 Alejandra Freire-Regatillo1,2,3
Alejandra Freire-Regatillo1,2,3 Pilar Argente-Arizón1,2,3
Pilar Argente-Arizón1,2,3 Jesús Argente1,2,3,4
Jesús Argente1,2,3,4 Luis Miguel García-Segura5,6
Luis Miguel García-Segura5,6 Julie A. Chowen1,3*
Julie A. Chowen1,3*
- 1Department of Endocrinology, Hospital Infantil Universitario Niño Jesús, Instituto de Investigación la Princesa, Madrid, Spain
- 2Department of Pediatrics, Facultad de Medicina, Universidad Autónoma de Madrid, Madrid, Spain
- 3Centro de Investigación Biomédica en Red: Fisiopatología de la Obesidad y Nutrición (CIBEROBN), Madrid, Spain
- 4IMDEA Food Institute, Campus of International Excellence (CEI) UAM + CSIC, Madrid, Spain
- 5Laboratory of Neuroactive Steroids, Department of Functional and Systems Neurobiology, Instituto Cajal, CSIC (Consejo Superior de Investigaciones Científicas), Madrid, Spain
- 6Centro de Investigación Biomédica en Red de Fragilidad y Envejecimiento Saludable (CIBERFES), Madrid, Spain
Although the brain is composed of numerous cell types, neurons have received the vast majority of attention in the attempt to understand how this organ functions. Neurons are indeed fundamental but, in order for them to function correctly, they rely on the surrounding “non-neuronal” cells. These different cell types, which include glia, epithelial cells, pericytes, and endothelia, supply essential substances to neurons, in addition to protecting them from dangerous substances and situations. Moreover, it is now clear that non-neuronal cells can also actively participate in determining neuronal signaling outcomes. Due to the increasing problem of obesity in industrialized countries, investigation of the central control of energy balance has greatly increased in attempts to identify new therapeutic targets. This has led to interesting advances in our understanding of how appetite and systemic metabolism are modulated by non-neuronal cells. For example, not only are nutrients and hormones transported into the brain by non-neuronal cells, but these cells can also metabolize these metabolic factors, thus modifying the signals reaching the neurons. The hypothalamus is the main integrating center of incoming metabolic and hormonal signals and interprets this information in order to control appetite and systemic metabolism. Hence, the factors transported and released from surrounding non-neuronal cells will undoubtedly influence metabolic homeostasis. This review focuses on what is known to date regarding the involvement of different cell types in the transport and metabolism of nutrients and hormones in the hypothalamus. The possible involvement of non-neuronal cells, in particular glial cells, in physiopathological outcomes of poor dietary habits and excess weight gain are also discussed.
Introduction
Our understanding of the neuronal circuits controlling metabolism has advanced in recent years and progress has been made in the development of potential treatments for obesity, particularly in specific monogenic forms of obesity (1). However, the brain is not composed of neurons alone; other cell types actually outnumber these electrically excitable nerve cells and participate in and/or modulate all neuronal functions. In the hypothalamus, this includes the participation of non-neuronal cells in the modulation of neuronal circuits controlling appetite and metabolism.
Non-neuronal cells in the central nervous system (CNS), including glia, epithelial cells, pericytes, and endothelia, perform a wide spectrum of functions throughout the brain. Many of these functions are common in each brain area, although the specific outcomes are at least in part dependent on the neuronal circuits that are affected by their actions. Moreover, within each class of non-neuronal cell type there are generalized subclassifications that, although quite incomplete, indicate diverse functional states. There are also specialized cell types found only in specific areas of the brain. One important example that will be discussed in greater detail is tanycytes, specialized glial cells found lining the third ventricle and in close proximity to the neuroendocrine hypothalamus. The fact that there is wide heterogeneity within each non-neuronal cell type has become increasingly clear; however, we currently do not have the tools available to sufficiently distinguish between these subpopulations and this has clearly hindered advances in this field.
With the explosion in the prevalence of obesity that has occurred almost worldwide, investigation in the area of metabolic control has become a priority. This has led to an increase in our understanding of how non-neuronal cell types participate in the neuroendocrine control of appetite and energy expenditure, as well as in the response to increased weight gain and the development of secondary complications. Here, we have briefly outlined the different types of non-neuronal brain cells and some of their functions, both in general and those that are specific to the hypothalamus and metabolic circuits.
Classification of Non-Neuronal Cells in the Brain
Astroglial Cells
Astrocytes were the first class of glial cells to be described (2) and they are also the most studied. One example of this is that a search of the word “astrocyte” in the PubMed Central database obtains approximately 48,000 results; typing “microglia” or “oligodendrocyte” receives less than 30,000 returns in either case. Astroglia are also the most abundant cell type in the CNS and were first thought to only constitute the physical and metabolic support for neuronal function (2). We now know that they are much more than just “neuron helpers” (3). Astrocytes do indeed transport nutrients and metabolic factors across the blood–brain barrier (BBB) and release them to the extracellular fluid where they can be used by neurons and other glial cells (4, 5) (Figure 1). However, it is now clear that this supply of energy substrates to other cell types is regulated with astrocytes responding to metabolic changes in order to maintain brain homeostasis (6–9). Astrocytes are also the only glial cells known to store energy through glycogenesis (10). In the synaptic cleft, they reuptake neurotransmitters and also can release gliotransmitters, forming part of what is called the “tripartite synapse” (11, 12). At the level of the BBB, astrocytes are involved in the formation and maintenance of some of the barrier properties (13) and can regulate vasodilatation, thus controlling the flow of blood-borne substances (14, 15).
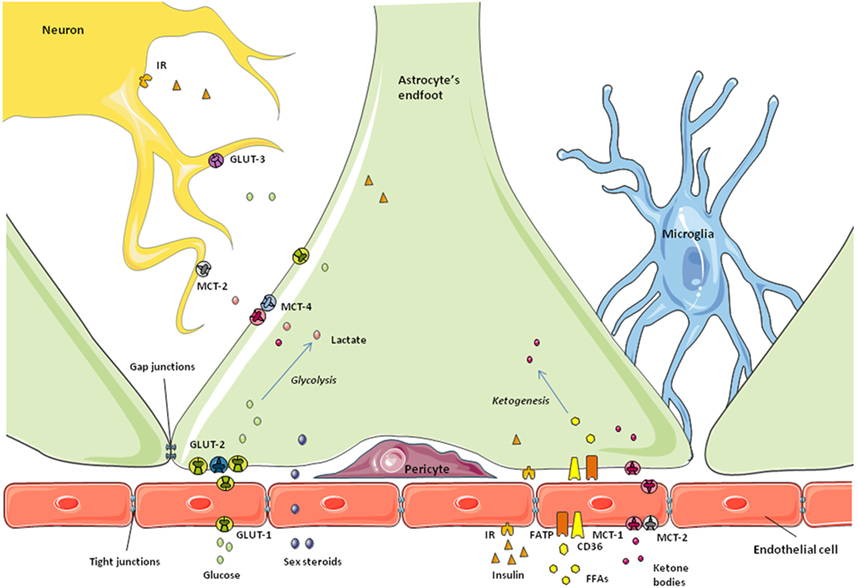
Figure 1. Schematic representation of the blood–brain barrier. Astrocytic endfeet surround the microvessels and take up the nutrients and metabolic factors coming from the bloodstream. Endothelial cells at this level express specific receptors and transporters and restrict the passage of small molecules to the brain due to the tight junctions between them. Depending on the metabolic state, nutrients and factors are processed by astrocytes to control their access to neurons and maintain brain homeostasis.
Astrocytes are connected by gap junctions in their plasma membranes, which enable direct transport of small molecules between cells. Initially, it was thought that these channels allowed passive diffusion of substances; however, the transport through gap junctions is tightly regulated (16, 17). One important function of these gap junctions is the rapid transmission of calcium waves within the glial network, resulting in a form of non-neuronal signal transmission (18).
When employing classical labeling methods, astrocytes appear to have a star-shaped morphology, although two different forms, protoplasmic and fibrous, can be distinguished. The first are mainly found close to synapses and blood vessels, whereas the latter are frequently found within the white matter (19–21). The morphology of these glial cells also changes in respect to their functional or activational state. The fact that astrocytes differentially express certain proteins (e.g., receptors, enzymes, channels, etc.) depending on the brain area and the physiological or pathophysiological conditions to which they are subjected raises questions regarding the current definition and classification of astroglial cells (22). Growing evidence indicates that astrocytes are vastly heterogeneous (23–28). For example, Matthias and colleagues reported that within the hippocampus subsets of GFAP expressing cells expressed either glutamate transporters or glutamate receptors (23). Moreover, astrocytes throughout the brain differentially express connexins (24) and GABA and glutamate receptors (26) and different astrocyte populations are reported to differentially support developmental functions and synapse formation (28, 29). Thus, our understanding of the functions of astrocytes is advancing, but much is yet to be learned. Indeed, we are only now beginning to have the tools to understand the grand diversity of these glial cells.
Microglia
Microglial cells constitute the bulk of the immune system in the brain. There have been different systems suggested for the classification of microglia, with most engaging morphological features. The most general classification includes an amoeboid form, characteristic of early development, and a ramified form or “resting” microglia and reactive microglia (30–32). The phenotype of reactive microglia is defined by changes in morphology, to short and thick projections, and the release of factors like cytokines, nitric oxide, and reactive oxygen species (30, 31, 33, 34). This “activation” or change in phenotype can occur in response to brain damage, toxic substances, or injury due to harmful conditions like obesity or a high fat diet (HFD) (35, 36) and when this state is sustained, it can lead to a pathological chronic state of reactive microgliosis (37). However, the division that separates resting and reactive microglia has become more diffuse as we learn more about these cells (38).
One of the main functions of microglia is to “clean” the CNS by phagocytosis of cellular debris, foreign matter, and other wastes (39). In this manner, they participate in development and synaptic plasticity (40–42). They can also release gliotransmitters and metabolic factors, contributing to maintain brain homeostasis (38, 43). Importantly, as part of the immune system, microglial cells respond to injury and harmful factors, including fatty acids, by releasing cytokines and to infection by presenting antigens to T-cells (35, 39, 43).
Oligodendrocytes
Oligodendrocyte projections wrap neuronal axons, forming the myelin sheaths in the CNS. To date, no direct link between these cells and systemic metabolic function has been verified, although some studies connecting metabolic signals with changes in myelination or oligodendrocyte survival suggest at least an indirect relationship with metabolism (44–48). However, it has been recently shown that oligodendrocyte precursors (NG2 glia) in the median eminence are important for the function of leptin receptor-expressing neurons, whose dendritic processes they contact (49).
Tanycytes
These specialized ependymal-like glial cells lining the ventral and ventrolateral part of the third ventricle (Figure 2) are proving to be very interesting as we know more about them. From dorsal to ventral, they are classified as subtypes α1, α2, β1, and β2. They are polarized cells: on the ventricle-side they express numerous receptors and transporters in their membrane and can be ciliated (not β2 tanycytes); and on the opposite side they present a long process that projects into the hypothalamic parenchyma or the median eminence (50). The β2 tanycytes can be found close to the median eminence, a subhypothalamic circumventricular organ. Capillaries on the median eminence are fenestrated, making the BBB permeable to many substances (50–52). The long processes of β2-tanycytes project into these fenestrated vessels, forming a blood–CSF barrier (BCSFB). The tight junctions between them, in addition to the specific transporters that they express, allow them to control the entry of many substances into the hypothalamus (53). They can also regulate the permeability of this barrier at this level of the brain, by the release of vascular endothelial growth factor-A in response to metabolic changes (54) and possibly by other mechanisms (55). Although astrocytes are the major cells expressing gap junctions, tanycytes also express these structures and can also produce calcium wave signaling (56). Tanycytes also possess stem cell properties (57) and participate in glutamate recycling (58), nutrient sensing (59, 60), and the conversion of thyroid hormones (TH) (61).
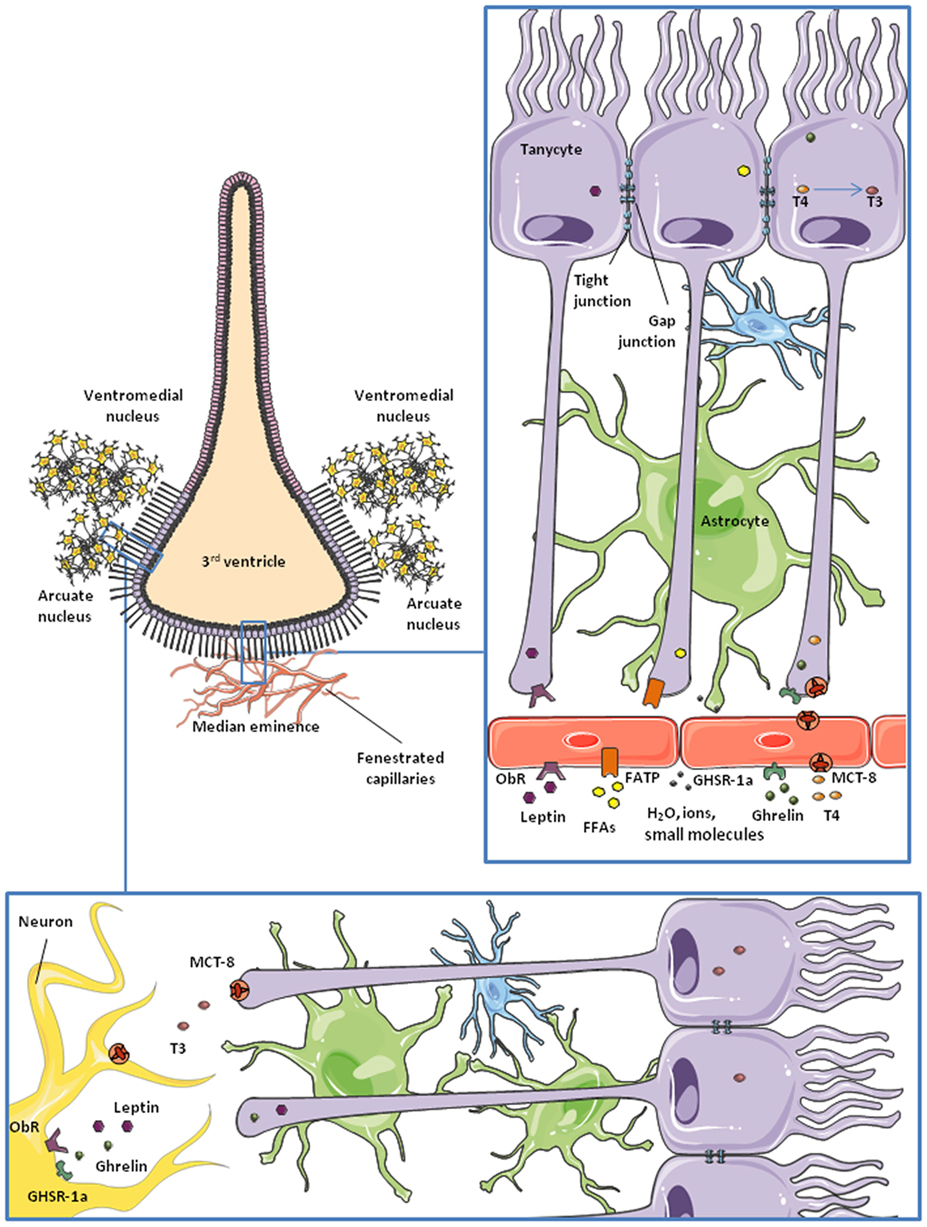
Figure 2. Schematic representation of tanycytes lining the third ventricle. Microvessels of the median eminence are fenestrated, so they allow water, ions, and small molecules to freely enter the brain. β2-tanycytes present tight junctions between them, forming a blood–CSF barrier. They take up nutrients and factors from the microvessels and control their access to the cerebrospinal fluid (CSF) and the rest of the brain. This is probably the main route for some hormones and nutrients to the hypothalamus. Also, tanycytes are communicated through gap junctions, so some molecules can be transported to lateral β1 and α-tanycytes and gain access to the arcuate nucleus and the ventromedial nucleus of the hypothalamus.
Pericytes
Pericytes are contractile cells surrounding the blood vessels (62, 63). In addition to their ability to modify blood flow due to their contractibility, brain pericytes have multiple roles in the development and maintenance of the BBB (64, 65), including macrophage-like functions and characteristics (66–68), angiogenic properties (69), and a role in neuroinflammation (70). Indeed, in response to brain injury, there is evidence that pericytes change to a microglia-like phenotype (68, 71), migrate to the brain parenchyma (72), and are involved in scar formation (73), antigen presentation (74), and the release of inflammatory factors (75, 76). Pericytes are also reported to be multipotential stem cells in the CNS (77). However, the identity of these stem cells is still a subject of controversy (78), due to the lack of reliable pericyte markers (79).
Endothelial Cells
Endothelial cells, along with pericytes, form the walls of the microvessels, taking part in the transport of metabolites through the BBB (80). The particularities of BBB endothelial cells, described below, allow for a strict control of the passage of substances from the blood into the CNS.
Epithelial Cells/Ependymocytes
In the CNS, epithelial cells can be found in the choroid plexus and lining the ventricles. They secrete the cerebrospinal fluid (CSF) that fills the ventricles and factors involved in neurogenesis and brain development (81–84). Epithelial cells of the CNS also express transporters for glucose, amino acids, and other molecules (85–87), as well as receptors for hormones such as sex steroids (88–90) and leptin (91). Moreover, they form a type of BCSFB due to the tight junctions between them (92). Ependymal cells are epithelial cells lining the ventricles. Their polarized organization and beating of numerous cilia are important for the movement of CSF (93, 94). They also possess precursor properties and, together with tanycytes, form the hypothalamic neurogenic niche (95).
Functions of Non-Neuronal Cells
Transport of Metabolic Signals into and within the Hypothalamus
The transport of nutrients and other metabolic signals is one of the best studied functions of non-neuronal cells in the nervous system. At the physiological level, nutrients from the diet, hormones, and other substances are delivered to all tissues through the bloodstream. However, due to its exceptional importance and vulnerability, the CNS protects its homeostasis by carefully controlling what can and cannot enter from the circulation. This function is carried out by the BBB, which is formed by specialized glia, pericytes, and endothelial cells expressing transporters, receptors, and sensors that allow them to select the information and nutrients accessing the nervous tissue (80) (Figure 1). As nutrients and metabolic signals are also found in the CSF, there is a BCSFB, formed by ependymal cells and tanycytes, in the third ventricle (50, 96, 97) (Figure 2). The distribution of tight junction proteins between tanycytes at this level is important in determining the permeability of the barrier, being lower at the median eminence, where there are fenestrated capillaries and higher next to the arcuate nucleus (98).
The first checkpoint for any substance to cross the BBB into the CNS is the endothelial cell, the bricks forming the capillary walls (Figure 1). Endothelial cells in the BBB are phenotypically different from those of peripheral vessels and restrict the access of blood-borne substances to the extracellular fluid of the CNS (80, 99). To achieve this, these endothelial cells have tight junctions between them, reduced endocytosis, no fenestrations, and specific transporters and receptors, in addition to a large number of mitochondria (65). Thus, brain capillary endothelial cells broadly determine the barrier permeability. Surrounding these capillaries are the astrocytic endfeet, along with pericytes and microglia (Figure 1). These other cells also participate in the regulation of nutrient and hormone entry, and thus metabolic signaling, from the periphery (80, 99, 100). Astrocytes and other non-neuronal cells can detect changes in the concentrations of specific nutrients and the presence of other signals and react consequently to maintain brain homeostasis, as described below.
Glucose
Glucose, the main energy source of the CNS, enters the brain from the bloodstream crossing the BBB through specific transporters. As normal brain function depends on its glucose supply, this step is highly regulated. That is, the transport of glucose across the BBB adapts in response to cerebral energy demand in order to maintain glucose homeostasis in the brain. The facilitative glucose transporter (GLUT)-1 is largely responsible for glucose transport across the BBB. This protein is expressed in non-neuronal cells throughout the CNS, especially in astrocytes and endothelial cells of the BBB (101), as well as in tanycytes along the BCSFB (50). However, GLUT-1 in endothelial cells is highly glycosylated, having a higher molecular weight than the isoform expressed in astrocytes and other glial cells (101, 102). As indicated in a recent review, some authors suggest different functional characteristics between the two forms of GLUT-1, although there is no consensus on this subject (103).
