
23. Boyer JK, Thanigaraj S, Schechtman KB, Perez JE. Prevalence of ventricular diastolic dysfunction in asymptomatic, normotensive patients with diabetes mellitus. Am J Cardiol (2004) 93(7):870–5. doi:10.1016/j.amjcard.2003.12.026
24. Shivalkar B, Dhondt D, Goovaerts I, Van Gaal L, Bartunek J, Van Crombrugge P, et al. Flow mediated dilatation and cardiac function in type 1 diabetes mellitus. Am J Cardiol (2006) 97(1):77–82. doi:10.1016/j.amjcard.2005.07.111
25. Carugo S, Giannattasio C, Calchera I, Paleari F, Gorgoglione MG, Grappiolo A, et al. Progression of functional and structural cardiac alterations in young normotensive uncomplicated patients with type 1 diabetes mellitus. J Hypertens (2001) 19(9):1675–80. doi:10.1097/00004872-200109000-00021
26. Nicolino A, Longobardi G, Furgi G, Rossi M, Zoccolillo N, Ferrara N, et al. Left ventricular diastolic filling in diabetes mellitus with and without hypertension. Am J Hypertens (1995) 8(4 Pt 1):382–9. doi:10.1016/0895-7061(95)00022-H
27. Di Bonito P, Cuomo S, Moio N, Sibilio G, Sabatini D, Quattrin S, et al. Diastolic dysfunction in patients with non-insulin-dependent diabetes mellitus of short duration. Diabet Med (1996) 13(4):321–4. doi:10.1002/(SICI)1096-9136(199604)13:4<321::AID-DIA3>3.3.CO;2-Z
28. Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord (2010) 11(1):31–9. doi:10.1007/s11154-010-9131-7
29. Vadlamudi RV, Rodgers RL, McNeill JH. The effect of chronic alloxan- and streptozotocin-induced diabetes on isolated rat heart performance. Can J Physiol Pharmacol (1982) 60(7):902–11. doi:10.1139/y82-127
30. El-Omar MM, Yang ZK, Phillips AO, Shah AM. Cardiac dysfunction in the Goto-Kakizaki rat. A model of type II diabetes mellitus. Basic Res Cardiol (2004) 99(2):133–41. doi:10.1007/s00395-004-0440-4
31. Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res (2006) 98(5):596–605. doi:10.1161/01.RES.0000207406.94146.c2
32. Severson DL. Diabetic cardiomyopathy: recent evidence from mouse models of type 1 and type 2 diabetes. Can J Physiol Pharmacol (2004) 82(10):813–23. doi:10.1139/y04-065
33. Bugger H, Abel ED. Rodent models of diabetic cardiomyopathy. Dis Model Mech (2009) 2(9–10):454–66. doi:10.1242/dmm.001941
34. Jackson CV, McGrath GM, Tahiliani AG, Vadlamudi RV, McNeill JH. A functional and ultrastructural analysis of experimental diabetic rat myocardium. Manifestation of a cardiomyopathy. Diabetes (1985) 34(9):876–83.
35. Dhalla NS, Pierce GN, Innes IR, Beamish RE. Pathogenesis of cardiac dysfunction in diabetes mellitus. Can J Cardiol (1985) 1(4):263–81.
36. Depre C, Young ME, Ying J, Ahuja HS, Han Q, Garza N, et al. Streptozotocin-induced changes in cardiac gene expression in the absence of severe contractile dysfunction. J Mol Cell Cardiol (2000) 32(6):985–96. doi:10.1006/jmcc.2000.1139
37. Rodrigues B, Cam MC, McNeill JH. Myocardial substrate metabolism: implications for diabetic cardiomyopathy. J Mol Cell Cardiol (1995) 27(1):169–79. doi:10.1016/S0022-2828(08)80016-8
38. Scognamiglio R, Avogaro A, Negut C, Piccolotto R, Vigili de Kreutzenberg S, Tiengo A. Early myocardial dysfunction in the diabetic heart: current research and clinical applications. Am J Cardiol (2004) 93(8A):17A–20A. doi:10.1016/j.amjcard.2003.11.004
39. Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab (2000) 279(5):E1104–13.
40. Barouch LA, Berkowitz DE, Harrison RW, O’Donnell CP, Hare JM. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation (2003) 108(6):754–9. doi:10.1161/01.CIR.0000083716.82622.FD
41. Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, et al. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes (2004) 53(9):2366–74. doi:10.2337/diabetes.53.9.2366
42. Zhao Y, Zhang L, Qiao Y, Zhou X, Wu G, Wang L, et al. Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy. PLoS One (2013) 8(9):e75927. doi:10.1371/journal.pone.0075927
43. Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes (2011) 60(6):1770–8. doi:10.2337/db10-0351
44. Kobayashi S, Liang Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim Biophys Acta (2015) 1852(2):252–61. doi:10.1016/j.bbadis.2014.05.020
45. Guay C, Roggli E, Nesca V, Jacovetti C, Regazzi R. Diabetes mellitus, a microRNA-related disease? Transl Res (2011) 157(4):253–64. doi:10.1016/j.trsl.2011.01.009
46. Kantharidis P, Wang B, Carew RM, Lan HY. Diabetes complications: the microRNA perspective. Diabetes (2011) 60(7):1832–7. doi:10.2337/db11-0082
47. Fernandez-Hernando C, Ramirez CM, Goedeke L, Suarez Y. microRNAs in metabolic disease. Arterioscler Thromb Vasc Biol (2013) 33(2):178–85. doi:10.1161/ATVBAHA.112.300144
49. Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res (2009) 81(3):412–9. doi:10.1093/cvr/cvn301
50. Lopaschuk GD, Belke DD, Gamble J, Itoi T, Schonekess BO. Regulation of fatty acid oxidation in the mammalian heart in health and disease. Biochim Biophys Acta (1994) 1213(3):263–76. doi:10.1016/0005-2760(94)00082-4
51. Saddik M, Lopaschuk GD. Myocardial triglyceride turnover and contribution to energy substrate utilization in isolated working rat hearts. J Biol Chem (1991) 266(13):8162–70.
52. Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res (1997) 33(2):243–57. doi:10.1016/S0008-6363(96)00245-3
53. Doria A, Nosadini R, Avogaro A, Fioretto P, Crepaldi G. Myocardial metabolism in type 1 diabetic patients without coronary artery disease. Diabet Med (1991) 8(Spec No):S104–7. doi:10.1111/j.1464-5491.1991.tb02168.x
54. Avogaro A, Nosadini R, Doria A, Fioretto P, Velussi M, Vigorito C, et al. Myocardial metabolism in insulin-deficient diabetic humans without coronary artery disease. Am J Physiol (1990) 258(4 Pt 1):E606–18.
55. Farhangkhoee H, Khan ZA, Mukherjee S, Cukiernik M, Barbin YP, Karmazyn M, et al. Heme oxygenase in diabetes-induced oxidative stress in the heart. J Mol Cell Cardiol (2003) 35(12):1439–48. doi:10.1016/j.yjmcc.2003.09.007
56. Cai L, Kang YJ. Oxidative stress and diabetic cardiomyopathy: a brief review. Cardiovasc Toxicol (2001) 1(3):181–93. doi:10.1385/CT:1:3:181
57. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature (2000) 404(6779):787–90. doi:10.1038/35008121
58. Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, et al. Inhibition of GAPDH activity by poly (ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest (2003) 112(7):1049–57. doi:10.1172/JCI18127
59. Mukhopadhyay P, Rajesh M, Cao Z, Horvath B, Park O, Wang H, et al. Poly (ADP-ribose) polymerase-1 is a key mediator of liver inflammation and fibrosis. Hepatology (2014) 59(5):1998–2009. doi:10.1002/hep.26763
60. Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C. The role of poly (ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes (2002) 51(2):514–21. doi:10.2337/diabetes.51.2.514
61. Pacher P, Szabo C. Role of poly(ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: endothelial dysfunction, as a common underlying theme. Antioxid Redox Signal (2005) 7(11–12):1568–80. doi:10.1089/ars.2005.7.1568
62. Camps M, Castello A, Munoz P, Monfar M, Testar X, Palacin M, et al. Effect of diabetes and fasting on GLUT-4 (muscle/fat) glucose-transporter expression in insulin-sensitive tissues. Heterogeneous response in heart, red and white muscle. Biochem J (1992) 282(Pt 3):765–72. doi:10.1042/bj2820765
63. Munoz P, Chillaron J, Camps M, Castello A, Furriols M, Testar X, et al. Evidence for posttranscriptional regulation of GLUT4 expression in muscle and adipose tissue from streptozotocin-induced diabetic and benfluorex-treated rats. Biochem Pharmacol (1996) 52(11):1665–73. doi:10.1016/S0006-2952(96)00506-0
64. Deng JY, Huang JP, Lu LS, Hung LM. Impairment of cardiac insulin signaling and myocardial contractile performance in high-cholesterol/fructose-fed rats. Am J Physiol Heart Circ Physiol (2007) 293(2):H978–87. doi:10.1152/ajpheart.01002.2006
65. Desrois M, Sidell RJ, Gauguier D, King LM, Radda GK, Clarke K. Initial steps of insulin signaling and glucose transport are defective in the type 2 diabetic rat heart. Cardiovasc Res (2004) 61(2):288–96. doi:10.1016/j.cardiores.2003.11.021
66. Armoni M, Harel C, Bar-Yoseph F, Milo S, Karnieli E. Free fatty acids repress the GLUT4 gene expression in cardiac muscle via novel response elements. J Biol Chem (2005) 280(41):34786–95. doi:10.1074/jbc.M502740200
67. Utriainen T, Takala T, Luotolahti M, Ronnemaa T, Laine H, Ruotsalainen U, et al. Insulin resistance characterizes glucose uptake in skeletal muscle but not in the heart in NIDDM. Diabetologia (1998) 41(5):555–9. doi:10.1007/s001250050946
68. Jagasia D, Whiting JM, Concato J, Pfau S, McNulty PH. Effect of non-insulin-dependent diabetes mellitus on myocardial insulin responsiveness in patients with ischemic heart disease. Circulation (2001) 103(13):1734–9. doi:10.1161/01.CIR.103.13.1734
69. Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, et al. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology (2005) 146(12):5341–9. doi:10.1210/en.2005-0938
70. Asayama K, Sandhir R, Sheikh FG, Hayashibe H, Nakane T, Singh I. Increased peroxisomal fatty acid beta-oxidation and enhanced expression of peroxisome proliferator-activated receptor-alpha in diabetic rat liver. Mol Cell Biochem (1999) 194(1–2):227–34. doi:10.1023/A:1006930513476
71. Kroetz DL, Yook P, Costet P, Bianchi P, Pineau T. Peroxisome proliferator-activated receptor alpha controls the hepatic CYP4A induction adaptive response to starvation and diabetes. J Biol Chem (1998) 273(47):31581–9. doi:10.1074/jbc.273.47.31581
