 Sebastiano Messineo1†
Sebastiano Messineo1† Anna Elisa Laria1†
Anna Elisa Laria1† Biagio Arcidiacono1
Biagio Arcidiacono1 Eusebio Chiefari1
Eusebio Chiefari1 Raúl M. Luque Huertas2
Raúl M. Luque Huertas2 Daniela P. Foti1*
Daniela P. Foti1* Antonio Brunetti1*
Antonio Brunetti1*
- 1Department of Health Sciences, University “Magna Græcia” of Catanzaro, Catanzaro, Italy
- 2Department of Cell Biology, Physiology and Immunology, Instituto Maimónides de Investigación Biomédica de Córdoba (IMIBIC), Hospital Universitario Reina Sofía (HURS), CIBERobn and ceiA3, University of Córdoba, Córdoba, Spain
The architectural transcription factor high-mobility group AT-hook 1 (HMGA1) is a chromatin regulator with implications in several biological processes, including tumorigenesis, inflammation, and metabolism. Previous studies have indicated a role for this factor in promoting the early stages of adipogenesis, while inhibiting adipocyte terminal differentiation, and decreasing fat mass. It has been demonstrated that hypoxia – through the hypoxia-inducible factor 1 (HIF-1) – plays a major role in triggering changes in the adipose tissue of the obese, leading to inhibition of adipocyte differentiation, adipose cell dysfunction, inflammation, insulin resistance, and type 2 diabetes. To examine the possible cooperation between HMGA1 and HIF-1, herein, we investigated the role of HMGA1 in the regulation of Visfatin and VEGF, two genes normally expressed in adipose cells, which are both responsive to hypoxia. We demonstrated that HMGA1 enhanced Visfatin and VEGF gene expression in human embryonic kidney (HEK) 293 cells in hypoxic conditions, whereas HMGA1 knockdown in differentiated 3T3-L1 adipocytes reduced these effects. Reporter gene analysis showed that Visfatin and VEGF transcriptional activity was increased by the addition of either HMGA1 or HIF-1 and even further by the combination of both factors. As demonstrated by chromatin immunoprecipitation in intact cells, HMGA1 directly interacted with the VEGF gene, and this interaction was enhanced in hypoxic conditions. Furthermore, as indicated by co-immunoprecipitation studies, HMGA1 and HIF-1 physically interacted with each other, supporting the notion that this association may corroborate a functional link between these factors. Therefore, our findings provide evidence for molecular cross-talk between HMGA1 and HIF-1, and this may be important for elucidating protein and gene networks relevant to obesity.
Introduction
Obesity is a pathological condition often associated with insulin resistance, type 2 diabetes, cardiovascular disease, and even certain types of cancers (1, 2). Because of its alarming rise in incidence, and consequences in health care, interest in understanding the pathophysiological role of adipose tissue has consistently increased in the last decades. A major, relatively recent advancement in this field has been the concept that adipose tissue is not just an inert reserve of lipids, but a source of biomolecules, collectively called adipocytokines, with important implications in insulin sensitivity, inflammation, and angiogenesis (3–5). As adipocytokines are differentially expressed in the adipose tissue of obese versus lean individuals, a pro-inflammatory, insulin resistant, pro-atherogenic pattern typically prevails in the obese state. For example, it has been recognized that, in obesity, hypertrophic adipocytes produce monocyte chemotactic protein-1 (MCP-1), a factor favoring the infiltration of macrophages into fat tissue, while these immune cells produce pro-inflammatory cytokines, including the tumor necrosis factor α (TNFα), which sustain, in turn, adipose cell dysfunction (6, 7). However, the initial events leading to these changes in adipose tissue are still poorly understood.
An important issue is that the adipose tissue from obese individuals may become poorly oxygenated as adipocytes become larger, and distant from the vasculature (8–12). Hypoxia-inducible factors (HIFs) are important mediators of cellular adaptive response to hypoxia and play a role in the regulation of genes implicated in anaerobic metabolism, cell growth and survival, angiogenesis, and immune response (13, 14). In this regard, a master regulator that mediates hypoxic response is the HIF-1, a heterodimer constituted by α and β subunits. While HIF-1β is a constitutively expressed subunit, HIF-1α is continuously translated and degraded through ubiquitination under normoxic conditions (15, 16). In contrast, in hypoxic conditions, HIF-1α accumulates in the cytosol, being subsequently translocated into the nucleus, where it dimerizes with the HIF-1β subunit. As a transcription factor, the HIF-1α/β dimer binds to hypoxia responsive elements located within O2-regulated genes (17, 18), modulating a wide range of adaptive responses at low oxygen conditions, including glucose utilization, angiogenesis, apoptosis, extracellular matrix remodeling, and inflammation (12–14, 19, 20). Several other transcription factors have been recognized to be implicated in this scenario, including NF-kB, C/EBP homologous protein (CHOP), and the cAMP response element binding protein (CREB), all of which may interact and cooperate with HIF-1, in particular in the context of inflammation (21–23).
As reported (9), over thousand genes change their expression in adipose tissue hypoxia. However, how HIF-1 acts in the regulation of fat tissue-specific target genes, and which molecular partners are involved, need to be further characterized. We have hypothesized that the high-mobility group protein AT-hook 1 (HMGA1) may be a player in the gene expression networks that involve HIF-1 in adipose tissue. HMGA1 is an architectural transcription factor that functions as a dynamic regulator of chromatin structure (24–26), and is implicated in a variety of biological processes, including development, tumorigenesis, inflammation, and metabolism (25, 27–30). Several evidences have demonstrated a role for HMGA1 in the development of adipose tissue, both in pathological and physiological conditions. HMGA (HMGA1 and HMGA2) gene rearrangements, due to chromosomal translocations, have been described in human benign neoplasias of mesenchimal origin, including lipomas (31), whereas a truncated form of HMGA1 has been reported to induce proliferation in 3T3-L1 preadipocytes (32). In a more physiological context, it has been reported that in vitro, in 3T3-L1 cells, HMGA1 is highly expressed during the first phases of adipose cell differentiation, while its deficit precludes terminal adipocyte conversion (27). Coherently, in vivo, in transgenic mice, HMGA1 overexpression in adipose tissue impairs adipogenesis and reduces fat mass by upregulating pre-adipocyte gene markers, and downregulating genes involved in adipocyte differentiation (33). At a molecular level, by binding to AT-rich sequences in the promoter region of the gene, HMGA1 modulates gene transcription either facilitating the binding of other transcription regulators to DNA, or interacting with other transcription factors that directly influence gene transcription (24, 34, 35). For example, HMGA1 has been shown to physically and/or functionally interact with NF-kB, PPARγ, and CEBP/β (36–38), which are abundantly expressed in adipose tissue. Previous findings indicate that hypoxia induces HMGA1 expression in vitro, and that this response is evolutionarily conserved (39, 40). However, up to now, a relationship between HMGA1 and HIF-1 has not been reported and the examination of this link could give further insights into our understanding of the regulation of genes involved in adipogenesis and obesity.
