 Monica Marzagalli1†
Monica Marzagalli1† Marina Montagnani Marelli1†
Marina Montagnani Marelli1† Lavinia Casati2
Lavinia Casati2 Fabrizio Fontana1
Fabrizio Fontana1 Roberta Manuela Moretti1
Roberta Manuela Moretti1 Patrizia Limonta1*
Patrizia Limonta1*
- 1Department of Pharmacological and Biomolecular Sciences, Università degli Studi di Milano, Milano, Italy
- 2Department of Medical Biotechnologies and Translational Medicine, Università degli Studi di Milano, Milano, Italy
Cutaneous melanoma is an aggressive tumor; its incidence has been reported to increase fast in the past decades. Melanoma is a heterogeneous tumor, with most patients harboring mutations in the BRAF or NRAS oncogenes, leading to the overactivation of the MAPK/ERK and PI3K/Akt pathways. The current therapeutic approaches are based on therapies targeting mutated BRAF and the downstream pathway, and on monoclonal antibodies against the immune checkpoint blockade. However, treatment resistance and side effects are common events of these therapeutic strategies. Increasing evidence supports that melanoma is a hormone-related cancer. Melanoma incidence is higher in males than in females, and females have a significant survival advantage over men. Estrogens exert their effects through estrogen receptors (ERα and ERβ) that affect cancer growth in an opposite way: ERα is associated with a proliferative action and ERβ with an anticancer effect. ERβ is the predominant ER in melanoma, and its expression decreases in melanoma progression, supporting its role as a tumor suppressor. Thus, ERβ is now considered as an effective molecular target for melanoma treatment. 17β-estradiol was reported to inhibit melanoma cells proliferation; however, clinical trials did not provide the expected survival benefits. In vitro studies demonstrate that ERβ ligands inhibit the proliferation of melanoma cells harboring the NRAS (but not the BRAF) mutation, suggesting that ERβ activation might impair melanoma development through the inhibition of the PI3K/Akt pathway. These data suggest that ERβ agonists might be considered as an effective treatment strategy, in combination with MAPK inhibitors, for NRAS mutant melanomas. In an era of personalized medicine, pretreatment evaluation of the expression of ER isoforms together with the concurrent oncogenic mutations should be considered before selecting the most appropriate therapeutic intervention. Natural compounds that specifically bind to ERβ have been identified. These phytoestrogens decrease the proliferation of melanoma cells. Importantly, these effects are unrelated to the oncogenic mutations of melanomas, suggesting that, in addition to their ERβ activating function, these compounds might impair melanoma development through additional mechanisms. A better identification of the role of ERβ in melanoma development will help increase the therapeutic options for this aggressive pathology.
Introduction
Human malignant melanoma is a very aggressive human cancer; its incidence has been found to increase faster than any other cancer during the past decades; importantly, it is one of the most frequent cancers in adolescents and young adults (1). Although it is less common than other malignancies of the skin, melanoma accounts for nearly 75% of skin cancer-related deaths (2).
The incidence rate of cutaneous melanoma is correlated with race; it has been consistently reported that White populations have a greater (10-fold) increase than Hispanian, Black, or Asian populations (3, 4). Based on this observation, the phenotype is considered one of the major risk factors; in particular, fair skin, red or blond hair, blue eyes, and freckles are classic presenting features of melanoma patients (5, 6). In addition to these host factors, the individual risk of developing melanoma also depends on factors such as sun exposure, family history, genetic factors, and their interaction (7).
Epidemiological studies have pointed out the relationship between intense ultraviolet-B (UVB) radiation exposure and melanoma development. In recent years, intense exposure to natural (suntans, sunburns) or artificial (indoor suntannings) UVB have increased in adolescents and young adults, and this seems to be correlated with the increased incidence of melanoma in these population subgroups (8, 9). Family history increases the personal risk of melanoma by three to eight times; this risk increases with the number of affected family members (10). Several genes have been implicated in the development of melanoma: genes involved in the MAPK pathway (RAS and BRAF, the most frequent mutations), CDKN2A [a cyclin-dependent kinase (CDK) gene] genes that are associated with nevi and pigmentation traits, such as MTAP, MC1R, and TYR (11–14).
Molecular Aspects of Melanoma Development and Progression
Cutaneous melanoma arises from the malignant transformation of melanocytes, the melanin producing cells of the skin. Melanin (i.e., brown/black eumelanin) is the photoprotective pigment that provides attenuation of UV radiations. In response to UV radiation, keratinocytes secrete factors that regulate melanocytes survival, differentiation, proliferation, and motility. Mutations in genes involved in the processes of melanoma development and progression are very frequently found in melanoma patients. Melanoma is now recognized as a very heterogeneous tumor; however, the majority of patients harbor driver oncogenic mutations at the level of genes encoding for proteins involved in the growth factor receptors signaling pathways (MAPK/ERK and PI3K/Akt) (15–17).
BRAF Mutations
The MAPK/ERK pathway includes the small G protein RAS and the kinases RAF, MEK, and ERK. BRAF is mutated in approximately 50% of melanomas; 80–90% of these activating mutations involve a single substitution of valine in position 600 with glutamic acid (V600E) (18). Additional, more rare, BRAF mutations include V600K (valine substituted with lysine) and V600D (valine substituted with aspartic acid) (19). BRAF mutations mimic phosphorylation on the regulatory domain of the protein; this leads to an enhanced kinase activity of the protein and activation of its downstream targets MEK and ERK. Activation of this pathway triggers the G1/S transition of the cell cycle through the synthesis of cyclin D1 and negative regulation of the cell cycle inhibitor p27 (20).
RAS Mutations
RAS was the first oncogene to be identified in melanomas (21). Mutations causing the constitutive activation of this small G protein lead to the hyperactivation of its two downstream pathways, the MAPK/ERK and PI3K pathways, involved in the control of both the proliferative and metastatic behavior of tumor cells. NRAS is the most frequently (about 20–30% of tumors) mutated isoform of the RAS family members in melanoma; very recently, an increase in NRAS mutant allele percentage during melanoma progression has been reported (22).
Other Genetic Mutations
KIT is a receptor with tyrosine kinase activity; it is involved in the development of melanocytes, controlling their proliferation, survival, and migration. It is coupled with the MAPK/ERK, PI3K/Akt, and JAK/STAT intracellular signaling pathways. The KIT receptor is mutated in approximately 15% of mucosal, acral, and chronic sun-damaged melanomas (23). The presence of KIT mutations is particularly interesting because they usually are mutually exclusive with NRAS and BRAF mutations and because of the availability of specific KIT kinase inhibitors in the clinic. Some KIT mutations are well characterized, others are still poorly described (24).
The PI3K/Akt signaling pathway is negatively regulated by PTEN, a tumor suppressor protein. Mutations as well as deletions of PTEN are found in approximately 30% of melanoma cell lines and are frequently associated with mutations in BRAF (24).
The CDKN2A is the primary familial high-risk melanoma susceptibility locus identified in families with different cases of melanoma. This gene encodes two suppressor proteins: p16 and p14, involved in the control of cell cycle progression. Specifically, p16 normally inhibits the CDKs, leading to G1/S cell cycle arrest. The second suppressor protein, p14 blocks the degradation of p53, leading to increased apoptosis. In the general population, the prevalence of CDKN2A mutations in primary melanomas is only 1.2%; however, germ-line mutations in this locus were reported in approximately 20–57% of families with at least three cases of melanomas (24, 25).
In addition to specific gene mutations, the status of DNA methylation of cutaneous melanoma has been extensively studied and shown to possess both prognostic and therapeutic relevance. Alterations of DNA methylation, histone modifications, and modified expression of microRNAs are well-established epigenetic mechanisms of cell neoplastic transformation. Melanoma cells present aberrant DNA methylation patterns with DNA hypermethylation at the level of CpG islands in the promoter of tumor suppressor genes (leading to their inactivation) and global DNA hypomethylation (contributing to genomic instability). Hypomethylation of specific genes was also reported, leading to the overexpression of normally silenced oncogenes (26, 27). Global DNA hypomethylation was shown to correlate with melanoma progression toward the most aggressive phase and with less favorable clinical outcomes (26, 28).
Current Therapies for Advanced Melanoma
Targeting Proliferative Pathways
The majority of melanomas are diagnosed in the early stage (in situ melanomas) and are treatable with surgical removal. On the other hand, the prognosis of highly aggressive, late stage melanoma is still poor (29). Prior to 2010, the systemic treatment of choice for advanced metastatic melanoma was limited to cytotoxic chemotherapy and traditional forms of immunotherapy (interleukin-2, IL-2; interferon α-2b, IFN α-2b). Alkylating agents, such as dacarbazine and its prodrug temozolomide, were approved in 1974 by the US FDA; however, the responses to this treatment were very short and less than 5% of patients could achieve a complete response (30, 31). Moreover, chemotherapy treatments were reported to be associated with severe side effects (32, 33) and a very rapid tumor relapse (34). In 1998, the US FDA approved IL-2 for the treatment of metastatic melanoma, based on clinical observations demonstrating sustained remissions in approximately 5–10% of patients (35). However, IL-2 therapy is associated with substantial toxicity, requiring its administration in an intensive care unit setting (36). A combination therapy of dacarbazine with IL-2 or IFN α-2b was reported to improve the progression-free survival but not the overall survival and to be associated with severe side effects (36, 37).
These disappointing results, together with the very quick advances in the dissection of the heterogeneity of melanomas (38) and the understanding of the molecular aspects of melanoma development and progression (24), stimulated the search for newer treatment strategies.
Thus, therapies were developed to specifically target (“targeted therapies”) melanomas harboring either the BRAF or the NRAS mutation (17, 39–42). Vemurafenib, the first targeted drug for melanoma, is a selective BRAF V600E inhibitor, approved by the FDA in August 2011. This drug inhibits the kinase domain of the mutated protein, decreasing cell proliferation through reduced activation of the downstream MAPK/ERK signaling pathway. Encouraging results were reported in phase I clinical trials, showing that vemurafenib was associated with a very good response rate (43); however, later phase trials underlined a short duration of response with a quick development of drug resistance, leading to only marginal patient benefit (44). Dabrafenib is a reversible ATP-competitive inhibitor of V600E- and V600K-mutant BRAF that was approved in 2013; however, the median progression-free survival in melanoma patients treated with dabrafenib was found to be shorter than that reported with vemurafenib (44–46).
It is now well documented that long-term exposure of melanomas to BRAF inhibitors is associated with a rapid development of drug resistance, and this is mainly linked to the rewiring of the MAPK/ERK signaling pathway (44). In this situation, RAS and MEK are elevated while tumor suppressors, such as PTEN, are decreased (47, 48), and BRAF/MEK inhibitor (trametinib and cobimetinib) combinations are now accepted as the standard treatment for resistant melanomas (49). The dabrafenib/trametinib and vemurafenib/cobimetinib combinations were approved by FDA in 2014 and 2015, respectively (17).
As mentioned above, NRAS is mutated in 20–30% of melanoma patients. Unfortunately, so far, the attempts to target mutated NRAS have not led to specific therapeutic strategies; for this reason, the current treatment options for these tumors are mainly focused on targeting the NRAS effector pathways with RAF/MEK and PI3K/Akt inhibitors, either as single agents or as combinations (50). The identification of novel compounds specifically targeting the NRAS-mutant signaling pathway represents a current emerging challenge for the treatment of these melanomas.
Targeting Immunity Pathways
The lack of the achievement of convincing results with targeted therapies stimulated the search of novel therapeutic approaches, leading to the resurgence of antibody-based immune therapies. Several immune targeted therapies implicate the development of recombinant, humanized, and monoclonal antibodies against specific proteins of the immune cells, such as CTLA-4 and PD-1 (37, 41). The cytotoxic T-lymphocyte antigen 4 (CTLA-4) is an inhibitor checkpoint receptor, expressed on the surface of T cells, that blocks T cell activation and helps regulate the balance between immune activation and tolerance. T cell activation is regulated by signals, provided by CD80 and CD86, located on the surface of antigen-presenting cells (APC), which bind to CD28 on the membrane of T cells. In this setting, T cells are stimulated to enter the cell cycle, differentiate, and produce cytokines (IL-2). In normal conditions, CTLA-4 replaces CD28 in the binding to CD80 and CD86, providing an autocrine regulatory mechanism for preventing uncontrolled T cell activation (51). The anti-CTLA-4 monoclonal antibody, ipilimumab, increases T cell activity and leads to tumor regression (37, 52). In 2011, the FDA approved ipilimumab for the treatment of advanced melanoma, despite its associated side effects related to autoimmune events (colitis) and a poor response rate in patients (53). Similar to CTLA-4, the programed death-1 (PD-1) receptor is a coinhibitory protein that is expressed on the surface of antigen-specific CD8+ T cell. The PD-1 ligands (PD-L1 and PD-L2) expressed by tumor cells interact with this receptor, thus triggering inhibitory signals in T cells and resulting in the protection of cancer against immune cell-mediated death (54). Nivolumab and pembrolizumab, two specific monoclonal antibodies against the PD-1 receptor, release the immune checkpoint blockade, thus inducing an anticancer effect (55).
