 Garrett Heinrich1,2
Garrett Heinrich1,2 Hilda E. Ghadieh3
Hilda E. Ghadieh3 Simona S. Ghanem3Harrison T. Muturi1Khadijeh Rezaei3
Simona S. Ghanem3Harrison T. Muturi1Khadijeh Rezaei3 Qusai Y. Al-Share3
Qusai Y. Al-Share3 Thomas A. Bowman3Deqiang Zhang4
Thomas A. Bowman3Deqiang Zhang4 Robert S. Garofalo5
Robert S. Garofalo5 Lei Yin4
Lei Yin4 Sonia M. Najjar1,2*
Sonia M. Najjar1,2*
- 1Department of Biomedical Sciences, Heritage College of Osteopathic Medicine, Ohio University, Athens, OH, USA
- 2Heritage College of Osteopathic Medicine, Diabetes Institute, Ohio University, Athens, OH, USA
- 3Center for Diabetes and Endocrine Research (CeDER), College of Medicine and Life Sciences, University of Toledo, Toledo, OH, USA
- 4Department of Molecular and Integrative Physiology, University of Michigan Medical School, Ann Arbor, MI, USA
- 5Yale Cancer Center, Office of Research Affairs, New Haven, CT, USA
The pathogenesis of human non-alcoholic fatty liver disease (NAFLD) remains unclear, in particular in the context of its relationship to insulin resistance and visceral obesity. Work on the carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) in mice has resolved some of the related questions. CEACAM1 promotes insulin clearance by enhancing the rate of uptake of the insulin-receptor complex. It also mediates a negative acute effect of insulin on fatty acid synthase activity. This positions CEACAM1 to coordinate the regulation of insulin and lipid metabolism. Fed a regular chow diet, global null mutation of Ceacam1 manifest hyperinsulinemia, insulin resistance, obesity, and steatohepatitis. They also develop spontaneous chicken-wire fibrosis, characteristic of non-alcoholic steatohepatitis. Reduction of hepatic CEACAM1 expression plays a significant role in the pathogenesis of diet-induced metabolic abnormalities, as bolstered by the protective effect of hepatic CEACAM1 gain-of-function against the metabolic response to dietary fat. Together, this emphasizes that loss of hepatic CEACAM1 links NAFLD to insulin resistance and obesity.
Physiologic Regulation of Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 (CEACAM1)
The CEACAM1 is a transmembrane glycoprotein that undergoes phosphorylation by the insulin receptor tyrosine kinase (1). Among insulin target tissues, CEACAM1 is predominantly expressed in the liver (2). This is consistent with its role in promoting insulin clearance, which occurs mostly in liver and to a lower extent in kidney. Consistent with the important role of the liver in regulating insulin and lipid metabolism, Ceacam1 transcription is coordinately regulated by insulin and fatty acids during fasting–refeeding conditions, with fatty acids at fasting repressing it via a mechanism depending on the peroxisome proliferator-activated receptor alpha (PPARα) (3, 4) and insulin inducing it in the first few hours of refeeding (3, 5).
CEACAM1 Promotes Insulin Clearance and Mediates an Acute Negative Effect of Insulin on Hepatic De Novo Lipogenesis
Insulin is released from pancreatic β-cells in a pulsatile manner (6). The acute rise of insulin in the portal vein causes phosphorylation and activation of the insulin receptor tyrosine kinase in the hepatocyte (7, 8). This, in turn, leads to phosphorylation of substrates, including CEACAM1 (1). Upon its phosphorylation, CEACAM1 promotes receptor-mediated insulin uptake into clathrin-coated pits/vesicles of the hepatocyte to be eventually degraded and cleared from the blood (9, 10). This process mediates the rapid extraction of ~50% of secreted insulin through its first pass into the liver.
Internalization of phosphorylated CEACAM1 as part of the insulin-receptor complex leads to its binding to fatty acid synthase (FASN) (11), a key enzyme that catalyzes the conversion of malonyl-CoA to palmitic acid during de novo lipogenesis. CEACAM1 association downregulates FASN enzymatic activity and restricts hepatic de novo lipogenesis, likely to protect the liver against the potential lipogenic effect of approximately twofold to threefold higher level of insulin in the portal than the systemic circulation (12). Thus, CEACAM1 phosphorylation by the insulin receptor in response to acute rise of insulin constitutes a key mechanism that underlies the maintenance of physiologic insulin levels, at the same time as mediating a suppressive acute effect of insulin on lipogenesis in liver. Combined, this restricts hepatic lipid production under normal physiologic conditions; assigning a major role for CEACAM1 in integrating the regulation of insulin and lipid metabolism in the hepatocyte. Under conditions of hyperinsulinemia, the pulsatility of insulin secretion is compromised (6), limiting insulin signaling in the hepatocyte, including CEACAM1 phosphorylation, and subsequently, the acute negative effect of insulin on FASN activity is removed to contribute to hyperinsulinemia-driven lipogenesis (11). This paradigm emphasizes the contrast between the previously unappreciated suppressive effect of acute insulin pulses on fatty acid synthesis and the well-recognized positive effect of chronically elevated levels of insulin on lipogenic genes’ expression by the coordinated action of sterol regulatory element-binding protein (SREBP1c) (13) and the upstream stimulatory factor 1 (14). Suppression of hepatic FASN activity by pulsatile insulin release proposes to include elevation in de novo lipogenesis as a manifest of hepatic insulin resistance in addition to increased hepatic glucose production (via glycogenolysis and gluconeogenesis) (8, 15).
