 Kimberley D. Bruce
Kimberley D. Bruce Andrea Zsombok
Andrea Zsombok Robert H. Eckel1
Robert H. Eckel1
- 1University of Colorado School of Medicine, Division of Endocrinology, Metabolism and Diabetes, Aurora, CO, USA
- 2Department of Physiology, School of Medicine, Tulane University, New Orleans, LA, USA
Metabolic disorders, particularly aberrations in lipid homeostasis, such as obesity, type 2 diabetes mellitus, and hypertriglyceridemia often manifest together as the metabolic syndrome (MetS). Despite major advances in our understanding of the pathogenesis of these disorders, the prevalence of the MetS continues to rise. It is becoming increasingly apparent that intermediary metabolism within the central nervous system is a major contributor to the regulation of systemic metabolism. In particular, lipid metabolism within the brain is tightly regulated to maintain neuronal structure and function and may signal nutrient status to modulate metabolism in key peripheral tissues such as the liver. There is now a growing body of evidence to suggest that fatty acid (FA) sensing in hypothalamic neurons via accumulation of FAs or FA metabolites may signal nutritional sufficiency and may decrease hepatic glucose production, lipogenesis, and VLDL-TG secretion. In addition, recent studies have highlighted the existence of liver-related neurons that have the potential to direct such signals through parasympathetic and sympathetic nervous system activity. However, to date whether these liver-related neurons are FA sensitive remain to be determined. The findings discussed in this review underscore the importance of the autonomic nervous system in the regulation of systemic metabolism and highlight the need for further research to determine the key features of FA neurons, which may serve as novel therapeutic targets for the treatment of metabolic disorders.
Introduction
Metabolic disorders, particularly aberrations in lipid homeostasis, such as obesity, type 2 diabetes mellitus (T2D), non-alcoholic fatty liver disease, and hypertriglyceridemia, often manifest together as the metabolic syndrome (MetS) (1). Despite major advances in our understanding of the pathogenesis of these disorders, the prevalence of the MetS continues to rise (2). Since MetS constitutes an increased risk to cardiovascular morbidity and mortality (3), a more detailed understanding of the common causes, and integration between these disorders of energy homeostasis, is necessary to identify novel therapeutic targets and interventions that may halt the development of severe metabolic disease.
It is becoming increasingly apparent that the central nervous system (CNS) is a major contributor to the regulation of systemic metabolism and lipid balance. In the CNS, the nutritional status of the body is constantly being surveyed and assessed by key energy-sensing regions of the brain, such as the hypothalamus. Key nuclei within the hypothalamus, such as the ventromedial nucleus (VMH), arcuate nucleus (ARC), dorsomedial hypothalamic nucleus (DMH), and the paraventricular nucleus (PVN), integrate signals to elicit peripheral responses, such as changes in feeding behavior, fuel mobilization, energy utilization, and energy storage (4). These nuclei detect both nutrients and nutritionally regulated endocrine factors, such as insulin (5), ghrelin (6), melanocortin (MC) (7), and leptin (8), in order to regulate feeding and energy balance. Here, in this review, we will focus on the mechanisms involved in lipid sensing in the brain and its emerging influence on systemic metabolism.
How Do Lipids Enter the Brain?
Lipids and lipid intermediates are essential components of the structure and function of the brain. In fact, the brain has the second highest lipid content behind adipose tissue, and brain lipids constitute 50% of the brain dry weight (9). However, unlike adipose tissue, which largely stores FAs as triglycerides for subsequent utilization and mobilization to other metabolic tissues, the brain is thought to mainly utilize acylated lipids to generate phospholipids for cell membranes (9). The FA composition of the brain is unique and is rich in long-chain polyunsaturated fatty acids (LC-PUFAs), particularly arachidonic acid (AA), eicosapentaenoic acid, and docosahexaenoic acid (DHA). Although some FAs can be synthesized de novo, essential FAs must be transported into the brain from the systemic circulation. And contrary to previously held theories, recent data suggest that this is a dynamic process, with up to 8% of LC-PUFAs actively being turned over daily and being replaced by plasma-derived FAs (10). Indeed, a number of studies have shown that FAs are able to cross the blood–brain barrier (BBB) and enter neurons. For example, radiolabeled FAs that are injected into the carotid artery of rats can be traced to neuronal cells (10). In addition, brain perfusion studies in rats have shown that radiolabeled palmitate is readily incorporated into cerebral phospholipids and neutral lipids and has a similar rate of uptake when delivered via whole rat plasma or a synthetic saline containing physiological levels of albumin. This suggests that FAs cross the BBB and that albumin may be important in this process (11). Even though these studies convincingly demonstrate FA uptake, they do not address the mechanism of transport.
How FAs enter the brain remains an unanswered fundamental question. Although we cannot rule out the possibility that FAs could passively diffuse across the BBB, several studies highlight the role of FA transporters in this process. The membrane localized FA transport proteins (FATP1 and FATP4) appear to be the predominant FA transport proteins expressed in the BBB based on human and mouse expression studies, whereas the FA translocase/CD36 plays a prominent role in the transport of FAs across human brain microvessel endothelial cells (12). Furthermore, cytosolic-localized fatty acid-binding protein 5 has an important function in FA transport across cultured brain microvascular cells (13). Although still under active investigation, is it plausible that some transporters may also exhibit specificity toward particular FAs. For example, the major facilitator superfamily d 2 a, which is exclusively expressed in the endothelium of the BBB, has been shown to selectively transport DHA in the form of the partially hydrolyzed phospholipid, lysophosphatidylcholine, otherwise known as lysolecithin (14).
Neuronal Uptake of Lipids and FAs
Neuronal uptake of FAs remains an important yet poorly understood process. However, it is possible that once FAs have traversed the BBB, FA transporters may also play a key role in facilitating FA uptake into neurons. For example, dissociated neurons from the VMH express both FATP1 and CD36 (15). Specifically, important studies using fura-2 calcium imaging and fluorometric imaging plate reader membrane potential dye, in addition to pharmacological manipulations have shown that while neurons of the VMH and ARC respond to oleic acid (OA, C18:1 n-9) (15, 16), this response is lost when CD36 is depleted in the VMH using an adeno-associated viral (AAV) vector expressing CD36 short hairpin RNA (17). Since CD38 is an established gustatory lipid sensor, it is also plausible that other lipid sensors involved in the chemoreception of long-chain fatty acids (LCFAs)/omega-3 fatty acids (ω-3 FAs), such as GPR120, are also involved in neuronal lipid sensing. However, although GPR120 is functionally active in immortalized hypothalamic neurons and mediates the anti-inflammatory actions of the ω-3 FA, DHA (18), its role in neuronal lipid sensing in vivo has not been determined. In addition, FABP3, which is localized in neurons, facilitates brain AA but not palmitic acid (C16:0) uptake and trafficking into specific brain lipid pools (19).
It is also becoming more widely accepted that neurons may receive metabolic support from glial cells in a variety of forms. The “astrocyte–neuron lactate shuttle” has been postulated, whereby astrocytes metabolize glucose to release lactate, which is then taken up by the neuronally expressed monocarboxylate transporter, providing a supplemental energy source for neurons (20). Moreover, lipid metabolism in astrocytes plays a key role in FA sensing, since FA oxidation is thought to occur predominantly in the astrocyte rather than the neuron. When the levels of FAs are increased, as in the case of a high-fat diet (HFD), astrocyte-mediated lipid oxidation results in elevated ketone levels in the brain. Conversely, changes in ketone abundance within energy-sensing regions of the hypothalamus are able to modify energy homeostasis. Specifically, reduced ketone production within the VMH and ARC signals a decrease in HFD intake, and ketone production can override glucose and FA sensing in VMH neurons (21, 22). Astrocytes may play a key role in the regulation of energy balance by sensing LCFAs as metabolic signals. For example, hypothalamic but not cortical astrocytes have a high capacity for the oxidation of LCFAs (23), and this flux may be AMP-activated protein kinase (AMPK) dependent. Both neurons of the VMH and astrocytes have been shown to express many of the FA transporters, including FATP1, FATP4, and CD36. Most recently, the fatty acid bind protein 7 (FABP7) was shown to be important for astrocyte–neuron lipid homeostasis. Mice lacking FABP7 develop neuropsychiatric disorders such as schizophrenia, which may at least in part be due to aberrant dendritic spine morphology, and decreased spine density compared to WT mice (24). Moreover, transplantation of WT astrocytes into FABP7 KO mice partially attenuated cognitive impairments (24). Once transported into the cell, LCFAs are esterified by acyl-CoA-binding protein (ACBP), a protein that is ubiquitously expressed in tissues with high lipid turnover. Interestingly, ACBP is expressed in the hypothalamus and may play a role in the LCFA sensing by hypothalamic astrocytes (25).
Astrocytes are also critical for the regulation of cholesterol homeostasis in the neuron. Cholesterol is an essential component of neuronal physiology during both development and adulthood and is independently tightly regulated in the brain, largely due to the existence of the BBB (26). Cholesterol depletion in neurons impairs vital functions, including synaptic vesicle exocytosis, neuronal activity, and neurotransmission and results in synaptic loss and neurodegeneration (27, 28). Clinically, deficits in cholesterol homeostasis in the CNS manifest as severe primary neurological disorders such as Neimann–Pick C disease and Parkinson’s disease (26). It is thought that astrocytes are a major site of lipoprotein synthesis and assembly in the brain (29). Of particular relevance is apolipoprotein E (ApoE), a 39-kDa protein that is highly expressed in the brain, surpassed only by hepatic ApoE production (30).
