 Wen-Jun Shen
Wen-Jun Shen Salman Azhar1,2
Salman Azhar1,2 Fredric B. Kraemer
Fredric B. Kraemer- 1The Division of Endocrinology, Stanford University, Stanford, CA, USA
- 2Geriatric Research, Education and Clinical Center, Veterans Affairs Palo Alto Health Care System, Palo Alto, CA, USA
The adrenal gland is one of the prominent sites for steroid hormone synthesis. Lipoprotein-derived cholesterol esters (CEs) delivered via SR-B1 constitute the dominant source of cholesterol for steroidogenesis, particularly in rodents. Adrenocorticotropic hormone (ACTH) stimulates steroidogenesis through downstream actions on multiple components involved in steroidogenesis. Both acute and chronic ACTH treatments can modulate SR-B1 function, including its transcription, posttranscriptional stability, phosphorylation and dimerization status, as well as the interaction with other protein partners, all of which result in changes in the ability of SR-B1 to mediate HDL-CE uptake and the supply of cholesterol for conversion to steroids. Here, we provide a review of the recent findings on the regulation of adrenal SR-B1 function by ACTH.
The adrenal gland, in addition to the gonads, is one of the prominent sites where steroid hormones are synthesized (1–5). Cholesterol is the common precursor for steroidogenesis, which involves the contribution from multiple enzymes and requires the conversion of cholesterol to pregnenolone as the initial step of a multistep process. Pregnenolone is subsequently metabolized to produce various biologically active steroids in a tissue-specific manner. In general, this process is thought to involve five major steps: (1) cholesterol acquisition through de novo synthesis and/or uptake from lipoproteins and stored as cholesterol esters (CEs) in lipid droplets (LDs), (2) cholesterol mobilization from CEs that are stored in LDs, (3) trafficking of cholesterol to the cytochrome P450 side-chain cleavage enzyme (P450scc, CYP11A1) at the inner mitochondrial membrane (IMM), following cholesterol trafficking to the outer mitochondrial membrane (OMM), (4) production of pregnenolone by CYP11A1 through cleavage of the cholesterol side-chain, and (5) efflux of pregnenolone from the mitochondria to the endoplasmic reticulum (ER), where enzymes convert it into intermediates that shuttle between mitochondria and ER to produce progestins, estrogens, androgens, glucocorticoids, or mineralocorticoids in a tissue-specific manner (2, 5).
The adrenal gland is a compound endocrine gland composed of two developmentally unrelated tissues, an outer layer of adrenal cortex and an inner layer of adrenal medulla. The adrenal cortex is the site of steroid hormone synthesis and produces three classes of steroid hormones. These are glucocorticoids (cortisol and corticosterone), mineralocorticoids (aldosterone), and androgens [androstenedione and dehydroepiandrosterone (DHEA)]. The cells of the adrenal zona glomerulosa, which is the outermost layer of the adrenal cortex, synthesize aldosterone in response to angiotensin II (1, 2), whereas the cells of the adrenal cortical zona fasciculata–reticularis produce cortisol, corticosterone, or androgens [androstenedione and DHEA/dehydroepiandrostenedione sulfate (DHEAS)] in response to adrenocorticotropic hormone (ACTH) stimulation. Humans synthesize cortisol, but because CYP17 is poorly expressed in the zona fasciculata in rats and mice, consequently, corticosterone is the dominant glucocorticoid produced in rodents. The adrenal steroidogenic pathways are illustrated in Figure 1.
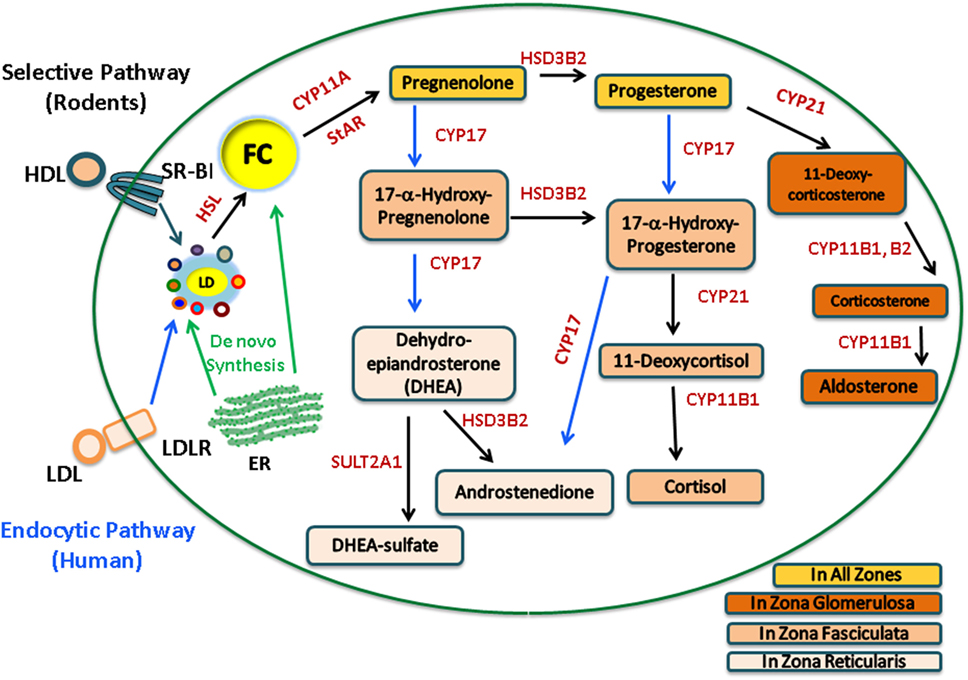
Figure 1. Human and rodent adrenal steroidogenic pathways. In the adrenal, upon stimulation, cholesterol esters (CE) from the LDL endocytic pathway (human) or the SR-B1 selective pathway (rodents) are hydrolyzed by hormone-sensitive lipase (HSL) to release free cholesterol (FC) as the common precursor for steroidogenesis. FC, which can also be synthesized de novo within the ER, traffics to the outer mitochondrial membrane and is then transported into the inner mitochondrial membrane by StAR, where it is cleaved by CYP11A to pregnenolone, a common precursor for all other steroid hormones. The adrenal cortex is the site of steroid hormone synthesis and different steroidogenic enzymes are expressed in cells located within different zones of the adrenal cortex, resulting in different classes of steroid hormones being released from different layers. The cells of the zona glomerulosa synthesize aldosterone, the cells of the zona fasciculata synthesize cortisol, and the cells of the zona reticularis produce androgens. In rodents, there is a very low level of CYP17; consequently, corticosterone is the dominant glucocorticoid produced.
All the steroid hormones synthesized within the adrenal cortex utilize cholesterol as the common precursor. For cells that produce polypeptide hormones, large amounts of mature hormones can be stored ready for rapid release; however, there is very little steroid hormone storage in steroidogenic cells. Therefore, upon stimulation, there is a rapid response from the steroidogenic cells to synthesize new steroids (3, 4), and with this a requirement for a constant supply of the precursor cholesterol to be converted to steroid hormones. The precursor cholesterol for steroidogenesis can be derived from a combination of sources (5–7): (1) de novo cellular cholesterol synthesis, (2) the mobilization of CEs stored in LDs, and (3) lipoprotein-derived CEs delivered through endocytic uptake, which is mediated by the LDL receptor or “selective” cellular uptake via the scavenger receptor, class B type 1 (SR-B1).
