 Justin T. Smith
Justin T. Smith Andrew D. Schneider
Andrew D. Schneider Karina M. Katchko
Karina M. Katchko Chawon Yun
Chawon Yun Erin L. Hsu
Erin L. Hsu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Endocrinol., 14 February 2017
Sec. Bone Research
Volume 8 - 2017 | https://doi.org/10.3389/fendo.2017.00022
This article is part of the Research TopicChemokines and BoneView all 7 articles
Chemokines play an important role in normal bone physiology and the pathophysiology of many bone diseases. The recent increased focus on the individual roles of this class of proteins in the context of bone has shown that members of the two major chemokine subfamilies—CC and CXC—support or promote the formation of new bone and the remodeling of existing bone in response to a myriad of stimuli. These chemotactic molecules are crucial in orchestrating appropriate cellular homing, osteoblastogenesis, and osteoclastogenesis during normal bone repair. Bone healing is a complex cascade of carefully regulated processes, including inflammation, progenitor cell recruitment, differentiation, and remodeling. The extensive role of chemokines in these processes and the known links between environmental contaminants and chemokine expression/activity leaves ample opportunity for disruption of bone healing by environmental factors. However, despite increased clinical awareness, the potential impact of many of these environmental factors on bone-related chemokines is still ill defined. A great deal of focus has been placed on environmental exposure to various endocrine disruptors (bisphenol A, phthalate esters, etc.), volatile organic compounds, dioxins, and heavy metals, though mainly in other tissues. Awareness of the impact of other less well-studied bone toxicants, such as fluoride, mold and fungal toxins, asbestos, and chlorine, is also reviewed. In many cases, the literature on these toxins in osteogenic models is lacking. However, research focused on their effects in other tissues and cell lines provides clues for where future resources could be best utilized. This review aims to serve as a current and exhaustive resource detailing the known links between several classes of high-interest environmental pollutants and their interaction with the chemokines relevant to bone healing.
Chemokines play an important role in normal bone physiology and the pathophysiology of many bone diseases. The recent increased focus on the individual roles of this class of proteins in the context of bone has shown that members of the two major chemokine subfamilies—CC and CXC—promote the formation of new bone and the remodeling of existing bone in response to a myriad of stimuli. These chemotactic molecules are crucial in orchestrating appropriate cellular homing, osteoblastogenesis, and osteoclastogenesis during normal bone repair. A recent review by Gilchrist and Stern provided a comprehensive assessment of the role key chemokines and receptors play in the regulation of bone healing (1). Several of these chemokines have received growing attention in recent literature and their mechanisms of action have been further defined.
Organizations and initiatives such as Physicians for Social Responsibility and the Collaborative on Health and the Environment recognize that environmental contaminants such as endocrine-disrupting chemicals (EDCs) are of serious concern with regard to bone health1 (2). A great deal of focus has been placed on environmental exposure to various endocrine disruptors (bisphenol A, phthalate esters, etc.), volatile organic compounds (VOCs), dioxins, and heavy metals. Awareness of the impact of other less well-studied bone toxicants, such as fluoride, mold and fungal toxins, asbestos, and chlorine, is also growing.
Bone healing is a complex cascade of carefully regulated processes, including inflammation, progenitor cell recruitment, differentiation, and remodeling. The extensive role of chemokines in these processes and the known links between environmental contaminants and chemokine expression and activity leaves ample opportunity for disruption of bone healing by environmental factors. However, despite increased clinical awareness, the potential impact of many of these environmental factors on bone-related chemokines is still ill defined. This review aims to serve as a current and exhaustive resource detailing the known links between several classes of high-interest environmental pollutants and their interaction with the chemokines relevant to bone healing. Areas where the literature is lacking and further research is prudent are also highlighted.
Endocrine-disrupting chemicals are of specific concern due to their exogenous influence over the endocrine system. These compounds exert their effects independent of biofeedback loops, leading to potentially harmful consequences. EDCs can be either natural or synthetic in origin. Natural EDCs consisting of organically produced compounds are out of the scope of this review and will not be discussed. Synthetic EDCs are commonly designed with another purpose in mind (e.g., pesticides or plasticizers), only to have their endocrine effects subsequently discovered. These effects have been observed as developmental anomalies in both invertebrate and aquatic species (3, 4). A summary of endocrine disruptors and their effects on bone-related chemokines is shown in Table 1.
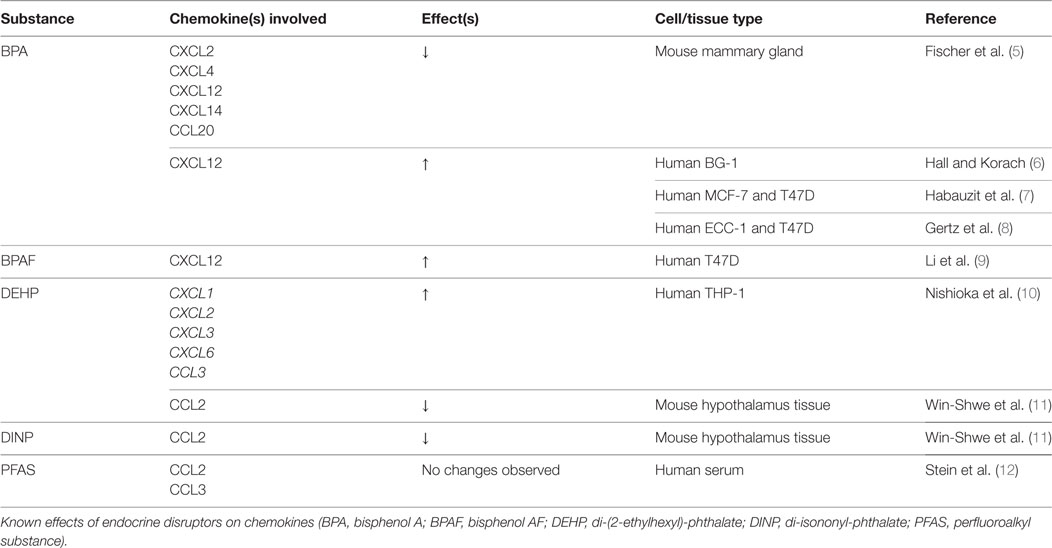
Table 1. Endocrine disruptors: chemokine changes.
Bisphenol A (BPA) is a chemical compound that has been used in commercial plastics and epoxy resins since the 1950s. The widespread use of BPA in manufacturing has led to its ubiquitous presence in industrialized environments (13). The primary method of exposure in humans is through the diet, mainly through drinking water in industrialized regions, as well as from food and drink storage or use containers (14). Specifically, BPA can leach into foods from the plastics or resins used during the manufacturing of such containers (e.g., water bottles, food cans, etc.). Up to 90% of the US population has detectable blood levels of BPA, which has been a growing public health concern (15). Thus far, chronic exposure has been linked to cancer, metabolic disorders, and a range of reproductive and cardiovascular diseases (16, 17).
Bisphenol A functions as an endocrine disruptor through binding to ERα and ERβ with roughly 1/1,000th the affinity of estradiol (18, 19). Because estradiol can attenuate osteoclastogenesis and induce osteoblast differentiation (20, 21), Hwang et al. recently examined the effects of BPA on osteoclast formation and osteoblast differentiation in vitro (22). The authors found that BPA directly inhibited both osteoclastogenesis and osteoblastogenesis, and increased apoptosis in both types of progenitors. BPA suppressed RANK expression in differentiating osteoclasts and RunX2 and Osterix expression in preosteoblasts. Wnt/β-catenin signaling and bone nodule formation was also reduced. These authors demonstrate that BPA promotes apoptosis of osteoclasts and osteoblasts through MAPK cascades and death receptor pathways in a dose-dependent manner (22).
Despite the effects of BPA on osteoblasts and osteoclasts, our search criteria yielded no studies examining BPA-induced chemokine expression changes in osteogenic models. However, given its endocrine-disrupting status, several studies have examined its impact on chemokines in reproductive tissues such as mouse mammary, breast cancer, and ovarian cancer cells (5–8). Significant decreases in CXCL2, CXCL4, CXCL14, and CCL20 have been observed in these various tissues with results that suggest BPA may act through pathways independent of estrogen. Additionally, the related compound, Bisphenol AF (BPAF), has demonstrated upregulation of CXCL12 in T47D ERα-positive cancer cell lines (23). Considering the critical role of the CXCL12/CXCR4 axis on bone regeneration, the known effects of BPA and BPAF on tissues other than bone make clear the importance of evaluating their effects in the context of bone healing.
Phthalate esters comprise another group of EDCs that are used to increase the flexibility of polymers, such as polyvinyl chloride (PVC)-based products (e.g., toys, food wraps, medical devices, and flooring). Similar to BPA, humans are exposed to phthalates through ingestion, inhalation, and physical contact on a daily basis (24, 25). Two particular phthalates, di-(2-ethylhexyl) phthalate (DEHP) and di-isononyl phthalate (DINP), have been examined in the context of the chemokine expression and bone formation (10, 11).
DEHP is cytotoxic to neonatal rat calvarial osteoblasts at high doses (1,000 µM), while inducing proliferation at low doses (10 µM) (26). In that system, osteogenic differentiation was inhibited at high doses of DEHP, with reduced RunX2 and ALP expression and decreased collagen and mineralization staining (26).
Inflammation is an important early step in the bone healing cascade. Chemokines play a critical role in recruitment of macrophages and progenitor cells to the site of injury, where they remove necrotic tissue and initiate the regenerative process. However, this process is tightly regulated, and chronic inflammatory diseases are associated with systemic bone loss (27). Even subclinical inflammation has been shown to increase fracture risk and alter bone remodeling (28). Nishioka et al. examined the effects of DEHP on the production of inflammatory cytokines in activated macrophage-like THP-1 cells. A 3-h exposure to 200 µM DEHP significantly induced CXCL1, CXCL2, CXCL3, CXCL6, and CCL3 transcripts (10). PTH induces CXCL1 expression in osteoblasts, which then attracts osteoclasts via CXCR2 (29). Interestingly, CXCL1 acts as a chemoattractant for osteoclast precursors without promoting osteoclastic differentiation, acting as a “primer” for additional factors (29). That said, it is reasonable to speculate that increased CXCL1 expression by macrophages recruited during bone repair could also prove chemotactic for osteoclasts. Further research should examine whether DEHP influences CXCL1 secretion in osteoblasts in a similar manner.
Human osteoblasts express CCL2, which is an important chemoattractant for monocytes and macrophages (30). Additionally, CCL2 is critical for osteoclastogenesis, and its absence results in inhibition of osteoclast formation (31). In studies examining the hypothalami of DEHP- and DINP-treated male mice, Win-Shwe et al. observed decreases in CCL2 and TNF-α expression in the DINP-exposed groups (11). Considering the important role that CCL2 plays on osteoclastogenesis, further investigation to the effects of DINP in bone models should be explored.
Perfluoroalkyl substances have been widely used as protective coatings in water- and stain-resistant clothing, furnishings, and non-stick home goods for over 60 years (32). These chemicals have been classified as EDCs based on their hormonal and metabolic actions (33, 34). PFASs are ubiquitous in the environment, and detectable amounts are found in humans worldwide (35). Examination of the U.S. National Health and Nutritional Examination Survey (NHANES) from 1999 to 2008 showed that four PFASs were found in 95% of the U.S. population: perfluorooctanoic acid (PFOA), perfluorooctane sulfonic acid (PFOS), perfluorohexane sulfonic acid (PFHxS), and perfluorononanoic acid (PFNA) (36, 37). No research has been conducted to examine the effects of PFASs on chemokine production. However, there is evidence justifying further investigation into the mechanisms of PFAS action in osteogenic models. In a representative sample of the U.S. population collected from the NHANES 2009–2010, serum PFAS levels were inversely correlated with bone mineral density (BMD) of the femur and lumbar spine (32). Most of the associations were limited to women, despite men having higher serum PFAS levels. In general, postmenopausal women had stronger associations with lower BMD than their premenopausal counterparts. Osteoporosis was associated with exposure to PFOA, PFNA, and PFHxS in women (32).
Organotins are environmental contaminants considered to be obesogens because they activate peroxisome proliferator-activated receptor γ (PPARγ), the primary regulator of adipogenesis (38, 39). These compounds are broadly used as pesticides, catalytic agents, plastic stabilizers, and antifouling agents (40). The actions of organotins on skeletal development have been examined in animal models (41, 42). Tsukamoto et al. demonstrated that mouse fetuses exposed to tributyltin chloride (TBT) showed reduced calcification of the supraoccipital bone (41). In vitro, TBT dose-dependently inhibited differentiation of primary rat calvarial osteoblasts, with reduced ALP activity, mineral deposition rate, and osteocalcin expression levels.
Despite these data, few studies have been published examining the effects of organotins on chemokine expression. Schutte et al. examined chemokine expression after implanting an organotin-stabilized PVC cage implant in Sprague-Dawley rats (43). They demonstrated a long-term (56 days postimplantation) 10-fold increase of CCL3, a pro-inflammatory chemokine responsible for increasing osteoclast motility (44). CCL2 demonstrated a significant increase on postoperative day 1, but then returned to normal levels. Given the endocrine activity associated with these compounds and their known effects on skeletal development, the mechanisms by which this class of compounds affects osteogenesis should be clarified.
In much of the industrialized world, individuals spend most of their time indoors. Indoor organic contaminants are classified by their volatility, dubbing them VOCs. These compounds are released from paints, adhesives, construction materials, cleaners, tobacco smoke, and carpets (45, 46). Because of the numerous sources found indoors, many of these compounds are present at concentrations significantly higher than outside (47). Elevated concentrations of VOCs are associated with asthma and respiratory diseases in children and adults (48–52). Table 2 summarizes the effects of the VOCs described below on chemokines involved in bone repair.
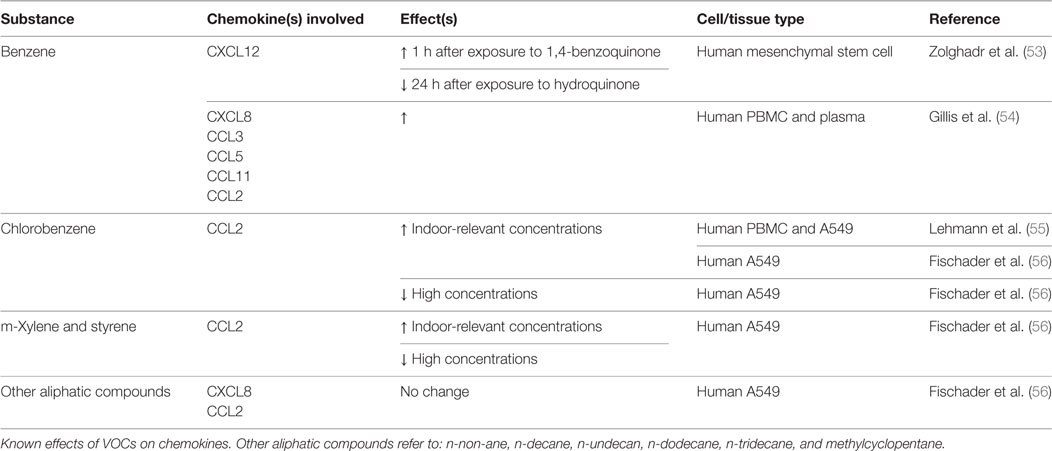
Table 2. Volatile organic compounds (VOCs): chemokine changes.
