 Azizun Nessa
Azizun Nessa Sofia A. Rahman
Sofia A. Rahman Khalid Hussain
Khalid Hussain- Genetics and Genomic Medicine Programme, Department of Paediatric Endocrinology, UCL Institute of Child Health, Great Ormond Street Hospital for Children NHS, London, UK
Under normal physiological conditions, pancreatic β-cells secrete insulin to maintain fasting blood glucose levels in the range 3.5–5.5 mmol/L. In hyperinsulinemic hypoglycemia (HH), this precise regulation of insulin secretion is perturbed so that insulin continues to be secreted in the presence of hypoglycemia. HH may be due to genetic causes (congenital) or secondary to certain risk factors. The molecular mechanisms leading to HH involve defects in the key genes regulating insulin secretion from the β-cells. At this moment, in time genetic abnormalities in nine genes (ABCC8, KCNJ11, GCK, SCHAD, GLUD1, SLC16A1, HNF1A, HNF4A, and UCP2) have been described that lead to the congenital forms of HH. Perinatal stress, intrauterine growth retardation, maternal diabetes mellitus, and a large number of developmental syndromes are also associated with HH in the neonatal period. In older children and adult’s insulinoma, non-insulinoma pancreatogenous hypoglycemia syndrome and post bariatric surgery are recognized causes of HH. This review article will focus mainly on describing the molecular mechanisms that lead to unregulated insulin secretion.
Introduction
Hyperinsulinemic hypoglycemia (HH) is biochemically characterized by the unregulated secretion of insulin from the pancreatic β-cells in the presence of low blood glucose levels. Under normal physiological conditions, β-cells synthesize, store, and secrete insulin in a precisely controlled manner so that the fasting blood glucose level is kept within a narrow range of 3.5–5.5 mmol/L (1).
The unregulated insulin secretion promotes glucose uptake into the skeletal muscles, liver, and adipose tissue, exacerbating the hypoglycemia further. Simultaneously, glucose production (via gluconeogenesis and glycogenolysis), lipolysis, and ketogenesis are all inhibited. In addition to the actions of insulin in inhibiting glucose production, there are also defects in the glucose counter regulatory hormones such as cortisol and glucagon (2–4). The lack of glucose (the brains primary source of fuel) accompanied by the deprivation of alternative substrates in the form of ketone bodies and lactate increase the risk of brain damage in these patients (5).
The genetic mechanisms that lead to HH involve defects in the key pathways regulating insulin secretion from pancreatic β-cells. These genetic defects perturb the normal physiological mechanisms of glucose-regulated insulin secretion, leading to inappropriate and unregulated insulin secretion. The unregulated insulin secretion leads to severe hypoglycaemia. The gene defects, which lead to congenital HH can be broadly categorised into two groups, channelopathies and metabolopathies.
The most severe forms of HH occur in the neonatal period (however, it can also present in the infancy, childhood, and in adults) and are usually congenital HH (CHH). HH may also occur due to secondary causes and these are summarized in Table 1 (6). The genetic causes of CHH are listed in Table 1.

Table 1. An outline of different causes of HH.
The most severe forms of CHH are due to defects in the genes (ABCC8 and KCNJ11), which encode for the sulfonylurea receptor protein (SUR1) and inwardly rectifying potassium channels (Kir6.2) proteins, respectively, of the potassium sensitive KATP channel (7, 8). These defects are referred to as channelopathies (9). The coassembly of four SUR1 and four Kir6.2 subunits forms a single ATP-sensitive potassium channels (KATP) (Figure 1). The second category, metabolopathies, constitutes the majority of the other genes known to cause CHH.
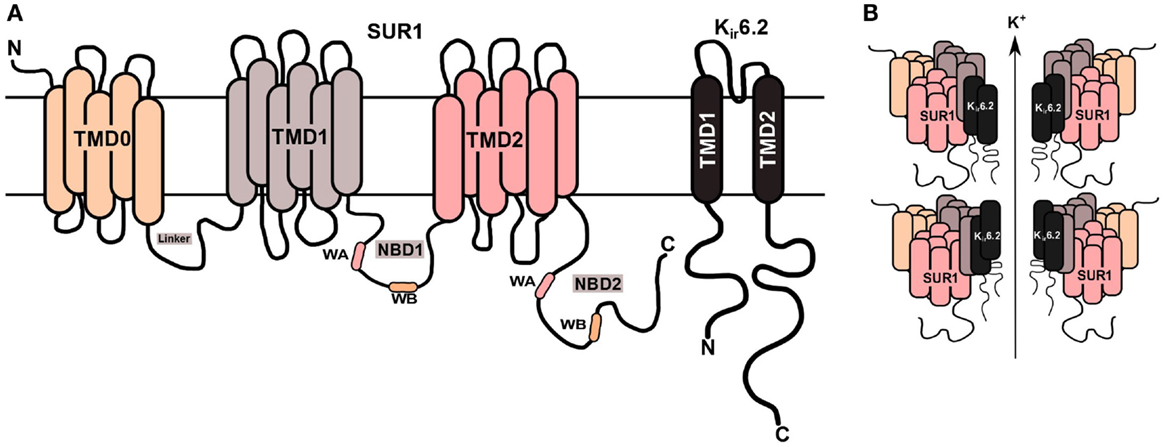
Figure 1. KATP channel structure on β-cell membrane. (A) The SUR1 subunit is made up of three transmembrane domains (TMD0, TMD1, and TMD2) and two nucleotide-binding domains (NBD1/NBD2), which face the cytoplasm. The NBD’s harbor the Walker A (WA) and Walker B (WB) motifs. Kir6.2 is the pore-forming subunit, containing two membrane-spanning domains, connected by an extracellular pore-forming region and cytoplasmic –NH2 and –COOH terminal domains. (B) Illustration of the predicted octameric structure of KATP channels, comprising four Kir6.2 and four SUR1 proteins.
In this review, we first review in detail the molecular basis of the ATP sensitive potassium KATP channel function, and then outline the molecular pathways that lead to CHH due to defects in these channels and second we describe the biochemical causes of HH due to defects in the non-KATP channel genes.
Channelopathies: Background to ABC Proteins
The transportation of ions, molecules, and various other proteins across a membrane is of paramount importance to the functioning of all living cells. A simple example is of the E. coli cell which transports ~106 glucose molecules per second into the cell in order to meet its physiological requirements (10). The movement of these essential (and non-essential) molecules is mediated by membrane-bound transporter proteins. There are 48 ATP-binding cassette (ABC) transporters present in humans (11). In the human genome, there are seven sub-groups of the ABC transporter family, involved in a diverse range of functions. This includes multidrug resistance, cholesterol and lipid transport, mitochondrial iron homeostasis, and also the ATP-dependent regulation of ion channels, which include the cystic fibrosis transmembrane conductance regulator and the sulfonylurea urea receptor. Many disease phenotypes arise as a result of mutations in the ABC genes, such as cystic fibrosis, diabetes, and CHH (12). The ABCC8 gene stands for ATP-Binding Cassette, Sub-Family C, Member 8, and it codes for the SUR1 protein, which is a major example of an ABC protein.
All ABC proteins including SUR1 harbor two major domains, which consists of the transmembrane domains (TMD) and the nucleotide-binding domains (NBD). The TMD, situated in the cell membrane, is primarily composed of α-cell-helices. The NBD’s, however, are always found within the cytoplasm of the cell. A distinguishing feature of ABC proteins is the presence of highly conserved sequence motifs, such as the Walker A (WA) and Walker B (WB) motif. The former is a characteristic phosphate-binding loop, and the latter is Magnesium [Mg2+-binding site (13)]. Another striking feature is the Q-loop, which is positioned in between the WA and WB motif, known to associate with the gamma phosphate through a water bond. These three motifs combine together to form the NBD.
Other characteristic feature of ABC proteins is the “switch” region, which contains a histidine loop (H-Loop). It has been hypothesized that it polarizes the water molecule for hydrolysis. In ABC proteins, the variation arises within the TMD’s, which can vary among the different ABC proteins. This provides the membrane protein with its ability to sense changes in different nucleotides or other ions and compounds (12).
The ABCC8 Gene
The ABCC8 gene is located within the human genome on chromosome 11p15.1. This gene has 39 exons (14), translating into either a 1581 or 1582 amino acid protein (15), which has a molecular weight of 176.9 kDa (16). The three transmembrane domains in SUR1 are TMD0, TMD1, and TMD2 (17). There are also two cytosolic nucleotide-binding domains (NBD1 and NBD2) within a single SUR1 transmembrane protein. SUR1 can combine ATP hydrolysis to transport substrates, however within the KATP channel complex it is primarily role is not to transport substrates; instead it regulates the activity of the Kir6.2 subunit. SUR1 also confers the KATP channel with its sensitivity and response to sulfonylureas such as glibenclamide (18) and channel activators such a diazoxide, which is commonly used to treat medically responsive CHH (19).
SUR1: The Walker A, Walker B, and Nucleotide-Binding Domain
The WA and WB motifs contain the consensus sequence for nucleotide hydrolysis, making them one of the most prominent features of SUR1 proteins (20). A flexible loop structure is formed at the WA motif, which contains the sequence Gly–X–Gly–X–Gly. This is followed by a lysine residue, which is predicted to bind to the α-cell-phosphate of ATP. A hydrophobic β sheet is formed at the WB motif, which has an adjacent aspartic acid residue, and this has been postulated to interact with the Mg2+ ion of MgATP (13). It has been predicted that the WA and WB motifs in KATP channels are able to interact with the phosphoryl group, and then co-ordinate the Mg2+ in the MgATP complex (21). There is a third motif, which situated in between the WA and WB, and this region has been denoted as a “linker,” which transduces the effect of nucleotide binding and hydrolysis (22).
The WA and WB sequence motif is contained in both NBD1 and NBD2 of SUR1 (20), and this motif can sense the levels of intracellular nucleotides within pancreatic β-cells. The key nucleotide is ATP, which is produced in large quantities after a meal when cell metabolism is high. ATP has an inhibitory effect on KATP channels, although MgATP can have the opposite effect. The hydrolysis of MgATP is believed to involve a conformational change at the NBD’s, which in turn generates two nucleotide binding sites known an ATP-binding site 1 (ABS1) and 2 (ABS2), believed to mediate the hydrolysis of MgATP. Azido-ATP labelling experiments demonstrate that the binding of nucleotides to NBD2 stabilizes the binding of ATP to NBD1 (23). It has been shown that NBD1 and NBD2 can be coimmunoprecipitated (24). A soluble NBD1–GFP construct can be taken to the cell membrane by TMD2–NBD2 construct when they co-expressed in insect cells (25).