SR-B1 has been shown to be a HDL receptor and can mediate selective uptake of lipoprotein (HDL)-derived CEs both in vitro and in vivo (8–11). In the selective CE uptake pathway that is mediated by SR-B1, CE-rich lipoproteins bind on the cell surface and deliver the CEs from the hydrophobic core of the lipoproteins to the inside of the cells. The lipoprotein particles remain intact at the cell surface and can be further recycled to deliver more CEs to the cells (12). In contrast, CEs delivered via LDL receptor-mediated lipoprotein uptake are hydrolyzed by lysosomal acid lipase, releasing unesterified free cholesterol (FC) from lysosomes that traffics to the ER and plasma membrane (PM) and is then available to traffic to mitochondria (13, 14). CEs delivered via SR-B1 appear to be incorporated directly into LDs (15, 16) and must be hydrolyzed to FC before being used in steroidogenesis. Upon ACTH treatment, adrenal CE stores within LDs are rapidly depleted (17) through the action of hormone-sensitive lipase (HSL), the major neutral cholesteryl ester hydrolase expressed in the adrenal gland (18). This newly released FC from stored LDs is the preferred source of cholesterol. Following LD depletion, lipoprotein-derived CEs delivered via SR-B1 become the dominant source of cholesterol for steroidogenesis in rodents (19–21).
Adrenal fasciculata–reticularis cell steroidogenesis is under the regulation of tropic hormone ACTH and is subject to both acute (14, 22–25) and chronic regulation (14, 26–29). ACTH binds to its G protein-coupled receptors, leading to the activation of adenylate cyclase, which generates cAMP and activates cAMP-dependent protein kinase (PKA) (30–33). Stimulation of the cAMP–PKA signaling cascade exerts both acute and chronic effects on the regulation of steroid hormone production (Figure 2). On the other hand, angiotensin (AII) stimulates aldosterone biosynthesis in adrenal glomerulosa cells, and its actions are primarily mediated by the protein kinase C signaling cascade, whereas potassium can also stimulate aldosterone production through Ca2+-calmodulin-dependent kinase (34). Both acute and chronic ACTH treatments can modulate SR-B1, including its expression levels as well as its phosphorylation status, dimerization, and the interaction with other protein partners, all of which result in changes of SR-B1 function. Here, we aim at providing a review of the most recent findings relevant to these aspects.
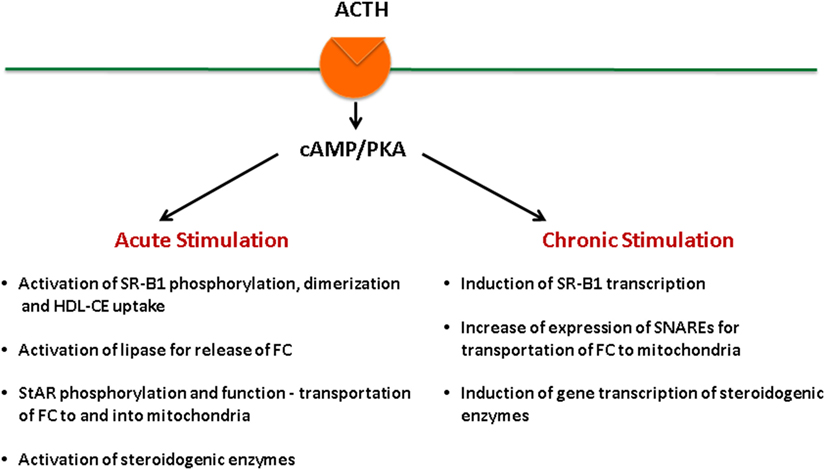
Figure 2. ACTH regulation of steroidogenesis in the adrenal.
ACTH Stimulation Regulates the Expression of SR-B1
Initial studies established a functional correlation between SR-B1 expression, HDL-CE uptake, and the ability of steroidogenic cells to produce steroid hormones (8, 35–40). Also, in both lower vertebrates (i.e., turtle) and the fruit fly, there is an increase in SR-B1 expression that correlates with the peak cholesterol flux required during their developmental stages (41). Follow-up studies revealed that the bulk of the cholesterol for steroidogenesis is provided from the selective uptake pathway, which is mediated by SR-B1.
Both rodent and human adrenals express exceptionally high levels of SR-B1; indeed, the highest expression level of SR-B1 per gram of tissue has been reported for rodent adrenals (42, 43). In vivo treatment of rats and mice and in vitro treatment of cultured rodent adrenocortical cells with ACTH increased SR-B1 expression both at the mRNA level and that of SR-B1 protein (8, 44). In one of our recent studies, the level of SR-B1 protein in rat adrenals shows a trend for increased SR-B1 expression as early as 1 h after treatment with ACTH (45). Similar stimulation of SR-B1 expression and regulation by ACTH was also demonstrated for cultured human adrenal cells (8, 43, 46, 47). Indeed, low levels of SR-B1 mRNA expression seen in normal adrenal tissue adjacent to adenomas causing Cushing’s syndrome, where plasma ACTH levels are reduced, is consistent with the notion that the regulation of human SR-B1 is possibly similar to that reported for rodents (43).
Much of the regulation of SR-B1 expression is at the level of transcriptional control. The promoter region of both human and rat SR-B1 contains binding sites for steroidogenic factor-1 (SF-1) (48), which is one of the major transcription factors involved in cAMP regulation of the SR-B1 gene (49). In addition, the rat promoter has two sterol-responsive elements (SREs) that can bind sterol-responsive element-binding protein (SREBP)-1a and regulate SR-B1 gene expression in response to altered intracellular sterol levels (50).
Further studies demonstrated that the promoter region of SR-B1 contains sites that can bind both positive and negative regulators. For the expression of SR-B1 in liver and adipose tissue, transcription factors, such as the liver X receptors α and β (LXRα and LXRβ) and the peroxisome proliferator-activated receptor α (PPARα), have been shown to positively regulate the expression of the human SR-B1 gene in response to oxysterols and fibrates, respectively (51, 52). Other positive regulators include the liver receptor homolog 1 (LRH-1) (53) as well as the estrogen receptors α and β (ERα and ERβ), which bind to three different estrogen-responsive elements on the rat SR-B1 promoter and regulate its activity in response to estrogens (54). The negative regulators of SR-B1 include the nuclear receptor dorsal-sensitive sex adrenal hypoplasia congenital critical region on the X chromosome gene 1 (DAX-1), a protein that plays an important role in adrenal development (49), the Yin Yang 1 (YY1) transcription factor, which represses the activity of the SR-B1 promoter by inhibiting the binding of SREBP-1a (55), and the pregnane X receptor, which represses the human SR-B1 promoter activity in response to the pregnane X receptor agonists rifampicin and lithocholic acid (56). Most of the binding sites for these transcription factors reside within the 2.2 kb proximal 5′-flanking region of the SR-B1 promoter.