Mutating CEACAM1 in Liver Causes Insulin Resistance and Non-Alcoholic Steatohepatitis (NASH)
Mice with liver-specific inactivation (L-SACC1) or with global null mutation of Ceacam1 (Cc1−/−) exhibit impairment in insulin clearance leading to chronic hyperinsulinemia and systemic insulin resistance (owing to downregulation of insulin receptor expression) (16–18). They also exhibit elevated lipid production in liver and redistribution to the white adipose tissue to be stored; thus, contributing to visceral obesity and increased release of free fatty acid (FFA) and adipokines (19).
Mutant Ceacam1 mice also develop inflammation in liver, in part due to the loss of the anti-inflammatory effect of CEACAM1 (20), apoptosis, and oxidative stress. Additionally, they manifest chicken-wire bridging fibrosis, a characteristic feature of NASH, even when fed a standard chow diet, making them rare mouse models of spontaneous fibrosis on the C57BL/6J genetic background. The underlying mechanisms of fibrosis in Ceacam1 mutants are the subject of intense investigations in our laboratories.
Dietary Fat Reduces Hepatic CEACAM1 Expression in C57BL/6J Mice
In uncomplicated obesity with low-grade insulin resistance, FFA are mobilized from white adipose tissue mainly to the liver to be removed by β-oxidation (21). This is supported by experimental evidence in rodents showing occurrence within few days of the initiation of high-fat intake as a result of dysregulated hypothalamic control in the adipose tissue (22). While this early lipolysis occurs in the absence of insulin resistance in the adipose tissue, the released FFA can rapidly initiate hepatic insulin resistance (23), in part by activating PKCδ-mediated pathways (24). As the nutritional burden persists, hepatic lipotoxicity develops in response to progressively compromised β-oxidation relative to re-esterification. Concomitantly, hepatic insulin resistance progresses into systemic insulin resistance to be manifested in peripheral tissues, including the white adipose tissue with ensuing advancement of a pro-inflammatory state (25).
Recent reports from our laboratories show that high-fat diet progressively reduces hepatic CEACAM1 level in C57BL/6J mice until it reaches >50% after 3 weeks, at which point, insulin clearance is impaired and hyperinsulinemia develops with attendant hepatic insulin resistance and steatohepatitis (26). Consistent with the key role for CEACAM1 in diet-induced insulin resistance and hepatosteatosis, adenoviral-mediated redelivery of wild-type, but not phosphorylation-defective CEACAM1 to the liver, completely reverses these metabolic abnormalities even while maintaining mice on a high-fat diet (27), demonstrating a causative role for the decrease in hepatic CEACAM1 level in sustaining diet-induced systemic insulin resistance and hepatic steatosis. That impairment of insulin clearance plays a significant role in hepatic insulin resistance in response to high-fat diet has recently been demonstrated in Asian men (28). Using a two-step hyperinsulinemic-euglycemic clamp, Bakker et al. (28) showed that in contrast to age- and sex-matched Caucasians, young and healthy South Asian men develop impairment of insulin clearance as well as hepatic insulin resistance in the absence of other metabolic alterations in skeletal muscle and white adipose tissue following 5 days of a high-fat Western diet intake. Several other studies in humans (28) as well as dogs (29) have supported the findings that defective hepatic insulin clearance is implicated in diet-induced insulin resistance.