72. Huang TH, Yang Q, Harada M, Uberai J, Radford J, Li GQ, et al. Salacia oblonga root improves cardiac lipid metabolism in Zucker diabetic fatty rats: modulation of cardiac PPAR-alpha-mediated transcription of fatty acid metabolic genes. Toxicol Appl Pharmacol (2006) 210(1–2):78–85. doi:10.1016/j.taap.2005.07.020
73. Memon RA, Tecott LH, Nonogaki K, Beigneux A, Moser AH, Grunfeld C, et al. Up-regulation of peroxisome proliferator-activated receptors (PPAR-alpha) and PPAR-gamma messenger ribonucleic acid expression in the liver in murine obesity: troglitazone induces expression of PPAR-gamma-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology (2000) 141(11):4021–31. doi:10.1210/endo.141.11.7771
74. Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J (1998) 329(Pt 1):191–6. doi:10.1042/bj3290191
75. Holness MJ, Sugden MC. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans (2003) 31(Pt 6):1143–51. doi:10.1042/bst0311143
76. Sykiotis GP, Papavassiliou AG. Serine phosphorylation of insulin receptor substrate-1: a novel target for the reversal of insulin resistance. Mol Endocrinol (2001) 15(11):1864–9. doi:10.1210/me.15.11.1864
77. Tanti JF, Jager J. Cellular mechanisms of insulin resistance: role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr Opin Pharmacol (2009) 9(6):753–62. doi:10.1016/j.coph.2009.07.004
78. Kim JK, Fillmore JJ, Sunshine MJ, Albrecht B, Higashimori T, Kim DW, et al. PKC-theta knockout mice are protected from fat-induced insulin resistance. J Clin Invest (2004) 114(6):823–7. doi:10.1172/JCI200422230
79. Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science (2001) 293(5535):1673–7. doi:10.1126/science.1061620
80. Malfitano C, de Souza Junior AL, Carbonaro M, Bolsoni-Lopes A, Figueroa D, de Souza LE, et al. Glucose and fatty acid metabolism in infarcted heart from streptozotocin-induced diabetic rats after 2 weeks of tissue remodeling. Cardiovasc Diabetol (2015) 14:149. doi:10.1186/s12933-015-0308-y
81. Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature (1992) 358(6389):771–4. doi:10.1038/358771a0
82. Motojima K, Passilly P, Peters JM, Gonzalez FJ, Latruffe N. Expression of putative fatty acid transporter genes are regulated by peroxisome proliferator-activated receptor alpha and gamma activators in a tissue- and inducer-specific manner. J Biol Chem (1998) 273(27):16710–4. doi:10.1074/jbc.273.27.16710
83. Schroeder MA, Cochlin LE, Heather LC, Clarke K, Radda GK, Tyler DJ. In vivo assessment of pyruvate dehydrogenase flux in the heart using hyperpolarized carbon-13 magnetic resonance. Proc Natl Acad Sci U S A (2008) 105(33):12051–6. doi:10.1073/pnas.0805953105
84. Huang B, Wu P, Popov KM, Harris RA. Starvation and diabetes reduce the amount of pyruvate dehydrogenase phosphatase in rat heart and kidney. Diabetes (2003) 52(6):1371–6. doi:10.2337/diabetes.52.6.1371
85. Wu P, Sato J, Zhao Y, Jaskiewicz J, Popov KM, Harris RA. Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase isoenzyme 4 in rat heart. Biochem J (1998) 329(Pt 1):197–201. doi:10.1042/bj3290197
86. Brandt JM, Djouadi F, Kelly DP. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor alpha. J Biol Chem (1998) 273(37):23786–92. doi:10.1074/jbc.273.37.23786
87. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev (2010) 90(1):207–58. doi:10.1152/physrev.00015.2009
88. Huynh K, Kiriazis H, Du XJ, Love JE, Jandeleit-Dahm KA, Forbes JM, et al. Coenzyme Q10 attenuates diastolic dysfunction, cardiomyocyte hypertrophy and cardiac fibrosis in the db/db mouse model of type 2 diabetes. Diabetologia (2012) 55(5):1544–53. doi:10.1007/s00125-012-2495-3
89. Huynh K, Kiriazis H, Du XJ, Love JE, Gray SP, Jandeleit-Dahm KA, et al. Targeting the upregulation of reactive oxygen species subsequent to hyperglycemia prevents type 1 diabetic cardiomyopathy in mice. Free Radic Biol Med (2013) 60:307–17. doi:10.1016/j.freeradbiomed.2013.02.021
90. Gorlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: a mutual interplay. Redox Biol (2015) 6:260–71. doi:10.1016/j.redox.2015.08.010
91. Helou C, Marier D, Jacolot P, Abdennebi-Najar L, Niquet-Leridon C, Tessier FJ, et al. Microorganisms and Maillard reaction products: a review of the literature and recent findings. Amino Acids (2014) 46(2):267–77. doi:10.1007/s00726-013-1496-y
92. Maillard LC. Action of amino acids on sugars. Formation of melanoidins in a methodical way. Compt Rend (1912) 154:66–8.