Benzene is a colorless, flammable organic liquid that can volatize to vapors at room temperatures. Used as an industrial chemical in the manufacturing of other compounds, it is also a component of crude oil, gasoline, and cigarette smoke (57, 58). Benzene was among the first compounds identified as a Group I known human carcinogen by the International Agency for Research on Cancer (59). The role of benzene in the development of acute myeloid leukemia and other myelodysplastic syndromes is well established (60–63). Human exposure to benzene occurs through inhalation and dermal absorption as well as ingestion of contaminated food and water (64). It is widely agreed that the toxicity of benzene results from its metabolism to reactive intermediates. In the liver, benzene is metabolized by CYP2E1 to phenol, which undergoes hydroxylation to hydroquinone, catechol, and 1,2,4-benzenetriol (65). Catechol and hydroquinone persist in bone marrow, where they are oxidized to 1,2-benzoquinone and 1,4-benzoquinone by myeloperoxidase (54, 66). The mechanism(s) by which these metabolites influence carcinogenesis have not been fully clarified (67).
While the effects of benzene and its metabolites on the immune and hematopoietic systems are well established, the influence of these compounds on bone development and skeletal remodeling is less fully understood. Zolghadr et al. examined the effects of benzene, hydroquinone, and 1,4-benzoquinone on human-derived mesenchymal stem cells (MSCs) (53). They examined cell viability, apoptosis, and expression of genes relevant to hematopoiesis and skeletal remodeling. At the lowest concentrations tested, all three compounds increased cell division rate after only 24 h, and the effects were attributed to cell cycle control defects (68). Interestingly, RunX2 expression was significantly upregulated (up to eightfold) by all three chemicals, as was Wnt5a, which is a non-canonical Wnt ligand. Simultaneously, Dkk1 expression was induced, which inhibits canonical Wnt signaling by interacting with the Wnt co-receptors, LRP-5/6. The authors hypothesized that the increase in Dkk expression was a response to induction of RunX2 and canonical Wnt signaling.
CXCL12, along with its receptor, CXCR4, plays a critical role in mesenchymal and hematopoietic stem cell recruitment, as well as BMP-mediated osteoblastic differentiation (69, 70). A major regulator of BMSC growth, CXCL12 expression is thought to decrease acutely after osteoblastic lineage commitment (71, 72), suggesting that its role is critical in the early stages of osteogenesis (69). In human MSC, CXCL12 expression was upregulated after short-term (1 h) exposure to 1,4-benzoquinone, but was downregulated after 24 h exposure to hydroquinone. The dysregulation of the Wnt and CXCL12 signaling pathways could have pronounced implications on bone regeneration. Further research needs to be conducted to define the upstream mechanisms regulating the effects of benzene metabolites on the CXCL12/CXCR4 axis.
Chlorobenzene is one of the most widely used chlorinated benzenes (55). It functions as a solvent for many substances, such as paints, adhesives, waxes, and polishes, and it is also commonly used in the dry-cleaning industry (73). Chlorobenzene inhalation can lead to irritation of the eyes and respiratory tract, drowsiness, loss of coordination, CNS depression, and loss of consciousness (73, 74). Additionally, epidemiological studies suggest that exposure to chlorobenzene is associated with allergic sensitizations and Th2-primed T cell immunity (75).
Chlorobenzene has been shown to induce CCL2, a chemokine critical for osteoclast formation, production at indoor-relevant concentrations in TNF-α-primed alveolar A549 cells (55, 56). Additional research has shown that the mechanism through which chlorobenzene induces CCL2 is through the NF-kB and MAPK pathways (76). Despite the CCL2 connection, however, no studies have attempted to correlate exposure with impaired osteoclastic differentiation, osteogenesis, or bone healing.
Similar to chlorobenzene, m-Xylene and styrene have been shown to induce CCL2 production, albeit in A549 cells. Still, dose-dependent alterations in osteoclast precursor recruitment and differentiation are worth investigating, which could lead to impaired bone remodeling and healing.
Fischader et al. additionally examined the effects of several aliphatic compounds (n-non-ane, n-decane, n-undecan, n-dodecane, n-tridecane, and methylcyclopentane) on A549 cells (56). Neither the single aliphatic compounds nor a mixture of the group demonstrated any effect on cytokine/chemokine release.
Dioxins and dioxin-like compounds (Benzo[a]pyrene, polychlorinated dibenzofurans, non-ortho-substituted, and mono-ortho-substituted pentachlorobiphenyls) compose a group of highly toxic environmental pollutants. Dioxins are formed as unintentional by-products of industrial manufacturing, when chlorine-based compounds are burned in the presence of hydrocarbons. Other important sources are waste-burning incinerators and backyard burning. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) was the major toxic contaminant in Agent Orange, the defoliant used extensively during the Vietnam War. Since dioxins and dioxin-like compounds are lipophilic, they bioaccumulate through the food chain. As such, the major source of dioxin exposure is the diet, the bulk coming from meat and dairy products, as well as fish. The half-life of TCDD is estimated as 7–10 years in humans.
TCDD causes a multitude of adverse health effects, including immune suppression, cancer (e.g., non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, and multiple myeloma), reproductive and developmental toxicity, cognitive function, and skin disorders (77–79). It has been shown to inhibit cell migration and osteogenic differentiation in vitro (80), alter normal bone phenotype (80–87), and inhibit bone healing in vivo (85, 88, 89). TCDD primarily exerts its deleterious effects through the aryl hydrocarbon receptor (AhR) pathway (83). Upon activation, this receptor acts as a transcription factor to modulate the expression of a large battery of genes, such as the Cytochrome P450 1 (CYP1) family, members of which are responsible for the Phase I biotransformation of hydrophobic xenobiotics. The Ahr can work through genomic (via binding to dioxin response elements in promoter/enhancer regions of dioxin-responsive genes) or non-genomic means. A great deal of evidence suggests Ahr cross-talk with NFKB signaling (90, 91), estrogen receptor signaling, and hypoxia-inducible factors (92–95).
With regard to immunomodulation, emerging data link AhR expression/activation with chronic diseases such as degenerative arthritis. Ahr activation promotes secretion of inflammatory cytokines, leading to local bone loss, inhibition of osteoblast proliferation and differentiation, and causes osteoporosis in mice (81, 88). Studies also suggest that the AhR acts as a negative regulator of stem cell proliferation (96), which may play a role on monocyte recruitment during bone remodeling. AhR has been shown to induce Th17 cell differentiation through a miRNA-mediated process (97–100). Th17 cells play a major role in the pathology of rheumatoid arthritis (RA), and the Ahr has been implicated in RA development through this mechanism. The downstream effects of Ahr-induced immunomodulation are excessive osteoclastic differentiation and inflammation (93, 101–103). AhR activation has also been tied to a diminished capacity for bone regeneration and healing (89, 104). The notion of excessive osteoclast proliferation by AhR ligands is supported by the impaired osteoclastogenesis and increased bone mass that is seen in AhR knockout mice (83).
The most toxic and well-studied dioxin and AhR ligand is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). TCDD was first shown to downregulate CXCL12 and CXCR4 expression in breast and ovarian cancer cells (23). A study conducted by Casado et al. demonstrated diminished CXCL12 migration of LSK cells exposed to TCDD with increased cell surface expression of CXCR4 (80). However, levels of CXCR4 mRNA were not induced, suggesting that AhR activation resulted in either upregulation of posttranslation modification or downregulation of degradation of the receptor.
Uncleaved osteopontin (OPN) is associated with cell adhesion, facilitating osteoclast attachment to bone matrix (105). On the other hand, cleaved OPN is chemotactic for neutrophils and leukocytes. OPN knockout bone marrow cells showed reduced migratory capacity in vivo (80), and antibody blockade of the binding domain reduced cell migration as well as PTH-induced osteoclast bone resorption (106). TCDD exposure reproduced the effects of antibody blockade, with a similar inhibition of OPN-induced cell migration (80). Our group has shown that TCDD inhibits BMP-2-mediated bone regeneration and spine fusion in the rat, with only a partial recovery of fusion capacity after cessation of exposure for a period of four half-lives (T1/2 for TCDD is 19 days in the rat) (89).
Vogel et al. demonstrated upregulation of CCL2 in the liver, thymus, kidney, adipose tissue, and heart of C57/BL6 mice injected with TCDD (107). Other in vitro studies in cell lines of various sources also suggest a direct correlation between CCL2 and TCDD exposure. Upregulation of CCL1, CXCL1, CXCL13, and CXCL8 (IL-8) has been observed as well (91, 108–110), while CCL5 was downregulated in mouse CD4+ T-cells ex vivo (111). The induction of IL-8 has been proposed to be as a result of NF-kB cross-talk through RelB activation (90, 91, 112). The upregulation of IL-8 and CCL2 by TCDD corresponds with findings of decreased expression in AhR-KO mice spleen (96). Notably, there was a 15-fold increase in CCR2 (the receptor for CCL2) expression in AhR-KO mice. These mice also demonstrated a >2-fold increase in CXCR5 and CCL20 (96). This supports the notion that the hyperactivated AhR may act as a negative regulator of BMSC proliferation and trafficking.
Dioxin-like compounds include other polycyclic aromatic hydrocarbons, such as Benzo[a]pyrene (BaP), polychlorinated dibenzofurans, and non-ortho-substituted or mono-ortho-substituted pentachlorobiphenyls (PCBs). Scant evidence can be found on the effects of these dioxin-like compounds on bone healing or any associated chemokine alterations. PCB-118 (2,3′,4,4′,5-pentachlorobiphenyl) exposure correlates with lower BMD (84) and induction of osteoclastic activity in vivo (113). No studies have found any associations with chemokine expression changes and PCB-118. Three additional environmental dioxin-like toxicants, PCB-126 (3,3′,4,4′,5-pentachlorobiphenyl), PCB-77 (3,3′,4,4′-tetrachlorobiphenyl), and BaP, have been shown to upregulate expression of CCL2 and CXCL13 in both in vitro and in vivo models, the latter of which contributes to osteoblast development (114–119). BaP has also been shown to reduce CCL5/RANTES mRNA expression during osteoclast formation in human keratinocytes (118). Considering that CCL5 promotes osteoclast development and chemotaxis (44), further work is needed to identify any direct effects of BaP on CCL5 expression in developing osteoclasts. Table 3 reviews the changes in bone repair chemokines by TCDD and the other dioxin-like compounds found in current literature.
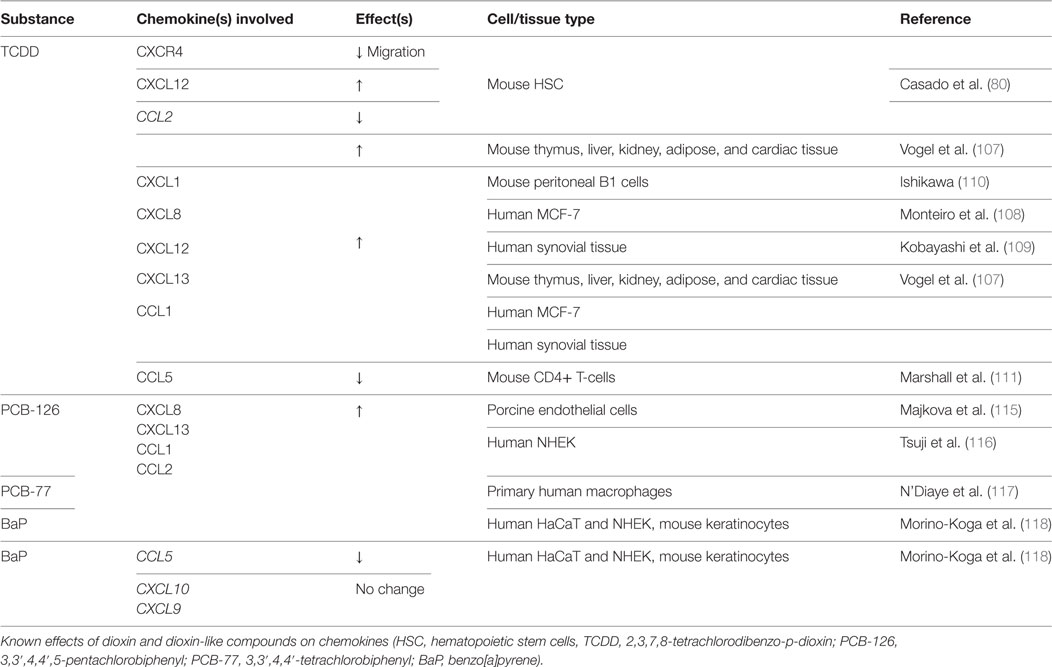
Table 3. Dioxin and dioxin-like compounds: chemokine changes.
We previously found that TCDD exposure inhibits spine fusion in rats (89). Subsequent work has shown that similar to breast and ovarian cancer cells, TCDD downregulates CXCL12 and CXCR4 expression in differentiating primary rat BMSC (our own unpublished data and Figures 1A,B). Concordant with this, BMSC chemotaxis toward CXCL12 was also reduced after TCDD treatment (unpublished data and Figure 1C). Since no such studies have investigated the effects of dioxin-like compounds on the CXCR4/CXCL12 axis, we performed similar work to examine the effects of these compounds on CXCL12 and CXCR4 expression in primary rat BMSC grown in both standard and osteogenic conditions.
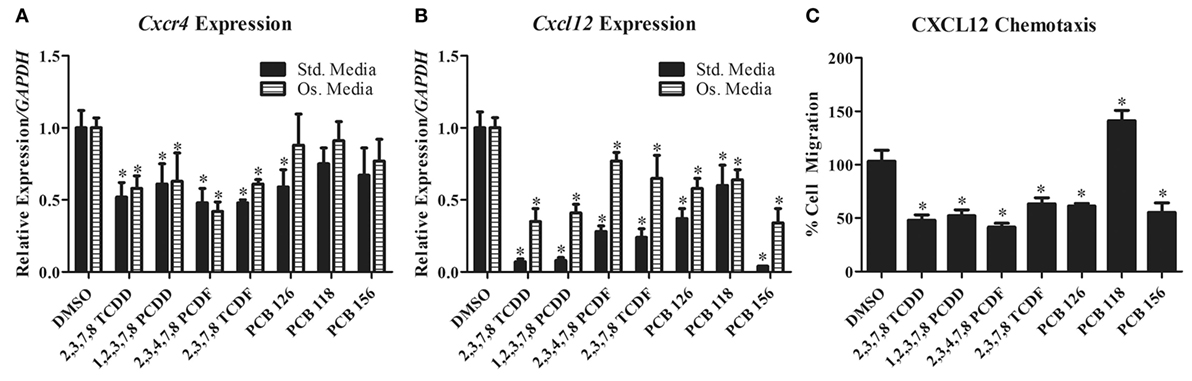
Figure 1. (A,B) Cxcr4 and Cxcl12 gene expression changes in primary rat BMSC grown in either standard or osteogenic media and treated with TCDD and dioxin-like compounds. (C) Chemotaxis rate of BMSC (toward 200 ng/mL of recombinant CXCL12) pretreated with TCDD and dioxin-like compounds. *p < 0.05 relative to DMSO vehicle control-treated cells. 2,3,7,8-TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; 1,2,3,7,8-PCDD, 1,2,3,7,8-pentachlorodibenzo-p-dioxin; 2,3,4,7,8-PCDD, 2,3,4,7,8-petachlorodibenzofuran; 2,3,7,8-TCDF, 2,3,7,8-tetrachlorodibenzofuran; PCB-126, 3,3′,4,4′,5-pentachlorobiphenyl; PCB-118, 2,3′,4,4′,5-pentachlorociphenyl; PCB-156, 2,3,3′,4,4′,5-hexachlorobiphenyl.