Changes in glucose concentration are rapidly detected in the hypothalamus, which adapts to such variations and emits a response to maintain glucose homeostasis not only in the brain, but also systemically as glucose-sensing neurons in the hypothalamus send signals to the autonomous nervous system, reaching peripheral organs such as the pancreas or the liver (104–107). There is more than one mechanism for central glucose sensing and different cell types are involved in this essential task (107–110). Two populations of glucose-sensing neurons have been identified: glucose-excited and glucose-inhibited neurons (GE and GI, respectively) (111) and glial cells also participate in these important glucose-sensing mechanisms. Astrocyte endfeet express GLUT-2 which, in addition to its transport functions, participates in glucose sensing (110, 112). This GLUT is highly expressed in tanycytes along the BCSFB (109), with these specialized glial cells also participating in glucose-sensing processes. In addition to expressing GLUT-2, astrocytes and tanycytes express sodium glucose transporter (SGLT)-1, glucokinase (GCK), and KATP channels (110), proteins that are all known to be involved in glucose-sensing mechanisms. Indeed, the classical mechanism for glucose sensing in pancreatic β-cells requires glucose uptake through GLUT-2 in rodents or GLUT-1 in humans, GCK, and activation of ATP-sensitive K+ channels (112, 113). This system shares some similarities with glucose-sensing pathways in astrocytes and tanycytes.
One proposed model for glucose sensing in tanycytes involves glucose entering the cell through GLUT-2 and phosphorylation by GCK. Subsequently, glucose-6-phosphate undergoes glycolysis, producing pyruvate and, through the action of lactate dehydrogenase, lactate. Lactate is transported to the extracellular space by monocarboxylate transporter (MCT)-4 or MCT-1, and then taken up by neurons through MCT-2 (109). Depending on the kind of neuron, GE or GI, an excitatory or inhibitory signal will be produced in the hypothalamus and sent to other brain areas and the autonomic nervous system (108). Tanycytes can also respond rapidly to glucose and other inputs by producing calcium waves, a process requiring ATP release and autocrine signaling through purinergic P2Y receptors (56, 59). The precise mechanisms involved in this tanycytic response are not yet fully elucidated, but it constitutes a possible model for tanycyte–neuron interaction.
Glucose sensing in astrocytes involves a similar process. Indeed, according to the “astrocyte-neuron lactate shuttle” hypothesis proposed by Pellerin and Magistretti over two decades ago (4), lactate from glucose or glycogen metabolism released by astrocytes is not only used by neurons as an energy source but can also signal energy availability to glucose-sensing neurons. Glucose transport into astrocytes is facilitated by GLUT-2 or occurs through gap junctions in a passive manner (112, 114–116). This glucose can be metabolized or stored as glycogen. However, it is still debated as to whether astrocytes secrete only lactate or also glucose to the extracellular fluid to act on glucose-sensing neurons and to be used as fuel (112). Moreover, astrocytes and tanycytes can respond to an increase in glucose or to other signals (i.e., some neurotransmitters) by secreting endozepines, anorexigenic peptides that act on hypothalamic neurons to maintain energy homeostasis (107, 117) and that also participate in unsaturated long-chain fatty acid metabolism in astrocytes (118).
The precise mechanisms of glucose transport and sensing in the hypothalamus are yet to be fully elucidated. For example, SGLT, an active sodium co-transporter, is reported to be involved in glucose sensing in the ventromedial nucleus of the rodent hypothalamus (119), although it is not clear whether this sensing occurs in glucose responsive neurons or in astrocytes. By using genetically engineered mouse models, García-Cáceres and collaborators recently demonstrated that insulin signaling in astrocytes plays a role in the regulation of systemic glucose homeostasis. Specific ablation of the insulin receptor (IR) in astrocytes was shown to impair their uptake of glucose and the ability to correctly respond to changes in glycemia (120). Other studies suggest a role of leptin in increasing (121) or ghrelin in reducing (122) glucose uptake by astrocytes, which might also affect glucose sensing. It thus appears that the transport of glucose by astrocytes is highly regulated by diverse nutrient and hormonal signals.
Ketone Bodies
Monocarboxylates are molecules with one carboxylate group; some examples with metabolic functions include not only lactate, but also pyruvate and ketones, all of which can be used by neurons as an alternative energy source in addition to acting as metabolic signals (123–126). The brain expresses MCTs-1, -2, and -4, with MCT-1 being found in endothelial and ependymal cells, as well as in astrocyte endfeet at the BBB (127, 128). MCT-2 is expressed in endothelial cells, but not in astrocytes, whereas MCT-4 appears to be specific for astrocytes (58, 129–131). Ketone bodies and other monocarboxylates from the bloodstream cross the BBB through specific MCTs present in both the luminal and abluminal sides of the endothelial cells (132, 133). Astrocyte endfeet not only takes up monocarboxylates through MCT-1 (132–134), but these glial cells are also able to synthesize ketone bodies from fatty acid β-oxidation and secrete them as an energy source for neurons and other glial cells (Figure 1). Tanycytes have also been suggested to transport lactate through MCT-2 in a photoperiodic model of Siberian hamster (58). These authors found that MCT-2 and the glutamate transporter GLAST were decreased during a short photoperiod, which could indicate a change in seasonal neurotransporter supply. In the rat brain, tanycytes were shown to express functional MCT-1 and MCT-4 in an anatomically specific manner (135), suggesting that these glial cells may also participate in lactate transport to neurons.
Regulation of the transport and production of ketone bodies in the brain is important in metabolic control as hypothalamic sensing of these monocarboxylates also participates in the regulation of food intake (126, 136). Indeed, after the initial HFD-induced hyperphagia, there is a reduction in food intake that is reported to be mediated, at least in part, by ketone body signaling to hypothalamic neurons. These ketone bodies are synthesized by hypothalamic astrocytes as products of fatty acid metabolism (136, 137).
Lipids
Lipid sensing in the hypothalamus is necessary for the correct regulation of energy balance (138). There are lipid sensing neurons that are excited or inhibited by fatty acids, depending on the type of neuron and also the metabolic state, i.e., fasting versus overfeeding (139). Although the role of glial cells in this process is not fully understood, astrocytes are the primary lipid metabolizers in the CNS. They also express proteins related to lipid sensing, such as transporter CD36 and peroxisome proliferator-activated receptor gamma, an important lipid-activated nuclear receptor that regulates transcription of numerous genes, including some involved in lipid metabolism (140, 141). In addition, astrocytic production of ketone bodies from fatty acids and their release to neurons could be one way by which an excess of fatty acids is signaled to metabolic neuronal circuits. Recent evidence suggests an increase in fatty acid β-oxidation in hypothalamic astrocytes from obese mice fed a HFD, as well as a role for tanycytes in restricting the passage of saturated fatty acids into the hypothalamus (142).
Although the brain produces lipids, it also has mechanisms to transport them from the bloodstream, but how they go through the BBB is not yet fully understood. Short and medium chain fatty acids appear to enter the CNS by simple diffusion through the plasma membrane (143). In contrast, long chain fatty acids (>12 carbons) need transporters to cross the BBB (144), with several fatty acid transport proteins (FATP) and fatty acid binding proteins (FABP) having been identified (145). In vitro studies indicate that FATP-1, FATP-4, and FABP-5 are the major isoforms expressed in microvessel endothelial cells and the gray matter of the human brain (145, 146). When the fatty acid translocase (FAT) CD36 is knocked-out in mice (CD36−/−), the uptake of monounsaturated fatty acids is significantly decreased, with no effect on polyunsaturated fatty acid uptake (147). In the CNS, CD36 is expressed in endothelial cells, microglial cells, astrocytes, and in ventromedial hypothalamic neurons (148–151). Although it is not the most highly expressed FATP, studies indicate that CD36 is responsible for fatty acid sensing in the hypothalamus and is thus important for the control of energy homeostasis (136, 137, 152).
In addition to the passage of free fatty acids through the BBB, lipids can also enter or exit the CNS as lipoproteins. This process is mediated by apolipoprotein E (ApoE) interacting with lipoprotein receptors (153). In the CNS, ApoE is expressed in astrocytes and tanycytes and its levels are upregulated by both leptin and TH (154, 155), with this process being involved in the regulation of food intake and energy balance (156).
Hormone Transport and Signaling
Leptin
Leptin is an anorexigenic hormone that exerts part of its effects by inhibiting orexigenic neurons and activating anorexigenic neurons in the hypothalamus (157–159). It also has a role in the regulation of systemic lipid and glucose metabolism (160, 161). The leptin (or obesity) receptor (ObR), which has six isoforms, is highly expressed in brain endothelial cells, astrocytes and tanycytes (162–165), and endothelial and astroglial cells have been studied in attempt to unravel the mechanisms of leptin transport into the brain (163, 166). However, González-Carter and colleagues have recently reported that, in a human in vitro BBB model, leptin–ObR interaction is not necessary for the transport of this hormone across the BBB. They propose that lipoprotein receptor-related protein-2, expressed in endothelial cells at the BBB, is responsible for the passage of leptin across the BBB (167). Increasing evidence points to the BCSFB as the main pathway for entry of leptin into, at least, the hypothalamus (165, 168) (Figure 2).
The median eminence, a circumventricular organ close to the third ventricle, is the first site in the brain reached by blood-borne leptin (165). After an intraperitoneal leptin injection, there is a 1–2 h lag between the activation of leptin signaling pathways in the ventral and dorsal nuclei of the hypothalamus. This time-lag disappears when leptin is administered intracerebroventricularly, instead of intraperitoneally (169), suggesting that leptin transport from blood to the CSF is an important step in the action of this hormone in the brain and that it involves a delay in circulating changes reaching central target sites. Moreover, this process appears to be a finely regulated step in the control of energy balance as tanycytes act as “gatekeepers” for the passage of leptin into the mediobasal hypothalamus. Evidence suggests that leptin is first taken up by tanycyte processes in contact with the fenestrated capillaries at the median eminence (165) and that this uptake requires the activation of ObRb and the internalization of leptin by clathrin-coated vesicles (165). According to research carried out by Vincent Prevot and his team, this process involves signal transducer and activator of transcription (STAT)-3, protein kinase B (PKB)/Akt, and extracellular signal regulated kinase (ERK) phosphorylation, but is janus kinase-2 independent (165). Leptin is then transported toward the tanycyte cell body and, finally, released to the CSF and hypothalamic parenchyma (Figure 2) employing an ERK-dependent pathway (165). By using STAT-3 phosphorylation as an indicator of leptin signaling (170–172), Balland and collaborators reported that neutralization of leptin in the CSF impairs leptin signaling in mediobasal hypothalamic neurons (165), supporting the idea of the blood–CSF–hypothalamic gateway for leptin entry into the brain.
Taking into account the above mentioned studies, it appears that both endothelial cells and tanycytes contribute to the transport of leptin through the BBB and between different brain regions (163, 166, 167). In contrast, there is no clear evidence of the involvement of astrocytes in leptin transport, but a number of studies demonstrate that leptin signaling in astrocytes is important for energy homeostasis (173, 174).
Leptin transport into the brain is modulated by conditions including obesity and fasting, as well as metabolic factors. Obesity associated to HFD intake is reported to induce central leptin resistance. There are two main mechanisms or levels of leptin resistance suggested to occur: impairment of leptin transport into the brain (165) and reduction in the central response to leptin (175). Mice exposed long term to a HFD develop leptin resistance only when high levels of plasma leptin are reached (176). This suggests that hyperleptinemia is at least one of the causes of diet-induced leptin resistance. In addition, hypothalamic inflammation associated with diet-induced obesity could contribute to leptin resistance by altering the cellular networks and molecular pathways that control energy homeostasis (177). Nevertheless, recent studies suggest that leptin resistance does not imply a loss of responsiveness to endogenous leptin, but rather that there is a threshold above which exogenous leptin barely increases the response to leptin (178, 179). Glucose and insulin are reported to increase the transport of leptin across the BBB (180), while an increase in circulating triglycerides could impair leptin transport across the BBB (181). The latter suggests a possible mechanism for the reported reduction in leptin transport into the brain during fasting (182).
Ghrelin
Ghrelin is an orexigenic hormone produced and secreted in the stomach (183). It has similar targets as leptin in the CNS and also plays an important, but opposite, role in energy balance (184). There are two forms of ghrelin, acylated and unacylated, depending on the post-translational acylation with octanoic or decanoic acid (183, 185). This modification occurs mainly in the stomach, but there is evidence that it can also take place in target tissues (186). The acylated form of ghrelin exerts the majority of the metabolic effects of this hormone in the CNS and it binds more efficiently to the ghrelin receptor than the unacylated form (187). This receptor, also called the growth hormone secretagogue receptor 1a, is widely expressed in the hypothalamus (188). The mechanism underlying the passage of ghrelin across the BBB is not yet fully understood, but recent studies indicate that ghrelin possibly uses a similar route as leptin into the brain (189), i.e., through tanycytes in contact with the median eminence (Figure 2). Other studies indicate that this process is carried out by saturable transporters, at least for the acylated form, whereas transport of des-acyl ghrelin is not saturable (190). Entry of acylated ghrelin into the CNS is increased by serum triglycerides and fasting and is decreased in obese mice (191), in contrast with leptin transport. Diet-induced obesity is reported to impair the hypothalamic response to peripherally or centrally administered ghrelin (192). The role of unacylated ghrelin on metabolism is largely unknown, but an increasing number of studies reveal that des-acyl ghrelin has similar and opposite functions as the acylated form (193–196).
Insulin
Insulin is a pancreatic hormone directly involved in glucose metabolism and homeostasis. Within the brain, it acts to increase energy expenditure and reduce food intake and energy storage (197). Insulin binds to its receptor in the plasma membranes of endothelial cells at the BBB and is internalized following a saturable pathway (198, 199). Recent studies have shown that IRs in astrocytes are involved in the entry of this hormone into the CNS (120). Also, as mentioned above, insulin signaling in astrocytes is necessary for the regulation of systemic glucose levels (120). Insulin transport into the brain is enhanced by satiation hormones like cholecystokinin (200). Although estradiol is known to impair insulin’s actions in the brain, its effects appear to be unrelated to insulin transport (201). Some studies show that leptin increases insulin sensitivity in the hypothalamus at the molecular level (202), while others have found that leptin impairs insulin signaling in the brain (203). This discrepancy could be a matter of the experimental model employed, but further research is needed to understand the relationship between the effects of leptin and insulin at the level of the CNS. Leptin shares some signaling pathways with insulin, but the effects of these two hormones are not entirely parallel, as they exert opposite effects in some hypothalamic neurons (204). Saturated fatty acids induce insulin resistance in the hypothalamus (205), as has been previously described in peripheral tissues (206).
Sex Steroids
As hydrophobic molecules, estrogens, androgens, and progesterone can enter the brain by simple diffusion (207). Moreover, steroids are synthesized in the brain (208). These neurosteroids are produced in the CNS either from brain-borne cholesterol or from peripherally synthesized steroid precursors, like pregnenolone, deoxycorticosterone, and testosterone (209). The enzymes necessary for this synthesis are found in non-neuronal cells, including astrocytes, tanycytes, ependymal cells, and oligodendrocytes (210, 211), and in some neurons (212). As steroid hormones are known to regulate neurosteroid metabolism in glial cells (213–217) and also the expression levels of steroid receptors in the hypothalamus (218, 219), neurosteroids could have paracrine/autocrine functions within the brain.
Steroids and neurosteroids exert neuroprotective effects in the brain following brain injury, neurological disease, or inflammation (220–227). The expression of aromatase, the enzyme that catalyzes the conversion of testosterone into estradiol, is stimulated in reactive astrocytes after brain injury as a neuroprotective measure (228–230). Both microglial cells and astrocytes play an important role in the neuroprotective functions of steroids (231), as sex steroids diminish microglia reactivity (232–234) and astrocyte production of proinflammatory molecules (235–238).
Sex steroids, but specially estrogens, modulate energy homeostasis at the hypothalamic level decreasing food intake (239–241), increasing energy expenditure (242), and modulating the sensitivity to other metabolic hormones (243, 244). Their effect differs depending on the neuronal population (245, 246), but with an overall anorectic effect (247–249), although the underlying mechanisms are not yet fully understood. While nuclear estrogen receptors (ERs) are involved, especially ER α (247, 250–252), evidence indicates that estrogen responsive G-coupled membrane receptors can also regulate these effects (253, 254). The apparently contradictory results in the literature regarding the mechanism of action of estrogens on metabolism indicate a complex system for estrogens’ function in energy homeostasis, where the different ERs could be acting in combination (255). Moreover, the mechanisms of action used by estrogens in metabolic control could be sexually dimorphic (256). The involvement of neurosteroids in energy homeostasis remains unknown.
Thyroid Hormones
The role of TH in increasing the metabolic rate has been known for more than a century (257). The involvement of these hormones in the control of energy homeostasis at the central level is a more recent discovery (258, 259). They promote lipogenesis at the level of the hypothalamus, which eventually leads to brown adipose tissue thermogenesis (259) and blockage of TH signaling in the hypothalamus reverts this process, leading to weight gain without an increase in feeding (259). Clinical studies and animal models with a pathological excess of TH synthesis and secretion (hyperthyroidism) have shed light on TH action in the hypothalamus and control of energy balance (260). Most hyperthyroid patients have an increased appetite and food intake and decreased body weight (261). Moreover, these same symptoms that are observed in animal models of hyperthyroidism are associated with the upregulation of orexigenic neuropeptides AgRP and NPY and downregulation of anorexigenic neuropeptides derived from POMC in the arcuate nucleus (259). There is evidence that TH are involved in brain inflammation, promoting survival, and processes growth in microglial cells and also in astrocytes (262–264). TH are also involved in systemic glucose homeostasis and insulin sensing (265, 266).
The thyroid gland produces and secretes mainly tetraiodo-l-thyronine or thyroxine (T4), which is generally transformed into triiodo-l-thyronine (T3) through deiodination at the level of peripheral tissues (267). Thus, deiodinase enzyme expression in peripheral tissues is important for the control of TH actions (268), as they catalyze the transformation of T4 into T3 and of both hormones into reverse T3 (rT3) and 3,5-diiodo-l-thyronine (T2), respectively (269). These two last forms are usually considered inactive, although in the last few years new roles have been proposed for them and other non-classical TH (270).
Thyroid hormones enter the hypothalamus mainly through MCT-8 (271) and organic anion transporting polypeptide-1C1 (272) in rodents (273). These transporters are expressed in endothelial cells of the BBB and epithelial cells of the choroid plexus (274), besides neurons and glial cells of the hypothalamus (275–277). Tanycytes act as gatekeepers for TH at the BBB (61) (Figure 2). These cells express the enzyme deiodinase II (DII) (278–280), catalyzing the formation of hormone T3 from the prohormone T4. Tanycytes uptake T4 from the capillaries and release T3 to the extracellular space in the hypothalamus, where this hormone can exert its central actions (258, 259, 281) (Figure 2). Modulation of deiodinase expression is a key point in TH homeostasis. For example, DII expression in tanycytes is promoted in fasting conditions (282). DII-expressing tanycytes are in direct contact with AgRP/NPY-expressing neurons of the arcuate nucleus. Upregulation of DII results in an increased production of T3, which activates AgRP/NPY neurons and, therefore, feeding behavior (283, 284). Tanycytes also express deiodinase III (DIII), which deiodinates T4 into reverse T3 which is biologically inactive, and T3 into T2. TH is important in the adaptation to different photoperiods in seasonal animals were, for example, there is a decrease in food intake and body weight during short photoperiods. The study of hypothalamic metabolism of TH during photoperiodic changes in seasonal mammals has shown that the there is an upregulation of DII during periods of long days, which would increase the levels of T3. In Siberian hamsters an upregulation of DIII in tanycytes has been shown to occur during short photoperiods, lowering active T3 levels (285). The retinoic acid pathway in tanycytes appears to be similarly regulated by photoperiodicity and also leads to modifications in energy expenditure (286, 287).
Thyroid hormone signaling usually occurs through nuclear thyroid receptors α and β (288) that function as transcription factors modulating gene expression (289). TH can also exert rapid non-genomic actions through membrane-associated receptors (290, 291). This signaling pathway could mediate TH effects on vasodilatation (292) and has been shown to be involved in neuronal excitability in the hippocampus (293, 294).
Centrally, THs control their own homeostasis in various ways, with non-neuronal cells having an important role, i.e., regulation of deiodinase expression (278) and inactivation of thyroid releasing hormone (295). Other hormones involved in metabolic control can enhance the secretion, synthesis, or sensing of TH, including leptin (296–298) and sex steroids (299–301).
Metabolism of Nutrients
Glucose
Perivascular astrocytes take-up blood-borne glucose that then undergoes glycolysis or glycogenesis (112). Lactate produced from glucose or glycogen metabolism in these cells is released to the extracellular space and enters neurons to be used as energy, constituting their primary energy source as suggested by some studies (302, 303). However, the question about the identity of the main energy source for neurons—lactate or glucose—is still debated. Tanycytes can metabolize and sense glucose in a similar manner (109).