The decrease in hepatic CEACAM1 by high-fat diet is attributed to lipolysis-derived FFA, in agreement with reducing hepatic CEACAM1 levels by intralipid–heparin infusion (24) and the negative effect of FFA on insulin clearance (30, 31). The underlying mechanism of CEACAM1 repression by FFA is via PPARα activation (4). In the presence of normoinsulinemia, this provides a positive feedback mechanism on fatty acid β-oxidation as it limits the negative effect of CEACAM1 on FASN activity (11) and subsequently, reduces malonyl-CoA-mediated inhibition of fatty acids translocation to the mitochondria (3). When CEACAM1 level is reduced by >50%, hepatic insulin clearance fails and chronic hyperinsulinemia develops, causing hepatic insulin resistance, at least in part by downregulating insulin receptors in the hepatocyte (32, 33) and triggering de novo lipogenesis by activating SREBP1c-mediated transcription of lipogenic genes (13), including acetyl-CoA carboxylase (ACC), a limiting enzyme in lipid biosynthesis. Elevation in ACC level (and activity) induces malonyl-CoA level, which in turn, inhibits fatty acid transport to the mitochondria and β-oxidation. Potentially contributing to the downregulation of β-oxidation under hyperinsulinemic conditions is the maintenance of insulin-stimulated phosphorylation and inactivation of Foxa2-mediated suppression of the transcription of genes involved in fatty acid β-oxidation (34, 35). Collectively, this limits fatty acid β-oxidation while promoting de novo lipogenesis, leading to hepatosteatosis. With the loss of the potential counter-regulatory anti-inflammatory function of CEACAM1, this causes a more robust change in the inflammatory milieu of the liver and steatohepatitis develops. Together, the data identify reduction in CEACAM1 expression as a novel molecular underpinning of the integrated regulation of lipid oxidation and hepatic insulin resistance (gluconeogenesis) by FFA mobilization from white adipose tissue (36–38).
Reduced Hepatic CEACAM1 Levels Causes Obesity by Contributing to Energy Imbalance
High-fat diet represses hepatic CEACAM1 levels to impair insulin clearance and cause hyperinsulinemia that in turn, drives increased hepatic lipid production and output to the white adipose depot for storage (39). This is consistent with the well-accepted association of hyperinsulinemia and liver steatosis with high plasma Apolipoprotein B levels and visceral obesity in humans and rodents (40–45). Together with visceral obesity, sustained hyperinsulinemia reduces glucose transporter 4-mediated glucose transport to cause insulin resistance in adipose tissue (46), as supported by hyperinsulinemic-euglycemic clamp analysis in Ceacam1 mutants (16–18, 47) and in the diet-induced model (26).
Consistent with the finding that reduction of hepatic CEACAM1 plays a critical role in diet-induced altered metabolic response, transgenic protection of hepatic CEACAM1 in L-CC1 mice prevents hyperinsulinemia, insulin resistance, and hepatosteatosis in response to high-fat diet (26). It also limits the size of adipocytes and total fat mass by countering the negative effect of high-fat diet on energy expenditure and spontaneous physical activity (26). Similarly, adenoviral-redelivery of wild-type CEACAM1 in the liver protects energy balance against high-fat intake, thereby reversing the gain in body weight and visceral adiposity (27). Given that CEACAM1 is not detected in the adipocyte at the protein level (2), it is likely that the gain-of-function of hepatic CEACAM1 drives this positive effect on energy expenditure and adipose tissue biology (limited adipocyte size, fibrosis, and inflammation) (27, 39). The beneficial effect of hepatic CEACAM1 gain-of-function on insulin response in white adipose tissue could be mediated, at least in part, by the rise in plasma FGF21 (48, 49) that induces the locomotor activity (50) and energy expenditure (51, 52).
Both L-SACC1 and Cc1−/− mutant mice display visceral obesity and a higher body mass than their wild-type counterparts (16–18). Visceral obesity, which is partly caused by elevated hepatic lipid production and redistribution to white adipose tissue (19), leads to hyperleptinemia, which could in turn, alter response to leptin and cause energy imbalance. Consistently, global Cc1−/− null mice develop elevated production and secretion of leptin from their expanded while adipose depot in addition to increased total fat mass and obesity resulting from hyperphagia and reduced spontaneous physical activity (53). In addition to leptin resistance, hyperinsulinemia also contributes to the obesity phenotype in these mice, at least in part, by inducing hypothalamic FASN level and activity (53), which in turn, causes hyperphagia (54) and lower physical activity (55, 56). Together, this demonstrates that altered CEACAM1-dependent insulin clearance pathways drive hyperinsulinemia-mediated link of hepatic steatosis to visceral obesity and increased total fat mass.