In order to overcome both drug resistance development and side effects, the challenge for basic researchers and clinicians will be the development of novel combination therapies (immune/immune or immune/target specific therapies) for the achievement of both long-term and safe responses (56–58).
Moreover, highlighting additional molecular pathways involved in melanoma growth and progression is urgently foreseen to help increase the development of novel targeted therapeutic strategies for this aggressive pathology.
The Sex Hormone Milieu and Melanoma
Although melanoma is classically considered a non-hormone-related cancer, growing evidence supports a direct correlation between sex hormones levels (estrogens, in particular) and melanoma growth and progression (59, 60). Epidemiological analyses pointed out a significant divergence in melanoma incidence between sexes in the past three decades. The Surveillance Epidemiology and End Results (SEER) data indicate that, during this period, melanoma incidence was nearly twofold higher in males than in females (61). Gender differences are also observed regarding the age-dependent onset of melanoma, with slightly higher rates in women aged 20–45 that decrease after the age of 45 years. On the other hand, in males, melanoma incidence progressively increases after 45–50 years of age and dramatically increase in men aged 50–85 (61). Moreover, a significant disparity has also been noted in the prognosis of this tumor between males and females, with women having a significant survival advantage over men (62–64). Finally, studies performed in a melanoma fish model (Xiphophorus couchianus) showed a twofold lower incidence of melanoma in females than in males after acute exposure to UVB irradiation, and this was accompanied by a sex-specific molecular genetic response (65).
A meta-analysis by Gandini et al. (66) summarizes the evidence pointing out the interaction between endogenous and exogenous estrogens, taking into account natural reproductive factors (menarche, fertility, parity, pregnancy, and menopausal status) and the use of oral contraceptives or hormonal replacement therapy.
Clinical studies documented that melanocytic nevi grow and darken during pregnancy. Melanoma neoplasms are thicker during pregnancy than those diagnosed outside of pregnancy (67), and this is in line with the observation that diethylstilbestrol enhances melanomagenesis in mouse B16 melanoma cells (68). However, it is still unclear whether melanoma patients during pregnancy have worsened outcomes with respect to non-pregnant patients (67, 69, 70). Similar contrasting results were reported on the incidence of melanoma during menopause (71).
A number of studies have focused on the possible link between sex steroid assumption and melanoma development. Most of these studies pointed out that the use of exogenous female hormones (either as oral contraceptives or as hormonal replacement) do not contribute to increased risk of cutaneous melanoma (66, 72). Finally, Auriemma et al. (73) recently investigated the possible mole modifications in women undergoing controlled ovarian stimulation (COS) for assisted reproduction technologies. The conclusion raised by these authors is that the results obtained do not support a causal relation between the supraphysiological hormone levels stimulation and worsening of clinical features of moles.
Another issue that needs to be solved is the expression/activity of aromatase in melanoma tissues. It seems that the skin has its own capacity to produce steroids (including estrogens) (59). In line with this, Santen and coworkers reported that the aromatase enzyme is expressed in melanoma tissues (74); however, no correlation was found in this paper between the expression of this enzyme and clinical outcomes. It must be underlined that this study did not have any follow-up; moreover, the first generation aromatase inhibitor aminoglutethimide was found to be ineffective in reducing melanoma progression (75). Further studies performed with newer aromatase inhibitors will likely help clarify this issue.
In conclusion, although contrasting results were so far reported on the relationship between estrogen levels and melanoma risk, the influence of the endocrine status on the development of melanoma is now well accepted. Based on these observations, de Giorgi and coworkers suggested that cutaneous melanoma should be considered as a hormone-related tumor (76), although this conclusion is mainly supported by the results from the studies analyzing the estrogen receptor (ER), particularly the ERβ, status of melanomas.
Estrogen Receptors α and β: Opposite Roles in Cancer
Estrogens exert their biological effects through two ER, ERα and ERβ, members of the nuclear receptor superfamily of transcription factors (77). ERα was cloned in 1985, and it was considered the only receptor responsible for estrogens action (78). ERβ was cloned from a rat prostate cDNA library in 1996, and this opened the way to the discovery of the human counterpart (79, 80). Both ERs are nuclear receptors, which can form either homo- or heterodimers and, upon activation, translocate into the nucleus to bind with coregulatory proteins and control the transcription of target genes through the binding to specific ERE regions (81–84).
The two receptors are encoded by two different genes (ESR1 and ESR2) that are located on chromosomes 6 and 14, respectively. They share the same general structure, characterized by three (independent, but interacting) functional domains: the N-terminal domain (NTD or A/B) containing a transactivation domain and a domain responsible for the recruitment of coactivators/corepressors; the DNA-binding domain (DBD or C) containing zinc fingers, necessary for the binding of the receptors to the estrogen response elements in the promoter region of target genes; the ligand-binding domain (LBD or D/E/F) with a ligand-dependent transactivating function. This LBD domain is also responsible for the binding to co-regulatory and chaperone proteins (77). The two ERs share about 97% similarity in the DBD, 59% in LBD, and only 16% in their NTD (77, 85). The differences in the LBD are responsible for the shape of the ligand-binding pocket and, based on this observation, specific ligands for each receptor subtype have been designed and synthesized (86, 87). At the same time, the same ligand may have different binding affinity for ERα or ERβ subtypes.
It has been widely reported that the endogenous hormone estradiol binds to both receptors with a similar binding affinity and the same transactivational activity in different cell types (88, 89). Thus, the specific effects of endogenous estradiol on ERα and ERβ activation largely depend on the different cell contexts and specifically on the recruitment of cell type-specific coactivators/corepressors as well as chromatin remodeling proteins (90). In melanoma cells, the binding affinity of estradiol for ERα and ERβ has not been specifically evaluated, but it is expected to be the same as in other cell types. However, it must be underlined that the majority of the studies so far performed pointed out a prevalence of expression of ERβ in these cells (see below).
Synthetic or natural ligands bind to ERα or ERβ with different affinities according to their chemical structure (88). Antiestrogens, such as tamoxifen, raloxifene, and ICI-164,384 are partial agonists/antagonists of ERα, depending on the target tissue, and are referred to as selective estrogen receptor modulators (SERMs). Different natural compounds [genistein, apigenin, liquiritigenin (LQ), silibinin, and silymarin] were reported to bind with higher affinity and, therefore, to specifically activate the ERβ subtype; for this reason, they are referred to as estrogen receptor subtype agonists (ERSAs) (86, 91, 92). Thus, in a given cell, the different binding affinity and transactivational activity of synthetic or natural ligands on the two ER subtypes seem to depend on the ERα/ERβ ratio as well as on the specific cell context (88, 90, 93). The full-length ERβ protein, also named ERβ1, is generated from 8 exons and includes 530 aminoacids. After ligand binding, ERβ1 can form either homodimers or ERα/ERβ1 heterodimers, which bind to ERE sequences on DNA; such interactions contribute in modulating ERβ1/ERα activity, in a specific cell context (94). In addition to ERβ1, four alternative splice variants of ERβ (ERβ2, ERβ3, ERβ4, and ERβ5) have been identified (95–97). All these variants have a truncated C-terminus and, for this reason, no ligands have been found for these forms; moreover, they are unable to form homodimers (97, 98). However, ERβ2 and ERβ5 can form heterodimers with ERβ1 isoform or with ERα, thus inhibiting their binding to ERE elements on DNA (96, 97). ERβ5 was reported to have a stable expression during the process of carcinogenesis, in contrast to ERβ1 and ERβ2 (99).
It is now clear that ERα and ERβ are associated with different activities, according to their specific tissue distribution (77, 94, 100). Increasing evidence supports a relationship between the perturbation of estrogen signaling and cancer initiation, promotion, and progression. Specifically, the variation of the ratio ERα/ERβ in tumor tissues supports the notion that the two ER subtypes have different functions in cancer biology and therapy. Overall, it is now well accepted that ERα contributes to tumorigenesis by stimulating cell proliferation, while ERβ is endowed with a significant antitumor activity. Thus, both synthetic and natural ERβ ligands may interfere with the mechanisms of tumor growth either by activating ERβ or by interfering with the tumor activity of ERα through ERα/ERβ heterodimers formation (77, 94, 101) (Figure 1).
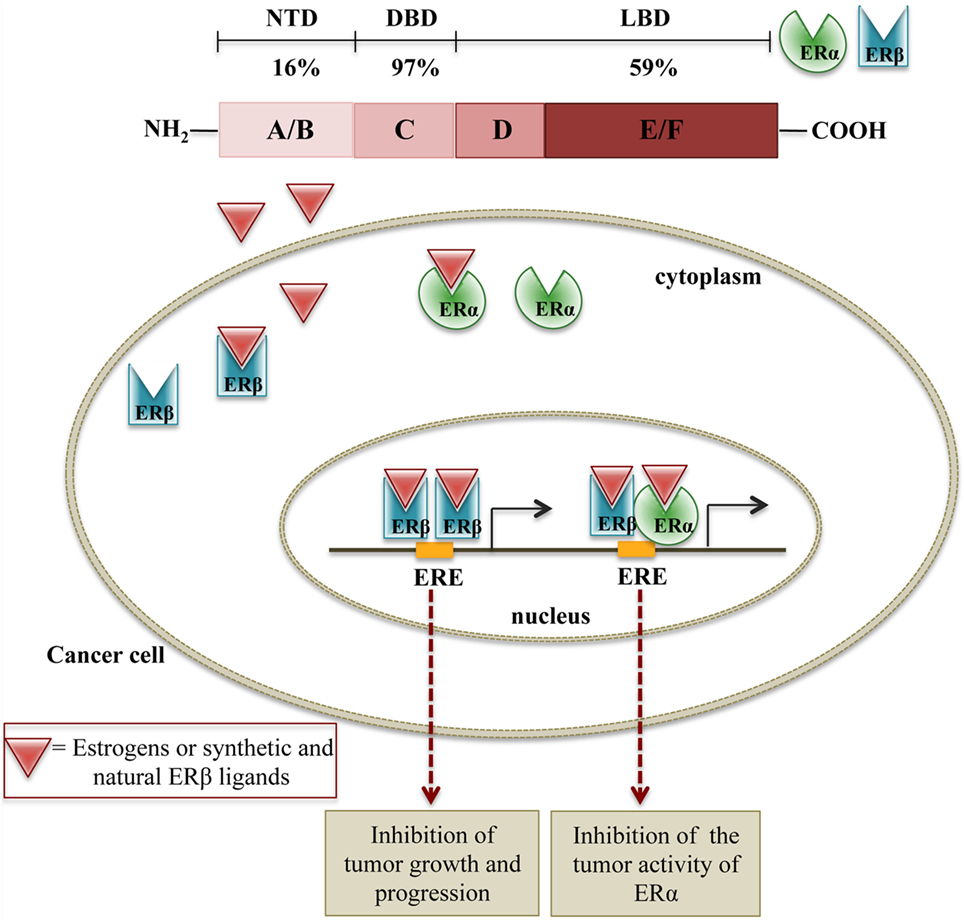
Figure 1. Schematic representation of the molecular mechanisms of ERβ activation on cell growth. In cancer cells expressing both ERβ and ERα, ERβ ligands can either induce the formation of ERβ:ERβ homodimers, inhibiting cell proliferation or ERβ:ERα heterodimers, counteracting the proliferative activity of ERα.