A select group of dioxins and dioxin-like compounds were chosen based on representative classification (dioxin, furan, or PCB) as well as the compound’s toxic equivalence within its class according to the World Health Organization (120). Northwestern University Institutional Animal Care and Use Committee (IACUC) approval was obtained prior to animal procedures and bone marrow stromal cell harvest. All experimental procedures were conducted in accordance with the recommendations of the IACUC. Rat BMSC were treated under standard or osteogenic conditions with various concentrations determined to activate the Ahr without inducing cell death (Table S1 in Supplementary Material). Total RNA was isolated using TRIzol (Invitrogen) and mRNA expression of CXCL12 and CXCR4 were analyzed using qPCR (Figures 1A,B). For chemotaxis assays, cells were counted by three blinded, independent reviewers (Figure 1C). Groups were compared using ANOVA and unpaired t-tests post hoc, with significance of p < 0.05.
CXCR4 expression was significantly decreased in primary rat BMSC grown under both standard and osteogenic conditions for all but three of the treatment groups (PCB-118, -126, and -156). PCB-126 showed a significant decrease of CXCR4 expression only under standard conditions. PCB-118 and -156 showed no changes in expression in either conditions. CXCL12 was significantly reduced for all treatment groups in both conditions. Additionally, we evaluated the capacity of BMSC to migrate toward a CXCL12 gradient after treatment with these compounds. Chemotaxis was significantly reduced in cells pretreated with all but PCB-118, which was likely a result of a lack of CXCR4 downregulation by PCB-118. Interestingly however, PCB-156 had no significant impact on CXCR4 expression, yet CXCL12-induced chemotaxis was significantly decreased.
There is a growing appreciation for the deleterious effects of dioxin-like compounds on bone. As the physiologic role of the AhR pathway becomes better understood, so too will AhR-mediated changes in chemotactic signaling. Further research is prudent to better understand the mechanisms of dioxin-induced inhibition of bone healing.
Another major source of chemokine-altering factors is circulating metal ions. Metal ions such as Mg, Fe, Cu, Zn, Mn, and Co are crucial for normal cellular functions but are toxic at elevated levels (121). These and other more exotic metals are being more keenly studied under the context of the progressive wear of metal prosthetic implants as well as other sources of environmental exposure.
Environmental metal pollution has long been a topic of interest to toxicologists, given the growth of industry and technology, which has led to supraphysiologic metal exposure in humans. Of particular interest are metals that are commonly encountered in developed or developing nations through food, drinking water, air, and soil. Lead, cadmium, strontium, and lithium have all been shown to affect levels of chemokines related to bone healing. Tungsten, arsenic, iron, boron, and mercury have been shown to impact bone homeostasis and healing potential, albeit with lacking evidence of alterations in chemokine expression levels (Table 4).
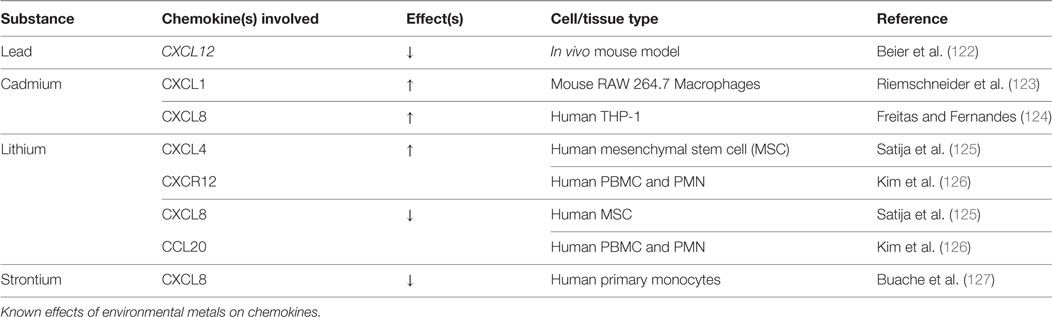
Table 4. Environmental metals: chemokine changes.
Despite the discovery of elevated lead levels in the drinking water in Flint, Michigan and other areas, the rate of lead toxicity as a result of environmental exposure has drastically improved since the 1970s. At that time, three-fourths of Americans had blood lead levels above 10 μg/dL, which had been the upper limit for categorization as lead poisoning (128, 129). Today, lead levels above 5 μg/dL warrant concern, according to the 2012 CDC update (130). Given the fact that bone acts as the main repository for lead (90–95%) and the body burden persists throughout life (lead t1/2 = 20 years), an extensive amount of research has evaluated its impact on bone health (131). Both basic and clinical studies suggest that lead has a significant impact on both bone growth/development and mature bone homeostasis. Similarly, lead exposure correlates with greater fracture risk and bone disease (25, 132, 133). Thus far, a dose-dependent response to lead accumulation in bone has been observed, causing osteopenia and poor BMD (133–136). However, it has been observed that low levels of lead cause increased BMD. The threshold that dictates the shift from increasing BMD to decreased BMD is still unknown (122, 137).
The only known study to have investigated chemokine fluctuations with exposure to lead is mouse tibia fracture study by Beier et al. This group found a ~50% reduction in CXCL12 mRNA expression 10 days postoperative, which was after 52 total days of lead exposure (15–18 μg/dL). Treatment with the GSK-3β inhibitor, 6-bromoindirubin-3′-oxime (BIO), rescued the inhibitory effect on CXCL12. BIO treatment also increased β-catenin staining to control levels (138), suggesting Wnt involvement. Indeed, BIO has been shown to induce CXCL12 expression in mouse tibial fracture callus (139). The Beier study suggests that lead may in part impact fracture repair by reducing CXCL12/CXCR4-mediated progenitor cell recruitment to the site of injury. No other group has attempted to investigate the mechanisms by which lead inhibits bone healing or alters chemokine expression.
Cadmium has long been recognized as a health hazard. It was originally described in Japan as Itai-Itai “ouch-ouch” disease in 1955, so named for the pain from osteomalacia and frequent long bone fractures, which occur secondarily to the ingestion of cadmium-contaminated rice (140). Today, the greatest source of cadmium comes from food (cereals/vegetables/potatoes) and tobacco smoke (141, 142). The toxicity of cadmium is in part due to its resemblance to metals such as calcium, iron, and zinc. Early kidney damage and osteoporosis have been the most widely studied consequences of cadmium exposure (141, 143). Long-term environmental exposure has been shown to cause osteoporosis with subsequent increases in fragility fractures (144–148).
Cadmium-induced reduction in BMD has been linked to both renal proximal tubule damage, osteoblast toxicity, and stimulation of osteoclastic bone resorption. Very few studies have attempted to uncover the direct mechanisms of cadmium-induced bone loss. Thus far, cadmium exposure has been shown to increase RANKL production with no change in OPG, increase prostaglandin E2, and trigger apoptosis in osteoblasts (149, 150).
In terms of chemokines and chemotaxis, Riemschneider et al. reported a twofold increase in secretion of CXCL1 in macrophage RAW 264.7 cells with exposure to 10 µM Cadmium. Similarly, they found reduced levels of IL-6 and IL-10 (123). Cadmium-mediated activation of NF-kB has also been reported, with a subsequent increase in IL-8, IL-6, IL-1β, and TNF-α (124). Together, this pattern suggests increased recruitment of preosteoclastic cells, which may contribute to observed decreases in BMD. Similarly, bone regeneration may be negatively affected by a dysregulated inflammatory response, since inflammatory signaling has a crucial role in optimal bone healing (151). Furthermore, Papa et al. showed that cadmium induced the destruction of the osteoblast cytoskeleton actin network, which is critical for cell polarity, motility, and chemotaxis (152). The actin depolymerization that is seen with cadmium exposure may also affect osteoblastic chemotaxis via the CXCL12/CXCR4 axis, which again is crucial in early bone formation (153, 154). Further research should be directed toward elucidating the mechanisms by which cadmium alters the chemotactic responses of osteoblasts/osteoclasts.
Lithium has long been used as a psychoactive drug in the treatment of various mood disorders. Recently, lithium has been recognized with growing concern as an environmental contaminant, due to greatly increased use of lithium ion batteries and alloys by industry and consumers. The most significant source of non-medical human exposure to lithium is thought to be through drinking water (155–157).
The effects of lithium on osteogenic differentiation vary according to cell system, state of differentiation, medium composition, passage number, and cell density (125). Lithium has been shown to inhibit Smad 1/5/8 phosphorylation in MC3T3-E1 preosteoblasts and murine myoblastic C2C12 cells, with a subsequent reduction in BMP-2-induced ALP activity independent of Wnt signaling (158). On the other hand, numerous studies have shown that lithium exposure induces a pro-bone formation phenotype, originating from osteoblastic activation and osteoclastic inhibition (159–163), with simultaneous antiadipogenic effects (125). Generally, Wnt/β-catenin pathway activation has been linked to most of the direct effects of lithium on bone homeostasis. Lithium inhibits GSK-3β, thereby preventing degradation of β-catenin (164). Enhanced nuclear β-catenin activity favors osteogenic differentiation of BMSC, reduced CXCL12 expression, and increased ALP activity in human MSC, although ALP activity was reduced relative to controls at higher doses (125). Expression levels of the pro-osteoclastic chemokines, IL-7, IL-8, and CCL20 were also reduced after hMSC exposure to lithium (125).
In the context of bone healing under chronic environmental lithium exposure, dysregulation of Wnt signaling—tight temporal control of which is critical for osteogenesis to proceed normally—may disturb the natural exit from the proliferative phase and completion of the differentiation program (165). Still, it is possible that precisely timed administration of lithium may promote osteoblastogenesis and inhibit osteoclastogenesis to result in overall improved bone healing (166). Further research into dosing and timing of lithium administration is required to validate lithium as a novel therapy for bone regeneration.
Strontium appears to prevent the age-related transition of BMSC lineage commitment from osteoblasts to adipocytes via NFATc/Maf and Wnt signaling (167). It is also being used as a therapeutic agent to prevent postmenapausal osteoporosis (168, 169). The only investigation of strontium-mediated chemokine changes was conducted by Bauche et al., in which LPS-stimulated monocytes exposed to strontium revealed diminished levels of IL-8 production. They also found IL-6 and TNF-α levels to be reduced, suggesting that strontium may have anti-inflammatory properties in addition to a potential osteoblastic lineage redirection (127).
Excess iron has been shown to generate reactive oxygen species (ROS) via the Fenton reaction (170). In addition to the potential ROS-induced cytotoxicity, ROS have been shown to antagonize Wnt signaling in osteoblast precursors by utilizing the limited pool of β-catenin for FoxO transcription, rather than of T-cell factor-mediated transcription (171). This may in turn lead to decreased bone formation, although this theory has yet to be examined in an in vivo model. Other evidence suggests that iron-ROS trigger osteoclastic bone resorption (172), which may increase the risk of osteoporosis (173). Despite these links to adverse effects on bone, modification of chemokine expression by excess iron has not been investigated.
Minimal research has been conducted on the effects of tungsten, arsenic, strontium, or mercury on bone; even less attention has been paid to chemokine expression changes. Tungsten has been shown to inhibit osteoblast differentiation of MSC in vitro (174). Arsenic was also shown to inhibit differentiation of osteoblasts, which occurred via ERK signaling. In vivo, arsenic exposure resulted in decreased BMD and altered bone microstructure in the rat (174, 175). Exposure to mercury occurs primarily through human consumption of fish (176), which has been shown to cause decreased activity of osteoclasts, with little, if any increased activity of osteoblasts (177). Some studies have suggested that high blood levels of mercury may in fact lower the risk of postmenopausal osteoporosis (178, 179). However, we found no studies investigating mercury’s effects on chemokine expression. Further research is needed to better understand the mechanisms of action of these toxicants.
Aseptic periprosthetic osteolysis is one of the most common causes of long-term prosthetic joint failure. Reports have cited failure rates as high as 56% at 6.5 years postoperative (180–182). Constant friction through the joint surface of metal-on-metal (MoM) and polyethylene-on-metal prostheses generates non-biodegradable particulate debris. In addition to polyethylene particles, metal ions such as titanium (Ti), titanium-6%/aluminum-4%/vanadium (Ti6Al4V) alloy, zirconium (Zr) oxide, Zr alloy, cobalt, cobalt chrome alloy, cobalt nickel chrome alloy, and cobalt chrome molybdenum (CoCrMo) alloy are produced from wear. Elevated concentrations of these metals have been measured in periprosthetic tissue, serum, urine, and in distant organs (liver, lymph nodes, spleen) (183–186).
Metal-on-metal implants became very popular in the late 1980s, with over one million implanted in the US to date (187). Because they are thought to deteriorate more slowly, MoM implants are used increasingly in young patients. However, elevated serum levels of metal ions, adverse reactions to metal debris, aseptic lymphocyte-dominated vasculitis, pseudotumors, and metal hypersensitivity are thought to lead to early loosening (188–199).
Metal-on-metal implants were initially thought to cause an immune reaction solely through the macrophage response; however, recent studies suggest that systemic metal ions, organometallic protein complexes, and particulate debris can also be immunoreactive (200). The mechanisms leading to pathologic bone resorption surrounding MoM implants are poorly understood. Numerous inflammatory cytokines and chemokines have been shown to be upregulated in peri-implant tissues of these patients, leading to a state of chronic inflammation and osteoclast activation/proliferation (201). Drynda et al. sought to clarify how circulating metal ions affect the CXCL12/CXCR4 axis and determine the impact this may have in BMSC homing/differentiation. Cultures of MG-63 (early-stage) and SaOs-2 (late-stage) osteoblast-like cells were exposed to Cobalt (Co), CoNiCrMo alloy (Nickel containing), or CoCrMo alloy (Nickel free). CXCL12/CXCR4 protein expression was dose-dependently activated in both cell lines with all metal exposures (202). Upregulation of CXCL12 in preosteoclasts has been associated with increased osteoclastic activity in several bone diseases, which may participate in eventual periprosthetic osteolysis (203–205). When AMD3100 was administered to block the interaction of CXCL12 with CXCR4, a partial reduction in TNF-α expression was observed. The same group also recovered periprosthetic tissue from patients undergoing revision for MoM aseptic loosening and found CXCR4 to be upregulated. Others have also reported similar increases in CXCR4 and TNF-α in vivo after exposure to Ti ions (206). Although no direct correlation can be drawn from either of these studies, it is possible that TNF-α may act as a trigger for CXCR4 upregulation.
Ti has been shown to increase CCL17 (TARC), CCL22 (MDC), and CCR4 expression (207). Cadosch et al. found that secretion of pro-inflammatory TNF-α, IL-6, and IL-1a/β was increased after Ti exposure, thereby leading to either direct osteoclast precursor activation or indirect activation through RANKL/M-CSF secretion by osteoblasts. RANKL activation also increased osteoclastic expression of CCL22 (208, 209), which lead to increased recruitment of CCR4+ osteoclast precursors. Correspondingly, there was an increase in CCL17 expression in hFOB 1.19 fetal osteoblastic cells and human osteoclasts. CCL17 functions in chemotactic recruitment of osteoclast precursors, likely through NF-kB activation (207).
Recent studies suggest that CCR4 is expressed in both Th2 and Th17 cells, and microscopic analysis of periprosthetic tissue reveals an observed increase in Th17 cell number (210). These periprosthetic Th17 cells may have been recruited and deposited through CCL22+ CCL17/CCR4-mediated chemotaxis and arrest (210). Furthermore, these deposited Th17 cells promote osteolysis through an IL-17-dependent increase in RANKL. Collectively, this is suggestive of a Ti-induced vicious cycle of osteoclastogenesis and inflammation (207).