The KCNJ11 Gene
In the vertebrate genome, there are two genes coding for Kir proteins, this includes the KCNJ8 and KCNJ11 genes which encode for the Kir6.1 and Kir6.2 proteins, respectively. The KCNJ11 gene is also located on chromosome 11p15.1. However, in contrast to the ABCC8 gene, the KCNJ11 gene only contains 1 exon, which translates into a 390 amino acid protein with a molecular weight of 43.5 kDa (14). A distinctive feature of Kir proteins is that they enable positive current to flow inwards into the cell, despite there being equal concentrations of K+ ions either side of the membrane. Kir6.2, however, has its own unique properties, the first being that it is not a strong inward rectifier (26); nevertheless its origins are also from a large super-family of Kir proteins, namely Kir1–Kir7 (27, 28). In the KATP channel complex, the Kir6.2 proteins functions as the pore of the channel, allowing K+ ions to flow through. Kir6.2 is composed of two transmembrane domains, which are linked by an extracellular pore-forming region. The –NH2 and –COOH terminal domains are located within the cytoplasmic side of the protein.
Histological Characterization of CHH
Three histological forms of CHH currently exist; focal CHH, diffuse CHH, and atypical CHH. Focal CHH is characterized by pathological pancreatic β-cells localizing to one or more specific locations in the pancreas. In contrast, in diffuse CHH, there are abnormal islet cells throughout the pancreatic tissue (29, 30). Although clinical presentation of both these types of CHH appears to be similar, their molecular mechanisms are different.
Atypical CHH are usually described as cases that are histologically difficult to categorize and are on general due to morphological mosaicism in a portion of the pancreas.
Focal CHH
The focal lesion has abnormal islet cells, whereas the surrounding pancreatic areas seem histologically normal, but islets are reported to have small nuclei. The non-focal area islet cells have a reduction in cytoplasmic mass as well as diminished proinsulin production (30–33).
Focal CHH appears to be a result of two events; (1) a paternally inherited KATP channel mutation and (2) the loss of the corresponding maternal allele located within the focal area. This results in an imbalance in the expression of various imprinted genes. These include the expression of insulin-like growth factor 2 (IGF-2) (34) and tumor suppressor genes H19 and cyclin-dependent kinase inhibitor 1C (CDKN1C). These genes have been shown to promote β-cell hyperplasia and are expressed paternally and maternally, respectively.
In a few cases, paternal allele duplication of chromosome 11 has also reported to cause focal CHH (35). However, the majority of focal CHH due to heterozygous paternally inherited mutation in the ABCC8 or KCNJ11 gene account for almost 30–40% of all CHH cases (36). The imbalanced 11p15 region interrupts pancreatic β-cell gene expression, which leads to focal islet hyperplasia.
Focal CHH is usually confirmed by a fluorine-18 dihydroxyphenylalanine-positron emission tomography (18F-DOPA-PET) scanning. This scan can identify the location of one or more focal lesions with diagnostic accuracy (37). Surgical resection of the lesion usually resolves HH.
Diffuse CHH
The diffuse pancreas affects all the pancreatic β-cells, but islet morphology is preserved (32). Affected islets cells appear with large hyperchromatic nuclei and an abundant mass of cytoplasm (29–33). Patients with diffuse CHH either have a homozygous recessive or a compound heterozygous mutation in their KATP channel genes. This form of CHH accounts for 60–70% of all CHH cases. Patients are mainly medically unresponsive, and hence, require a “near total pancreatectomy.” However, this often leads to patients having life-long diabetes and exocrine insufficiency.
Atypical CHH
Histologically, atypical CHH are categorized when the pancreatic morphology neither fits the focal nor diffuse CHH category (34, 38). The islets have been described to be enlarged or shrunken, and atypical CHH still remains poorly defined. Variability in management of atypical CHH occurs because some cases have been cured with a surgical resection while others also required medical therapy. 18F-DOPA-PET scanning usually used to recognize between focal and diffuse CHH has failed to identify atypical CHH (34).
To date, no associated mutations have been reported for atypical CHH, with the exception of one patient with an ABCC8 nonsense mutation (p.Gln54X) (38). Other studies have described the heterogeneous expression of hexokinase 1 of the pancreatic β-cells in atypical pathology (39, 40). Furthermore, a recent study has also implicated an abnormal postprandial incretin response in two cases of atypical CHH of unknown genetic cause (40).
KATP Channels and Insulin Secretion
In 1983, Noma and colleagues identified K+ currents from tissue extracted from heart muscle cells, when they were induced with metabolic poisons or under hypoxic conditions (41). During the period of 1983–1988, there was a vast amount of research done on understanding the basic electrophysiological properties of KATP channels. As a result, the expression of KATP channels has been observed in many tissues, including cardiac muscle, skeletal muscle, smooth muscle, pancreatic β-cells, and neurons (41–45). KATP channel currents have now also been identified in the rough endoplasmic reticulum of rat hepatocytes (46).
However, it was not until the mid-1990s that recombinant in vitro functional work had advanced in the field of KATP channels. It was soon realized that the lone expression of Kir proteins was not enough to generate KATP channel currents in cells. This lead to the discovery of the SUR protein and the functional reconstitution of KATP channels (14). The coexpression of the proteins leads to the channel being sensitive to nucleotides and sulfonylureas.
Several studies carried out in isolated pancreatic β-cells have shown the functional significance of KATP channels as they play a major role in glucose homeostasis. These studies have used electrophysiological and radioactive techniques to elucidate the effect of sulfonylureas, potassium channel openers, and nucleotides on pancreatic β-cells (47–52).
The key regulators of KATP channels are intracellular nucleotides such as ATP and ADP (Figure 2). This has a direct link to cellular metabolism as the production and consumption of ATP has a direct impact on the activity of KATP channels. A rise in the intracellular concentration of ATP changes the physical conformation of KATP channels from an open to a closed state. The closure of KATP channels in the β-cell membrane changes the resting membrane potential (−70 mV) from being hyperpolarized to being depolarized (−50 mV), which in turn triggers the activation of voltage-gated calcium channels (53). The influx of Ca2+ ions into the β-cell enhances the exocytosis of insulin, which ultimately aims to restore blood glucose levels (54).
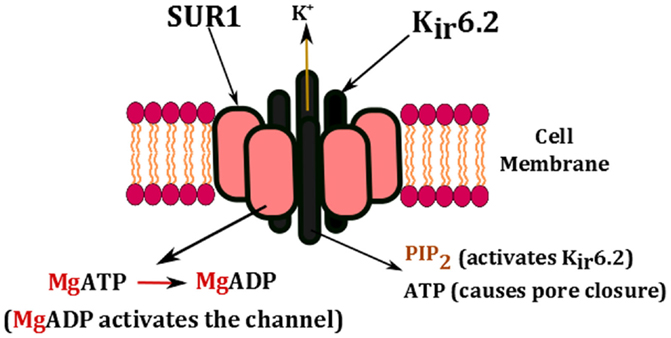
Figure 2. Nucleotide regulation of KATP channels. The channel is activated by the presence of PIP2 and the conversion of MgATP to MgADP. High concentrations of ATP block the pore and cause channel closure.
Gating Mechanisms in KATP Channels
The gating properties of KATP channels are affected by many different ligands. ATP has the ability to block the channel both in the presence and absence of Mg2+ ions, whereas PIP2 can activate the channel (Figure 2) (55–57). It has been suggested that there may be two gates in the permeation pathway. It has been observed that there is in fact ligand-independent gating, which indicates that the gate may be in the selectivity filter. Experimental observations suggest that KATP channels may harbor two independent types of gates in the permeation pathway (58). This includes:
(1) Ligand independent: fast gating, controlled by changes in membrane potential, this suggests that there is a selectivity filter, something which allows K+ through irrespective of all metabolic changes.
(2) Ligand dependent: slow gating, modulated by membrane PIP2 and nucleotides.
Experiments studying the kinetics of KATP channels reveal that un-liganded subunits are quite unstable. This means that subunits, which are not bound by either ATP or PIP2, are unstable, but once bound, they are mutually excluded. Hence, the subunits will be bound by one ligand or another at any one time.
Wild type (WT) KATP channel subunits are bound to PIP2 in the absence of ATP, which is why the channels are in the open state around ~90% of the time (59). But for the channel to be active, all four subunits must be in the open state. However, binding of ATP to the un-liganded subunit will trap it into the closed state. In WT subunits, the association constant for ATP is around ~10 μM. In energized cells, the concentration of ATP is two to three magnitudes higher; therefore, it is likely that each WT subunit is bound by ATP around ~90% of the time. Locking any one subunit in the closed conformation is enough to induce channel closure (60, 61). So apart from extreme conditions, the probability of the channel being in the open state is <0.14, which means that without the stimulatory effects of Mg2+ nucleotides the channel will almost always remain closed. PIP2 has the effect of “pulling” the channels to open, whereas ATP does the opposite and tries to “push” the channels to close.
Previous studies have shown that ATP destabilizes the channel open state and overall it enhances channel closure (62–65). In earlier studies, it was understood that the interaction of one ATP molecule with the open state resulted in channel closure. However, there are four Kir6.2 subunits in every KATP channel complex, therefore there are four ATP inhibitory binding sites and so up to four ATP molecules can bind to the open state. The binding of four ATP molecules can either work together in concert to induce channel closure or they can act independently (66). To this date, the relationship between the number of ATP molecules bound and the closure of the Kir6.2 pore is not well understood. Hence, it is debatable on whether the KATP channel, which is an octameric complex, acts under a concerted gating mechanism using all four subunits, or whether there are transitional steps involving each subunit.
Nucleotide Regulation of KATP Channels
The changing concentrations of ATP, ADP, GTP, and GDP regulate the opening and closing of the KATP channels. Adenine nucleotides, such as ATP and ADP, are able to interact with Mg2+ ions in a stimulatory manner; however, in the absence of Mg2+ ions, the non-hydrolytic binding of ATP takes precedence over KATP channels and promotes channel closure. Mg2+ ions are one of the most vital elements in the human body; hence, they are present at all times inside cells (67). Therefore, in order for the KATP channels to close, there must be significantly higher levels of ATP than MgADP.
MgADP can stimulate channel activity by direct binding, or by the binding and hydrolysis of MgATP (68). This can be seen in Figure 2. ATP binds to the cytosolic domain of Kir6.2. In a study by Craig et al. (66), they examined the mechanism of inhibition by ATP, and they achieved this by removing Mg2+ from the solutions, which enables them to study ATP interactions in isolation from nucleotides (66). The single channel kinetics of KATP channels show bursts of short openings and closings, which are separated by long, non-conducting interbursts intervals. Once ATP is added however, it causes a notable reduction in bursts duration, and a significantly longer duration of the closed states. Hence, ATP interacts with all these states, and ATP ends the burst state and aids the start of the long closed state.