93. Zhang Q, Ames JM, Smith RD, Baynes JW, Metz TO. A perspective on the Maillard reaction and the analysis of protein glycation by mass spectrometry: probing the pathogenesis of chronic disease. J Proteome Res (2009) 8(2):754–69. doi:10.1021/pr800858h
94. Helou C, Gadonna-Widehem P, Robert N, Branlard G, Thebault J, Librere S, et al. The impact of raw materials and baking conditions on Maillard reaction products, thiamine, folate, phytic acid and minerals in white bread. Food Funct (2016) 7(6):2498–507. doi:10.1039/c5fo01341k
95. Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation (2006) 114(6):597–605. doi:10.1161/CIRCULATIONAHA.106.621854
96. Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Babaei-Jadidi R, et al. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem J (2003) 375(Pt 3):581–92. doi:10.1042/bj20030763
97. Aragno M, Mastrocola R, Medana C, Catalano MG, Vercellinatto I, Danni O, et al. Oxidative stress-dependent impairment of cardiac-specific transcription factors in experimental diabetes. Endocrinology (2006) 147(12):5967–74. doi:10.1210/en.2006-0728
98. Basta G, Schmidt AM, De Caterina R. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res (2004) 63(4):582–92. doi:10.1016/j.cardiores.2004.05.001
99. Fang ZY, Prins JB, Marwick TH. Diabetic cardiomyopathy: evidence, mechanisms, and therapeutic implications. Endocr Rev (2004) 25(4):543–67. doi:10.1210/er.2003-0012
100. Lapolla A, Piarulli F, Sartore G, Ceriello A, Ragazzi E, Reitano R, et al. Advanced glycation end products and antioxidant status in type 2 diabetic patients with and without peripheral artery disease. Diabetes Care (2007) 30(3):670–6. doi:10.2337/dc06-1508
101. Yoshida N, Okumura K, Aso Y. High serum pentosidine concentrations are associated with increased arterial stiffness and thickness in patients with type 2 diabetes. Metabolism (2005) 54(3):345–50. doi:10.1016/j.metabol.2004.09.014
102. Berg TJ, Snorgaard O, Faber J, Torjesen PA, Hildebrandt P, Mehlsen J, et al. Serum levels of advanced glycation end products are associated with left ventricular diastolic function in patients with type 1 diabetes. Diabetes Care (1999) 22(7):1186–90. doi:10.2337/diacare.22.7.1186
103. Wu MS, Liang JT, Lin YD, Wu ET, Tseng YZ, Chang KC. Aminoguanidine prevents the impairment of cardiac pumping mechanics in rats with streptozotocin and nicotinamide-induced type 2 diabetes. Br J Pharmacol (2008) 154(4):758–64. doi:10.1038/bjp.2008.119
104. Norton GR, Candy G, Woodiwiss AJ. Aminoguanidine prevents the decreased myocardial compliance produced by streptozotocin-induced diabetes mellitus in rats. Circulation (1996) 93(10):1905–12. doi:10.1161/01.CIR.93.10.1905
105. Liu J, Masurekar MR, Vatner DE, Jyothirmayi GN, Regan TJ, Vatner SF, et al. Glycation end-product cross-link breaker reduces collagen and improves cardiac function in aging diabetic heart. Am J Physiol Heart Circ Physiol (2003) 285(6):H2587–91. doi:10.1152/ajpheart.00516.2003
106. Van Linthout S, Seeland U, Riad A, Eckhardt O, Hohl M, Dhayat N, et al. Reduced MMP-2 activity contributes to cardiac fibrosis in experimental diabetic cardiomyopathy. Basic Res Cardiol (2008) 103(4):319–27. doi:10.1007/s00395-008-0715-2
107. Westermann D, Rutschow S, Jager S, Linderer A, Anker S, Riad A, et al. Contributions of inflammation and cardiac matrix metalloproteinase activity to cardiac failure in diabetic cardiomyopathy: the role of angiotensin type 1 receptor antagonism. Diabetes (2007) 56(3):641–6. doi:10.2337/db06-1163
108. Kranstuber AL, Del Rio C, Biesiadecki BJ, Hamlin RL, Ottobre J, Gyorke S, et al. Advanced glycation end product cross-link breaker attenuates diabetes-induced cardiac dysfunction by improving sarcoplasmic reticulum calcium handling. Front Physiol (2012) 3:292. doi:10.3389/fphys.2012.00292
109. Bidasee KR, Zhang Y, Shao CH, Wang M, Patel KP, Dincer UD, et al. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes (2004) 53(2):463–73. doi:10.2337/diabetes.53.2.463
110. Santulli G, Marks AR. Essential roles of intracellular calcium release channels in muscle, brain, metabolism, and aging. Curr Mol Pharmacol (2015) 8(2):206–22. doi:10.2174/1874467208666150507105105
111. Santulli G, Pagano G, Sardu C, Xie W, Reiken S, D’Ascia SL, et al. Calcium release channel RyR2 regulates insulin release and glucose homeostasis. J Clin Invest (2015) 125(11):4316. doi:10.1172/JCI84937
112. Schulze PC, Drosatos K, Goldberg IJ. Lipid use and misuse by the heart. Circ Res (2016) 118(11):1736–51. doi:10.1161/CIRCRESAHA.116.306842
113. Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J Lipid Res (2009) 50(Suppl):S86–90. doi:10.1194/jlr.R800085-JLR200
114. An D, Rodrigues B. Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol (2006) 291(4):H1489–506. doi:10.1152/ajpheart.00278.2006