Interestingly, the majority of Ti particle-induced osteolysis is associated with CXCR2, which is the receptor for IL-8 (211, 212). This receptor can be found on macrophages, osteoclasts, osteoblasts, and neutrophils (213), where ligand binding promotes neutrophil attraction, osteoclastic differentiation, and bone resorption (214). Silencing CXCR2 mRNA reduced Ti-induced bone resorption rates in a mouse calvarial defect model by suppressing osteoclastogenesis indirectly through osteoblastic downregulation of RANKL (215). Several other groups have observed Ti-induced upregulation of CXCL8 (IL-8), CCL5 (RANTES), CCL3 (MIP-1α), and CCL2 (MCP-1) as a function of time-dependent NF-kB activation (216–220). Of these, IL-8 and CCL2 are strong chemoattractants for monocytes/macrophages/osteoclasts and neutrophils (221), which recruit bone-resorbing cells to induce periprosthetic osteolysis. Similar results were found with cobalt exposure in cultured human osteoblasts (222–224). Dalal et al. found that a CoCrMo alloy produced the greatest inflammatory response in comparison to Ti, Zr oxide, or Zr alloy, with a 100-fold increase (>2,000 pg/mL) of IL-8, a 30-fold increase of IL-6, and a 15-fold increase of TNF-α levels (224).
Metal particle-induced osteolysis is largely due to excessive osteoclast/inflammatory cell recruitment, osteoclastic activation, and inhibition of osteoblast activity. This occurs locally through phagocytosis and/or by the metal ions triggering toll-like receptors (214). These processes (is this true?) are mediated primarily by chemokines (225). Thus far, CXCL12/CXCR4, CCL17/CCL22/CCR4, IL-8/CXCR2, CCL5, CCL3, and CCL2 have all been implicated as mediators of prosthetic metal-induced bone destruction (Table 5). More research is needed to identify the chemokines responsible for recruitment of osteoclasts and Th17 cells, which in turn contribute to osteolysis and aseptic loosening. This understanding would provide for targeted approach to early diagnosis and treatment of prosthetic-induced loosening.
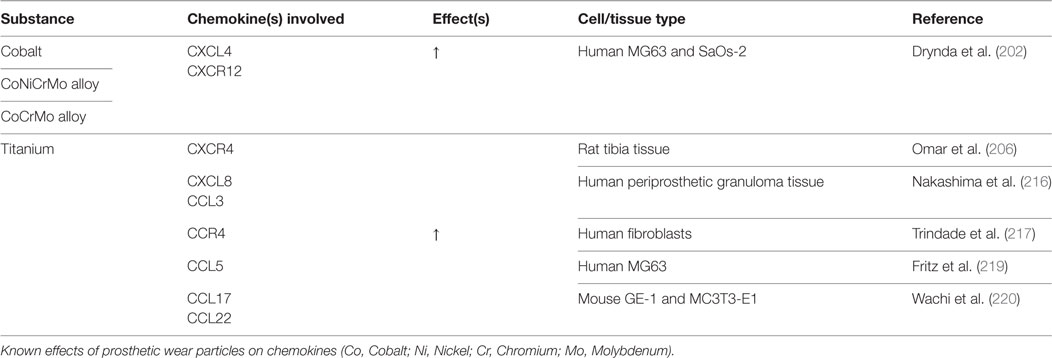
Table 5. Prosthetic wear particles: chemokine changes.
Fluoride is found naturally and as an additive in tap water. It is well known for its ability to reduce the rate of dental caries. In low doses, fluoride increases osteoblast proliferation (226) and inhibits osteoclastic bone resorption (226, 227). Some clinical evidence suggests that fluoride may improve BMD (228), although other studies suggest that this more dense bone may in fact be more brittle and prone to fracture (226).
With high dose or prolonged exposure time, skeletal fluorosis and other toxic side effects may ensue. Skeletal fluorosis occurs secondarily to an increase in bone density, through a supraphysiologic increase of the growth of osteophytes. This results in symptoms and disability similar to what is observed in osteoarthritic patients. A safe level of fluoride in drinking water has proven difficult to establish. Many factors, including climate, air temperature, patient age, duration of exposure, and dietary calcium intake may play a role in the variability of doses reported in the literature, as each of these can alter fluoride bioavailability (229, 230). In an attempt to maintain safe doses of fluoride in drinking water, the EPA established and enforces a maximum allowable fluoride concentration of 4 mg/L; however, given recent experimental findings, the EPA now suggests maintaining fluoride levels at less than 2 mg/L to prevent possible deleterious effects.
Rat osteoblasts undergo apoptosis upon exposure to sodium fluoride (NaF) at doses of 500 µM and higher (231). At lower doses however, fluoride has positive effects on Wnt signaling, which leads to upregulation of osteoblastic differentiation makers, including ALP, COL1A1, osteonectin, and RunX2 in rat primary osteoblasts (232). Fluoride was also found to increase phosphorylation and inhibition of GSK-3β, resulting in prolonged activation of the Wnt pathway. Other groups have found COL1A1 and COL1A2 mRNA levels to be induced in rat osteoblasts after 24-h NaF exposure, although by 72 h, expression decreases in a dose-dependent fashion (231).
In addition to its direct effects on osteoblasts, fluoride has also been linked to anti-osteoclastic activity, with reduced cathepsin K, IL-1B, MMP-9, and TRAP activity upon RANKL- and M-CSF-induced osteoclastogenesis (233). In IL-1B-induced gingival inflammation, NaF was strongly anti-inflammatory, downregulating IL-1B, IL-8, and TNF-α expression at doses that did not induce apoptosis (234); however, the effects of fluoride on inflammation appear to depend on route of administration and are tissue specific (235–237). The inflammatory effects for fluoride in the context of bone have not been studied.
Fluoride may play a more direct role in calcium metabolism and bone turnover. In MC3T3E1 preosteoblasts, low-dose fluoride increased free calcium ion in the cell culture supernatant and inhibited PTH and PTHrP expression, with opposite effects on Ca++ and PTHrP at high NaF doses. In vivo, the same group found that NaF reduced serum calcium levels, and the effect of fluoride on PTHrP depended on whether the rats received standard or low calcium diet (238). These results suggest that high fluoride ingestion causes hypocalcemia by upregulating calcitonin through PTH and PTHrP secretion (238). Fluoride treatment also increased RANKL and OPG expression, hypothetically promoting osteoclast activation. Further research regarding chemokine signaling after fluoride exposure may explain the dose-dependent effects of fluoride on skeletal health.
Fungi and molds are common naturally occurring environmental contaminants. Toxic effects related to mold exposure are most commonly caused by mycotoxins and the various VOCs they produce. A study by Hokeness et al. evaluated the effects of two common VOCs ((E)-2-octenal and oct-1-en-3-ol) on BMSC viability (239). Both VOCs were cytotoxic, and the effect was determined to be secondary to alterations to membrane fluidity. The authors postulated that this could lead to breakdown of the immune system, but did not comment on the potential impact this could have on bone. However, membrane fluidity is known to affect osteoblastic differentiation of MSC (240).
The active compound from the mushroom Cordyceps militaris, cordycepin, is used medicinally for anti-inflammatory and chemotherapeutic purposes. Both C. militaris extract (CME) and isolated cordycepin have been shown to reduce RANKL-mediated osteoclastogenesis in vitro. In an in vivo murine model, cordycepin reduced LPS-induced inflammatory bone loss. Further studies will need to be conducted to determine whether cordycepin treatment is suitable in humans for the prevention of bone loss.
Peripheral blood mononuclear cells from mold-exposed workers expressed higher levels of eotaxin, INF-α, IL-1α, IL-12 p40, IL-12 p70, IP-10, PDGF-AA, TNF-β, and VEGF when exposed to a panel of common mold toxins ex vivo relative to cells from control patients (241). Asthmatic patients had a significant difference in chemokine expression, regardless of mold exposure. When exposed to aflatoxin B1 in vitro, polymorphonuclear leukocytes harvested from peripheral blood demonstrated decreased IL-8, CXCL1, and CXCL2 (242). While these studies demonstrate a link between exposure to fungal toxins and chemokines, it is unclear whether this has an impact on bone health. Subsequent research will be necessary to determine if fungal extracts and toxins are related to bone regeneration through chemokine mediation.
Asbestos has a well-established role in inflammation and disease of respiratory tissues (243). Asbestos-derived ROS have been shown to activate TGF-β in lung epithelial tissue (244). CXCR3 levels have been reported to be decreased in CD4+ T-cells, inhibiting chemotaxis and potentially antitumor immunity in patients with asbestos-related lung disease (245). Other systemic markers of inflammation including IgE, IgA, IL-6, IL-8, and ICAM-1 have also been found to be elevated in asbestos-exposed persons (246). Upregulation of gremlin plays a role in asbestos-induced pulmonary fibrosis, which has been linked to decreased BMP and increased TGF-β signaling locally (247). No literature connecting asbestos to BMP, IL-8, or other chemokine levels in the context of bone were found. Further studies will be needed to determine if asbestos affects growth factor signaling in the bone, or has any impact on bone-related chemokines.
While chlorinated compounds have been studied, no research examining the relationship between chlorine itself and bone-related chemokines has been conducted. Chlorinated and fluorinated hydroxyapatite-based biomaterials are currently under development. The addition of chlorine and fluorine to these scaffolds creates an acidic environment that simulates the osteoclastic conditions necessary for bone resorption and remodeling while simultaneously increasing apatite formation in vitro (248). Further studies will be necessary to determine if chlorine impacts bone healing through chemokine modulation, which may enhance or inhibit the osseointegration of chlorinated biomaterials.
Environmental contaminants are ubiquitous in today’s world. Over the last several decades, public awareness that consumer goods are not necessarily safe has grown exponentially. Unfortunately, toxicological research cannot possibly keep pace with the rate at which new compounds are introduced into consumer goods, and a higher level of attention to the issue is prudent. Relatively little consideration has been given to the effects of environmental contaminants on bone and other musculoskeletal tissues, with even less focus on mechanisms of action. Considering the scale and impact of musculoskeletal disease and disorders on global health as well as the associated financial burden on the healthcare system, the MSK system should be a subject of major mechanistic toxicological research efforts.
There is a paucity of literature on the relationship between the majority of the environmental compounds reviewed here and chemokines relevant to bone. Many of the compounds discussed in this review warrant further research. Given their direct effects on PPARγ and adipogenic differentiation, organotins are of particular interest. Another subgroup of EDCs, the phthalate esters, have been shown to influence CXCL1 and CCL2 in other tissues. Given the known roles of CXCL1 and CCL2 in osteoclastogenesis, this could prove a high yield topic of study. The effect of prosthetic metals and periprosthetic wear particles may have on chemokines and bone regeneration is growing in clinical importance and represents another area of future focus. Finally, research on the effects of dioxins and dioxin-like compounds on bone-related chemokines will also continue to be a critical area of study, as their presence in cigarette smoke impacts such a large population.
The important roles that chemokines play in bone homeostasis and repair has become clear. Several therapeutics targeting chemokine receptors are now FDA-approved, albeit outside the context of MSK disease (1). A much more thorough understanding of the mechanisms by which environmental toxins adversely affect bone is critical to developing more effective, targeted approaches to mitigate these effects.
JS designed, drafted, edited manuscript, and interpreted basic science work. AS designed, drafted, edited manuscript, and interpreted basic science work. KK designed, drafted, edited manuscript, and interpreted basic science work. CY designed, performed original basic science work, and interpreted results. EH did experimental design and data interpretation and designed, directed, and edited manuscript.
EH reports: consulting (immediate family member): Stryker, Bacterin, Graftys, Globus, AONA, Synthes, Spinesmith, SI Bone, Relievant, Ceramtec, Medtronic, Pioneer, Bioventus, and LifeNet. Speaking and/or teaching arrangements (immediate family member): AONA. Trips/trave (immediate family member): Stryker, Pioneer Surgical, Medtronic, Bioventus, and AONA. Board of Directors (immediate family member): Lumbar Spine Research Society and Cervical Spine Research Society. Scientific Advisory Board (immediate family member): Bioventus. JS, AS, KK, and CY report no relevant disclosures. None of the above disclosures are relevant to this paper. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Dr. Paula Stern for her guidance in composing this review.
Funding for the original data was provided in part by the Orthopaedics Research and Education Foundation.
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fendo.2017.00022/full#supplementary-material.