Until 1997, it was widely accepted that the prime location for ATP inhibition was on the SUR1 subunit of the KATP channel complex, possibly within the NBD’s (69). However, the discovery that a truncated Kir6.2 protein could produce a homomeric channel (70), and function similar to a KATP channel, made it clear that the ATP inhibition actually affected the Kir6.2 subunit.
KATP Channel Mutations Causing HH
Inactivating mutations in any region of the ABCC8 and KCNJ11 genes account for the vast majority of mutations known to date. Mutations in these genes cause the most severe form of CHH that is typically medically unresponsive to therapy with diazoxide. The condition presents in the neonatal period with severe hypoglycemia, which is difficult to control with oral feeds and will require the infusion of concentrated intravenous dextrose. Both recessive and dominant mutations can have a major negative impact on many aspects of KATP channel function, regulation, and processing. Ultimately, this leads to persistent depolarization of the pancreatic β-cell membrane, which leads to unregulated insulin secretion (71). A recessive mutation observed in >90% of Ashkenazi Jewish population is p.Phe1388del (ABCC8) (9), which is known to cause severe CHH (14, 15). The p.Val187Asp and p.Glu1506Lys mutations in ABCC8 are largely found in the Finnish populations, which are also known to cause CHH. A schematic representation of known mechanisms of KATP channel defects has been shown in Figure 3. This section will describe some of these known mechanisms.
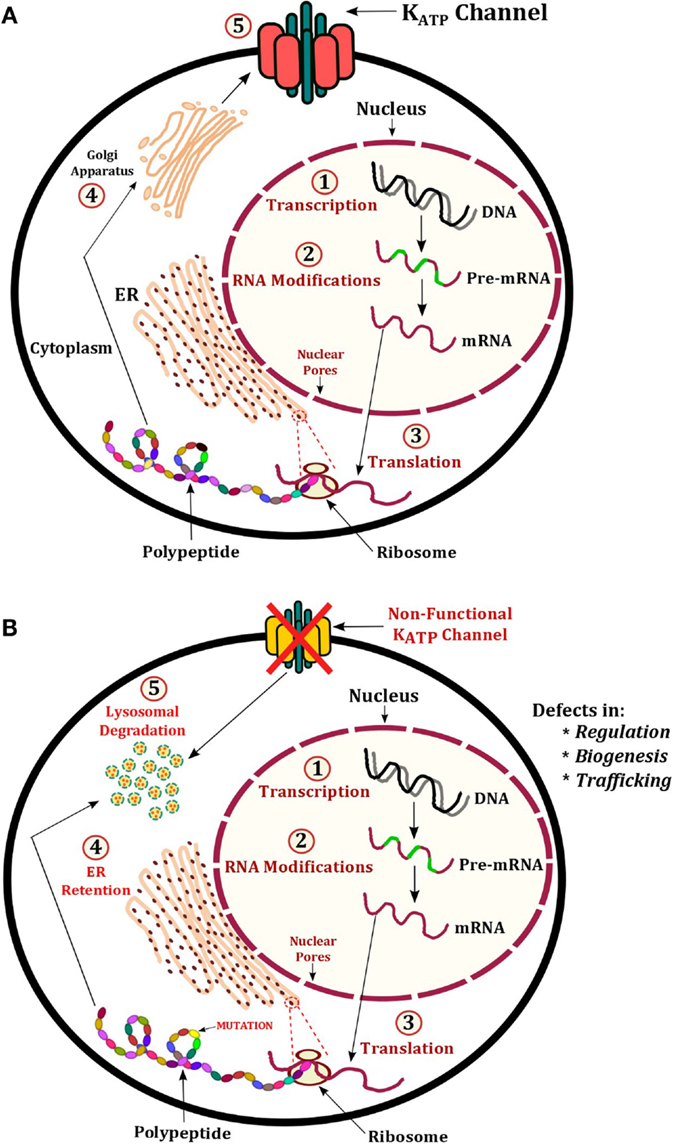
Figure 3. Illustration of KATP channel protein production and regulation. (A) Normal production of KATP channels involves transcription of ABCC8 and KCNJ11 to produce pre-mRNA, this undergoes modification to become mature mRNA. The mRNA exits the nucleus and is translated into a protein on ribosomes embedded on the ER. The polypeptide(s) fold into the tertiary structure and enter the Golgi apparatus for post-translational modifications. Vesicles containing the fully assembled KATP channel proteins are then expressed at the membrane. (B) Mechanisms of CHI include defects in regulation, biogenesis, and trafficking. In these cases, the defective KATP channel may undergo protein degradation in lysosomes.
Mechanism 1: Recessive KATP Channel Mutations
The membrane expression of KATP channels is vital to its role in maintaining the resting membrane potential of pancreatic β-cells and insulin secretion. In order for KATP channels to traffic to the plasma membrane, the SUR1 and Kir6.2 subunits must first assemble into their tertiary protein structure. This allows the endoplasmic reticulum retention signal known as RKR (Arg–Lys–Arg) present in both subunits to be shielded, enabling successful migration of the KATP channel complex from the endoplasmic reticulum to the plasma membrane (Figure 3) (72, 73). The following ABCC8 mutations cause trafficking defects including: p.Arg1437Gln, p.Phe1388del, p.Arg1394His, and p.Leu1544Pro. The KCNJ11 mutation p.His259Arg produces a non-functional KATP channel with impaired trafficking to the cell surface.
Within the SUR1 subunit, the RKR signal is located on the N-terminal of NBD2, and in Kir6.2, it is on the C-terminal. Removal of the last 36 amino acid residues in Kir6.2, which contains the RKR signal results in the membrane expression of a homotetrameric Kir6.2 structure, which has unique properties unlike that of KATP channels (70). There are prominent differences between WT KATP channels and homotetrameric Kir6.2 channels. For example, the half-maximal inhibition concentration (IC50) by ATP is ~10-fold higher in homotetrameric Kir6.2 channels compared to WT channels.
The SUR1 and Kir6.2 subunits of the KATP channel complex are sensitive to stimulation via Mg2+ bound nucleotides (MgATP, MgADP). A key function of the SUR1 subunit is to regulate the conductance of the Kir6.2 protein within the channel complex. Recessive mutations have been shown to affect the conductance of the KATP channel, and this includes the ABCC8 mutations p.Thr1139Met and p.Arg1215Glu (74). These mutants display a loss of ADP-dependent gating, which causes constitutive inhibition of KATP channels by ATP. The p.Arg1420Cys is another ABCC8 mutation, which generates a KATP channel that has a fivefold reduced affinity for ATP and ADP (75). In addition, the ability of MgATP and MgADP to stabilize ATP binding at NBD1 is also affected. The p.Arg1420Cys mutation acts as a hindrance to the transduction of a conformational change at NBD2 and thereby its ability to stabilize ATP binding at NBD1.
Mechanism 2: Dominant Mutations in ABCC8 and KCNJ11
The p.Glu1506Lys mutation was the first dominant mutation identified in the ABCC8 gene, which caused CHH in seven related patients (76). The diagnosis was based on the presence of inappropriately high levels of insulin, non-ketotic hypoglycemia, and a raised demand for glucose in order to prevent hypoglycemia. Mutations in the KCNJ11 gene causing diazoxide responsive CHH have also been reported. Patients with a dominant mutation in ABCC8 or KCNJ11 have a milder form of CHH and are mostly responsive to medical therapy (77), thus avoiding a pancreatectomy. The age of presentation is usually later than those with the recessive condition. Dominant mutations in ABCC8 and KCNJ11 demonstrate normal assembly with their respective WT partner and normal trafficking of assembled channels to the plasma membrane (78). However, dominant mutations causing diazoxide unresponsive CHH can generate KATP channels, which are retained in the endoplasmic reticulum.
Functional Consequences of Dominant Diazoxide Responsive CHH
Thornton et al. (79) identified a novel tri-nucleotide deletion in the ABCC8 gene, which caused the loss of serine 1387 (79). Experiments were conducted in COSm6 cells using human Kir6.2 and SUR1, which were transfected by electroporation. The rubidium (Rb86+) flux assay using metabolic inhibitors (2-deoxy glucose and oligomycin) and diazoxide revealed that when the p.Ser1387del mutation was expressed alone, it showed no response to metabolic inhibitors, or even to lower ATP concentrations, hence the mutation was disease causing. However, when the p.Ser1387del was expressed with WT SUR1, it generated the same level of activation as the pure WT channels. This is in contrast to the p.Glu1506Lys mutation, which only exhibited partial response to channel activators when expressed with WT SUR1 (76). The p.Ser1387del mutant, however, does reach the plasma membrane. Hence, the cause of CHH may be due to the lack of active KATP channels at the membrane.
Dominant Mutation Carriers Developing Diabetes
A striking feature of dominant mutations is that dominant mutations carriers have the tendency to develop diabetes later in life (76). This was the case with five mothers of CHH patients who had developed gestational diabetes, and two of them experienced hypoglycemia during infancy. The proposed theory suggests that β-cell apoptosis, may be what is causing diabetes later in life (80). It has been claimed that as CHH is linked with β-cell depolarization, enhanced Ca2+ entry into the cell and overall increase in intracellular Ca2+ concentrations, then ultimately this increased energy demand required to transport the additional Ca2+ into the cell may be contributing to β-cell apoptosis. Therefore, the use of diazoxide may relieve the cell and delay the onset of β-cell apoptosis, and hinder the development of diabetes, as diazoxide reduces the influx of Ca2+.
However, there is controversy as to whether the dominant mutations predispose to diabetes later in life (79). Two large cohorts of dominant mutation carriers in different centers have shown opposing results (76, 78). Huopio et al. (76) saw that six out of eight mothers developed impaired glucose tolerance during pregnancy. However, Pinney et al. (78) had a larger group and did not see a positive trend toward diabetes in mutation carries. Only 3 out of 13 women had gestational diabetes. It has been argued that Sur1 KO mice or Kir6.2 KO mice do not demonstrate β-cell apoptosis or develop diabetes (78). And so there are clearly other pathways, which act to maintain insulin secretion independently of KATP channels. Therefore, the glucose intolerance in both dominant and recessive mutations is the result of defective KATP channels, and not a sign of developing diabetes in the future (78).
Kapoor et al. (81), however, showed that the main phenotype linked with dominant ABCC8 and KCNJ11 mutations is quite varied (81). It ranges from asymptomatic macrosomia to persistent HH in childhood. For adults who are mutation carriers, these mutations may be a potential cause of dominantly inherited early onset diabetes mellitus.