Apolipoprotein E-containing lipoproteins have been extensively studied and are known to have several major functions. For example, ApoE-containing high-density lipoprotein (HDL)-like lipoproteins are secreted by astrocytes and are taken up into neurons via low-density lipoprotein receptors. This transfer of key lipids and cholesterol facilitates axonal extension and neuronal survival and requires the presence of sphingomyelin in the ApoE-containing lipoprotein particle (31, 32). ApoE does not only play a major role in glia-neuronal lipid metabolism but also acts as a ligand for multiple receptors in neurons, which interact with a number of downstream physiological processes (33). In particular, a number of recent studies have shown that brain ApoE is an important regulator of peripheral energy homeostasis. In rats, intracerebroventricular (ICV) ApoE injections significantly decreased food intake, whereas infusion of ApoE antiserum stimulated feeding, therefore suggesting that ApoE may be important satiety factor in the hypothalamus (34). In support, exogenous ApoE treatment has been shown to activate phosphatidylinositol-3-kinase (PI3K)/Akt signaling, and PI3K inhibition by LY294002 attenuates both ApoE-induced signaling and satiation (35).
Apolipoprotein E has three major isoforms (ApoE2, ApoE3, and ApoE4), which have varying effects on lipid homeostasis and neuronal function. While both ApoE2 and ApoE3 preferentially associate with phospholipid rich HDL-like particles, ApoE4 prefers large triglyceride-rich VLDL particles (36). Interestingly, ApoE4 has been repeatedly implicated in the pathogenesis of Alzheimer’s disease (AD) and may bind to the Aβ peptide leading to impair Aβ clearance (37). Current hypothesis suggests that the proteolytic cleavage products of ApoE4 may have a central role in AD pathology, since AD patients have markedly increased levels of these cleavage products compared to controls (38).
These findings suggest that lipoprotein metabolism in the CNS may be key to lipid homeostasis and nutrient sensing; however, to date, the molecular mechanisms underlying these processes remain largely unknown. Nonetheless, we have recently shown that lipoprotein lipase (LPL), the rate-limiting enzyme in the hydrolysis of lipoprotein-derived FAs, may facilitate the uptake of FAs into dissociated hypothalamic neurons (39). Moreover, mice with a specific neuronal LPL deficiency exhibit a defect in nutrient sensing and become hyperphagic, relatively inactive and obsessed compared to WT control mice (40). Interestingly, these mice are also polyunsaturated fatty acid (PUFA) deficient in the hypothalamus (40), and in the hippocampus, where LPL deficiency is also associated with alterations in learning and memory and synaptic function (41). In further support, a specific deletion of LPL in the hippocampus also leads to increased weight gain and decreased activity via a ceramide-dependent pathway (42). We have also shown that while mice lacking pan-neuronal LPL develop obesity, this obesity is not exacerbated on an HF diet, or rescued by a PUFA enriched diet, further highlighting the importance of LPL in lipid sensing and body weight regulation (43). These data highlight the potential role of LPL as a mechanistic link between brain lipid uptake, neuronal lipoprotein metabolism, and nutrient sensing; however, the precise role of LPL in lipid sensing in both neurons and glia requires further investigation.
Hypothalamic FA Metabolism
The evidence for facilitated FA uptake into neurons is limited, which may be due to the long-held view that neurons do not derive much of their energy supply from lipids. However, neurons do express many molecular components of lipid catabolism pathways, suggesting that lipid utilization is a critical process to neuronal function. For example, neurons of the VMH express enzymes involved in the intracellular metabolism of FAs, including long-chain acyl-CoA synthase (ACS), carnitine palmitoyltransferase-1a and 1c (CPT-1a and 1c), and uncoupling protein-2 (UCP2), and enzymes involved in de novo lipogenesis, such as fatty acid synthase (FAS) (15). In addition, the nuclear receptor peroxisome proliferator-activated receptor (PPARγ), a key factor in lipid metabolism that can be activated by endogenous lipid ligands to promote adipogenesis and insulin sensitivity, is predominantly expressed in neurons of the hypothalamus (44), including the ARC.
Activation of these intracellular metabolic pathways in hypothalamic cells in response to FAs provides further support of the role of lipid metabolism and sensing in hypothalamic neurons. For example, UCP2, which increases proton leak from the respiratory electron transport chain, may act as a metabolic switch from glucose metabolism to mitochondrial FA oxidation in hypothalamic NPY/AgRP neurons during fasting (45). Interestingly, UCP2 likely mediates the actions of ghrelin, which is increased upon fasting, on the activation of NPY/AgRP neurons. While ghrelin increases palmitate-induced mitochondrial respiration, this is not observed in UCP2−/− mice. Moreover, NPY/AgRP neurons of UCP2−/− mice do not show the ghrelin-induced increase in mitochondrial biogenesis (45). In normal circumstances, increased FA oxidation increases reactive oxygen species (ROS), which are then scavenged by UCP2 via enhanced proton leak. However, in UCP2−/− mice, these ROS levels remain increased, supporting the hypothesis that ghrelin-triggered ROS production promotes UCP2 activity and mRNA expression to further promote ROS scavenging (45). These studies also suggest that ROS may be involved in neuronal lipid metabolism and activation. In further support, suppression of ROS activates NPY/AgRP neurons to promote feeding, whereas activation of ROS activates proopiomelanocortin (POMC) neurons to reduce feeding. Although further studies are warranted, it is likely that neuronal ROS accumulation may intrinsically link neuronal substrate metabolism to feeding behavior and systemic energy balance (46).
AMP-activated protein kinase may also act as a cellular energy sensor within neurons to link neuronal lipid metabolism to systemic lipid metabolism and energy balance. AMPK is widely expressed in the ARC, PVN, and VMH of the hypothalamus and is able to sense intracellular energy status by the AMP/ATP ratio and the level of adipokines (e.g., leptin and ghrelin) [see Ref. (47) for a comprehensive review]. Activated AMPK responds to the cellular energy status and can switch cellular metabolism toward catabolic processes that produce ATP and away from anabolic processes that consume ATP (48). In response to glucose, hypothalamic AMPK activity is inhibited leading to the activation of acetyl-CoA carboxylase (ACC) and the generation of malonyl-CoA from glucose-derived acetyl-CoA, with downstream effects on food intake (described in more detail below) (49). Similar to peripheral tissues, malonyl-CoA is thought to inhibit CPT-1a and LCFA oxidation in the brain (49). Specifically, glucose inhibits palmitate oxidation via AMPK in hypothalamic neurons (23). Hypothalamic AMPK may also regulate downstream lipid metabolism through brown adipose tissue (BAT) thermogenesis. A number of recent studies have suggested that hypothalamic AMPK is involved in the autonomic regulation of BAT thermogenesis, specifically the SNS. For example, central administration of 3,3′,5′-triiodothyronine (T3) within the VMH stimulates a thermogenic response associated with decreased AMPK activity in the VMH and elevated sympathetic firing in BAT (50). Whether this effect is observed following neuronal glucose (or lipid) sensing remains to be seen. Nonetheless, this interesting topic has recently been reviewed in detail (51).
AMP-activated protein kinase also plays a key role in the hypothalamic response to leptin in the context of high-fat feeding. While leptin results in reduced food intake and reduced hypothalamic AMPK activity, these effects were not observed in diet-induced obese mice (52). Hypothalamic phospho-AMPK is also modulated by the FA composition of the diet and is dependent on brain region and metabolic status (53). Interestingly, AMPK regulates the activity of a number of enzymes involved in the synthesis of complex lipids that are critical for optimal brain function and metabolism. For example, long-term AMPK stimulation blunted FA-mediated induction of serine palmitoyl transferase and the synthesis of ceramides de novo and has thus been shown to protect against fatty acid-mediated apoptosis in the astrocyte (54). Similarly, AMPK is highly expressed in neurons due to their high-energy demands and can promote neuronal survival during periods of glucose deprivation (55). Recently, neuronal AMPK has also been implicated in the pathogenesis of AD. AMPK activity can reduce sphingomyelin levels, inhibit Aβ generation, and reduce amyloid precursor protein (APP) distribution in lipid rafts, whereas deletion of AMPKα2 increases sphingomyelin and APP distribution in lipid rafts (56).
There is growing evidence to suggest that the accumulation of FA metabolites may signal nutrient status and thus may be critical to central lipid sensing, and the modulation of systemic metabolism. For example, upon entry into the neuron, LCFAs are esterified to LCFA-CoA by ACS. It is thought that this accumulation of FA derived LCFA-CoA triggers a lipid-sensing mechanism to inhibit hepatic glucose production (HGP) and to maintain systemic glucose homeostasis (57). In support, direct inhibition of ACS in the hypothalamus disrupts the accumulation of hypothalamic LCFA-CoA, and in turn disrupts the inhibitory effect on hepatic gluconeogenesis, resulting in dysregulated glucose production (58).