In steroidogenic tissues, SR-B1 expression was shown to be upregulated by ACTH (8, 44, 46, 57–59). As mentioned above, SF-1 was shown to mediate the regulation of SR-B1 gene expression through the cAMP–PKA pathway by binding to the promoter region of SR-B1. The SF-1 binding site in the human SR-B1 promoter (5′-CCAAGGCT-3′) resides 77 bp upstream of the transcription start site, and the SF-1 binding site (5′-TCAAGGCC-3′) in the rat SR-B1 promoter is located at −645 bp upstream of the translation start site. These two sites share 75% identity (48, 49). Both the human and rat SR-B1 promoters were shown to be active in mouse adrenocortical tumor Y1 cells, and mutagenesis analysis confirmed the involvement of SF-1 in regulating their promoter activity. The SF-1 binding motif in the rat SR-B1 promoter was shown to be involved in both basal and cAMP-induced regulation of SR-B1 gene expression. Further analysis of the functional domains in SF-1 revealed that both amino acids 448–461 and phosphorylation at Ser430 by PKA are involved in regulating the binding to the consensus sequence in the rat SR-B1 promoter. A recent report showed that when glucocorticoid levels are elevated, SR-B1 expression can also be inhibited by feedback regulation by glucocorticoid (60). In corticosterone-insufficient corticotrophin-releasing hormone knockout Crh (−/−) mice, there is an increase of SR-B1 mRNA levels in adrenal, and oral administration of corticosterone inhibited SR-B1 gene expression. Further studies reveal that the glucocorticoid receptor (GR) can suppress SR-B1 promoter activity. The region between −201 and −62 of the human SR-B1 promoter was shown to contain putative binding sites for transcriptional repressors that are involved in mediating glucocorticoid regulation of SR-B1 expression. However, examination into the mechanism of suppression suggested that GR suppression of SR-B1 in adrenal cells occurs through an indirect mechanism since no direct binding of GR to the SR-B1 promoter was observed. This was the first report showing that by suppressing SR-B1-mediated HDL cholesterol uptake, steroidogenic tissues maintain steroid hormone homeostasis when the endogenous levels of glucocorticoids are elevated.
Recently, in the search for the cellular and molecular mechanisms involved in the regulation of SR-B1 expression and function in steroidogenic cells, we demonstrated that two microRNAs, miRNA-125a and miRNA-455, can bind to specific sites in the 3′ UTR of SR-B1 mRNA and regulate the expression of SR-B1 (61). The expression of miRNA-125a and miRNA-455 is detected in steroidogenic tissue/cells, including adrenal, primary ovarian granulosa cells, and model Leydig cell lines. Both ACTH and cAMP downregulate the expression of miRNA-125a and miRNA-455. When either miRNA-125a or miRNA-455 is overexpressed or inhibited, the amount of SR-B1 protein expressed on the cell surface is decreased or increased, respectively, leading to SR-B1-mediated selective HDL uptake and SR-B1-supported steroid hormone synthesis being inhibited and stimulated, respectively. Therefore, our findings suggest that miRNA-125a and miRNA-455, in response to ACTH stimulation, act as SR-B1 attenuators to negatively regulate SR-B1 expression and SR-B1-mediated selective delivery of lipoprotein cholesterol in steroidogenic cells and, consequently, inhibition of SR-B1-supported steroidogenesis.
Modulation of SR-B1 Protein Function by ACTH
SR-B1 facilitates HDL-CE selective uptake in two separate independent steps: binding of the lipid-rich lipoprotein to the extracellular domain (ECD) of SR-B1 and the delivery of the CEs from the hydrophobic core of the lipoprotein to the PM (62, 63). A specialized cell surface structure, termed “microvillar channels,” was reported to be induced by SR-B1 and shown to facilitate selective lipid transfer to inside the cell (64–67). Studies using electron microscopy demonstrated the presence of microvillar membrane domains in rat ovarian luteal, testicular Leydig, and adrenocortical cells. These domains form channels at the PM and various lipoproteins, including HDL, get trapped within the channels. Immunostaining using specific antibodies for SR-B1 revealed that SR-B1 is preferentially localized on these domains (65, 66). SR-B1 was also shown to be able to facilitate the formation of specific lipid rafts, hence change the properties of the PM and, in turn, affect the flux of free cholesterol (67). These lipid rafts were indicated to be necessary for the formation of the membrane microvillar channels, which are considered a trap for HDL particles for enhancing the efficiency of the selective uptake of HDL-CE. Interestingly, expression of SR-B1 and microvilli are under hormonal regulation; ACTH treatment increases, whereas dexamethasone treatment decreases, SR-B1 expression. Furthermore, the steady-state levels of adrenal microvilli are dictated by SR-B1. In response to SR-B1 deficiency, mouse adrenal microvilli become disorganized, and microvillar channels show a disrupted appearance along with substantially reduced binding of HDL particles to the cell surface (64–67).
In addition, experiments utilizing mutational analysis of SR-B1 or chimeras of CD36/SR-B1 have demonstrated that high affinity binding of lipoproteins to the ECD of SR-B1 is important, but not sufficient to mediate efficient lipid uptake (68). However, at the same time the ECD of SR-B1 does influence the efficient transfer of lipid by SR-B1 (69). This dichotomy is highlighted by the findings that some chemicals can increase lipoprotein binding to SR-B1 while actually blocking lipid transfer (70). Further studies show that the dominant characteristic of lipoprotein binding to SR-B1 involves protein–protein interactions between ligand and receptor. Many of the CE donors (HDL, apoA-I/phospholipid bilayer disks, and lipid-free apoA-I) for SR-B1 all share class A amphipathic helices that could be the structural feature to which SR-B1 is binding (68, 69). In addition, SR-B1 occurs as a multimeric complex with itself or other membrane proteins on the cell surface to facilitate lipid transfer, and the ECD of SR-B1 is essential for efficient CE transfer. ACTH has been shown to induce changes in the oligomeric status as well as protein interaction of SR-B1 and hence modulates SR-B1 protein function.
ACTH Stimulation Modulates the Oligomerization of SR-B1
In response to ACTH stimulation, SR-B1 changes its oligomeric status to facilitate CE uptake (for simplicity, here, we use the term dimerization to include the multiple forms of the SR-B1 protein; i.e., dimers and higher order oligomers). In one of the earliest direct demonstrations of protein–protein interactions involving SR-B1, SR-B1 was shown to exist as homodimers in PMs isolated from rat adrenals stimulated with 17α-ethinyl estradiol (70). Subsequently, dimeric and higher order oligomeric forms of SR-B1 were shown to exist in all cells and tissues that display HDL-CE selective uptake activity (65, 71, 72). In normal rat adrenal tissue, SR-B1 exists primarily in a monomeric form with some dimer formation. Upon ACTH stimulation, there is a significant increase in the dimerization/oligomerization of SR-B1 along with increased selective CE uptake. On the other hand, dexamethasone-mediated suppression of ACTH leads to dramatic loss of SR-B1, SR-B1 dimers/oligomers, and HDL-CE selective uptake activity. When combined with the substantial architectural alterations of the cell surface as related to microvillar formation, these findings indicate that SR-B1 dimer/oligomer formation appears to have significant implications for the expression of the functional properties of SR-B1.
Studies in adrenal cells and other steroidogenic tissues have established a strong correlation between the levels of SR-B1 dimers and enhanced HDL-CE selective uptake activity. Co-immunoprecipitation with differentially epitope-tagged SR-B1s further confirmed that SR-B1 can exist as homodimers (71). In addition, in both native steroidogenic cell lines (endogenous) and in a heterologous insect cell overexpressing SR-B1, dimers/oligomers of SR-B1 were seen when cross-linking agents were added to the cell lysates (65). When cellular extracts from SR-B1 transfected HEK-293 cells or ACTH-treated Y1-BS1 cells were analyzed by size-exclusion chromatography and sucrose density centrifugation, a significant portion of SR-B1 eluted at peaks that correlate with the size of dimeric and oligomeric forms of SR-B1. Immunoelectron microscopy was used as an independent means for confirming the homodimerization of SR-B1. For these experiments, differentially epitope-tagged-SR-B1 proteins were co-expressed in HEK-293 cells, and the epitope-tagged proteins were subsequently immunostained and identified using two differently sized gold particles. The observed mixing and clustering of gold particles suggested that the proteins were localized to the same regions of the cell and that many of the gold particles were in extremely close proximity, i.e., within a distance for protein–protein interactions, as detected by fluorescent resonance energy transfer (FRET) technique. Similar results were obtained when Y1-BS1 mouse adrenocortical cells were transfected with differentially epitope-tagged-SR-B1 constructs. Interestingly, transfection of Y1-BS1 cells with SR-B1 in these experiments resulted in substantial architectural changes with the formation of microvillar structures. Gold-labeled secondary antibodies localized SR-B1 to these sites and revealed substantial dimer formation of this protein – shown by close contact between gold particles (71, 72).