Electrophysiology of KATP Channels in CHH
Studies involving the patch-clamp technique have shown that there are very few or almost no KATP channels on the surface on pancreatic β-cells from patients with diffuse, medically unresponsive CHH (71). The β-cells are depolarized, with variably high levels of intracellular Ca2+ in the presence of low glucose concentrations. The mutated KATP channels also exhibit no change in Ca2+ concentrations upon application of high glucose concentrations, tolbutamide, or diazoxide (82).
Diazoxide Enhances Insulin Secretion via Unknown Mechanism
The collection of diseased pancreatic tissue from CHH patients who undergo surgery is a complicated process. The subsequent study of such tissue is made more problematic due to the challenging prospect of collecting healthy control tissue. Henquin et al. (82) collected suitable controls for comparison studies, and they have shown that diazoxide often caused an unexpected increase in insulin secretion (82). The results could not be considered as an artifact as pinacidil, which is also another KATP channel opener, was also reported to have increased the secretion of insulin. It is known that higher concentrations of diazoxide reduce the production of ATP by affecting the mitochondrial membrane potential, but this does not explain why more insulin is released from focal and diffuse tissue as less ATP should in fact decrease the secretion of insulin.
Correction of Defective KATP Channels in Diseased Pancreatic Tissue
Powell and colleagues performed studies on pancreatic tissue obtained from CHH patients who underwent a pancreatectomy, and demonstrated the rescue of KATP channels (83). Inside-out patch-clamp recordings revealed that defective KATP channels had a normal response to diazoxide and ADP, when incubated at 25°C for 16 h. Whereas at 37°C, the defective KATP channels only showed between 0 and 10% activity compared to control cells. This work was based on the ABCC7 gene, which encodes the cystic fibrosis transmembrane regulator (CFTR), where much research has been done to enhance the trafficking of CFTR to the cell membrane. This research may now be an invaluable tool to enhance the treatment of CHH, by rescuing the defective KATP channels (83).
Metabolopathies
Metabolopathies is a term used to describe metabolic defects in seven genes (GCK, SCHAD, GLUD1, SLC16A1, HNF1A, HNF4A, and UCP2), which cause rare forms of CHH (84, 85). These genetic defects will be reviewed in this section.
GLUD1: Glutamate Dehydrogenase-1
The GLUD1 gene encodes for the glutamate dehydrogenase-1 (GDH) enzyme, which is expressed in the mitochondrial matrix. Amino acids are an essential source of fuel, which can be used as substitutes for glucose to generate ATP. The amino acid glutamate is interchangeably converted to α-ketoglutarate by the GDH enzyme, which is channeled into the Krebs cycle to generate ATP. The rising concentration of ATP triggers the depolarization of pancreatic β-cells through the closure of KATP channels, which culminates in the secretion of insulin.
The GDH enzyme is known to harbor two structural features, which are known to regulate its function. The first is the binding of leucine to the enzyme active site, and the second is an allosteric inhibitory binding site know to associate with nucleotides such as ATP and GTP (86). Dominantly inherited and de novo mutations in the GLUD1 are known to cause CHH. The majority of mutations have been identified in exon 11 and 12 of the GLUD1 gene (87); although, there are also reports of mutations in exon 6 and 7 (88). These activating mutations in the GLUD1 gene desensitize the GDH enzyme to inhibition through ATP and GTP (89), causing re-current activation through the amino acid leucine. This leads to the oxidation of glutamate to α-ketoglutarate, which triggers an increase in the ratio of ATP/ADP, which subsequently enhances the secretion of insulin.
A striking feature of GLUD1–CHH patients is their sensitivity to protein, in particular to the amino acid leucine, as it has been noted that they can develop re-current symptomatic hypoglycemia after consuming a protein rich meal. Hence, GLUD1–CHH is also known as Hyperinsulinism-hyperammonemia syndrome (HI/HA). The HI/HA may due to renal ammoniagenesis (90). GLUD1–CHH patients present with high levels (two to three times the normal) of plasma ammonia; however, they do not develop the symptoms associated with hyperammonemia. These patients do, however, experience episodes of epilepsy, which suggests that this condition may directly affect the brain. The overall medical management of the disease relies on the use of the drug diazoxide, which is effective for GLUD1–CHH patients.
GCK: Glucokinase (Hexokinase-4)
The enzyme, glucokinase, plays a fundamental role in “sensing” glucose in cells (91, 92). In pancreatic β-cells, the uptake of glucose occurs through an islet-specific glucose transporter, which is encoded by the GLUT2 gene. Glucose enters the cell through glucose transporters embedded in cell membranes. Once inside the cell, a single molecule of glucose is chemically modified using the enzyme glucokinase, which phosphorylates it and converts it to glucose-6-phosphate. This in essence “traps” the glucose molecule inside the cell, and it can no longer exit the cell membrane. This enzyme provides a mechanism by which cells are able to store glucose and reduce the amount of glucose which is circulating in the bloodstream.
Dominant mutations in the GCK gene alter the enzyme’s affinity for glucose. The penultimate effect is on insulin secretion, which becomes unregulated due to the increasing concentration of glucose inside the β-cells. Thus dominant gain-of-function mutations in GCK cause CHH (93). A total of 15 mutations have been identified in this gene associated with CHH, which target various sites across the GCK gene (94). There are many other mutations associated with MODY2.
Patients presenting with GCK–CHH can have a variable phenotype to one another, as everything from the birth weight to the age of presentation to the severity of the disease is dissimilar (78). Although there are cases of patients responding to drugs such as diazoxide, others have required surgery to control the hypoglycemia.
HADH: Short Chain L3-Hydroxyacyl-CoA Dehydrogenase
The oxidation of fatty acids and amino acids generates an alternative source of fuel that can be used to generate ATP. This is an essential process, which is highly used during fasting periods when the body is deprived of energy in form of dietary carbohydrates. The HADH gene is located on chromosome 4q22-26, and it codes for the enzyme Short chain L3-Hydroxyacyl-CoA dehydrogenase (SCHAD), which is involved in β-oxidation. This enzyme is expressed in many tissues, with high concentrations in the islet of Langerhans (95). However, its prime activity is specifically in the mitochondria, where it catalyzes the oxidation of different sized L3-hydroxacyl-CoA to their respective 3-ketoacyl-CoAs. The reaction requires the use of one NAD+ cofactor, and generates a single NADH as a product.
Autosomal recessive mutations in HADH are a rare cause of CHH (96), which leads to a reduction in the levels of the SCHAD enzyme. As a result, there is an accumulation of medium and short-chain fatty acids, which are not broken down properly, and therefore cannot enter the metabolic pathway leading to ATP production, which ultimately affects the KATP channel pathway. Other targets of HADH inactivity can effect G protein-coupled receptor GPR40 (97) as well play a role in the inhibition of carnitine palmitoyltransferase 1 (CPT-1) (98). HADH is known to be regulated by transcription factors such as FOXA2, which may play a role in the differentiation process of pancreatic β-cells in mice (99).
The clinical presentation of HADH–CHH is also a varied among patients with the disease. The patients are largely from consanguineous families, and the age of disease onset is unpredictable, with cases presenting at birth with severe CHH, as well other late onset with mild hypoglycemia. The first case of adult HH due to a HADH mutation has been reported, where the hypoglycemia became milder as the patient grew older (100).
Abnormal levels of hydroxybutyryl-acylcarnitine in blood and 3-hydroxyglutaric acid in urine are usually found in HADH–CHH patients. However, all cases have been known to be responsive to diazoxide, although patients may still suffer from a hypoglycemic episode after a protein-rich meal. Examples of HADH mutations include IVS6-2a > g. This is a splice site mutation generating, an enzyme which has ~90% reduction is activity in comparison to controls. The p.Met188Val mutant, however, had mild effects on enzyme activity, which originated in a patient with normal levels of acylcarnitines and organic acids.
It has been reported that some HADH–CHH patients are also leucine sensitive (101), however, unlike GLUD1–CHH patients, they do not present with hyperammonemia. The mechanism of leucine action in HADH–CHH patients is clearly different to GLUD1–CHH. A protein–protein interaction between SCHAD and GDH had been demonstrated in the liver (86). Furthermore, a hadh KO mouse model developed by Li et al. (86) had previously shown that leucine loading leads to a hyperinsulinemic response (86). With this in mind, the direct link between SCHAD and GDH in patient lymphoblast cells was first reported by Heslegrave et al. (101). It has been postulated that the regulation of the GDH enzyme may be affected by the ratio of active SCHAD to GDH in pancreatic β-cells. SCHAD may be an inhibitor of GDH, which suppresses the secretion of insulin via leucine stimulation (102). Loss of protein–protein interaction may cause a mild increase in activation of GDH (86). Patients with HADH mutations do have lower levels of SCHAD, although this is not the case in all patients, which may be the cause of the varying phenotypes seen in these patients. Further studies are needed to understand the mechanism of this interaction.
HNF4A: Hepatocyte Nuclear Factor 4 Alpha
The HNF4A gene belongs to the nuclear hormone receptor superfamily. This gene harbors two distinct promoter regions known as P1 and P2. The transcription of the latter promoter is enhanced in the pancreatic β-cells, where it regulates key genes involved in glucose-stimulated insulin secretion (103, 104). Genes known to be regulated by HNF4A include glucose transporter-2, glycolytic enzymes, and enzymes involved in mitochondrial metabolism (104). A gene of significant interest is KCNJ11, which is also regulated by HNF4A. It has been hypothesized that HNF4A may down regulate the expression of the KCNJ11 by up to 60%, although the precise mechanism is not known (105). The decreased expression was observed in mice using the conditional Cre-loxP-based inactivation system, which was used to delete the HNF4A gene. However conflicting evidence has shown by other researchers where the deletion of HNF4A had no effect on the activity or expression of KCNJ11 (106, 107).
The peroxisome proliferator-activated receptor alpha (PPARα) is a transcription factor which may be an alternative target of HNF4A activity. PPARα is also involved in regulating the expression of enzymes involved in fatty acid oxidation, and during a fast PPARα, KO mice have been shown to develop HH (108). The association between the two transcription factors is based on the fact that there are reduced levels of PPARα in β-cells lacking HNF4A (105). Such a reduction of PPARα can lead to a reduction in the levels of β-oxidation, leading to the accumulation of lipids such as malonyl-CoA within the cytoplasm. This in turn can inhibit the enzyme CPT-1, which can raise the concentration of cytosolic long-chain acyl-CoA, terminating with the increased production of insulin.
Only a small fraction of patients with dominant inactivating HNF4A mutations actually go on to develop CHH (106, 109–111), and hence the penetrance of the disease is quite low. However, these patient’s do go on to develop maturity onset diabetes type 1 (MODY1), a form diabetes which is inherited in an autosomal dominant fashion (106). Another prominent feature in HNF4A–CHH patients is macrosomia. In comparison to family members, the HNF4A–CHH patients had a median increase in birth weight by 790 g in one cohort (106) and 751 g in another (109). This increase in birth weight is similar to patients with KATP channel mutations.