115. Harmon CM, Abumrad NA. Binding of sulfosuccinimidyl fatty acids to adipocyte membrane proteins: isolation and amino-terminal sequence of an 88-kD protein implicated in transport of long-chain fatty acids. J Membr Biol (1993) 133(1):43–9. doi:10.1007/BF00231876
116. Habets DD, Coumans WA, Voshol PJ, den Boer MA, Febbraio M, Bonen A, et al. AMPK-mediated increase in myocardial long-chain fatty acid uptake critically depends on sarcolemmal CD36. Biochem Biophys Res Commun (2007) 355(1):204–10. doi:10.1016/j.bbrc.2007.01.141
117. Guerre-Millo M, Rouault C, Poulain P, Andre J, Poitout V, Peters JM, et al. PPAR-alpha-null mice are protected from high-fat diet-induced insulin resistance. Diabetes (2001) 50(12):2809–14. doi:10.2337/diabetes.50.12.2809
118. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev (2005) 85(3):1093–129. doi:10.1152/physrev.00006.2004
119. Song S, Attia RR, Connaughton S, Niesen MI, Ness GC, Elam MB, et al. Peroxisome proliferator activated receptor alpha (PPARalpha) and PPAR gamma coactivator (PGC-1alpha) induce carnitine palmitoyltransferase IA (CPT-1A) via independent gene elements. Mol Cell Endocrinol (2010) 325(1–2):54–63. doi:10.1016/j.mce.2010.05.019
120. Rakhshandehroo M, Knoch B, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res (2010) 2010: 612089. doi:10.1155/2010/612089
121. Hinkle PC. P/O ratios of mitochondrial oxidative phosphorylation. Biochim Biophys Acta (2005) 1706(1–2):1–11. doi:10.1016/j.bbabio.2004.09.004
122. Woldegiorgis G, Yousufzai SY, Shrago E. Studies on the interaction of palmitoyl coenzyme A with the adenine nucleotide translocase. J Biol Chem (1982) 257(24):14783–7.
123. Shrago E, Woldegiorgis G, Ruoho AE, DiRusso CC. Fatty acyl CoA esters as regulators of cell metabolism. Prostaglandins Leukot Essent Fatty Acids (1995) 52(2–3):163–6. doi:10.1016/0952-3278(95)90016-0
124. Shug AL, Shrago E, Bittar N, Folts JD, Koke JR. Acyl-CoA inhibition of adenine nucleotide translocation in ischemic myocardium. Am J Physiol (1975) 228(3):689–92.
125. Nishida K, Otsu K. Inflammation and metabolic cardiomyopathy. Cardiovasc Res (2017) 113:389–98. doi:10.1093/cvr/cvx012
126. Ray I, Mahata SK, De RK. Obesity: an immunometabolic perspective. Front Endocrinol (2016) 7:157. doi:10.3389/fendo.2016.00157
127. Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol (2015) 15(2):104–16. doi:10.1038/nri3793
128. Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation (2007) 115(25):3213–23. doi:10.1161/CIRCULATIONAHA.106.679597
129. Frieler RA, Mortensen RM. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation (2015) 131(11):1019–30. doi:10.1161/CIRCULATIONAHA.114.008788
130. Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res (2015) 116(7):1254–68. doi:10.1161/CIRCRESAHA.116.302317
131. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res (2016) 119(1):91–112. doi:10.1161/CIRCRESAHA.116.303577
132. Min W, Bin ZW, Quan ZB, Hui ZJ, Sheng FG. The signal transduction pathway of PKC/NF-kappa B/c-fos may be involved in the influence of high glucose on the cardiomyocytes of neonatal rats. Cardiovasc Diabetol (2009) 8:8. doi:10.1186/1475-2840-8-8
133. Nan WQ, Shan TQ, Qian X, Ping W, Bing GA, Ying LL. PPARalpha agonist prevented the apoptosis induced by glucose and fatty acid in neonatal cardiomyocytes. J Endocrinol Invest (2011) 34(4):271–5. doi:10.1007/BF03347084
134. Fuentes-Antras J, Ioan AM, Tunon J, Egido J, Lorenzo O. Activation of toll-like receptors and inflammasome complexes in the diabetic cardiomyopathy-associated inflammation. Int J Endocrinol (2014) 2014:847827. doi:10.1155/2014/847827
135. Rojas A, Delgado-Lopez F, Gonzalez I, Perez-Castro R, Romero J, Rojas I. The receptor for advanced glycation end-products: a complex signaling scenario for a promiscuous receptor. Cell Signal (2013) 25(3):609–14. doi:10.1016/j.cellsig.2012.11.022
136. Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes (2013) 62(1):194–204. doi:10.2337/db12-0420
137. Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS One (2014) 9(8):e104771. doi:10.1371/journal.pone.0104771
138. Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol (2014) 15(8):738–48. doi:10.1038/ni.2919
139. Butts B, Gary RA, Dunbar SB, Butler J. The importance of NLRP3 inflammasome in heart failure. J Card Fail (2015) 21(7):586–93. doi:10.1016/j.cardfail.2015.04.014
140. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science (2013) 339(6116):161–6. doi:10.1126/science.1230719
141. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med (2007) 204(12):3037–47. doi:10.1084/jem.20070885
142. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity (2014) 40(1):91–104. doi:10.1016/j.immuni.2013.11.019