Estrogen Receptor β and Melanoma
ERα is the main ER in human skin; however, this receptor does not seem to play any role in the pathophysiology of melanoma precursor lesions or melanomas. On the other hand, ERβ has been reported to be the predominant ER subtype in melanocytic lesions; its distribution and levels of expression are different in the different classifications of the lesions. In 2006, Schmidt and coworkers investigated the expression of ERα and ERβ in benign nevi, dysplastic nevi with mild/moderate/severe cytological atypia, lentigo malignas, and melanomas with different depth (Clark) and thickness (Breslow) (102). They found that ERβ, but not ERα, was the predominant ER in both benign and malignant lesions and that ERβ expression levels also correlated with the tumor microenvironment. In line with these observations, it was reported that melanocytic nevi and malignant melanomas are both positive for ERβ, while they are negative for ERα (103). de Giorgi and coworkers investigated the expression of ERα and ERβ in human melanoma tissues (104). They found that both receptors are expressed at the mRNA level, but only the ERβ protein was present in these tissues. Analyzing melanoma cases into two groups according to Breslow thickness (104), these authors observed that the levels of both ERβ mRNA and protein were lower in thicker and more invasive tumors. According to these authors, these data support a protective role for ERβ in the metastatic process of melanoma cells (104, 105). An opposite correlation between ERβ levels and Breslow thickness was also reported by Schmidt et al. (102). Moreover, ERβ expression was found to be lower in tumor tissues compared with the adjacent healthy skin. Men showed lower levels of ERβ than women in both melanoma and healthy tissues, in agreement with sex differences in melanoma survival (106). ERβ has been found to be expressed in melanomas of pregnant women more frequently than in men and also a trend to a higher expression in women than in men has been reported. This indicates that ERβ might explain the generally favorable prognosis of melanoma in women (107). ERβ expression has been recently shown to be downregulated in aggressive, metastatic melanomas, suggesting its possible utility as a marker for metastatic potential and for prognosis in malignant melanomas (108). Finally, a polymorphism at the AluI restriction site was identified in a high proportion of melanoma, suggesting that the polymorphism of this receptor could be related to a higher susceptibility to the development of this tumor (109).
In line with these clinical observations, we recently demonstrated that ERβ, but not ERα, is the ER expressed in human melanoma cell lines, harboring different genetic mutations (A375, BLM, WM115, and WM1552) (110). Moreover, the pattern of expression of the different ERβ isoforms was similar in BLM (NRAS-mutant, BRAF-wild type) and WM115 (BRAF V600D-mutant) melanoma cells: ERβ1 and ERβ5 were found to be expressed at similar levels while ERβ2 showed a higher level of expression. On the other hand, in A375 (BRAF V600E-mutant) cells, both ERβ2 and ERβ5 were expressed at higher levels than ERβ1 (110).
Taken together, these data indicate that ERβ is expressed in melanoma cells, and its levels of expression negatively correlate with melanoma growth and progression, further supporting the notion that this receptor might be endowed with an antitumor activity in melanoma. Similar data have been previously reported for different types of tumors, both related and unrelated to the reproductive system (111–119).
Antitumor Activity of ERβ in Melanoma
Despite the extensive clinical observations reporting the expression of ERβ in melanoma tissues and its negative correlation with tumor progression, the data so far available on the direct antitumor effects of ERβ ligands are still scanty.
As mentioned above, both genomic and non-genomic signaling pathways mediate the effects of ERs activation. The genomic effects of ERs activation are mediated by receptor dimerization and binding to ERE on DNA while interacting with transcriptional coactivators, to regulate gene transcription. The non-genomic effects of ER ligands are mediated by two main signaling pathways: RAS/RAF/MEK/ERK and PI3K/Akt. Specifically, the PI3K/Akt pathway is well known to mediate the antitumor activity of ERβ in several types of cancers (94, 120). As discussed, some of the proteins involved in these signaling cascades are mutated in the majority of melanoma. Thus, both the ERs and the MAPK and PI3K pathways might represent molecular targets to counteract melanoma growth and progression.
Although extensive epidemiological studies clearly indicate that ERβ is the main ER subtype to be expressed in melanoma, the data so far available on the possible effect of estrogens on the growth and progression of this tumor are still scanty. Sarti and coworkers reported that 17β-estradiol exerts a significant inhibitory activity on the proliferation of the human SK-Mel 23 melanoma cell line, expressing ERβ, but not ERα (referred to as “type II estrogen binding site” by these authors) (121). The sex hormone 17β-estradiol was also shown to reduce the invasive behavior of human melanoma cells lacking the ERα receptor (122). In vitro and in vivo antitumor effects on melanoma were also reported for 2-methoxyestradiol, an endogenous metabolite of estradiol; however, it must be pointed out that the antitumor activity of this compound was not found to be mediated by ERs (both α and β) activation (123, 124).
The ER antagonist tamoxifen was first shown to induce cell death in human malignant melanoma cells, possibly through inactivation of the IGF-I receptor (125). This antiestrogen was also reported to reduce tumor cell metastatic behavior in the mouse melanoma cell line B16BL6 (126). However, later clinical studies did not confirm these promising results from in vitro studies. The effects of chemotherapy with and without tamoxifen for the treatment of aggressive melanoma were compared in different clinical trials. These studies reported that co-treatment with tamoxifen may provide improvements in response rates, although it is often accompanied by increased toxicity and no survival benefit (60, 127–131). It has been proposed that this is due to the fact that tamoxifen may decrease cell proliferation when it binds to ERα while it may increase cell proliferation when it binds to ERβ (60). Thus, the antitumor vs. prosurvival effects of tamoxifen likely depend on the different ERα/ERβ ratios in a given tissue (105). In line with this observation, low levels of expression of ERβ were shown to correlate with tamoxifen resistance in breast cancer cells (132, 133).
In a recent paper, we showed that ERβ, but not ERα, is the ER expressed in human melanoma cell lines, harboring different genetic mutations. We demonstrated that, in BLM (BRAF-wild type, NRAS-mutant) melanoma cells, ERβ agonists [17β-estradiol, diarylpropionitrile (DPN), KB1] significantly inhibited cell proliferation, and this effect was abrogated by an ER antagonist. ERβ activation triggered the translocation of the receptor from the cytoplasmic to the nuclear compartment and its transcriptional activity. The activity of ERβ agonists was accompanied by an altered expression of the proteins involved in the G1/S transition of the cell cycle (decreased levels of cyclin D1 and cyclin D3, and increased expression of p27, a CDK inhibitor); the apoptosis pathway was not involved in this activity (no change in the cleaved, active form of caspase-3). Importantly, we reported an aberrant global DNA hypomethylation in BLM cells, indicative of genome instability; ERβ activation reverted this hypomethylation status (110). Taken together, these data demonstrate that, in NRAS-mutant melanoma cells, ERβ is associated with an antitumor activity, by causing cell cycle arrest and through the regulation of cell cycle-associated proteins; similar observations were previously reported in different types of cancers expressing this receptor (94, 101, 134–140). Surprisingly, in our paper, we could also show that ERβ agonists were ineffective in reducing the proliferation of A375 and WM1552 (V600E BRAF-mutant) melanoma cells, expressing the ER isoform. We speculated that the different effects of ERβ ligands in these cell lines might be related to their specific oncogenic mutation status. Actually, as mentioned above, NRAS and BRAF mutations are the most frequent oncogenic mutations found in melanoma. NRAS mutations have been reported to be associated with increased activation of the two main downstream pathways: PI3K/Akt and MEK/ERK. On the other hand, in melanoma cells harboring BRAF mutations, only the MEK/ERK signaling cascade was shown to be overactivated. In agreement with our data, ERβ ligands have been previously shown to exert their antitumor effects through inactivation of RAS as well as of the PI3K/Akt pathway in some cancer cells (94, 141, 142). Moreover, Wang and coworkers (143) have recently reported an inverse correlation between the expression of ERβ and the activity of the PI3K/Akt pathway in aggressive, triple-negative, breast cancer. Chen et al. (141) demonstrated that, in breast cancer cells, the isoflavone calycosin activates ERβ, and this is followed by a decreased activity of the PI3K/Akt pathway; on the other hand, calycosin did not affect the activity of the MAPK/ERK cascade. Based on these observations, it seems possible to conclude that ERβ activation might significantly reduce the growth of cutaneous melanoma cells harboring the NRAS mutation, possibly through the inhibition of the PI3K/Akt signaling pathway (Figure 2). On the other hand, ERβ agonists will not be able to reduce the proliferation of melanoma cells carrying the BRAF (V600E) mutation, which is associated with the overactivation of the MEK/ERK signaling cascade.
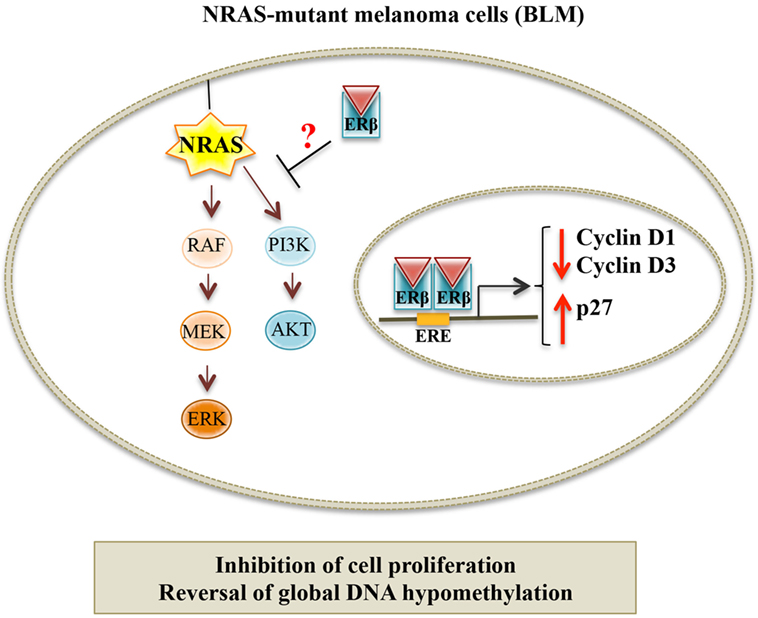
Figure 2. Proposed model for the targeting of NRAS-mutant melanoma by ERβ ligands. In NRAS-mutant melanoma cells (BLM), ERβ agonists trigger genomic effects at the nuclear level by modulating the expression of cell cycle-related proteins and by reversing the global hypomethylation status of these cells. Moreover, it is hypothesized that activated ERβ might also exert non-genomic effects, by interfering with the PI3K/Akt signaling pathway, as previously described for different cancer cell lines expressing this receptor.
These data suggest that, in melanoma patients harboring the NRAS mutation, ERβ might represent an effective molecular target for personalized therapeutic interventions. For instance, these interventions might be based on an ERβ agonist given either alone or in combination with specific inhibitors of the MEK cascade (i.e., trametinib and cobimetinib), with the aim to block the activity of both the PI3K/Akt and the MEK/ERK signaling pathways. In an era of personalized medicine, it is suggested that a pretreatment evaluation of the ER isoforms expression in each melanoma patient, together with the concurrent oncogenic mutations, should be considered in order to anticipate the response of melanoma patients to novel therapeutic strategies (50).
Natural ERβ Ligands and Melanoma
Phytoestrogens are natural, plant-derived, estrogenic compounds that preferentially bind to ERβ than ERα (91, 144). These compounds have been suggested as possible effective agents in the prevention of several diseases, such as menopausal symptoms, osteoporosis, and cardiac diseases (145). They have also been reported to exert antitumor activity on different types of cancers, based on their high affinity binding to ERβ and their ability to increase the expression of this receptor subtype (115, 119, 138, 146–148) (Table 1).
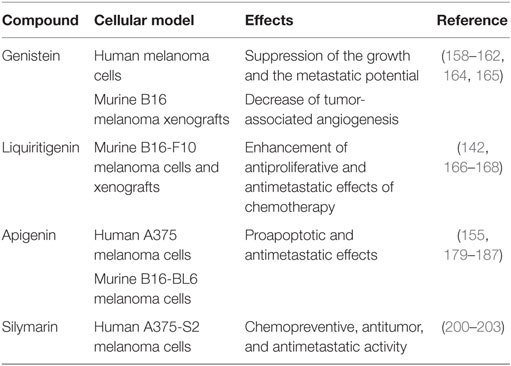
Table 1. Natural ERβ ligands and melanoma.
Thus, the antitumor activity of phytoestrogens largely depends on the subtype of ER expressed in a given tissue, as well as its levels of expression and the circulating steroid hormones milieu (149, 150). As discussed for estrogens, phytoestrogens can exert their effects through both genomic and non-genomic (by affecting different intracellular signaling pathways) activities (151). These effects include alterations of tyrosine kinase pathways, antioxidant effects, and epigenetic mechanisms, and also through recruitment of coregulators endowed with chromatin binding activities, thus underlying the relevance of epigenetic mechanisms in estrogen signaling (152).
Flavonoids are a large subclass of polyphenolic compounds present in many vegetables and medicinal herbs (153); some of these compounds have been demonstrated to play a potential role in the management of different types of tumors with little side effects since they interfere with cancer progression by regulating cell proliferation, apoptosis, invasion, and metastasis (154, 155).
The phytoestrogens genistein and daidzein are isoflavones, found in soybean, one of the most important food components in Asian diet (156, 157). These compounds received considerable attention based on epidemiological studies demonstrating that the soybean-containing diets were associated with a lower incidence of particular cancers in Asian population (158). Genistein interacts with both ERα and ERβ but has higher affinity for ERβ (138, 159).