1. Gilchrist A, Stern PH. Chemokines and bone. Clin Rev Bone Miner Metabol (2015) 13(2):61–82. doi: 10.1007/s12018-015-9184-y
2. Brantigan JW, Steffee AD, Lewis ML, Quinn LM, Persenaire JM. Lumbar interbody fusion using the Brantigan I/F cage for posterior lumbar interbody fusion and the variable pedicle screw placement system: two-year results from a Food and Drug Administration investigational device exemption clinical trial. Spine (2000) 25(11):1437–46. doi:10.1097/00007632-200006010-00017
3. Crain DA, Eriksen M, Iguchi T, Jobling S, Laufer H, LeBlanc GA, et al. An ecological assessment of bisphenol-A: evidence from comparative biology. Reprod Toxicol (2007) 24(2):225–39. doi:10.1016/j.reprotox.2007.05.008
4. Elango A, Shepherd B, Chen TT. Effects of endocrine disrupters on the expression of growth hormone and prolactin mRNA in the rainbow trout pituitary. Gen Comp Endocrinol (2006) 145(2):116–27. doi:10.1016/j.ygcen.2005.08.003
5. Fischer C, Mamillapalli R, Goetz LG, Jorgenson E, Ilagan Y, Taylor HS. Bisphenol A (BPA) exposure in utero leads to immunoregulatory cytokine dysregulation in the mouse mammary gland: a potential mechanism programming breast cancer risk. Horm Cancer (2016) 7(4):241–51. doi:10.1007/s12672-016-0254-5
6. Hall JM, Korach KS. Endocrine disrupting chemicals promote the growth of ovarian cancer cells via the ER-CXCL12-CXCR4 signaling axis. Mol Carcinog (2013) 52(9):715–25. doi:10.1002/mc.21913
7. Habauzit D, Boudot A, Kerdivel G, Flouriot G, Pakdel F. Development and validation of a test for environmental estrogens: checking xeno-estrogen activity by CXCL12 secretion in BREAST CANCER CELL LINES (CXCL-test). Environ Toxicol (2010) 25(5):495–503. doi:10.1002/tox.20594
8. Gertz J, Reddy TE, Varley KE, Garabedian MJ, Myers RM. Genistein and bisphenol A exposure cause estrogen receptor 1 to bind thousands of sites in a cell type-specific manner. Genome Res (2012) 22(11):2153–62. doi:10.1101/gr.135681.111
9. Li M, Han X, Gao W, Chen F, Shao B. Bisphenol AF stimulates transcription and secretion of C-X-C chemokine ligand 12 to promote proliferation of cultured T47D breast cancer cells. Toxicology (2015) 338:30–6. doi:10.1016/j.tox.2015.09.007
10. Nishioka J, Iwahara C, Kawasaki M, Yoshizaki F, Nakayama H, Takamori K, et al. Di-(2-ethylhexyl) phthalate induces production of inflammatory molecules in human macrophages. Inflamm Res (2012) 61(1):69–78. doi:10.1007/s00011-011-0390-x
11. Win-Shwe TT, Yanagisawa R, Koike E, Nitta H, Takano H. Expression levels of neuroimmune biomarkers in hypothalamus of allergic mice after phthalate exposure. J Appl Toxicol (2013) 33(10):1070–8. doi:10.1002/jat.2835
12. Stein CR, Ge Y, Wolff MS, Ye X, Calafat AM, Kraus T, et al. Perfluoroalkyl substance serum concentrations and immune response to FluMist vaccination among healthy adults. Environ Res (2016) 149:171–8. doi:10.1016/j.envres.2016.05.020
13. Rubin BS. Bisphenol A: an endocrine disruptor with widespread exposure and multiple effects. J Steroid Biochem Mol Biol (2011) 127(1–2):27–34. doi:10.1016/j.jsbmb.2011.05.002
14. Kang JH, Kondo F, Katayama Y. Human exposure to bisphenol A. Toxicology (2006) 226(2–3):79–89. doi:10.1016/j.tox.2006.06.009
15. Calafat AM, Ye X, Wong LY, Reidy JA, Needham LL. Exposure of the U.S. population to bisphenol A and 4-tertiary-octylphenol: 2003-2004. Environ Health Perspect (2008) 116(1):39–44. doi:10.1289/ehp.10753
16. Rochester JR. Bisphenol A and human health: a review of the literature. Reprod Toxicol (2013) 42:132–55. doi:10.1016/j.reprotox.2013.08.008
17. De Coster S, van Larebeke N. Endocrine-disrupting chemicals: associated disorders and mechanisms of action. J Environ Public Health (2012) 2012:713696. doi:10.1155/2012/713696
18. Matthews J, Celius T, Halgren R, Zacharewski T. Differential estrogen receptor binding of estrogenic substances: a species comparison. J Steroid Biochem Mol Biol (2000) 74(4):223–34. doi:10.1016/S0960-0760(00)00126-6
19. Kuiper GGJM, Lemmen JG, Carlsson B, Corton JC, Safe SH, Van Der Saag PT, et al. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor β. Endocrinology (1998) 139(10):4252–63. doi:10.1210/endo.139.10.6216
20. Srivastava S, Toraldo G, Weitzmann MN, Cenci S, Ross FP, Pacifici R. Estrogen decreases osteoclast formation by down-regulating receptor activator of NF-κB ligand (RANKL)-induced JNK activation. J Biol Chem (2001) 276(12):8836–40. doi:10.1074/jbc.M010764200
21. Kousteni S, Almeida M, Han L, Bellido T, Jilka RL, Manolagas SC. Induction of osteoblast differentiation by selective activation of kinase-mediated actions of the estrogen receptor. Mol Cell Biol (2007) 27(4):1516–30. doi:10.1128/MCB.01550-06
22. Hwang JK, Min KH, Choi KH, Hwang YC, Jeong IK, Ahn KJ, et al. Bisphenol A reduces differentiation and stimulates apoptosis of osteoclasts and osteoblasts. Life Sci (2013) 93(9–11):367–72. doi:10.1016/j.lfs.2013.07.020
23. Hsu EL, Yoon D, Choi HH, Wang F, Taylor RT, Chen N, et al. A proposed mechanism for the protective effect of dioxin against breast cancer. Toxicol Sci (2007) 98(2):436–44. doi:10.1093/toxsci/kfm125
24. Schettler T. Human exposure to phthalates via consumer products. Int J Androl (2006) 29(1):134–9; discussion 81–5. doi:10.1111/j.1365-2605.2005.00567.x
25. Ignasiak Z, Slawinska T, Rozek K, Little BB, Malina RM. Lead and growth status of school children living in the copper basin of south-western Poland: differential effects on bone growth. Ann Hum Biol (2006) 33(4):401–14. doi:10.1080/03014460600730752
26. Bhat FA, Ramajayam G, Parameswari S, Vignesh RC, Karthikeyan S, Senthilkumar K, et al. Di 2-ethyl hexyl phthalate affects differentiation and matrix mineralization of rat calvarial osteoblasts – in vitro. Toxicol In Vitro (2013) 27(1):250–6. doi:10.1016/j.tiv.2012.09.003
27. Hardy R, Cooper MS. Bone loss in inflammatory disorders. J Endocrinol (2009) 201(3):309–20. doi:10.1677/JOE-08-0568
28. Schett G, Kiechl S, Weger S, Pederiva A, Mayr A, Petrangeli M, et al. High-sensitivity c-reactive protein and risk of nontraumatic fractures in the bruneck study. Arch Intern Med (2006) 166(22):2495–501. doi:10.1001/archinte.166.22.2495
29. Onan D, Allan EH, Quinn JM, Gooi JH, Pompolo S, Sims NA, et al. The chemokine Cxcl1 is a novel target gene of parathyroid hormone (PTH)/PTH-related protein in committed osteoblasts. Endocrinology (2009) 150(5):2244–53. doi:10.1210/en.2008-1597
30. Williams SR, Jiang Y, Cochran D, Dorsam G, Graves DT. Regulated expression of monocyte chemoattractant protein-1 in normal human osteoblastic cells. Am J Physiol (1992) 263(1 Pt 1):C194–9.
31. Khan UA, Hashimi SM, Bakr MM, Forwood MR, Morrison NA. CCL2 and CCR2 are essential for the formation of osteoclasts and foreign body giant cells. J Cell Biochem (2016) 117(2):382–9. doi:10.1002/jcb.25282
32. Khalil N, Chen A, Lee M, Czerwinski SA, Ebert JR, DeWitt JC, et al. Association of perfluoroalkyl substances, bone mineral density, and osteoporosis in the U.S. population in NHANES 2009–2010. Environ Health Perspect (2016) 124(1):81–7. doi:10.1289/ehp.1307909
33. White SS, Fenton SE, Hines EP. Endocrine disrupting properties of perfluorooctanoic acid. J Steroid Biochem Mol Biol (2011) 127(1–2):16–26. doi:10.1016/j.jsbmb.2011.03.011
34. Lin CY, Chen PC, Lin YC, Lin LY. Association among serum perfluoroalkyl chemicals, glucose homeostasis, and metabolic syndrome in adolescents and adults. Diabetes Care (2009) 32(4):702–7. doi:10.2337/dc08-1816
35. Fromme H, Tittlemier SA, Volkel W, Wilhelm M, Twardella D. Perfluorinated compounds – exposure assessment for the general population in Western countries. Int J Hyg Environ Health (2009) 212(3):239–70. doi:10.1016/j.ijheh.2008.04.007
36. Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Tully JS, Needham LL. Serum concentrations of 11 polyfluoroalkyl compounds in the U.S. population: data from the national health and nutrition examination survey (NHANES). Environ Sci Technol (2007) 41(7):2237–42. doi:10.1021/es062686m
37. Kato K, Wong LY, Jia LT, Kuklenyik Z, Calafat AM. Trends in exposure to polyfluoroalkyl chemicals in the U.S. population: 1999-2008. Environ Sci Technol (2011) 45(19):8037–45. doi:10.1021/es1043613
38. Baker AH, Watt J, Huang CK, Gerstenfeld LC, Schlezinger JJ. Tributyltin engages multiple nuclear receptor pathways and suppresses osteogenesis in bone marrow multipotent stromal cells. Chem Res Toxicol (2015) 28(6):1156–66. doi:10.1021/tx500433r
39. Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev (1994) 8(10):1224–34. doi:10.1101/gad.8.10.1224
40. Fent K. Ecotoxicology of organotin compounds. Crit Rev Toxicol (1996) 26(1):1–117. doi:10.3109/10408449609089891
41. Tsukamoto Y, Ishihara Y, Miyagawa-Tomita S, Hagiwara H. Inhibition of ossification in vivo and differentiation of osteoblasts in vitro by tributyltin. Biochem Pharmacol (2004) 68(4):739–46. doi:10.1016/j.bcp.2004.04.020
42. Sarpa M, De-Carvalho RR, Delgado IF, Paumgartten FJR. Developmental toxicity of triphenyltin hydroxide in mice. Regul Toxicol Pharmacol (2007) 49(1):43–52. doi:10.1016/j.yrtph.2007.05.006
43. Schutte RJ, Xie L, Klitzman B, Reichert WM. In vivo cytokine-associated responses to biomaterials. Biomaterials (2009) 30(2):160–8. doi:10.1016/j.biomaterials.2008.09.026
44. Yu X, Huang Y, Collin-Osdoby P, Osdoby P. CCR1 chemokines promote the chemotactic recruitment, RANKL development, and motility of osteoclasts and are induced by inflammatory cytokines in osteoblasts. J Bone Miner Res (2004) 19(12):2065–77. doi:10.1359/jbmr.040910
45. Cooke TF. Indoor air pollutants a literature review. Rev Environ Health (1991) 9(3):137–60. doi:10.1515/REVEH.1991.9.3.137
46. Samet JM, Marbury MC, Spengler JD. Health effects and sources of indoor air pollution. Part II. Am Rev Respir Dis (1988) 137(1):221–42. doi:10.1164/ajrccm/137.1.221
47. Sexton K, Adgate JL, Mongin SJ, Pratt GC, Ramachandran G, Stock TH, et al. Evaluating differences between measured personal exposures to volatile organic compounds and concentrations in outdoor and indoor air. Environ Sci Technol (2004) 38(9):2593–602. doi:10.1021/es030607q
48. Diez U, Kroeßner T, Rehwagen M, Richter M, Wetzig H, Schulz R, et al. Effects of indoor painting and smoking on airway symptoms in atopy risk children in the first year of life results of the LARS-study. Int J Hyg Environ Health (2000) 203(1):23–8. doi:10.1078/S1438-4639(04)70004-8
49. Ware JH, Spengler JD, Neas LM, Samet JM, Wagner GR, Coultas D, et al. Respiratory and irritant health effects of ambient volatile organic compounds: the kanawha county health study. Am J Epidemiol (1993) 137(12):1287–301.
50. Rumchev K, Spickett J, Bulsara M, Phillips M, Stick S. Association of domestic exposure to volatile organic compounds with asthma in young children. Thorax (2004) 59(9):746–51. doi:10.1136/thx.2003.013680
51. Wieslander G, Norbäck D, Björnsson E, Janson C, Boman G. Asthma and the indoor environment: the significance of emission of formaldehyde and volatile organic compounds from newly painted indoor surfaces. Int Arch Occup Environ Health (1997) 69(2):115–24. doi:10.1007/s004200050125
52. Pappas GP, Herbert RJ, Henderson W, Koenig J, Stover B, Barnhart S. The respiratory effects of volatile organic compounds. Int J Occup Environ Health (2000) 6(1):1–8. doi:10.1179/oeh.2000.6.1.1
53. Zolghadr F, Sadeghizadeh M, Amirizadeh N, Hosseinkhani S, Nazem S. How benzene and its metabolites affect human marrow derived mesenchymal stem cells. Toxicol Lett (2012) 214(2):145–53. doi:10.1016/j.toxlet.2012.08.015
54. Gillis B, Gavin IM, Arbieva Z, King ST, Jayaraman S, Prabhakar BS. Identification of human cell responses to benzene and benzene metabolites. Genomics (2007) 90(3):324–33. doi:10.1016/j.ygeno.2007.05.003
55. Lehmann I, Roder-Stolinski C, Nieber K, Fischader G. In vitro models for the assessment of inflammatory and immuno-modulatory effects of the volatile organic compound chlorobenzene. Exp Toxicol Pathol (2008) 60(2–3):185–93. doi:10.1016/j.etp.2008.01.009
56. Fischader G, Roder-Stolinski C, Wichmann G, Nieber K, Lehmann I. Release of MCP-1 and IL-8 from lung epithelial cells exposed to volatile organic compounds. Toxicol In Vitro (2008) 22(2):359–66. doi:10.1016/j.tiv.2007.09.015
57. Minciullo PL, Navarra M, Calapai G, Gangemi S. Cytokine network involvement in subjects exposed to benzene. J Immunol Res (2014) 2014:937987. doi:10.1155/2014/937987
58. Vermeulen R, Lan Q, Zhang L, Gunn L, McCarthy D, Woodbury RL, et al. Decreased levels of CXC-chemokines in serum of benzene-exposed workers identified by array-based proteomics. Proc Natl Acad Sci U S A (2005) 102(47):17041–6. doi:10.1073/pnas.0508573102
59. WHO. Some industrial chemicals and dyestuffs. IARC Monogr Eval Carcinog Risk Chem Hum (1982) 29:1–398.
60. Aksoy M. Malignancies due to occupational exposure to benzene. Am J Ind Med (1985) 7(5–6):395–402. doi:10.1002/ajim.4700070506
61. Austin H, Delzell E, Cole P. Benzene and leukemia. A review of the literature and a risk assessment. Am J Epidemiol (1988) 127(3):419–39.