In the present study, we investigated two known HIF-1 target genes: the vascular endothelial growth factor (VEGF) gene and the Visfatin gene, both of which are normally expressed in adipose tissue (41–44). We show that HMGA1 physically interacts with HIF-1, and this interaction is required for proper transcription of these genes.
Materials and Methods
Cell Cultures
HEK-293 cells and 3T3-L1 mouse fibroblasts were cultured in Dulbecco’s modified Eagle medium (DMEM) (Sigma Aldrich) supplemented with 10% fetal bovine serum, 2 mM glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml), in a humidified 37°C, 5% CO2 incubator. Differentiation of 3T3-L1 fibroblasts into adipocytes was induced as described previously (45). Hypoxic conditions were obtained by placing cells for 24 h at 2% O2 in a hypoxic chamber (New Brunswick Galaxy 48R, Eppendorf), or by chemical induction with 100 μM cobalt chloride (CoCl2).
Plasmids, Small Interfering RNA, and Transient Transfection
Reporter plasmids were as follows: p2025-Visfatin-Luc (a gift from A. Fukuhara, Osaka University, Osaka, Japan) and VEGF 2.6-Luc (a gift from N. Sheehy, University College Dublin, Dublin, Ireland). Expression plasmids were as follows: pcDNA3/HA-HMGA1 (32) and pcDNA3.1/HA-HIF-1α (a gift from G.L. Semenza, Johns Hopkins University, Baltimore, USA). For gene silencing experiments, a pool of three target-specific 20–25 nt siRNAs targeting mouse HMGA1 (Santa Cruz Biotech) was used. In all knockdown experiments, cells were transfected with 250 pmol of HMGA1 siRNA in six-well plates, and incubated without further treatment for 72 h before being used in subsequent analyses. Cell transient transfections were carried out using the Lipofectamine 2000 method (Invitrogen), and luciferase activity was assayed 48 h later, using the dual-luciferase reporter assay system (Promega).
Real-Time PCR
For quantitative RT-PCR (qRT-PCR), total cellular RNA was extracted from 3T3-L1 cells using the RNAqueous-4PCR kit (Ambion), subjected to DNase treatment, and cDNAs were synthesized from 1 μg of total RNA using the RETROscript first strand synthesis kit (Ambion). Primers for mouse HMGA1, Visfatin, VEGF, and Rps9 were designed according to sequences from the GenBank database. A real-time thermocycler (Eppendorf Mastercycler ep realplex ES) was used to perform qRT-PCR. SYBR Green fluorescence was measured, and relative quantification was made against the Rps9 cDNA used as an internal standard. All PCR reactions were carried out in triplicates.
Western Blot and Co-Immunoprecipitation Studies
Western blots were performed in nuclear extracts from 3T3-L1 and HEK-293 cells in both normoxic or hypoxic conditions. The antibodies used for these studies were: anti-HIF-1α (NovusBio), anti-Sp1, anti-HMGA1, and anti-Lamin A/C (SantaCruz) polyclonal antibodies. For co-immunoprecipitation studies, 200 μg of nuclear extracts from 3T3-L1 adipocytes or HEK-293 cells were mixed overnight with 5 μg of anti-HMGA1 antibody, as reported previously (38). Protein A Sepharose beads (GE Healthcare) were added for 90 min with rotation at 4°C. Antibody coupled protein A beads were washed twice with phosphate-buffered saline (PBS), proteins dissolved in Laemmli buffer and analyzed by SDS-PAGE and immunoblotting, using the polyclonal HIF-1α-specific antibody.
Chromatin Immunoprecipitation
Chromatin Immunoprecipitation was performed in 3T3-L1 cells as described previously (35). As soon as the cells reached full differentiation, hypoxia was induced by treating cells with O2 2%, 24 h. Then, cells were washed with PBS and fresh DMEM was added. DNA–protein complexes were cross-linked by adding formaldehyde for 10 min at room temperature, followed by blocking with glycine for 2 min. Cells were washed twice with cold PBS and lysed on ice using SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris pH 8). Chromatin samples were sonicated on ice and the formaldehyde-fixed DNA–protein complexes were immunoprecipitated with anti-HMGA1 antibody, and sequence-specific primers for the mouse Vegf (and Visfatin) gene promoters were used for PCR amplification of immunoprecipitated DNA, using PCR ready-to-go beads (GE Healthcare). For the mouse Vegf gene, Vegf-specific primers (for 5′-GCT CTC TCT GAC CGG TCT CT-3′; rev 5′-GCA GAC TAT TCA GCG GAC TCA-3′) amplified a 270 bp region from −922 to −652 bp upstream of the ATG start site, which encompasses the consensus site for HMGA1. PCR products were electrophoretically resolved on 1.5% agarose gel and stained with ethidium bromide staining solution. To improve the reliability of comparisons within multiple experiments, we performed also qRT-PCR to amplify ChIP-ed DNA samples and to compare the amount of each with total input DNA used for each immunoprecipitation. Primers used were as above, while dissociation curves were analyzed to verify the quality of the amplicon and to exclude the presence of primer dimers.
Statistical Analysis
All calculations were performed with SPSS 20.0 statistical software (SPSS Inc.). The non-parametric Mann–Whitney test was used for comparisons of data with a control, as foreseen in the Dunnett test approach (46). Results are shown as mean ± SE. A p-value <0.05 (two-tailed) was considered significant.