HNF1A: Hepatocyte Nuclear Factor 1 Alpha
The HNF1A gene codes for a transcription factor, which is known to enhance the expression of liver specific genes, as well as genes expressed in the pancreas (112). Heterozygous, loss of function mutations in this gene are known to cause CHH. HNF1A–CHH patients present with milder fetal macrosomia, and are known to develop MODY3 as they age. The severity of the CHH is also less compared to patients with HNF4A–CHH. It is currently unknown as to why there is switch from CHH to MODY3 in these patients. HNF1A–CHH patients are responsive to drugs such as diazoxide. For the first time, Rozenkova and colleagues have demonstrated the functional impact of HNF1A mutations, which cause CHH. DNA-binding studies revealed that the p.Asn62Lysfs*93 and p.Leu254Gln HNF1A mutations reduced the transcriptional activity of the protein by 2.9 and 22%, respectively (113). The overall incidence of HNF1A–CHH is very low, and this could be due to the fact that some patients escape detection as their phenotype is very mild.
SLC16A1: Solute Carrier Family 16, Member 1
The SLC16A1 gene encodes for a transporter protein known as monocarboxylate transporter 1 (MCT1). Its function is to regulate the movement of lactate and pyruvate into the pancreatic β-cell. Under normal physiological conditions, lactate and pyruvate are present in low concentrations in the β-cell as they are potent insulin secretagogues. This transporter protein is not normally expressed (disinhibited) in pancreatic β-cells, but gain-of-function mutations in the promoter region of the gene enable its expression in β-cells, this allowing increased pyruvate into the β-cell and eventually leading to CHH. A unique feature about these patients is that the onset of the disease is exercise induced. Hypoglycemia was observed in these patients after around 30–45 min of strenuous exercise. These features were prominent in Finnish cohort of 13 patients (114). In these patients, lactate is produced during anaerobic exercise, and this is fed into the Krebs cycle together with pyruvate, which generates ATP and triggers insulin release through the KATP channel pathway. Most patients are responsive to medical management using diazoxide, although this is ineffective after anaerobic exercise, hence strenuous exercise is not tolerated by these patients (115).
UCP2: Uncoupling Protein 2
The UCP2 gene codes for a mitochondrial carrier protein largely expressed in both pancreatic α- and β-cells. The structure of the UCP2 protein entails six transmembrane domains (116) and one nucleotide-binding domain. It has been hypothesized that this gene provides protection against oxidative stress during fatty acid metabolism. In vivo studies observing UCP2 KO mice have shown that these mice develop hyperinsulinism, which suggests that UCP2 is involved in the regulation of insulin secretion. In animal models, it has also been shown that the overexpression of ucp2 can cause diabetes. Hence there is a clear involvement of UCP2 in glucose physiology. It has been hypothesized that the UCP2 protein may be involved in regulating the leak of protons across the inner mitochondrial membrane (117). This would lead to the uncoupling of mitochondrial oxidative metabolism from ATP synthesis, which ultimately reduces the ATP yield for substrate oxidation.
González-Barroso et al. (118) described the first two cases of CHH due to mutations in the UCP2 gene (118). The age of presentation in these two patients was later around ~8 months. Insulinoma and yeast cells have been used to further understand the link between UCP2 and HH. Studies have revealed that mutations in UCP2 have a loss-of-function effect on the protein, which enhances insulin secretion, whereas the wild type (WT) UCP2 protein inhibits insulin secretion (118). Further studies are warranted in order to establish a clear mechanism of UCP2-induced HH.
Insulin Receptor Gene Mutation
A report of a novel heterozygous mutation in the insulin receptor gene has been causative of hypoglycemia in patients with severe HH (119). The autosomal dominant change in the insulin receptor gene (p.Arg1174Gln) has been described to cause postprandial hyperinsulinemic hypoglycemia (PPHH) (119). PPHH occurs after a few hours of eating a meal. PPHH is secondary to the unregulated insulin release due to the meal ingested.
The hyperinsulinism seen in these patients appear to be due to a reduction in the clearance of insulin rather than the increased release of insulin, since C-peptide concentrations are within normal ranges.
Non-Genetic Causes of HH
Postprandial Hyperinsulinemic Hypoglycemia
Other non-genetic causes of PPHH also exist (Table 1):
Dumping Syndrome
Dumping syndrome is an example of PPHH, which occurs after Nissen’s fundoplication (120). Ingestion of carbohydrate-rich foods is absorbed into the small intestine, which in turn cause increased glucose absorption, hyperglycemia, rapid release of insulin, and overall results in hypoglycemia. The molecular mechanisms of this is thought to be due to abnormally exaggerated levels of glucagon-like peptide-1 (GLP-1), which is known to contribute to the HH (121).
Where dumping syndrome is suspected, a mixed meal or oral glucose tolerance test (OGTT) is performed. A current diagnostic criteria for dumping syndrome is a decrease of more than 6 mmol/L between peak and nadir blood glucose levels (122).
Insulin Autoimmune Syndrome
Insulin autoimmune syndrome is also known as Hirata disease, which is a rare cause of hypoglycemia characterized by high levels of endogenous insulin autoantibodies. This condition has the absence of pathological islets and is in the absence of exposure to exogenous insulin (123). Insulin autoimmunity affects mainly adults of 40 years and over.
In this condition, post meal or after a glucose load, the raised blood glucose levels cause insulin secretion but hyperglycemia occurs. This is due to the insulin antibodies binding to insulin and reducing the bioavailability to its receptor and this in turn leads to a reduction in its action. After a few hours, the blood glucose levels decrease with corresponding insulin concentrations, and the bound insulin is released from the antibodies, resulting in the hypoglycemia (124).
Bariatric Surgery
Obese individuals with comorbidities such a type-2 diabetes mellitus are causing a rise in invasive gastric bypass procedures (125). Prior to weight-loss, these patients have shown a resolution of their hyperglycemia. The theory for this mechanism is that the food bypasses the upper gastrointestinal tract and is passed along to the lower gut, here the insulinotropic hormone; GLP-1 is hyperstimulated (126). However, a serious side effect appears to be an increase in gastric bypass patients presenting with PPHH and nesidioblastosis (127).
Non-Insulinoma Pancreatogenous Hypoglycemia Syndrome
Postprandial neuroglycopenia characterizes the non-insulinoma pancreatogenous hypoglycemia syndrome (NIPHS) (128). The genetic cause of NIPHS remains to be found, these patients are negative for prolonged fasts and show islet hypertrophy and evidence of nesidioblastosis in the absence of insulinoma. However, studies are yet to identify if the increased β-cell proliferation production as well as pancreatic differentiation factors are contributing agents (129). In the absence of KATP channel mutations, some of these patients are surprisingly responsive to diazoxide (130), while the unresponsive patients require a partial pancreatectomy.
Insulinoma
In adults, insulinomas are the most common cause of endogenous HH with an incidence of four per million (131). Predominantly of benign origin, these pancreatic tumors secrete insulin and are usually small and <2 mm in diameter. Initially, hypoglycemia was thought to show only either on fasting or post-exercise; however, it has also presented post meal (132). In suspected cases, current guidelines recommend the measurement of plasma glucose, insulin, C-peptide, proinsulin, β-hydroxybutyrate, insulin antibodies, and circulating oral hypoglycemic agents during the hypoglycemic episode (133).
Conclusion
Hyperinsulinemic hypoglycemia is characterized by the dysregulation of insulin secretion from pancreatic β-cells. The most severe forms of congenital hyperinsulinemic hypoglycemia are due to molecular defects in the genes ABCC8 and KCNJ11. Milder forms of hyperinsulinemic hypoglycemia may occur due to molecular defects in several other genes, which play a key role in the regulation of insulin secretion. Currently genetic abnormalities are only found in about 50% of patients presenting with hyperinsulinemic hypoglycemia. Understanding the molecular mechanisms of dysregulated insulin secretion in those patients with no genetic etiology will provide novel insights into pancreatic β-cell function.
Author Contributions
In this review article, AN wrote the following parts: (1) introduction (wrote the majority of this section, except for the first three paragraphs), (2) KATP channels and insulin secretion (wrote the entire section), (3) metabolopathies (wrote the entire section, except the part on “insulin receptor gene mutation”), and (4) figures (generated all the three figures). SR wrote the following parts: (1) introduction (wrote the starting paragraphs), (2) histological characterisation of CHH, (3) metabolopathies (section listed below): (a) insulin receptor gene mutation, (4) non-genetic causes of HH (wrote the entire section), (5) conclusion, and (6) tables (generated Table 1). KH wrote the following parts: (1) abstract, (2) read through the entire review article and corrected for grammar, and (3) also contributed to the style and structure of the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Güemes M, Rahman SA, Hussain K. What is a normal blood glucose? Arch Dis Child (2015). doi:10.1136/archdischild-2015-308336
2. Aynsley-Green A, Hussain K, Hall J, Saudubray JM, Nihoul-Fékété C, De Lonlay-Debeney P, et al. Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal Ed (2000) 82(2):F98–107. doi:10.1136/fn.82.2.F98
3. Hussain K, Bryan J, Christesen HT, Brusgaard K, Aguilar-Bryan L. Serum glucagon counterregulatory hormonal response to hypoglycemia is blunted in congenital hyperinsulinism. Diabetes (2005) 54(10):2946–51. doi:10.2337/diabetes.54.10.2946
4. Hussain K, Hindmarsh P, Aynsley-Green A. Neonates with symptomatic hyperinsulinemic hypoglycemia generate inappropriately low serum cortisol counterregulatory hormonal responses. J Clin Endocrinol Metab (2003) 88(9):4342–7. doi:10.1210/jc.2003-030135
5. Meissner T, Wendel U, Burgard P, Schaetzle S, Mayatepek E. Long-term follow-up of 114 patients with congenital hyperinsulinism. Eur J Endocrinol (2003) 149(1):43–51. doi:10.1530/eje.0.1490043
6. Kapoor RR, Flanagan SE, James C, Shield J, Ellard S, Hussain K. Hyperinsulinaemic hypoglycaemia. Arch Dis Child (2009) 94(6):450–7. doi:10.1136/adc.2008.148171
7. Thomas P, Ye Y, Lightner E. Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Hum Mol Genet (1996) 5(11):1809–12. doi:10.1093/hmg/5.11.1809
8. Thomas PM, Cote GJ, Wohllk N, Haddad B, Mathew PM, Rabl W, et al. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science (1995) 268(5209):426–9. doi:10.1126/science.7716548
9. Dunne MJ, Cosgrove KE, Shepherd RM, Aynsley-Green A, Lindley KJ. Hyperinsulinism in infancy: from basic science to clinical disease. Physiol Rev (2004) 84(1):239–75. doi:10.1152/physrev.00022.2003
10. Phillips R, Quake SR. The biological frontier of physics. Phys Today (2006) 59(5):38–43. doi:10.1063/1.2216960
11. Dean M, Annilo T. Evolution of the ATP-binding cassette (ABC) transporter superfamily in verterbrates. Annu Rev Genomics Hum Genet (2005) 6(1):123–42. doi:10.1146/annurev.genom.6.080604.162122
12. Rees DC, Johnson E, Lewinson O. ABC transporters: the power to change. Nat Rev Mol Cell Biol (2009) 10(3):218–27. doi:10.1038/nrm2646
13. Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J (1982) 1(8):945–51.