This isoflavone was first shown to suppress the growth and also the metastatic potential of human melanoma cells, both in vitro and in vivo, with or without the induction of differentiation, dendritic formation, etc. (160–162).
It is well known that phytoestrogens undergo glycosidic binding to carbohydrates to form molecules that cannot be easily absorbed. For this reason, the glycosidic binding must be broken up by enzymes (glycosidases) present in the gut, produced by the intestinal microflora, that transform the glycosidic forms in the corresponding “aglicones” that are very easily absorbed (163). However, it has also been reported that genistin, as other flavonoid glycosides, is partly absorbed without previous cleavage and does not need to be hydrolized to be biologically active. In line with these observations, Russo and coworkers (158) reported that genistin exerts an inhibitory effect of human melanoma cells and inhibited ultraviolet (UV) light-induced oxidative DNA damage. Moreover, genistin and daidzin (the glycosidic forms of the two isoflavones) showed a protective effect on DNA damage; only genistin, but not daidzin, was able to counteract the proliferation of human melanoma cells (M14). These data suggest that not only the isoflavone aglycons but also the corresponding glycosides may trigger significant antitumor effects in melanoma cells. Genistein, at non-cytotoxic concentrations, was reported to reduce the growth as well as the motility of mouse B16 melanoma cells, through modulation of the activity of enzymes involved in the degradation of the extracellular matrix [i.e., urokinase-type plasminogen activator (uPA)]. Moreover, in vivo i.p. administration of genistein was found to decrease tumor-associated angiogenesis in nude mice-bearing mouse melanoma xenografts (B16); similar results were obtained with a soybean-based diet (164, 165).
The flavanone LQ is extracted from conventional herbal medicine-glycyrrhizae and is considered a highly selective ERβ agonist (166). In a recent paper, Shi and coworkers (167) investigated whether LQ may potentiate the antimetastatic effect of chemotherapeutic drugs [cis-diamine dichloroplatinum (CDDP)] in murine B16F10 melanoma cells. It was found that LQ significantly potentiates the anti-migratory/anti-invasive effects of CDDP, and this is mediated by downregulation of the PI3K/Akt pathway. LQ also enhanced the CDDP-induced suppression of lung metastasis formation in nude mice bearing B16F10 xenografts.
It has been reported that LQ displays only 20% greater binding affinity for ERβ than ERα (166); however, recent data demonstrate that different molecular mechanisms, in addition to its ability to bind ERβ, can account for its activity. In particular, it has been recently shown that the LQ/ERβ complex is able to recruit ERβ coactivators and that it specifically binds to ERβ responsive elements in the promoter region of target genes (159, 166, 168).
As a further support to the notion that ERβ is involved in LQ antitumor activity, data from the literature demonstrate that, in temozolomide-resistant U138 glioma cells, LQ treatment sensitizes cancer cells to drug-induced inhibition of cell proliferation and significantly increases ERβ expression. Moreover, LQ treatment significantly inhibits the activity of the PI3K/Akt pathway, and this effect is completely reversed by ERβ knockdown (142).
On the other hand, LQ is known to have a lower affinity for ERβ than estradiol (169); this raises the question of the dose of this compound that should be necessary to exert a physiological/therapeutic effect. Although further studies are required to specifically answer this question, it must be underlined that LQ has been reported to inhibit tumor growth in preclinical mouse models of gliomas and human cervical cancers (138, 170, 171).
Liquiritigenin is a component of licorice roots, and licorice root extracts are utilized by menopausal women as dietary supplements to fight menopausal symptoms as an alternative to classical pharmacological interventions (172, 173). In addition to LQ and iso-liquiritigenin, licorice extracts contain a series of different compounds, such as glycyrrhizin, glabridin, glabrene, calycosin, methoxychalcone, vestitol, and glycycoumarin. Glycyrrhizin, when consumed at high doses and for a long period, is associated with serious side effects, such as hypertension. Therefore, attention has been recently given to the other components of licorice root extracts with the aim to investigate their possible interaction with the ER system. Most of these compounds have been reported to behave as SERMs, specifically acting as ERα agonists in some cell types while behaving as ERα antagonists in other cell contexts (169, 174, 175).
Whether the components of licorice root extracts might exert antiproliferative effects on cancer cells, and particularly on melanoma cells, by interfering with the estrogenic system is still unclear. To this purpose, it must be recalled that ERβ, and not ERα, is the predominant ER subtype in melanocytic lesions (see above). The novel flavonoid isoangustone A has been shown to inhibit the proliferation of human melanoma cells through blockade of cell-cycle progression and downregulation of the PI3K/Akt pathway (176); glycyrrhizin seems to protect human melanoma cells from UVB irradiations (177); more recently, the licorice component licochalcone A has been reported to reduce the growth of different cancer cell lines, including melanoma cells (178). However, the possible involvement of ERβ in the antitumor activity of these compounds still needs to be investigated.
The flavone apigenin is another phytoestrogen that received growing attention as a potential chemopreventive agent and suppressor of cancer growth (155, 179–182). In A375 human melanoma cells, apigenin was reported to induce apoptosis through accumulation of reactive oxygen species in mitochondria, upregulation of Bax, caspase 3, 9, and PARP and downregulation of Bcl-2, thus triggering the mitochondrial apoptotic pathway (183). Apigenin was also shown to reduce the metastatic potential of melanoma cells in vitro (184) as well as in vivo in a preclinical model of mice injected with B16-BL6 murine melanoma cells (185). In addition, the antimetastatic activity of this compound in murine B16F10 melanoma cells has been demonstrated to be mediated by suppression of signal transducer and activator of transcription 3 (STAT3) phosphorylation and downregulation of STAT3 target genes MMP-2, MMP-9, VEGF, and Twist1, known to be involved in the metastatic behavior of cancer cells (186). Moreover, apigenin was found to overcome resistance to the anticancer agent TRAIL (tumor necrosis factor-related apoptosis-inducing ligand), a very promising compound that kills different tumor cells while sparing normal tissues, but is very often associated with development of resistance. Apigenin inhibits the expression of the p53 antagonist murine double minute 2 (Mdm2), thus increasing p53 levels and, consequently, upregulating the TRAIL receptor 2, the p53 target gene (187). However, whether ERβ might be involved in the antitumor activity of apigenin in melanoma is still unclear. On the other hand, this phytoestrogen has been reported to exert its antitumor activity in breast and prostate cancer cells through activation of ERβ (180); interestingly, in prostate cancer cells, apigenin induces apoptosis by selectively inhibiting proteasomal activity, thus rescuing ERβ from degradation and, therefore, increasing its intracellular levels (188).
Apigenin has also been shown to cross-react with progesterone receptors (189). This compound induces apoptosis and blocks the medroxyprogesterone acetate-dependent growth of aggressive breast cancer xenograft tumors (190–193). On the other hand, the expression/activity of the progesterone receptor and the possible cross-reaction of apigenin with this receptor in melanoma cells still need to be clarified. Progesterone receptors have been shown to be expressed in both conjunctival nevi and melanoma specimens. However, the rate of progesterone receptors was not found to correlate with the disease course; progesterone has been reported to inhibit the proliferation of melanoma cells, but this effect was found not to be mediated by the progesterone receptor (194, 195). Silymarin, extracted from Silybum marianum, is a mixture of four flavolignans (silybinin, isosilybinin, silydianin, and silychristin) and the isoflavone taxifolin. Since the first demonstration that silymarin selectively binds to ERβ (but not to ERα), this mixture was considered an ERβ specific ligand (196). The effects of silymarin were analyzed on ultraviolet light (UV)-induced cell apoptosis in human A375-S2 melanoma cells. It was found that this flavonoid prevents UV irradiation-induced apoptosis of these cells, through the activation of the Akt and SIRT1 pathways (197–199). On the other hand, silymarin has been recently shown to inihibit melanoma cell growth both in vitro and in vivo (200, 201) and to induce cell cycle arrest in melanoma cells directly targeting the MEK1/2 pathway (202). Moreover, Vaid and coworkers (203) investigated the effects of silymarin on the metastatic properties of human melanoma cell lines. They reported that this polyphenolic flavonoid significantly inhibits the migratory/invasive behavior of these cells, and this antimetastatic behavior is mediated by the Wnt/β-catenin pathway. Taken together, these observations strongly support the notion that the natural ERβ agonist silymarin may exert both a chemopreventive and an antitumor activity in melanoma cells. Similar observations were previously reported for experimental models of colon carcinogenesis (204).
It must be underlined that silymarin can also bind the androgen receptor, and it activates the same molecular pathways involved in the ERβ signaling, specifically in prostate cancer cells (205, 206). However, the expression and the possible role of this receptor in skin carcinogenesis is still a matter of debate (60, 107, 207, 208). Moreover, to the authors’ knowledge, no data are so far available in the literature on the possible role of androgen receptors in the antitumor activity of silymarin in melanoma.
Given the antiproliferative/proapoptotic activity of phytoestrogens in melanoma, as well as in different types of tumors, and based on their high-binding affinity to the ERβ subtype, further studies are needed to definitely confirm the role of ERβ in the antitumor activity of these natural compounds. Results from these studies will likely open the way to novel, nutraceutical-based, chemopreventive and therapeutic (i.e., combinatorial) options for this aggressive pathology.
Conclusion
Increasing evidence supports a close relationship between the sex hormone (i.e., estrogens and ERs) milieu and melanoma growth and progression. Clinical studies have pointed out that ERβ, but not ERα, is the predominant ER subtype in melanoma tissues, and its levels of expression are downregulated during melanoma progression toward the most aggressive phases. Considering that ERβ is the ER subtype widely shown to be associated with an anticancer effect in tumors in which it is expressed, these clinical observations strongly support the notion that ERβ might be considered as a possible molecular target for the development of therapeutic strategies for melanoma. However, the data so far available on the direct antitumor effects of ERβ ligands in melanoma are still scanty. Clinical studies reported that the ER antagonist tamoxifen provides only variable improvements in the rates of response of melanoma patients to chemotherapy. In vitro studies demonstrate that ERβ agonists can impair melanoma cell proliferation, but this depends on the genetic mutational status (NRAS vs. BRAF) of melanoma cells. Taken together, these data strongly support the notion that evaluation of the oncogenic mutation (BRAF vs. NRAS) together with the expression of the ER subtype (ERβ vs. ERα) in melanoma patients should be taken into consideration when considering the most specific therapeutic approach to be applied. Moreover, based on the promising experimental and preclinical data so far available, we believe that a better identification of the molecular mechanisms of the antitumor activity of natural ERβ ligands will likely improve the treatment strategies for melanoma patients.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication. MMM and MM evaluated the paper and designed the figures. PL wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
The reviewer JC and handling editor declared their shared affiliation, and the handling editor states that the process nevertheless met the standards of a fair and objective review.