Results
Hypoxia and HMGA1 Increase Visfatin and VEGF Gene Expression
To test the hypothesis that HMGA1 could play a functional role in the transcriptional regulation of genes activated in hypoxic conditions, we first examined two genes, VEGF and Visfatin, which are both naturally expressed in adipose cells, and regulated by hypoxia. HMGA1 and HIF-1 consensus binding sites within the regulatory region of both these genes were identified by using the TRANSFAC database searched with MatInspector as informatic support (version 8.1, Genomatix, http://www.genomatix.de/). As shown in Figure 1, bioinformatic analysis predicted putative binding sites for HIF-1 and HMGA1 nuclear proteins in both human and mouse VEGF and Visfatin gene promoters.
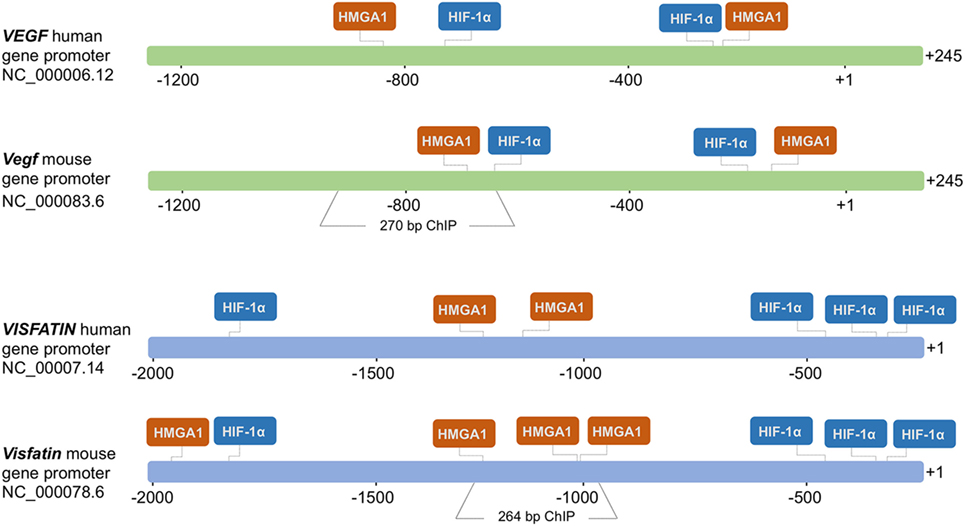
Figure 1. Schematic representation of the human and mouse VEGF and Visfatin promoter regions. HMGA1- and HIF-1α-binding sites are shown in each region. The numbers indicate positions in base pairs relative to the transcriptional start site (+1). Mouse Vegf and Visfatin promoter regions encompassing binding sites for HMGA1 and used in ChIP are indicated.
We next tested Visfatin and VEGF gene expression, and their response to hypoxia, in murine differentiated 3T3-L1 adipocytes, which express high levels of HMGA1 protein, and in human HEK-293 cells, a cell line ideally suited for studying the effects of HMGA1 on transcription since it does not express appreciable amounts of HMGA1. As shown by qRT-PCR, Visfatin and VEGF cDNAs were detectable in these cell lines in normal growth conditions, whereas both genes were further activated in cells under hypoxic conditions, as obtained by incubating cells in 2% O2 or when exposed to chemical hypoxia with CoCl2 (Figure 2A). When HMGA1 was overexpressed into HEK-293 cells, the expression of both VEGF and Visfatin genes increased in both normoxic and hypoxic conditions (Figure 2B). Conversely, knockdown of endogenous HMGA1 with HMGA1-specific siRNA inhibited Vegf and Visfatin gene expression in 3T3-L1 adipocytes in normoxic and hypoxic conditions (Figure 2C), thereby indicating a role for HMGA1 in the regulation of these hypoxia-inducible genes, and suggesting that HMGA1 is required for full activation of these genes during hypoxia.
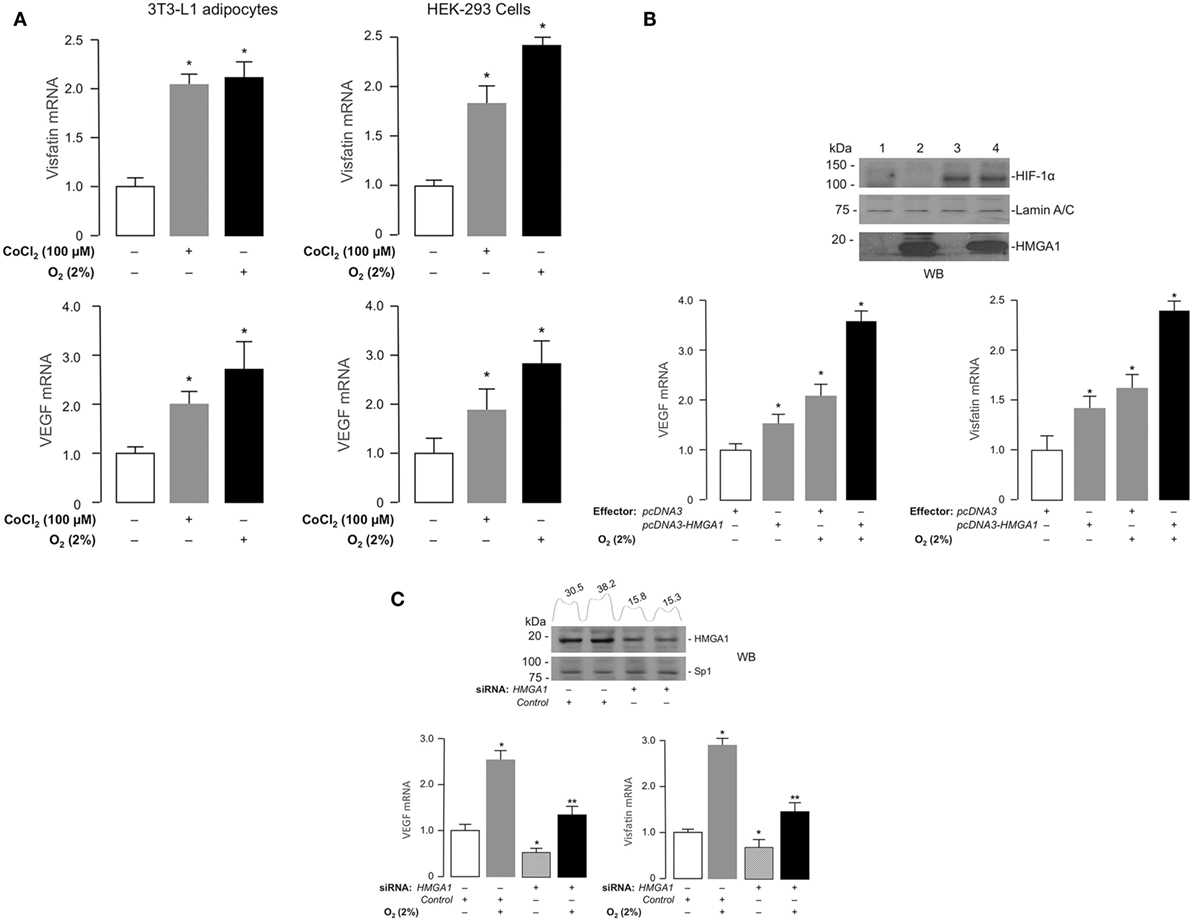
Figure 2. Effects of hypoxia and HMGA1 on Visfatin and VEGF gene expression. (A) 3T3-L1 adipocytes (left) and HEK-293 cells (right) were treated with CoCl2 or incubated under hypoxic conditions (O2 2%) for 24 h. Visfatin and VEGF mRNA were measured by RT-PCR. Results are the means ± SE of triplicates from three independent experiments. Data are shown as fold of increment versus controls (white bars), which are assigned a value of 1. *p < 0.05 versus controls. (B) HEK-293 cells were transfected with HMGA1 plasmid (1 μg) under normoxic or hypoxic (O2 2%) conditions. After 48 h, VEGF and Visfatin mRNA were measured