14. Inagaki N, Gonoi T, Clement JP, Namba N, Inazawa J, Gonzalez G, et al. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science (1995) 270(5239):1166–70. doi:10.1126/science.270.5239.1166
15. Nestorowicz A, Wilson BA, Schoor KP, Inoue H, Glaser B, Landau H, et al. Mutations in the sulfonylurea receptor gene are associated with familial hyperinsulinism in Ashkenazi Jews. Hum Mol Genet (1996) 5(11):1813–22. doi:10.1093/hmg/5.11.1813
16. Aguilar-Bryan L, Nelson DA, Vu QA, Humphrey MB, Boyd AE. Photoaffinity labeling and partial purification of the beta cell sulfonylurea receptor using a novel, biologically active glyburide analog. J Biol Chem (1990) 265(14):8218–24.
17. Aguilar-Bryan L, Clement JP, Gonzalez G, Kunjilwar K, Babenko A, Bryan J. Toward understanding the assembly and structure of KATP channels. Physiol Rev (1998) 78(1):227–45.
18. Gribble FM, Reimann F. Sulphonylurea action revisited: the post-cloning era. Diabetologia (2003) 46(7):875–91. doi:10.1007/s00125-003-1143-3
19. Ashcroft FM, Gribble FM. New windows on the mechanism of action of K(ATP) channel openers. Trends Pharmacol Sci (2000) 21(11):439–45. doi:10.1016/S0165-6147(00)01563-7
20. Gribble FM, Tucker SJ, Ashcroft FM. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO J (1997) 16(6):1145–52. doi:10.1093/emboj/16.6.1145
21. Schlichting I, Almo SC, Rapp G, Wilson K, Petratos K, Lentfer A, et al. Time-resolved X-ray crystallographic study of the conformational change in Ha-Ras p21 protein on GTP hydrolysis. Nature (1990) 345(6273):309–15. doi:10.1038/345309a0
22. Dean M, Allikmets R. Complete characterization of the human ABC gene family. J Bioenerg Biomembr (2001) 33(6):475–9. doi:10.1023/a:1012823120935
23. Matsuo M, Kioka N, Amachi T, Ueda K. ATP binding properties of the nucleotide-binding folds of SUR1. J Biol Chem (1999) 274:37479–82. doi:10.1074/jbc.274.52.37479
24. Hough E, Mair L, Mackenzie W, Sivaprasadarao A. Expression, purification, and evidence for the interaction of the two nucleotide-binding folds of the sulphonylurea receptor. Biochem Biophys Res Commun (2002) 294(1):191–7. doi:10.1016/S0006-291X(02)00454-0
25. Mikhailov MV, Ashcroft SJ. Interactions of the sulfonylurea receptor 1 subunit in the molecular assembly of beta-cell K(ATP) channels. J Biol Chem (2000) 275(5):3360–4. doi:10.1074/jbc.275.5.3360
26. Doupnik CA, Davidson N, Lester HA. The inward rectifier potassium channel family. Curr Opin Neurobiol (1995) 5(3):268–77. doi:10.1016/0959-4388(95)80038-7
27. Ruppersberg JP. Intracellular regulation of inward rectifier K+ channels. Pflügers Arch (2000) 441(1):1–11. doi:10.1007/s004240000380
28. Krapivinsky G, Medina I, Eng L, Krapivinsky L, Yang Y, Clapham DE. A novel inward rectifier K+ channel with unique pore properties. Neuron (1998) 20(5):995–1005. doi:10.1016/S0896-6273(00)80480-8
29. Sempoux C, Guiot Y, Jaubert F, Rahier J. Focal and diffuse forms of congenital hyperinsulinism: the keys for differential diagnosis. Endocr Pathol (2004) 15(3):241–6. doi:10.1385/ep:15:3:241
30. Goossens A, Gepts W, Saudubray JM, Bonnefont JP, Nihoul F, Heitz PU, et al. Diffuse and focal nesidioblastosis. A clinicopathological study of 24 patients with persistent neonatal hyperinsulinemic hypoglycemia. Am J Surg Pathol (1989) 13(9):766–75. doi:10.1097/00000478-198909000-00006
31. Rahier J, Falt K, Muntefering H, Becker K, Gepts W, Falkmer S. The basic structural lesion of persistent neonatal hypoglycaemia with hyperinsulinism: deficiency of pancreatic D cells or hyperactivity of B cells? Diabetologia (1984) 26(4):282–9. doi:10.1007/BF00283651
32. Rahier J, Guiot Y, Sempoux C. Persistent hyperinsulinaemic hypoglycaemia of infancy: a heterogeneous syndrome unrelated to nesidioblastosis. Arch Dis Child Fetal Neonatal Ed (2000) 82(2):F108–12. doi:10.1136/fn.82.2.F108
33. Sempoux C, Guiot Y, Lefevre A, Nihoul-Fekete C, Jaubert F, Saudubray JM, et al. Neonatal hyperinsulinemic hypoglycemia: heterogeneity of the syndrome and keys for differential diagnosis. J Clin Endocrinol Metab (1998) 83(5):1455–61. doi:10.1210/jc.83.5.1455
34. Sempoux C, Capito C, Bellanné-Chantelot C, Verkarre V, de Lonlay P, Aigrain Y, et al. Morphological mosaicism of the pancreatic islets: a novel anatomopathological form of persistent hyperinsulinemic hypoglycemia of infancy. J Clin Endocrinol Metabol (2011) 96(12):3785–93. doi:10.1210/jc.2010-3032
35. Damaj L, le Lorch M, Verkarre V, Werl C, Hubert L, Nihoul-Fekete C, et al. Chromosome 11p15 paternal isodisomy in focal forms of neonatal hyperinsulinism. J Clin Endocrinol Metab (2008) 93(12):4941–7. doi:10.1210/jc.2008-0673
36. Rahier J, Sempoux C, Fournet JC, Poggi§ F, Brunelle F, Nihoul-Fekete C, et al. Partial or near-total pancreatectomy for persistent neonatal hyperinsulinaemic hypoglycaemia: the pathologist’s role. Histopathology (1998) 32(1):15–9. doi:10.1046/j.1365-2559.1998.00326.x
37. Otonkoski T, Näntö-Salonen K, Seppänen M, Veijola R, Huopio H, Hussain K, et al. Noninvasive diagnosis of focal hyperinsulinism of infancy with [18F]-DOPA positron emission tomography. Diabetes (2006) 55(1):13–8. doi:10.2337/diabetes.55.01.06.db05-1128
38. Hussain K, Flanagan SE, Smith VV, Ashworth M, Day M, Pierro A, et al. An ABCC8 gene mutation and mosaic uniparental isodisomy resulting in atypical diffuse congenital hyperinsulinism. Diabetes (2008) 57(1):259–63. doi:10.2337/db07-0998
39. Henquin J-C, Sempoux C, Marchandise J, Godecharles S, Guiot Y, Nenquin M, et al. Congenital hyperinsulinism caused by hexokinase I expression or glucokinase-activating mutation in a subset of β-cells. Diabetes (2013) 62(5):1689–96. doi:10.2337/db12-1414
40. Shi Y, Avatapalle HB, Skae MS, Padidela R, Newbould M, Rigby L, et al. Increased plasma incretin concentrations identifies a subset of patients with persistent congenital hyperinsulinism without KATP channel gene defects. J Pediatr (2015) 166(1):191–4. doi:10.1016/j.jpeds.2014.09.019
41. Noma A. ATP-regulated K+ channels in cardiac muscle. Nature (1983) 305(5930):147–8. doi:10.1038/305147a0
42. Spruce AE, Standen NB, Stanfield PR. Voltage-dependent ATP-sensitive potassium channels of skeletal muscle membrane. Nature (1985) 316(6030):736–8. doi:10.1038/316736a0
43. Cook DL, Hales CN. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature (1984) 311(5983):271–3. doi:10.1038/311271a0
44. Standen N, Quayle J, Davies N, Brayden J, Huang Y, Nelson M. Hyperpolarizing vasodilators activate ATP-sensitive K+ channels in arterial smooth muscle. Science (1989) 245(4914):177–80. doi:10.1126/science.2501869
45. Ashcroft FM. Adenosine 5’-triphosphate-sensitive potassium channels. Annu Rev Neurosci (1988) 11(1):97–118. doi:10.1146/annurev.ne.11.030188.000525
46. Salari S, Ghasemi M, Fahanik-Babaei J, Saghiri R, Sauve R, Eliassi A. Evidence for a KATP channel in rough endoplasmic reticulum (rerKATP channel) of rat hepatocytes. PLoS One (2015) 10(5):e0125798. doi:10.1371/journal.pone.0125798
47. Rorsman P, Trube G. Glucose dependent K+ channels in pancreatic beta-cells are regulated by intracellular ATP. Pflugers Arch (1985) 405(4):305–9. doi:10.1007/BF00595682
48. Henquin JC. Tolbutamide stimulation and inhibition of insulin release: studies of the underlying ionic mechanisms in isolated rat islets. Diabetologia (1980) 18(2):151–60. doi:10.1007/BF00290493
49. Henquin JC. The potassium permeability of pancreatic islet cells: mechanisms of control and influence on insulin release. Horm Metab Res (1980) 10:66–73.
50. Meissner HP. Electrical characteristics of the beta-cells in pancreatic islets. J Physiol (Paris) (1976) 72(2):757–67.
51. Trube G, Rorsman P, Ohno-Shosaku T. Opposite effects of tolbutamide and diazoxide on the ATP-dependent K+ channel in mouse pancreatic beta-cells. Pflugers Arch (1986) 407(5):493–9. doi:10.1007/BF00657506
52. Ashcroft FM, Kakei M. ATP-sensitive K+ channels in rat pancreatic beta-cells: modulation by ATP and Mg2+ ions. J Physiol (1989) 416:349–67. doi:10.1113/jphysiol.1989.sp017765
53. Rajan AS, Aguilar-Bryan L, Nelson DA, Nichols CG, Wechsler SW, Lechago J, et al. Sulfonylurea receptors and ATP-sensitive K+ channels in clonal pancreatic alpha cells. Evidence for two high affinity sulfonylurea receptors. J Biol Chem (1993) 268(20):15221–8.