Glucose storage as glycogen in astrocytes provides a way to guarantee energy release to neurons when it is needed, i.e., when faced with a raise in neuronal activity (304), by production of lactate from glycogenolysis. Several factors can regulate glycogen production and utilization in astrocytes, with insulin, insulin-like growth factor (IGF)-1 (305, 306), and leptin (203, 307) increasing their production of glycogen. More recently, ghrelin has been reported to possibly promote glycogenolysis in hypothalamic neurons (122).
Lipids and Ketone Bodies
It has been suggested that some fatty acids, like erucic acid (308, 309), suffer metabolic changes as they cross the BBB, whereas others do not (310, 311). Studies indicate that lipoproteins are hydrolyzed as they cross the BBB by the enzyme lipoprotein lipase associated to the membrane of endothelial cells (312–316).
In the absence of glucose and when glycogen stores are exhausted, such as in fasting conditions, astrocytes increase their uptake and utilization of fatty acids (136, 317, 318), which enter the mitochondria through carnitine palmitoyltransferase-1 to undergo β-oxidation (319). In the mitochondria, the enzymes 3-hydroxy-3-methylglutaryl-CoA synthase and lyase (320–322) transform fatty acids into β-hydroxybutyrate, a ketone body (323). Ketone bodies produced from this metabolic pathway are used by astrocytes themselves for fuel or secreted to be used by neurons and other glial cells (318).
Neurogenesis
Glial cells were first reported to participate in neurogenesis during development (324–326), but it later became apparent that they are also involved in this process in adulthood (327). In the developing hypothalamus of the rat, the birth of metabolically important neurons occurs between embryonic days 10.5 and 18.5 (328–330). Environmental changes during this period, including nutritional and hormonal disturbances, can modulate the normal process of hypothalamic neurogenesis and have an impact on later neuroendocrine function (330–332). For example, HFD intake by pregnant dams stimulates the proliferation, differentiation, and migration of orexigenic neuronal precursors and increases the density of orexigenic neurons at the level of the paraventricular nucleus in the offspring. This increase in the number and density of appetite-stimulating neurons and orexigenic neuropeptide expression leads to increased appetite, body weight, and propensity to develop obesity later in life (331).
The clear demonstration, as well as its acceptance by the scientific community, of neurogenesis in the adult hypothalamus is relatively recent and there is still much to be learned. Tanycytes form part of the pool of neuroprogenitor cells in the hypothalamus and these precursors are capable of differentiating into not only neurons, but also astrocytes both during development and in the adult brain (333, 334). These specialized glial cells form an important neurogenic niche in the vicinity of the median eminence (333) and can proliferate and differentiate under basal conditions and when stimulated by growth factors such as IGF-1 (57, 95), fibroblast growth factor (FGF)-2 (335), FGF-10 (336, 337), or even vitamins, as tanycytes have been shown to express receptors, transporters, and other components of the vitamin A and C pathways (286, 287, 334, 338). FGF-10 positive tanycytes are reported to be important neural progenitors for NPY neurons in the arcuate nucleus, a function that may continue even during adulthood (337, 339). In addition, other isoforms of FGF are known to play a role in glucose homeostasis, inhibition of food intake, and body weight (340–343), with a possible involvement of glial cells (344–347). Although the generation of newborn neurons in the postnatal hypothalamus takes place at lower rates than during the embryonic period, it is physiologically relevant and has been shown to be regulated by diverse factors, including hormones and growth factors such as estradiol (332), FGF (335), and IGF-1 (348). Moreover, the nutritional status and dietary intake of an individual can modulate neurogenesis in hypothalamic metabolic circuits even in the adult animal (329, 333, 349–351).
The neurons composing the hypothalamic metabolic circuits experience a turnover rate such that approximately half of these cells are reported to be replaced between 4 and 12 weeks of age in mice (329). Diet-induced obesity suppresses this remodeling, at least in part, due to a decrease in actively proliferating cells in the hypothalamus with caloric restriction reversing this effect (329). Voluntary exercise is also reported to induce hypothalamic neurogenesis (352, 353). The effect of nutrient intake on the adult hypothalamus may be anatomically specific, as diets rich in fat are reported to inhibit neurogenesis in the mediobasal hypothalamic parenchyma (333), but to promote it in the median eminence in female mice (333). The enhanced neurogenesis that occurs at the level of the median eminence is suggested to be involved in the restoration of neurons that are damaged as a consequence of HFD intake (333); hence, impedance of this process could amplify the derogatory effects of poor dietary habits. There is tantalizing data indicating that hypothalamic neurogenesis in response to HFD differs between males and females (332, 333, 354), but whether this is involved in the sexually dimorphic metabolic response to HFD and weight gain requires further investigation.
Astrocytes are involved in the regulation of neuronal differentiation, proliferation, and synaptogenesis during development (3, 355). Microglia also actively participate in neurogenesis, both during development and adulthood, with most studies being performed in the hippocampus (356). Microglia not only phagocytize cells undergoing apoptosis in proliferative zones, but they also produce factors that can either inhibit or stimulate neuroprogenitor cells. The cross-talk between microglia and neuroprogenitor cells is an active area of investigation as this is a finely tuned process where these cells continuously interchange information (356). However, less is known regarding the role of astrocytes and microglia in neurogenesis in metabolic circuits of the adult animal.
Diet not only affects neurogenesis in the hypothalamus, but also in other brain areas such as the hippocampus, an area known to maintain active neurogenesis even in the adult (357). In the dentate gyrus of the hippocampus, HFD intake impairs neurogenesis (358) in addition to producing oxidative stress and lipid peroxidation (357). Palmitic acid (PA), a saturated fatty acid that is a major component of the majority of HFDs, was shown to reduce the proliferation of the neuroprogenitor cells (359) and the levels of brain-derived neurotrophic factor, indicating that PA-rich diets impair neurogenesis in the hippocampus. Caloric restriction and exercise increase neurogenesis in the hippocampus (350, 360, 361) and this has been associated with the anti-depressive effects of exercise (360).
Synaptogenesis, Synaptic Plasticity, and Synaptic Transmission
Astrocytes, in addition to participating in neuronal proliferation and differentiation, also regulate synaptogenesis during development (3, 355). In the hypothalamus, the neonatal and early prenatal hormonal and nutritional environments can affect the synaptic connectivity of metabolic circuits (189, 362). Astroglial coverage of neuronal cell surfaces has been shown to be inversely correlated with the number of synaptic inputs to their somas, with this astroglial ensheathment/synaptic input arrangement being physiologically relevant in the neuroendocrine hypothalamus (363–367). Thus, changes in astrocyte numbers or morphology in the hypothalamus might be expected to modify synaptic inputs both during development and in later life.
The generation and maturation of astrocytes is not fully complete until the third postnatal week in rodents (368, 369), so variations in the physiological levels of specific metabolic hormones or signals during early life could affect the development of these cells. For example, neonatal overnutrition and modifications in leptin levels or signaling affect the number and morphology of astrocytes in the arcuate nucleus in adulthood (121, 173, 370). The leptin peak that takes place between postnatal days 5 and 13 in rodents is essential not only for neuronal outgrowth and maturation, but also astrogenesis (368, 371, 372) and astrocyte development (373, 374). The timing and magnitude of this leptin surge can be modified by nutrition (371, 375, 376), as well as other conditions such as stress (377) and is one mechanism by which these early environmental influences can have long-term effects on metabolism.
Maternal dietary intake and body weight during gestation and lactation can also influence metabolic circuit formation in the offspring, including the astroglial ensheathment/synaptic input arrangement. For example, newborns from mothers fed a HFD during gestation and lactation have increased astroglial ensheathment of POMC neurons that is associated with a decrease in the resting mini inhibitory post synaptic currents of these neurons (121). The response of these POMC neurons to changes in glucose concentrations was also shown to be modified (121). Hence, alterations in the early nutritional environment could imply the modification of the appropriate development of neuron–glial interaction of metabolic circuits and therefore affect long-term metabolism.
Microglia are involved in synaptogenesis throughout the brain (378, 379); however, there is little information regarding the specific effects of microglia on the development of the synaptic interactions of metabolic circuits. These glial cells have been shown to have an active role in the sexual differentiation of behavior and masculinization of the brain (380), suggesting that they may indeed be important for the development of endocrine circuits and possibly the sexual differentiation of some of these systems.
Modifications in the synaptic connectivity of metabolic circuits occur in postnatal life in response to metabolic and hormonal signals (241, 381–384) and are most likely involved in the adaptation to changes in energy inputs/conditions in attempt to maintain metabolic homeostasis, with astroglia participating in these synaptic rearrangements. HFD induced-obesity is associated with an increase in the glial coverage of both POMC and NPY cell bodies in the arcuate nucleus, which is coincident with a decrease in the number of synaptic inputs to the perikarya of these neurons (384). However, there is a decline in stimulatory inputs to NPY neurons and of inhibitory inputs to POMC neurons (384), which would result in an overall decline in orexigenic signaling. When first given a HFD, rodents experience a phase of hyperphagia that is normally followed by an attenuation of this rise in energy intake. The levels of polysialic acid (PSA) are rapidly increased in the arcuate nucleus in response to HFD (385). This cell-surface glycogen can attach to cell membrane proteins to weaken cell–cell interactions and facilitate synaptic reorganization (386). If PSA is enzymatically removed from neural-cell adhesion molecule (NCAM) in the hypothalamus, HFD induced modifications in metabolic circuits can be blocked and the adaptation to HFD-induced hyperphagia attenuated (385). In addition, studies in photoperiodic models have shown that PSA and NCAM levels in tanycytes are reduced during short photoperiods in conjunction with vimentin levels, modulating the plasticity for tanycyte connections with arcuate neurons (387).
Diverse hormonal/metabolic signals could be involved in these structural modifications, including leptin. This hormone rapidly induces synaptic changes in metabolic circuits (381), with some of these effects being mediated through astrocytes. These glial cells express different isoforms of ObR (163, 164), with the expression of this receptor being increased in astrocytes of obese rodents (163). Leptin can modify astrocyte morphology, inducing changes in the length and number of primary astrocytic projections and astroglial coverage of hypothalamic neurons (173, 388). The lack of leptin signaling due to the knock-out of this receptor in astrocytes changes synaptic inputs to POMC and NPY neurons, resulting in modifications in the function of these metabolic neurons and rendering the animals less susceptible to the effects of leptin (173). However, it remains unclear as to the mechanisms involved in the changes in neuronal/glial interactions, including identification of the initial step that triggers these morphological modifications.
Astrocytes modulate neuronal transmission by controlling glutamate concentrations in the synaptic cleft, which also plays an important role in preventing excitotoxicity (389). Leptin and ghrelin modulate glutamate uptake by these glial cells (121, 122) and could thus affect stimulatory signaling in metabolic circuits through this mechanism. Astrocytes also actively participate in synaptic transmission and plasticity by releasing gliotransmitters, including adenosine, ATP, d-serine, glutamate, and tumor necrosis factor α that directly activate postsynaptic receptors and by altering neurotransmitter release from presynaptic neuronal elements to induce short-term plasticity and to modulate synaptic efficacy (12, 390–393). Adenosine release by astrocytes inhibits the firing rate of AgRP neurons and food intake, modifying the response to metabolic hormones such as ghrelin (394).
Inflammatory Response
The inflammatory response to infection, foreign substances, mechanical damage, or any situation that could damage neurons is one of the best studied functions of glial cells (231, 395–398). However, the description of hypothalamic inflammation in obesity, as well as its association with the development of secondary complications, is more recent. In 2005, the group of Licio Velloso reported that inflammatory pathways were activated in the hypothalamus in HFD-induced obese rats (399). This same group went on to demonstrate that this hypothalamic inflammation was involved in the disruption of systemic glucose homeostasis (35). Numerous studies have since reported the link between hypothalamic inflammation and obesity-related comorbidities (36, 400–404). Hypothalamic inflammation is reported to be associated with the development of insulin resistance and type 2 diabetes (405) and increased cell death in the hypothalamus (329). Most studies analyzing hypothalamic inflammation have employed HFD-induced obesity models and suggest that dietary factors are involved in at least part of the inflammatory response. Indeed, hypothalamic inflammation is reported to occur even before an increase in adiposity or systemic inflammation are detected (36) and central administration of saturated fatty acids directly activates inflammatory signaling mechanisms in the hypothalamus (35, 406). However, increased weight gain can occur in response to genetic, epigenetic, and excess energy intake that is not due to increased fat consumption and the hypothalamic inflammatory/gliosis response differs depending on the underlying cause of weight gain (121, 195, 407, 408). These differential responses are most likely the result of dietary signals and the changes in metabolic signals associated to weight gain acting on both microglia and astrocytes. Sex may also be a factor, as the hypothalamic inflammatory response to chronic HFD-intake is reported to differ between males and females, with males being more susceptible (409). This could result from the greater rise in PA levels in the CNS of male mice compared to females, even though there is no sex difference in circulating fatty acid levels (410).
Inhibition of hypothalamic inflammation is reported to blunt or block the development of obesity-associated complications (400, 403) and dietary restriction can reverse central inflammatory processes (411–415). Exercise also protects against HFD-induced hypothalamic inflammation (416).
Microglia in Hypothalamic Inflammation
Microglia, the innate immune cells of the CNS, are the first line of defense in response to foreign substances (417, 418) and are activated in response to saturated fat consumption (36, 403, 408, 419). Indeed, these glial cells are suggested to dictate the inflammation that occurs in response to saturated fats (419). Microglia can also be activated when weight gain is due to excess intake of a normal diet and due to high fat intake, (402), indicating that not only dietary signals are involved. Leptin stimulates the release of inflammatory cytokines from microglia (420), suggesting that hyperleptinemia could be involved in microglial activation in obese subjects.
Astrocytes in Hypothalamic Inflammation
Astrocytes also respond to HFD intake (36, 384, 421) and can be directly activated in vitro by fatty acids (408, 409, 422). Hyperleptinemia associated with weight gain may also participate in the activation of glia in situation of obesity (121, 173, 388, 408, 423). Indeed, ob/ob mice, which are dramatically obese due to the genetic lack of leptin, do not exhibit astrogliosis and leptin-induced weight loss actually increases astrocytic profiles in the hypothalamus of these animals (408). However, we have found that in some situations of increased weight gain, such as increased carbohydrate intake in the form of sucrose, astrocytic markers may actually be decreased (407).
The astrogliosis response to HFD differs between males and females, as does the in vitro response to PA (409). The protective effects of estrogens are exerted through ERα in astrocytes (424), with estrogens protecting against PA activation of astrocytes in vitro (409). Morselli et al demonstrated that HFD-intake reduces hypothalamic ERα levels in males, but not in females, which may be involved in the decreased protection against diet-induced obesity in males.
Astrocytes have also been implicated in determining the preference for a HFD, with this mechanism involving cannabinoid receptor 1 (CB1) (425, 426). The intake of a HFD induces the preference for this type of diet and this appears to involve the production of endocannabinoids in the hypothalamus (426). Leptin signaling in astrocytes is regulated by CB1, with disruption of CB1 in these glial cells resulting in the inability of leptin to regulate glycogen storage (307) and thus possibly affecting central energy storage and glucose sensing/signaling.
Conclusion
It is clear that non-neuronal cells are fundamental for the correct functioning of metabolic circuits, beginning with the essential process of regulating the nutrients and signals that reach these neurons. These cells are not only involved with the development, maintenance, and protection of their neuronal neighbors, but participate in all aspects of neuronal function (summarized in Figure 3). In the hypothalamus, numerous studies have shown how non-neuronal cells play an active role in the control of metabolism and in the pathological outcomes of poor metabolic control. Although the advances in laboratory techniques and genetically engineered animal models have increased our knowledge in this field, there is yet much to be learned regarding the mechanisms involved. Studies directed at developing markers to further identify different populations or subclasses of glial cells are of great importance in order to better understand the vast roles that these cells play in the different physiological functions controlled by the CNS. This increased knowledge will also hopefully add to our understanding of pathophysiological processes and future targets for treatments, including that of obesity and its secondary complications.
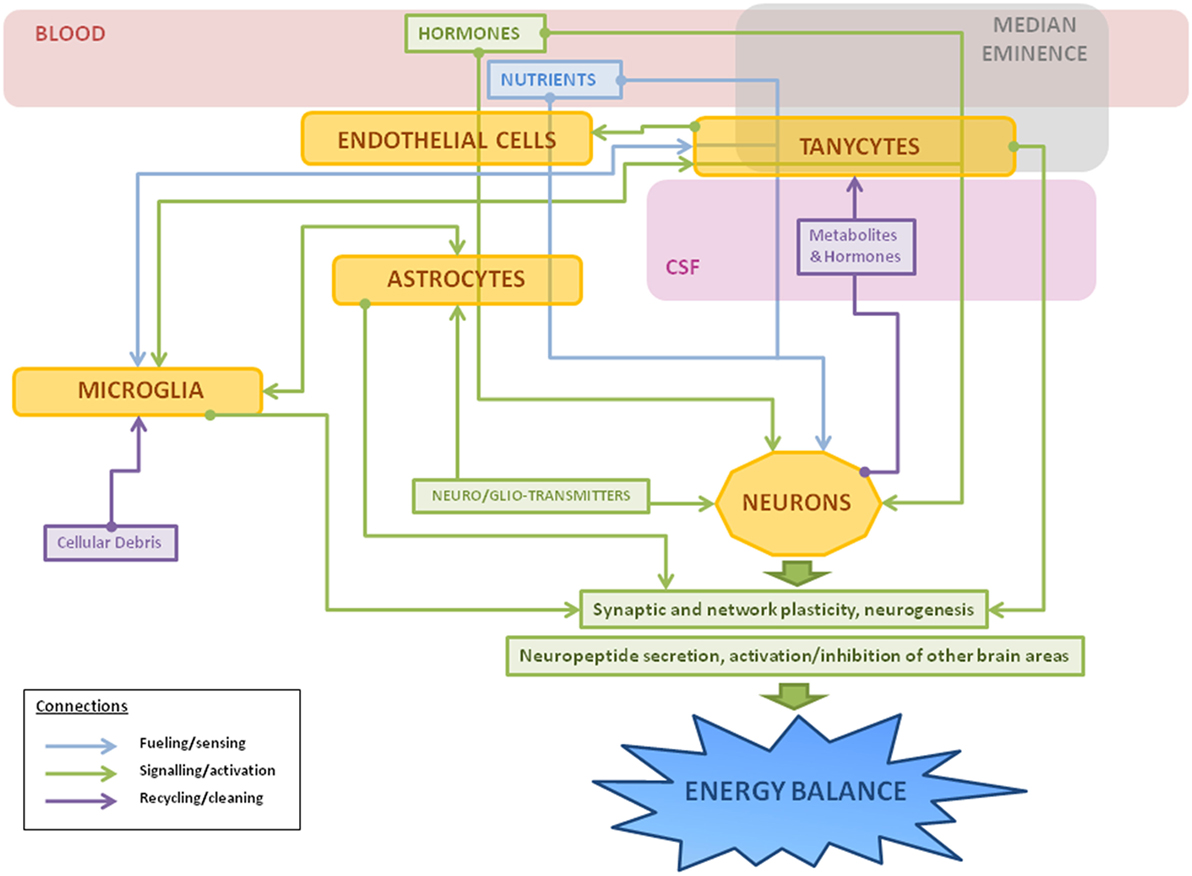
Figure 3. Schematic representation of the main roles of hypothalamic non-neuronal cells in metabolism. Thin arrows represent the different connections between cells: fueling and sensing of nutrients (in blue); signaling through gliotransmitters, neurotransmitters, or other factors (in green); and recycling of molecules or cleaning of cellular debris (in purple). Hormones and nutrients from the bloodstream pass through one or more types of non-neuronal cells before reaching neurons, sometimes suffering metabolic changes during the process. In response to a metabolic imbalance (excess of saturated fatty acids, hyperleptinemia, etc), microglial cells change to an activated state, releasing inflammatory factors, such as cytokines, and activating astrocytes as a neuroprotective measure. If the insult continues, it can lead to gliosis, hypothalamic inflammation, and neuronal damage.
Author Contributions
All authors have contributed to the writing and editing of this review. Figures were designed and made by AF-R.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors want to acknowledge Servier Medical Art for their PowerPoint image bank, which has been used as a source for the elaboration of the figures on this manuscript.
Funding
The authors are funded by grants from the Spanish Ministry of Science and Innovation (BFU2014-51836-C2-2 to JAC and BFU2014-51836-C2-1 to LG-S), Spanish Ministry of Education, Culture and Sports (university training grant FPU13/00909 to AF-R), Fondo de Investigación Sanitaria (PI-1302195, PI-1600485, and CIBEROBN to JA and CIBERFES to LG-S) and Fondos FEDER.