Funding
This work was supported by Fondazione Banca del Monte di Lombardia, Comitato Emme Rouge Onlus, and PRIN 2010–2011.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin (2015) 65:5–29. doi:10.3322/caac.21254
2. Higgins HW II, Lee KC, Galan A, Leffell DJ. Melanoma in situ: part I. Epidemiology, screening, and clinical features. J Am Acad Dermatol (2015) 73:181–90. doi:10.1016/j.jaad.2015.04.014
3. Geller AC, Clapp RW, Sober AJ, Gonsalves L, Mueller L, Christiansen CL, et al. Melanoma epidemic: an analysis of six decades of data from the Connecticut Tumor Registry. J Clin Oncol (2013) 31:4172–8. doi:10.1200/JCO.2012.47.3728
4. Kamath S, Miller KA, Cockburn MG. Current data on risk factor estimates does not explain the difference in rates of melanoma between Hispanics and non-Hispanic whites. J Skin Cancer (2016) 2016:2105250. doi:10.1155/2016/2105250
5. Rees JL. Genetics of hair and skin color. Annu Rev Genet (2003) 37:67–90. doi:10.1146/annurev.genet.37.110801.143233
6. Berwick M, Buller DB, Cust A, Gallagher R, Lee TK, Meyskens F, et al. Melanoma epidemiology and prevention. Cancer Treat Res (2016) 167:17–49. doi:10.1007/978-3-319-22539-5_2
7. Russak JE, Rigel DS. Risk factors for the development of primary cutaneous melanoma. Dermatol Clin (2012) 30:363–8. doi:10.1016/j.det.2012.05.002
8. Dore JF, Chignol MC. Tanning salons and skin cancer. Photoch Photobio Sci (2012) 11:30–7. doi:10.1039/C1PP05186E
9. Whiteman DC, Whiteman CA, Green AC. Childhood sun exposure as a risk factor for melanoma: a systematic review of epidemiologic studies. Cancer Causes Control (2001) 12:69–82. doi:10.1023/A:1008980919928
10. Florell SR, Boucher KM, Garibotti G, Astle J, Kerber R, Mineau G, et al. Population-based analysis of prognostic factors and survival in familial melanoma. J Clin Oncol (2005) 23:7168–77. doi:10.1200/JCO.2005.11.999
11. Clark WH Jr, Reimer RR, Greene M, Ainsworth AM, Mastrangelo MJ. Origin of familial malignant melanomas from heritable melanocytic lesions. ‘The B-K mole syndrome’. Arch Dermatol (1978) 114:732–8. doi:10.1001/archderm.114.5.732
12. Holland EA, Schmid H, Kefford RF, Mann GJ. CDKN2A (P16(INK4a)) and CDK4 mutation analysis in 131 Australian melanoma probands: effect of family history and multiple primary melanomas. Genes Chromosomes Cancer (1999) 25:339–48. doi:10.1002/(SICI)1098-2264(199908)25:4<339::AID-GCC5>3.0.CO;2-H
13. Haluska FG, Tsao H, Wu H, Haluska FS, Lazar A, Goel V. Genetic alterations in signaling pathways in melanoma. Clin Cancer Res (2006) 12:2301s–7s. doi:10.1158/1078-0432.CCR-05-2518
14. Gudbjartsson DF, Sulem P, Stacey SN, Goldstein AM, Rafnar T, Sigurgeirsson B, et al. ASIP and TYR pigmentation variants associate with cutaneous melanoma and basal cell carcinoma. Nat Genet (2008) 40:886–91. doi:10.1038/ng.161
16. Zhang T, Dutton-Regester K, Brown KM, Hayward NK. The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res (2016) 29:266–83. doi:10.1111/pcmr.12459
17. Wellbrock C, Arozarena I. The complexity of the ERK/MAP-kinase pathway and the treatment of melanoma skin cancer. Front Cell Dev Biol (2016) 4:33. doi:10.3389/fcell.2016.00033
18. Flaherty KT, McArthur G. BRAF, a target in melanoma: implications for solid tumor drug development. Cancer (2010) 116:4902–13. doi:10.1002/cncr.25261
19. Heinzerling L, Kuhnapfel S, Meckbach D, Baiter M, Kaempgen E, Keikavoussi P, et al. Rare BRAF mutations in melanoma patients: implications for molecular testing in clinical practice. Br J Cancer (2013) 108:2164–71. doi:10.1038/bjc.2013.143
20. Uzdensky AB, Demyanenko SV, Bibov MY. Signal transduction in human cutaneous melanoma and target drugs. Curr Cancer Drug Targets (2013) 13:843–66. doi:10.2174/1568009611313080004
21. Albino AP, Nanus DM, Mentle IR, Cordon-Cardo C, McNutt NS, Bressler J, et al. Analysis of ras oncogenes in malignant melanoma and precursor lesions: correlation of point mutations with differentiation phenotype. Oncogene (1989) 4:1363–74.
22. Funck-Brentano E, Helias-Rodzewicz Z, Longvert C, Mokhtari K, Saiag P, Emile JF. Increase in NRAS mutant allele percentage during metastatic melanoma progression. Exp Dermatol (2016) 25:472–4. doi:10.1111/exd.13001
23. Espinosa E, Berrocal A, Lopez Martin JA, Gonzalez Cao M, Cerezuela P, Mayordomo JI, et al. Advances in cutaneous melanoma. Clin Transl Oncol (2012) 14:325–32. doi:10.1007/s12094-012-0804-4
24. Timar J, Vizkeleti L, Doma V, Barbai T, Raso E. Genetic progression of malignant melanoma. Cancer Metastasis Rev (2016) 35:93–107. doi:10.1007/s10555-016-9613-5
25. Berwick M, Orlow I, Hummer AJ, Armstrong BK, Kricker A, Marrett LD, et al. The prevalence of CDKN2A germ-line mutations and relative risk for cutaneous malignant melanoma: an international population-based study. Cancer Epidemiol Biomarkers Prev (2006) 15:1520–5. doi:10.1158/1055-9965.EPI-06-0270
26. Ecsedi SI, Hernandez-Vargas H, Lima SC, Herceg Z, Adany R, Balazs M. Transposable hypomethylation is associated with metastatic capacity of primary melanomas. Int J Clin Exp Pathol (2013) 6:2943–8.
27. Besaratinia A, Tommasi S. Epigenetics of human melanoma: promises and challenges. J Mol Cell Biol (2014) 6:356–67. doi:10.1093/jmcb/mju027
28. Ecsedi S, Hernandez-Vargas H, Lima SC, Vizkeleti L, Toth R, Lazar V, et al. DNA methylation characteristics of primary melanomas with distinct biological behaviour. PLoS One (2014) 9:e96612. doi:10.1371/journal.pone.0096612
29. Ascierto PA, Chiarion-Sileni V, Muggiano A, Mandala M, Pimpinelli N, Del Vecchio M, et al. Interferon alpha for the adjuvant treatment of melanoma: review of international literature and practical recommendations from an expert panel on the use of interferon. J Chemother (2014) 26:193–201. doi:10.1179/1973947813Y.0000000154
30. Middleton MR, Grob JJ, Aaronson N, Fierlbeck G, Tilgen W, Seiter S, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol (2000) 18:158–66.
31. Danson SJ, Middleton MR. Temozolomide: a novel oral alkylating agent. Expert Rev Anticancer Ther (2001) 1:13–9. doi:10.1586/14737140.1.1.13
32. Teimouri F, Nikfar S, Abdollahi M. Efficacy and side effects of dacarbazine in comparison with temozolomide in the treatment of malignant melanoma: a meta-analysis consisting of 1314 patients. Melanoma Res (2013) 23:381–9. doi:10.1097/CMR.0b013e3283649a97
33. Ma C, Armstrong AW. Severe adverse events from the treatment of advanced melanoma: a systematic review of severe side effects associated with ipilimumab, vemurafenib, interferon alfa-2b, dacarbazine and interleukin-2. J Dermatolog Treat (2014) 25:401–8. doi:10.3109/09546634.2013.813897
34. Wu S, Singh RK. Resistance to chemotherapy and molecularly targeted therapies: rationale for combination therapy in malignant melanoma. Curr Mol Med (2011) 11:553–63. doi:10.2174/156652411800615153
35. Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol (1999) 17:2105–16.
36. Dunki-Jacobs EM, Callender GG, McMasters KM. Current management of melanoma. Curr Probl Surg (2013) 50:351–82. doi:10.1067/j.cpsurg.2013.04.001
37. Rotte A, Bhandaru M, Zhou Y, McElwee KJ. Immunotherapy of melanoma: present options and future promises. Cancer Metastasis Rev (2015) 34:115–28. doi:10.1007/s10555-014-9542-0
38. Romano E, Schwartz GK, Chapman PB, Wolchock JD, Carvajal RD. Treatment implications of the emerging molecular classification system for melanoma. Lancet Oncol (2011) 12:913–22. doi:10.1016/S1470-2045(10)70274-6
39. Russo A, Ficili B, Candido S, Pezzino FM, Guarneri C, Biondi A, et al. Emerging targeted therapies for melanoma treatment (review). Int J Oncol (2014) 45:516–24. doi:10.3892/ijo.2014.2481
40. DePeralta DK, Boland GM. Melanoma: advances in targeted therapy and molecular markers. Ann Surg Oncol (2015) 22:3451–8. doi:10.1245/s10434-015-4702-1
41. Tang T, Eldabaje R, Yang L. Current status of biological therapies for the treatment of metastatic melanoma. Anticancer Res (2016) 36:3229–41.
42. Wong DJ, Ribas A. Targeted therapy for melanoma. Cancer Treat Res (2016) 167:251–62. doi:10.1007/978-3-319-22539-5_10
43. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med (2011) 364:2507–16. doi:10.1056/NEJMoa1103782
44. Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med (2012) 366:707–14. doi:10.1056/NEJMoa1112302
45. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet (2012) 380:358–65. doi:10.1016/S0140-6736(12)60868-X
46. Banzi M, De Blasio S, Lallas A, Longo C, Moscarella E, Alfano R, et al. Dabrafenib: a new opportunity for the treatment of BRAF V600-positive melanoma. Onco Targets Ther (2016) 9:2725–33. doi:10.2147/OTT.S75104
47. Sullivan RJ, Flaherty KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer (2013) 49:1297–304. doi:10.1016/j.ejca.2012.11.019
48. Lo RS, Shi H. Detecting mechanisms of acquired BRAF inhibitor resistance in melanoma. Methods Mol Biol (2014) 1102:163–74. doi:10.1007/978-1-62703-727-3_10
49. Welsh SJ, Rizos H, Scolyer RA, Long GV. Resistance to combination BRAF and MEK inhibition in metastatic melanoma: where to next? Eur J Cancer (2016) 62:76–85. doi:10.1016/j.ejca.2016.04.005
50. Vu HL, Aplin AE. Targeting mutant NRAS signaling pathways in melanoma. Pharmacol Res (2016) 107:111–6. doi:10.1016/j.phrs.2016.03.007
51. Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med (1996) 183:2541–50. doi:10.1084/jem.183.6.2541
52. Nakamura K, Okuyama R. Immunotherapy for advanced melanoma: current knowledge and future directions. J Dermatol Sci (2016) 83:87–94. doi:10.1016/j.jdermsci.2016.05.009
53. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364:2517–26. doi:10.1056/NEJMoa1104621
54. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi:10.1038/nrc3239
55. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515:568–71. doi:10.1038/nature13954
56. Atkins MB, Larkin J. Immunotherapy combined or sequenced with targeted therapy in the treatment of solid tumors: current perspectives. J Natl Cancer Inst (2016) 108:djv414. doi:10.1093/jnci/djv414
57. Davey RJ, van der Westhuizen A, Bowden NA. Metastatic melanoma treatment: combining old and new therapies. Crit Rev Oncol Hematol (2016) 98:242–53. doi:10.1016/j.critrevonc.2015.11.011
58. Singh BP, Salama AK. Updates in therapy for advanced melanoma. Cancers (Basel) (2016) 8:17. doi:10.3390/cancers8010017
59. Janik ME, Belkot K, Przybylo M. Is oestrogen an important player in melanoma progression? Contemp Oncol (Pozn) (2014) 18:302–6. doi:10.5114/wo.2014.43938
60. Mitkov M, Joseph R, Copland J III. Steroid hormone influence on melanomagenesis. Mol Cell Endocrinol (2015) 417:94–102. doi:10.1016/j.mce.2015.09.020
61. Mitchell DL, Fernandez AA, Garcia R, Paniker L, Lin K, Hanninen A, et al. Acute exposure to ultraviolet-B radiation modulates sex steroid hormones and receptor expression in the skin and may contribute to the sex bias of melanoma in a fish model. Pigment Cell Melanoma Res (2014) 27:408–17. doi:10.1111/pcmr.12213
62. Joosse A, de Vries E, Eckel R, Nijsten T, Eggermont AM, Holzel D, et al. Gender differences in melanoma survival: female patients have a decreased risk of metastasis. J Invest Dermatol (2011) 131:719–26. doi:10.1038/jid.2010.354
63. Joosse A, Collette S, Suciu S, Nijsten T, Lejeune F, Kleeberg UR, et al. Superior outcome of women with stage I/II cutaneous melanoma: pooled analysis of four European Organisation for Research and Treatment of Cancer phase III trials. J Clin Oncol (2012) 30:2240–7. doi:10.1200/JCO.2011.38.0584
64. Gamba CS, Clarke CA, Keegan TH, Tao L, Swetter SM. Melanoma survival disadvantage in young, non-Hispanic white males compared with females. JAMA Dermatol (2013) 149:912–20. doi:10.1001/jamadermatol.2013.4408
65. Boswell W, Boswell M, Titus J, Savage M, Lu Y, Shen J, et al. Sex-specific molecular genetic response to UVB exposure in Xiphophorus maculatus skin. Comp Biochem Physiol C Toxicol Pharmacol (2015) 178:76–85. doi:10.1016/j.cbpc.2015.07.007
66. Gandini S, Iodice S, Koomen E, Di Pietro A, Sera F, Caini S. Hormonal and reproductive factors in relation to melanoma in women: current review and meta-analysis. Eur J Cancer (2011) 47:2607–17. doi:10.1016/j.ejca.2011.04.023
67. Travers RL, Sober AJ, Berwick M, Mihm MC Jr, Barnhill RL, Duncan LM. Increased thickness of pregnancy-associated melanoma. Br J Dermatol (1995) 132:876–83. doi:10.1111/j.1365-2133.1995.tb16942.x