Further investigations concentrated on the contribution of the cysteine residues in the ECD of SR-B1 either independently or in cooperation with the C-terminal domain on SR-B1 dimerization. SR-B1 contains a total of eight cysteine (C) residues (C21, C251, C280, C321, C323, C334, C384, and C470) and six of them are located in the ECD. Mutagenesis studies showed that C280, C321, C323, and C334 residues in the ECD are necessary for preserving normal SR-B1 (HDL) binding activity, selective CE uptake, and/or cell surface expression. Interestingly, mutation of any of these four cysteine residues to serine resulted in a robust induction of SR-B1 dimer formation, but, in contrast to normal SR-B1, these SR-B1 mutants lost their ability to mediate HDL-CE selective uptake. These results indicate that these cysteine residues are most likely essential for optimal HDL binding and selective CE uptake (73).
ACTH Stimulation Regulates SR-B1 Interaction with Accessory Proteins
Adrenocorticotropic hormone treatment activates the cAMP–PKA signaling cascade, and we have recently shown that the expression of salt-inducible kinase 1 (SIK1), a serine/threonine kinase that belongs to the stress- and energy-sensing AMPK family of kinases, is also rapidly induced in Y1 adrenal cells in response to ACTH via the cAMP–PKA signaling cascade. Previously, it had been suggested that an increased level of SIK1 expression inhibits adrenal steroidogenesis by repressing the cAMP-dependent transcription of steroidogenic proteins, CYP11A1 and StAR, by attenuating CREB transcriptional activity (74). In contrast, we showed that SIK1 stimulates adrenal steroidogenesis by modulating the selective HDL-CE transport activity of SR-B1. Overexpression of SIK1 increases cAMP-stimulated and SR-B1-mediated selective HDL-BODIPY-CE uptake in cell lines without impacting SR-B1 protein levels, whereas knockdown of SIK1 attenuated cAMP-stimulated selective HDL-BODIPY-CE uptake. SIK1 forms a complex with SR-B1 by interacting with its cytoplasmic C-terminal domain, and in vitro kinase activity measurements indicate that SIK1 can phosphorylate the C-terminal domain of SR-B1. Among potential phosphorylation sites, SIK1-catalyzed phosphorylation of Ser496 is critical for SIK1 stimulation of the selective CE transport activity of SR-B1. Mutational studies further demonstrated that both the intact catalytic activity of SIK1 and its PKA-catalyzed phosphorylation are essential for SIK1 stimulation of SR-B1 activity. Finally, overexpression of SIK1 caused time-dependent increases in SR-B1-mediated and HDL-supported steroid production in Y1 cells; however, these effects were lost with knockdown of SR-B1. It should be noted that, as opposed to these stimulatory effects of SIK1 on SR-B1 function, we confirmed that SIK1 does suppress CREB activity, which could contribute to the inhibition of steroidogenesis under some conditions. Taken together, these studies establish a role for SIK1 in the positive regulation of selective HDL-CE transport function of SR-B1 and steroidogenesis and suggest a potential mechanism for SIK1 signaling in modulating SR-B1-mediated selective CE uptake and associated steroidogenesis.
Many studies have also indicated that accessory proteins are crucial for the proper cellular expression of SR-B1 and SR-B1-mediated HDL-CE transport as well as other functions (75–83). For example, it has been shown that PDZK1/NHERF3 regulates hepatic SR-B1 stability and steady-state protein levels. Interestingly, PDZK1/NHERF3 is neither expressed nor essential for SR-B1 abundance or its cellular localization in steroidogenic cells of the adrenal gland, ovary, and testis (77). Recently, we have shown that two other NHERF family members, NHERF1 and NHERF2, negatively regulate the expression and function of SR-B1 in steroidogenic cells of the adrenal and gonads in response to ACTH (84). Specifically, we showed that ACTH treatment decreases NHERF1 and NHERF2 protein levels in rat adrenals and increases SR-B1 function. Co-immunoprecipitation, colocalization, bimolecular fluorescence complementation, and mutational analysis all indicated that NHERF1 and NHERF2 form complexes with SR-B1 protein and, as a result, inhibit SR-B1-mediated selective CE transport and steroidogenesis. Moreover, we demonstrated that the structural components required for NHERF1/2 to interact with SR-B1 included an intact COOH-terminal PDZ recognition motif (EAKL) in SR-B1 as well as the PDZ1 or PDZ2 domain of NHERF1, the PDZ2 domain of NHERF2, or the MERM domains of NHERF1/2. The de novo synthesis of SR-B1 was also inhibited by both NHERF1 and NHERF2 (84). In contrast to NHERF1 and NHERF2, NHERF4 had no effect on selective HDL-CE uptake or steroidogenesis. Altogether, these experiments demonstrated that NHERF1 and NHERF2 bind SR-B1 and negatively regulate SR-B1 expression, selective CE transport, and steroidogenesis (84).
ACTH and Regulation of SR-B1 Under Pathophysiological Conditions
Since ACTH tightly regulates SR-B1 gene transcription, it should be expected that any pathophysiological conditions that affect ACTH levels would also impact SR-B1 gene expression in an identical manner. However, there is the possibility that altered SR-B1 function may represent an adaptive response to cope with stressful conditions. Indeed, results from studies of mice that underwent chronic psychosocial stress exhibited an exaggerated adrenal corticosterone response along with elevated SR-B1 protein levels (85). On the other hand, since adrenal cholesterol uptake is required for the production of anti-inflammatory glucocorticoids, gene deletion of functional SR-B1 in adrenals results in impaired steroid synthesis (86). Other studies using SR-B1 knockout mice demonstrated that SR-B1 can protect mice against endotoxemia (87). There is an uncontrolled robust inflammatory cytokine response in the SR-B1 deficient animals, and they exhibit higher lethality when challenged with LPS-doses that induce endotoxic shock. Furthermore, these animals also exhibit a dysregulated adrenal glucocorticoid-mediated stress response to fasting. In addition, fasting-induced elevated levels of serum ACTH were the consequence of adrenal glucocorticoid insufficiency in SR-B1 knockout mice (86). Finally, when SR-B1 was only selectively deleted in adrenocortical cells in a tissue-specific manner, the animals had impaired rates of glucocorticoid secretion in response to stress, especially when they were subjected to an endotoxin challenge; these animals with SR-B1 ablated only in adrenocortical cells showed enhanced local and systematic inflammatory response, blunted activation of atrophy genes in skeletal muscle, and have a high incidence of mortality (88). In the setting of acute stress, where the release of corticosterone in rodents or cortisol in humans peaks rapidly and declines quickly, it is likely that posttranslational regulation of SR-B1, such as protein phosphorylation and dimerization, could be more important in the regulation of steroidogenesis than transcriptional control. In contrast, under chronic stress conditions, mechanisms regulating SR-B1 at the transcriptional level and/or through miRNAs would appear to be more relevant.