References
1. Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med (1999) 341(12):879–84. doi:10.1056/NEJM199909163411204
2. Young JZ. The concept of neuroglia. Ann NY Acad Sci (1991) 633(1):1–18. doi:10.1111/j.1749-6632.1991.tb15590.x
3. Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci (2003) 26(10):523–30. doi:10.1016/j.tins.2003.08.008
4. Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A (1994) 91(22):10625–9. doi:10.1073/pnas.91.22.10625
5. Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood–brain barrier. Nat Rev Neurosci (2006) 7(1):41–53. doi:10.1038/nrn1824
6. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol (2010) 119(1):7–35. doi:10.1007/s00401-009-0619-8
7. Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte–neuron metabolic cooperation. Cell Metab (2011) 14(6):724–38. doi:10.1016/j.cmet.2011.08.016
8. Parpura V, Heneka MT, Montana V, Oliet SHR, Schousboe A, Haydon PG, et al. Glial cells in (patho)physiology. J Neurochem (2012) 121(1):4–27. doi:10.1111/j.1471-4159.2012.07664.x
9. Teschemacher AG, Gourine AV, Kasparov S. A role for astrocytes in sensing the brain microenvironment and neuro-metabolic integration. Neurochem Res (2015) 40(12):2386–93. doi:10.1007/s11064-015-1562-9
10. Cataldo AM, Broadwell RD. Cytochemical identification of cerebral glycogen and glucose-6-phosphatase activity under normal and experimental conditions. II. Choroid plexus and ependymal epithelia, endothelia and pericytes. J Neurocytol (1986) 15(4):511–24. doi:10.1007/BF01611733
11. Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci (1999) 22(5):208–15. doi:10.1016/S0166-2236(98)01349-6
12. Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci (2009) 32(8):421–31. doi:10.1016/j.tins.2009.05.001
13. Janzer RC, Raff MC. Astrocytes induce blood–brain barrier properties in endothelial cells. Nature (1987) 325(6101):253–7. doi:10.1038/325253a0
14. Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci (2003) 6(1):43–50. doi:10.1038/nn980
15. MacVicar BA, Newman EA. Astrocyte regulation of blood flow in the brain. Cold Spring Harb Perspect Biol (2015) 7(5):a020388. doi:10.1101/cshperspect.a020388
16. Söhl G, Willecke K. Gap junctions and the connexin protein family. Cardiovasc Res (2004) 62(2):228–32. doi:10.1016/j.cardiores.2003.11.013
17. Theis M, Söhl G, Eiberger J, Willecke K. Emerging complexities in identity and function of glial connexins. Trends Neurosci (2005) 28(4):188–95. doi:10.1016/j.tins.2005.02.006
18. Scemes E, Giaume C. Astrocyte calcium waves: what they are and what they do. Glia (2006) 54(7):716–25. doi:10.1002/glia.20374
19. Ramón y Cajal S. Textura Del Sistema Nervioso Del Hombre y De Los Vertebrados. Madrid: Nicolás Moya (1899).
20. Miller RH, Raff MC. Fibrous and protoplasmic astrocytes are biochemically and developmentally distinct. J Neurosci (1984) 4(2):585.
21. Privat A, Rataboul P. Fibrous and protoplasmic astrocytes. In: Federoff S, Vernadakis A, editors. Astrocytes. Development, Morphology, and Regional Specialization of Astrocytes. Orlando, FL: Academic Press (2007). p. 105–29.
22. Hu X, Yuan Y, Wang D, Su Z. Heterogeneous astrocytes: active players in CNS. Brain Res Bull (2016) 125:1–18. doi:10.1016/j.brainresbull.2016.03.017
23. Matthias K, Kirchhoff F, Seifert G, Hüttmann K, Matyash M, Kettenmann H, et al. Segregated expression of AMPA-type glutamate receptors and glutamate transporters defines distinct astrocyte populations in the mouse hippocampus. J Neurosci (2003) 23(5):1750–8.
24. Degen J, Dublin P, Zhang J, Dobrowolski R, Jokwitz M, Karram K, et al. Dual reporter approaches for identification of Cre efficacy and astrocyte heterogeneity. FASEB J (2012) 26(11):4576–83. doi:10.1096/fj.12-207183
25. Rusnakova V, Honsa P, Dzamba D, Ståhlberg A, Kubista M, Anderova M. Heterogeneity of astrocytes: from development to injury—single cell gene expression. PLoS One (2013) 8(8):e69734. doi:10.1371/journal.pone.0069734
26. Höft S, Griemsmann S, Seifert G, Steinhäuser C. Heterogeneity in expression of functional ionotropic glutamate and GABA receptors in astrocytes across brain regions: insights from the thalamus. Philos Trans R Soc B Biol Sci (2014) 369(1654):20130602. doi:10.1098/rstb.2013.0602
27. Sosunov AA, Wu X, Tsankova NM, Guilfoyle E, McKhann GM, Goldman JE. Phenotypic heterogeneity and plasticity of isocortical and hippocampal astrocytes in the human brain. J Neurosci (2014) 34(6):2285–98. doi:10.1523/JNEUROSCI.4037-13.2014
28. John Lin CC, Yu K, Hatcher A, Huang TW, Lee HK, Carlson J, et al. Identification of diverse astrocyte populations and their malignant analogs. Nat Neurosci (2017) 20(3):396–405. doi:10.1038/nn.4493
29. Buosi AS, Matias I, Araujo APB, Batista C, Gomes FCA. Heterogeneity in synaptogenic profile of astrocytes from different brain regions. Mol Neurobiol (2017):1–12. doi:10.1007/s12035-016-0343-z
30. Ling EA, Penney D, Leblond CP. Use of carbon labeling to demonstrate the role of blood monocytes as precursors of the ‘ameboid cells’ present in the corpus callosum of postnatal rats. J Comp Neurol (1980) 193(3):631–57. doi:10.1002/cne.901930304
31. Dalmau I, Finsen B, Tønder N, Zimmer J, González B, Castellano B. Development of microglia in the prenatal rat hippocampus. J Comp Neurol (1997) 377(1):70–84. doi:10.1002/(SICI)1096-9861(19970106)377:1<70:AID-CNE7>3.0.CO;2-G
32. Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front Cell Neurosci (2013) 7:45. doi:10.3389/fncel.2013.00045
33. Delgado R, Carlin A, Airaghi L, Demitri MT, Meda L, Galimberti D, et al. Melanocortin peptides inhibit production of proinflammatory cytokines and nitric oxide by activated microglia. J Leukoc Biol (1998) 63(6):740–5.
34. Gonçalves J, Ribeiro CF, Malva JO, Silva AP. Protective role of neuropeptide Y Y2 receptors in cell death and microglial response following methamphetamine injury. Eur J Neurosci (2012) 36(9):3173–83. doi:10.1111/j.1460-9568.2012.08232.x
35. Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci (2009) 29(2):359–70. doi:10.1523/JNEUROSCI.2760-08.2009
36. Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest (2012) 122(1):153–62. doi:10.1172/JCI59660
37. Block ML, Hong JS. Chronic microglial activation and progressive dopaminergic neurotoxicity. Biochem Soc Trans (2007) 35(5):1127–32. doi:10.1042/BST0351127
38. Gertig U, Hanisch UK. Microglial diversity by responses and responders. Front Cell Neurosci (2014) 8:101. doi:10.3389/fncel.2014.00101
40. Batchelor PE, Liberatore GT, Wong JYF, Porritt MJ, Frerichs F, Donnan GA, et al. Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J Neurosci (1999) 19(5):1708–16.
41. Batchelor PE, Porritt MJ, Martinello P, Parish CL, Liberatore GT, Donnan GA, et al. Macrophages and microglia produce local trophic gradients that stimulate axonal sprouting toward but not beyond the wound edge. Mol Cell Neurosci (2002) 21(3):436–53. doi:10.1006/mcne.2002.1185
42. Zhong Y, Zhou LJ, Ren WJ, Xin WJ, Li YY, Zhang T, et al. The direction of synaptic plasticity mediated by C-fibers in spinal dorsal horn is decided by Src-family kinases in microglia: the role of tumor necrosis factor-α. Brain Behav Immun (2010) 24(6):874–80. doi:10.1016/j.bbi.2010.01.007
43. Elkabes S, DiCicco-Bloom EM, Black IB. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J Neurosci (1996) 16(8):2508–21.
44. Hashimoto R, Udagawa J, Kagohashi Y, Matsumoto A, Hatta T, Otani H. Direct and indirect effects of neuropeptide Y and neurotrophin 3 on myelination in the neonatal brains. Brain Res (2011) 1373:55–66. doi:10.1016/j.brainres.2010.12.027
45. Amaral AI, Meisingset TW, Kotter MR, Sonnewald U. Metabolic aspects of neuron-oligodendrocyte-astrocyte interactions. Front Endocrinol (2013) 4:54. doi:10.3389/fendo.2013.00054
46. Hashimoto R, Matsumoto A, Udagawa J, Hioki K, Otani H. Effect of leptin administration on myelination in ob/ob mouse cerebrum after birth. Neuroreport (2013) 24(1):22–9. doi:10.1097/WNR.0b013e32835ba875
47. Lee JY, Yune TY. Ghrelin inhibits oligodendrocyte cell death by attenuating microglial activation. Endocrinol Metab (Seoul) (2014) 29(3):371–8. doi:10.3803/EnM.2014.29.3.371
48. Peferoen L, Kipp M, Valk P, Noort JM, Amor S. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology (2014) 141(3):302–13. doi:10.1111/imm.12163
49. Djogo T, Robins SC, Schneider S, Kryzskaya D, Liu X, Mingay A, et al. Adult NG2-glia are required for median eminence-mediated leptin sensing and body weight control. Cell Metab (2016) 23(5):797–810. doi:10.1016/j.cmet.2016.04.013
50. Rodriguez EM, Blazquez JL, Pastor FE, Pelaez B, Peña P, Peruzzo B, et al. Hypothalamic tanycytes: a key component of brain–endocrine interaction. Int Rev Cytol (2005) 247:89–164. doi:10.1016/S0074-7696(05)47003-5
51. Ciofi P, Garret M, Lapirot O, Lafon P, Loyens A, Prevot V, et al. Brain–endocrine interactions: a microvascular route in the mediobasal hypothalamus. Endocrinology (2009) 150(12):5509–19. doi:10.1210/en.2009-0584
52. Miyata S. New aspects in fenestrated capillary and tissue dynamics in the sensory circumventricular organs of adult brains. Front Neurosci (2015) 9:390. doi:10.3389/fnins.2015.00390
53. Langlet F, Mullier A, Bouret SG, Prevot V, Dehouck B. Tanycyte-like cells form a blood–cerebrospinal fluid barrier in the circumventricular organs of the mouse brain. J Comp Neurol (2013) 521(15):3389–405. doi:10.1002/cne.23355
54. Langlet F, Levin BE, Luquet S, Mazzone M, Messina A, Dunn-Meynell AA, et al. Tanycytic VEGF-A boosts blood–hypothalamus barrier plasticity and access of metabolic signals to the arcuate nucleus in response to fasting. Cell Metab (2013) 17(4):607–17. doi:10.1016/j.cmet.2013.03.004
55. Langlet F. Tanycytes: a gateway to the metabolic hypothalamus. J Neuroendocrinol (2014) 26(11):753–60. doi:10.1111/jne.12191
56. Orellana JA, Sáez PJ, Cortés-Campos C, Elizondo RJ, Shoji KF, Contreras-Duarte S, et al. Glucose increases intracellular free Ca(2+) in tanycytes via ATP released through connexin 43 hemichannels. Glia (2012) 60(1):53–68. doi:10.1002/glia.21246
57. Rojczyk-Gołębiewska E, Pałasz A, Wiaderkiewicz R. Hypothalamic subependymal niche: a novel site of the adult neurogenesis. Cell Mol Neurobiol (2014) 34(5):631–42. doi:10.1007/s10571-014-0058-5
58. Nilaweera K, Herwig A, Bolborea M, Campbell G, Mayer CD, Morgan PJ, et al. Photoperiodic regulation of glycogen metabolism, glycolysis, and glutamine synthesis in tanycytes of the Siberian hamster suggests novel roles of tanycytes in hypothalamic function. Glia (2011) 59(11):1695–705. doi:10.1002/glia.21216
59. Frayling C, Britton R, Dale N. ATP-mediated glucosensing by hypothalamic tanycytes. J Physiol (2011) 589(9):2275–86. doi:10.1113/jphysiol.2010.202051
60. Bolborea M, Dale N. Hypothalamic tanycytes: potential roles in the control of feeding and energy balance. Trends Neurosci (2013) 36(2):91–100. doi:10.1016/j.tins.2012.12.008
61. Barrett P, Ebling FJP, Schuhler S, Wilson D, Ross AW, Warner A, et al. Hypothalamic thyroid hormone catabolism acts as a gatekeeper for the seasonal control of body weight and reproduction. Endocrinology (2007) 148(8):3608–17. doi:10.1210/en.2007-0316
62. Rouget C. Note sur le developpement de la tunique contractile des vaisseaux. C R Acad Sci (1874) 79:559–62.
63. Dore SE. On the contractility and nervous supply of the capillaries. Br J Dermatol (1923) 35(11):398–404. doi:10.1111/j.1365-2133.1923.tb09069.x
64. Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood–brain barrier. Nature (2010) 468(7323):557–61. doi:10.1038/nature09522
65. Dalvi S, On N, Nguyen H, Pogorzelec M, Miller DW, Hatch GM. The blood–brain-barrier: regulation of fatty acid and drug transport. In: Heinbockel DT, editor. Neurochemistry. InTech (2014). Available from: http://www.intechopen.com/books/neurochemistry/the-blood-brain-barrier-regulation-of-fatty-acid-and-drug-transport
66. Balabanov R, Washington R, Wagnerova J, Dore-Duffy P. CNS microvascular pericytes express macrophage-like function, cell surface integrin αM, and macrophage marker ED-2. Microvasc Res (1996) 52(2):127–42. doi:10.1006/mvre.1996.0049
67. Thomas WE. Brain macrophages: on the role of pericytes and perivascular cells. Brain Res Brain Res Rev (1999) 31(1):42–57. doi:10.1016/S0165-0173(99)00024-7
68. Guillemin GJ, Brew BJ. Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol (2004) 75(3):388–97. doi:10.1189/jlb.0303114
69. Watanabe S, Morisaki N, Tezuka M, Fukuda K, Ueda S, Koyama N, et al. Cultured retinal pericytes stimulate in vitro angiogenesis of endothelial cells through secretion of a fibroblast growth factor-like molecule. Atherosclerosis (1997) 130(1–2):101–7. doi:10.1016/S0021-9150(96)06050-9
70. Jansson D, Rustenhoven J, Feng S, Hurley D, Oldfield RL, Bergin PS, et al. A role for human brain pericytes in neuroinflammation. J Neuroinflammation (2014) 11:104–104. doi:10.1186/1742-2094-11-104
71. Özen I, Deierborg T, Miharada K, Padel T, Englund E, Genové G, et al. Brain pericytes acquire a microglial phenotype after stroke. Acta Neuropathol (2014) 128(3):381–96. doi:10.1007/s00401-014-1295-x
72. Dore-Duffy P, Owen C, Balabanov R, Murphy S, Beaumont T, Rafols JA. Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc Res (2000) 60(1):55–69. doi:10.1006/mvre.2000.2244
73. Göritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisén J. A pericyte origin of spinal cord scar tissue. Science (2011) 333(6039):238. doi:10.1126/science.1203165
74. Balabanov R, Beaumont T, Dore-Duffy P. Role of central nervous system microvascular pericytes in activation of antigen-primed splenic T-lymphocytes. J Neurosci Res (1999) 55(5):578–87. doi:10.1002/(SICI)1097-4547(19990301)55:5<578:AID-JNR5>3.0.CO;2-E
75. Kovac A, Erickson MA, Banks WA. Brain microvascular pericytes are immunoactive in culture: cytokine, chemokine, nitric oxide, and LRP-1 expression in response to lipopolysaccharide. J Neuroinflammation (2011) 8:139–139. doi:10.1186/1742-2094-8-139
76. Alcendor DJ, Charest AM, Zhu WQ, Vigil HE, Knobel SM. Infection and upregulation of proinflammatory cytokines in human brain vascular pericytes by human cytomegalovirus. J Neuroinflammation (2012) 9:95. doi:10.1186/1742-2094-9-95
77. Dore-Duffy P, Katychev A, Wang X, Van Buren E. CNS microvascular pericytes exhibit multipotential stem cell activity. J Cereb Blood Flow Metab (2006) 26(5):613–24. doi:10.1038/sj.jcbfm.9600272
78. Souza LE, Malta TM, Kashima Haddad S, Covas DT. Mesenchymal stem cells and pericytes: to what extent are they related? Stem Cells Dev (2016) 25(24):1843–52. doi:10.1089/scd.2016.0109
79. Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell (2011) 21(2):193–215. doi:10.1016/j.devcel.2011.07.001
80. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis (2010) 37(1):13–25. doi:10.1016/j.nbd.2009.07.030
81. Thouvenot E, Lafon-Cazal M, Demettre E, Jouin P, Bockaert J, Marin P. The proteomic analysis of mouse choroid plexus secretome reveals a high protein secretion capacity of choroidal epithelial cells. Proteomics (2006) 6(22):5941–52. doi:10.1002/pmic.200600096
82. Parada C, Gato A, Bueno D. All-trans retinol and retinol-binding protein from embryonic cerebrospinal fluid exhibit dynamic behaviour during early central nervous system development. Neuroreport (2008) 19(9):945–50. doi:10.1097/WNR.0b013e3283021c94
83. Lehtinen MK, Zappaterra MW, Chen X, Yang YJ, Hill A, Lun M, et al. The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron (2011) 69(5):893–905. doi:10.1016/j.neuron.2011.01.023
84. Marques F, Sousa JC, Coppola G, Gao F, Puga R, Brentani H, et al. Transcriptome signature of the adult mouse choroid plexus. Fluids Barriers CNS (2011) 8:10–10. doi:10.1186/2045-8118-8-10
85. Spector R. Micronutrient homeostasis in mammalian brain and cerebrospinal fluid. J Neurochem (1989) 53(6):1667–74. doi:10.1111/j.1471-4159.1989.tb09229.x
86. Spector R, Johanson CE. Micronutrient and urate transport in choroid plexus and kidney: implications for drug therapy. Pharm Res (2006) 23(11):2515–24. doi:10.1007/s11095-006-9091-5
87. Saunders NR, Dziegielewska KM, Møllgård K, Habgood MD, Wakefield MJ, Lindsay H, et al. Influx mechanisms in the embryonic and adult rat choroid plexus: a transcriptome study. Front Neurosci (2015) 9:123. doi:10.3389/fnins.2015.00123
88. Hong-Goka BC, Chang FF. Estrogen receptors α and β in choroid plexus epithelial cells in Alzheimer’s disease. Neurosci Lett (2004) 360(3):113–6. doi:10.1016/j.neulet.2004.01.075
89. Quadros PS, Pfau JL, Wagner CK. Distribution of progesterone receptor immunoreactivity in the fetal and neonatal rat forebrain. J Comp Neurol (2007) 504(1):42–56. doi:10.1002/cne.21427
90. Alves CH, Gonçalves I, Socorro S, Baltazar G, Quintela T, Santos CRA. Androgen receptor is expressed in murine choroid plexus and downregulated by 5α-dihydrotestosterone in male and female mice. J Mol Neurosci (2009) 38(1):41–9. doi:10.1007/s12031-008-9157-4
91. Mitchell SE, Nogueiras R, Morris A, Tovar S, Grant C, Cruickshank M, et al. Leptin receptor gene expression and number in the brain are regulated by leptin level and nutritional status. J Physiol (2009) 587(14):3573–85. doi:10.1113/jphysiol.2009.173328
92. Wolburg H, Paulus W. Choroid plexus: biology and pathology. Acta Neuropathol (2010) 119(1):75–88. doi:10.1007/s00401-009-0627-8
93. Del Bigio MR. Ependymal cells: biology and pathology. Acta Neuropathol (2010) 119(1):55–73. doi:10.1007/s00401-009-0624-y
94. Ohata S, Alvarez-Buylla A. Planar organization of multiciliated ependymal (E1) cells in the brain ventricular epithelium. Trends Neurosci (2016) 39(8):543–51. doi:10.1016/j.tins.2016.05.004
95. Mirzadeh Z, Merkle FT, Soriano-Navarro M, Garcia-Verdugo JM, Alvarez-Buylla A. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell (2008) 3(3):265–78. doi:10.1016/j.stem.2008.07.004
96. Johanson CE, Stopa EG, McMillan PN. The blood–cerebrospinal fluid barrier: structure and functional significance. In: Nag S, editor. The Blood–Brain and Other Neural Barriers: Reviews and Protocols. Totowa, NJ: Humana Press (2011). p. 101–31.