68. Jian D, Jiang D, Su J, Chen W, Hu X, Kuang Y, et al. Diethylstilbestrol enhances melanogenesis via cAMP-PKA-mediating up-regulation of tyrosinase and MITF in mouse B16 melanoma cells. Steroids (2011) 76:1297–304. doi:10.1016/j.steroids.2011.06.008
69. Daryanani D, Plukker JT, De Hullu JA, Kuiper H, Nap RE, Hoekstra HJ. Pregnancy and early-stage melanoma. Cancer (2003) 97:2248–53. doi:10.1002/cncr.11321
70. O’Meara AT, Cress R, Xing G, Danielsen B, Smith LH. Malignant melanoma in pregnancy. A population-based evaluation. Cancer (2005) 103:1217–26. doi:10.1002/cncr.20925
71. Durvasula R, Ahmed SM, Vashisht A, Studd JW. Hormone replacement therapy and malignant melanoma: to prescribe or not to prescribe? Climacteric (2002) 5:197–200. doi:10.1080/713605225
72. Tang JY, Spaunhurst KM, Chlebowski RT, Wactawski-Wende J, Keiser E, Thomas F, et al. Menopausal hormone therapy and risks of melanoma and nonmelanoma skin cancers: women’s health initiative randomized trials. J Natl Cancer Inst (2011) 103:1469–75. doi:10.1093/jnci/djr333
73. Auriemma M, Di Nicola M, Varrati S, Carbone A, Pamio A, Capo A, et al. Mole modifications following controlled ovarian stimulation for assisted reproduction technologies. J Eur Acad Dermatol Venereol (2015) 29:1913–7. doi:10.1111/jdv.13065
74. Santen RJ, Santner SJ, Harvey HA, Lipton A, Simmonds M, Feil PD, et al. Marked heterogeneity of aromatase activity in human malignant melanoma tissue. Eur J Cancer Clin Oncol (1988) 24:1811–6. doi:10.1016/0277-5379(88)90090-9
75. Block S, Bonneterre J, Adenis A, Pion JM, Demaille A. Aminoglutethimide in malignant melanoma. A phase II study. Am J Clin Oncol (1992) 15:260–1. doi:10.1097/00000421-199206000-00016
76. de Giorgi V, Gori A, Alfaioli B, Papi F, Grazzini M, Rossari S, et al. Influence of sex hormones on melanoma. J Clin Oncol (2011) 29:e94–5. doi:10.1200/JCO.2010.33.1876
77. Jia M, Dahlman-Wright K, Gustafsson JA. Estrogen receptor alpha and beta in health and disease. Best Pract Res Clin Endocrinol Metab (2015) 29:557–68. doi:10.1016/j.beem.2015.04.008
78. Walter P, Green S, Greene G, Krust A, Bornert JM, Jeltsch JM, et al. Cloning of the human estrogen receptor cDNA. Proc Natl Acad Sci U S A (1985) 82:7889–93. doi:10.1073/pnas.82.23.7889
79. Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A (1996) 93:5925–30. doi:10.1073/pnas.93.12.5925
80. Enmark E, Pelto-Huikko M, Grandien K, Lagercrantz S, Lagercrantz J, Fried G, et al. Human estrogen receptor beta-gene structure, chromosomal localization, and expression pattern. J Clin Endocrinol Metab (1997) 82:4258–65. doi:10.1210/jc.82.12.4258
81. Hall JM, McDonnell DP. The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology (1999) 140:5566–78. doi:10.1210/endo.140.12.7179
82. Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology (2006) 147:4831–42. doi:10.1210/en.2006-0563
83. Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev (2007) 87:905–31. doi:10.1152/physrev.00026.2006
84. Williams C, Edvardsson K, Lewandowski SA, Strom A, Gustafsson JA. A genome-wide study of the repressive effects of estrogen receptor beta on estrogen receptor alpha signaling in breast cancer cells. Oncogene (2008) 27:1019–32. doi:10.1038/sj.onc.1210712
85. Pettersson K, Gustafsson JA. Role of estrogen receptor beta in estrogen action. Annu Rev Physiol (2001) 63:165–92. doi:10.1146/annurev.physiol.63.1.165
86. Nilsson S, Gustafsson JA. Estrogen receptors: therapies targeted to receptor subtypes. Clin Pharmacol Ther (2011) 89:44–55. doi:10.1038/clpt.2010.226
87. Paterni I, Granchi C, Katzenellenbogen JA, Minutolo F. Estrogen receptors alpha (ERalpha) and beta (ERbeta): subtype-selective ligands and clinical potential. Steroids (2014) 90:13–29. doi:10.1016/j.steroids.2014.06.012
88. Belcher SM, Zsarnovszky A. Estrogenic actions in the brain: estrogen, phytoestrogens, and rapid intracellular signaling mechanisms. J Pharmacol Exp Ther (2001) 299:408–14.
89. Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor-beta potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem (2001) 44:4230–51. doi:10.1021/jm010254a
90. Nilsson S, Koehler KF, Gustafsson JA. Development of subtype-selective oestrogen receptor-based therapeutics. Nat Rev Drug Discov (2011) 10:778–92. doi:10.1038/nrd3551
91. Leitman DC, Paruthiyil S, Vivar OI, Saunier EF, Herber CB, Cohen I, et al. Regulation of specific target genes and biological responses by estrogen receptor subtype agonists. Curr Opin Pharmacol (2010) 10:629–36. doi:10.1016/j.coph.2010.09.009
92. Minutolo F, Macchia M, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor beta ligands: recent advances and biomedical applications. Med Res Rev (2011) 31:364–442. doi:10.1002/med.20186
93. Matthews J, Celius T, Halgren R, Zacharewski T. Differential estrogen receptor binding of estrogenic substances: a species comparison. J Steroid Biochem Mol Biol (2000) 74:223–34. doi:10.1016/S0960-0760(00)00126-6
94. Dey P, Barros RP, Warner M, Strom A, Gustafsson JA. Insight into the mechanisms of action of estrogen receptor beta in the breast, prostate, colon, and CNS. J Mol Endocrinol (2013) 51:T61–74. doi:10.1530/JME-13-0150
95. Moore JT, McKee DD, Slentz-Kesler K, Moore LB, Jones SA, Horne EL, et al. Cloning and characterization of human estrogen receptor beta isoforms. Biochem Biophys Res Commun (1998) 247:75–8. doi:10.1006/bbrc.1998.8738
96. Ogawa S, Inoue S, Watanabe T, Orimo A, Hosoi T, Ouchi Y, et al. Molecular cloning and characterization of human estrogen receptor betacx: a potential inhibitor ofestrogen action in human. Nucleic Acids Res (1998) 26:3505–12. doi:10.1093/nar/26.15.3505
97. Leung YK, Mak P, Hassan S, Ho SM. Estrogen receptor (ER)-beta isoforms: a key to understanding ER-beta signaling. Proc Natl Acad Sci U S A (2006) 103:13162–7. doi:10.1073/pnas.0605676103
98. Leung YK, Lam HM, Wu S, Song D, Levin L, Cheng L, et al. Estrogen receptor beta2 and beta5 are associated with poor prognosis in prostate cancer, and promote cancer cell migration and invasion. Endocr Relat Cancer (2010) 17:675–89. doi:10.1677/ERC-09-0294
99. Girault I, Andrieu C, Tozlu S, Spyratos F, Bieche I, Lidereau R. Altered expression pattern of alternatively spliced estrogen receptor beta transcripts in breast carcinoma. Cancer Lett (2004) 215:101–12. doi:10.1016/j.canlet.2004.05.006
100. Bottner M, Thelen P, Jarry H. Estrogen receptor beta: tissue distribution and the still largely enigmatic physiological function. J Steroid Biochem Mol Biol (2014) 139:245–51. doi:10.1016/j.jsbmb.2013.03.003
101. Thomas C, Gustafsson JA. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer (2011) 11:597–608. doi:10.1038/nrc3093
102. Schmidt AN, Nanney LB, Boyd AS, King LE Jr, Ellis DL. Oestrogen receptor-beta expression in melanocytic lesions. Exp Dermatol (2006) 15:971–80. doi:10.1111/j.1600-0625.2006.00502.x
103. Ohata C, Tadokoro T, Itami S. Expression of estrogen receptor beta in normal skin, melanocytic nevi and malignant melanomas. J Dermatol (2008) 35:215–21. doi:10.1111/j.1346-8138.2008.00447.x
104. de Giorgi V, Mavilia C, Massi D, Gozzini A, Aragona P, Tanini A, et al. Estrogen receptor expression in cutaneous melanoma: a real-time reverse transcriptase-polymerase chain reaction and immunohistochemical study. Arch Dermatol (2009) 145:30–6. doi:10.1001/archdermatol.2008.537
105. de Giorgi V, Gori A, Grazzini M, Rossari S, Scarfi F, Corciova S, et al. Estrogens, estrogen receptors and melanoma. Expert Rev Anticancer Ther (2011) 11:739–47. doi:10.1586/era.11.42
106. de Giorgi V, Gori A, Gandini S, Papi F, Grazzini M, Rossari S, et al. Oestrogen receptor beta and melanoma: a comparative study. Br J Dermatol (2013) 168:513–9. doi:10.1111/bjd.12056
107. Zhou JH, Kim KB, Myers JN, Fox PS, Ning J, Bassett RL, et al. Immunohistochemical expression of hormone receptors in melanoma of pregnant women, nonpregnant women, and men. Am J Dermatopathol (2014) 36:74–9. doi:10.1097/DAD.0b013e3182914c64
108. Spyropoulos C, Melachrinou M, Vasilakos P, Tzorakoleftherakis E. Expression of estrogen receptors in melanoma and sentinel lymph nodes; a “female” clinical entity or a possible treatment modality? Eur J Gynaecol Oncol (2015) 36:123–30.
109. de Giorgi V, Sestini S, Gori A, Mazzotta C, Grazzini M, Rossari S, et al. Polymorphisms of estrogen receptors: risk factors for invasive melanoma – a prospective study. Oncology (2011) 80:232–7. doi:10.1159/000328321
110. Marzagalli M, Casati L, Moretti RM, Montagnani Marelli M, Limonta P. Estrogen receptor beta agonists differentially affect the growth of human melanoma cell lines. PLoS One (2015) 10:e0134396. doi:10.1371/journal.pone.0134396
111. Kawai H, Ishii A, Washiya K, Konno T, Kon H, Yamaya C, et al. Estrogen receptor alpha and beta are prognostic factors in non-small cell lung cancer. Clin Cancer Res (2005) 11:5084–9. doi:10.1158/1078-0432.CCR-05-0200
112. Haring J, Skrzypczak M, Stegerer A, Lattrich C, Weber F, Gorse R, et al. Estrogen receptor beta transcript variants associate with oncogene expression in endometrial cancer. Int J Mol Med (2012) 29:1127–36. doi:10.3892/ijmm.2012.929
113. Barzi A, Lenz AM, Labonte MJ, Lenz HJ. Molecular pathways: estrogen pathway in colorectal cancer. Clin Cancer Res (2013) 19:5842–8. doi:10.1158/1078-0432.CCR-13-0325
114. Yakimchuk K, Jondal M, Okret S. Estrogen receptor alpha and beta in the normal immune system and in lymphoid malignancies. Mol Cell Endocrinol (2013) 375:121–9. doi:10.1016/j.mce.2013.05.016
115. Christoforou P, Christopoulos PF, Koutsilieris M. The role of estrogen receptor beta in prostate cancer. Mol Med (2014) 20:427–34. doi:10.2119/molmed.2014.00105
116. Haldosen LA, Zhao C, Dahlman-Wright K. Estrogen receptor beta in breast cancer. Mol Cell Endocrinol (2014) 382:665–72. doi:10.1016/j.mce.2013.08.005
117. Omoto Y, Iwase H. Clinical significance of estrogen receptor beta in breast and prostate cancer from biological aspects. Cancer Sci (2015) 106:337–43. doi:10.1111/cas.12613
118. Kyriakidis I, Papaioannidou P. Estrogen receptor beta and ovarian cancer: a key to pathogenesis and response to therapy. Arch Gynecol Obstet (2016) 293:1161–8. doi:10.1007/s00404-016-4027-8
119. Williams C, DiLeo A, Niv Y, Gustafsson JA. Estrogen receptor beta as target for colorectal cancer prevention. Cancer Lett (2016) 372:48–56. doi:10.1016/j.canlet.2015.12.009
120. Renoir JM, Marsaud V, Lazennec G. Estrogen receptor signaling as a target for novel breast cancer therapeutics. Biochem Pharmacol (2013) 85:449–65. doi:10.1016/j.bcp.2012.10.018
121. Sarti MS, Visconti MA, Castrucci AM. Biological activity and binding of estradiol to SK-Mel 23 human melanoma cells. Braz J Med Biol Res (2004) 37:901–5. doi:10.1590/S0100-879X2004000600016
122. Richardson B, Price A, Wagner M, Williams V, Lorigan P, Browne S, et al. Investigation of female survival benefit in metastatic melanoma. Br J Cancer (1999) 80:2025–33. doi:10.1038/sj.bjc.6690637
123. Dobos J, Timar J, Bocsi J, Burian Z, Nagy K, Barna G, et al. In vitro and in vivo antitumor effect of 2-methoxyestradiol on human melanoma. Int J Cancer (2004) 112:771–6. doi:10.1002/ijc.20473
124. Ireson CR, Chander SK, Purohit A, Perera S, Newman SP, Parish D, et al. Pharmacokinetics and efficacy of 2-methoxyoestradiol and 2-methoxyoestradiol-bis-sulphamate in vivo in rodents. Br J Cancer (2004) 90:932–7. doi:10.1038/sj.bjc.6601591
125. Kanter-Lewensohn L, Girnita L, Girnita A, Dricu A, Olsson G, Leech L, et al. Tamoxifen-induced cell death in malignant melanoma cells: possible involvement of the insulin-like growth factor-1 (IGF-1) pathway. Mol Cell Endocrinol (2000) 165:131–7. doi:10.1016/S0303-7207(00)00253-7
126. Matsuoka H, Tsubaki M, Yamazoe Y, Ogaki M, Satou T, Itoh T, et al. Tamoxifen inhibits tumor cell invasion and metastasis in mouse melanoma through suppression of PKC/MEK/ERK and PKC/PI3K/Akt pathways. Exp Cell Res (2009) 315:2022–32. doi:10.1016/j.yexcr.2009.04.009
127. Falkson CI, Ibrahim J, Kirkwood JM, Coates AS, Atkins MB, Blum RH. Phase III trial of dacarbazine versus dacarbazine with interferon alpha-2b versus dacarbazine with tamoxifen versus dacarbazine with interferon alpha-2b and tamoxifen in patients with metastatic malignant melanoma: an Eastern Cooperative Oncology Group study. J Clin Oncol (1998) 16:1743–51.