In metabolic tissues, intracellular LCFA-CoAs enter the mitochondria via CPT-1, where they are then subject to FA β-oxidation. Importantly, the liver isoform of CPT-1, CPT-1a, is prevalent in the hypothalamus, and inhibition of hypothalamic CPT-1a causes an increase in intracellular LCFA-CoA, which triggers a satiation signal, leading to reduced systemic glucose production and food intake (59). However, the neurocircuitry is complex since the role of CPT-1a may vary between hypothalamic nuclei. For example, VMH-selective overexpression of CPT-1a causes over feeding, a phenotype which can be reversed with the CPT-1 specific inhibitor etomoxir (60). In support, long-term overexpression of permanently activated CPT-1a using a viral AAV vector injected into the VMH of rats, leads to hyperghrelinemia, increased food intake, increased body weight, hyperglycemia, and insulin resistance (61). In contrast, CPT-1a expression in the ARC does not have the same effect on the central regulation of feeding (60). Interestingly, the brain also expresses a neuron-specific isoform of CPT-1, CPT-1c, which is found in the endoplasmic reticulum (ER) of key energy-sensing nuclei of the hypothalamus (ARC) and has also been repeatedly implicated in the modulation of systemic metabolism (62–64). Specifically, CPT-1c KO mice have reduced body weight and food intake compared to control mice (62, 63). CPT-1c does not have a typical acyltransferase activity, and thus its precise molecular function is less well understood. Nonetheless, data from recent studies suggest that the orexigenic action of ghrelin is associated with increased hypothalamic (C18:0) ceramide levels, an effect that is blunted in CPT-1c KO mice (65). While the mechanism linking increased ceramide levels to energy balance remain an active area of research, recent studies have demonstrated that ceramides induce hypothalamic lipotoxicity and ER stress, which leads to sympathetic inhibition, reduced BAT thermogenesis, weight gain, and hepatic steatosis (66). In addition, recent metabolomic analysis of the brains of CPT-1c KO mice shows reduced levels of oxidized glutathione, suggesting that CPT-1c may play a role in neuronal oxidative metabolism (67). In addition, these mice show suppressed endocannabinoid levels, which offers an alternative yet consistent mechanism to account for the suppressed food intake observed in CPT-1c KO mice (67). In addition, to their established role in appetite modulation, endogenous endocannabinoids may serve as functional neuromodulatory lipids, derived from neuronal phospholipids, which once secreted undergo lipid catabolism within glial cells. A detailed description of endocannabinoid signaling is beyond the scope of this manuscript but has been previously reviewed in depth (68).
CPT-1 has been suggested to act downstream of malonyl-CoA in the hypothalamic control of feeding (60). This is particularly pertinent to FA metabolism in hypothalamic neurons since CPT-1 activity is inhibited by malonyl-CoA (69), and thus elevated malonyl-CoA leads to an accumulation of LCFA-CoA (70), which has been previously referred to as a satiety signal. Indeed, manipulation of the key enzymes involved in malonyl-CoA metabolism, including ACC, FAS, and malonyl-CoA decarboxylase (MCD), have all been shown to have major effects on food intake and peripheral metabolism (71). For example, ACC, which catalyzes the carboxylation of acetyl-CoA to malonyl-CoA, is expressed in the ARC of the hypothalamus, where it is increased following central leptin treatment resulting in elevated malonyl-CoA and reduced food intake (72). Similarly, inhibition of FAS activity by central administration of the pharmacological FAS inhibitor C75 has been shown to reduce food intake (73) and to elevate malonyl-CoA levels (74). MCD, which is important for malonyl-CoA degradation, is also important for regulating lipid intermediates and systemic metabolism. Overexpression of MCD in the mediobasal hypothalamus (MBH) chronically reduces malonyl-CoA levels and causes rapid increases in food intake and weight gain (75). In addition, MBH expression depleted both malonyl-CoA and LCFA-CoA and was sufficient to induce hepatic insulin resistance in the presence of hyperlipidemia (75). These studies begin to highlight the importance of hypothalamic sensing of circulating lipids in the maintenance of hepatic metabolism and systemic glucose homeostasis. However, the physiological and molecular mechanisms that integrate the brain–liver axis remain unresolved and an active area of investigation.
Hypothalamic Lipid Sensing and Hepatic Metabolism
Early studies by Obici and colleagues, where OA was administered into the brain via ICV injection, were among the first to demonstrate the profound central effects of LCFAs on peripheral metabolism. Interestingly, the short chain FA octanoic acid (C8) did not have the same effect. Moreover, these studies showed that central OA administration could lower plasma insulin and glucose levels under basal physiological conditions. To determine the mode of blood glucose reduction, pancreatic euglycemic clamps were performed during ICV infusion of OA. ICV OA administration leads to a marked decline in glucose production compared to basal levels (59). However, when clamps were performed in the presence of ICV OA and sulfonylurea, a potent inhibitor of neuronal KATP channels, the profound effect of central OA on HGP was blunted, suggesting that OA acutely enhances hepatic insulin action via the activation of KATP channels in the hypothalamus (59). Further studies support this notion and have shown that pharmacological activation of KATP channels in the hypothalamus (via hypothalamic diazoxide administration) suppresses HGP (76). These observations, taken together with the findings from CPT-1 inhibition studies, strongly suggest that the accumulation of LCFA-CoAs is able to suppress HGP (70). Recent studies have suggested that this system may actually be even more complex and have shown that the effect of LCFAs on hepatic metabolism may be FA specific. For example, while bilateral infusion of OA into the MBH results in a significant reduction in HGP, palmitic acid (PA, C16:0) has a lesser effect, and linoleic acid has no effect compared to vehicle control (LA, C18:2 n-6) (77). These data suggest that mono-unsaturated fatty acids are a more potent suppressor than saturated fatty acids or PUFAs; however, the mechanisms underlying these differential effects of FAs on hepatic glucose metabolism remain to be determined.
In addition to the central regulation of HGP, hypothalamic nutrient sensing may also be a key regulator of hepatic lipid homeostasis. The liver maintains lipid homeostasis through tightly coordinated synthesis and secretion of triglyceride-rich lipoproteins (VLDL-TG), lipogenesis, and FA oxidation. Interestingly, infusion of the orexigenic neuropeptide Y directly into the third ventricle of the hypothalamus has been shown to increase hepatic VLDL-TG secretion, which may be part of the physiological response to fasting when lipids become the main energy source and NPY neurons in the ARC of the hypothalamus are activated (78). Thus, VLDL-TG secretion may be a response by the autonomic nervous system (ANS) to mobilize lipids during a period of relative nutrient deficiency. In addition, sympathetic denervation prevented the increase in the VLDL-TG secretion in the fasted state, whereas total denervation, or parasympathetic denervation did not, suggesting that the central regulation of hepatic lipid mobilization during fasting may be largely mediated through the sympathetic nervous system (78). In further support, sympathetic hepatic denervation also prevented the stimulatory effect of NPY on VLDL-TG secretion (78). This is in contrast to glucose sensing in the MBH (79), and glycine in the dorsal vagal complex (DVC) (80), which is thought to signal sufficient nutrient status and inhibit VLDL-TG secretion, possibly through the parasympathetic nervous system.
In addition to NPY, a number of studies have also shown that MC expressing neurons of the hypothalamus may also regulate hepatic lipogenesis and TG metabolism. Central administration of MTII, a synthetic MC3/4 receptor agonist, has been shown to reduce hepatic lipogenic gene expression in mice (81) and decrease hepatic TG content in rats (82), suggesting that increased MC signaling can inhibit hepatic VLDL-TG production. In support of this notion, central administration of an MC3/4 receptor antagonist, markedly increased liver TG content in rats, strongly suggesting that hepatic lipogenesis was increased (7).
Melanin-concentrating hormone (MCH) is also involved in the neuronal circuits that modify autonomic outflow to the liver and white adipose tissue. MCH-deficient mice are hyperphagic and lean when fed a normal diet (83), are resistant to age associated insulin resistance (84), and when fed an HFD they are resistant to obesity and hepatic steatosis (85). Recent key studies have also shown that genetic activation of MCH specifically in the LH triggers hepatic lipid accumulation and lipoprotein uptake (86).
This neurocircuitry is relevant to the pathogenesis of obesity, since numerous models of obesity and diabetes are characterized by elevated NPY. In a recent study, ICV administration of NPY or a selective NPY Y1 receptor agonist was shown to robustly elevate key genes involved in MUFA synthesis, such as stearol-CoA desatrate-1, and PL remodeling, such as ribosylation factor-1 (ARF-1) and lipin-1 (87). Importantly, these effects were attenuated following sympathetic denervation of the liver, supporting a model in which central NPY modifies hepatic PL and VLDL via the sympathetic signaling to the liver (87). Although these findings suggest that central lipid sensing may be implicated in the regulation of hepatic lipid homeostasis (see Figure 1), the direct mechanisms remain elusive. Nonetheless, a number of recent studies highlight the role of central FAs in hepatic lipid homeostasis. For example, central administration of PA resulted in impaired leptin signaling and pro-inflammatory response in the MBH and PVN (88). Furthermore, this was coupled with blunted leptin-induced changes in hepatic gluconeogenesis, glucose transportation, and lipogenesis (88). In a recent report, Yue and colleagues have shown that infusion of OA directly into the MBH activates a PKC-δ to KATP channel axis, which suppresses VLDL-TG secretion in rats (89). Moreover, this signaling requires DVC and hepatic innervation, highlighting a novel MBD-DVC neurocircuitry that mediates MBH FA sensing and hepatic lipid homeostasis (89). These findings are one step closer to the development of novel therapies that lower VLDL-TG secretion and restore lipid homeostasis in metabolic disorders; however, there is considerably more to learn regarding differential function of other hypothalamic neurons and their role in the autonomic regulation of hepatic metabolism.