Concluding Remarks
The mechanisms underlying the pathogenesis of non-alcoholic fatty liver disease (NAFLD) in humans remain unclear (57) and whether insulin resistance plays a role in NAFLD has been debated, owing to the lack of appropriate animal models that replicate all features of the human disease and its progression to NASH (58, 59). As summarized in this review, our laboratory has demonstrated in the last couple of decades that loss in hepatic CEACAM1 expression and its defective phosphorylation impair insulin clearance and subsequently, play a pivotal role in insulin resistance, fatty liver disease, and obesity (Figure 1) (9, 10, 16–19, 25, 27, 39, 53, 60, 61). Demonstration of a role for impaired insulin clearance in insulin resistance in human disease is emerging (62–65). In this regard, compromised hepatic insulin extraction has been shown to constitute a risk factor for obesity (66, 67), type 2 diabetes (68), metabolic syndrome (65, 69), and fatty liver disease (70). The study by Lee (71) showing a marked decline in hepatic CEACAM1 levels in patients with high-grade fatty liver and obesity coupled with our mechanistic studies demonstrating that redelivering CEACAM1 to the liver reverses diet-induced insulin resistance, fatty liver, and visceral obesity (27) emphasizes a critical role for CEACAM1 in metabolic control. Of note, while our studies show that reduction of hepatic CEACAM1 causes insulin resistance, hepatosteatosis, and visceral obesity, they also show that diet-induced visceral obesity represses hepatic CEACAM1 to cause fat accumulation in liver and insulin resistance (3, 26, 27). Further emphasizing the metabolic role of hepatic CEACAM1, liver-specific overexpression of CEACAM1 curbs the metabolic abnormalities caused by high-fat diet and prevents insulin resistance and hepatosteatosis (26). Similarly, adenoviral-mediated redelivery of CEACAM1 to the liver reverses diet-induced metabolic derangement (27). Collectively, this positions the loss of hepatic CEACAM1 expression (and its resulting hyperinsulinemia and insulin resistance) on the crossroad of the pathogenesis of NAFLD and obesity.
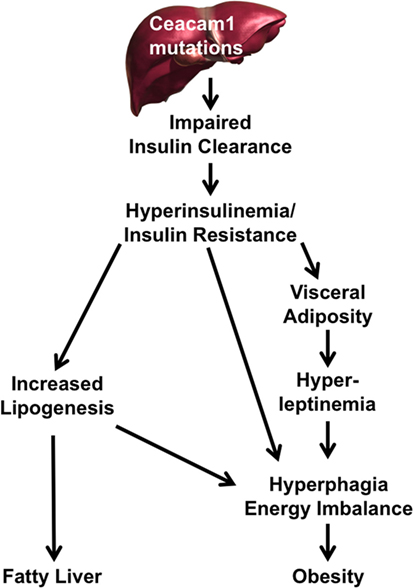
Figure 1. A pivotal role for carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) reduction in the pathogenesis of fatty liver disease and obesity. Reduction or mutation of Ceacam1 in the liver results in decreased insulin clearance from the portal circulation. Reduced clearance leads to hyperinsulinemia followed by insulin resistance (owing to downregulation of the insulin receptor) and increased hepatic lipogenesis. Elevation in hepatic lipogenesis leads to lipid redistribution to the while adipose depot to increase visceral adiposity. This leads to hyperleptinemia, which along with hyperinsulinemia, increases food intake and energy imbalance, further exacerbating obesity. Hyperinsulinemia drives hepatic lipogenesis and fat accumulation in liver.
Author Contributions
GH wrote a first draft of the manuscript. HG, SG, HM, KR, QA-S, TB, DZ contributed to the writing. RG and LY reviewed the manuscript. SN was responsible for revising the manuscript. SN had full access to all the data of the study and takes responsibility for the integrity and accuracy of data analysis and the decision to submit and publish the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from the NIH: R01 DK054254, R01 DK083850, and R01 HL112248 (to SN) and K99/R00-DK077449 and R01 DK099593 (to LY). It was also supported by a fund from the Heritage College of Osteopathic Medicine (to GH). The work was also supported by fellowships from the Middle-East Diabetes Research Center (to HG and SG).