Some of the adrenal steroid hormones, i.e., glucocorticoids, display robust daily variations in circulation under the circadian control by ACTH, and their rhythmic activity is considered to play important roles in whole body health and disease (89, 90). Ablation of one of the genes involved in circadian control, BMAL1, results in loss of circadian regulation of glucocorticoids. The BMAL1-deficient animals showed impaired response to ACTH regulation of adrenal function and downregulation of genes involved in cholesterol transport, such as StAR and LDLR, in adrenals (89). There is a well-accepted concept of the adrenal clock, with evidence of differential responses between males and females (91). While studies have shown a circadian variation in the expression of several genes involved in regulating the steroid production, no publications have specifically examined whether SR-B1 expression and/or function displays a circadian rhythm, but this is likely in view of the known regulation of SR-B1 by ACTH.
In summary, through binding to its G protein-coupled receptors, leading to the activation of adenylate cyclase, which generates cAMP and activates PKA, ACTH exerts tight regulation of SR-B1 function in the adrenal at three different levels. As illustrated in Figure 3, increased cAMP–PKA signaling can increase the phosphorylation of transcription factors, such as SF1, and stimulate promoter activity of SR-B1. ACTH can increase the expression of miRNA-125 and miRNA-455, which can bind to the 3′ UTR of SR-B1 and result in suppressed expression and function of SR-B1 protein. Increased cAMP–PKA signaling can induce oligomerization of SR-B1 as well as stimulate phosphorylation of SIK1, which, in turn, increases the phosphorylation of SR-B1 and results in increased function of SR-B1 protein. Meanwhile, ACTH can also induce the expression of NHERF1 and NHERF2, both of which can bind to SR-B1 and negatively regulate SR-B1 function. Altered SR-B1 function may represent an adaptive response to cope with stressful conditions, as demonstrated by ablation of SR-B1 resulting in disturbed anti-inflammatory glucocorticoid homeostasis and impaired steroid synthesis.
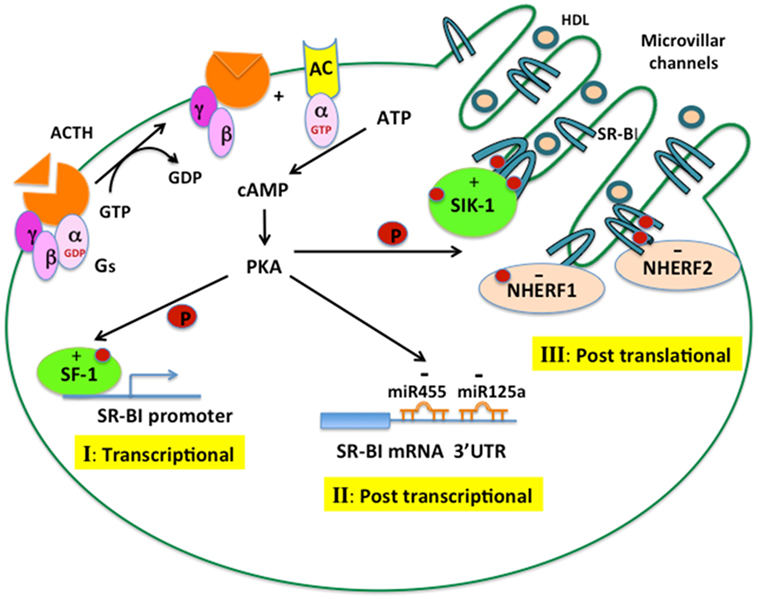
Figure 3. ACTH regulation of SR-B1 in the adrenal. ACTH binds to its G protein-coupled receptor, leading to the activation of adenylate cyclase, which generates cAMP and activates cAMP-dependent protein kinase (PKA). The cAMP–PKA signaling cascade can regulate SR-B1 expression and function at different levels. (I) Transcriptional control: PKA increases the phosphorylation of transcription factors, such as SF1, leading to increased promoter activity of SR-B1. (II) Posttranscriptional control: PKA increases the expression of miRNA-125a and miRNA-455, which can bind to the 3′ UTR of SR-B1 mRNA and negatively regulate SR-B1 expression. (III) Posttranslational control: PKA induces the oligomerization of SR-B1 and stimulates an interaction with SIK1, leading to phosphorylation of SR-B1, both events resulting in increased SR-B1 protein function. PKA can also increase the interaction of SR-B1 with NHERF1 and NHERF2, which negatively regulate SR-B1 function.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Office of Research and Development, Medical Service, Department of Veterans Affairs and by the National Institutes of Health, NHLBI, grant 2R01HL33881.
References
1. McKenna T, Fearon U, Clarke D, Cunningham S. A critical review of the origin and control of adrenal androgens. Baillieres Clin Obstet Gynecol (1997) 11:229–48. doi:10.1016/S0950-3552(97)80035-1
2. Miller W. Androgen biosynthesis from cholesterol to DHEA. Mol Cell Endocrinol (2002) 198:7–14. doi:10.1016/S0303-7207(02)00363-5
3. Davis WW, Garren LD. On the mechanism of action of adrenocorticotropic hormone. The inhibitory site of cycloheximide in the pathway of steroid biosynthesis. J Biol Chem (1968) 243:5153–7.
4. Garren LD, Ney RL, Davis WW. Studies on the role of protein synthesis in the regulation of corticosterone production by adrenocorticotropic hormone in vivo. Proc Natl Acad Sci U S A (1965) 53:1443–50. doi:10.1073/pnas.53.6.1443
5. Andersen JM, Dietschy JM. Relative importance of high and low density lipoproteins in the regulation of cholesterol synthesis in the adrenal gland, ovary, and testis of the rat. J Biol Chem (1978) 253:9024–32.
6. Gwynne JT, Strauss JF III. The role of lipoproteins in steroidogenesis and cholesterol metabolism in steroidogenic glands. Endocr Rev (1982) 3:299–329. doi:10.1210/edrv-3-3-299
7. Kraemer FB. Adrenal cholesterol utilization. Mol Cell Endocrinol (2007) 26(5–266):42–5. doi:10.1016/j.mce.2006.12.001
8. Rigotti A, Edelmann E, Seifert P, Iqbal S, DeMattos R, Temel R, et al. Regulation by adrenocorticotropic hormone of the in vivo expression of scavenger receptor class B type I (SR-BI), a high density lipoprotein receptor, in steroidogenic cells of the murine adrenal gland. J Biol Chem (1996) 271:33545–9. doi:10.1074/jbc.271.52.33545
9. Connelly M, Klein S, Azhar S, Abumrad N. Comparison of class B scavenger receptors, CD36, and scavenger receptor BI (SR-BI), shows that both receptors mediate high-density lipoprotein-cholesteryl ester selective uptake bu SR-BI exhibits a unique enhancement of cholesteryl ester uptake. J Biol Chem (1999) 274:41–7. doi:10.1074/jbc.274.1.41
10. Stangl H, Cao G, Wyne KL, Hobbs HH. Scavenger receptor, class B, type I-dependent stimulation of cholesterol esterification by high density lipoproteins, low density lipoproteins, and nonlipoprotein cholesterol. J Biol Chem (1998) 273:31002–8. doi:10.1074/jbc.273.47.31002
11. Swarnakar S, Temel RE, Connelly MA, Azhar S, Williams DL. Scavenger receptor class B, type I, mediates selective uptake of low density lipoprotein cholesteryl ester. J Biol Chem (1999) 274:29733–9. doi:10.1074/jbc.274.42.29733
12. Simpson E, Lauber M, Demeter M, Means G, Mahendroo M, Kilgore M, et al. Regulation of expression of the genes encoding steroidogenic enzymes in the ovary. J Steroid Biochem Mol Biol (1992) 41:409–13. doi:10.1016/0960-0760(92)90366-Q
13. Faust JR, Goldstein JL, Brown MS. Receptor-mediated uptake of low density lipoprotein and utilization of its cholesterol for steroid synthesis in cultured mouse adrenal cells. J Biol Chem (1977) 252:4861–71.