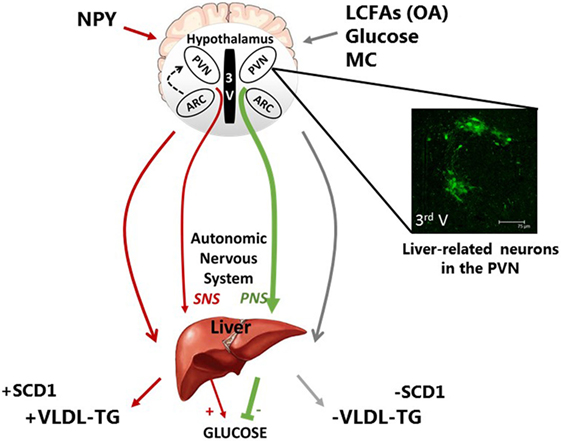
Figure 1. Schematic representation of the neuronal regulation of hepatic carbohydrate and lipid metabolism. Neurons of discrete hypothalamic nuclei including the paraventricular nucleus (PVN) and arcuate nucleus (ARC) within the mediobasal hypothalamus respond to neuronal inputs to regulate hepatic glucose and very low density lipoprotein (VLDL-TG) metabolism. The accumulation or application of long-chain fatty acids (LCFAs), such as oleic acid (OA), glucose, and melanocortin (MC) signaling act through the autonomic nervous system to signal nutritional plenty a reduced requirement for energy substrate mobilization, by inhibiting VLDL-TG secretion and hepatic glucose production (HGP). This is at least in part due to reduced stearol-CoA desatrate-1 (SCD1) expression. In contrast, hypothalamic NPY is indicative of hunger and nutritional deficiency and can increase energy substrate mobilization via an increase in VLDL-TG and SCD1 expression and HGP. The features of these fatty acid and nutritionally sensitive neurons are unclear; however, pseudorabies virus-152 labeling supports the notion that liver-related neurons in the PVN exist and may be involved in this autonomic regulation of hepatic metabolism.
Liver-Related Neurons in the Hypothalamus
Recent findings support the existence of liver-related neurons in key hypothalamic nuclei; therefore, we can speculate that these liver-related neurons are the link between central lipid sensing and hepatic lipid metabolism. The control of hepatic functions by the ANS is well known. In general, beside the abovementioned examples, sympathetic stimulation of the liver enhances endogenous glucose production and glycogenolysis, while the parasympathetic nerves are responsible for inhibiting glucose production and promoting glucose storage (90–92). Preganglionic neurons are located in the spinal cord and brainstem, respectively, and transmit the information through the sympathetic and parasympathetic nerves. These autonomic motor neurons receive information from preautonomic neurons, which are located in higher brain areas and crucial for integration of brain signals (93, 94). Therefore, identifying the location of liver-related neurons and determining their cellular and molecular properties would be crucial to the understanding of brain–liver circuit.
Within the hypothalamus, the ARC, VMH, DMH, LH, and PVN are well recognized for their involvement in the regulation of a variety of metabolic functions of the body, and in particular the control of hepatic metabolism (95–101). Electrical and chemical stimulation have been feasible methods to establish direct connections between the hypothalamus and sympathetic neurons in the spinal cord or parasympathetic neurons in the brainstem and to demonstrate the importance of the autonomic control in hepatic functions (98, 102–105). On the other hand, establishing the location of premotor inputs to the sympathetic and parasympathetic motor neurons has been more challenging. Anterograde and retrograde tracers in combination with histochemical studies provided valuable information on the connections between the brain and liver (106), and the development of retrograde viral tracers opened new avenues to dissect the brain–liver pathway. Currently, transsynaptic neurotropic viruses are very valuable tools for identification of synaptic connections and neural networks. Among the neurotropic viruses, pseudorabies viruses (PRVs) are often used for circuit analysis and revealing organization of the nervous system (107–110). PRVs, such as PRV-152, an attenuated viral strain driving the expression of EGFP, are reliable and effective transsynaptic tracers, and numerous publications reported consistent organ-specific labeling of neurons (111–115). The spread of PRV-152 is strictly retrograde across synapses, and the virus is not capable of assembling in axons or glia; therefore, labeling of neurons not specific to the liver is unlikely, as has been shown (110, 116–118).
Polysynaptic neural connections between the brain and liver were identified in rodents using PRVs (118–120). PRV labeling was observed in the spinal cord of rats 3 days following inoculation, whereas at this time point no labeling was detected in parasympathetic nuclei (119). Labeled neurons in the dorsal motor nucleus of the vagus (DMV) were observed 4–5 days after inoculation of the liver. At this time point, liver-related neurons were also detected in the brainstem including the ventrolateral medulla, NTS, raphe pallidus, and few neurons were identified in the hypothalamic PVN and LH (119). Longer survival time provided labeling of liver-related neurons in nuclei connected to the PVN including the medial preoptic area, anterior hypothalamic area, and ARC. PRV-labeled cells were also present in the VMH, suprachiasmatic nuclei, central amygdala, and bed nucleus of stria terminalis (119). This study demonstrated that PRV provides reliable identification of polysynaptic sympathetic and parasympathetic pathways to the liver (113, 115, 118, 120).
Despite the identification of liver-related neurons in the CNS, labeling with PRV does not distinguish between sympathetic and parasympathetic liver-related neurons in higher brain areas. In order to identify sympathetic- or parasympathetic liver-related hypothalamic neurons in rats, PRV inoculation was combined with hepatic sympathectomy or parasympathectomy (121, 122). Following sympathetic denervation of the liver, parasympathetic liver-related neurons were identified in the brainstem DMV and nucleus ambiguus (122). Medium length survival time (~4 days) resulted in labeling of additional brainstem areas (e.g., NTS, area postrema) and hypothalamic nuclei including PVN, LH, DMH, ARC, and others (122). Retrograde labeling following parasympathectomy identified pre-sympathetic liver-related neurons in the PVN, medial preoptic area, anterior hypothalamic area, DMH, ARC, VMH, and SCN (121). These studies revealed that preautonomic PVN neurons project either to sympathetic or parasympathetic division, and the segregation of the neurons exists in other hypothalamic areas including LH and SCN suggesting functional specialization of preautonomic neurons controlling liver function. The segregation of the autonomic divisions was also supported by the observation that stimulation of PVN resulted in hyperglycemia largely due to sympathetic activation of the liver (121). On the other hand, we have to note, that viral injection into peripheral organs causes infection of nerve terminals innervating both vascular and non-vascular tissues (123). PRV injected into the liver is taken up by nerve endings near hepatocytes and sinusoidal cells, and we cannot distinguish between neurons innervating liver function or hepatic vasculature at the injection site. Therefore, it is likely that the PRV-labeled pre-sympathetic neurons contribute to both vasomotor and non-vasomotor sympathetic innervation of the liver.
Our current knowledge regarding the role and cellular properties of liver-related hypothalamic neurons is somewhat limited. Earlier studies suggested that the VMH is involved in the sympathetic control of the liver, whereas the LH plays role in the parasympathetic control of the liver (91, 124, 125). The PVN was also shown as an important, integrative center for the regulation of sympathetic and parasympathetic pathways to the liver (121, 126–128). Hypothalamic action of metabolic signals including insulin, leptin, and FAs has been shown to control glucose homeostasis (58, 59, 76, 129–133); however, the cellular properties, the phenotype, or the involved neural circuits underlying the central control of hepatic functions are less defined.
Hypothalamic nuclei are heterogeneous containing different types of neurons, which are able to control multiple organ systems; therefore, identification of pathway-related neurons is crucial. Our laboratory used the retrograde PRV tracing technique, discussed above, to identify liver-related neurons in the PVN and to reveal synaptic properties of liver-related neurons in control and hyperglycemic conditions (113). The studies determined that liver-related neurons receive transient receptor potential vanilloid type 1 (TRPV1) containing inputs. TRPV1 is a non-selective cation channel and has been linked to the development and progression of type 1 diabetes mellitus and T2D (134), and recent reviews have discussed its role in diabetes mellitus and obesity in further detail (135, 136). The TRPV1-dependent excitation of liver-related PVN neurons was diminished in a hyperglycemic, insulin-deficient mouse model. Both in vivo and in vitro insulin replacement restored the TRPV1-dependent excitatory neurotransmission via PI3-kinase, PKC, and/or TRPV1 trafficking (113). Similarly, TRPV1 was shown to play role in the regulation of excitatory neurotransmission to motor neurons in the DMV and stomach-related neurons in the PVN (137, 138). Furthermore, potential interaction between leptin signaling and TRPV1 has been proposed in the brainstem (114). In addition, there is limited information about the neurochemical phenotype of liver-related neurons. A study by Stanley and coworkers identified liver-related hypothalamic neurons using PRV and showed that a subpopulation of liver-related neurons co-localized with oxytocin and CRH in the mouse PVN (118). In the LH, a subset of MCH and orexin neurons was shown to be liver related. In the ARC, a subpopulation of POMC neurons but not NPY-expressing neurons were labeled with PRV, indicating that they are part of the brain–liver pathway (118).
Despite the scientific advances in our understanding of liver-related neurons, their precise role in the autonomic regulation of hepatic lipid metabolism remains to be determined. Although there is clear evidence that increased sympathetic outflow to the liver may increase VLDL-TG production, resulting in increased systemic FA availability, it remains to be determined whether the liver-related neurons of the hypothalamus are indeed FA sensitive, or indeed whether the FA-sensing neurons of the hypothalamus are liver related. It is plausible to suggest that liver-related neurons that direct hepatic triglyceride metabolism may express key lipid processing factors involved in lipid transport, e.g., CD36, FATP1 and/or TG metabolism, e.g., LPL. However, a more detailed understanding of these fundamental autonomic processes is needed in order to identify novel therapeutic targets that may halt the development of lipid-related disorders.