54. Shimono D, Fujimoto S, Mukai E, Takehiro M, Nabe K, Radu RG, et al. ATP enhances exocytosis of insulin secretory granules in pancreatic islets under Ca2+ depleted condition. Diabetes Res Clin Pract (2005) 69(3):216–23. doi:10.1016/j.diabres.2005.01.010
55. Shyng S-L, Nichols CG. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science (1998) 282(5391):1138–41. doi:10.1126/science.282.5391.1138
56. Suh B-C, Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys (2008) 37(1):175–95. doi:10.1146/annurev.biophys.37.032807.125859
57. Ribalet B, John SA, Xie L-H, Weiss JN. ATP-sensitive K+ channels: regulation of bursting by the sulphonylurea receptor, PIP2 and regions of Kir6.2. J Physiol (2006) 571(2):303–17. doi:10.1113/jphysiol.2005.100719
58. Proks P, Capener CE, Jones P, Ashcroft FM. Mutations within the P-loop of Kir6.2 modulate the intraburst kinetics of the ATP-sensitive potassium channel. J Gen Physiol (2001) 118:341–53. doi:10.1085/jgp.118.4.341
59. Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature (2006) 440(7083):470–6. doi:10.1038/nature04711
60. Enkvetchakul D, Loussouarn G, Makhina E, Shyng SL, Nichols CG. The kinetic and physical basis of KATP channel gating: toward a unified molecular understanding. Biophys J (2000) 78:2334–48. doi:10.1016/S0006-3495(00)76779-8
61. Markworth E, Schwanstecher C, Schwanstecher M. ATP4- mediates closure of pancreatic beta-cell ATP-sensitive potassium channels by interaction with 1 of 4 identical sites. Diabetes (2000) 49:1413–8. doi:10.2337/diabetes.49.9.1413
62. Gillis KD, Gee WM, Hammoud A, McDaniel ML, Falke LC, Misler S. Effects of sulfonamides on a metabolite-regulated ATPi-sensitive K+ channel in rat pancreatic B-cells. Am J Physiol (1989) 257(6 Pt 1):C1119–27.
63. Nichols CG, Lederer WJ, Cannell MB. ATP dependence of KATP channel kinetics in isolated membrane patches from rat ventricle. Biophys J (1991) 60(5):1164–77. doi:10.1016/S0006-3495(91)82152-X
64. Trapp S, Proks P, Tucker SJ, Ashcroft FM. Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP. J Gen Physiol (1998) 112(3):333–49. doi:10.1085/jgp.112.3.333
65. Li L, Geng X, Drain P. Open state destabilization by atp occupancy is mechanism speeding burst exit underlying KATP channel inhibition by ATP. J Gen Physiol (2002) 119(1):105–16. doi:10.1085/jgp.119.1.105
66. Craig TJ, Ashcroft FM, Proks P. How ATP Inhibits the open KATP channel. J Gen Physiol (2008) 132(1):131–44. doi:10.1085/jgp.200709874
67. Ebel H, Günther T. Magnesium metabolism: a review. J Clin Chem Clin Biochem (1980) 18(5):257–70.
68. Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev (1999) 20(2):101–35. doi:10.1210/er.20.2.101
69. Shyng S, Nichols CG. Octameric stoichiometry of the KATP channel complex. J Gen Physiol (1997) 110:655–64. doi:10.1085/jgp.110.6.655
70. Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature (1997) 387(6629):179–83. doi:10.1038/387179a0
71. Charlotte Kane RMS, Squires PE, Johnson PR, James RF, Milla PJ, Aynsley-Green A, et al. Loss of functional KATP channels in pancreatic beta−cells causes persistent hyperinsulinemic hypoglycemia of infancy. Nat Med (1996) 2:1344–7. doi:10.1038/nm1296-1344
72. Zerangue N, Schwappach B, Jan YN, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neuron (1999) 22(3):537–48. doi:10.1016/S0896-6273(00)80708-4
73. Neagoe I, Schwappach B. Pas de deux in groups of four – the biogenesis of KATP channels. J Mol Cell Cardiol (2005) 38:887–94. doi:10.1016/j.yjmcc.2004.11.023
74. Shyng SL, Ferrigni T, Shepard JB, Nestorowicz A, Glaser B, Permutt MA, et al. Functional analyses of novel mutations in the sulfonylurea receptor 1 associated with persistent hyperinsulinemic hypoglycemia of infancy. Diabetes (1998) 47(7):1145–51. doi:10.2337/diabetes.47.7.1145
75. Matsuo M, Trapp S, Tanizawa Y, Kioka N, Amachi T, Oka Y, et al. Functional analysis of a mutant sulfonylurea receptor, SUR1-R1420C, that is responsible for persistent hyperinsulinemic hypoglycemia of infancy. J Biol Chem (2000) 275(52):41184–91. doi:10.1074/jbc.M006503200
76. Huopio H, Reimann F, Ashfield R, Komulainen J, Lenko H-L, Rahier J, et al. Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. J Clin Invest (2000) 106(7):897–906. doi:10.1172/JCI9804
77. MacMullen CM, Zhou Q, Snider KE, Tewson PH, Becker SA, Aziz AR, et al. Diazoxide-unresponsive congenital hyperinsulinism in children with dominant mutations of the β-cell sulfonylurea receptor SUR1. Diabetes (2011) 60(6):1797–804. doi:10.2337/db10-1631
78. Pinney SE, MacMullen C, Becker S, Lin Y-W, Hanna C, Thornton P, et al. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J Clin Invest (2008) 118(8):2877–86. doi:10.1172/JCI35414
79. Thornton PS, MacMullen C, Ganguly A, Ruchelli E, Steinkrauss L, Crane A, et al. Clinical and molecular characterization of a dominant form of congenital hyperinsulinism caused by a mutation in the high-affinity sulfonylurea receptor. Diabetes (2003) 52(9):2403–10. doi:10.2337/diabetes.52.9.2403
80. Glaser B, Ryan F, Donath M, Landau H, Stanley CA, Baker L, et al. Hyperinsulinism caused by paternal-specific inheritance of a recessive mutation in the sulfonylurea-receptor gene. Diabetes (1999) 48(8):1652–7. doi:10.2337/diabetes.48.8.1652
81. Kapoor RR, Flanagan SE, James CT, McKiernan J, Thomas AM, Harmer SC, et al. Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant ABCC8/KCNJ11 mutations. Diabetologia (2011) 54(10):2575–83. doi:10.1007/s00125-011-2207-4
82. Henquin JC, Nenquin M, Sempoux C, Guiot Y, Bellanné-Chantelot C, Otonkoski T, et al. In vitro insulin secretion by pancreatic tissue from infants with diazoxide-resistant congenital hyperinsulinism deviates from model predictions. J Clin Invest (2011) 121(10):3932–42. doi:10.1172/JCI58400
83. Powell PD, Bellanné-Chantelot C, Flanagan SE, Ellard S, Rooman R, Hussain K, et al. In vitro recovery of ATP-sensitive potassium channels in β-cells from patients with congenital hyperinsulinism of infancy. Diabetes (2011) 60(4):1223–8. doi:10.2337/db10-1443
84. Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K. Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur J Endocrinol (2013) 168(4):557–64. doi:10.1530/eje-12-0673
85. Snider KE, Becker S, Boyajian L, Shyng S-L, MacMullen C, Hughes N, et al. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metabol (2013) 98(2):E355–63. doi:10.1210/jc.2012-2169
86. Li C, Najafi H, Daikhin Y, Nissim IB, Collins HW, Yudkoff M, et al. Regulation of leucine-stimulated insulin secretion and glutamine metabolism in isolated rat islets. J Biol Chem (2003) 278(5):2853–8. doi:10.1074/jbc.M210577200
87. Stanley C, Fang J, Kutyna K, Hsu B, Ming J, Glaser B, et al. Molecular basis and characterization of the hyperinsulinism/hyperammonemia syndrome: predominance of mutations in exons 11 and 12 of the glutamate dehydrogenase gene. Diabetes (2000) 49:667–73. doi:10.2337/diabetes.49.4.667
88. Santer R, Kinner M, Passarge M, Superti-Furga A, Mayatepek E, Meissner T, et al. Novel missense mutations outside the allosteric domain of glutamate dehydrogenase are prevalent in European patients with the congenital hyperinsulinism-hyperammonemia syndrome. Hum Genet (2001) 108(1):66–71. doi:10.1007/s004390000432
89. Stanley CA, Lieu YK, Hsu BYL, Burlina AB, Greenberg CR, Hopwood NJ, et al. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med (1998) 338(19):1352–7. doi:10.1056/NEJM199805073381904
90. Treberg J, Clow K, Greene K, Brosnan M, Brosnan J. Systemic activation of glutamate dehydrogenase increases renal ammoniagenesis: implications for the hyperinsulinism/hyperammonemia syndrome. Am J Physiol Endocrinol Metab (2010) 298:E1219–25. doi:10.1152/ajpendo.00028.2010
91. Johnson JH, Newgard CB, Milburn JL, Lodish HF, Thorens B. The high Km glucose transporter of islets of Langerhans is functionally similar to the low affinity transporter of liver and has an identical primary sequence. J Biol Chem (1990) 265(12):6548–51.
92. Thorens B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia (2014) 58(2):221–32. doi:10.1007/s00125-014-3451-1
93. Glaser B, Kesavan P, Heyman M, Davis E, Cuesta A, Buchs A, et al. Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med (1998) 338(4):226–30. doi:10.1056/NEJM199801223380404
94. Matschinsky FM. Regulation of pancreatic β-cell glucokinase: from basics to therapeutics. Diabetes (2002) 51(Suppl 3):S394–404. doi:10.2337/diabetes.51.2007.S394
95. Agren A, Borg K, Brolin S, Carlman J, Lundqvist G. Hydroxyacyl CoA dehydrogenase, an enzyme important in fat metabolism in different cell types in the islets of Langerhans. Diabete Metab (1977) 3:169–72.