by RT-PCR. Results are the means ± SE of triplicates from three independent experiments. Data are shown as fold of increment versus controls (white bars), which are assigned a value of 1. *p < 0.05 versus controls. Representative Western blots (WB) of HIF-1α and HMGA1 are shown for each experimental condition. Lanes: 1, control (pcDNA3 empty vector); 2, pcDNA3–HMGA1 effector vector; 3, pcDNA3 empty vector plus hypoxia; 4, pcDNA3–HMGA1 effector vector plus hypoxia. Lamin A/C, control of protein loading (C) 3T3-L1 adipocytes were incubated in the absence or presence of siRNA targeting HMGA1 (250 pmol) under normoxic or hypoxic (O2 2%) conditions. VEGF and Visfatin mRNAs were measured by RT-PCR. Results are the means ± SE of triplicates from three independent experiments. *p < 0.05 versus controls (white bars); **p < 0.05 versus hypoxic conditions in the presence of scrambled siRNA (control, gray bars). Representative WBs of HMGA1 and Sp1 (this latter used as a control) from nuclear extracts of 3T3-L1 cells, either untreated or treated with siRNA targeting HMGA1, are shown. Densitometric slot blot analysis, using the ImageJ software program, is shown for HMGA1. Numbers on the peaks are the size of the corresponding slot as a percentage of the total size of the slots.
Transcriptional Regulation of Visfatin and VEGF Genes by HMGA1 and HIF-1
To explore the possibility that Visfatin and VEGF genes were transcriptionally regulated by a functional cooperation between HIF-1 and HMGA1, we transiently transfected HEK-293 cells with luciferase reporter plasmids containing the full-length sequence of the mouse Visfatin or the human VEGF gene promoter, in the absence or presence of recombinant plasmids expressing either HMGA1, HIF-1α, or both. As shown in Figure 3, forced expression of HMGA1 or HIF-1α in HEK-293 cells induced Visfatin and VEGF gene promoters, while concomitant overexpression of HMGA1 and HIF-1α yielded a synergistic increase in both Visfatin and VEGF promoter activities, thus indicating that, in hypoxic conditions, HMGA1 is required for full transactivation of these genes by HIF-1.
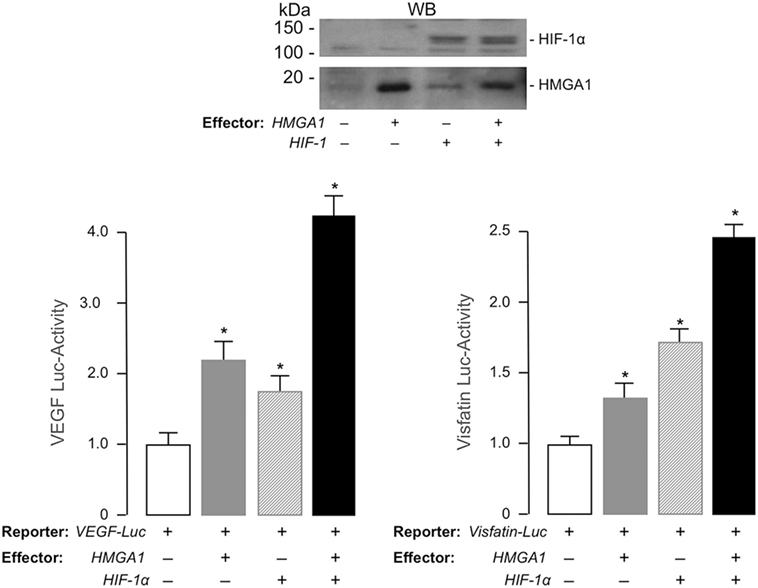
Figure 3. Functional activity of HMGA1 and HIF-1α on VEGF and Visfatin gene transcription. HEK-293 cells were cotransfected with 1 μg of human VEGF-Luc or 100 ng of mouse Visfatin-Luc reporter plasmid, in the absence or presence of effector vectors for HMGA1 and HIF-1α (1 μg each), either alone or in combination. Data represent the means ± SE for three separate experiments. Values are expressed relative to the VEGF- and Visfatin-Luc activities obtained in transfections with the reporter vector alone (control, white bars), which is assigned an arbitrary value of 1. *p < 0.05 versus control. Representative WBs of HMGA1 and HIF-1α from nuclear extracts of HEK-293 cells, either untransfected or transfected with human HMGA1 or HIF-1α expression vectors, are shown.