Summary, Implications, and Interventions
The findings summarized in this review highlight the role of central lipid sensing in the regulation of systemic metabolism, including food intake, body weight, and hepatic glucose and lipid metabolism. Since increased HGP is a key factor in the development of glucose intolerance, understanding the neuronal mechanisms that drive the central regulation of HGP may be key to the development of novel therapeutic strategies that prevent hyperglycemia and the development of type 2 diabetes. In addition, our increased understanding of the hypothalamic control of TG metabolism highlights the potential utility of specifically modulating sympathetic nervous system activity toward the liver reduces VLDL-TG secretion and combat hypertriglyceridemia (139). The clinical relevance of this strategy is highlighted by a recent north European human cohort, in which elevated sympathetic nervous system activity was associated with features of the MetS, such as elevated circulated VLDL-TG (140). While current interventions for hypertriglyceridemia are aimed at reducing circulating FAs by increasing FA uptake from the plasma, an alternative approach to effectively lower TG could be though decreasing VLDL-TG production by the liver. This is the case with GLP-1 receptor agonists, which have been shown to decrease both hepatic lipogenesis (141) and VLDL-TG production (142). Based on the literature outlined in this review, interventions targeting specific liver-related or FA-sensitive neurons in of the hypothalamus could have a similar impact on systemic metabolism, and we recommend that the identification of the features of these neuronal populations should be the subject of intensive research focus.
Author Contributions
KB, AZ, and RE wrote and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was in part supported by the grant awarded to AZ (NIH R01 DK099598) with RE as a consultant and to RE (NIH R01 DK089309).
References
1. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet (2005) 365:1415–28. doi: 10.1016/S0140-6736(05)66378-7
2. Aguilar M, Bhuket T, Torres S, Liu B, Wong RJ. Prevalence of the metabolic syndrome in the United States, 2003-2012. JAMA (2015) 313:1973–4. doi:10.1001/jama.2015.4260
3. Isomaa B, Almgren P, Tuomi T, Forsen B, Lahti K, Nissen M, et al. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care (2001) 24:683–9. doi:10.2337/diacare.24.4.683
4. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature (2006) 443:289–95. doi:10.1038/nature05026
5. Koch L, Wunderlich FT, Seibler J, Konner AC, Hampel B, Irlenbusch S, et al. Central insulin action regulates peripheral glucose and fat metabolism in mice. J Clin Invest (2008) 118:2132–47. doi:10.1172/JCI31073
6. Theander-Carrillo C, Wiedmer P, Cettour-Rose P, Nogueiras R, Perez-Tilve D, Pfluger P, et al. Ghrelin action in the brain controls adipocyte metabolism. J Clin Invest (2006) 116:1983–93. doi:10.1172/JCI25811
7. Nogueiras R, Wiedmer P, Perez-Tilve D, Veyrat-Durebex C, Keogh JM, Sutton GM, et al. The central melanocortin system directly controls peripheral lipid metabolism. J Clin Invest (2007) 117:3475–88. doi:10.1172/JCI31743
8. Asilmaz E, Cohen P, Miyazaki M, Dobrzyn P, Ueki K, Fayzikhodjaeva G, et al. Site and mechanism of leptin action in a rodent form of congenital lipodystrophy. J Clin Invest (2004) 113:414–24. doi:10.1172/JCI200419511
9. Hamilton JA, Hillard CJ, Spector AA, Watkins PA. Brain uptake and utilization of fatty acids, lipids and lipoproteins: application to neurological disorders. J Mol Neurosci (2007) 33:2–11. doi:10.1007/s12031-007-0060-1
10. Rapoport SI, Chang MC, Spector AA. Delivery and turnover of plasma-derived essential PUFAs in mammalian brain. J Lipid Res (2001) 42:678–85.
11. Smith QR, Nagura H. Fatty acid uptake and incorporation in brain: studies with the perfusion model. J Mol Neurosci (2001) 16:167–72; discussion 215–21. doi:10.1385/JMN:16:2-3:167
12. Woods SC, Porte D Jr, Bobbioni E, Ionescu E, Sauter JF, Rohner-Jeanrenaud F, et al. Insulin: its relationship to the central nervous system and to the control of food intake and body weight. Am J Clin Nutr (1985) 42:1063–71.
13. Mitchell RW, On NH, Del Bigio MR, Miller DW, Hatch GM. Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J Neurochem (2011) 117:735–46. doi:10.1111/j.1471-4159.2011.07245.x
14. Nguyen LN, Ma D, Shui G, Wong P, Cazenave-Gassiot A, Zhang X, et al. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature (2014) 509:503–6. doi:10.1038/nature13241
15. Le Foll C, Irani BG, Magnan C, Dunn-Meynell AA, Levin BE. Characteristics and mechanisms of hypothalamic neuronal fatty acid sensing. Am J Physiol Regul Integr Comp Physiol (2009) 297:R655–64. doi:10.1152/ajpregu.00223.2009
16. Le Foll C, Irani BG, Magnan C, Dunn-Meynell A, Levin BE. Effects of maternal genotype and diet on offspring glucose and fatty acid-sensing ventromedial hypothalamic nucleus neurons. Am J Physiol Regul Integr Comp Physiol (2009) 297:R1351–7. doi:10.1152/ajpregu.00370.2009
17. Le Foll C, Dunn-Meynell A, Musatov S, Magnan C, Levin BE. FAT/CD36: a major regulator of neuronal fatty acid sensing and energy homeostasis in rats and mice. Diabetes (2013) 62:2709–16. doi:10.2337/db12-1689
18. Wellhauser L, Belsham DD. Activation of the omega-3 fatty acid receptor GPR120 mediates anti-inflammatory actions in immortalized hypothalamic neurons. J Neuroinflammation (2014) 11:60. doi:10.1186/1742-2094-11-60
19. Murphy EJ, Owada Y, Kitanaka N, Kondo H, Glatz JF. Brain arachidonic acid incorporation is decreased in heart fatty acid binding protein gene-ablated mice. Biochemistry (2005) 44:6350–60. doi:10.1021/bi047292r
20. Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, et al. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia (2007) 55:1251–62. doi:10.1002/glia.20528
21. Le Foll C, Dunn-Meynell AA, Miziorko HM, Levin BE. Regulation of hypothalamic neuronal sensing and food intake by ketone bodies and fatty acids. Diabetes (2014) 63:1259–69. doi:10.2337/db13-1090
22. Le Foll C, Dunn-Meynell AA, Miziorko HM, Levin BE. Role of VMH ketone bodies in adjusting caloric intake to increased dietary fat content in DIO and DR rats. Am J Physiol Regul Integr Comp Physiol (2015) 308:R872–8. doi:10.1152/ajpregu.00015.2015
23. Taib B, Bouyakdan K, Hryhorczuk C, Rodaros D, Fulton S, Alquier T. Glucose regulates hypothalamic long-chain fatty acid metabolism via AMP-activated kinase (AMPK) in neurons and astrocytes. J Biol Chem (2013) 288:37216–29. doi:10.1074/jbc.M113.506238
24. Ebrahimi M, Yamamoto Y, Sharifi K, Kida H, Kagawa Y, Yasumoto Y, et al. Astrocyte-expressed FABP7 regulates dendritic morphology and excitatory synaptic function of cortical neurons. Glia (2016) 64:48–62. doi:10.1002/glia.22902
25. Bouyakdan K, Taib B, Budry L, Zhao S, Rodaros D, Neess D, et al. A novel role for central ACBP/DBI as a regulator of long-chain fatty acid metabolism in astrocytes. J Neurochem (2015) 133:253–65. doi:10.1111/jnc.13035
26. Zhang J, Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell (2015) 6:254–64. doi:10.1007/s13238-014-0131-3
27. Linetti A, Fratangeli A, Taverna E, Valnegri P, Francolini M, Cappello V, et al. Cholesterol reduction impairs exocytosis of synaptic vesicles. J Cell Sci (2010) 123:595–605. doi:10.1242/jcs.060681
28. Liu Q, Trotter J, Zhang J, Peters MM, Cheng H, Bao J, et al. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J Neurosci (2010) 30:17068–78. doi:10.1523/JNEUROSCI.4067-10.2010
29. Wang H, Eckel RH. What are lipoproteins doing in the brain? Trends Endocrinol Metab (2014) 25:8–14. doi:10.1016/j.tem.2013.10.003
30. Linton MF, Gish R, Hubl ST, Butler E, Esquivel C, Bry WI, et al. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J Clin Invest (1991) 88:270–81. doi:10.1172/JCI115288
31. Matsuo M, Campenot RB, Vance DE, Ueda K, Vance JE. Involvement of low-density lipoprotein receptor-related protein and ABCG1 in stimulation of axonal extension by apoE-containing lipoproteins. Biochim Biophys Acta (2011) 1811:31–8. doi:10.1016/j.bbalip.2010.10.004