14. Miller WL, Bose HS. Early steps in steroidogenesis: intracellular cholesterol trafficking thematic review series: genetics of human lipid diseases. J Lipid Res (2011) 52:2111–35. doi:10.1194/jlr.R016675
15. Connelly MA, Kellner-Weibel G, Rothblat GH, Williams DL. SR-BI-directed HDL-cholesteryl ester hydrolysis. J Lipid Res (2003) 44:331–41. doi:10.1194/jlr.M200186-JLR200
16. Kraemer FB, Shen WJ, Harada K, Patel S, Osuga J, Ishibashi S, et al. Hormone-sensitive lipase is required for high-density lipoprotein cholesteryl ester-supported adrenal steroidogenesis. Mol Endocrinol (2004) 18:549–57. doi:10.1210/me.2003-0179
17. Vahouny GV, Chanderbhan R, Hinds R, Hodges VA, Treadwell CR. ACTH-induced hydrolysis of cholesteryl esters in rat adrenal cells. J Lipid Res (1978) 19:570–7.
18. Kraemer FB, Shen WJ, Natu V, Patel S, Osuga J, Ishibashi S, et al. Adrenal neutral cholesteryl hydrolase: identification, subcellular distribution and sex differences. Endocrinology (2002) 143:801–6. doi:10.1210/endo.143.3.8693
19. Kraemer FB, Shen WJ, Patel S, Osuga J, Ishibashi S, Azhar S. The LDL receptor is not necessary for acute adrenal steroidogenesis in mouse adrenocortical cells. Am J Physiol Endocrinol Metab (2007) 292:E408–12. doi:10.1152/ajpendo.00428.2006
20. Verschoor-Klootwyk AH, Verchoor L, Azhar S, Reaven GM. Role of exogenous cholesterol in regulation of adrenal steroidogenesis in the rat. J Biol Chem (1982) 257:7666–71.
21. Xie C, Richardson JA, Turley SD, Dietschy JM. Cholesterol substrate pools and steroid hormone levels are normal in the face of mutational inactivation of NPC1 protein. J Lipid Res (2006) 47:953–63. doi:10.1194/jlr.M500534-JLR200
22. Pon LA, Hartigan JA, Orme-Johnson NR. Acute ACTH regulation of adrenal corticosteroid biosynthesis. Rapid accumulation of a phosphoprotein. J Biol Chem (1986) 261:13309–16.
23. Pon LA, Orme-Johnson NR. Acute stimulation of corpus luteum cells by gonadotrophin or adenosine 3′,5′-monophosphate causes accumulation of a phosphoprotein concurrent with acceleration of steroid synthesis. Endocrinology (1988) 123:1942–8. doi:10.1210/endo-123-4-1942
24. Epstein L, Orme-Johnson N. Regulation of steroid hormone biosynthesis: identification of precursors of a phosphoprotein targeted to the mitochondrion in stimulated rat adrenal cortex cells. J Biol Chem (1991) 266:19739–45.
25. Stocco D, Clark B. Regulation of the acute production of steroids in steroidogenic cells. Endocr Rev (1996) 17:221–44. doi:10.1210/er.17.3.221
26. Miller WL. Molecular biology of steroid hormone synthesis. Endocr Rev (1988) 9:295–318. doi:10.1210/edrv-9-3-295
27. Simpson ER, Waterman MR. Regulation of the synthesis of steroidogenic enzymes in adrenal cortical cells by ACTH. Annu Rev Physiol (1988) 50:427–40. doi:10.1146/annurev.ph.50.030188.002235
28. Payne AH, Youngblood GL, Sha L, Burgos-Trinidad M, Hammond SH. Hormonal regulation of steroidogenic enzyme gene expression in Leydig cells. J Steroid Biochem Mol Biol (1992) 43:895–906. doi:10.1016/0960-0760(92)90317-C
29. LaVoie H, King S. Transcriptional regulation of steroidogenic genes: STARD1, CYP11A1 and HSD3B. Exp Biol Med (Maywood) (2009) 234:880–907. doi:10.3181/0903-MR-97
30. JM M. The role of cyclic AMP in gonadal arteroidogenesis. Biol Reprod (1976) 14:30–53. doi:10.1095/biolreprod14.1.30
31. Simpson E, Waternan M. Regulation by ACTH of steroid hormone biosynthesis in the adrenal cortex. Can J Biochem Cell Biol (1983) 61:692–707. doi:10.1139/o83-088
32. Sanorn B, Heindel J, Robinson G. The role of cyclic nucleotides in the reproductive processes. Ann Rev Physiol (1980) 42:37–57. doi:10.1146/annurev.ph.42.030180.000345
33. Strauss J, Golos T, Silavin S, Soto E, Takagi K. Involvement of cyclic AMP in the functions of granulosa and luteal cells: regulation of steroidogenesis. Prog Clin Biol Res (1988) 267:177–200.
34. Spat A, Hunyady L. Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev (2004) 84:489–539. doi:10.1152/physrev.00030.2003
35. Reaven E, Spicher M, Azhar S. Microvillar channels: a unique plasma membrane compartment for concentrating lipoproteins on the surface of rat adrenal cortical cells. J Lipid Res (1989) 30:1551–60.
36. Plump AS, Erickson SK, Weng W, Partin JS, Breslow JL, Williams DL. Apolipoprotein A-I is required for cholesteryl ester accumulation in steroidogenic cells and for normal adrenal steroid production. J Clin Invest (1996) 97:2660–71. doi:10.1172/JCI118716
37. Landschulz K, Pathak R, Rigotti A, Krieger M, Hobb H. Regulation of scavenger receptor, class B, type I, a high density lipoprotein receptor, in liver and steroidogenic tissues of the rat. J Clin Invest (1996) 98:984–5. doi:10.1172/JCI118883
38. Azhar S, Nomoto A, Leers-Sucheta S, Reaven E. Simultaneous induction of an HDL receptor protein (SR-BI) and the selective uptake of HDL-cholesteryl esters in a physiologically relevant steroiodgenic cell model. J Lipid Res (1998) 39:1616–28.
39. Gwynne JT, Hughes T, Hess B. Characterization of high density lipoprotein binding activity in rat adrenocortical cells. J Lipid Res (1984) 25:1059–71.
40. Gwynne JT, Hess B. The role of high density lipoproteins in rat adrenal cholesterol metabolism and steroidogenesis. J Biol Chem (1980) 255:10875–83.