97. Brinker T, Stopa E, Morrison J, Klinge P. A new look at cerebrospinal fluid circulation. Fluids Barriers CNS (2014) 11:10–10. doi:10.1186/2045-8118-11-10
98. Mullier A, Bouret SG, Prevot V, Dehouck B. Differential distribution of tight junction proteins suggests a role for tanycytes in blood-hypothalamus barrier regulation in the adult mouse brain. J Comp Neurol (2010) 518(7):943–62. doi:10.1002/cne.22273
99. Alvarez JI, Katayama T, Prat A. Glial influence on the blood–brain barrier. Glia (2013) 61(12):1939–58. doi:10.1002/glia.22575
100. Hampl R, Bicikova M, Sosvorova L. Hormones and the blood–brain barrier. Horm Mol Biol Clin Investig (2015) 21(3):159–64. doi:10.1515/hmbci-2014-0042
101. Morgello S, Uson RR, Schwartz EJ, Haber RS. The human blood–brain barrier glucose transporter (GLUT1) is a glucose transporter of gray matter astrocytes. Glia (1995) 14(1):43–54. doi:10.1002/glia.440140107
102. Sivitz W, Desautel S, Walker PS, Pessin JE. Regulation of the glucose transporter in developing rat brain. Endocrinology (1989) 124(4):1875–80. doi:10.1210/endo-124-4-1875
103. Patching SG. Glucose transporters at the blood–brain barrier: function, regulation and gateways for drug delivery. Mol Neurobiol (2016) 54(2):1046–77. doi:10.1007/s12035-015-9672-6
104. Borg WP, Sherwin RS, During MJ, Borg MA, Shulman GI. Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes (1995) 44(2):180. doi:10.2337/diab.44.2.180
105. Borg MA, Sherwin RS, Borg WP, Tamborlane WV, Shulman GI. Local ventromedial hypothalamus glucose perfusion blocks counterregulation during systemic hypoglycemia in awake rats. J Clin Invest (1997) 99(2):361–5. doi:10.1172/JCI119165
106. Lam TK, Gutierrez-Juarez R, Pocai A, Rossetti L. Regulation of blood glucose by hypothalamic pyruvate metabolism. Science (2005) 309(5736):943. doi:10.1126/science.1112085
107. Tonon MC, Lanfray D, Castel H, Vaudry H, Morin F. Hypothalamic glucose-sensing: role of glia-to-neuron signaling. Horm Metab Res (2013) 45(13):955–9. doi:10.1055/s-0033-1355357
108. Routh VH, Hao L, Santiago AM, Sheng Z, Zhou C. Hypothalamic glucose sensing: making ends meet. Front Syst Neurosci (2014) 8:236. doi:10.3389/fnsys.2014.00236
109. Elizondo-Vega R, Cortes-Campos C, Barahona MJ, Oyarce KA, Carril CA, Garcia-Robles MA. The role of tanycytes in hypothalamic glucosensing. J Cell Mol Med (2015) 19(7):1471–82. doi:10.1111/jcmm.12590
110. Steinbusch L, Labouebe G, Thorens B. Brain glucose sensing in homeostatic and hedonic regulation. Trends Endocrinol Metab (2015) 26(9):455–66. doi:10.1016/j.tem.2015.06.005
111. Song Z, Levin BE, McArdle JJ, Bakhos N, Routh VH. Convergence of pre- and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes (2001) 50(12):2673–81. doi:10.2337/diabetes.50.12.2673
112. Leloup C, Allard C, Carneiro L, Fioramonti X, Collins S, Penicaud L. Glucose and hypothalamic astrocytes: more than a fueling role? Neuroscience (2016) 323:110–20. doi:10.1016/j.neuroscience.2015.06.007
113. Schuit FC, Huypens P, Heimberg H, Pipeleers DG. Glucose sensing in pancreatic β-cells: a model for the study of other glucose-regulated cells in gut, pancreas, and hypothalamus. Diabetes (2001) 50(1):1–11. doi:10.2337/diabetes.50.1.1
114. Leloup C, Orosco M, Serradas P, Nicolaïdis S, Pénicaud L. Specific inhibition of GLUT2 in arcuate nucleus by antisense oligonucleotides suppresses nervous control of insulin secretion. Brain Res Mol Brain Res (1998) 57(2):275–80. doi:10.1016/S0169-328X(98)00097-7
115. Marty N, Dallaporta M, Foretz M, Emery M, Tarussio D, Bady I, et al. Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J Clin Invest (2005) 115(12):3545–53. doi:10.1172/JCI26309
116. Harris AL. Connexin channel permeability to cytoplasmic molecules. Prog Biophys Mol Biol (2007) 94(1–2):120–43. doi:10.1016/j.pbiomolbio.2007.03.011
117. Lanfray D, Arthaud S, Ouellet J, Compere V, Do Rego JL, Leprince J, et al. Gliotransmission and brain glucose sensing: critical role of endozepines. Diabetes (2013) 62(3):801–10. doi:10.2337/db11-0785
118. Bouyakdan K, Taïb B, Budry L, Zhao S, Rodaros D, Neess D, et al. A novel role for central ACBP/DBI as a regulator of long-chain fatty acid metabolism in astrocytes. J Neurochem (2015) 133(2):253–65. doi:10.1111/jnc.13035
119. Fan X, Chan O, Ding Y, Zhu W, Mastaitis J, Sherwin R. Reduction in SGLT1 mRNA expression in the ventromedial hypothalamus improves the counterregulatory responses to hypoglycemia in recurrently hypoglycemic and diabetic rats. Diabetes (2015) 64(10):3564–72. doi:10.2337/db15-0022
120. García-Cáceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell (2016) 166(4):867–80. doi:10.1016/j.cell.2016.07.028
121. Fuente-Martin E, García-Cáceres C, Granado M, de Ceballos ML, Sanchez-Garrido MA, Sarman B, et al. Leptin regulates glutamate and glucose transporters in hypothalamic astrocytes. J Clin Invest (2012) 122(11):3900–13. doi:10.1172/JCI64102
122. Fuente-Martin E, García-Cáceres C, Argente-Arizon P, Diaz F, Granado M, Freire-Regatillo A, et al. Ghrelin regulates glucose and glutamate transporters in hypothalamic astrocytes. Sci Rep (2016) 6:23673. doi:10.1038/srep23673
123. Bouzier-Sore AK, Merle M, Magistretti PJ, Pellerin L. Feeding active neurons: (re)emergence of a nursing role for astrocytes. J Physiol Paris (2002) 96(3–4):273–82. doi:10.1016/S0928-4257(02)00016-5
124. Pellerin L. Lactate as a pivotal element in neuron–glia metabolic cooperation. Neurochem Int (2003) 43(4–5):331–8. doi:10.1016/S0197-0186(03)00020-2
125. Kokorovic A, Cheung GW, Rossetti L, Lam TK. Hypothalamic sensing of circulating lactate regulates glucose production. J Cell Mol Med (2009) 13(11–12):4403–8. doi:10.1111/j.1582-4934.2008.00596.x
126. Carneiro L, Geller S, Fioramonti X, Hebert A, Repond C, Leloup C, et al. Evidence for hypothalamic ketone body sensing: impact on food intake and peripheral metabolic responses in mice. Am J Physiol Endocrinol Metab (2016) 310(2):E103–15. doi:10.1152/ajpendo.00282.2015
127. Pierre K, Pellerin L. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem (2005) 94(1):1–14. doi:10.1111/j.1471-4159.2005.03168.x
128. Nalecz KA. Solute carriers in the blood–brain barrier: safety in abundance. Neurochem Res (2016). doi:10.1007/s11064-016-2030-x
129. Bergersen L, Waerhaug O, Helm J, Thomas M, Laake P, Davies AJ, et al. A novel postsynaptic density protein: the monocarboxylate transporter MCT2 is co-localized with d-glutamate receptors in postsynaptic densities of parallel fiber-Purkinje cell synapses. Exp Brain Res (2001) 136(4):523–34. doi:10.1007/s002210000600
130. Bergersen L, Rafiki A, Ottersen OP. Immunogold cytochemistry identifies specialized membrane domains for monocarboxylate transport in the central nervous system. Neurochem Res (2002) 27(1):89–96. doi:10.1023/A:1014806723147
131. Rafiki A, Boulland JL, Halestrap AP, Ottersen OP, Bergersen L. Highly differential expression of the monocarboxylate transporters MCT2 and MCT4 in the developing rat brain. Neuroscience (2003) 122(3):677–88. doi:10.1016/j.neuroscience.2003.08.040
132. Gerhart DZ, Enerson BE, Zhdankina OY, Leino RL, Drewes LR. Expression of monocarboxylate transporter MCT1 by brain endothelium and glia in adult and suckling rats. Am J Physiol Endocrinol Metab (1997) 273(1):E207.
133. Gerhart DZ, Enerson BE, Zhdankina OY, Leino RL, Drewes LR. Expression of the monocarboxylate transporter MCT2 by rat brain glia. Glia (1998) 22(3):272–81. doi:10.1002/(SICI)1098-1136(199803)22:3<272:AID-GLIA6>3.0.CO;2-7
134. Leino RL, Gerhart DZ, Drewes LR. Monocarboxylate transporter (MCT1) abundance in brains of suckling and adult rats: a quantitative electron microscopic immunogold study. Brain Res Dev Brain Res (1999) 113(1–2):47–54. doi:10.1016/S0165-3806(98)00188-6
135. Cortes-Campos C, Elizondo RJ, Carril CA, Martínez F, Boric K, Nualart F, et al. MCT2 expression and lactate influx in anorexigenic and orexigenic neurons of the arcuate nucleus. PLoS One (2013) 8(4):e62532. doi:10.1371/journal.pone.0062532
136. Le Foll C, Dunn-Meynell AA, Miziorko HM, Levin BE. Regulation of hypothalamic neuronal sensing and food intake by ketone bodies and fatty acids. Diabetes (2014) 63(4):1259–69. doi:10.2337/db13-1090
137. Le Foll C, Dunn-Meynell AA, Miziorko HM, Levin BE. Role of VMH ketone bodies in adjusting caloric intake to increased dietary fat content in DIO and DR rats. Am J Physiol Regul Integr Comp Physiol (2015) 308(10):R872–8. doi:10.1152/ajpregu.00015.2015
138. Lopez M, Tovar S, Vazquez MJ, Nogueiras R, Senaris R, Dieguez C. Sensing the fat: fatty acid metabolism in the hypothalamus and the melanocortin system. Peptides (2005) 26(10):1753–8. doi:10.1016/j.peptides.2004.11.025
139. Magnan C, Levin BE, Luquet S. Brain lipid sensing and the neural control of energy balance. Mol Cell Endocrinol (2015) 418(Pt 1):3–8. doi:10.1016/j.mce.2015.09.019
140. Cristiano L, Cimini A, Moreno S, Ragnelli AM, Paola Ceru M. Peroxisome proliferator-activated receptors (PPARs) and related transcription factors in differentiating astrocyte cultures. Neuroscience (2005) 131(3):577–87. doi:10.1016/j.neuroscience.2004.11.008
141. Heneka MT, Landreth GE. PPARs in the brain. Biochim Biophys Acta (2007) 1771(8):1031–45. doi:10.1016/j.bbalip.2007.04.016
142. Hofmann K, Lamberz C, Piotrowitz K, Offermann N, But D, Scheller A, et al. Tanycytes and a differential fatty acid metabolism in the hypothalamus. Glia (2016) 65(2):231–49. doi:10.1002/glia.23088
143. Kamp F, Guo W, Souto R, Pilch PF, Corkey BE, Hamilton JA. Rapid flip-flop of oleic acid across the plasma membrane of adipocytes. J Biol Chem (2003) 278(10):7988–95. doi:10.1074/jbc.M206648200
144. Levin VA. Relationship of octanol/water partition coefficient and molecular weight to rat brain capillary permeability. J Med Chem (1980) 23(6):682–4. doi:10.1021/jm00180a022
145. Mitchell RW, On NH, Del Bigio MR, Miller DW, Hatch GM. Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J Neurochem (2011) 117(4):735–46. doi:10.1111/j.1471-4159.2011.07245.x
146. Mitchell RW, Edmundson CL, Miller DW, Hatch GM. On the mechanism of oleate transport across human brain microvessel endothelial cells. J Neurochem (2009) 110(3):1049–57. doi:10.1111/j.1471-4159.2009.06199.x
147. Song BJ, Elbert A, Rahman T, Orr SK, Chen CT, Febbraio M, et al. Genetic ablation of CD36 does not alter mouse brain polyunsaturated fatty acid concentrations. Lipids (2010) 45(4):291–9. doi:10.1007/s11745-010-3398-z
148. Greenwalt DE, Mather IH. Characterization of an apically derived epithelial membrane glycoprotein from bovine milk, which is expressed in capillary endothelia in diverse tissues. J Cell Biol (1985) 100(2):397–408. doi:10.1083/jcb.100.2.397
149. Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC. Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia (2002) 40(2):195–205. doi:10.1002/glia.10148
150. Le Foll C, Irani BG, Magnan C, Dunn-Meynell AA, Levin BE. Characteristics and mechanisms of hypothalamic neuronal fatty acid sensing. Am J Physiol Regul Integr Comp Physiol (2009) 297(3):R655–64. doi:10.1152/ajpregu.00223.2009
151. Bao Y, Qin L, Kim E, Bhosle S, Guo H, Febbraio M, et al. CD36 is involved in astrocyte activation and astroglial scar formation. J Cereb Blood Flow Metab (2012) 32(8):1567–77. doi:10.1038/jcbfm.2012.52
152. Le Foll C, Dunn-Meynell A, Musatov S, Magnan C, Levin BE. FAT/CD36: a major regulator of neuronal fatty acid sensing and energy homeostasis in rats and mice. Diabetes (2013) 62(8):2709–16. doi:10.2337/db12-1689
153. Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM. Apolipoprotein E associated with astrocytic glia of the central nervous system and with non-myelinating glia of the peripheral nervous system. J Clin Invest (1985) 76:1501–13. doi:10.1172/JCI112130
154. Shen L, Tso P, Wang DQH, Woods SC, Davidson WS, Sakai R, et al. Up-regulation of apolipoprotein E by leptin in the hypothalamus of mice and rats. Physiol Behav (2009) 98(1–2):223–8. doi:10.1016/j.physbeh.2009.05.013
155. Roman C, Fuior EV, Trusca VG, Kardassis D, Simionescu M, Gafencu AV. Thyroid hormones upregulate apolipoprotein E gene expression in astrocytes. Biochem Biophys Res Commun (2015) 468(1–2):190–5. doi:10.1016/j.bbrc.2015.10.132
156. Shen L, Tso P, Woods SC, Clegg DJ, Barber KL, Carey K, et al. Brain apolipoprotein E: an important regulator of food intake in rats. Diabetes (2008) 57(8):2092–8. doi:10.2337/db08-0291
157. Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Invest (1996) 98(5):1101–6. doi:10.1172/JCI118891
158. Wang Q, Bing C, Al-Barazanji K, Mossakowaska DE, Wang X, McBay DL, et al. Interactions between leptin and hypothalamic neuropeptide Y neurons in the control of food intake and energy homeostasis in the rat. Diabetes (1997) 46(3):335–41. doi:10.2337/diab.46.3.335
159. Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature (2001) 411(6836):480–4. doi:10.1038/35078085
160. Kamohara S, Burcelin R, Halaas JL, Friedman JM, Charron MJ. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature (1997) 389(6649):374–7. doi:10.1038/38717
161. Frühbeck G, Aguado M, Gómez-Ambrosi J, Martínez JA. Lipolytic effect of in vivo leptin administration on adipocytes of lean and ob/ob mice, but not db/db mice. Biochem Biophys Res Commun (1998) 250(1):99–102. doi:10.1006/bbrc.1998.9277
162. Bjørbaek C, Elmquist JK, Michl P, Ahima RS, van Bueren A, McCall AL, et al. Expression of leptin receptor isoforms in rat brain microvessels. Endocrinology (1998) 139(8):3485–91. doi:10.1210/endo.139.8.6154
163. Pan W, Hsuchou H, Tu H, Kastin AJ. Developmental changes of leptin receptors in cerebral microvessels: unexpected relation to leptin transport. Endocrinology (2008) 149(3):877–85. doi:10.1210/en.2007-0893
164. Hsuchou H, He Y, Kastin AJ, Tu H, Markadakis EN, Rogers RC, et al. Obesity induces functional astrocytic leptin receptors in hypothalamus. Brain (2009) 132(4):889–902. doi:10.1093/brain/awp029
165. Balland E, Dam J, Langlet F, Caron E, Steculorum S, Messina A, et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab (2014) 19(2):293–301. doi:10.1016/j.cmet.2013.12.015
166. Hsuchou H, Kastin AJ, Tu H, Markadakis EN, Stone KP, Wang Y, et al. Effects of cell-type specific leptin receptor mutation on leptin transport across the BBB. Peptides (2011) 32(7):1392–9. doi:10.1016/j.peptides.2011.05.011
167. Gonzalez-Carter D, Goode AE, Fiammengo R, Dunlop IE, Dexter DT, Porter AE. Inhibition of leptin-ObR interaction does not prevent leptin translocation across a human blood–brain barrier model. J Neuroendocrinol (2016) 28(6). doi:10.1111/jne.12392
168. Zlokovic BV, Jovanovic S, Miao W, Samara S, Verma S, Farrell CL. Differential regulation of leptin transport by the choroid plexus and blood–brain barrier and high affinity transport systems for entry into hypothalamus and across the blood-cerebrospinal fluid barrier. Endocrinology (2000) 141(4):1434–41. doi:10.1210/endo.141.4.7435
169. Faouzi M, Leshan R, Björnholm M, Hennessey T, Jones J, Münzberg H. Differential accessibility of circulating leptin to individual hypothalamic sites. Endocrinology (2007) 148(11):5414–23. doi:10.1210/en.2007-0655
170. Baumann H, Morella KK, White DW, Dembski M, Bailon PS, Kim H, et al. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc Natl Acad Sci U S A (1996) 93(16):8374–8. doi:10.1073/pnas.93.16.8374
171. Munzberg H, Myers MGJ. Molecular and anatomical determinants of central leptin resistance. Nat Neurosci (2005) 8(5):566–70. doi:10.1038/nn1454
172. Kwon O, Kim KW, Kim M. Leptin signalling pathways in hypothalamic neurons. Cell Mol Life Sci (2016) 73(7):1457–77. doi:10.1007/s00018-016-2133-1
173. Kim JG, Suyama S, Koch M, Jin S, Argente-Arizon P, Argente J, et al. Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat Neurosci (2014) 17(7):908–10. doi:10.1038/nn.3725
174. Wang Y, Hsuchou H, He Y, Kastin AJ, Pan W. Role of astrocytes in leptin signaling. J Mol Neurosci (2015) 56(4):829–39. doi:10.1007/s12031-015-0518-5
175. Balland E, Cowley MA. New insights in leptin resistance mechanisms in mice. Front Neuroendocrinol (2015) 39:59–65. doi:10.1016/j.yfrne.2015.09.004
176. Knight ZA, Hannan KS, Greenberg ML, Friedman JM. Hyperleptinemia is required for the development of leptin resistance. PLoS One (2010) 5(6):e11376. doi:10.1371/journal.pone.0011376
177. de Git KC, Adan RA. Leptin resistance in diet-induced obesity: the role of hypothalamic inflammation. Obes Rev (2015) 16(3):207–24. doi:10.1111/obr.12243
178. Ottaway N, Mahbod P, Rivero B, Norman LA, Gertler A, D’Alessio DA, et al. Diet-induced obese mice retain endogenous leptin action. Cell Metab (2015) 21(6):877–82. doi:10.1016/j.cmet.2015.04.015
179. Flak JN, Myers MG Jr. CNS mechanisms of leptin action. Mol Endocrinol (2016) 30(1):3–12. doi:10.1210/me.2015-1232
180. Kastin AJ, Akerstrom V. Glucose and insulin increase the transport of leptin through the blood–brain barrier in normal mice but not in streptozotocin-diabetic mice. Neuroendocrinology (2001) 73(4):237–42. doi:10.1159/000054640
181. Banks WA, Coon AB, Robinson SM, Moinuddin A, Shultz JM, Nakaoke R, et al. Triglycerides induce leptin resistance at the blood–brain barrier. Diabetes (2004) 53(5):1253. doi:10.2337/diabetes.53.5.1253