32. Hayashi H. Lipid metabolism and glial lipoproteins in the central nervous system. Biol Pharm Bull (2011) 34:453–61. doi:10.1248/bpb.34.453
33. Beffert U, Stolt PC, Herz J. Functions of lipoprotein receptors in neurons. J Lipid Res (2004) 45:403–9. doi:10.1194/jlr.R300017-JLR200
34. Shen L, Tso P, Woods SC, Clegg DJ, Barber KL, Carey K, et al. Brain apolipoprotein E: an important regulator of food intake in rats. Diabetes (2008) 57:2092–8. doi:10.2337/db08-0291
35. Shen L, Wang DQ, Tso P, Jandacek RJ, Woods SC, Liu M. Apolipoprotein E reduces food intake via PI3K/Akt signaling pathway in the hypothalamus. Physiol Behav (2011) 105:124–8. doi:10.1016/j.physbeh.2011.04.018
36. Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol Dis (2014) 72 Pt A:3–12. doi:10.1016/j.nbd.2014.08.025
37. Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med (2011) 3:89ra57. doi:10.1126/scitranslmed.3002156
38. Jones PB, Adams KW, Rozkalne A, Spires-Jones TL, Hshieh TT, Hashimoto T, et al. Apolipoprotein E: isoform specific differences in tertiary structure and interaction with amyloid-beta in human Alzheimer brain. PLoS One (2011) 6:e14586. doi:10.1371/journal.pone.0014586
39. Libby AE, Wang H, Mittal R, Sungelo M, Potma E, Eckel RH. Lipoprotein lipase is an important modulator of lipid uptake and storage in hypothalamic neurons. Biochem Biophys Res Commun (2015) 465:287–92. doi:10.1016/j.bbrc.2015.08.026
40. Wang H, Astarita G, Taussig MD, Bharadwaj KG, DiPatrizio NV, Nave KA, et al. Deficiency of lipoprotein lipase in neurons modifies the regulation of energy balance and leads to obesity. Cell Metab (2011) 13:105–13. doi:10.1016/j.cmet.2010.12.006
41. Yu T, Taussig MD, DiPatrizio NV, Astarita G, Piomelli D, Bergman BC, et al. Deficiency of lipoprotein lipase in neurons decreases AMPA receptor phosphorylation and leads to neurobehavioral abnormalities in mice. PLoS One (2015) 10:e0135113. doi:10.1371/journal.pone.0135113
42. Picard A, Rouch C, Kassis N, Moulle VS, Croizier S, Denis RG, et al. Hippocampal lipoprotein lipase regulates energy balance in rodents. Mol Metab (2014) 3:167–76. doi:10.1016/j.molmet.2013.11.002
43. Wang H, Taussig MD, DiPatrizio NV, Bruce K, Piomelli D, Eckel RH. Obesity development in neuron-specific lipoprotein lipase deficient mice is not responsive to increased dietary fat content or change in fat composition. Metabolism (2016) 65:987–97. doi:10.1016/j.metabol.2016.01.015
44. Sarruf DA, Yu F, Nguyen HT, Williams DL, Printz RL, Niswender KD, et al. Expression of peroxisome proliferator-activated receptor-gamma in key neuronal subsets regulating glucose metabolism and energy homeostasis. Endocrinology (2009) 150:707–12. doi:10.1210/en.2008-0899
45. Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, et al. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature (2008) 454:846–51. doi:10.1038/nature07181
46. Diano S, Liu ZW, Jeong JK, Dietrich MO, Ruan HB, Kim E, et al. Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nat Med (2011) 17:1121–7. doi:10.1038/nm.2421
47. Lopez M, Nogueiras R, Tena-Sempere M, Dieguez C. Hypothalamic AMPK: a canonical regulator of whole-body energy balance. Nat Rev Endocrinol (2016) 12:421–32. doi:10.1038/nrendo.2016.67
48. Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol (2007) 8:774–85. doi:10.1038/nrm2249
49. Wolfgang MJ, Cha SH, Sidhaye A, Chohnan S, Cline G, Shulman GI, et al. Regulation of hypothalamic malonyl-CoA by central glucose and leptin. Proc Natl Acad Sci U S A (2007) 104:19285–90. doi:10.1073/pnas.0709778104
50. Lopez M, Varela L, Vazquez MJ, Rodriguez-Cuenca S, Gonzalez CR, Velagapudi VR, et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat Med (2010) 16:1001–8. doi:10.1038/nm.2207
51. Lopez M. EJE PRIZE 2017: hypothalamic AMPK: a golden target against obesity? Eur J Endocrinol (2017). doi:10.1530/EJE-16-0927
52. Martin TL, Alquier T, Asakura K, Furukawa N, Preitner F, Kahn BB. Diet-induced obesity alters AMP kinase activity in hypothalamus and skeletal muscle. J Biol Chem (2006) 281:18933–41. doi:10.1074/jbc.M512831200
53. Gomez-Pinilla F, Ying Z. Differential effects of exercise and dietary docosahexaenoic acid on molecular systems associated with control of allostasis in the hypothalamus and hippocampus. Neuroscience (2010) 168:130–7. doi:10.1016/j.neuroscience.2010.02.070
54. Blazquez C, Geelen MJ, Velasco G, Guzman M. The AMP-activated protein kinase prevents ceramide synthesis de novo and apoptosis in astrocytes. FEBS Lett (2001) 489:149–53. doi:10.1016/S0014-5793(01)02089-0
55. Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci (2001) 17:45–58. doi:10.1385/JMN:17:1:45
56. Won JS, Im YB, Kim J, Singh AK, Singh I. Involvement of AMP-activated-protein-kinase (AMPK) in neuronal amyloidogenesis. Biochem Biophys Res Commun (2010) 399:487–91. doi:10.1016/j.bbrc.2010.07.081
57. Yue JT, Lam TK. Lipid sensing and insulin resistance in the brain. Cell Metab (2012) 15:646–55. doi:10.1016/j.cmet.2012.01.013
58. Lam TK, Pocai A, Gutierrez-Juarez R, Obici S, Bryan J, Aguilar-Bryan L, et al. Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nat Med (2005) 11:320–7. doi:10.1038/nm1201
59. Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci (2002) 5:566–72. doi:10.1038/nn0602-861
60. Gao S, Serra D, Keung W, Hegardt FG, Lopaschuk GD. Important role of ventromedial hypothalamic carnitine palmitoyltransferase-1a in the control of food intake. Am J Physiol Endocrinol Metab (2013) 305:E336–47. doi:10.1152/ajpendo.00168.2013
61. Mera P, Mir JF, Fabrias G, Casas J, Costa AS, Malandrino MI, et al. Long-term increased carnitine palmitoyltransferase 1A expression in ventromedial hypothalamus causes hyperphagia and alters the hypothalamic lipidomic profile. PLoS One (2014) 9:e97195. doi:10.1371/journal.pone.0097195
62. Wolfgang MJ, Kurama T, Dai Y, Suwa A, Asaumi M, Matsumoto S, et al. The brain-specific carnitine palmitoyltransferase-1c regulates energy homeostasis. Proc Natl Acad Sci U S A (2006) 103:7282–7. doi:10.1073/pnas.0602205103
63. Wolfgang MJ, Cha SH, Millington DS, Cline G, Shulman GI, Suwa A, et al. Brain-specific carnitine palmitoyl-transferase-1c: role in CNS fatty acid metabolism, food intake, and body weight. J Neurochem (2008) 105:1550–9. doi:10.1111/j.1471-4159.2008.05255.x
64. Lane MD, Wolfgang M, Cha SH, Dai Y. Regulation of food intake and energy expenditure by hypothalamic malonyl-CoA. Int J Obes (2008) 32 Suppl 4:S49–54. doi:10.1038/ijo.2008.123
65. Ramirez S, Martins L, Jacas J, Carrasco P, Pozo M, Clotet J, et al. Hypothalamic ceramide levels regulated by CPT1C mediate the orexigenic effect of ghrelin. Diabetes (2013) 62:2329–37. doi:10.2337/db12-1451
66. Contreras C, Gonzalez-Garcia I, Martinez-Sanchez N, Seoane-Collazo P, Jacas J, Morgan DA, et al. Central ceramide-induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell Rep (2014) 9:366–77. doi:10.1016/j.celrep.2014.08.057
67. Lee J, Wolfgang MJ. Metabolomic profiling reveals a role for CPT1c in neuronal oxidative metabolism. BMC Biochem (2012) 13:23. doi:10.1186/1471-2091-13-23
68. Pazos MR, Nunez E, Benito C, Tolon RM, Romero J. Functional neuroanatomy of the endocannabinoid system. Pharmacol Biochem Behav (2005) 81:239–47. doi:10.1016/j.pbb.2005.01.030
69. McGarry JD, Mannaerts GP, Foster DW. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J Clin Invest (1977) 60:265–70. doi:10.1172/JCI108764
70. Caspi L, Wang PY, Lam TK. A balance of lipid-sensing mechanisms in the brain and liver. Cell Metab (2007) 6:99–104. doi:10.1016/j.cmet.2007.07.005
71. Gao S, Moran TH, Lopaschuk GD, Butler AA. Hypothalamic malonyl-CoA and the control of food intake. Physiol Behav (2013) 122:17–24. doi:10.1016/j.physbeh.2013.07.014
72. Gao S, Kinzig KP, Aja S, Scott KA, Keung W, Kelly S, et al. Leptin activates hypothalamic acetyl-CoA carboxylase to inhibit food intake. Proc Natl Acad Sci U S A (2007) 104:17358–63. doi:10.1073/pnas.0708385104