HMGA1 and HIF-1 Protein–Protein Interaction and ChIP
The above results were further supported by protein–protein interaction experiments in whole 3T3-L1 adipocytes and HEK-293 cells, in both of which hypoxia was induced by either oxygen-deprivation in hypoxia-chamber or by the chemical hypoxia-mimicking agent CoCl2. While confirming that the HIF-1α protein product was indeed induced in nuclear extracts from 3T3-L1 cells in response to hypoxia (Figure 4A), we investigated whether HMGA1 and HIF-1 proteins physically interacted with each other in the context of the intact cell by performing co-immunoprecipitation studies. As shown in Figure 4A, immunoprecipitation of HMGA1 in nuclear extracts from 3T3-L1 adipocytes exposed to normoxia or hypoxia, followed by Western blot analysis for HIF-1α, revealed a protein-specific band, which migrated in a position corresponding to the size of HIF-1α, whose intensity considerably increased with hypoxia. HMGA1-HIF1 protein–protein interaction in hypoxia was substantiated in co-immunoprecipitation experiments using HMGA1 overexpressing HEK-293 cells, a cell line that normally produces barely amounts of HMGA1 (Figure 4A). Overall, these results indicate that HMGA1 and HIF-1 physically interact in vivo in intact cells, thereby suggesting that this step may represent an important prerequisite for the functional interplay between HMGA1 and HIF-1 in hypoxia.
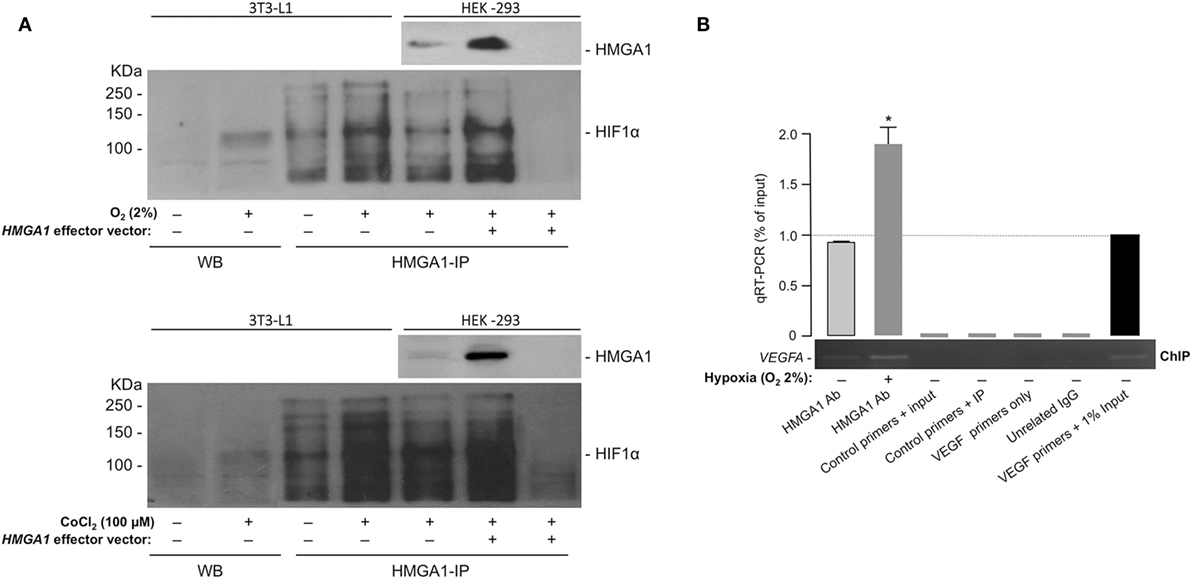
Figure 4. HMGA1 and HIF-1 protein–protein interaction and ChIP. (A) HIF-1α protein expression as shown by WB from 3T3-L1 adipocytes nuclear extracts in both normoxic or hypoxic (O2 2%, upper panel; CoCl2 100 μM, lower panel) conditions. Immunoprecipitation of HIF-1α from 3T3-L1 adipocytes and HEK-293 cell nuclear extracts under normoxic and hypoxic conditions, by using an anti-HMGA1 antibody (HMGA1-IP), followed by immunoblotting with an anti-HIF-1α specific antibody is shown in both panels. A negative control of immunoprecipitation (in the absence of HMGA1 antibody) is shown in the last right lane of each panel. Representative immunoblots are shown for each condition. Immunoprecipitations of HMGA1 followed by immunoblot analyses with the anti-HMGA1 antibody are shown in hypoxic HEK-293 cells, either untransfected or transfected with the HMGA1 effector vector. (B) ChIP analysis of the mouse Vegf promoter gene in 3T3-L1 adipocytes under normoxic and hypoxic (O2 2%) conditions, using the anti-HMGA1-specific antibody (Ab). A representative ChIP assay is shown, together with qRT-PCR of ChIP-ed samples. Results are the mean ± SE for three independent experiments. p < 0.05 versus control (slashed bar).
These studies were complemented by ChIP in differentiated 3T3-L1 adipocytes. We found that HMGA1 bound to the endogenous VEGFA chromosomal locus, and that this binding was enhanced after incubation of cultured cells in hypoxic conditions (2% O2) (Figure 4B). Of note, no binding of HMGA1 to the Visfatin gene was observed in parallel experiments with 3T3-L1 cells (not shown), suggesting that HMGA1 may influence Visfatin gene transcription through alternative mechanisms, not necessarily involving direct binding of HMGA1 to DNA. A similar behavior has been reported for other genes, such as the Leptin and the interferon beta (IFN-β) genes (27, 47).
Discussion
Cellular oxygen sensing is known to be mostly mediated by specific nuclear transcription factors, among which HIF-1 occupies a pivotal position. How hypoxia, through HIF-1, impacts, at the molecular level, fat tissue-specific target genes and gene networks, is only partly understood. It is plausible that in most cases, HIF-1 modulates the transcriptional activity of target genes by regulating and/or interacting with other known ubiquitous or tissue-specific nuclear factors. In this regard, in adipose tissue, HIF-1 prevents adipogenesis by inhibiting PPARγ2 and C/EBPβ gene expression (48). In addition, HIF-1 is regulated by and interacts with NF-kB, a transcription factor that plays a pivotal role in the regulation of the inflammatory and immune responses (49).