143. Pinto AR, Paolicelli R, Salimova E, Gospocic J, Slonimsky E, Bilbao-Cortes D, et al. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS One (2012) 7(5):e36814. doi:10.1371/journal.pone.0036814
144. Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH, et al. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation (1998) 98(7):699–710. doi:10.1161/01.CIR.98.7.699
145. Saxena A, Dobaczewski M, Rai V, Haque Z, Chen W, Li N, et al. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am J Physiol Heart Circ Physiol (2014) 307(8):H1233–42. doi:10.1152/ajpheart.00328.2014
146. Zouggari Y, Ait-Oufella H, Bonnin P, Simon T, Sage AP, Guerin C, et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med (2013) 19(10):1273–80. doi:10.1038/nm.3284
147. Choi JH, Do Y, Cheong C, Koh H, Boscardin SB, Oh YS, et al. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J Exp Med (2009) 206(3):497–505. doi:10.1084/jem.20082129
148. Hettinger J, Richards DM, Hansson J, Barra MM, Joschko AC, Krijgsveld J, et al. Origin of monocytes and macrophages in a committed progenitor. Nat Immunol (2013) 14(8):821–30. doi:10.1038/ni.2638
149. Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, et al. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat Immunol (2011) 12(8):778–85. doi:10.1038/ni.2063
150. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity (2013) 38(1):79–91. doi:10.1016/j.immuni.2012.12.001
151. Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol (2006) 7(3):311–7. doi:10.1038/ni1309
152. Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, et al. CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res (2005) 96(8):881–9. doi:10.1161/01.RES.0000163017.13772.3a
153. Zhou L, Azfer A, Niu J, Graham S, Choudhury M, Adamski FM, et al. Monocyte chemoattractant protein-1 induces a novel transcription factor that causes cardiac myocyte apoptosis and ventricular dysfunction. Circ Res (2006) 98(9):1177–85. doi:10.1161/01.RES.0000220106.64661.71
154. Frangogiannis NG, Dewald O, Xia Y, Ren G, Haudek S, Leucker T, et al. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation (2007) 115(5):584–92. doi:10.1161/CIRCULATIONAHA.106.646091
155. Monnerat G, Alarcon ML, Vasconcellos LR, Hochman-Mendez C, Brasil G, Bassani RA, et al. Macrophage-dependent IL-1beta production induces cardiac arrhythmias in diabetic mice. Nat Commun (2016) 7:13344. doi:10.1038/ncomms13344
156. Shiraishi M, Shintani Y, Shintani Y, Ishida H, Saba R, Yamaguchi A, et al. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest (2016) 126(6):2151–66. doi:10.1172/JCI85782
157. Cordero-Reyes AM, Youker KA, Trevino AR, Celis R, Hamilton DJ, Flores-Arredondo JH, et al. Full expression of cardiomyopathy is partly dependent on B-cells: a pathway that involves cytokine activation, immunoglobulin deposition, and activation of apoptosis. J Am Heart Assoc (2016) 5(1):e002484. doi:10.1161/JAHA.115.002484
158. Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, et al. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol (2016) 13(3):167–79. doi:10.1038/nrcardio.2015.169
159. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133(5):775–87. doi:10.1016/j.cell.2008.05.009
160. Rudensky AY. Regulatory T cells and Foxp3. Immunol Rev (2011) 241(1):260–8. doi:10.1111/j.1600-065X.2011.01018.x
161. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155(3):1151–64.
162. Tschope C, Walther T, Escher F, Spillmann F, Du J, Altmann C, et al. Transgenic activation of the kallikrein-kinin system inhibits intramyocardial inflammation, endothelial dysfunction and oxidative stress in experimental diabetic cardiomyopathy. FASEB J (2005) 19(14):2057–9. doi:10.1096/fj.05-4095fje
163. Westermann D, Van Linthout S, Dhayat S, Dhayat N, Schmidt A, Noutsias M, et al. Tumor necrosis factor-alpha antagonism protects from myocardial inflammation and fibrosis in experimental diabetic cardiomyopathy. Basic Res Cardiol (2007) 102(6):500–7. doi:10.1007/s00395-007-0673-0
164. Westermann D, Walther T, Savvatis K, Escher F, Sobirey M, Riad A, et al. Gene deletion of the kinin receptor B1 attenuates cardiac inflammation and fibrosis during the development of experimental diabetic cardiomyopathy. Diabetes (2009) 58(6):1373–81. doi:10.2337/db08-0329
165. Rajesh M, Mukhopadhyay P, Batkai S, Mukhopadhyay B, Patel V, Hasko G, et al. Xanthine oxidase inhibitor allopurinol attenuates the development of diabetic cardiomyopathy. J Cell Mol Med (2009) 13(8B):2330–41. doi:10.1111/j.1582-4934.2008.00564.x
166. Rajesh M, Batkai S, Kechrid M, Mukhopadhyay P, Lee WS, Horvath B, et al. Cannabinoid 1 receptor promotes cardiac dysfunction, oxidative stress, inflammation, and fibrosis in diabetic cardiomyopathy. Diabetes (2012) 61(3):716–27. doi:10.2337/db11-0477