73. Loftus TM, Jaworsky DE, Frehywot GL, Townsend CA, Ronnett GV, Lane MD, et al. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science (2000) 288:2379–81. doi:10.1126/science.288.5475.2379
74. Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, et al. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem (2004) 279:12005–8. doi:10.1074/jbc.C300557200
75. He W, Lam TK, Obici S, Rossetti L. Molecular disruption of hypothalamic nutrient sensing induces obesity. Nat Neurosci (2006) 9:227–33. doi:10.1038/nn1626
76. Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, et al. Hypothalamic K(ATP) channels control hepatic glucose production. Nature (2005) 434:1026–31. doi:10.1038/nature03439
77. Ross RA, Rossetti L, Lam TK, Schwartz GJ. Differential effects of hypothalamic long-chain fatty acid infusions on suppression of hepatic glucose production. Am J Physiol Endocrinol Metab (2010) 299:E633–9. doi:10.1152/ajpendo.00190.2010
78. Bruinstroop E, Pei L, Ackermans MT, Foppen E, Borgers AJ, Kwakkel J, et al. Hypothalamic neuropeptide Y (NPY) controls hepatic VLDL-triglyceride secretion in rats via the sympathetic nervous system. Diabetes (2012) 61:1043–50. doi:10.2337/db11-1142
79. Lam TK, Gutierrez-Juarez R, Pocai A, Bhanot S, Tso P, Schwartz GJ, et al. Brain glucose metabolism controls the hepatic secretion of triglyceride-rich lipoproteins. Nat Med (2007) 13:171–80. doi:10.1038/nm1540
80. Yue JT, Mighiu PI, Naples M, Adeli K, Lam TK. Glycine normalizes hepatic triglyceride-rich VLDL secretion by triggering the CNS in high-fat fed rats. Circ Res (2012) 110:1345–54. doi:10.1161/CIRCRESAHA.112.268276
81. Leckstrom A, Lew PS, Poritsanos NJ, Mizuno TM. Central melanocortin receptor agonist reduces hepatic lipogenic gene expression in streptozotocin-induced diabetic mice. Life Sci (2011) 88:664–9. doi:10.1016/j.lfs.2011.01.026
82. Wiedmer P, Chaudhary N, Rath M, Yi CX, Ananthakrishnan G, Nogueiras R, et al. The HPA axis modulates the CNS melanocortin control of liver triacylglyceride metabolism. Physiol Behav (2012) 105:791–9. doi:10.1016/j.physbeh.2011.10.019
83. Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature (1998) 396:670–4. doi:10.1038/25341
84. Jeon JY, Bradley RL, Kokkotou EG, Marino FE, Wang X, Pissios P, et al. MCH-/- mice are resistant to aging-associated increases in body weight and insulin resistance. Diabetes (2006) 55:428–34. doi:10.2337/diabetes.55.02.06.db05-0203
85. Wang Y, Ziogas DC, Biddinger S, Kokkotou E. You deserve what you eat: lessons learned from the study of the melanin-concentrating hormone (MCH)-deficient mice. Gut (2010) 59:1625–34. doi:10.1136/gut.2010.210526
86. Imbernon M, Beiroa D, Vazquez MJ, Morgan DA, Veyrat-Durebex C, Porteiro B, et al. Central melanin-concentrating hormone influences liver and adipose metabolism via specific hypothalamic nuclei and efferent autonomic/JNK1 pathways. Gastroenterology (2013) 144:636.e–49.e. doi:10.1053/j.gastro.2012.10.051
87. Rojas JM, Bruinstroop E, Printz RL, Alijagic-Boers A, Foppen E, Turney MK, et al. Central nervous system neuropeptide Y regulates mediators of hepatic phospholipid remodeling and very low-density lipoprotein triglyceride secretion via sympathetic innervation. Mol Metab (2015) 4:210–21. doi:10.1016/j.molmet.2015.01.004
88. Cheng L, Yu Y, Szabo A, Wu Y, Wang H, Camer D, et al. Palmitic acid induces central leptin resistance and impairs hepatic glucose and lipid metabolism in male mice. J Nutr Biochem (2015) 26:541–8. doi:10.1016/j.jnutbio.2014.12.011
89. Yue JT, Abraham MA, LaPierre MP, Mighiu PI, Light PE, Filippi BM, et al. A fatty acid-dependent hypothalamic-DVC neurocircuitry that regulates hepatic secretion of triglyceride-rich lipoproteins. Nat Commun (2015) 6:5970. doi:10.1038/ncomms6970
90. Nonogaki K. New insights into sympathetic regulation of glucose and fat metabolism. Diabetologia (2000) 43:533–49. doi:10.1007/s001250051341
91. Shimazu T. Innervation of the liver and glucoregulation: roles of the hypothalamus and autonomic nerves. Nutrition (1996) 12:65–6. doi:10.1016/S0899-9007(96)90348-2
92. Uyama N, Geerts A, Reynaert H. Neural connections between the hypothalamus and the liver. Anat Rec A Discov Mol Cell Evol Biol (2004) 280:808–20. doi:10.1002/ar.a.20086
93. Saper CB, Loewy AD, Swanson LW, Cowan WM. Direct hypothalamo-autonomic connections. Brain Res (1976) 117:305–12. doi:10.1016/0006-8993(76)90738-1
94. Swanson LW, Sawchenko PE. Paraventricular nucleus: a site for the integration of neuroendocrine and autonomic mechanisms. Neuroendocrinology (1980) 31:410–7. doi:10.1159/000123111
95. Sandoval DA, Obici S, Seeley RJ. Targeting the CNS to treat type 2 diabetes. Nat Rev Drug Discov (2009) 8:386–98. doi:10.1038/nrd2874
96. Schwartz MW, Seeley RJ, Tschop MH, Woods SC, Morton GJ, Myers MG, et al. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature (2013) 503:59–66. doi:10.1038/nature12709
97. Kalsbeek A, Bruinstroop E, Yi CX, Klieverik LP, La Fleur SE, Fliers E. Hypothalamic control of energy metabolism via the autonomic nervous system. Ann N Y Acad Sci (2010) 1212:114–29. doi:10.1111/j.1749-6632.2010.05800.x
98. Yi CX, la Fleur SE, Fliers E, Kalsbeek A. The role of the autonomic nervous liver innervation in the control of energy metabolism. Biochim Biophys Acta (2010) 1802:416–31. doi:10.1016/j.bbadis.2010.01.006
99. O’Hare JD, Zsombok A. Brain-liver connections: role of the preautonomic PVN neurons. Am J Physiol Endocrinol Metab (2016) 310:E183–9. doi:10.1152/ajpendo.00302.2015
100. Bruinstroop E, Fliers E, Kalsbeek A. Hypothalamic control of hepatic lipid metabolism via the autonomic nervous system. Best Pract Res Clin Endocrinol Metab (2014) 28:673–84. doi:10.1016/j.beem.2014.05.001
101. Puschel GP. Control of hepatocyte metabolism by sympathetic and parasympathetic hepatic nerves. Anat Rec A Discov Mol Cell Evol Biol (2004) 280:854–67. doi:10.1002/ar.a.20091
102. Lawrence D, Pittman QJ. Interaction between descending paraventricular neurons and vagal motor neurons. Brain Res (1985) 332:158–60. doi:10.1016/0006-8993(85)90399-3
103. Rogers RC, Nelson DO. Neurons of the vagal division of the solitary nucleus activated by the paraventricular nucleus of the hypothalamus. J Auton Nerv Syst (1984) 10:193–7. doi:10.1016/0165-1838(84)90057-2
104. Yamashita H, Inenaga K, Koizumi K. Possible projections from regions of paraventricular and supraoptic nuclei to the spinal cord: electrophysiological studies. Brain Res (1984) 296:373–8. doi:10.1016/0006-8993(84)90077-5
105. Cailotto C, La Fleur SE, Van Heijningen C, Wortel J, Kalsbeek A, Feenstra M, et al. The suprachiasmatic nucleus controls the daily variation of plasma glucose via the autonomic output to the liver: are the clock genes involved? Eur J Neurosci (2005) 22:2531–40. doi:10.1111/j.1460-9568.2005.04439.x
106. Rogers RC, Hermann GE. Central connections of the hepatic branch of the vagus nerve: a horseradish peroxidase histochemical study. J Auton Nerv Syst (1983) 7:165–74. doi:10.1016/0165-1838(83)90044-9
107. Banfield BW, Kaufman JD, Randall JA, Pickard GE. Development of pseudorabies virus strains expressing red fluorescent proteins: new tools for multisynaptic labeling applications. J Virol (2003) 77:10106–12. doi:10.1128/JVI.77.18.10106-10112.2003
108. Boldogkoi Z, Reichart A, Toth IE, Sik A, Erdelyi F, Medveczky I, et al. Construction of recombinant pseudorabies viruses optimized for labeling and neurochemical characterization of neural circuitry. Brain Res Mol Brain Res (2002) 109:105–18. doi:10.1016/S0169-328X(02)00546-6
109. Card JP. Exploring brain circuitry with neurotropic viruses: new horizons in neuroanatomy. Anat Rec (1998) 253:176–85. doi:10.1002/(SICI)1097-0185(199812)253:6<176::AID-AR6>3.0.CO;2-W
110. Card JP, Enquist LW. Transneuronal circuit analysis with pseudorabies viruses. Curr Protoc Neurosci (2014) 68:1.5.1–39; editorial board, Jacqueline N. Crawley … [et al.]. doi:10.1002/0471142301.ns0105s68