41. Duggan A, Marie R, Callard I. Expression of SR-BI (scavenger receptor class B type I) in turtle (Chrysemys picta) tissues and other nonmammalian vertebrates. J Exp Zool (2002) 292:430–4. doi:10.1002/jez.10067
42. Acton S, Rigotti A, Landschultz K, Xu S, Hobs H, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science (1996) 271:518–20. doi:10.1126/science.271.5248.518
43. Liu J, Voutilainen R, Heikkila P, Kahri AI. Ribonucleic acid expression of the CLA-1 gene, a human homolog to mouse high density lipoprotein receptor SR-BI, in human adrenal tumors and cultured adrenal cells. J Clin Endocrinol Metab (1997) 82:2522–7. doi:10.1210/jcem.82.6.3968
44. Wang N, Weng W, Breslow JL, Tall AR. Scavenger receptor BI (SR-BI) is up-regulated in adrenal gland in apolipoprotein A-I and hepatic lipase knock-out mice as a response to depletion of cholesterol stores. In vivo evidence that SR-BI is a functional high density lipoprotein receptor under feedback control. J Biol Chem (1996) 271:21001–4.
45. Hu Z, Hu J, Shen WJ, Kraemer FB, Azhar S. A novel role of salt-inducible kinase 1 (SIK1) in the post-translational regulation of scavenger receptor class B type 1 activity. Biochemistry (2015) 54:6917–30. doi:10.1021/acs.biochem.5b00147
46. Sun Y, Wang N, Tall AR. Regulation of adrenal scavenger receptor-BI expression by ACTH and cellular cholesterol pools. J Lipid Res (1999) 40:1799–805.
47. Martin G, Pilon A, Albert C, Valle M, Hum DW, Fruchart JC, et al. Comparison of expression and regulation of the high-density lipoprotein receptor SR-BI and the low-density lipoprotein receptor in human adrenocortical carcinoma NCI-H295 cells. Eur J Biochem (1999) 261:481–91. doi:10.1046/j.1432-1327.1999.00296.x
48. Cao G, Garcia CK, Wyne KL, Schultz RA, Parker KL, Hobbs HH. Structure and localization of the human gene encoding SR-BI/CLA-1. Evidence for transcriptional control by steroidogenic factor 1. J Biol Chem (1997) 272:33068–76. doi:10.1074/jbc.272.52.33068
49. Lopez D, Sandhoff TW, McLean MP. Steroidogenic factor-1 mediates cyclic 3′,5′-adenosine monophosphate regulation of the high density lipoprotein receptor. Endocrinology (1999) 140:3034–44. doi:10.1210/en.140.7.3034
50. Lopez D, McLean MP. Sterol regulatory element-binding protein-1a binds to cis elements in the promoter of the rat high density lipoprotein receptor SR-BI gene. Endocrinology (1999) 140:5669–81. doi:10.1210/endo.140.12.7220
51. Malerod L, Juvet LK, Hanssen-Bauer A, Eskild W, Berg T. Oxysterol-activated LXRalpha/RXR induces hSR-BI-promoter activity in hepatoma cells and preadipocytes. Biochem Biophys Res Commun (2002) 299:916–23. doi:10.1016/S0006-291X(02)02760-2
52. Lopez D, McLean MP. Activation of the rat scavenger receptor class B type I gene by PPARalpha. Mol Cell Endocrinol (2006) 251:67–77. doi:10.1016/j.mce.2006.02.011
53. Schoonjans K, Annicotte JS, Huby T, Botrugno OA, Fayard E, Ueda Y, et al. Liver receptor homolog 1 controls the expression of the scavenger receptor class B type I. EMBO Rep (2002) 3:1181–7. doi:10.1093/embo-reports/kvf238
54. Lopez D, Sanchez MD, Shea-Eaton W, McLean MP. Estrogen activates the high-density lipoprotein receptor gene via binding to estrogen response elements and interaction with sterol regulatory element binding protein-1A. Endocrinology (2002) 143:2155–68. doi:10.1210/endo.143.6.8855
55. Shea-Eaton W, Lopez D, McLean MP. Yin yang 1 protein negatively regulates high-density lipoprotein receptor gene transcription by disrupting binding of sterol regulatory element binding protein to the sterol regulatory element. Endocrinology (2001) 142:49–58. doi:10.1210/endo.142.4.8075
56. Sporstol M, Tapia G, Malerod L, Mousavi SA, Berg T. Pregnane X receptor-agonists down-regulate hepatic ATP-binding cassette transporter A1 and scavenger receptor class B type I. Biochem Biophys Res Commun (2005) 331:1533–41. doi:10.1016/j.bbrc.2005.04.071
57. Temel RE, Trigatti B, DeMattos RB, Azhar S, Krieger M, Williams DL. Scavenger receptor class B, type I (SR-BI) is the major route for the delivery of high density lipoprotein cholesterol to the steroidogenic pathway in cultured mouse adrenocortical cells. Proc Natl Acad Sci U S A (1997) 94:13600–5. doi:10.1073/pnas.94.25.13600
58. Cao G, Zhao L, Stangl H, Hasegawa T, Richardson J, Parker K, et al. Developmental and hormonal regulation of murine scavenger receptor, class B, type 1. Mol Endocrinol (1999) 13:1460–73. doi:10.1210/mend.13.9.0346
59. Vieira-van Bruggen D, Kalkman I, van Gent T, van Tol A, Jansen H. Induction of adrenal scavenger receptor BI and increased high density lipoprotein-cholesteryl ether uptake by in vivo inhibition of hepatic lipase. J Biol Chem (1998) 273:32038–41. doi:10.1074/jbc.273.48.32038
60. Mavridou S, Venihaki M, Rassouli O, Tsatsanis C, Kardassis D. Feedback inhibition of human scavenger receptor class B type I gene expression by glucocorticoid in adrenal and ovarian cells. Endocrinology (2010) 151:3214–24. doi:10.1210/en.2009-1302
61. Hu Z, Shen W, Kraemer F, Azhar S. MicroRNAs 125a and 455 repress lipoprotein-supported steroidogenesis by targeting scavenger receptor class B type I in steroidogenic cells. Mol Cell Biol (2012) 32:5035–45. doi:10.1128/MCB.01002-12
62. Rigotti A, Miettinen H, Krieger M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr Rev (2003) 24:357–87. doi:10.1210/er.2001-0037
63. Gu X, Trigatti B, Xu S, Acton S, Babitt J, Krieger M. The efficient cellular uptake of high density lipoprotein lipids via scavenger receptor class B type I requires not only receptor-mediated surface binding but also receptor-specific lipid transfer mediated by its extracellular domain. J Biol Chem (1998) 273:26338–48. doi:10.1074/jbc.273.41.26338
64. Reaven E, Zhan L, Nomoto A, Leers-Sucheta S, Azhar S. Expression and microvillar localization of scavenger receptor class B, type I (SR-BI) and selective cholesteryl ester uptake in Leydig cells from rat testis. J Lipid Res (2000) 41:343–56.
65. Reaven E, Leers-Sucheta S, Nomoto A, Azhar S. Expression of scavenger receptor class B type 1 (SR-BI) promotes microvillar channel formation and selective cholesteryl ester transport in a heterologous reconstituted system. Proc Natl Acad Sci U S A (2001) 98:1613. doi:10.1073/pnas.98.4.1613
66. Williams DL, Wong JS, Hamilton RL. SR-BI is required for microvillar channel formation and the localization of HDL particles to the surface of adrenocortical cells in vivo. J Lipid Res (2002) 43:544–9.