128. Agarwala SS, Ferri W, Gooding W, Kirkwood JM. A phase III randomized trial of dacarbazine and carboplatin with and without tamoxifen in the treatment of patients with metastatic melanoma. Cancer (1999) 85:1979–84. doi:10.1002/(SICI)1097-0142(19990501)85:9<1979::AID-CNCR15>3.0.CO;2-G
129. Chapman PB, Einhorn LH, Meyers ML, Saxman S, Destro AN, Panageas KS, et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol (1999) 17:2745–51.
130. Lens MB, Reiman T, Husain AF. Use of tamoxifen in the treatment of malignant melanoma. Cancer (2003) 98:1355–61. doi:10.1002/cncr.11644
131. Beguerie JR, Xingzhong J, Valdez RP. Tamoxifen vs. non-tamoxifen treatment for advanced melanoma: a meta-analysis. Int J Dermatol (2010) 49:1194–202. doi:10.1111/j.1365-4632.2010.04529.x
132. Feun L, Marini A, Moffat F, Savaraj N, Hurley J, Mazumder A. Cyclosporine A, alpha-lnterferon and interleukin-2 following chemotherapy with BCNU, DTIC, cisplatin, and tamoxifen: a phase II study in advanced melanoma. Cancer Invest (2005) 23:3–8. doi:10.1081/CNV-46368
133. Shaaban AM, Jarvis C, Moore F, West C, Dodson A, Foster CS. Prognostic significance of estrogen receptor Beta in epithelial hyperplasia of usual type with known outcome. Am J Surg Pathol (2005) 29:1593–9. doi:10.1097/01.pas.0000184807.38037.75
134. Hartman J, Edvardsson K, Lindberg K, Zhao C, Williams C, Strom A, et al. Tumor repressive functions of estrogen receptor beta in SW480 colon cancer cells. Cancer Res (2009) 69:6100–6. doi:10.1158/0008-5472.CAN-09-0506
135. Pinton G, Thomas W, Bellini P, Manente AG, Favoni RE, Harvey BJ, et al. Estrogen receptor beta exerts tumor repressive functions in human malignant pleural mesothelioma via EGFR inactivation and affects response to gefitinib. PLoS One (2010) 5:e14110. doi:10.1371/journal.pone.0014110
136. Dondi D, Piccolella M, Biserni A, Della Torre S, Ramachandran B, Locatelli A, et al. Estrogen receptor beta and the progression of prostate cancer: role of 5alpha-androstane-3beta,17beta-diol. Endocr Relat Cancer (2010) 17:731–42. doi:10.1677/ERC-10-0032
137. Marzioni M, Torrice A, Saccomanno S, Rychlicki C, Agostinelli L, Pierantonelli I, et al. An oestrogen receptor beta-selective agonist exerts anti-neoplastic effects in experimental intrahepatic cholangiocarcinoma. Dig Liver Dis (2012) 44:134–42. doi:10.1016/j.dld.2011.06.014
138. Sareddy GR, Nair BC, Gonugunta VK, Zhang QG, Brenner A, Brann DW, et al. Therapeutic significance of estrogen receptor beta agonists in gliomas. Mol Cancer Ther (2012) 11:1174–82. doi:10.1158/1535-7163.MCT-11-0960
139. Ruddy SC, Lau R, Cabrita MA, McGregor C, McKay BC, Murphy LC, et al. Preferential estrogen receptor beta ligands reduce Bcl-2 expression in hormone-resistant breast cancer cells to increase autophagy. Mol Cancer Ther (2014) 13:1882–93. doi:10.1158/1535-7163.MCT-13-1066
140. Yakimchuk K, Hasni MS, Guan J, Chao MP, Sander B, Okret S. Inhibition of lymphoma vascularization and dissemination by estrogen receptor beta agonists. Blood (2014) 123:2054–61. doi:10.1182/blood-2013-07-517292
141. Chen J, Hou R, Zhang X, Ye Y, Wang Y, Tian J. Calycosin suppresses breast cancer cell growth via ERbeta-dependent regulation of IGF-1R, p38 MAPK and PI3K/Akt pathways. PLoS One (2014) 9:e91245. doi:10.1371/journal.pone.0091245
142. Liu X, Wang L, Chen J, Ling Q, Wang H, Li S, et al. Estrogen receptor beta agonist enhances temozolomide sensitivity of glioma cells by inhibiting PI3K/AKT/mTOR pathway. Mol Med Rep (2015) 11:1516–22. doi:10.3892/mmr.2014.2811
143. Wang J, Zhang C, Chen K, Tang H, Tang J, Song C, et al. ERbeta1 inversely correlates with PTEN/PI3K/AKT pathway and predicts a favorable prognosis in triple-negative breast cancer. Breast Cancer Res Treat (2015) 152:255–69. doi:10.1007/s10549-015-3467-3
144. Paruthiyil S, Cvoro A, Zhao X, Wu Z, Sui Y, Staub RE, et al. Drug and cell type-specific regulation of genes with different classes of estrogen receptor beta-selective agonists. PLoS One (2009) 4:e6271. doi:10.1371/journal.pone.0006271
145. Knight DC, Eden JA. A review of the clinical effects of phytoestrogens. Obstet Gynecol (1996) 87:897–904.
146. Hartman J, Strom A, Gustafsson JA. Estrogen receptor beta in breast cancer – diagnostic and therapeutic implications. Steroids (2009) 74:635–41. doi:10.1016/j.steroids.2009.02.005
147. Patisaul HB, Jefferson W. The pros and cons of phytoestrogens. Front Neuroendocrinol (2010) 31:400–19. doi:10.1016/j.yfrne.2010.03.003
148. Hartman J, Strom A, Gustafsson JA. Current concepts and significance of estrogen receptor beta in prostate cancer. Steroids (2012) 77:1262–6. doi:10.1016/j.steroids.2012.07.002
149. Riggs BL, Hartmann LC. Selective estrogen-receptor modulators – mechanisms of action and application to clinical practice. N Engl J Med (2003) 348:618–29. doi:10.1056/NEJMra022219
150. Gong P, Madak-Erdogan Z, Li J, Cheng J, Greenlief CM, Helferich W, et al. Transcriptomic analysis identifies gene networks regulated by estrogen receptor alpha (ERalpha) and ERbeta that control distinct effects of different botanical estrogens. Nucl Recept Signal (2014) 12:e001. doi:10.1621/nrs.12001
151. Pilsakova L, Riecansky I, Jagla F. The physiological actions of isoflavone phytoestrogens. Physiol Res (2010) 59:651–64.
152. Vrtacnik P, Ostanek B, Mencej-Bedrac S, Marc J. The many faces of estrogen signaling. Biochem Med (Zagreb) (2014) 24:329–42. doi:10.11613/BM.2014.035
153. Testai L. Flavonoids and mitochondrial pharmacology: a new paradigm for cardioprotection. Life Sci (2015) 135:68–76. doi:10.1016/j.lfs.2015.04.017
154. Romagnolo DF, Selmin OI. Flavonoids and cancer prevention: a review of the evidence. J Nutr Gerontol Geriatr (2012) 31:206–38. doi:10.1080/21551197.2012.702534
155. Liu-Smith F, Meyskens FL. Molecular mechanisms of flavonoids in melanin synthesis and the potential for the prevention and treatment of melanoma. Mol Nutr Food Res (2016) 60:1264–74. doi:10.1002/mnfr.201500822
156. Ganai AA, Farooqi H. Bioactivity of genistein: a review of in vitro and in vivo studies. Biomed Pharmacother (2015) 76:30–8. doi:10.1016/j.biopha.2015.10.026
157. Russo M, Russo GL, Daglia M, Kasi PD, Ravi S, Nabavi SF, et al. Understanding genistein in cancer: the “good” and the “bad” effects: a review. Food Chem (2016) 196:589–600. doi:10.1016/j.foodchem.2015.09.085
158. Russo A, Cardile V, Lombardo L, Vanella L, Acquaviva R. Genistin inhibits UV light-induced plasmid DNA damage and cell growth in human melanoma cells. J Nutr Biochem (2006) 17:103–8. doi:10.1016/j.jnutbio.2005.05.011
159. Sareddy GR, Vadlamudi RK. Cancer therapy using natural ligands that target estrogen receptor beta. Chin J Nat Med (2015) 13:801–7. doi:10.1016/S1875-5364(15)30083-2
160. Kiguchi K, Constantinou AI, Huberman E. Genistein-induced cell-differentiation and protein-linked DNA strand breakage in human-melanoma cells. Cancer Commun (1990) 2:271–8.
161. Rauth S, Kichina J, Green A. Inhibition of growth and induction of differentiation of metastatic melanoma cells in vitro by genistein: chemosensitivity is regulated by cellular p53. Br J Cancer (1997) 75:1559–66. doi:10.1038/bjc.1997.268
162. Miyazaki K. Novel approach for evaluation of estrogenic and anti-estrogenic activities of genistein and daidzein using B16 melanoma cells and dendricity assay. Pigment Cell Res (2004) 17:407–12. doi:10.1111/j.1600-0749.2004.00167.x
163. Otieno DO, Shah NP. Endogenous beta-glucosidase and beta-galactosidase activities from selected probiotic micro-organisms and their role in isoflavone biotransformation in soymilk. J Appl Microbiol (2007) 103:910–7. doi:10.1111/j.1365-2672.2007.03438.x
164. Farina HG, Pomies M, Alonso DF, Gomez DE. Antitumor and antiangiogenic activity of soy isoflavone genistein in mouse models of melanoma and breast cancer. Oncol Rep (2006) 16:885–91.
165. Danciu C, Borcan F, Bojin F, Zupko I, Dehelean C. Effect of the isoflavone genistein on tumor size, metastasis potential and melanization in a B16 mouse model of murine melanoma. Nat Prod Commun (2013) 8:343–6.