References
1. Najjar SM, Philippe N, Suzuki Y, Ignacio GA, Formisano P, Accili D, et al. Insulin-stimulated phosphorylation of recombinant pp120/HA4, an endogenous substrate of the insulin receptor tyrosine kinase. Biochemistry (1995) 34:9341–9. doi:10.1021/bi00029a009
2. Najjar SM. Regulation of insulin action by CEACAM1. Trends Endocrinol Metab (2002) 13:240–5. doi:10.1016/S1043-2760(02)00608-2
3. Ramakrishnan SK, Khuder SS, Al-Share QY, Russo L, Abdallah SL, Patel PR, et al. PPARalpha (peroxisome proliferator-activated receptor alpha) activation reduces hepatic CEACAM1 protein expression to regulate fatty acid oxidation during fasting-refeeding transition. J Biol Chem (2016) 291:8121–9. doi:10.1074/jbc.M116.714014
4. Ramakrishnan SK, Russo L, Ghanem SS, Patel PR, Oyarce AM, Heinrich G, et al. Fenofibrate decreases insulin clearance and insulin secretion to maintain insulin sensitivity. J Biol Chem (2016) 291(46):23915–24. doi:10.1074/jbc.M116.745778
5. Najjar S, Boisclair Y, Nabih Z, Philippe N, Imai Y, Suzuki Y, et al. Cloning and characterization of a functional promoter of the rat pp120 gene, encoding a substrate of the insulin receptor tyrosine kinase. J Biol Chem (1996) 271:8809–17. doi:10.1074/jbc.271.15.8809
6. Matveyenko AV, Liuwantara D, Gurlo T, Kirakossian D, Dalla Man C, Cobelli C, et al. Pulsatile portal vein insulin delivery enhances hepatic insulin action and signaling. Diabetes (2012) 61:2269–79. doi:10.2337/db11-1462
7. White MF. The IRS-signalling system: a network of docking proteins that mediate insulin action. Mol Cell Biochem (1998) 182:3–11. doi:10.1023/A:1006806722619
8. Haeusler RA, Accili D. The double life of Irs. Cell Metab (2008) 8:7–9. doi:10.1016/j.cmet.2008.06.010
9. Formisano P, Najjar SM, Gross CN, Philippe N, Oriente F, Kern-Buell CL, et al. Receptor-mediated internalization of insulin. Potential role of pp120/HA4, a substrate of the insulin receptor kinase. J Biol Chem (1995) 270:24073–7. doi:10.1074/jbc.270.41.24073
10. Choice CV, Howard MJ, Poy MN, Hankin MH, Najjar SM. Insulin stimulates pp120 endocytosis in cells co-expressing insulin receptors. J Biol Chem (1998) 273:22194–200. doi:10.1074/jbc.273.35.22194
11. Najjar SM, Yang Y, Fernstrom MA, Lee SJ, Deangelis AM, Rjaily GA, et al. Insulin acutely decreases hepatic fatty acid synthase activity. Cell Metab (2005) 2:43–53. doi:10.1016/j.cmet.2005.06.001
12. Ward GM, Walters JM, Aitken PM, Best JD, Alford FP. Effects of prolonged pulsatile hyperinsulinemia in humans. Enhancement of insulin sensitivity. Diabetes (1990) 39:501–7. doi:10.2337/diab.39.4.501
13. Osborne TF. Sterol regulatory element-binding proteins (SREBPs): key regulators of nutritional homeostasis and insulin action. J Biol Chem (2000) 275:32379–82. doi:10.1074/jbc.R000017200
14. Wong RH, Chang I, Hudak CS, Hyun S, Kwan HY, Sul HS. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell (2009) 136:1056–72. doi:10.1016/j.cell.2008.12.040
15. Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab (2008) 7:95–6. doi:10.1016/j.cmet.2007.12.009
16. Park SY, Cho YR, Kim HJ, Hong EG, Higashimori T, Lee SJ, et al. Mechanism of glucose intolerance in mice with dominant negative mutation of CEACAM1. Am J Physiol Endocrinol Metab (2006) 291:E517–24. doi:10.1152/ajpendo.00077.2006
17. Poy MN, Yang Y, Rezaei K, Fernstrom MA, Lee AD, Kido Y, et al. CEACAM1 regulates insulin clearance in liver. Nat Genet (2002) 30:270–6. doi:10.1038/ng840
18. DeAngelis AM, Heinrich G, Dai T, Bowman TA, Patel PR, Lee SJ, et al. Carcinoembryonic antigen-related cell adhesion molecule 1: a link between insulin and lipid metabolism. Diabetes (2008) 57:2296–303. doi:10.2337/db08-0379
19. Ghosh S, Kaw M, Patel PR, Ledford KJ, Bowman TA, McLnerney MF, et al. Mice with null mutation of Ceacam1 develop nonalcoholic steatohepatitis. Hepat Med (2010) 2010:69–78. doi:10.2147/HMER.S8902
20. Nagaishi T, Pao L, Lin SH, Iijima H, Kaser A, Qiao SW, et al. SHP1 phosphatase-dependent T cell inhibition by CEACAM1 adhesion molecule isoforms. Immunity (2006) 25:769–81. doi:10.1016/j.immuni.2006.08.026