67. Peng Y, Akmentin W, Connelly M, Lund-Katz S, Phillips M, Williams D. Scavenger receptor BI (SR-BI) clustered on microvillar extensions suggests that this plasma membrane domain is a way station for cholesterol trafficking between cells and high-density lipoprotein. Mol Biol Cell (2004) 15:384–96. doi:10.1091/mbc.E03-06-0445
68. Bocharov A, Baranova I, Vishnyakova T, Remaley A, Csako G, Thomas F, et al. Targeting of scavenger receptor class B type I by synthetic amphipathic alpha-helical-containing peptides blocks lipopolysaccharide (LPS) uptake and LPS-induced pro-inflammatory cytokine responses in THP-1 monocyte cells. J Biol Chem (2004) 279:36072–82. doi:10.1074/jbc.M314264200
69. Thuahnai S, Lund-Katz S, Anantharamaiah G, Williams D, Phillips M. A quantitative analysis of apolipoprotein binding to SR-BI: multiple binding sites for lipid-free and lipid-associated apolipoproteins. J Lipid Res (2003) 44:1132–42. doi:10.1194/jlr.M200429-JLR200
70. Azhar S, Nomoto A, Reaven E. Hormonal regulation of adrenal microvillar channel formation. J Lipid Res (2002) 43:861–71.
71. Reaven E, Nomoto A, Cortez Y, Azhar S. Consequences of over-expression of rat scavenger receptor, SR-BI, in an adrenal cell model. Nutr Metab (Lond) (2006) 3:43. doi:10.1186/1743-7075-3-43
72. Reaven E, Cortez Y, Leers-Sucheta S, Nomoto A, Azhar S. Dimerization of the scavenger receptor class B type I: formation, function, and localization in diverse cells and tissues. J Lipid Res (2004) 45:513–28. doi:10.1194/jlr.M300370-JLR200
73. Hu J, Zhang Z, Shen W, Nomoto A, Azhar S. Differential roles of cysteine residues in the cellular trafficking, dimerization, and function of the high-density lipoprotein receptor, SR-BI. Biochemistry (2011) 50:10860–75. doi:10.1021/bi201264y
74. Spiga F, Liu Y, Aguilera G, Lightman SL. Temporal effect of adrenocorticotrophic hormone on adrenal glucocorticoid steroidogenesis: involvement of the transducer of regulated cyclic AMP-response element-binding protein activity. J Neuroendocrinol (2011) 23:136–42. doi:10.1111/j.1365-2826.2010.02096.x
75. Ikemoto M, Arai H, Feng D, Tanaka K, Aoki J, Dohmae N, et al. Identification of a PDZ-domain-containing protein that interacts with the scavenger receptor class B type I. Proc Natl Acad Sci U S A (2000) 97:6538–43. doi:10.1073/pnas.100114397
76. Silver D. A carboxyl-terminal PDZ-interacting domain of scavenger receptor B, type I is essential for cell surface expression in liver. J Biol Chem (2002) 277:34042–7. doi:10.1074/jbc.M206584200
77. Kocher O, Yesilaltay A, Cirovic C, Pal R, Rigotti A, Krieger M. Targeted disruption of the PDZK1 gene in mice causes tissue-specific depletion of the high density lipoprotein receptor scavenger receptor class B type I and altered lipoprotein metabolism. J Biol Chem (2003) 278:52820–5. doi:10.1074/jbc.M310482200
78. Komori H, Arai H, Kashima T, Huby T, Kita T, Ueda Y. Coexpression of CLA-1 and human PDZK1 in murine liver modulates HDL cholesterol metabolism. Arterioscler Thromb Vasc Biol (2008) 28:1298–303. doi:10.1161/ATVBAHA.108.165845
79. Robichaud J, Francis G, Vance D. A role for hepatic scavenger receptor class B, type I in decreasing high density lipoprotein levels in mice that lack phosphatidylethanolamine N-methyltransferase. J Biol Chem (2008) 283:35496–506. doi:10.1074/jbc.M807433200
80. Zhu W, Saddar S, Seetharam D, Chambliss K, Longoria C, Silver D, et al. The scavenger receptor class B type I adaptor protein PDZK1 maintains endothelial monolayer integrity. Circ Res (2008) 102(4):480–7. doi:10.1161/CIRCRESAHA.107.159079
81. Fenske S, Yesilaltay A, Pal R, Daniels K, Barker C, Quiñones V, et al. Normal hepatic cell surface localization of the high density lipoprotein receptor, scavenger receptor class B, type I, depends on all four PDZ domains of PDZK1. J Biol Chem (2009) 284:5797–806. doi:10.1074/jbc.M808211200
82. Eyre N, Drummer H, Beard M. The SR-BI partner PDZK1 facilitates hepatitis C virus entry. PLoS Pathog (2010) 6:e1001130. doi:10.1371/journal.ppat.1001130
83. Kocher O, Krieger M. Role of the adaptor protein PDZK1 in controlling the HDL receptor SR-BI. Curr Opin Lipidol (2009) 20:236–41. doi:10.1097/MOL.0b013e32832aee82
84. Hu Z, Hu J, Zhang Z, Shen W, Yun C, Berlot C, et al. Regulation of expression and function of scavenger receptor class B, type I (SR-BI) by Na+/H+ exchanger regulatory factors (NHERFs). J Biol Chem (2013) 2013(288):11416–35. doi:10.1074/jbc.M112.437368
85. Fuchsl AM, Uschold-Schmidt N, Reber SO. Chronic psychosocial stress in male mice causes an up-regulation of scavenger receptor class B type 1 protein in the adrenal glands. Stress (2013) 16:461–8. doi:10.3109/10253890.2013.793303
86. Hoekstra M, Meurs I, Koenders M, Out R, Hildebrand RB, Kruijt JK, et al. Absence of HDL cholesteryl ester uptake in mice via SR-BI impairs an adequate adrenal glucocorticoid-mediated stress response to fasting. J Lipid Res (2008) 49:738–45. doi:10.1194/jlr.M700475-JLR200
87. Cai L, Ji A, de Beer FC, Tannock LR, van der Westhuyzen DR. SR-BI protects against endotoxemia in mice through its roles in glucocorticoid production and hepatic clearance. J Clin Invest (2008) 118:364–75. doi:10.1172/JCI31539
88. Gilibert S, Galle-Treger L, Moreau M, Saint-Charles F, Costa S, Ballaire R, et al. Adrenocortical scavenger receptor class B type I deficiency exacerbates endotoxic shock and precipitates sepsis-induced mortality in mice. J Immunol (2014) 193:817–26. doi:10.4049/jimmunol.1303164
89. Leliavski A, Shostak A, Husse J, Oster H. Impaired glucocorticoid production and response to stress in Arntl-deficient male mice. Endocrinology (2014) 155:133–42. doi:10.1210/en.2013-1531
90. Kil IS, Lee SK, Ryu KW, Woo HA, Hu MC, Bae SH, et al. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol Cell (2012) 46:584–94. doi:10.1016/j.molcel.2012.05.030
Keywords: adrenal, cholesterol, ACTH, SR-B1
Citation: Shen W-J, Azhar S and Kraemer FB (2016) ACTH Regulation of Adrenal SR-B1. Front. Endocrinol. 7:42. doi: 10.3389/fendo.2016.00042
Received: 29 February 2016; Accepted: 01 May 2016;
Published: 13 May 2016
Edited by:
Antoine Martinez, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Tamas Kozicz, Radboud University Medical Center, NetherlandsFrédéric Gachon, Nestlé Institute of Health Sciences, Switzerland
Copyright: © 2016 Shen, Azhar and Kraemer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fredric B. Kraemer, ZmJrJiN4MDAwNDA7c3RhbmZvcmQuZWR1