166. Mersereau JE, Levy N, Staub RE, Baggett S, Zogovic T, Chow S, et al. Liquiritigenin is a plant-derived highly selective estrogen receptor beta agonist. Mol Cell Endocrinol (2008) 283:49–57. doi:10.1016/j.mce.2007.11.020
167. Shi H, Wu Y, Wang Y, Zhou M, Yan S, Chen Z, et al. Liquiritigenin potentiates the inhibitory effects of cisplatin on invasion and metastasis via downregulation MMP-2/9 and PI3 K/AKT signaling pathway in B16F10 melanoma cells and mice model. Nutr Cancer (2015) 67:761–70. doi:10.1080/01635581.2015.1037962
168. Jiang Y, Gong P, Madak-Erdogan Z, Martin T, Jeyakumar M, Carlson K, et al. Mechanisms enforcing the estrogen receptor beta selectivity of botanical estrogens. FASEB J (2013) 27:4406–18. doi:10.1096/fj.13-234617
169. Boonmuen N, Gong P, Ali Z, Chittiboyina AG, Khan I, Doerge DR, et al. Licorice root components in dietary supplements are selective estrogen receptor modulators with a spectrum of estrogenic and anti-estrogenic activities. Steroids (2016) 105:42–9. doi:10.1016/j.steroids.2015.11.006
170. Liu Y, Xie S, Wang Y, Luo K, Wang Y, Cai Y. Liquiritigenin inhibits tumor growth and vascularization in a mouse model of HeLa cells. Molecules (2012) 17:7206–16. doi:10.3390/molecules17067206
171. Cvoro A, Paruthiyil S, Jones JO, Tzagarakis-Foster C, Clegg NJ, Tatomer D, et al. Selective activation of estrogen receptor-beta transcriptional pathways by an herbal extract. Endocrinology (2007) 148:538–47. doi:10.1210/en.2006-0803
172. Hajirahimkhan A, Simmler C, Yuan Y, Anderson JR, Chen SN, Nikolic D, et al. Evaluation of estrogenic activity of licorice species in comparison with hops used in botanicals for menopausal symptoms. PLoS One (2013) 8:e67947. doi:10.1371/journal.pone.0067947
173. Kao TC, Wu CH, Yen GC. Bioactivity and potential health benefits of licorice. J Agric Food Chem (2014) 62:542–53. doi:10.1021/jf404939f
174. Somjen D, Knoll E, Vaya J, Stern N, Tamir S. Estrogen-like activity of licorice root constituents: glabridin and glabrene, in vascular tissues in vitro and in vivo. J Steroid Biochem Mol Biol (2004) 91:147–55. doi:10.1016/j.jsbmb.2004.04.008
175. Simmler C, Pauli GF, Chen SN. Phytochemistry and biological properties of glabridin. Fitoterapia (2013) 90:160–84. doi:10.1016/j.fitote.2013.07.003
176. Song NR, Lee E, Byun S, Kim JE, Mottamal M, Park JH, et al. Isoangustone A, a novel licorice compound, inhibits cell proliferation by targeting PI3K, MKK4, and MKK7 in human melanoma. Cancer Prev Res (Phila) (2013) 6:1293–303. doi:10.1158/1940-6207.CAPR-13-0134
177. Rossi T, Benassi L, Magnoni C, Ruberto AI, Coppi A, Baggio G. Effects of glycyrrhizin on UVB-irradiated melanoma cells. In Vivo (2005) 19:319–22.
178. Park SY, Kim EJ, Choi HJ, Seon MR, Lim SS, Kang YH, et al. Anti-carcinogenic effects of non-polar components containing licochalcone A in roasted licorice root. Nutr Res Pract (2014) 8:257–66. doi:10.4162/nrp.2014.8.3.257
179. Afaq F, Adhami VM, Ahmad N, Mukhtar H. Botanical antioxidants for chemoprevention of photocarcinogenesis. Front Biosci (2002) 7:d784–92. doi:10.2741/afaq
180. Mak P, Leung YK, Tang WY, Harwood C, Ho SM. Apigenin suppresses cancer cell growth through ERbeta. Neoplasia (2006) 8:896–904. doi:10.1593/neo.06538
181. Uzarska M, Czajkowski R, Schwartz RA, Bajek A, Zegarska B, Drewa T. Chemoprevention of skin melanoma: facts and myths. Melanoma Res (2013) 23:426–33. doi:10.1097/CMR.0000000000000016
182. Pal HC, Hunt KM, Diamond A, Elmets CA, Afaq F. Phytochemicals for the management of melanoma. Mini Rev Med Chem (2016) 16:953–79. doi:10.2174/1389557516666160211120157
183. Das S, Das J, Samadder A, Boujedaini N, Khuda-Bukhsh AR. Apigenin-induced apoptosis in A375 and A549 cells through selective action and dysfunction of mitochondria. Exp Biol Med (Maywood) (2012) 237:1433–48. doi:10.1258/ebm.2012.012148
184. Spoerlein C, Mahal K, Schmidt H, Schobert R. Effects of chrysin, apigenin, genistein and their homoleptic copper(II) complexes on the growth and metastatic potential of cancer cells. J Inorg Biochem (2013) 127:107–15. doi:10.1016/j.jinorgbio.2013.07.038
185. Piantelli M, Rossi C, Iezzi M, La Sorda R, Iacobelli S, Alberti S, et al. Flavonoids inhibit melanoma lung metastasis by impairing tumor cells endothelium interactions. J Cell Physiol (2006) 207:23–9. doi:10.1002/jcp.20510
186. Cao HH, Chu JH, Kwan HY, Su T, Yu H, Cheng CY, et al. Inhibition of the STAT3 signaling pathway contributes to apigenin-mediated anti-metastatic effect in melanoma. Sci Rep (2016) 6:21731. doi:10.1038/srep21731
187. Ding J, Polier G, Kohler R, Giaisi M, Krammer PH, Li-Weber M. Wogonin and related natural flavones overcome tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) protein resistance of tumors by down-regulation of c-FLIP protein and up-regulation of TRAIL receptor 2 expression. J Biol Chem (2012) 287:641–9. doi:10.1074/jbc.M111.286526
188. Singh V, Sharma V, Verma V, Pandey D, Yadav SK, Maikhuri JP, et al. Apigenin manipulates the ubiquitin-proteasome system to rescue estrogen receptor-beta from degradation and induce apoptosis in prostate cancer cells. Eur J Nutr (2015) 54:1255–67. doi:10.1007/s00394-014-0803-z
189. Toh MF, Sohn J, Chen SN, Yao P, Bolton JL, Burdette JE. Biological characterization of non-steroidal progestins from botanicals used for women’s health. Steroids (2012) 77:765–73. doi:10.1016/j.steroids.2012.03.013
190. Rosenberg RS, Grass L, Jenkins DJ, Kendall CW, Diamandis EP. Modulation of androgen and progesterone receptors by phytochemicals in breast cancer cell lines. Biochem Biophys Res Commun (1998) 248:935–9. doi:10.1006/bbrc.1998.8977
191. Mafuvadze B, Benakanakere I, Hyder SM. Apigenin blocks induction of vascular endothelial growth factor mRNA and protein in progestin-treated human breast cancer cells. Menopause (2010) 17:1055–63. doi:10.1097/gme.0b013e3181dd052f
192. Mafuvadze B, Benakanakere I, Lopez Perez FR, Besch-Williford C, Ellersieck MR, Hyder SM. Apigenin prevents development of medroxyprogesterone acetate-accelerated 7,12-dimethylbenz(a)anthracene-induced mammary tumors in Sprague-Dawley rats. Cancer Prev Res (Phila) (2011) 4:1316–24. doi:10.1158/1940-6207.CAPR-10-0382
193. Mafuvadze B, Liang Y, Besch-Williford C, Zhang X, Hyder SM. Apigenin induces apoptosis and blocks growth of medroxyprogesterone acetate-dependent BT-474 xenograft tumors. Horm Cancer (2012) 3:160–71. doi:10.1007/s12672-012-0114-x
194. Ramaraj P, Cox JL. In vitro effect of progesterone on human melanoma (BLM) cell growth. Int J Clin Exp Med (2014) 7:3941–53.
195. Leder DC, Brown JR, Ramaraj P. In-vitro rescue and recovery studies of human melanoma (BLM) cell growth, adhesion and migration functions after treatment with progesterone. Int J Clin Exp Med (2015) 8:12275–85.
196. Seidlova-Wuttke D, Becker T, Christoffel V, Jarry H, Wuttke W. Silymarin is a selective estrogen receptor beta (ERbeta) agonist and has estrogenic effects in the metaphysis of the femur but no or antiestrogenic effects in the uterus of ovariectomized (ovx) rats. J Steroid Biochem Mol Biol (2003) 86:179–88. doi:10.1016/S0960-0760(03)00270-X
197. Li LH, Wu LJ, Zhou B, Wu Z, Tashiro S, Onodera S, et al. Silymarin prevents UV irradiation-induced A375-S2 cell apoptosis. Biol Pharm Bull (2004) 27:1031–6. doi:10.1248/bpb.27.1031
198. Li LH, Wu LJ, Tashiro SI, Onodera S, Uchiumi F, Ikejima T. The roles of Akt and MAPK family members in silymarin’s protection against UV-induced A375-S2 cell apoptosis. Int Immunopharmacol (2006) 6:190–7. doi:10.1016/j.intimp.2005.08.003
199. Li LH, Wu LJ, Tashiro SI, Onodera S, Uchiumi F, Ikejima T. Activation of the SIRT1 pathway and modulation of the cell cycle were involved in silymarin’s protection against UV-induced A375-S2 cell apoptosis. J Asian Nat Prod Res (2007) 9:245–52. doi:10.1080/10286020600604260
200. Li LH, Wu LJ, Jiang YY, Tashiro S, Onodera S, Uchiumi F, et al. Silymarin enhanced cytotoxic effect of anti-Fas agonistic antibody CH11 on A375-S2 cells. J Asian Nat Prod Res (2007) 9:593–602. doi:10.1080/10286020600882502
201. Vaid M, Singh T, Prasad R, Katiyar SK. Silymarin inhibits melanoma cell growth both in vitro and in vivo by targeting cell cycle regulators, angiogenic biomarkers and induction of apoptosis. Mol Carcinog (2015) 54:1328–39. doi:10.1002/mc.22208
202. Lee MH, Huang Z, Kim DJ, Kim SH, Kim MO, Lee SY, et al. Direct targeting of MEK1/2 and RSK2 by silybin induces cell-cycle arrest and inhibits melanoma cell growth. Cancer Prev Res (Phila) (2013) 6:455–65. doi:10.1158/1940-6207.CAPR-12-0425
203. Vaid M, Prasad R, Sun Q, Katiyar SK. Silymarin targets beta-catenin signaling in blocking migration/invasion of human melanoma cells. PLoS One (2011) 6:e23000. doi:10.1371/journal.pone.0023000
204. Barone M, Tanzi S, Lofano K, Scavo MP, Guido R, Demarinis L, et al. Estrogens, phytoestrogens and colorectal neoproliferative lesions. Genes Nutr (2008) 3:7–13. doi:10.1007/s12263-008-0081-6
205. Deep G, Oberlies NH, Kroll DJ, Agarwal R. Isosilybin B causes androgen receptor degradation in human prostate carcinoma cells via PI3K-Akt-Mdm2-mediated pathway. Oncogene (2008) 27:3986–98. doi:10.1038/onc.2008.45
206. Deep G, Gangar SC, Oberlies NH, Kroll DJ, Agarwal R. Isosilybin A induces apoptosis in human prostate cancer cells via targeting Akt, NF-kappaB, and androgen receptor signaling. Mol Carcinog (2010) 49:902–12. doi:10.1002/mc.20670
207. Allil PA, Visconti MA, Castrucci AM, Isoldi MC. Photoperiod and testosterone modulate growth and melanogenesis of s91 murine melanoma. Med Chem (2008) 4:100–5. doi:10.2174/157340608783789185
Keywords: melanoma, ERβ, BRAF, NRAS, oncogenic mutations, targeted therapy, ERβ ligands, phytoestrogens
Citation: Marzagalli M, Montagnani Marelli M, Casati L, Fontana F, Moretti RM and Limonta P (2016) Estrogen Receptor β in Melanoma: From Molecular Insights to Potential Clinical Utility. Front. Endocrinol. 7:140. doi: 10.3389/fendo.2016.00140
Received: 29 July 2016; Accepted: 12 October 2016;
Published: 26 October 2016
Edited by:
Krzysztof Reiss, LSU Health Sciences Center New Orleans, USAReviewed by:
Dario Giuffrida, Istituto Oncologico del Mediterraneo, ItalyJudy Sue Crabtree, LSU Health Sciences Center New Orleans, USA
Copyright: © 2016 Marzagalli, Montagnani Marelli, Casati, Fontana, Moretti and Limonta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrizia Limonta, cGF0cml6aWEubGltb250YUB1bmltaS5pdA==
†Monica Marzagalli and Marina Montagnani Marelli contributed equally to this work.