21. Groop LC, Saloranta C, Shank M, Bonadonna RC, Ferrannini E, DeFronzo RA. The role of free fatty acid metabolism in the pathogenesis of insulin resistance in obesity and noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab (1991) 72:96–107. doi:10.1210/jcem-72-1-96
22. Scherer T, Lindtner C, Zielinski E, O’Hare J, Filatova N, Buettner C. Short term voluntary overfeeding disrupts brain insulin control of adipose tissue lipolysis. J Biol Chem (2012) 287:33061–9. doi:10.1074/jbc.M111.307348
23. Kabir M, Catalano KJ, Ananthnarayan S, Kim SP, Van Citters GW, Dea MK, et al. Molecular evidence supporting the portal theory: a causative link between visceral adiposity and hepatic insulin resistance. Am J Physiol Endocrinol Metab (2005) 288:E454–61. doi:10.1152/ajpendo.00203.2004
24. Pereira S, Park E, Mori Y, Haber CA, Han P, Uchida T, et al. FFA-induced hepatic insulin resistance in vivo is mediated by PKCδ, NADPH oxidase, and oxidative stress. Am J Physiol Endocrinol Metab (2014) 307:E34–46. doi:10.1152/ajpendo.00436.2013
25. Najjar SM, Russo L. CEACAM1 loss links inflammation to insulin resistance in obesity and non-alcoholic steatohepatitis (NASH). Semin Immunopathol (2014) 36:55–71. doi:10.1007/s00281-013-0407-3
26. Al-Share QY, DeAngelis AM, Lester SG, Bowman TA, Ramakrishnan SK, Abdallah SL, et al. Forced hepatic overexpression of CEACAM1 curtails diet-induced insulin resistance. Diabetes (2015) 64:2780–90. doi:10.2337/db14-1772
27. Russo L, Ghadieh HE, Ghanem SS, Al-Share QY, Smiley ZN, Gatto-Weis C, et al. Role for hepatic CEACAM1 in regulating fatty acid metabolism along the adipocyte-hepatocyte axis. J Lipid Res (2016) 57(12):2163–75. doi:10.1194/jlr.M072066
28. Bakker LEH, van Schinkel LD, Guigas B, Streefland TCM, Jonker JT, van Klinken JB, et al. A 5-day high-fat, high-calorie diet impairs insulin sensitivity in healthy, young South Asian men but not in Caucasian men. Diabetes (2014) 63:248–58. doi:10.2337/db13-0696
29. Mittelman SD, Van Citters GW, Kim SP, Davis DA, Dea MK, Hamilton-Wessler M, et al. Longitudinal compensation for fat-induced insulin resistance includes reduced insulin clearance and enhanced beta-cell response. Diabetes (2000) 49:2116–25. doi:10.2337/diabetes.49.12.2116
30. Wiesenthal SR, Sandhu H, McCall RH, Tchipashvili V, Yoshii H, Polonsky K, et al. Free fatty acids impair hepatic insulin extraction in vivo. Diabetes (1999) 48:766–74. doi:10.2337/diabetes.48.4.766
31. Svedberg J, Stromblad G, Wirth A, Smith U, Bjorntorp P. Fatty acids in the portal vein of the rat regulate hepatic insulin clearance. J Clin Invest (1991) 88:2054–8. doi:10.1172/JCI115534
32. Shanik MH, Xu Y, Skrha J, Dankner R, Zick Y, Roth J. Insulin resistance and hyperinsulinemia: is hyperinsulinemia the cart or the horse? Diabetes Care (2008) 31(Suppl 2):S262–8. doi:10.2337/dc08-s264
33. Cook JR, Langlet F, Kido Y, Accili D. Pathogenesis of selective insulin resistance in isolated hepatocytes. J Biol Chem (2015) 290:13972–80. doi:10.1074/jbc.M115.638197
34. Wolfrum C, Asilmaz E, Luca E, Friedman JM, Stoffel M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature (2004) 432:1027–32. doi:10.1038/nature03047
35. Weickert MO, Pfeiffer AF. Signalling mechanisms linking hepatic glucose and lipid metabolism. Diabetologia (2006) 49:1732–41. doi:10.1007/s00125-006-0295-3
36. Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell (2015) 160:745–58. doi:10.1016/j.cell.2015.01.012
37. Titchenell PM, Quinn WJ, Lu M, Chu Q, Lu W, Li C, et al. Direct hepatocyte insulin signaling is required for lipogenesis but is dispensable for the suppression of glucose production. Cell Metab (2016) 23:1154–66. doi:10.1016/j.cmet.2016.04.022
38. Groop LC, Bonadonna RC, Shank M, Petrides AS, DeFronzo RA. Role of free fatty acids and insulin in determining free fatty acid and lipid oxidation in man. J Clin Invest (1991) 87:83–9. doi:10.1172/JCI115005
39. Lester SG, Russo L, Ghanem SS, Khuder SS, DeAngelis AM, Esakov EL, et al. Hepatic CEACAM1 over-expression protects against diet-induced fibrosis and inflammation in white adipose tissue. Front Endocrinol (2015) 6:116. doi:10.3389/fendo.2015.00116
40. Elam MB, Wilcox HG, Cagen LM, Deng X, Raghow R, Kumar P, et al. Increased hepatic VLDL secretion, lipogenesis, and SREBP-1 expression in the corpulent JCR:LA-cp rat. J Lipid Res (2001) 42:2039–48.
41. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest (2002) 109:1125–31. doi:10.1172/JCI0215593
42. Ginsberg HN, Zhang YL, Hernandez-Ono A. Regulation of plasma triglycerides in insulin resistance and diabetes. Arch Med Res (2005) 36:232–40. doi:10.1016/j.arcmed.2005.01.005
43. Matikainen N, Manttari S, Westerbacka J, Vehkavaara S, Lundbom N, Yki-Jarvinen H, et al. Postprandial lipemia associates with liver fat content. J Clin Endocrinol Metab (2007) 92:3052–9. doi:10.1210/jc.2007-0187
44. Vine DF, Takechi R, Russell JC, Proctor SD. Impaired postprandial apolipoprotein-B48 metabolism in the obese, insulin-resistant JCR:LA-cp rat: increased atherogenicity for the metabolic syndrome. Atherosclerosis (2007) 190:282–90. doi:10.1016/j.atherosclerosis.2006.03.013
45. Biddinger SB, Hernandez-Ono A, Rask-Madsen C, Haas JT, Aleman JO, Suzuki R, et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab (2008) 7:125–34. doi:10.1016/j.cmet.2007.11.013
46. Gonzalez E, Flier E, Molle D, Accili D, McGraw TE. Hyperinsulinemia leads to uncoupled insulin regulation of the GLUT4 glucose transporter and the FoxO1 transcription factor. Proc Natl Acad Sci U S A (2011) 108:10162–7. doi:10.1073/pnas.1019268108
47. Xu E, Dubois MJ, Leung N, Charbonneau A, Turbide C, Avramoglu RK, et al. Targeted disruption of carcinoembryonic antigen-related cell adhesion molecule 1 promotes diet-induced hepatic steatosis and insulin resistance. Endocrinology (2009) 150:3503–12. doi:10.1210/en.2008-1439
48. Emanuelli B, Vienberg SG, Smyth G, Cheng C, Stanford KI, Arumugam M, et al. Interplay between FGF21 and insulin action in the liver regulates metabolism. J Clin Invest (2014) 124:515–27. doi:10.1172/JCI67353
49. Owen BM, Ding X, Morgan DA, Coate KC, Bookout AL, Rahmouni K, et al. FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab (2014) 20(4):670–7. doi:10.1016/j.cmet.2014.07.012
50. Cornu M, Oppliger W, Albert V, Robitaille AM, Trapani F, Quagliata L, et al. Hepatic mTORC1 controls locomotor activity, body temperature, and lipid metabolism through FGF21. Proc Natl Acad Sci U S A (2014) 111:11592–9. doi:10.1073/pnas.1412047111
51. Rosenbaum M, Leibel RL. Leptin: a molecule integrating somatic energy stores, energy expenditure and fertility. Trends Endocrinol Metab (1998) 9:117–24. doi:10.1016/S1043-2760(98)00028-9
52. Choi MS, Kim YJ, Kwon EY, Ryoo JY, Kim SR, Jung UJ. High-fat diet decreases energy expenditure and expression of genes controlling lipid metabolism, mitochondrial function and skeletal system development in the adipose tissue, along with increased expression of extracellular matrix remodelling- and inflammation-related genes. Br J Nutr (2015) 113:867–77. doi:10.1017/S0007114515000100
53. Heinrich G, Russo L, Castaneda TR, Pfeiffer V, Ghadieh HE, Ghanem SS, et al. Leptin resistance contributes to obesity in mice with null mutation of carcinoembryonic antigen-related cell adhesion molecule 1. J Biol Chem (2016) 291:11124–32. doi:10.1074/jbc.M116.716431
54. Cha SH, Hu Z, Lane MD. Long-term effects of a fatty acid synthase inhibitor on obese mice: food intake, hypothalamic neuropeptides, and UCP3. Biochem Biophys Res Commun (2004) 317:301–8. doi:10.1016/j.bbrc.2004.03.026
55. Chakravarthy MV, Zhu Y, Lopez M, Yin L, Wozniak DF, Coleman T, et al. Brain fatty acid synthase activates PPARalpha to maintain energy homeostasis. J Clin Invest (2007) 117:2539–52. doi:10.1172/JCI31183
56. Gao S, Lane MD. Effect of the anorectic fatty acid synthase inhibitor C75 on neuronal activity in the hypothalamus and brainstem. Proc Natl Acad Sci U S A (2003) 100:5628–33. doi:10.1073/pnas.1031698100
57. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science (2011) 332:1519–23. doi:10.1126/science.1204265