62. Baan R, Grosse Y, Straif K, Secretan B, El Ghissassi F, Bouvard V, et al. A review of human carcinogens – part F: chemical agents and related occupations. Lancet Oncol (2009) 10(12):1143–4. doi:10.1016/S1470-2045(09)70358-4
63. McHale CM, Zhang L, Smith MT. Current understanding of the mechanism of benzene-induced leukemia in humans: implications for risk assessment. Carcinogenesis (2012) 33(2):240–52. doi:10.1093/carcin/bgr297
64. Wilbur S, Wohlers D, Paikoff S, Keith LS, Faroon O. ATSDR evaluation of potential for human exposure to benzene. Toxicol Ind Health (2008) 24(5–6):399–442. doi:10.1177/0748233708095772
65. Snyder R, Witz G, Goldstein BD. The toxicology of benzene. Environ Health Perspect (1993) 100:293–306. doi:10.1289/ehp.93100293
66. Rickert DE, Baker TS, Bus JS, Barrow CS, Irons RD. Benzene disposition in the rat after exposure by inhalation. Toxicol Appl Pharmacol (1979) 49(3):417–23. doi:10.1016/0041-008X(79)90441-1
67. Atkinson TJ. A review of the role of benzene metabolites and mechanisms in malignant transformation: summative evidence for a lack of research in nonmyelogenous cancer types. Int J Hyg Environ Health (2009) 212(1):1–10. doi:10.1016/j.ijheh.2007.09.013
68. Giuliano M, Stellavato A, Cammarota M, Lamberti M, Miraglia N, Sannolo N, et al. Effects of low concentrations of benzene on human lung cells in vitro. Toxicol Lett (2009) 188(2):130–6. doi:10.1016/j.toxlet.2009.03.018
69. Zhu W, Boachie-Adjei O, Rawlins BA, Frenkel B, Boskey AL, Ivashkiv LB, et al. A novel regulatory role for stromal-derived factor-1 signaling in bone morphogenic protein-2 osteogenic differentiation of mesenchymal C2C12 cells. J Biol Chem (2007) 282(26):18676–85. doi:10.1074/jbc.M610232200
70. Ratajczak MZ, Kim CH, Abdel-Latif A, Schneider G, Kucia M, Morris AJ, et al. A novel perspective on stem cell homing and mobilization: review on bioactive lipids as potent chemoattractants and cationic peptides as underappreciated modulators of responsiveness to SDF-1 gradients. Leukemia (2012) 26(1):63–72. doi:10.1038/leu.2011.242
71. Kortesidis A, Zannettino A, Isenmann S, Shi S, Lapidot T, Gronthos S. Stromal-derived factor-1 promotes the growth, survival, and development of human bone marrow stromal stem cells. Blood (2005) 105(10):3793–801. doi:10.1182/blood-2004-11-4349
72. Tamura M, Sato MM, Nashimoto M. Regulation of CXCL12 expression by canonical Wnt signaling in bone marrow stromal cells. Int J Biochem Cell Biol (2011) 43(5):760–7. doi:10.1016/j.biocel.2011.01.021
73. Willhite CC, Book SA. Toxicology update: chlorobenzene. J Appl Toxicol (1990) 10(4):307–10. doi:10.1002/jat.2550100414
75. Lehmann I, Rehwagen M, Diez U, Seiffart A, Rolle-Kampczyk U, Richter M, et al. Enhanced in vivo IgE production and T cell polarization toward the type 2 phenotype in association with indoor exposure to VOC: results of the LARS study. Int J Hyg Environ Health (2001) 204(4):211–21. doi:10.1078/1438-4639-00100
76. Roder-Stolinski C, Fischader G, Oostingh GJ, Eder K, Duschl A, Lehmann I. Chlorobenzene induces the NF-kappa B and p38 MAP kinase pathways in lung epithelial cells. Inhal Toxicol (2008) 20(9):813–20. doi:10.1080/08958370802020810
77. Frumkin H. Agent orange and cancer: an overview for clinicians. CA Cancer J Clin (2003) 53(4):245–55. doi:10.3322/canjclin.53.4.245
78. Sorg O. AhR signalling and dioxin toxicity. Toxicol Lett (2014) 230(2):225–33. doi:10.1016/j.toxlet.2013.10.039
79. Nguyen NT, Hanieh H, Nakahama T, Kishimoto T. The roles of aryl hydrocarbon receptor in immune responses. Int Immunol (2013) 25(6):335–43. doi:10.1093/intimm/dxt011
80. Casado FL, Singh KP, Gasiewicz TA. Aryl hydrocarbon receptor activation in hematopoietic stem/progenitor cells alters cell function and pathway-specific gene modulation reflecting changes in cellular trafficking and migration. Mol Pharmacol (2011) 80(4):673–82. doi:10.1124/mol.111.071381
81. Wejheden C, Brunnberg S, Larsson S, Lind PM, Andersson G, Hanberg A. Transgenic mice with a constitutively active aryl hydrocarbon receptor display a gender-specific bone phenotype. Toxicol Sci (2010) 114(1):48–58. doi:10.1093/toxsci/kfp284
82. Brankovic J, Jovanovski S, Jevnikar P, Hofmeister A, Reininger-Gutmann B, Jan J, et al. Alterations in geometry, biomechanics, and mineral composition of juvenile rat femur induced by nonplanar PCB-155 and/or planar PCB-169. Environ Toxicol (2016). doi:10.1002/tox.22309
83. Yu TY, Pang WJ, Yang GS. Aryl hydrocarbon receptors in osteoclast lineage cells are a negative regulator of bone mass. PLoS One (2015) 10(1):e0117112. doi:10.1371/journal.pone.0117112
84. Hodgson S, Thomas L, Fattore E, Lind PM, Alfven T, Hellström L, et al. Bone mineral density changes in relation to environmental PCB exposure. Environ Health Perspect (2008) 116(9):1162–6. doi:10.1289/ehp.11107
85. Herlin M, Finnila MA, Zioupos P, Aula A, Risteli J, Miettinen HM, et al. New insights to the role of aryl hydrocarbon receptor in bone phenotype and in dioxin-induced modulation of bone microarchitecture and material properties. Toxicol Appl Pharmacol (2013) 273(1):219–26. doi:10.1016/j.taap.2013.09.002
86. Hermsen SA, Larsson S, Arima A, Muneoka A, Ihara T, Sumida H, et al. In utero and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) affects bone tissue in rhesus monkeys. Toxicology (2008) 253(1–3):147–52. doi:10.1016/j.tox.2008.09.005
87. Lind PM, Wejheden C, Lundberg R, Alvarez-Lloret P, Hermsen SA, Rodriguez-Navarro AB, et al. Short-term exposure to dioxin impairs bone tissue in male rats. Chemosphere (2009) 75(5):680–4. doi:10.1016/j.chemosphere.2008.12.024
88. Yu H, Du Y, Zhang X, Sun Y, Li S, Dou Y, et al. The aryl hydrocarbon receptor suppresses osteoblast proliferation and differentiation through the activation of the ERK signaling pathway. Toxicol Appl Pharmacol (2014) 280(3):502–10. doi:10.1016/j.taap.2014.08.025
89. Hsu EL, Sonn K, Kannan A, Bellary S, Yun C, Hashmi S, et al. Dioxin exposure impairs BMP-2-mediated spinal fusion in a rat arthrodesis model. J Bone Joint Surg Am (2015) 97(12):1003–10. doi:10.2106/JBJS.N.01311
90. Vogel CF, Khan EM, Leung PS, Gershwin ME, Chang WL, Wu D, et al. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: a role for nuclear factor-kappaB. J Biol Chem (2014) 289(3):1866–75. doi:10.1074/jbc.M113.505578
91. Vogel CF, Sciullo E, Li W, Wong P, Lazennec G, Matsumura F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol Endocrinol (2007) 21(12):2941–55. doi:10.1210/me.2007-0211
92. Mulero-Navarro S, Fernandez-Salguero PM. New trends in Aryl hydrocarbon receptor biology. Front Cell Dev Biol (2016) 4:45. doi:10.3389/fcell.2016.00045
93. Nguyen NT, Nakahama T, Nguyen CH, Tran TT, Le VS, Chu HH, et al. Aryl hydrocarbon receptor antagonism and its role in rheumatoid arthritis. J Exp Pharmacol (2015) 7:29–35. doi:10.2147/JEP.S63549
94. Swedenborg E, Pongratz I. AhR and ARNT modulate ER signaling. Toxicology (2010) 268(3):132–8. doi:10.1016/j.tox.2009.09.007
95. Mandl M, Depping R. Hypoxia-inducible aryl hydrocarbon receptor nuclear translocator (ARNT) (HIF-1beta): is it a rare exception? Mol Med (2014) 20:215–20. doi:10.2119/molmed.2014.00032
96. Singh KP, Garrett RW, Casado FL, Gasiewicz TA. Aryl hydrocarbon receptor-null allele mice have hematopoietic stem/progenitor cells with abnormal characteristics and functions. Stem Cells Dev (2011) 20(5):769–84. doi:10.1089/scd.2010.0333
97. Veldhoen M, Hirota K, Christensen J, O’Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med (2009) 206(1):43–9. doi:10.1084/jem.20081438
98. Van Voorhis M, Fechner JH, Zhang X, Mezrich JD. The aryl hydrocarbon receptor: a novel target for immunomodulation in organ transplantation. Transplantation (2013) 95(8):983–90. doi:10.1097/TP.0b013e31827a3d1d
99. Nguyen NT, Nakahama T, Le DH, Van Son L, Chu HH, Kishimoto T. Aryl hydrocarbon receptor and kynurenine: recent advances in autoimmune disease research. Front Immunol (2014) 5:551. doi:10.3389/fimmu.2014.00551
100. Nakahama T, Hanieh H, Nguyen NT, Chinen I, Ripley B, Millrine D, et al. Aryl hydrocarbon receptor-mediated induction of the microRNA-132/212 cluster promotes interleukin-17-producing T-helper cell differentiation. Proc Natl Acad Sci U S A (2013) 110(29):11964–9. doi:10.1073/pnas.1311087110
101. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6(11):1123–32. doi:10.1038/ni1254
102. Won HY, Lee JA, Park ZS, Song JS, Kim HY, Jang SM, et al. Prominent bone loss mediated by RANKL and IL-17 produced by CD4+ T cells in TallyHo/JngJ mice. PLoS One (2011) 6(3):e18168. doi:10.1371/journal.pone.0018168
103. Fumoto T, Takeshita S, Ito M, Ikeda K. Physiological functions of osteoblast lineage and T cell-derived RANKL in bone homeostasis. J Bone Miner Res (2014) 29(4):830–42. doi:10.1002/jbmr.2096
104. Kung MH, Yukata K, O’Keefe RJ, Zuscik MJ. Aryl hydrocarbon receptor-mediated impairment of chondrogenesis and fracture healing by cigarette smoke and benzo(a)pyrene. J Cell Physiol (2012) 227(3):1062–70. doi:10.1002/jcp.22819
105. Gravallese EM. Osteopontin: a bridge between bone and the immune system. J Clin Invest (2003) 112(2):147–9. doi:10.1172/JCI200319190
106. Yamamoto N, Sakai F, Kon S, Morimoto J, Kimura C, Yamazaki H, et al. Essential role of the cryptic epitope SLAYGLR within osteopontin in a murine model of rheumatoid arthritis. J Clin Invest (2003) 112(2):181–8. doi:10.1172/JCI17778
107. Vogel CF, Nishimura N, Sciullo E, Wong P, Li W, Matsumura F. Modulation of the chemokines KC and MCP-1 by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice. Arch Biochem Biophys (2007) 461(2):169–75. doi:10.1016/j.abb.2007.01.015
108. Monteiro P, Gilot D, Le Ferrec E, Rauch C, Lagadic-Gossmann D, Fardel O. Dioxin-mediated up-regulation of aryl hydrocarbon receptor target genes is dependent on the calcium/calmodulin/CaMKIalpha pathway. Mol Pharmacol (2008) 73(3):769–77. doi:10.1124/mol.107.043125
109. Kobayashi S, Okamoto H, Iwamoto T, Toyama Y, Tomatsu T, Yamanaka H, et al. A role for the aryl hydrocarbon receptor and the dioxin TCDD in rheumatoid arthritis. Rheumatology (Oxford) (2008) 47(9):1317–22. doi:10.1093/rheumatology/ken259
110. Ishikawa S. Children’s immunology, what can we learn from animal studies (3): Impaired mucosal immunity in the gut by 2,3,7,8-tetraclorodibenzo-p-dioxin (TCDD): a possible role for allergic sensitization. J Toxicol Sci (2009) 34(Suppl 2):S349–61. doi:10.2131/jts.34.SP349
111. Marshall NB, Vorachek WR, Steppan LB, Mourich DV, Kerkvliet NI. Functional characterization and gene expression analysis of CD4+ CD25+ regulatory T cells generated in mice treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Immunol (2008) 181(4):2382–91. doi:10.4049/jimmunol.181.4.2382
112. Vogel CF, Li W, Wu D, Miller JK, Sweeney C, Lazennec G, et al. Interaction of aryl hydrocarbon receptor and NF-kappaB subunit RelB in breast cancer is associated with interleukin-8 overexpression. Arch Biochem Biophys (2011) 512(1):78–86. doi:10.1016/j.abb.2011.05.011
113. Yachiguchi K, Matsumoto N, Haga Y, Suzuki M, Matsumura C, Tsurukawa M, et al. Polychlorinated biphenyl (118) activates osteoclasts and induces bone resorption in goldfish. Environ Sci Pollut Res Int (2014) 21(10):6365–72. doi:10.1007/s11356-012-1347-5
114. Li X, Liu H, Qin L, Tamasi J, Bergenstock M, Shapses S, et al. Determination of dual effects of parathyroid hormone on skeletal gene expression in vivo by microarray and network analysis. J Biol Chem (2007) 282(45):33086–97. doi:10.1074/jbc.M705194200
115. Majkova Z, Smart E, Toborek M, Hennig B. Up-regulation of endothelial monocyte chemoattractant protein-1 by coplanar PCB77 is caveolin-1-dependent. Toxicol Appl Pharmacol (2009) 237(1):1–7. doi:10.1016/j.taap.2009.02.016
116. Tsuji G, Takahara M, Uchi H, Takeuchi S, Mitoma C, Moroi Y, et al. An environmental contaminant, benzo(a)pyrene, induces oxidative stress-mediated interleukin-8 production in human keratinocytes via the aryl hydrocarbon receptor signaling pathway. J Dermatol Sci (2011) 62(1):42–9. doi:10.1016/j.jdermsci.2010.10.017
117. N’Diaye M, Le Ferrec E, Lagadic-Gossmann D, Corre S, Gilot D, Lecureur V, et al. Aryl hydrocarbon receptor- and calcium-dependent induction of the chemokine CCL1 by the environmental contaminant benzo[a]pyrene. J Biol Chem (2006) 281(29):19906–15. doi:10.1074/jbc.M601192200
118. Morino-Koga S, Uchi H, Tsuji G, Takahara M, Kajiwara J, Hirata T, et al. Reduction of CC-chemokine ligand 5 by aryl hydrocarbon receptor ligands. J Dermatol Sci (2013) 72(1):9–15. doi:10.1016/j.jdermsci.2013.04.031
119. Lisignoli G, Toneguzzi S, Grassi F, Piacentini A, Tschon M, Cristino S, et al. Different chemokines are expressed in human arthritic bone biopsies: IFN-gamma and IL-6 differently modulate IL-8, MCP-1 and rantes production by arthritic osteoblasts. Cytokine (2002) 20(5):231–8. doi:10.1006/cyto.2002.2006
120. Van den Berg M, Birnbaum LS, Denison M, De Vito M, Farland W, Feeley M, et al. The 2005 World Health Organization reevaluation of human and mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol Sci (2006) 93(2):223–41. doi:10.1093/toxsci/kfl055
121. Lakatos B, Szentmihalyi K, Vinkler P, Balla J, Balla G. [The role of essential metal ions in the human organism and their oral supplementation to the human body in deficiency states]. Orv Hetil (2004) 145(25):1315–9.
122. Beier EE, Holz JD, Sheu TJ, Puzas JE. Elevated lifetime lead exposure impedes osteoclast activity and produces an increase in bone mass in adolescent mice. Toxicol Sci (2016) 149(2):277–88. doi:10.1093/toxsci/kfv234
123. Riemschneider S, Herzberg M, Lehmann J. Subtoxic doses of cadmium modulate inflammatory properties of murine RAW 264.7 macrophages. Biomed Res Int (2015) 2015:295303. doi:10.1155/2015/295303
124. Freitas M, Fernandes E. Zinc, cadmium and nickel increase the activation of NF-kappaB and the release of cytokines from THP-1 monocytic cells. Metallomics (2011) 3(11):1238–43. doi:10.1039/c1mt00050k
125. Satija NK, Sharma D, Afrin F, Tripathi RP, Gangenahalli G. High throughput transcriptome profiling of lithium stimulated human mesenchymal stem cells reveals priming towards osteoblastic lineage. PLoS One (2013) 8(1):e55769. doi:10.1371/journal.pone.0055769
126. Kim HK, Kim JE, Chung J, Park KH, Han KS, Cho HI. Lithium down-regulates the expression of CXCR4 in human neutrophils. J Trace Elem Med Biol (2007) 21(3):204–9. doi:10.1016/j.jtemb.2007.03.001
127. Buache E, Velard F, Bauden E, Guillaume C, Jallot E, Nedelec JM, et al. Effect of strontium-substituted biphasic calcium phosphate on inflammatory mediators production by human monocytes. Acta Biomater (2012) 8(8):3113–9. doi:10.1016/j.actbio.2012.04.045
128. Mahaffey KR, Annest JL, Roberts J, Murphy RS. National estimates of blood lead levels: United States, 1976-1980: association with selected demographic and socioeconomic factors. N Engl J Med (1982) 307(10):573–9. doi:10.1056/NEJM198209023071001
129. CDC. Preventing Lead Poisoning in Young Children. Atlanta, GA: Centers for Disease Control (1991).
130. CDC. Prevent Lead Poisoning in Young Children. Atlanta, GA: Centers for Disease Control (2012).
131. Wedeen RP. Removing lead from bone: clinical implications of bone lead stores. Neurotoxicology (1992) 13(4):843–52.
132. Jackson LW, Cromer BA, Panneerselvamm A. Association between bone turnover, micronutrient intake, and blood lead levels in pre- and postmenopausal women, NHANES 1999-2002. Environ Health Perspect (2010) 118(11):1590–6. doi:10.1289/ehp.1002158
133. Khalil N, Cauley JA, Wilson JW, Talbott EO, Morrow L, Hochberg MC, et al. Relationship of blood lead levels to incident nonspine fractures and falls in older women: the study of osteoporotic fractures. J Bone Miner Res (2008) 23(9):1417–25. doi:10.1359/jbmr.080404
134. Campbell JR, Auinger P. The association between blood lead levels and osteoporosis among adults – results from the third national health and nutrition examination survey (NHANES III). Environ Health Perspect (2007) 115(7):1018–22. doi:10.1289/ehp.9716
135. Nash D, Magder LS, Sherwin R, Rubin RJ, Silbergeld EK. Bone density-related predictors of blood lead level among peri- and postmenopausal women in the United States: the Third National Health and Nutrition Examination Survey, 1988–1994. Am J Epidemiol (2004) 160(9):901–11. doi:10.1093/aje/kwh296
136. Beier EE, Maher JR, Sheu TJ, Cory-Slechta DA, Berger AJ, Zuscik MJ, et al. Heavy metal lead exposure, osteoporotic-like phenotype in an animal model, and depression of Wnt signaling. Environ Health Perspect (2013) 121(1):97–104. doi:10.1289/ehp.1205374
137. Campbell JR, Rosier RN, Novotny L, Puzas JE. The association between environmental lead exposure and bone density in children. Environ Health Perspect (2004) 112(11):1200–3. doi:10.1289/ehp.6555
138. Beier EE, Sheu T-J, Dang D, Holz JD, Ubayawardena R, Babij P, et al. Heavy metal ion regulation of gene expression: mechanisms by which lead inhibits osteoblastic bone-forming activity through modulation of the Wnt/β-catenin signaling pathway. J Biol Chem (2015) 290(29):18216–26. doi:10.1074/jbc.M114.629204
139. Beier EE, Sheu TJ, Buckley T, Yukata K, O’Keefe R, Zuscik MJ, et al. Inhibition of beta-catenin signaling by Pb leads to incomplete fracture healing. J Orthop Res (2014) 32(11):1397–405. doi:10.1002/jor.22677
140. Hagino N, Kono M. A study on the cause of Itai-itai disease. Proc 17th Mtg Japanese Society of Clinical Surgeons. (1955).