High-mobility group AT-hook 1 shares with HIF-1 similar effects on adipogenesis and inflammation by inhibiting adipose cell terminal differentiation and by promoting the expression of proinflammatory cytokines. For example, an involvement of both HMGA1 and HIF-1 has been reported for the regulation of the Leptin and Pref-1 genes in the early phase of adipogenesis (9, 27, 33). On the other hand, cooperation among molecular partners common to HMGA1 and HIF-1, such as NF-kB, has been reported for the upregulation of genes involved in the immune response/inflammation, including IL-6 (9, 50), IFN-β (36, 51), and several cytokine and adhesion molecule genes (9, 52). Furthermore, in the context of the cyclooxygenase-2 (COX-2) gene promoter, hypoxia has been shown to induce HMGA1 as part of a hypoxia-induced enhanceosome that encompasses, among others, the NF-kB transcription factor, and helps to promote transcription of COX-2 (39). Despite of these findings, however, no evidence of a link between HIF-1 and HMGA1 has been previously searched and identified.
Herein, we were interested to investigate whether HIF-1 cooperates with HMGA1 in the functional regulation of adipose tissue-specific gene expression. Using VEGF and Visfatin as established HIF-1-target genes in 3T3-L1 adipocytes, we demonstrate that HMGA1 functionally interacts with HIF-1, thereby inducing transcriptional activation of both target genes. Our findings indicate that hypoxia-induced transcription of VEGF and Visfatin requires HMGA1 for maximal transactivation of these genes by HIF-1, and that repression of HMGA1 expression adversely affects HIF-1 function. Using ChIP, we demonstrated that HMGA1 can directly interact with the endogenous VEGF promoter. Intriguingly, this was not the case for the Visfatin gene, for which no HMGA1–DNA binding was observed in ChIP assays, although multiple putative binding sites for HMGA1 were detected within the Visfatin promoter. One possibility for the functional interrelationship between HIF-1 and HMGA1 in the context of the Visfatin gene, during hypoxia, is that HMGA1 may act through protein–protein interaction mechanism, without direct binding to DNA. This is the case, for example, of the Leptin gene (27) and IFN-β gene (47), for which HMGA1 strongly potentiates transactivation by C/EBPβ and NF-kB, respectively, through direct protein–protein contacts independent of DNA. Alternatively, HMGA1 could recruit component(s) of the basal transcription machinery toward protein–DNA complexes, thereby promoting transcription, with little, if any, specificity for the putative target DNA sequence (53). In support of these non-canonical mechanisms, physical association between HIF-1 and HMGA1 has been observed in our study, and this association may be a prerequisite for the HIF-1 effects in hypoxia.
While in adipose tissue the cooperation between HIF-1 and HMGA1 may coherently recapitulate their parallel roles in adipogenesis and inflammation, other important biological effects, such as those on insulin action and glucose metabolism, in which HIF-1 and HMGA1 act in an apparently divergent manner (i.e., insulin resistance versus insulin sensitivity), deserve future searches and explanations. Studies in adipose tissue and cells have shown a role of hypoxia in insulin resistance (9, 20) and in the impairment of insulin action (54), while adipose tissue-specific disruption of HIF-1 in mice fed with high-fat diet improves insulin sensitivity (55). In contrast, HMGA1 plays a positive role in insulin biosynthesis and action (29, 35, 56), as well as in the transcription of a number of glucose metabolism-related genes (29, 30, 38, 57). Thus, whereas HMGA1 gene defects associate with insulin resistance and diabetes in humans and mice (29, 58, 59), HMGA1 overexpression in adipose tissue prevents insulin resistance in mice (33). Therefore, studies aimed at investigating cross-talks between HMGA1, HIF-1, and their molecular partners at the level of gene transcription may help explaining the divergent role of these factors in insulin response.
Overall, we believe our findings may contribute to elucidate the molecular mechanisms underlying hypoxia-induced gene regulation and expression in adipose cells. The identification of gene expression networks involved in adipose cell dysfunction represents a challenging field of research that may improve our knowledge on the pathophysiology of obesity and obesity-related disorders.
Author Contributions
SM, AEL, and BA performed the experiments; DF conceived the work and wrote the first draft of the manuscript; EC and RH supervised PCR and immunoblot experiments and critically read the manuscript; AB critically revised and edited the manuscript. All the authors have approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer (ZM) and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
We thank Drs. A. Fukuhara (Osaka University, Osaka, Japan), N. Sheehy (University College Dublin, Dublin, Ireland), and G. L. Semenza (Johns Hopkins University, Baltimore, USA) for providing plasmids. Dr. SM and Dr. BA are supported as postdoctoral researchers by a grant from Regione Calabria (PO Calabria FSE 2007–2013). This work was supported by MIUR (protocol 2004062059-002 Italy) to AB, and by Instituto de Salud Carlos III (PI13-00651) to RH.
References
2. Obesity: preventing and managing the global epidemic. Report of a WHO consultation. World Health Organ Tech Rep Ser (2000) 894:i–xii,1–253.