58. Lau JK, Zhang X, Yu J. Animal models of non-alcoholic fatty liver disease: current perspectives and recent advances. J Pathol (2017) 241:36–44. doi:10.1002/path.4829
59. Anstee QM, Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol (2006) 87:1–16. doi:10.1111/j.0959-9673.2006.00465.x
60. Heinrich G, Ghosh S, Deangelis AM, Schroeder-Gloeckler JM, Patel PR, Castaneda TR, et al. Carcinoembryonic antigen-related cell adhesion molecule 2 controls energy balance and peripheral insulin action in mice. Gastroenterology (2010) 139:644–52. doi:10.1053/j.gastro.2010.03.056
61. Lee SJ, Heinrich G, Fedorova L, Al-Share QY, Ledford KJ, Fernstrom MA, et al. Development of nonalcoholic steatohepatitis in insulin-resistant liver-specific S503A carcinoembryonic antigen-related cell adhesion molecule 1 mutant mice. Gastroenterology (2008) 135:2084–95. doi:10.1053/j.gastro.2008.08.007
62. Dankner R, Chetrit A, Shanik MH, Raz I, Roth J. Basal-state hyperinsulinemia in healthy normoglycemic adults is predictive of type 2 diabetes over a 24-year follow-up: a preliminary report. Diabetes Care (2009) 32:1464–6. doi:10.2337/dc09-0153
63. Pories WJ, Dohm GL. Diabetes: have we got it all wrong? Hyperinsulinism as the culprit: surgery provides the evidence. Diabetes Care (2012) 35:2438–42. doi:10.2337/dc12-0684
64. Corkey BE. Banting lecture 2011: hyperinsulinemia: cause or consequence? Diabetes (2012) 61:4–13. doi:10.2337/db11-1483
65. Marini MA, Frontoni S, Succurro E, Arturi F, Fiorentino TV, Sciacqua A, et al. Differences in insulin clearance between metabolically healthy and unhealthy obese subjects. Acta Diabetol (2014) 51:257–61. doi:10.1007/s00592-013-0511-9
66. Meistas MT, Margolis S, Kowarski AA. Hyperinsulinemia of obesity is due to decreased clearance of insulin. Am J Physiol Endocrinol Metab (1983) 245:E155–9.
67. Jones CN, Abbasi F, Carantoni M, Polonsky KS, Reaven GM. Roles of insulin resistance and obesity in regulation of plasma insulin concentrations. Am J Physiol Endocrinol Metab (2000) 278:E501–8.
68. Lee CC, Haffner SM, Wagenknecht LE, Lorenzo C, Norris JM, Bergman RN, et al. Insulin clearance and the incidence of type 2 diabetes in Hispanics and African Americans: the IRAS family study. Diabetes Care (2013) 36:901–7. doi:10.2337/dc12-1316
69. Pivovarova O, Bernigau W, Bobbert T, Isken F, Mohlig M, Spranger J, et al. Hepatic insulin clearance is closely related to metabolic syndrome components. Diabetes Care (2013) 36:3779–85. doi:10.2337/dc12-1203
70. Finucane FM, Sharp SJ, Hatunic M, Sleigh A, De Lucia Rolfe E, Aihie Sayer A, et al. Liver fat accumulation is associated with reduced hepatic insulin extraction and beta cell dysfunction in healthy older individuals. Diabetol Metab Syndr (2014) 6:43. doi:10.1186/1758-5996-6-43
Keywords: insulin clearance, insulin resistance, lipogenesis, fatty liver oxidation, lipolysis, NAFLD, visceral obesity
Citation: Heinrich G, Ghadieh HE, Ghanem SS, Muturi HT, Rezaei K, Al-Share QY, Bowman TA, Zhang D, Garofalo RS, Yin L and Najjar SM (2017) Loss of Hepatic CEACAM1: A Unifying Mechanism Linking Insulin Resistance to Obesity and Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 8:8. doi: 10.3389/fendo.2017.00008
Received: 04 November 2016; Accepted: 10 January 2017;
Published: 26 January 2017
Edited by:
Amiya Prasad Sinha-Hikim, Charles R. Drew University of Medicine and Science, USAReviewed by:
Guillermo Romero, University of Pittsburgh, USAYves Combarnous, Centre National de la Recherche Scientifique (CNRS), France
Copyright: © 2017 Heinrich, Ghadieh, Ghanem, Muturi, Rezaei, Al-Share, Bowman, Zhang, Garofalo, Yin and Najjar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sonia M. Najjar, bmFqamFyQG9oaW8uZWR1