141. Olsson IM, Bensryd I, Lundh T, Ottosson H, Skerfving S, Oskarsson A. Cadmium in blood and urine – impact of sex, age, dietary intake, iron status, and former smoking – association of renal effects. Environ Health Perspect (2002) 110(12):1185–90. doi:10.1289/ehp.021101185
142. McElroy JA, Shafer MM, Trentham-Dietz A, Hampton JM, Newcomb PA. Urinary cadmium levels and tobacco smoke exposure in women age 20-69 years in the United States. J Toxicol Environ Health A (2007) 70(20):1779–82. doi:10.1080/15287390600754953
143. Jarup L, Akesson A. Current status of cadmium as an environmental health problem. Toxicol Appl Pharmacol (2009) 238(3):201–8. doi:10.1016/j.taap.2009.04.020
144. Abbas S, Khan K, Khan MP, Nagar GK, Tewari D, Maurya SK, et al. Developmental exposure to As, Cd, and Pb mixture diminishes skeletal growth and causes osteopenia at maturity via osteoblast and chondrocyte malfunctioning in female rats. Toxicol Sci (2013) 134(1):207–20. doi:10.1093/toxsci/kft093
145. Brzoska MM, Majewska K, Moniuszko-Jakoniuk J. Bone mineral density, chemical composition and biomechanical properties of the tibia of female rats exposed to cadmium since weaning up to skeletal maturity. Food Chem Toxicol (2005) 43(10):1507–19. doi:10.1016/j.fct.2005.04.008
146. Brzoska MM, Moniuszko-Jakoniuk J. Low-level lifetime exposure to cadmium decreases skeletal mineralization and enhances bone loss in aged rats. Bone (2004) 35(5):1180–91. doi:10.1016/j.bone.2004.07.010
147. Brzoska MM, Moniuszko-Jakoniuk J. Effect of chronic exposure to cadmium on the mineral status and mechanical properties of lumbar spine of male rats. Toxicol Lett (2005) 157(2):161–72. doi:10.1016/j.toxlet.2005.01.015
148. Yuan G, Lu H, Yin Z, Dai S, Jia R, Xu J, et al. Effects of mixed subchronic lead acetate and cadmium chloride on bone metabolism in rats. Int J Clin Exp Med (2014) 7(5):1378–85.
149. Duranova H, Martiniakova M, Omelka R, Grosskopf B, Bobonova I, Toman R. Changes in compact bone microstructure of rats subchronically exposed to cadmium. Acta Vet Scand (2014) 56:64. doi:10.1186/s13028-014-0064-0
150. Chen X, Zhu G, Gu S, Jin T, Shao C. Effects of cadmium on osteoblasts and osteoclasts in vitro. Environ Toxicol Pharmacol (2009) 28(2):232–6. doi:10.1016/j.etap.2009.04.010
151. Mountziaris PM, Spicer PP, Kasper FK, Mikos AG. Harnessing and modulating inflammation in strategies for bone regeneration. Tissue Eng Part B Rev (2011) 17(6):393–402. doi:10.1089/ten.TEB.2011.0182
152. Papa V, Bimonte VM, Wannenes F, D’Abusco AS, Fittipaldi S, Scandurra R, et al. The endocrine disruptor cadmium alters human osteoblast-like Saos-2 cells homeostasis in vitro by alteration of Wnt/beta-catenin pathway and activation of caspases. J Endocrinol Invest (2015) 38(12):1345–56. doi:10.1007/s40618-015-0380-x
153. Go YM, Orr M, Jones DP. Actin cytoskeleton redox proteome oxidation by cadmium. Am J Physiol Lung Cell Mol Physiol (2013) 305(11):L831–43. doi:10.1152/ajplung.00203.2013
154. Shahnazari M, Chu V, Wronski TJ, Nissenson RA, Halloran BP. CXCL12/CXCR4 signaling in the osteoblast regulates the mesenchymal stem cell and osteoclast lineage populations. FASEB J (2013) 27(9):3505–13. doi:10.1096/fj.12-225763
155. Bluml V, Regier MD, Hlavin G, Rockett IR, Konig F, Vyssoki B, et al. Lithium in the public water supply and suicide mortality in Texas. J Psychiatr Res (2013) 47(3):407–11. doi:10.1016/j.jpsychires.2012.12.002
156. Concha G, Broberg K, Grander M, Cardozo A, Palm B, Vahter M. High-level exposure to lithium, boron, cesium, and arsenic via drinking water in the Andes of northern Argentina. Environ Sci Technol (2010) 44(17):6875–80. doi:10.1021/es1010384
157. Kabacs N, Memon A, Obinwa T, Stochl J, Perez J. Lithium in drinking water and suicide rates across the East of England. Br J Psychiatry (2011) 198(5):406–7. doi:10.1192/bjp.bp.110.088617
158. Li J, Khavandgar Z, Lin SH, Murshed M. Lithium chloride attenuates BMP-2 signaling and inhibits osteogenic differentiation through a novel WNT/GSK3-independent mechanism. Bone (2011) 48(2):321–31. doi:10.1016/j.bone.2010.09.033
159. Vestergaard P, Rejnmark L, Mosekilde L. Reduced relative risk of fractures among users of lithium. Calcif Tissue Int (2005) 77(1):1–8. doi:10.1007/s00223-004-0258-y
160. Zamani A, Omrani GR, Nasab MM. Lithium’s effect on bone mineral density. Bone (2009) 44(2):331–4. doi:10.1016/j.bone.2008.10.001
161. Wilting I, de Vries F, Thio BM, Cooper C, Heerdink ER, Leufkens HG, et al. Lithium use and the risk of fractures. Bone (2007) 40(5):1252–8. doi:10.1016/j.bone.2006.12.055
162. Clement-Lacroix P, Ai M, Morvan F, Roman-Roman S, Vayssiere B, Belleville C, et al. Lrp5-independent activation of Wnt signaling by lithium chloride increases bone formation and bone mass in mice. Proc Natl Acad Sci U S A (2005) 102(48):17406–11. doi:10.1073/pnas.0505259102
163. Kugimiya F, Kawaguchi H, Ohba S, Kawamura N, Hirata M, Chikuda H, et al. GSK-3beta controls osteogenesis through regulating Runx2 activity. PLoS One (2007) 2(9):e837. doi:10.1371/journal.pone.0000837
164. Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol (1996) 6(12):1664–8. doi:10.1016/S0960-9822(02)70790-2
165. Marcellini S, Henriquez JP, Bertin A. Control of osteogenesis by the canonical Wnt and BMP pathways in vivo: cooperation and antagonism between the canonical Wnt and BMP pathways as cells differentiate from osteochondroprogenitors to osteoblasts and osteocytes. Bioessays (2012) 34(11):953–62. doi:10.1002/bies.201200061
166. Arioka M, Takahashi-Yanaga F, Sasaki M, Yoshihara T, Morimoto S, Hirata M, et al. Acceleration of bone regeneration by local application of lithium: Wnt signal-mediated osteoblastogenesis and Wnt signal-independent suppression of osteoclastogenesis. Biochem Pharmacol (2014) 90(4):397–405. doi:10.1016/j.bcp.2014.06.011
167. Saidak Z, Hay E, Marty C, Barbara A, Marie PJ. Strontium ranelate rebalances bone marrow adipogenesis and osteoblastogenesis in senescent osteopenic mice through NFATc/Maf and Wnt signaling. Aging Cell (2012) 11(3):467–74. doi:10.1111/j.1474-9726.2012.00804.x
168. O’Donnell S, Cranney A, Wells GA, Adachi JD, Reginster JY. Strontium ranelate for preventing and treating postmenopausal osteoporosis. Cochrane Database Syst Rev (2006) 3:Cd005326. doi:10.1002/14651858.CD005326.pub2
169. Reginster JY, Brandi ML, Cannata-Andia J, Cooper C, Cortet B, Feron JM, et al. The position of strontium ranelate in today’s management of osteoporosis. Osteoporos Int (2015) 26(6):1667–71. doi:10.1007/s00198-015-3109-y
170. Kalinowski DS, Richardson DR. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol Rev (2005) 57(4):547–83. doi:10.1124/pr.57.4.2
171. Almeida M, Han L, Martin-Millan M, O’Brien CA, Manolagas SC. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor- to forkhead box O-mediated transcription. J Biol Chem (2007) 282(37):27298–305. doi:10.1074/jbc.M702811200
172. Jia P, Xu YJ, Zhang ZL, Li K, Li B, Zhang W, et al. Ferric ion could facilitate osteoclast differentiation and bone resorption through the production of reactive oxygen species. J Orthop Res (2012) 30(11):1843–52. doi:10.1002/jor.22133
173. Chen B, Li GF, Shen Y, Huang XI, Xu YJ. Reducing iron accumulation: a potential approach for the prevention and treatment of postmenopausal osteoporosis. Exp Ther Med (2015) 10(1):7–11. doi:10.3892/etm.2015.2484
174. Bolt AM, Grant MP, Wu TH, Flores Molina M, Plourde D, Kelly AD, et al. Tungsten promotes sex-specific adipogenesis in the bone by altering differentiation of bone marrow-resident mesenchymal stromal cells. Toxicol Sci (2016) 150(2):333–46. doi:10.1093/toxsci/kfw008
175. Wu CT, Lu TY, Chan DC, Tsai KS, Yang RS, Liu SH. Effects of arsenic on osteoblast differentiation in vitro and on bone mineral density and microstructure in rats. Environ Health Perspect (2014) 122(6):559–65. doi:10.1289/ehp.1307832
176. World Health Organization WHO. Guidance for Identifying Populations at Risk from Mercury Poisoning. Geneva, Switzerland: World Health Organization (2008).
177. Yachiguchi K, Sekiguchi T, Nakano M, Hattori A, Yamamoto M, Kitamura K, et al. Effects of inorganic mercury and methylmercury on osteoclasts and osteoblasts in the scales of the marine teleost as a model system of bone. Zoolog Sci (2014) 31(5):330–7. doi:10.2108/zs130265
178. Cho GJ, Park HT, Shin JH, Hur JY, Kim SH, Lee KW, et al. The relationship between blood mercury level and osteoporosis in postmenopausal women. Menopause (2012) 19(5):576–81. doi:10.1097/gme.0b013e3182377294
179. Pollack AZ, Mumford SL, Wactawski-Wende J, Yeung E, Mendola P, Mattison DR, et al. Bone mineral density and blood metals in premenopausal women. Environ Res (2013) 120:76–81. doi:10.1016/j.envres.2012.06.001
180. Barouk P, Maynou C, Hildebrand HF, Aubertin F, Breme J, Cassagnaud X, et al. [High incidence of total hip arthroplasty aseptic loosening with ion-coated titanium femoral heads]. Rev Chir Orthop Reparatrice Appar Mot (2004) 90(1):26–32. doi:10.1016/S0035-1040(04)70003-3
181. Khan M, Osman K, Green G, Haddad FS. The epidemiology of failure in total knee arthroplasty: avoiding your next revision. Bone Joint J (2016) 98-b(1 Suppl A):105–12. doi:10.1302/0301-620X.98B1.36293
183. Savarino L, Granchi D, Ciapetti G, Cenni E, Nardi Pantoli A, Rotini R, et al. Ion release in patients with metal-on-metal hip bearings in total joint replacement: a comparison with metal-on-polyethylene bearings. J Biomed Mater Res (2002) 63(5):467–74. doi:10.1002/jbm.10299
184. Urban RM, Jacobs JJ, Tomlinson MJ, Gavrilovic J, Black J, Peoc’h M. Dissemination of wear particles to the liver, spleen, and abdominal lymph nodes of patients with hip or knee replacement. J Bone Joint Surg Am (2000) 82(4):457–76. doi:10.2106/00004623-200004000-00002
185. Hartmann A, Hannemann F, Lutzner J, Seidler A, Drexler H, Gunther KP, et al. Metal ion concentrations in body fluids after implantation of hip replacements with metal-on-metal bearing – systematic review of clinical and epidemiological studies. PLoS One (2013) 8(8):e70359. doi:10.1371/journal.pone.0070359
186. Bradberry SM, Wilkinson JM, Ferner RE. Systemic toxicity related to metal hip prostheses. Clin Toxicol (Phila) (2014) 52(8):837–47. doi:10.3109/15563650.2014.944977
188. Bozic KJ, Browne J, Dangles CJ, Manner PA, Yates AJ Jr, Weber KL, et al. Modern metal-on-metal hip implants. J Am Acad Orthop Surg (2012) 20(6):402–6. doi:10.5435/JAAOS-20-06-402
189. Delaunay C, Petit I, Learmonth ID, Oger P, Vendittoli PA. Metal-on-metal bearings total hip arthroplasty: the cobalt and chromium ions release concern. Orthop Traumatol Surg Res (2010) 96(8):894–904. doi:10.1016/j.otsr.2010.05.008
190. Browne JA, Bechtold CD, Berry DJ, Hanssen AD, Lewallen DG. Failed metal-on-metal hip arthroplasties: a spectrum of clinical presentations and operative findings. Clin Orthop Relat Res (2010) 468(9):2313–20. doi:10.1007/s11999-010-1419-0
191. Mann BS, Whittingham-Jones PM, Shaerf DA, Nawaz ZS, Harvie P, Hart AJ, et al. Metal-on-metal bearings, inflammatory pseudotumours and their neurological manifestations. Hip Int (2012) 22(2):129–36. doi:10.5301/HIP.2012.9185
192. Haddad FS, Thakrar RR, Hart AJ, Skinner JA, Nargol AV, Nolan JF, et al. Metal-on-metal bearings: the evidence so far. J Bone Joint Surg Br (2011) 93(5):572–9. doi:10.1302/0301-620X.93B4.26429
193. Shetty VD, Villar RN. Development and problems of metal-on-metal hip arthroplasty. Proc Inst Mech Eng H J Eng Med (2006) 220(2):371–7. doi:10.1243/095441105X63264
194. MacDonald SJ. Metal-on-metal total hip arthroplasty: the concerns. Clin Orthop Relat Res (2004) 429:86–93. doi:10.1097/01.blo.0000150309.48474.8b
195. Fabi D, Levine B, Paprosky W, Della Valle C, Sporer S, Klein G, et al. Metal-on-metal total hip arthroplasty: causes and high incidence of early failure. Orthopedics (2012) 35(7):e1009–16. doi:10.3928/01477447-20120621-12
196. Gonzalez MH, Carr R, Walton S, Mihalko WM. The evolution and modern use of metal-on-metal bearings in total hip arthroplasty. Instr Course Lect (2011) 60:247–55.