182. Kastin AJ, Akerstrom V. Fasting, but not adrenalectomy, reduces transport of leptin into the brain. Peptides (2000) 21(5):679–82. doi:10.1016/S0196-9781(00)00195-9
183. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature (1999) 402(6762):656–60. doi:10.1038/45230
184. Horvath TL, Diano S, Sotonyi P, Heiman ML, Tschöp M. Minireview: ghrelin and the regulation of energy balance—a hypothalamic perspective. Endocrinology (2001) 142(10):4163–9. doi:10.1210/endo.142.10.8490
185. Hosoda H, Kojima M, Mizushima T, Shimizu S, Kangawa K. Structural divergence of human ghrelin: identification of multiple ghrelin-derived molecules produced by post-translational processing. J Biol Chem (2003) 278(1):64–70. doi:10.1074/jbc.M205366200
186. Gahete MD, Cordoba-Chacon J, Salvatori R, Castano JP, Kineman RD, Luque RM. Metabolic regulation of ghrelin O-acyl transferase (GOAT) expression in the mouse hypothalamus, pituitary, and stomach. Mol Cell Endocrinol (2010) 317(1–2):154–60. doi:10.1016/j.mce.2009.12.023
187. Staes E, Absil PA, Lins L, Brasseur R, Deleu M, Lecouturier N, et al. Acylated and unacylated ghrelin binding to membranes and to ghrelin receptor: towards a better understanding of the underlying mechanisms. Biochim Biophys Acta (2010) 1798(11):2102–13. doi:10.1016/j.bbamem.2010.07.002
188. Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol (2006) 494(3):528–48. doi:10.1002/cne.20823
189. Collden G, Balland E, Parkash J, Caron E, Langlet F, Prevot V, et al. Neonatal overnutrition causes early alterations in the central response to peripheral ghrelin. Mol Metab (2015) 4(1):15–24. doi:10.1016/j.molmet.2014.10.003
190. Banks WA, Tschop M, Robinson SM, Heiman ML. Extent and direction of ghrelin transport across the blood–brain barrier is determined by its unique primary structure. J Pharmacol Exp Ther (2002) 302(2):822–7. doi:10.1124/jpet.102.034827
191. Banks WA, Burney BO, Robinson SM. Effects of triglycerides, obesity, and starvation on ghrelin transport across the blood–brain barrier. Peptides (2008) 29(11):2061–5. doi:10.1016/j.peptides.2008.07.001
192. Briggs DI, Enriori PJ, Lemus MB, Cowley MA, Andrews ZB. Diet-induced obesity causes ghrelin resistance in arcuate NPY/AgRP neurons. Endocrinology (2010) 151(10):4745–55. doi:10.1210/en.2010-0556
193. Toshinai K, Yamaguchi H, Sun Y, Smith RG, Yamanaka A, Sakurai T, et al. Des-acyl ghrelin induces food intake by a mechanism independent of the growth hormone secretagogue receptor. Endocrinology (2006) 147(5):2306–14. doi:10.1210/en.2005-1357
194. Delhanty PJ, Neggers SJ, van der Lely AJ. Des-acyl ghrelin: a metabolically active peptide. Endocr Dev (2013) 25:112–21. doi:10.1159/000346059
195. García-Cáceres C, Fuente-Martin E, Diaz F, Granado M, Argente-Arizon P, Frago LM, et al. The opposing effects of ghrelin on hypothalamic and systemic inflammatory processes are modulated by its acylation status and food intake in male rats. Endocrinology (2014) 155(8):2868–80. doi:10.1210/en.2014-1074
196. Stevanovic DM, Grefhorst A, Themmen APN, Popovic V, Holstege J, Haasdijk E, et al. Unacylated ghrelin suppresses ghrelin-induced neuronal activity in the hypothalamus and brainstem of male rats. PLoS One (2014) 9(5):e98180. doi:10.1371/journal.pone.0098180
197. Begg DP, Woods SC. The central insulin system and energy balance. Handb Exp Pharmacol (2012) 209:111–29. doi:10.1007/978-3-642-24716-3_5
198. Banks WA, Jaspan JB, Huang W, Kastin AJ. Transport of insulin across the blood–brain barrier: saturability at euglycemic doses of insulin. Peptides (1997) 18(9):1423–9. doi:10.1016/S0196-9781(97)00231-3
199. Meijer RI, Gray S, Aylor K, Barrett EJ. Pathways for insulin access to the brain: the role of the microvascular endothelial cell. Am J Physiol Heart Circ Physiol (2016) 311(5):H1132–8. doi:10.1152/ajpheart.00081.2016
200. May AA, Liu M, Woods SC, Begg DP. CCK increases the transport of insulin into the brain. Physiol Behav (2016) 165:392–7. doi:10.1016/j.physbeh.2016.08.025
201. May AA, Bedel ND, Shen L, Woods SC, Liu M. Estrogen and insulin transport through the blood–brain barrier. Physiol Behav (2016) 163:312–21. doi:10.1016/j.physbeh.2016.05.019
202. Koch CE, Augustine RA, Steger J, Ganjam GK, Benzler J, Pracht C, et al. Leptin rapidly improves glucose homeostasis in obese mice by increasing hypothalamic insulin sensitivity. J Neurosci (2010) 30(48):16180–7. doi:10.1523/JNEUROSCI.3202-10.2010
203. Sartorius T, Heni M, Tschritter O, Preissl H, Hopp S, Fritsche A, et al. Leptin affects insulin action in astrocytes and impairs insulin-mediated physical activity. Cell Physiol Biochem (2012) 30(1):238–46. doi:10.1159/000339060
204. Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest (2005) 115(4):951–8. doi:10.1172/JCI200524301
205. Benoit SC, Kemp CJ, Elias CF, Abplanalp W, Herman JP, Migrenne S, et al. Palmitic acid mediates hypothalamic insulin resistance by altering PKC-theta subcellular localization in rodents. J Clin Invest (2009) 119(9):2577–89. doi:10.1172/JCI36714
206. Boden G, Cheung P, Stein TP, Kresge K, Mozzoli M. FFA cause hepatic insulin resistance by inhibiting insulin suppression of glycogenolysis. Am J Physiol Endocrinol Metab (2002) 283(1):E12. doi:10.1152/ajpendo.00429.2001
207. Pardridge WM. New approaches to drug delivery through the blood–brain barrier. Trends Biotechnol (1994) 12(6):239–45. doi:10.1016/0167-7799(94)90123-6
208. Baulieu EE, Robel P. Neurosteroids: a new brain function? J Steroid Biochem Mol Biol (1990) 37(3):395–403. doi:10.1016/0960-0760(90)90490-C
209. Reddy DS. Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog Brain Res (2010) 186:113–37. doi:10.1016/B978-0-444-53630-3.00008-7
210. Jung-Testas I, Baulieu EE. Steroid hormone receptors and steroid action in rat glial cells of the central and peripheral nervous system. J Steroid Biochem Mol Biol (1998) 65(1–6):243–51. doi:10.1016/S0960-0760(97)00191-X
211. Mensah-Nyagan AG, Do-Rego JL, Beaujean D, Luu-The V, Pelletier G, Vaudry H. Neurosteroids: expression of steroidogenic enzymes and regulation of steroid biosynthesis in the central nervous system. Pharmacol Rev (1999) 51(1):63.
212. Agís-Balboa RC, Pinna G, Zhubi A, Maloku E, Veldic M, Costa E, et al. Characterization of brain neurons that express enzymes mediating neurosteroid biosynthesis. Proc Natl Acad Sci U S A (2006) 103(39):14602–7. doi:10.1073/pnas.0606544103
213. Mouriec K, Gueguen MM, Manuel C, Percevault F, Thieulant ML, Pakdel F, et al. Androgens upregulate cyp19a1b (aromatase b) gene expression in the brain of zebrafish (Danio rerio) through estrogen receptors. Biol Reprod (2009) 80(5):889–96. doi:10.1095/biolreprod.108.073643
214. Chen C, Kuo J, Wong A, Micevych P. Estradiol modulates translocator protein (TSPO) and steroid acute regulatory protein (StAR) via protein kinase a (PKA) signaling in hypothalamic astrocytes. Endocrinology (2014) 155(8):2976–85. doi:10.1210/en.2013-1844
215. Wicher G, Norlin M. Estrogen-mediated regulation of steroid metabolism in rat glial cells; effects on neurosteroid levels via regulation of CYP7B1-mediated catalysis. J Steroid Biochem Mol Biol (2015) 145:21–7. doi:10.1016/j.jsbmb.2014.09.022
216. Xing L, Esau C, Trudeau VL. Direct regulation of aromatase B expression by 17beta-estradiol and dopamine D1 receptor agonist in adult radial glial cells. Front Neurosci (2015) 9:504. doi:10.3389/fnins.2015.00504
217. Yilmaz MB, Zhao H, Brooks DC, Fenkci IV, Imir-Yenicesu G, Attar E, et al. Estrogen receptor alpha (Esr1) regulates aromatase (Cyp19a1) expression in the mouse brain. Neuro Endocrinol Lett (2015) 36(2):178–82.
218. Jung-Testas I, Renoir JM, Gasc JM, Baulieu EE. Estrogen-inducible progesterone receptor in primary cultures of rat glial cells. Exp Cell Res (1991) 193(1):12–9. doi:10.1016/0014-4827(91)90532-Y
219. Liu X, Shi H. Regulation of estrogen receptor alpha expression in the hypothalamus by sex steroids: implication in the regulation of energy homeostasis. Int J Endocrinol (2015) 2015:949085. doi:10.1155/2015/949085
220. Wise PM. Estradiol exerts neuroprotective actions against ischemic brain injury: insights derived from animal models. Endocrine (2003) 21(1):11–5. doi:10.1385/ENDO:21:1:11
221. Vegeto E, Benedusi V, Maggi A. Estrogen anti-inflammatory activity in brain: a therapeutic opportunity for menopause and neurodegenerative diseases. Front Neuroendocrinol (2008) 29(4):507–19. doi:10.1016/j.yfrne.2008.04.001
222. Gold SM, Voskuhl RR. Estrogen and testosterone therapies in multiple sclerosis. Prog Brain Res (2009) 175:239–51. doi:10.1016/S0079-6123(09)17516-7
223. Sarkaki AR, Khaksari Haddad M, Soltani Z, Shahrokhi N, Mahmoodi M. Time- and dose-dependent neuroprotective effects of sex steroid hormones on inflammatory cytokines after a traumatic brain injury. J Neurotrauma (2011) 30(1):47–54. doi:10.1089/neu.2010.1686
224. Day NL, Floyd CL, D’Alessandro TL, Hubbard WJ, Chaudry IH. 17β-Estradiol confers protection after traumatic brain injury in the rat and involves activation of G protein-coupled estrogen receptor 1. J Neurotrauma (2013) 30(17):1531–41. doi:10.1089/neu.2013.2854
225. De Nicola AF, Gonzalez-Deniselle MC, Garay L, Meyer M, Gargiulo-Monachelli G, Guennoun R, et al. Progesterone protective effects in neurodegeneration and neuroinflammation. J Neuroendocrinol (2013) 25(11):1095–103. doi:10.1111/jne.12043
226. Brotfain E, Gruenbaum SE, Boyko M, Kutz R, Zlotnik A, Klein M. Neuroprotection by estrogen and progesterone in traumatic brain injury and spinal cord injury. Curr Neuropharmacol (2016) 14(6):641–53. doi:10.2174/1570159X14666160309123554
227. Rahmani B, Ghasemi R, Dargahi L, Ahmadiani A, Haeri A. Neurosteroids; potential underpinning roles in maintaining homeostasis. Gen Comp Endocrinol (2016) 225:242–50. doi:10.1016/j.ygcen.2015.09.030
228. García-Segura LM, Wozniak A, Azcoitia I, Rodriguez JR, Hutchison RE, Hutchison JB. Aromatase expression by astrocytes after brain injury: implications for local estrogen formation in brain repair. Neuroscience (1999) 89(2):567–78. doi:10.1016/S0306-4522(98)00340-6
229. Azcoitia I, Sierra A, Veiga S, García-Segura LM. Aromatase expression by reactive astroglia is neuroprotective. Ann N Y Acad Sci (2003) 1007:298–305. doi:10.1196/annals.1286.028
230. Saldanha CJ, Duncan KA, Walters BJ. Neuroprotective actions of brain aromatase. Front Neuroendocrinol (2009) 30(2):106–18. doi:10.1016/j.yfrne.2009.04.016
231. Johann S, Beyer C. Neuroprotection by gonadal steroid hormones in acute brain damage requires cooperation with astroglia and microglia. J Steroid Biochem Mol Biol (2013) 137:71–81. doi:10.1016/j.jsbmb.2012.11.006
232. Kipp M, Karakaya S, Johann S, Kampmann E, Mey J, Beyer C. Oestrogen and progesterone reduce lipopolysaccharide-induced expression of tumour necrosis factor-α and interleukin-18 in midbrain astrocytes. J Neuroendocrinol (2007) 19(10):819–22. doi:10.1111/j.1365-2826.2007.01588.x
233. Lewis DK, Johnson AB, Stohlgren S, Simpson A, Sohrabji F. Effects of estrogen receptor agonists on regulation of the inflammatory response in astrocytes from young adult and middle-aged female rats. J Neuroimmunol (2008) 195(1–2):47–59. doi:10.1016/j.jneuroim.2008.01.006
234. Cerciat M, Unkila M, Garcia-Segura LM, Arevalo MA. Selective estrogen receptor modulators decrease the production of interleukin-6 and interferon-γ-inducible protein-10 by astrocytes exposed to inflammatory challenge in vitro. Glia (2010) 58(1):93–102. doi:10.1002/glia.20904
235. Bruce-Keller AJ, Keeling JL, Keller JN, Huang FF, Camondola S, Mattson MP. Antiinflammatory effects of estrogen on microglial activation. Endocrinology (2000) 141(10):3646–56. doi:10.1210/endo.141.10.7693
236. Drew PD, Chavis JA. Female sex steroids: effects upon microglial cell activation. J Neuroimmunol (2000) 111(1–2):77–85. doi:10.1016/S0165-5728(00)00386-6
237. Dimayuga FO, Reed JL, Carnero GA, Wang C, Dimayuga ER, Dimayuga VM, et al. Estrogen and brain inflammation: effects on microglial expression of MHC, costimulatory molecules and cytokines. J Neuroimmunol (2005) 161(1–2):123–36. doi:10.1016/j.jneuroim.2004.12.016
238. Vegeto E, Belcredito S, Ghisletti S, Meda C, Etteri S, Maggi A. The endogenous estrogen status regulates microglia reactivity in animal models of neuroinflammation. Endocrinology (2006) 147(5):2263–72. doi:10.1210/en.2005-1330
239. Czaja JA, Goy RW. Ovarian hormones and food intake in female guinea pigs and rhesus monkeys. Horm Behav (1975) 6(4):329–49. doi:10.1016/0018-506X(75)90003-3
240. Butera PC, Czaja JA. Intracranial estradiol in ovariectomized guinea pigs: effects on ingestive behaviors and body weight. Brain Res (1984) 322(1):41–8. doi:10.1016/0006-8993(84)91178-8
241. Gao Q, Mezei G, Nie Y, Rao Y, Choi CS, Bechmann I, et al. Anorectic estrogen mimics leptin’s effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat Med (2007) 13(1):89–94. doi:10.1038/nm1525
242. Musatov S, Chen W, Pfaff DW, Mobbs CV, Yang XJ, Clegg DJ, et al. Silencing of estrogen receptor α in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proc Natl Acad Sci U S A (2007) 104(7):2501–6. doi:10.1073/pnas.0610787104
243. Clegg DJ, Brown LM, Woods SC, Benoit SC. Gonadal hormones determine sensitivity to central leptin and insulin. Diabetes (2006) 55(4):978–87. doi:10.2337/diabetes.55.04.06.db05-1339
244. Clegg DJ, Brown LM, Zigman JM, Kemp CJ, Strader AD, Benoit SC, et al. Estradiol-dependent decrease in the orexigenic potency of ghrelin in female rats. Diabetes (2007) 56(4):1051–8. doi:10.2337/db06-0015
245. Xu Y, Nedungadi TP, Zhu L, Sobhani N, Irani BG, Davis KE, et al. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab (2011) 14(4):453–65. doi:10.1016/j.cmet.2011.08.009
246. Smith AW, Ronnekleiv OK, Kelly MJ. Gq-mER signaling has opposite effects on hypothalamic orexigenic and anorexigenic neurons. Steroids (2014) 81:31–5. doi:10.1016/j.steroids.2013.11.007
247. Geary N, Asarian L, Korach KS, Pfaff DW, Ogawa S. Deficits in E2-dependent control of feeding, weight gain, and cholecystokinin satiation in ER-α null mice. Endocrinology (2001) 142(11):4751–7. doi:10.1210/endo.142.11.8504
248. Asarian L, Geary N. Cyclic estradiol treatment normalizes body weight and restores physiological patterns of spontaneous feeding and sexual receptivity in ovariectomized rats. Horm Behav (2002) 42(4):461–71. doi:10.1006/hbeh.2002.1835
249. Asarian L, Geary N. Modulation of appetite by gonadal steroid hormones. Philos Trans R Soc Lond B Biol Sci (2006) 361(1471):1251–63. doi:10.1098/rstb.2006.1860
250. Liang YQ, Akishita M, Kim S, Ako J, Hashimoto M, Iijima K, et al. Estrogen receptor beta is involved in the anorectic action of estrogen. Int J Obes Relat Metab Disord (2002) 26(8):1103–9. doi:10.1038/sj.ijo.0802054
251. Roesch DM. Effects of selective estrogen receptor agonists on food intake and body weight gain in rats. Physiol Behav (2006) 87(1):39–44. doi:10.1016/j.physbeh.2005.08.035
252. Santollo J, Katzenellenbogen BS, Katzenellenbogen JA, Eckel LA. Activation of ERα is necessary for estradiol’s anorexigenic effect in female rats. Horm Behav (2010) 58(5):872–7. doi:10.1016/j.yhbeh.2010.08.012
253. Qiu J, Bosch MA, Tobias SC, Krust A, Graham SM, Murphy SJ, et al. A G-protein-coupled estrogen receptor is involved in hypothalamic control of energy homeostasis. J Neurosci (2006) 26(21):5649–55. doi:10.1523/JNEUROSCI.0327-06.2006
254. Roepke TA, Bosch MA, Rick EA, Lee B, Wagner EJ, Seidlova-Wuttke D, et al. Contribution of a membrane estrogen receptor to the estrogenic regulation of body temperature and energy homeostasis. Endocrinology (2010) 151(10):4926–37. doi:10.1210/en.2010-0573
255. Filardo EJ, Quinn JA, Bland KI, Frackelton AR. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol (2000) 14(10):1649–60. doi:10.1210/mend.14.10.0532
256. Davis KE, Carstens EJ, Irani BG, Gent LM, Hahner LM, Clegg DJ. Sexually dimorphic role of G protein-coupled estrogen receptor (GPER) in modulating energy homeostasis. Horm Behav (2014) 66(1):196–207. doi:10.1016/j.yhbeh.2014.02.004
257. Magnus-Levy A. Uber den respiratorischen Gaswechsel unter Einfluss der Thyroidea sowie unter verschiedenen pathologische Zustand. Berlin Klin Wochschr (1895) 32:650–2.
258. Sjögren M, Alkemade A, Mittag J, Nordström K, Katz A, Rozell B, et al. Hypermetabolism in mice caused by the central action of an unliganded thyroid hormone receptor α1. EMBO J (2007) 26(21):4535–45. doi:10.1038/sj.emboj.7601882
259. Lopez M, Varela L, Vazquez MJ, Rodriguez-Cuenca S, Gonzalez CR, Velagapudi VR, et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat Med (2010) 16(9):1001–8. doi:10.1038/nm.2207
260. De Leo S, Lee SY, Braverman LE. Hyperthyroidism. Lancet (2016) 388(10047):906–18. doi:10.1016/S0140-6736(16)00278-6
261. Ingbar SH. The thyroid gland. In: Wilson JD, Foster DW, editors. Williams Textbook of Endocrinology. Philadelphia, PA: W. B. Saunders Company (1985). p. 975–1170.
262. Lima FRS, Gervais A, Colin C, Izembart M, Neto VM, Mallat M. Regulation of microglial development: a novel role for thyroid hormone. J Neurosci (2001) 21(6):2028–38.