167. Rajesh M, Mukhopadhyay P, Batkai S, Patel V, Saito K, Matsumoto S, et al. Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. J Am Coll Cardiol (2010) 56(25):2115–25. doi:10.1016/j.jacc.2010.07.033
168. Ferdinandy P, Danial H, Ambrus I, Rothery RA, Schulz R. Peroxynitrite is a major contributor to cytokine-induced myocardial contractile failure. Circ Res (2000) 87(3):241–7. doi:10.1161/01.RES.87.3.241
169. Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res (2004) 94(12):1543–53. doi:10.1161/01.RES.0000130526.20854.fa
170. Li YY, Feng YQ, Kadokami T, McTiernan CF, Draviam R, Watkins SC, et al. Myocardial extracellular matrix remodeling in transgenic mice overexpressing tumor necrosis factor alpha can be modulated by anti-tumor necrosis factor alpha therapy. Proc Natl Acad Sci U S A (2000) 97(23):12746–51. doi:10.1073/pnas.97.23.12746
171. Chen Y, Ohmori K, Mizukawa M, Yoshida J, Zeng Y, Zhang L, et al. Differential impact of atorvastatin vs pravastatin on progressive insulin resistance and left ventricular diastolic dysfunction in a rat model of type II diabetes. Circ J (2007) 71(1):144–52. doi:10.1253/circj.71.144
172. Al-Attas OS, Al-Daghri NM, Al-Rubeaan K, da Silva NF, Sabico SL, Kumar S, et al. Changes in endotoxin levels in T2DM subjects on anti-diabetic therapies. Cardiovasc Diabetol (2009) 8:20. doi:10.1186/1475-2840-8-20
173. Elrashidy RA, Asker ME, Mohamed HE. Beneficial effects of pioglitazone against cardiovascular injury are enhanced by combination with aliskiren in a rat model of diabetic nephropathy. J Pharm Pharmacol (2012) 64(6):862–71. doi:10.1111/j.2042-7158.2012.01508.x
174. Singh M, Saini HK. Resident cardiac mast cells and ischemia-reperfusion injury. J Cardiovasc Pharmacol Ther (2003) 8(2):135–48. doi:10.1177/107424840300800207
175. Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. microRNA genes are transcribed by RNA polymerase II. EMBO J (2004) 23(20):4051–60. doi:10.1038/sj.emboj.7600385
176. van de Vrie M, Heymans S, Schroen B. microRNA involvement in immune activation during heart failure. Cardiovasc Drugs Ther (2011) 25(2):161–70. doi:10.1007/s10557-011-6291-y
177. Martin MM, Buckenberger JA, Jiang J, Malana GE, Nuovo GJ, Chotani M, et al. The human angiotensin II type 1 receptor +1166 A/C polymorphism attenuates microRNA-155 binding. J Biol Chem (2007) 282(33):24262–9. doi:10.1074/jbc.M701050200
178. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of bic/microRNA-155 for normal immune function. Science (2007) 316(5824):608–11. doi:10.1126/science.1139253
179. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A (2006) 103(33):12481–6. doi:10.1073/pnas.0605298103
180. Haasch D, Chen YW, Reilly RM, Chiou XG, Koterski S, Smith ML, et al. T cell activation induces a noncoding RNA transcript sensitive to inhibition by immunosuppressant drugs and encoded by the proto-oncogene, BIC. Cell Immunol (2002) 217(1–2):78–86. doi:10.1016/S0008-8749(02)00506-3
181. Leptidis S, El Azzouzi H, Lok SI, de Weger R, Olieslagers S, Kisters N, et al. A deep sequencing approach to uncover the miRNOME in the human heart. PLoS One (2013) 8(2):e57800. doi:10.1371/journal.pone.0057800
182. Zheng D, Ma J, Yu Y, Li M, Ni R, Wang G, et al. Silencing of miR-195 reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia (2015) 58(8):1949–58. doi:10.1007/s00125-015-3622-8
183. Baseler WA, Thapa D, Jagannathan R, Dabkowski ER, Croston TL, Hollander JM. miR-141 as a regulator of the mitochondrial phosphate carrier (Slc25a3) in the type 1 diabetic heart. Am J Physiol Cell Physiol (2012) 303(12):C1244–51. doi:10.1152/ajpcell.00137.2012
184. Kuwabara Y, Horie T, Baba O, Watanabe S, Nishiga M, Usami S, et al. microRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ Res (2015) 116(2):279–88. doi:10.1161/CIRCRESAHA.116.304707
185. Horie T, Ono K, Nishi H, Iwanaga Y, Nagao K, Kinoshita M, et al. microRNA-133 regulates the expression of GLUT4 by targeting KLF15 and is involved in metabolic control in cardiac myocytes. Biochem Biophys Res Commun (2009) 389(2):315–20. doi:10.1016/j.bbrc.2009.08.136
186. Lu H, Buchan RJ, Cook SA. microRNA-223 regulates Glut4 expression and cardiomyocyte glucose metabolism. Cardiovasc Res (2010) 86(3):410–20. doi:10.1093/cvr/cvq010
187. Chen S, Puthanveetil P, Feng B, Matkovich SJ, Dorn GW II, Chakrabarti S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J Cell Mol Med (2014) 18(3):415–21. doi:10.1111/jcmm.12218
188. Marchand A, Atassi F, Mougenot N, Clergue M, Codoni V, Berthuin J, et al. miR-322 regulates insulin signaling pathway and protects against metabolic syndrome-induced cardiac dysfunction in mice. Biochim Biophys Acta (2016) 1862(4):611–21. doi:10.1016/j.bbadis.2016.01.010