111. Cano G, Card JP, Sved AF. Dual viral transneuronal tracing of central autonomic circuits involved in the innervation of the two kidneys in rat. J Comp Neurol (2004) 471:462–81. doi:10.1002/cne.20040
112. Williams KW, Zsombok A, Smith BN. Rapid inhibition of neurons in the dorsal motor nucleus of the vagus by leptin. Endocrinology (2007) 148:1868–81. doi:10.1210/en.2006-1098
113. Gao H, Miyata K, Bhaskaran MD, Derbenev AV, Zsombok A. Transient receptor potential vanilloid type 1-dependent regulation of liver-related neurons in the paraventricular nucleus of the hypothalamus diminished in the type 1 diabetic mouse. Diabetes (2012) 61:1381–90. doi:10.2337/db11-0820
114. Zsombok A, Jiang Y, Gao H, Anwar IJ, Rezai-Zadeh K, Enix CL, et al. Regulation of leptin receptor-expressing neurons in the brainstem by TRPV1. Physiol Rep (2014) 2:e12160. doi:10.14814/phy2.12160
115. Anwar IJ, Miyata K, Zsombok A. Brain stem as target site for the metabolic side effects of olanzapine. J Neurophysiology (2016) 115:1389–98. doi:10.1152/jn.00387.2015
116. Aston-Jones G, Card JP. Use of pseudorabies virus to delineate multisynaptic circuits in brain: opportunities and limitations. J Neurosci Methods (2000) 103:51–61. doi:10.1016/S0165-0270(00)00295-8
117. Ch’ng TH, Spear PG, Struyf F, Enquist LW. Glycoprotein D-independent spread of pseudorabies virus infection in cultured peripheral nervous system neurons in a compartmented system. J Virol (2007) 81:10742–57. doi:10.1128/JVI.00981-07
118. Stanley S, Pinto S, Segal J, Perez CA, Viale A, DeFalco J, et al. Identification of neuronal subpopulations that project from hypothalamus to both liver and adipose tissue polysynaptically. Proc Natl Acad Sci U S A (2010) 107:7024–9. doi:10.1073/pnas.1002790107
119. la Fleur SE, Kalsbeek A, Wortel J, Buijs RM. Polysynaptic neural pathways between the hypothalamus, including the suprachiasmatic nucleus, and the liver. Brain Res (2000) 871:50–6. doi:10.1016/S0006-8993(00)02423-9
120. Zsombok A, Gao H, Miyata K, Issa A, Derbenev AV. Immunohistochemical localization of transient receptor potential vanilloid type 1 and insulin receptor substrate 2 and their co-localization with liver-related neurons in the hypothalamus and brainstem. Brain Res (2011) 1398:30–9. doi:10.1016/j.brainres.2011.04.048
121. Kalsbeek A, La Fleur S, Van Heijningen C, Buijs RM. Suprachiasmatic GABAergic inputs to the paraventricular nucleus control plasma glucose concentrations in the rat via sympathetic innervation of the liver. J Neurosci (2004) 24:7604–13. doi:10.1523/JNEUROSCI.5328-03.2004
122. Buijs RM, la Fleur SE, Wortel J, Van Heyningen C, Zuiddam L, Mettenleiter TC, et al. The suprachiasmatic nucleus balances sympathetic and parasympathetic output to peripheral organs through separate preautonomic neurons. J Comp Neurol (2003) 464:36–48. doi:10.1002/cne.10765
123. Cano G, Sved AF, Rinaman L, Rabin BS, Card JP. Characterization of the central nervous system innervation of the rat spleen using viral transneuronal tracing. J Comp Neurol (2001) 439:1–18. doi:10.1002/cne.1331
124. Shimazu T. Neuronal regulation of hepatic glucose metabolism in mammals. Diabetes Metab Rev (1987) 3:185–206. doi:10.1002/dmr.5610030109
125. Shimazu T, Fukuda A, Ban T. Reciprocal influences of the ventromedial and lateral hypothalamic nuclei on blood glucose level and liver glycogen content. Nature (1966) 210:1178–9. doi:10.1038/2101178a0
126. Yi CX, Sun N, Ackermans MT, Alkemade A, Foppen E, Shi J, et al. Pituitary adenylate cyclase-activating polypeptide stimulates glucose production via the hepatic sympathetic innervation in rats. Diabetes (2010) 59:1591–600. doi:10.2337/db09-1398
127. van den Hoek AM, van Heijningen C, Schroder-van der Elst JP, Ouwens DM, Havekes LM, Romijn JA, et al. Intracerebroventricular administration of neuropeptide Y induces hepatic insulin resistance via sympathetic innervation. Diabetes (2008) 57:2304–10. doi:10.2337/db07-1658
128. Klieverik LP, Janssen SF, van Riel A, Foppen E, Bisschop PH, Serlie MJ, et al. Thyroid hormone modulates glucose production via a sympathetic pathway from the hypothalamic paraventricular nucleus to the liver. Proc Natl Acad Sci U S A (2009) 106:5966–71. doi:10.1073/pnas.0805355106
129. Lam TK, Gutierrez-Juarez R, Pocai A, Rossetti L. Regulation of blood glucose by hypothalamic pyruvate metabolism. Science (2005) 309:943–7. doi:10.1126/science.1112085
130. Lam TK, Schwartz GJ, Rossetti L. Hypothalamic sensing of fatty acids. Nat Neurosci (2005) 8:579–84. doi:10.1038/nn1456
131. Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med (2002) 8:1376–82. doi:10.1038/nm1202-798
132. Pocai A, Morgan K, Buettner C, Gutierrez-Juarez R, Obici S, Rossetti L. Central leptin acutely reverses diet-induced hepatic insulin resistance. Diabetes (2005) 54:3182–9. doi:10.2337/diabetes.54.11.3182
133. Pocai A, Obici S, Schwartz GJ, Rossetti L. A brain-liver circuit regulates glucose homeostasis. Cell Metab (2005) 1:53–61. doi:10.1016/j.cmet.2004.11.001
134. Razavi R, Chan Y, Afifiyan FN, Liu XJ, Wan X, Yantha J, et al. TRPV1+ sensory neurons control beta cell stress and islet inflammation in autoimmune diabetes. Cell (2006) 127:1123–35. doi:10.1016/j.cell.2006.10.038
135. Zsombok A, Derbenev AV. TRP channels as therapeutic targets in diabetes and obesity. Pharmaceuticals (Basel) (2016) 9:E50. doi:10.3390/ph9030050
136. Derbenev AV, Zsombok A. Potential therapeutic value of TRPV1 and TRPA1 in diabetes mellitus and obesity. Semin Immunopathol (2016) 38:397–406. doi:10.1007/s00281-015-0529-x
137. Zsombok A, Bhaskaran MD, Gao H, Derbenev AV, Smith BN. Functional plasticity of central TRPV1 receptors in brainstem dorsal vagal complex circuits of streptozotocin-treated hyperglycemic mice. J Neurosci (2011) 31:14024–31. doi:10.1523/JNEUROSCI.2081-11.2011
138. Boychuk CR, Zsombok A, Tasker JG, Smith BN. Rapid glucocorticoid-induced activation of TRP and CB1 receptors causes biphasic modulation of glutamate release in gastric-related hypothalamic preautonomic neurons. Front Neurosci (2013) 7:3. doi:10.3389/fnins.2013.00003
139. Geerling JJ, Boon MR, Kooijman S, Parlevliet ET, Havekes LM, Romijn JA, et al. Sympathetic nervous system control of triglyceride metabolism: novel concepts derived from recent studies. J Lipid Res (2014) 55:180–9. doi:10.1194/jlr.R045013
140. Licht CM, Vreeburg SA, van Reedt Dortland AK, Giltay EJ, Hoogendijk WJ, DeRijk RH, et al. Increased sympathetic and decreased parasympathetic activity rather than changes in hypothalamic-pituitary-adrenal axis activity is associated with metabolic abnormalities. J Clin Endocrinol Metab (2010) 95:2458–66. doi:10.1210/jc.2009-2801
141. Nogueiras R, Perez-Tilve D, Veyrat-Durebex C, Morgan DA, Varela L, Haynes WG, et al. Direct control of peripheral lipid deposition by CNS GLP-1 receptor signaling is mediated by the sympathetic nervous system and blunted in diet-induced obesity. J Neurosci (2009) 29:5916–25. doi:10.1523/JNEUROSCI.5977-08.2009
142. Parlevliet ET, Wang Y, Geerling JJ, Schroder-Van der Elst JP, Picha K, O’Neil K, et al. GLP-1 receptor activation inhibits VLDL production and reverses hepatic steatosis by decreasing hepatic lipogenesis in high-fat-fed APOE*3-Leiden mice. PLoS One (2012) 7:e49152. doi:10.1371/journal.pone.0049152
Keywords: lipid metabolism, brain, liver, energy homeostasis, hypothalamus
Citation: Bruce KD, Zsombok A and Eckel RH (2017) Lipid Processing in the Brain: A Key Regulator of Systemic Metabolism. Front. Endocrinol. 8:60. doi: 10.3389/fendo.2017.00060
Received: 02 February 2017; Accepted: 17 March 2017;
Published: 04 April 2017
Edited by:
Hubert Vaudry, University of Rouen, FranceReviewed by:
Alexandre Benani, Centre national de la recherche scientifique (CNRS), FranceMiguel Lopez, Universidade de Santiago de Compostela, Spain
Christelle Le Foll, University of Zurich, Switzerland
Copyright: © 2017 Bruce, Zsombok and Eckel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kimberley D. Bruce, a2ltYmVybGV5LmJydWNlQHVjZGVudmVyLmVkdQ==