197. Catelas I, Wimmer MA. New insights into wear and biological effects of metal-on-metal bearings. J Bone Joint Surg Am (2011) 93(Suppl 2):76–83. doi:10.2106/JBJS.J.01877
198. Natu S, Sidaginamale RP, Gandhi J, Langton DJ, Nargol AV. Adverse reactions to metal debris: histopathological features of periprosthetic soft tissue reactions seen in association with failed metal on metal hip arthroplasties. J Clin Pathol (2012) 65(5):409–18. doi:10.1136/jclinpath-2011-200398
199. Willert HG, Buchhorn GH, Fayyazi A, Flury R, Windler M, Koster G, et al. Metal-on-metal bearings and hypersensitivity in patients with artificial hip joints. A clinical and histomorphological study. J Bone Joint Surg Am (2005) 87(1):28–36. doi:10.2106/JBJS.A.02039pp
200. Lohmann CH, Meyer H, Nuechtern JV, Singh G, Junk-Jantsch S, Schmotzer H, et al. Periprosthetic tissue metal content but not serum metal content predicts the type of tissue response in failed small-diameter metal-on-metal total hip arthroplasties. J Bone Joint Surg Am (2013) 95(17):1561–8. doi:10.2106/JBJS.L.01273
201. Shen Z, Crotti TN, McHugh KP, Matsuzaki K, Gravallese EM, Bierbaum BE, et al. The role played by cell-substrate interactions in the pathogenesis of osteoclast-mediated peri-implant osteolysis. Arthritis Res Ther (2006) 8(3):R70. doi:10.1186/ar1938
202. Drynda A, Singh G, Buchhorn GH, Awiszus F, Ruetschi M, Feuerstein B, et al. Metallic wear debris may regulate CXCR4 expression in vitro and in vivo. J Biomed Mater Res A (2015) 103(6):1940–8. doi:10.1002/jbm.a.35330
203. Zannettino AC, Farrugia AN, Kortesidis A, Manavis J, To LB, Martin SK, et al. Elevated serum levels of stromal-derived factor-1alpha are associated with increased osteoclast activity and osteolytic bone disease in multiple myeloma patients. Cancer Res (2005) 65(5):1700–9. doi:10.1158/0008-5472.CAN-04-1687
204. Xing Q, de Vos P, Faas MM, Ye Q, Ren Y. LPS promotes pre-osteoclast activity by up-regulating CXCR4 via TLR-4. J Dent Res (2011) 90(2):157–62. doi:10.1177/0022034510379019
205. Yu X, Collin-Osdoby P, Osdoby P. SDF-1 increases recruitment of osteoclast precursors by upregulation of matrix metalloproteinase-9 activity. Connect Tissue Res (2003) 44(Suppl 1):79–84. doi:10.1080/713713646
206. Omar O, Lenneras M, Svensson S, Suska F, Emanuelsson L, Hall J, et al. Integrin and chemokine receptor gene expression in implant-adherent cells during early osseointegration. J Mater Sci Mater Med (2010) 21(3):969–80. doi:10.1007/s10856-009-3915-x
207. Cadosch D, Gautschi OP, Chan E, Simmen HP, Filgueira L. Titanium induced production of chemokines CCL17/TARC and CCL22/MDC in human osteoclasts and osteoblasts. J Biomed Mater Res A (2010) 92(2):475–83. doi:10.1002/jbm.a.32390
208. Lean JM, Murphy C, Fuller K, Chambers TJ. CCL9/MIP-1gamma and its receptor CCR1 are the major chemokine ligand/receptor species expressed by osteoclasts. J Cell Biochem (2002) 87(4):386–93. doi:10.1002/jcb.10319
209. Nakamura ES, Koizumi K, Kobayashi M, Saitoh Y, Arita Y, Nakayama T, et al. RANKL-induced CCL22/macrophage-derived chemokine produced from osteoclasts potentially promotes the bone metastasis of lung cancer expressing its receptor CCR4. Clin Exp Metastasis (2006) 23(1):9–18. doi:10.1007/s10585-006-9006-1
210. Wooley PH, Nasser S, Fitzgerald RH Jr. The immune response to implant materials in humans. Clin Orthop Relat Res (1996) 326:63–70. doi:10.1097/00003086-199605000-00008
211. Lassus J, Waris V, Xu JW, Li TF, Hao J, Nietosvaara Y, et al. Increased interleukin-8 (IL-8) expression is related to aseptic loosening of total hip replacement. Arch Orthop Trauma Surg (2000) 120(5–6):328–32. doi:10.1007/s004020050475
212. Haleem-Smith H, Argintar E, Bush C, Hampton D, Postma WF, Chen FH, et al. Biological responses of human mesenchymal stem cells to titanium wear debris particles. J Orthop Res (2012) 30(6):853–63. doi:10.1002/jor.22002
213. Rose JJ, Foley JF, Murphy PM, Venkatesan S. On the mechanism and significance of ligand-induced internalization of human neutrophil chemokine receptors CXCR1 and CXCR2. J Biol Chem (2004) 279(23):24372–86. doi:10.1074/jbc.M401364200
214. Gu Q, Shi Q, Yang H. The role of TLR and chemokine in wear particle-induced aseptic loosening. J Biomed Biotechnol (2012) 2012:596870. doi:10.1155/2012/596870
215. Wang C, Liu Y, Wang Y, Li H, Zhang RX, He MS, et al. Adenovirus-mediated siRNA targeting CXCR2 attenuates titanium particle-induced osteolysis by suppressing osteoclast formation. Med Sci Monit (2016) 22:727–35. doi:10.12659/MSM.897243
216. Nakashima Y, Sun DH, Trindade MC, Chun LE, Song Y, Goodman SB, et al. Induction of macrophage C-C chemokine expression by titanium alloy and bone cement particles. J Bone Joint Surg Am (1999) 81(1):155–62. doi:10.1302/0301-620X.81B1.8884
217. Trindade MC, Schurman DJ, Maloney WJ, Goodman SB, Smith RL. G-protein activity requirement for polymethylmethacrylate and titanium particle-induced fibroblast interleukin-6 and monocyte chemoattractant protein-1 release in vitro. J Biomed Mater Res (2000) 51(3):360–8. doi:10.1002/1097-4636(20000905)51:3<360::AID-JBM9>3.0.CO;2-E
218. Fritz EA, Glant TT, Vermes C, Jacobs JJ, Roebuck KA. Titanium particles induce the immediate early stress responsive chemokines IL-8 and MCP-1 in osteoblasts. J Orthop Res (2002) 20(3):490–8. doi:10.1016/S0736-0266(01)00154-1
219. Fritz EA, Glant TT, Vermes C, Jacobs JJ, Roebuck KA. Chemokine gene activation in human bone marrow-derived osteoblasts following exposure to particulate wear debris. J Biomed Mater Res A (2006) 77(1):192–201. doi:10.1002/jbm.a.30609
220. Wachi T, Shuto T, Shinohara Y, Matono Y, Makihira S. Release of titanium ions from an implant surface and their effect on cytokine production related to alveolar bone resorption. Toxicology (2015) 327:1–9. doi:10.1016/j.tox.2014.10.016
221. Fuller K, Owens JM, Chambers TJ. Macrophage inflammatory protein-1 alpha and IL-8 stimulate the motility but suppress the resorption of isolated rat osteoclasts. J Immunol (1995) 154(11):6065–72.
222. Devitt BM, Queally JM, Vioreanu M, Butler JS, Murray D, Doran PP, et al. Cobalt ions induce chemokine secretion in a variety of systemic cell lines. Acta Orthop (2010) 81(6):756–64. doi:10.3109/17453674.2010.537806
223. Queally JM, Devitt BM, Butler JS, Malizia AP, Murray D, Doran PP, et al. Cobalt ions induce chemokine secretion in primary human osteoblasts. J Orthop Res (2009) 27(7):855–64. doi:10.1002/jor.20837
224. Dalal A, Pawar V, McAllister K, Weaver C, Hallab NJ. Orthopedic implant cobalt-alloy particles produce greater toxicity and inflammatory cytokines than titanium alloy and zirconium alloy-based particles in vitro, in human osteoblasts, fibroblasts, and macrophages. J Biomed Mater Res A (2012) 100(8):2147–58. doi:10.1002/jbm.a.34122
225. Athanasou NA. The pathobiology and pathology of aseptic implant failure. Bone Joint Res (2016) 5(5):162–8. doi:10.1302/2046-3758.55.BJR-2016-0086
226. Council NR. Fluoride in Drinking Water: A Scientific Review of EPA’s Standards. Washington, DC: The National Academies Press (2006). 530 p.
227. Pei J, Li B, Gao Y, Wei Y, Zhou L, Yao H, et al. Fluoride decreased osteoclastic bone resorption through the inhibition of NFATc1 gene expression. Environ Toxicol (2014) 29(5):588–95. doi:10.1002/tox.21784
228. Kleerekoper M, Balena R. Fluorides and osteoporosis. Annu Rev Nutr (1991) 11:309–24. doi:10.1146/annurev.nu.11.070191.001521
229. Jha SK, Mishra VK, Sharma DK, Damodaran T. Fluoride in the environment and its metabolism in humans. Rev Environ Contam Toxicol (2011) 211:121–42. doi:10.1007/978-1-4419-8011-3_4
230. Krishnamachari KA. Skeletal fluorosis in humans: a review of recent progress in the understanding of the disease. Prog Food Nutr Sci (1986) 10(3–4):279–314.
231. Yan X, Yan X, Morrison A, Han T, Chen Q, Li J, et al. Fluoride induces apoptosis and alters collagen I expression in rat osteoblasts. Toxicol Lett (2011) 200(3):133–8. doi:10.1016/j.toxlet.2010.11.005
232. Pan L, Shi X, Liu S, Guo X, Zhao M, Cai R, et al. Fluoride promotes osteoblastic differentiation through canonical Wnt/beta-catenin signaling pathway. Toxicol Lett (2014) 225(1):34–42. doi:10.1016/j.toxlet.2013.11.029
233. Bhawal UK, Lee HJ, Arikawa K, Shimosaka M, Suzuki M, Toyama T, et al. Micromolar sodium fluoride mediates anti-osteoclastogenesis in Porphyromonas gingivalis-induced alveolar bone loss. Int J Oral Sci (2015) 7(4):242–9. doi:10.1038/ijos.2015.28
234. Lee HJ, Choi CH. Anti-inflammatory effects of bamboo salt and sodium fluoride in human gingival fibroblasts – an in vitro study. Kaohsiung J Med Sci (2015) 31(6):303–8. doi:10.1016/j.kjms.2015.03.005
235. Hirano S, Ando M, Kanno S. Inflammatory responses of rat alveolar macrophages following exposure to fluoride. Arch Toxicol (1999) 73(6):310–5. doi:10.1007/s002040050624
236. Refsnes M, Becher R, Lag M, Skuland T, Schwarze PE. Fluoride-induced interleukin-6 and interleukin-8 synthesis in human epithelial lung cells. Hum Exp Toxicol (1999) 18(11):645–52. doi:10.1191/096032799678839572
237. Refsnes M, Thrane EV, Lag M, Thoresen GH, Schwarze PE. Mechanisms in fluoride-induced interleukin-8 synthesis in human lung epithelial cells. Toxicology (2001) 167(2):145–58. doi:10.1016/S0300-483X(01)00448-6
238. Wang Y, Duan XQ, Zhao ZT, Zhang XY, Wang H, Liu DW, et al. Fluoride affects calcium homeostasis by regulating parathyroid hormone, PTH-related peptide, and calcium-sensing receptor expression. Biol Trace Elem Res (2015) 165(2):159–66. doi:10.1007/s12011-015-0245-3
239. Hokeness K, Kratch J, Nadolny C, Aicardi K, Reid CW. The effects of fungal volatile organic compounds on bone marrow stromal cells. Can J Microbiol (2014) 60(1):1–4. doi:10.1139/cjm-2013-0708
240. Gonzalez-Garcia C, Moratal D, Oreffo RO, Dalby MJ, Salmeron-Sanchez M. Surface mobility regulates skeletal stem cell differentiation. Integr Biol (Camb) (2012) 4(5):531–9. doi:10.1039/c2ib00139j
241. Rosenblum Lichtenstein JH, Hsu YH, Gavin IM, Donaghey TC, Molina RM, Thompson KJ, et al. Environmental mold and mycotoxin exposures elicit specific cytokine and chemokine responses. PLoS One (2015) 10(5):e0126926. doi:10.1371/journal.pone.0126926
242. Ubagai T, Tansho S, Ito T, Ono Y. Influences of aflatoxin B1 on reactive oxygen species generation and chemotaxis of human polymorphonuclear leukocytes. Toxicol In Vitro (2008) 22(4):1115–20. doi:10.1016/j.tiv.2008.01.007
243. Hillerdal G, Henderson DW. Asbestos, asbestosis, pleural plaques and lung cancer. Scand J Work Environ Health (1997) 23(2):93–103. doi:10.5271/sjweh.186
244. Pociask DA, Sime PJ, Brody AR. Asbestos-derived reactive oxygen species activate TGF-beta1. Lab Invest (2004) 84(8):1013–23. doi:10.1038/labinvest.3700109
245. Maeda M, Nishimura Y, Hayashi H, Kumagai N, Chen Y, Murakami S, et al. Decreased CXCR3 expression in CD4+ T cells exposed to asbestos or derived from asbestos-exposed patients. Am J Respir Cell Mol Biol (2011) 45(4):795–803. doi:10.1165/rcmb.2010-0435OC
246. Ilavska S, Jahnova E, Tulinska J, Horvathova M, Dusinska M, Wsolova L, et al. Immunological monitoring in workers occupationally exposed to asbestos. Toxicology (2005) 206(2):299–308. doi:10.1016/j.tox.2004.09.004
247. Myllarniemi M, Lindholm P, Ryynanen MJ, Kliment CR, Salmenkivi K, Keski-Oja J, et al. Gremlin-mediated decrease in bone morphogenetic protein signaling promotes pulmonary fibrosis. Am J Respir Crit Care Med (2008) 177(3):321–9. doi:10.1164/rccm.200706-945OC
Keywords: chemokines, bone healing, environmental toxins, dioxin, metal, endocrine disruptors, volatile organic compound
Citation: Smith JT, Schneider AD, Katchko KM, Yun C and Hsu EL (2017) Environmental Factors Impacting Bone-Relevant Chemokines. Front. Endocrinol. 8:22. doi: 10.3389/fendo.2017.00022
Received: 01 November 2016; Accepted: 25 January 2017;
Published: 14 February 2017
Edited by:
Giacomina Brunetti, University of Bari, ItalyReviewed by:
Andy Sunters, Royal Veterinary College, UKCopyright: © 2017 Smith, Schneider, Katchko, Yun and Hsu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erin L. Hsu, ZXJpbmtoc3VAZ21haWwuY29t
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.