263. Nam SM, Kim YN, Yoo DY, Yi SS, Choi JH, Hwang IK, et al. Hypothyroidism affects astrocyte and microglial morphology in type 2 diabetes. Neural Regen Res (2013) 8(26):2458–67. doi:10.3969/j.issn.1673-5374.2013.26.007
264. Xu M, Iwasaki T, Shimokawa N, Sajdel-Sulkowska EM, Koibuchi N. The effect of low dose lipopolysaccharide on thyroid hormone-regulated actin cytoskeleton modulation and type 2 iodothyronine deiodinase activity in astrocytes. Endocr J (2013) 60(11):1221–30. doi:10.1507/endocrj.EJ13-0294
265. Klieverik LP, Janssen SF, van Riel A, Foppen E, Bisschop PH, Serlie MJ, et al. Thyroid hormone modulates glucose production via a sympathetic pathway from the hypothalamic paraventricular nucleus to the liver. Proc Natl Acad Sci U S A (2009) 106(14):5966–71. doi:10.1073/pnas.0805355106
266. Fliers E, Klieverik LP, Kalsbeek A. Novel neural pathways for metabolic effects of thyroid hormone. Trends Endocrinol Metab (2010) 21(4):230–6. doi:10.1016/j.tem.2009.11.008
267. Leonard JL, Koehrle J. Intracellular pathways of iodothyronine metabolism. In: Braverman LE, Utiger RD, editors. The Thyroid. Philadelphia: Lippincott Williams and Wilkins (2000). p. 136–73.
268. Bianco AC, Kim BW. Deiodinases: implications of the local control of thyroid hormone action. J Clin Invest (2006) 116(10):2571–9. doi:10.1172/JCI29812
269. Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev (2002) 23(1):38–89. doi:10.1210/edrv.23.1.0455
270. Senese R, Cioffi F, de Lange P, Goglia F, Lanni A. Thyroid: biological actions of ‘nonclassical’ thyroid hormones. J Endocrinol (2014) 221(2):R1–12. doi:10.1530/joe-13-0573
271. Friesema ECH, Ganguly S, Abdalla A, Fox JEM, Halestrap AP, Visser TJ. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem (2003) 278(41):40128–35. doi:10.1074/jbc.M300909200
272. Pizzagalli F, Hagenbuch B, Stieger B, Klenk U, Folkers G, Meier PJ. Identification of a novel human organic anion transporting polypeptide as a high affinity thyroxine transporter. Mol Endocrinol (2002) 16(10):2283–96. doi:10.1210/me.2001-0309
273. Mayerl S, Müller J, Bauer R, Richert S, Kassmann CM, Darras VM, et al. Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J Clin Invest (2014) 124(5):1987–99. doi:10.1172/JCI70324
274. Roberts LM, Woodford K, Zhou M, Black DS, Haggerty JE, Tate EH, et al. Expression of the thyroid hormone transporters monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood–brain barrier. Endocrinology (2008) 149(12):6251–61. doi:10.1210/en.2008-0378
275. Heuer H, Maier MK, Iden S, Mittag J, Friesema ECH, Visser TJ, et al. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology (2005) 146(4):1701–6. doi:10.1210/en.2004-1179
276. Alkemade A, Friesema ECH, Kalsbeek A, Swaab DF, Visser TJ, Fliers E. Expression of thyroid hormone transporters in the human hypothalamus. J Clin Endocrinol Metab (2011) 96(6):E967–71. doi:10.1210/jc.2010-2750
277. Müller J, Heuer H. Expression pattern of thyroid hormone transporters in the postnatal mouse brain. Front Endocrino (2014) 5:92. doi:10.3389/fendo.2014.00092
278. Tu HM, Kim SW, Salvatore D, Bartha T, Legradi G, Larsen PR, et al. Regional distribution of type 2 thyroxine deiodinase messenger ribonucleic acid in rat hypothalamus and pituitary and its regulation by thyroid hormone. Endocrinology (1997) 138(8):3359–68. doi:10.1210/endo.138.8.5318
279. Diano S, Naftolin F, Goglia F, Csernus V, Horvath TL. Monosynaptic pathway between the arcuate nucleus expressing glial type II iodothyronine 5′-deiodinase mRNA and the median eminence-projective TRH cells of the rat paraventricular nucleus. J Neuroendocrinol (1998) 10(10):731–42. doi:10.1046/j.1365-2826.1998.00204.x
280. Alkemade A, Friesema EC, Unmehopa UA, Fabriek BO, Kuiper GG, Leonard JL, et al. Neuroanatomical pathways for thyroid hormone feedback in the human hypothalamus. J Clin Endocrinol Metab (2005) 90(7):4322–34. doi:10.1210/jc.2004-2567
281. Martinez-Sanchez N, Alvarez CV, Ferno J, Nogueiras R, Dieguez C, Lopez M. Hypothalamic effects of thyroid hormones on metabolism. Best Pract Res Clin Endocrinol Metab (2014) 28(5):703–12. doi:10.1016/j.beem.2014.04.004
282. Coppola A, Meli R, Diano S. Inverse shift in circulating corticosterone and leptin levels elevates hypothalamic deiodinase type 2 in fasted rats. Endocrinology (2005) 146(6):2827–33. doi:10.1210/en.2004-1361
283. Coppola A, Liu ZW, Andrews Z, Paradis E, Roy MC, Friedman JM, et al. A central thermogenic-like mechanism in feeding regulation: an interplay between arcuate nucleus T3 and UCP2. Cell Metab (2007) 5(1):21–33. doi:10.1016/j.cmet.2006.12.002
284. Varela L, Martínez-Sánchez N, Gallego R, Vázquez MJ, Roa J, Gándara M, et al. Hypothalamic mTOR pathway mediates thyroid hormone-induced hyperphagia in hyperthyroidism. J Pathol (2012) 227(2):209–22. doi:10.1002/path.3984
285. Ebling FJP. Hypothalamic control of seasonal changes in food intake and body weight. Front Neuroendocrinol (2015) 37:97–107. doi:10.1016/j.yfrne.2014.10.003
286. Shearer KD, Goodman TH, Ross AW, Reilly L, Morgan PJ, McCaffery PJ. Photoperiodic regulation of retinoic acid signaling in the hypothalamus. J Neurochem (2010) 112(1):246–57. doi:10.1111/j.1471-4159.2009.06455.x
287. Helfer G, Ross AW, Russell L, Thomson LM, Shearer KD, Goodman TH, et al. Photoperiod regulates vitamin A and Wnt/β-catenin signaling in F344 rats. Endocrinology (2012) 153(2):815–24. doi:10.1210/en.2011-1792
288. Brent GA. Mechanisms of thyroid hormone action. J Clin Invest (2012) 122(9):3035–43. doi:10.1172/JCI60047
289. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev (2010) 31(2):139–70. doi:10.1210/er.2009-0007
290. Davis PJ, Davis FB. Nongenomic actions of thyroid hormone. Thyroid (1996) 6(5):497–504. doi:10.1089/thy.1996.6.497
291. Kalyanaraman H, Schwappacher R, Joshua J, Zhuang S, Scott BT, Klos M, et al. Nongenomic thyroid hormone signaling occurs through a plasma membrane-localized receptor. Sci Signal (2014) 7(326):ra48. doi:10.1126/scisignal.2004911
292. Hiroi Y, Kim HH, Ying H, Furuya F, Huang Z, Simoncini T, et al. Rapid nongenomic actions of thyroid hormone. Proc Natl Acad Sci U S A (2006) 103(38):14104–9. doi:10.1073/pnas.0601600103
293. Losi G, Garzon G, Puia G. Nongenomic regulation of glutamatergic neurotransmission in hippocampus by thyroid hormones. Neuroscience (2008) 151(1):155–63. doi:10.1016/j.neuroscience.2007.09.064
294. Caria MA, Dratman MB, Kow LM, Mameli O, Pavlides C. Thyroid hormone action: nongenomic modulation of neuronal excitability in the hippocampus. J Neuroendocrinol (2009) 21(2):98–107. doi:10.1111/j.1365-2826.2008.01813.x
295. Sanchez E, Vargas MA, Singru PS, Pascual I, Romero F, Fekete C, et al. Tanycyte pyroglutamyl peptidase II contributes to regulation of the hypothalamic-pituitary-thyroid axis through glial-axonal associations in the median eminence. Endocrinology (2009) 150(5):2283–91. doi:10.1210/en.2008-1643
296. Légrádi G, Emerson CH, Ahima RS, Flier JS, Lechan RM. Leptin prevents fasting-induced suppression of prothyrotropin-releasing hormone messenger ribonucleic acid in neurons of the hypothalamic paraventricular nucleus. Endocrinology (1997) 138(6):2569–76. doi:10.1210/endo.138.6.5209
297. Seoane LM, Carro E, Tovar S, Casanueva FF, Dieguez C. Regulation of in vivo TSH secretion by leptin. Regul Pept (2000) 92(1–3):25–9. doi:10.1016/S0167-0115(00)00145-2
298. Ortiga-Carvalho TM, Oliveira KJ, Soares BA, Pazos-Moura CC. The role of leptin in the regulation of TSH secretion in the fed state: in vivo and in vitro studies. J Endocrinol (2002) 174(1):121–5. doi:10.1677/joe.0.1740121
299. Ahlquist JA, Franklyn JA, Wood DF, Balfour NJ, Docherty K, Sheppard MC, et al. Hormonal regulation of thyrotrophin synthesis and secretion. Horm Metab Res Suppl (1987) 17:86–9.
300. Franklyn JA, Wood DF, Balfour NJ, Ramsden DB, Docherty K, Sheppard MC. Modulation by oestrogen of thyroid hormone effects on thyrotrophin gene expression. J Endocrinol (1987) 115(1):53–9. doi:10.1677/joe.0.1150053
301. Fitts JM, Klein RM, Powers CA. Comparison of tamoxifen effects on the actions of triiodothyronine or growth hormone in the ovariectomized-hypothyroid rat. J Pharmacol Exp Ther (1998) 286(1):392–402.
302. Wyss MT, Jolivet R, Buck A, Magistretti PJ, Weber B. In vivo evidence for lactate as a neuronal energy source. J Neurosci (2011) 31(20):7477–85. doi:10.1523/jneurosci.0415-11.2011
303. Bouzier-Sore AK, Pellerin L. Unraveling the complex metabolic nature of astrocytes. Front Cell Neurosci (2013) 7:179. doi:10.3389/fncel.2013.00179
304. Cruz NF, Dienel GA. High glycogen levels in brains of rats with minimal environmental stimuli: implications for metabolic contributions of working astrocytes. J Cereb Blood Flow Metab (2002) 22(12):1476–89. doi:10.1097/01.wcb.0000034362.37277.c0
305. Heni M, Hennige AM, Peter A, Siegel-Axel D, Ordelheide AM, Krebs N, et al. Insulin promotes glycogen storage and cell proliferation in primary human astrocytes. PLoS One (2011) 6(6):e21594. doi:10.1371/journal.pone.0021594
306. Muhič M, Vardjan N, Chowdhury HH, Zorec R, Kreft M. Insulin and insulin-like growth factor 1 (IGF-1) modulate cytoplasmic glucose and glycogen levels but not glucose transport across the membrane in astrocytes. J Biol Chem (2015) 290(17):11167–76. doi:10.1074/jbc.M114.629063
307. Bosier B, Bellocchio L, Metna-Laurent M, Soria-Gomez E, Matias I, Hebert-Chatelain E, et al. Astroglial CB(1) cannabinoid receptors regulate leptin signaling in mouse brain astrocytes. Mol Metab (2013) 2(4):393–404. doi:10.1016/j.molmet.2013.08.001
308. Golovko MY, Murphy EJ. Uptake and metabolism of plasma-derived erucic acid by rat brain. J Lipid Res (2006) 47(6):1289–97. doi:10.1194/jlr.M600029-JLR200
309. Murphy EJ. Blood–brain barrier and brain fatty acid uptake: role of arachidonic acid and PGE2. J Neurochem (2015) 135(5):845–8. doi:10.1111/jnc.13289
310. Rosenberger TA, Villacreses NE, Hovda JT, Bosetti F, Weerasinghe G, Wine RN, et al. Rat brain arachidonic acid metabolism is increased by a 6-day intracerebral ventricular infusion of bacterial lipopolysaccharide. J Neurochem (2004) 88(5):1168–78. doi:10.1046/j.1471-4159.2003.02246.x
311. Golovko MY, Faergeman NJ, Cole NB, Castagnet PI, Nussbaum RL, Murphy EJ. Alpha-synuclein gene deletion decreases brain palmitate uptake and alters the palmitate metabolism in the absence of α-synuclein palmitate binding. Biochemistry (2005) 44(23):8251–9. doi:10.1021/bi0502137
312. Shirai K, Saito Y, Yoshida SHO, Matsuoka N. Existence of lipoprotein lipase in rat brain microvessels. Tohoku J Exp Med (1986) 149(4):449–50. doi:10.1620/tjem.149.449
313. Vilaró S, Camps L, Reina M, Perez-Clausell J, Llobera M, Olivecrona T. Localization of lipoprotein lipase to discrete areas of the guinea pig brain. Brain Res (1990) 506(2):249–53. doi:10.1016/0006-8993(90)91258-I
314. Spector AA. Plasma free fatty acid and lipoproteins as sources of polyunsaturated fatty acid for the brain. J Mol Neurosci (2001) 16(2):159–65. doi:10.1385/JMN:16:2-3:159
315. Hamilton JA, Brunaldi K. A model for fatty acid transport into the brain. J Mol Neurosci (2007) 33:12–7. doi:10.1007/s12031-007-0050-3
316. Wang H, Eckel RH. Lipoprotein lipase in the brain and nervous system. Annu Rev Nutr (2012) 32:147–60. doi:10.1146/annurev-nutr-071811-150703
317. Edmond J, Robbins RA, Bergstrom JD, Cole RA, de Vellis J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J Neurosci Res (1987) 18(4):551–61. doi:10.1002/jnr.490180407
318. Le Foll C, Levin BE. Fatty acid-induced astrocyte ketone production and the control of food intake. Am J Physiol Regul Integr Comp Physiol (2016) 310(11):R1186–92. doi:10.1152/ajpregu.00113.2016
319. Edmond J, Higa TA, Korsak RA, Bergner EA, Lee WNP. Fatty acid transport and utilization for the developing brain. J Neurochem (1998) 70(3):1227–34. doi:10.1046/j.1471-4159.1998.70031227.x
320. Shah SN. Cytosolic 3-hydroxy-3-methyl glutaryl coenzyme a synthase in rat brain: properties and developmental change. Neurochem Res (1982) 7(11):1359–66. doi:10.1007/BF00966064
321. Cullingford TE, Dolphin CT, Bhakoo KK, Peuchen S, Canevari L, Clark JB. Molecular cloning of rat mitochondrial 3-hydroxy-3-methylglutaryl-CoA lyase and detection of the corresponding mRNA and of those encoding the remaining enzymes comprising the ketogenic 3-hydroxy-3-methylglutaryl-CoA cycle in central nervous system of suckling rat. Biochem J (1998) 329(Pt 2):373–81.
322. Hegardt FG. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: a control enzyme in ketogenesis. Biochem J (1999) 338(Pt 3):569–82. doi:10.1042/bj3380569
323. Auestad N, Korsak RA, Morrow JW, Edmond J. Fatty acid oxidation and ketogenesis by astrocytes in primary culture. J Neurochem (1991) 56(4):1376–86. doi:10.1111/j.1471-4159.1991.tb11435.x
324. Misson JP, Edwards MA, Yamamoto M, Caviness VS Jr. Identification of radial glial cells within the developing murine central nervous system: studies based upon a new immunohistochemical marker. Brain Res Dev Brain Res (1988) 44(1):95–108. doi:10.1016/0165-3806(88)90121-6
325. Hartfuss E, Galli R, Heins N, Götz M. Characterization of CNS precursor subtypes and radial glia. Dev Biol (2001) 229(1):15–30. doi:10.1006/dbio.2000.9962
326. Noctor SC, Flint AC, Weissman TA, Wong WS, Clinton BK, Kriegstein AR. Dividing precursor cells of the embryonic cortical ventricular zone have morphological and molecular characteristics of radial glia. J Neurosci (2002) 22(8):3161–73.
327. Song H, Stevens CF, Gage FH. Astroglia induce neurogenesis from adult neural stem cells. Nature (2002) 417(6884):39–44. doi:10.1038/417039a
328. Padilla SL, Carmody JS, Zeltser LM. Pomc-expressing progenitors give rise to antagonistic populations in hypothalamic feeding circuits. Nat Med (2010) 16(4):403–5. doi:10.1038/nm.2126
329. McNay DE, Briancon N, Kokoeva MV, Maratos-Flier E, Flier JS. Remodeling of the arcuate nucleus energy-balance circuit is inhibited in obese mice. J Clin Invest (2012) 122(1):142–52. doi:10.1172/JCI43134
330. Sousa-Ferreira L, de Almeida LP, Cavadas C. Role of hypothalamic neurogenesis in feeding regulation. Trends Endocrinol Metab (2014) 25(2):80–8. doi:10.1016/j.tem.2013.10.005
331. Chang GQ, Gaysinskaya V, Karatayev O, Leibowitz SF. Maternal high-fat diet and fetal programming: increased proliferation of hypothalamic peptide-producing neurons that increase risk for overeating and obesity. J Neurosci (2008) 28(46):12107–19. doi:10.1523/JNEUROSCI.2642-08.2008
332. Bless EP, Yang J, Acharya KD, Nettles SA, Vassoler FM, Byrnes EM, et al. Adult neurogenesis in the female mouse hypothalamus: estradiol and high fat diet alter the generation of newborn neurons expressing estrogen receptor α. eNeuro (2016) 3(4). doi:10.1523/ENEURO.0027-16.2016
333. Lee DA, Yoo S, Pak T, Salvatierra J, Velarde E, Aja S, et al. Dietary and sex-specific factors regulate hypothalamic neurogenesis in young adult mice. Front Neurosci (2014) 8:157. doi:10.3389/fnins.2014.00157
334. Oyarce KA, Bongarzone ER, Nualart F. Unconventional neurogenic niches and neurogenesis modulation by vitamins. J Stem Cell Res Ther (2014) 4(3):184. doi:10.4172/2157-7633.1000184
335. Robins SC, Stewart I, McNay DE, Taylor V, Giachino C, Goetz M, et al. α-Tanycytes of the adult hypothalamic third ventricle include distinct populations of FGF-responsive neural progenitors. Nat Commun (2013) 4:2049. doi:10.1038/ncomms3049
336. Hajihosseini MK, Langhe SD, Lana-Elola E, Morrison H, Sparshott N, Kelly R, et al. Localization and fate of Fgf10-expressing cells in the adult mouse brain implicate Fgf10 in control of neurogenesis. Mol Cell Neurosci (2008) 37(4):857–68. doi:10.1016/j.mcn.2008.01.008
337. Goodman TH, Hajihosseini MK. Hypothalamic tanycytes—masters and servants of metabolic, neuroendocrine, and neurogenic functions. Front Neurosci (2015) 9:387. doi:10.3389/fnins.2015.00387
338. García MA, Salazar K, Millán C, Rodríguez F, Montecinos H, Caprile T, et al. Sodium vitamin C cotransporter SVCT2 is expressed in hypothalamic glial cells. Glia (2005) 50(1):32–47. doi:10.1002/glia.20133
339. Haan N, Goodman TH, Nadji-Samiei A, Stratford CM, Rice R, Agha EE, et al. Fgf10-expressing tanycytes add new neurons to the appetite/energy-balance regulating centres of the postnatal and adult hypothalamus. J Neurosci (2013) 33(14):6170–80. doi:10.1523/JNEUROSCI.2437-12.2013
340. Bookout AL, de Groot MHM, Owen BM, Lee S, Gautron L, Lawrence HL, et al. FGF21 regulates circadian behavior and metabolism by acting on the nervous system. Nat Med (2013) 19(9):1147–52. doi:10.1038/nm.3249
341. Ryan KK, Kohli R, Gutierrez-Aguilar R, Gaitonde SG, Woods SC, Seeley RJ. Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats. Endocrinology (2013) 154(1):9–15. doi:10.1210/en.2012-1891
342. Liang Q, Zhong L, Zhang J, Wang Y, Bornstein SR, Triggle CR, et al. FGF21 maintains glucose homeostasis by mediating the cross talk between liver and brain during prolonged fasting. Diabetes (2014) 63(12):4064. doi:10.2337/db14-0541
343. Owen BM, Mangelsdorf DJ, Kliewer SA. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol Metab (2015) 26(1):22–9. doi:10.1016/j.tem.2014.10.002
344. Suzuki S, Li AJ, Akaike T, Imamura T. Intracerebroventricular infusion of fibroblast growth factor-1 increases Fos immunoreactivity in periventricular astrocytes in rat hypothalamus. Neurosci Lett (2001) 300(1):29–32. doi:10.1016/S0304-3940(01)01535-X
345. Kajitani N, Hisaoka-Nakashima K, Okada-Tsuchioka M, Hosoi M, Yokoe T, Morioka N, et al. Fibroblast growth factor 2 mRNA expression evoked by amitriptyline involves extracellular signal-regulated kinase-dependent early growth response 1 production in rat primary cultured astrocytes. J Neurochem (2015) 135(1):27–37. doi:10.1111/jnc.13247
346. Garré JM, Yang G, Bukauskas FF, Bennett MVL. FGF-1 triggers pannexin-1 hemichannel opening in spinal astrocytes of rodents and promotes inflammatory responses in acute spinal cord slices. J Neurosci (2016) 36(17):4785–801. doi:10.1523/JNEUROSCI.4195-15.2016
347. Pierozan P, Biasibetti H, Schmitz F, Ávila H, Parisi MM, Barbe-Tuana F, et al. Quinolinic acid neurotoxicity: differential roles of astrocytes and microglia via FGF-2-mediated signaling in redox-linked cytoskeletal changes. Biochim Biophys Acta (2016) 1863(12):3001–14. doi:10.1016/j.bbamcr.2016.09.014
348. Pérez-Martín M, Cifuentes M, Grondona JM, Bermúdez-Silva FJ, Arrabal PM, Pérez-Fígares JM, et al. Neurogenesis in explants from the walls of the lateral ventricle of adult bovine brain: role of endogenous IGF-1 as a survival factor. Eur J Neurosci (2003) 17(2):205–11. doi:10.1046/j.1460-9568.2003.02432.x
349. Migaud M, Batailler M, Segura S, Duittoz A, Franceschini I, Pillon D. Emerging new sites for adult neurogenesis in the mammalian brain: a comparative study between the hypothalamus and the classical neurogenic zones. Eur J Neurosci (2010) 32(12):2042–52. doi:10.1111/j.1460-9568.2010.07521.x
350. Lee DA, Bedont JL, Pak T, Wang H, Song J, Miranda-Angulo A, et al. Tanycytes of the hypothalamic median eminence form a diet-responsive neurogenic niche. Nat Neurosci (2012) 15(5):700–2. doi:10.1038/nn.3079
351. Bless EP, Reddy T, Acharya KD, Beltz BS, Tetel MJ. Oestradiol and diet modulate energy homeostasis and hypothalamic neurogenesis in the adult female mouse. J Neuroendocrinol (2014) 26(11):805–16. doi:10.1111/jne.12206
352. Borg ML, Lemus MB, Reichenbach A, Selathurai A, Oldfield BJ, Andrews ZB, et al. Hypothalamic neurogenesis is not required for the improved insulin sensitivity following exercise training. Diabetes (2014) 63(11):3647. doi:10.2337/db13-1762
353. Niwa A, Nishibori M, Hamasaki S, Kobori T, Liu K, Wake H, et al. Voluntary exercise induces neurogenesis in the hypothalamus and ependymal lining of the third ventricle. Brain Struct Funct (2016) 221(3):1653–66. doi:10.1007/s00429-015-0995-x
354. Blackshaw S, Lee DA, Pak T, Yoo S. Regulation of body weight and metabolism by tanycyte-derived neurogenesis in young adult mice. In: Pfaff D, Christen Y, editors. Stem Cells in Neuroendocrinology. Cham: Springer International Publishing (2016). p. 51–67.
355. Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci (2013) 14(5):311–21. doi:10.1038/nrn3484
356. Sierra A, Beccari S, Diaz-Aparicio I, Encinas JM, Comeau S, Tremblay ME. Surveillance, phagocytosis, and inflammation: how never-resting microglia influence adult hippocampal neurogenesis. Neural Plast (2014) 2014:610343. doi:10.1155/2014/610343
357. Park HR, Park M, Choi J, Park KY, Chung HY, Lee JY. A high-fat diet impairs neurogenesis: involvement of lipid peroxidation and brain-derived neurotrophic factor. Neurosci Lett (2010) 482(3):235–9. doi:10.1016/j.neulet.2010.07.046
358. Lindqvist A, Mohapel P, Bouter B, Frielingsdorf H, Pizzo D, Brundin P, et al. High-fat diet impairs hippocampal neurogenesis in male rats. Eur J Neurol (2006) 13(12):1385–8. doi:10.1111/j.1468-1331.2006.01500.x
359. Park HR, Kim JY, Park KY, Lee J. Lipotoxicity of palmitic acid on neural progenitor cells and hippocampal neurogenesis. Toxicol Res (2011) 27(2):103–10. doi:10.5487/TR.2011.27.2.103
360. Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA. Adult hippocampal neurogenesis buffers stress responses and depressive behavior. Nature (2011) 476(7361):458–61. doi:10.1038/nature10287
361. Nokia MS, Lensu S, Ahtiainen JP, Johansson PP, Koch LG, Britton SL, et al. Physical exercise increases adult hippocampal neurogenesis in male rats provided it is aerobic and sustained. J Physiol (2016) 594(7):1855–73. doi:10.1113/JP271552
362. Bouret SG. Nutritional programming of hypothalamic development: critical periods and windows of opportunity. Int J Obes Suppl (2012) 2(Suppl 2):S19–24. doi:10.1038/ijosup.2012.17
363. Perlmutter LS, Tweedle CD, Hatton GI. Neuronal/glial plasticity in the supraoptic dendritic zone: dendritic bundling and double synapse formation at parturition. Neuroscience (1984) 13(3):769–79. doi:10.1016/0306-4522(84)90095-2
364. Theodosis DT, Poulain DA. Evidence for structural plasticity in the supraoptic nucleus of the rat hypothalamus in relation to gestation and lactation. Neuroscience (1984) 11(1):183–93. doi:10.1016/0306-4522(84)90222-7
365. Theodosis DT, Chapman DB, Montagnese C, Poulain DA, Morris JF. Structural plasticity in the hypothalamic supraoptic nucleus at lactation affects oxytocin-, but not vasopressin-secreting neurones. Neuroscience (1986) 17(3):661–78. doi:10.1016/0306-4522(86)90038-2
366. Marzban F, Tweedle CD, Hatton GI. Reevaluation of the plasticity in the rat supraoptic nucleus after chronic dehydration using immunogold for oxytocin and vasopressin at the ultrastructural level. Brain Res Bull (1992) 28(5):757–66. doi:10.1016/0361-9230(92)90256-W
367. García-Segura LM, Chowen JA, Párducz A, Naftolin F. Gonadal hormones as promoters of structural synaptic plasticity: cellular mechanisms. Prog Neurobiol (1994) 44(3):279–307. doi:10.1016/0301-0082(94)90042-6
368. Bandeira F, Lent R, Herculano-Houzel S. Changing numbers of neuronal and non-neuronal cells underlie postnatal brain growth in the rat. Proc Natl Acad Sci U S A (2009) 106(33):14108–13. doi:10.1073/pnas.0804650106
369. Freeman MR. Specification and morphogenesis of astrocytes. Science (2010) 330(6005):774. doi:10.1126/science.1190928
370. Ahima RS, Kelly J, Elmquist JK, Flier JS. Distinct physiologic and neuronal responses to decreased leptin and mild hyperleptinemia. Endocrinology (1999) 140(11):4923–31. doi:10.1210/endo.140.11.7105
371. Ahima RS, Prabakaran D, Flier JS. Postnatal leptin surge and regulation of circadian rhythm of leptin by feeding. Implications for energy homeostasis and neuroendocrine function. J Clin Invest (1998) 101(5):1020–7. doi:10.1172/JCI1176
372. Rottkamp DM, Rudenko IA, Maier MT, Roshanbin S, Yulyaningsih E, Perez L, et al. Leptin potentiates astrogenesis in the developing hypothalamus. Mol Metab (2015) 4(11):881–9. doi:10.1016/j.molmet.2015.08.005
373. Udagawa J, Hashimoto R, Suzuki H, Hatta T, Sotomaru Y, Hioki K, et al. The role of leptin in the development of the cerebral cortex in mouse embryos. Endocrinology (2006) 147(2):647–58. doi:10.1210/en.2005-0791
374. Fisette A, Alquier T. AstroGenesis: and there was leptin on the sixth day. Mol Metab (2015) 4(11):755–7. doi:10.1016/j.molmet.2015.09.009
375. Yura S, Itoh H, Sagawa N, Yamamoto H, Masuzaki H, Nakao K, et al. Role of premature leptin surge in obesity resulting from intrauterine undernutrition. Cell Metab (2005) 1(6):371–8. doi:10.1016/j.cmet.2005.05.005
376. Kirk SL, Samuelsson AM, Argenton M, Dhonye H, Kalamatianos T, Poston L, et al. Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS One (2009) 4(6):e5870. doi:10.1371/journal.pone.0005870
377. Viveros MP, Díaz F, Mateos B, Rodríguez N, Chowen JA. Maternal deprivation induces a rapid decline in circulating leptin levels and sexually dimorphic modifications in hypothalamic trophic factors and cell turnover. Horm Behav (2010) 57(4–5):405–14. doi:10.1016/j.yhbeh.2010.01.009
378. Harry GJ, Kraft AD. Microglia in the developing brain: a potential target with lifetime effects. Neurotoxicology (2012) 33(2):191–206. doi:10.1016/j.neuro.2012.01.012
379. Sierra A, Abiega O, Shahraz A, Neumann H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci (2013) 7:6. doi:10.3389/fncel.2013.00006
380. Lenz KM, Nugent BM, Haliyur R, McCarthy MM. Microglia are essential to masculinization of brain and behavior. J Neurosci (2013) 33(7):2761. doi:10.1523/JNEUROSCI.1268-12.2013
381. Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, et al. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science (2004) 304(5667):110. doi:10.1126/science.1089459
382. Horvath TL, Gao XB. Input organization and plasticity of hypocretin neurons: possible clues to obesity’s association with insomnia. Cell Metab (2005) 1(4):279–86. doi:10.1016/j.cmet.2005.03.003
383. Gyengesi E, Liu ZW, D’Agostino G, Gan G, Horvath TL, Gao XB, et al. Corticosterone regulates synaptic input organization of POMC and NPY/AgRP neurons in adult mice. Endocrinology (2010) 151(11):5395–402. doi:10.1210/en.2010-0681
384. Horvath TL, Sarman B, García-Cáceres C, Enriori PJ, Sotonyi P, Shanabrough M, et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc Natl Acad Sci U S A (2010) 107(33):14875–80. doi:10.1073/pnas.1004282107
385. Benani A, Hryhorczuk C, Gouazé A, Fioramonti X, Brenachot X, Guissard C, et al. Food intake adaptation to dietary fat involves PSA-dependent rewiring of the arcuate melanocortin system in mice. J Neurosci (2012) 32(35):11970. doi:10.1523/JNEUROSCI.0624-12.2012
386. Bonfanti L, Theodosis DT. Polysialic acid and activity-dependent synapse remodeling. Cell Adh Migr (2009) 3(1):43–50. doi:10.4161/cam.3.1.7258
387. Bolborea M, Laran-Chich MP, Rasri K, Hildebrandt H, Govitrapong P, Simonneaux V, et al. Melatonin controls photoperiodic changes in tanycyte vimentin and neural cell adhesion molecule expression in the Djungarian hamster (Phodopus sungorus). Endocrinology (2011) 152(10):3871–83. doi:10.1210/en.2011-1039
388. García-Cáceres C, Fuente-Martín E, Burgos-Ramos E, Granado M, Frago LM, Barrios V, et al. Differential acute and chronic effects of leptin on hypothalamic astrocyte morphology and synaptic protein levels. Endocrinology (2011) 152(5):1809–18. doi:10.1210/en.2010-1252
389. Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia (2000) 32(1):1–14. doi:10.1002/1098-1136(200010)32:1<1:AID-GLIA10>3.0.CO;2-W
390. Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature (1994) 369(6483):744–7. doi:10.1038/369744a0
391. Araque A, Parpura V, Sanzgiri RP, Haydon PG. Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur J Neurosci (1998) 10(6):2129–42. doi:10.1046/j.1460-9568.1998.00221.x
392. Araque A, Sanzgiri RP, Parpura V, Haydon PG. Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J Neurosci (1998) 18(17):6822–9.
393. Halassa MM, Fellin T, Haydon PG. Tripartite synapses: roles for astrocytic purines in the control of synaptic physiology and behavior. Neuropharmacology (2009) 57(4):343–6. doi:10.1016/j.neuropharm.2009.06.031
394. Yang L, Qi Y, Yang Y. Astrocytes control food intake by inhibiting AGRP neuron activity via adenosine A1 receptors. Cell Rep (2015) 11(5):798–807. doi:10.1016/j.celrep.2015.04.002
395. Bélanger M, Magistretti PJ. The role of astroglia in neuroprotection. Dialogues Clin Neurosci (2009) 11(3):281–95.
396. Barreto GE, Gonzalez J, Torres Y, Morales L. Astrocytic-neuronal crosstalk: implications for neuroprotection from brain injury. Neurosci Res (2011) 71(2):107–13. doi:10.1016/j.neures.2011.06.004
397. Chen Z, Trapp BD. Microglia and neuroprotection. J Neurochem (2016) 136:10–7. doi:10.1111/jnc.13062
398. Jha MK, Lee WH, Suk K. Functional polarization of neuroglia: implications in neuroinflammation and neurological disorders. Biochem Pharmacol (2016) 103:1–16. doi:10.1016/j.bcp.2015.11.003
399. De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology (2005) 146(10):4192–9. doi:10.1210/en.2004-1520
400. Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKβ/NF-κB and ER stress link overnutrition to energy imbalance and obesity. Cell (2008) 135(1):61–73. doi:10.1016/j.cell.2008.07.043
401. Calegari VC, Torsoni AS, Vanzela EC, Araújo EP, Morari J, Zoppi CC, et al. Inflammation of the hypothalamus leads to defective pancreatic islet function. J Biol Chem (2011) 286(15):12870–80. doi:10.1074/jbc.M110.173021
402. Tapia-González S, García-Segura LM, Tena-Sempere M, Frago LM, Castellano JM, Fuente-Martín E, et al. Activation of microglia in specific hypothalamic nuclei and the cerebellum of adult rats exposed to neonatal overnutrition. J Neuroendocrinol (2011) 23(4):365–70. doi:10.1111/j.1365-2826.2011.02113.x
403. Milanski M, Arruda AP, Coope A, Ignacio-Souza LM, Nunez CE, Roman EA, et al. Inhibition of hypothalamic inflammation reverses diet-induced insulin resistance in the liver. Diabetes (2012) 61(6):1455–62. doi:10.2337/db11-0390
404. Morari J, Anhe GF, Nascimento LF, de Moura RF, Razolli D, Solon C, et al. Fractalkine (CX3CL1) is involved in the early activation of hypothalamic inflammation in experimental obesity. Diabetes (2014) 63(11):3770. doi:10.2337/db13-1495
405. Cai D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends Endocrinol Metab (2013) 24(1):40–7. doi:10.1016/j.tem.2012.11.003
406. Cheng L, Yu Y, Szabo A, Wu Y, Wang H, Camer D, et al. Palmitic acid induces central leptin resistance and impairs hepatic glucose and lipid metabolism in male mice. J Nutr Biochem (2015) 26(5):541–8. doi:10.1016/j.jnutbio.2014.12.011
407. Fuente-Martin E, García-Cáceres C, Díaz F, Argente-Arizon P, Granado M, Barrios V, et al. Hypothalamic inflammation without astrogliosis in response to high sucrose intake is modulated by neonatal nutrition in male rats. Endocrinology (2013) 154(7):2318–30. doi:10.1210/en.2012-2196
408. Gao Y, Tschop MH, Luquet S. Hypothalamic tanycytes: gatekeepers to metabolic control. Cell Metab (2014) 19(2):173–5. doi:10.1016/j.cmet.2014.01.008
409. Morselli E, Criollo A, Rodriguez-Navas C, Clegg DJ. Chronic high fat diet consumption impairs metabolic health of male mice. Inflamm Cell Signal (2014) 1(6):e561. doi:10.14800/ics.561
410. Morselli E, Frank AP, Palmer BF, Rodriguez-Navas C, Criollo A, Clegg DJ. A sexually dimorphic hypothalamic response to chronic high-fat diet consumption. Int J Obes (2016) 40(2):206–9. doi:10.1038/ijo.2015.114
411. Morgan TE, Xie Z, Goldsmith S, Yoshida T, Lanzrein AS, Stone D, et al. The mosaic of brain glial hyperactivity during normal ageing and its attenuation by food restriction. Neuroscience (1999) 89(3):687–99. doi:10.1016/S0306-4522(98)00334-0
412. MacDonald L, Radler ME, Paolini AG, Kent S. Calorie restriction attenuates LPS-induced sickness behavior and shifts hypothalamic signaling pathways to an anti-inflammatory bias. Am J Physiol Regul Integr Comp Physiol (2011) 301(1):R172. doi:10.1152/ajpregu.00057.2011
413. Loncarevic-Vasiljkovic N, Pesic V, Todorovic S, Popic J, Smiljanic K, Milanovic D, et al. Caloric restriction suppresses microglial activation and prevents neuroapoptosis following cortical injury in rats. PLoS One (2012) 7(5):e37215. doi:10.1371/journal.pone.0037215
414. Radler ME, Hale MW, Kent S. Calorie restriction attenuates lipopolysaccharide (LPS)-induced microglial activation in discrete regions of the hypothalamus and the subfornical organ. Brain Behav Immun (2014) 38:13–24. doi:10.1016/j.bbi.2013.11.014
415. Radler ME, Wright BJ, Walker FR, Hale MW, Kent S. Calorie restriction increases lipopolysaccharide-induced neuropeptide Y immunolabeling and reduces microglial cell area in the arcuate hypothalamic nucleus. Neuroscience (2015) 285:236–47. doi:10.1016/j.neuroscience.2014.11.014
416. Yi CX, Al-Massadi O, Donelan E, Lehti M, Weber J, Ress C, et al. Exercise protects against high-fat diet-induced hypothalamic inflammation. Physiol Behav (2012) 106(4):485–90. doi:10.1016/j.physbeh.2012.03.021
417. Glezer I, Simard AR, Rivest S. Neuroprotective role of the innate immune system by microglia. Neuroscience (2007) 147(4):867–83. doi:10.1016/j.neuroscience.2007.02.055
418. Kofler J, Wiley CA. Microglia: key innate immune cells of the brain. Toxicol Pathol (2011) 39(1):103–14. doi:10.1177/0192623310387619
419. Valdearcos M, Robblee MM, Benjamin DI, Nomura DK, Xu AW, Koliwad SK. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep (2014) 9(6):2124–38. doi:10.1016/j.celrep.2014.11.018
420. Pinteaux E, Inoue W, Schmidt L, Molina-Holgado F, Rothwell NJ, Luheshi GN. Leptin induces interleukin-1β release from rat microglial cells through a caspase 1 independent mechanism. J Neurochem (2007) 102(3):826–33. doi:10.1111/j.1471-4159.2007.04559.x
421. Pan W, Hsuchou H, Xu C, Wu X, Bouret SG, Kastin AJ. Astrocytes modulate distribution and neuronal signaling of leptin in the hypothalamus of obese A(vy) mice. J Mol Neurosci (2011) 43(3):478–84. doi:10.1007/s12031-010-9470-6
422. Gupta S, Knight AG, Gupta S, Keller JN, Bruce-Keller AJ. Saturated long chain fatty acids activate inflammatory signaling in astrocytes. J Neurochem (2012) 120(6):1060–71. doi:10.1111/j.1471-4159.2012.07660.x
423. Lafrance V, Inoue W, Kan B, Luheshi GN. Leptin modulates cell morphology and cytokine release in microglia. Brain Behav Immun (2010) 24(3):358–65. doi:10.1016/j.bbi.2009.11.003
424. Spence RD, Hamby ME, Umeda E, Itoh N, Du S, Wisdom AJ, et al. Neuroprotection mediated through estrogen receptor-α in astrocytes. Proc Natl Acad Sci U S A (2011) 108(21):8867–72. doi:10.1073/pnas.1103833108
425. Higuchi S, Irie K, Mishima S, Araki M, Ohji M, Shirakawa A, et al. The cannabinoid 1-receptor silent antagonist O-2050 attenuates preference for high-fat diet and activated astrocytes in mice. J Pharmacol Sci (2010) 112(3):369–72. doi:10.1254/jphs.09326SC
Keywords: hypothalamus, metabolism, energy, homeostasis, glia, inflammation
Citation: Freire-Regatillo A, Argente-Arizón P, Argente J, García-Segura LM and Chowen JA (2017) Non-Neuronal Cells in the Hypothalamic Adaptation to Metabolic Signals. Front. Endocrinol. 8:51. doi: 10.3389/fendo.2017.00051
Received: 05 January 2017; Accepted: 03 March 2017;
Published: 21 March 2017
Edited by:
Serge H. Luquet, Paris Diderot University, FranceReviewed by:
Matei Bolborea, University of Warwick, UKVincent Prevot, Institut national de la santé et de la recherche médicale (INSERM), France
Copyright: © 2017 Freire-Regatillo, Argente-Arizón, Argente, García-Segura and Chowen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie A. Chowen, anVsaWVhbm4uY2hvd2VuQHNhbHVkLm1hZHJpZC5vcmc=