3. Ahima RS, Flier JS. Adipose tissue as an endocrine organ. Trends Endocrinol Metab (2000) 11(8):327–32. doi:10.1016/S1043-2760(00)00301-5
4. Fasshauer M, Blüher M. Adipokines in health and disease. Trends Pharmacol Sci (2015) 36(7):461–70. doi:10.1016/j.tips.2015.04.014
5. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest (2011) 121(6):2111–7. doi:10.1172/JCI57132
6. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitasawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest (2006) 116(6):1494–505. doi:10.1172/JCI26498
7. Chen XH, Zhao YP, Xue M, Ji CB, Gao CL, Zhu JG, et al. TNF-alpha induces mitocondrial dysfunction in 3T3-L1 adipocytes. Mol Cell Endocrinol (2010) 328(1–2):63–9. doi:10.1016/j.mce.2010.07.005
8. Kabon B, Nagele A, Reddy D, Eagon C, Fleshman JW, Sessler DI, et al. Obesity decreases perioperative tissue oxygenation. Anesthesiology (2004) 100(2):274–80. doi:10.1097/00000542-200402000-00015
9. Trayhurn P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol Rev (2013) 93(1):1–21. doi:10.1152/physrev.00017.2012
10. Lin Q, Yun Z. The hypoxia-inducible factor pathway in adipocytes: the role of HIF-2 in adipose inflammation and hypertrophic cardiomyopathy. Front Endocrinol (2015) 6:36. doi:10.3389/fendo.2015.00039
11. Goossens GH, Blaak EE. Adipose tissue dysfunction and impaired metabolic health in human obesity: a matter of oxygen? Front Endocrinol (2015) 6:55. doi:10.3389/fendo.2015.00055
12. Ichiki T, Sunagawa K. Novel roles of hypoxia response system in glucose metabolism and obesity. Trends Cardiovasc Med (2014) 24(5):197–201. doi:10.1016/j.tcm.2014.03.004
13. Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med (2011) 365(6):537–47. doi:10.1056/NEJMra1011165
14. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity (2014) 41(4):518–28. doi:10.1016/j.immuni.2014.09.008
15. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA (1995) 92(12):5510–4. doi:10.1073/pnas.92.12.5510
16. Salceda S, Caro J. Hypoxia-inducible factor 1α (HIF-1α) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem (1997) 272(36):22642–7. doi:10.1074/jbc.272.36.22642
18. Brocato J, Chervona Y, Costa M. Molecular responses to hypoxia-inducible factor 1α and beyond. Mol Pharmacol (2014) 85(5):651–7. doi:10.1124/mol.113.089623
19. Dehne N, Brüne B. HIF-1 in the inflammatory microenvironment. Exp Cell Res (2009) 315(11):1791–7. doi:10.1016/j.yexcr.2009.03.019
20. Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, et al. Hypoxia-Inducible Factor 1α induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol (2009) 29(6):4467–83. doi:10.1128/MCB.00192-09
21. Van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem J (2008) 412(3):477–84. doi:10.1042/BJ20080476
22. Chou YT, Zhan G, Zhu Y, Fenik P, Panossian L, Li Y, et al. C/EBP homologous binding protein (CHOP) underlies neural injury in sleep apnoea model. Sleep (2013) 36(4):481–92. doi:10.5665/sleep.2528
23. Ruas JL, Poellinger L, Pereira T. Functional analysis of hypoxia-inducible factor-1 alpha-mediated transactivation. Identification of amino acid residues critical for transcriptional activation and/or interaction with CREB-binding protein. J Biol Chem (2002) 277(41):38723–30. doi:10.1074/jbc.M205051200
24. Reeves R, Beckerbauer L. HMGI/Y proteins: flexible regulators of transcription and chromatin structure. Biochim Biophys Acta (2001) 1519(1–2):13–29.
25. Liu F, Chau KY, Arlotta P, Ono SJ. The HMGI proteins: dynamic roles in gene activation, development, and tumorigenesis. Immunol Res (2001) 24(1):13–29. doi:10.1385/IR:24:1:13
26. Arnoldo L, Sgarra R, Chiefari E, Iiritano S, Arcidiacono B, Pegoraro S, et al. A novel mechanism of post-translational modulation of HMGA functions by the histone chaperone nucleophosmin. Sci Rep (2015) 5:8552. doi:10.1038/srep08552
27. Melillo RM, Pierantoni GM, Scala S, Battista S, Fedele M, Stella A, et al. Critical role of the HMGI(Y) proteins in adipocytic cell growth and differentiation. Mol Cell Biol (2001) 21(7):2485–95. doi:10.1128/MCB.21.7.2485-2495.2001
28. Schuldenfrei A, Belton A, Kowalski J, Talbot CC Jr, Di Cello F, Poh W, et al. HMGA1 drives stem cell, inflammatory pathway, and cell cycle progression genes during lymphoid tumorigenesis. BMC Genomics (2011) 12:549. doi:10.1186/1471-2164-12-549
29. Foti D, Chiefari E, Fedele M, Iuliano R, Brunetti L, Paonessa F, et al. Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in humans and mice. Nat Med (2005) 11(7):765–73. doi:10.1038/nm1254
30. Chiefari E, Paonessa F, Iiritano S, Le Pera I, Palmieri D, Brunetti G, et al. The cAMP-HMGA1-RBP4 system: a novel biochemical pathway for modulating glucose homeostasis. BMC Biol (2009) 7:24. doi:10.1186/1741-7007-7-24
31. Fedele M, Battista S, Manfioletti G, Croce CM, Giancotti V, Fusco A. Role of the high mobility group A proteins in human lipomas. Carcinogenesis (2001) 22(10):1583–91. doi:10.1093/carcin/22.10.1583
32. Pierantoni GM, Battista S, Pentimalli F, Fedele M, Visone R, Federico A, et al. A truncated HMGA1 gene induces proliferation of the 3T3-L1 pre-adipocytic cells: a model of human lipomas. Carcinogenesis (2003) 24(12):1861–9. doi:10.1093/carcin/bgg149
33. Arce-Cerezo A, García M, Rodríguez-Nuevo A, Crosa-Bonell M, Enguix N, Peró A, et al. HMGA1 overexpression in adipose tissue impairs adipogenesis and prevents diet-induced obesity and insulin resistance. Sci Rep (2015) 5:14487. doi:10.1038/srep14487
34. Thanos D, Maniatis T. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell (1995) 83(7):1091–100. doi:10.1016/0092-8674(95)90136-1