189. Chavali V, Tyagi SC, Mishra PK. Differential expression of dicer, miRNAs, and inflammatory markers in diabetic Ins2± Akita hearts. Cell Biochem Biophys (2014) 68(1):25–35. doi:10.1007/s12013-013-9679-4
190. Nandi SS, Zheng H, Sharma NM, Shahshahan HR, Patel KP, Mishra PK. Lack of miR-133a decreases contractility of diabetic hearts: a role for novel cross talk between tyrosine aminotransferase and tyrosine hydroxylase. Diabetes (2016) 65(10):3075–90. doi:10.2337/db16-0023
191. Costantino S, Paneni F, Luscher TF, Cosentino F. microRNA profiling unveils hyperglycaemic memory in the diabetic heart. Eur Heart J (2016) 37(6):572–6. doi:10.1093/eurheartj/ehv599
192. Hsu SH, Wang B, Kota J, Yu J, Costinean S, Kutay H, et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest (2012) 122(8):2871–83. doi:10.1172/JCI63539
193. Tsai WC, Hsu SD, Hsu CS, Lai TC, Chen SJ, Shen R, et al. microRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest (2012) 122(8):2884–97. doi:10.1172/JCI63455
194. Marquart TJ, Allen RM, Ory DS, Baldan A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc Natl Acad Sci U S A (2010) 107(27):12228–32. doi:10.1073/pnas.1005191107
195. Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, et al. microRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science (2010) 328(5985):1566–9. doi:10.1126/science.1189123
196. Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science (2010) 328(5985):1570–3. doi:10.1126/science.1189862
197. Horie T, Ono K, Horiguchi M, Nishi H, Nakamura T, Nagao K, et al. microRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (Srebp2) regulates HDL in vivo. Proc Natl Acad Sci U S A (2010) 107(40):17321–6. doi:10.1073/pnas.1008499107
198. el Azzouzi H, Leptidis S, Dirkx E, Hoeks J, van Bree B, Brand K, et al. The hypoxia-inducible microRNA cluster miR-199a approximately 214 targets myocardial PPARdelta and impairs mitochondrial fatty acid oxidation. Cell Metab (2013) 18(3):341–54. doi:10.1016/j.cmet.2013.08.009
199. Miki T, Yuda S, Kouzu H, Miura T. Diabetic cardiomyopathy: pathophysiology and clinical features. Heart Fail Rev (2013) 18(2):149–66. doi:10.1007/s10741-012-9313-3
200. Zhao F, Li B, Wei YZ, Zhou B, Wang H, Chen M, et al. microRNA-34a regulates high glucose-induced apoptosis in H9c2 cardiomyocytes. J Huazhong Univ Sci Technolog Med Sci (2013) 33(6):834–9. doi:10.1007/s11596-013-1207-7
201. Yu XY, Song YH, Geng YJ, Lin QX, Shan ZX, Lin SG, et al. Glucose induces apoptosis of cardiomyocytes via microRNA-1 and IGF-1. Biochem Biophys Res Commun (2008) 376(3):548–52. doi:10.1016/j.bbrc.2008.09.025
202. Feng B, Chen S, George B, Feng Q, Chakrabarti S. miR133a regulates cardiomyocyte hypertrophy in diabetes. Diabetes Metab Res Rev (2010) 26(1):40–9. doi:10.1002/dmrr.1054
203. Iribarren C, Karter AJ, Go AS, Ferrara A, Liu JY, Sidney S, et al. Glycemic control and heart failure among adult patients with diabetes. Circulation (2001) 103(22):2668–73. doi:10.1161/01.CIR.103.22.2668
204. Held C, Gerstein HC, Yusuf S, Zhao F, Hilbrich L, Anderson C, et al. Glucose levels predict hospitalization for congestive heart failure in patients at high cardiovascular risk. Circulation (2007) 115(11):1371–5. doi:10.1161/CIRCULATIONAHA.106.661405
205. Thrainsdottir IS, Aspelund T, Thorgeirsson G, Gudnason V, Hardarson T, Malmberg K, et al. The association between glucose abnormalities and heart failure in the population-based Reykjavik study. Diabetes Care (2005) 28(3):612–6. doi:10.2337/diacare.28.3.612
206. Castagno D, Baird-Gunning J, Jhund PS, Biondi-Zoccai G, MacDonald MR, Petrie MC, et al. Intensive glycemic control has no impact on the risk of heart failure in type 2 diabetic patients: evidence from a 37,229 patient meta-analysis. Am Heart J (2011) 162(5):938–48.e2. doi:10.1016/j.ahj.2011.07.030
Keywords: obesity, insulin resistance, inflammation, cardiomyopathy, innate and adaptive immunity, glucose metabolism, fat metabolism, miRNA
Citation: Mishra PK, Ying W, Nandi SS, Bandyopadhyay GK, Patel KK and Mahata SK (2017) Diabetic Cardiomyopathy: An Immunometabolic Perspective. Front. Endocrinol. 8:72. doi: 10.3389/fendo.2017.00072
Received: 02 February 2017; Accepted: 27 March 2017;
Published: 07 April 2017
Edited by:
Gaetano Santulli, Columbia University, USAReviewed by:
Sarah Costantino, University of Zurich, SwitzerlandLong Yang, New York Medical College, USA
Daniele Catalucci, Institute of Genetic and Biomedical Research (CNR), Italy
Antonio Paolo Beltrami, University of Udine, Italy
Copyright: © 2017 Mishra, Ying, Nandi, Bandyopadhyay, Patel and Mahata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paras K. Mishra, cGFyYXNrdW1hci5taXNocmFAdW5tYy5lZHU=;
Sushil K. Mahata, c21haGF0YUB1Y3NkLmVkdQ==