96. Clayton P, Eaton S, Aynsley-Green A, Edginton M, Hussain K, Krywawych S, et al. Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of beta-oxidation in insulin secretion. J Clin Invest (2001) 108:457–65. doi:10.1172/JCI200111294
97. Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, et al. Free fatty acids regulate insulin secretion from pancreatic [beta] cells through GPR40. Nature (2003) 422(6928):173–6. doi:10.1038/nature01478
98. Eaton S, Chatziandreou I, Krywawych S, Pen S, Clayton PT, Hussain K. Short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency associated with hyperinsulinism: a novel glucose–fatty acid cycle? Biochem Soc Trans (2003) 31(6):1137–9. doi:10.1042/bst0311137
99. Lantz KA, Vatamaniuk MZ, Brestelli JE, Friedman JR, Matschinsky FM, Kaestner KH. Foxa2 regulates multiple pathways of insulin secretion. J Clin Investig (2004) 114(4):512–20. doi:10.1172/JCI200421149
100. Babiker O, Flanagan SE, Ellard S, Girim HA, Hussain K, Senniappan S. Protein-induced hyperinsulinaemic hypoglycaemia due to a homozygous HADH mutation in three siblings of a Saudi family. J Pediatr Endocrinol Metab (2015) 28(9–10):1073–7. doi:10.1515/jpem-2015-0033
101. Heslegrave A, Kapoor R, Eaton S, Chadefaux B, Akcay T, Simsek E, et al. Leucine-sensitive hyperinsulinaemic hypoglycaemia in patients with loss of function mutations in 3-Hydroxyacyl-CoA Dehydrogenase. Orphanet J Rare Dis (2012) 7(1):25. doi:10.1186/1750-1172-7-25
102. Filling C, Keller B, Hirschberg D, Marschall H, Jornvall H, Bennett M, et al. Role of short-chain hydroxyacyl CoA dehydrogenases in SCHAD deficiency. Biochem Biophys Res Commun (2008) 368:6–11. doi:10.1016/j.bbrc.2007.10.188
103. Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science (2004) 303(5662):1378–81. doi:10.1126/science.1089769
104. Wang H, Maechler P, Antinozzi PA, Hagenfeldt KA, Wollheim CB. Hepatocyte nuclear factor 4α regulates the expression of pancreatic β-cell genes implicated in glucose metabolism and nutrient-induced insulin secretion. J Biol Chem (2000) 275(46):35953–9. doi:10.1074/jbc.M006612200
105. Gupta RK, Vatamaniuk MZ, Lee CS, Flaschen RC, Fulmer JT, Matschinsky FM, et al. The MODY1 gene HNF-4alpha regulates selected genes involved in insulin secretion. J Clin Invest (2005) 115(4):1006–15. doi:10.1172/JCI200522365
106. Pearson ER, Boj SF, Steele AM, Barrett T, Stals K, Shield JP, et al. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med (2007) 4(4):e118. doi:10.1371/journal.pmed.0040118
107. Miura A, Yamagata K, Kakei M, Hatakeyama H, Takahashi N, Fukui K, et al. Hepatocyte nuclear factor-4α is essential for glucose-stimulated insulin secretion by pancreatic β-cells. J Biol Chem (2006) 281(8):5246–57. doi:10.1074/jbc.M507496200
108. Gremlich S, Nolan C, Roduit R, Burcelin R, Peyot ML, Delghingaro-Augusto V, et al. Pancreatic islet adaptation to fasting is dependent on peroxisome proliferator-activated receptor alpha transcriptional up-regulation of fatty acid oxidation. Endocrinology (2005) 146(1):375–82. doi:10.1210/en.2004-0667
109. Fajans SS, Bell GI. Macrosomia and neonatal hypoglycaemia in RW pedigree subjects with a mutation (Q268X) in the gene encoding hepatocyte nuclear factor 4α (HNF4A). Diabetologia (2007) 50(12):2600–1. doi:10.1007/s00125-007-0833-7
110. Kapoor RR, Locke J, Colclough K, Wales J, Conn JJ, Hattersley AT, et al. Persistent hyperinsulinemic hypoglycemia and maturity-onset diabetes of the young due to heterozygous HNF4A mutations. Diabetes (2008) 57(6):1659–63. doi:10.2337/db07-1657
111. Flanagan SE, Kapoor RR, Mali G, Cody D, Murphy N, Schwahn B, et al. Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. Eur J Endocrinol (2010) 162(5):987–92. doi:10.1530/eje-09-0861
112. Thomas H, Jaschkowitz K, Bulman M, Frayling TM, Mitchell SMS, Roosen S, et al. A distant upstream promoter of the HNF-4α gene connects the transcription factors involved in maturity-onset diabetes of the young. Hum Mol Genet (2001) 10(19):2089–97. doi:10.1093/hmg/10.19.2089
113. Rozenkova K, Malikova J, Nessa A, Dusatkova L, Bjorkhaug L, Obermannova B, et al. High incidence of heterozygous ABCC8 and HNF1A mutations in czech patients with congenital hyperinsulinism. J Clin Endocrinol Metab (2015) 100(12):E1540–9. doi:10.1210/jc.2015-2763
114. Otonkoski T, Jiao H, Kaminen-Ahola N, Tapia-Paez I, Ullah MS, Parton LE, et al. Physical exercise–induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic β cells. Am J Hum Genet (2007) 81(3):467–74. doi:10.1086/520960
115. Meissner T, Otonkoski T, Feneberg R, Beinbrech B, Apostolidou S, Sipila I, et al. Exercise induced hypoglycaemic hyperinsulinism. Arch Dis Child (2001) 84(3):254–7. doi:10.1136/adc.84.3.254
116. Tu N, Chen H, Winnikes U, Reinert I, Marmann G, Pirke KM, et al. Structural organization and mutational analysis of the human uncoupling protein-2 (hUCP2) gene. Life Sci (1999) 64(3):l41–50.
117. Krauss S, Zhang C-Y, Lowell BB. A significant portion of mitochondrial proton leak in intact thymocytes depends on expression of UCP2. Proc Natl Acad Sci U S A (2002) 99(1):118–22. doi:10.1073/pnas.012410699
118. González-Barroso MM, Giurgea I, Bouillaud F, Anedda A, Bellanné-Chantelot C, Hubert L, et al. Mutations in UCP2 in congenital hyperinsulinism reveal a role for regulation of insulin secretion. PLoS One (2008) 3(12):e3850. doi:10.1371/journal.pone.0003850
119. Hojlund K, Hansen T, Lajer M, Henriksen JE, Levin K, Lindholm J, et al. A novel syndrome of autosomal-dominant hyperinsulinemic hypoglycemia linked to a mutation in the human insulin receptor gene. Diabetes (2004) 53(6):1592–8. doi:10.2337/diabetes.53.6.1592
120. Samuk I, Afriat R, Horne T, Bistritzer T, Barr J, Vinograd I. Dumping syndrome following Nissen fundoplication, diagnosis, and treatment. J Pediatr Gastroenterol Nutr (1996) 23(3):235–40. doi:10.1097/00005176-199610000-00006
121. Palladino AA, Sayed S, Levitt Katz LE, Gallagher PR, De Leon DD. Increased glucagon-like peptide-1 secretion and postprandial hypoglycemia in children after Nissen fundoplication. J Clin Endocrinol Metab (2009) 94(1):39–44. doi:10.1210/jc.2008-1263
122. Kneepkens CM, Fernandes J, Vonk RJ. Dumping syndrome in children. Diagnosis and effect of glucomannan on glucose tolerance and absorption. Acta Paediatr Scand (1988) 77(2):279–86. doi:10.1111/j.1651-2227.1988.tb10643.x
123. Basu A, Service FJ, Yu L, Heser D, Ferries LM, Eisenbarth G. Insulin autoimmunity and hypoglycemia in seven white patients. Endocr Pract (2005) 11(2):97–103. doi:10.4158/EP.11.2.97
124. Lupsa BC, Chong AY, Cochran EK, Soos MA, Semple RK, Gorden P. Autoimmune forms of hypoglycemia. Medicine (2009) 88(3):141–53. doi:10.1097/MD.0b013e3181a5b42e
125. Chandarana K, Batterham RL. Shedding pounds after going under the knife: metabolic insights from cutting the gut. Nat Med (2012) 18(5):668–9. doi:10.1038/nm.2748
126. Yousseif A, Emmanuel J, Karra E, Millet Q, Elkalaawy M, Jenkinson AD, et al. Differential effects of laparoscopic sleeve gastrectomy and laparoscopic gastric bypass on appetite, circulating acyl-ghrelin, peptide YY3-36 and active GLP-1 levels in non-diabetic humans. Obes Surg (2014) 24(2):241–52. doi:10.1007/s11695-013-1066-0
127. Service GJ, Thompson GB, Service FJ, Andrews JC, Collazo-Clavell ML, Lloyd RV. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med (2005) 353(3):249–54. doi:10.1056/NEJMoa043690
128. Service FJ, Natt N, Thompson GB, Grant CS, van Heerden JA, Andrews JC, et al. Noninsulinoma pancreatogenous hypoglycemia: a novel syndrome of hyperinsulinemic hypoglycemia in adults independent of mutations in Kir6.2 and SUR1 genes. J Clin Endocrinol Metab (1999) 84(5):1582–9. doi:10.1210/jc.84.5.1582
129. Won JG, Tseng HS, Yang AH, Tang KT, Jap TS, Lee CH, et al. Clinical features and morphological characterization of 10 patients with noninsulinoma pancreatogenous hypoglycaemia syndrome (NIPHS). Clin Endocrinol (2006) 65(5):566–78. doi:10.1111/j.1365-2265.2006.02629.x
130. Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest (2005) 115(8):2047–58. doi:10.1172/jci25495
131. Service FJ, McMahon MM, O’Brien PC, Ballard DJ. Functioning insulinoma – incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc (1991) 66(7):711–9. doi:10.1016/S0025-6196(12)62083-7
132. Kar P, Price P, Sawers S, Bhattacharya S, Reznek RH, Grossman AB. Insulinomas may present with normoglycemia after prolonged fasting but glucose-stimulated hypoglycemia. J Clin Endocrinol Metab (2006) 91(12):4733–6. doi:10.1210/jc.2006-1430
Keywords: hyperinsulinemic hypoglycemia, congenital hyperinsulinism, glucose, insulin, KATP channels
Citation: Nessa A, Rahman SA and Hussain K (2016) Hyperinsulinemic Hypoglycemia – The Molecular Mechanisms. Front. Endocrinol. 7:29. doi: 10.3389/fendo.2016.00029
Received: 14 December 2015; Accepted: 21 March 2016;
Published: 31 March 2016
Edited by:
José C. Moreno, Institute for Medical and Molecular Genetics-INGEMM, SpainReviewed by:
Mars Skae, Royal Manchester Children’s Hospital, UKGuiomar Perez De Nanclares, Osakidetza, Spain
Copyright: © 2016 Nessa, Rahman and Hussain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Khalid Hussain, a2hhbGlkLmh1c3NhaW5AdWNsLmFjLnVr