35. Arcidiacono B, Iiritano S, Chiefari E, Brunetti FS, Gu G, Foti DP, et al. Cooperation between HMGA1, PDX-1, and MafA is essential for glucose-induced insulin transcription in pancreatic beta cells. Front Endocrinol (2014) 5:237. doi:10.3389/fendo.2014.00237
36. Thanos D, Maniatis T. The high mobility group protein I(Y) is required for NF-kappa B-dependent virus induction of the human IFN-beta gene. Cell (1992) 71(5):777–89. doi:10.1016/0092-8674(92)90554-P
37. Bloch M, Prock A, Paonessa F, Benz V, Bähr IN, Herbst L, et al. High-mobility group A1 protein: a new coregulator of peroxisome proliferator-activated receptor-γ-mediated transrepression in the vasculature. Circ Res (2012) 110(3):394–405. doi:10.1161/CIRCRESAHA.111.253658
38. Foti D, Iuliano R, Chiefari E, Brunetti A. A nucleoprotein complex containing Sp1, C/EBP beta, and HMGI-Y controls human insulin receptor gene transcription. Mol Cell Biol (2003) 23(8):2720–32. doi:10.1128/MCB.23.8.2720-2732.2003
39. Ji YS, Xu Q, Schmedtje JF Jr. Hypoxia induces high-mobility-group protein I(Y) and transcription of the cyclooxygenase-2 gene in human vascular endothelium. Circ Res (1998) 83(3):295–304. doi:10.1161/01.RES.83.3.295
40. Moussavi Nik SH, Newman M, Lardelli M. The response of HMGA1 to changes in oxygen availability is evolutionarily conserved. Exp Cell Res (2011) 317(11):1503–12. doi:10.1016/j.yexcr.2011.04.004
41. Ferrara N. Vascular endothelial growth factor. Arterioscler Thromb Vasc Biol (2009) 29(6):789–91. doi:10.1161/ATVBAHA.108.179663
42. Voros G, Maquoi E, Demeulemeester D, Clerx N, Collen D, Lijnen HR. Modulation of angiogenesis during adipose tissue development in murine models of obesity. Endocrinology (2005) 146(10):4545–54. doi:10.1210/en.2005-0532
43. Luk T, Malam Z, Marshall JC. Pre-B cell colony-enhancing factor (PBEF)/visfatin: a novel mediator of innate immunity. J Leukoc Biol (2008) 83(4):804–16. doi:10.1189/jlb.0807581
44. Segawa K, Fukuhara A, Hosogai N, Morita K, Okuno Y, Tanaka M, et al. Visfatin in adipocytes is upregulated by hypoxia through HIF-1alpha-dependent mechanism. Biochem Biophys Res Commun (2006) 349(3):875–82. doi:10.1016/j.bbrc.2006.07.083
45. Jiang MS, Lane MD. Sequential repression and activation of the CCAAT enhancer-binding protein-α (C/EBPα) gene during adipogenesis. Proc Natl Acad Sci USA (2000) 97(23):12519–23. doi:10.1073/pnas.220426097
46. Dunnett CW. A multiple comparison procedure for comparing several treatments with a control. J Am Stat Assoc (1955) 50:1096–121. doi:10.1080/01621459.1955.10501294
47. Zhang XM, Verdine GL. A small region in HMGI(Y) is critical for cooperation with NF-kappaB on DNA. J Biol Chem (1999) 274(29):20235–43. doi:10.1074/jbc.274.29.20235
48. Yun Z, Maecker HL, Johnson RS, Giaccia A. Inhibition of PPARg2 expression by the HIF-1- regulated gene DEC1/Stra13: a mechanism for regulation of adipogenesis by hypoxia. Dev Cell (2002) 2:331–41. doi:10.1016/S1534-5807(02)00131-4
49. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature (2008) 453:807–11. doi:10.1038/nature06905
50. Chen CA, Chang JT, Ho YF, Shyu AB. MiR-26 down-regulates TNF-α/NF-κB signaling and IL-6 expression by silencing HMGA1 and MALT1. Nucleic Acids Res (2016) 44(8):3772–87. doi:10.1093/nar/gkw205
51. Wobben R, Hüsecken Y, Lodewick C, Gibbert K, Fandrey J, Winning S. Role of hypoxia inducible factor-1α for interferon synthesis in mouse dendritic cells. Biol Chem (2013) 394(4):495–505. doi:10.1515/hsz-2012-0320
52. Carvajal IM, Baron RM, Perrella MA. High-mobility group-I/Y proteins: potential role in the pathophysiology of critical illnesses. Crit Care Med (2002) 30(1 Suppl):S36–42. doi:10.1097/00003246-200201001-00005
53. Bustin M. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol Cell Biol (1999) 19(8):5237–46. doi:10.1128/MCB.19.8.5237
54. Regazzetti C, Peraldi P, Grémeaux T, Najem-Lendom R, Ben-Sahra I, Cormont M, et al. Hypoxia decreases insulin signaling pathways in adipocytes. Diabetes (2009) 58(1):95–103. doi:10.2337/db08-0457
55. Jiang C, Qu A, Matsubara T, Chanturiya T, Jou W, Gavrilova O, et al. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes (2011) 60(10):2484–95. doi:10.2337/db11-0174
56. Chiefari E, Nevolo MT, Arcidiacono B, Maurizio E, Nocera A, Iiritano S, et al. HMGA1 is a novel downstream nuclear target of the insulin receptor signalling pathway. Sci Rep (2012) 2:251. doi:10.1038/srep00251
57. Iiritano S, Chiefari E, Ventura V, Arcidiacono B, Possidente K, Nocera A, et al. The HMGA1-IGF-I/IGFBP system: a novel pathway for modulating glucose uptake. Mol Endocrinol (2012) 26(9):1578–89. doi:10.1210/me.2011-1379
58. Chiefari E, Tanyolaç S, Paonessa F, Pullinger CR, Capula C, Iiritano S, et al. Functional variants of the HMGA1 gene and type 2 diabetes mellitus. JAMA (2011) 305(9):903–12. doi:10.1001/jama.2011.207
Keywords: hypoxia, HMGA1, HIF-1, obesity, Visfatin, VEGF, adipocytes, gene transcription regulation
Citation: Messineo S, Laria AE, Arcidiacono B, Chiefari E, Luque Huertas RM, Foti DP and Brunetti A (2016) Cooperation between HMGA1 and HIF-1 Contributes to Hypoxia-Induced VEGF and Visfatin Gene Expression in 3T3-L1 Adipocytes. Front. Endocrinol. 7:73. doi: 10.3389/fendo.2016.00073
Received: 08 March 2016; Accepted: 14 June 2016;
Published: 27 June 2016
Edited by:
Nicholas Michael Morton, University of Edinburgh, UKReviewed by:
Justin J. Rochford, University of Aberdeen, UKZoi Michailidou, University of Edinburgh, UK
Copyright: © 2016 Messineo, Laria, Arcidiacono, Chiefari, Luque Huertas, Foti and Brunetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela P. Foti, Zm90aUB1bmljei5pdA==;
Antonio Brunetti, YnJ1bmV0dGlAdW5pY3ouaXQ=
†These authors contributed equally to this work.