 Simona Ronchetti
Simona Ronchetti Graziella Migliorati
Graziella Migliorati Carlo Riccardi
Carlo Riccardi- Section of Pharmacology, Department of Medicine, University of Perugia, Perugia, Italy
Glucocorticoid-induced leucine zipper (GILZ) is a dexamethasone-inducible gene that mediates glucocorticoid (GC) actions in a variety of cell types, including many cells of immune system. In particular, GILZ can control T cell activities, such as activation and differentiation, mainly through its ability to homo- and hetero-dimerize with partner proteins, such as NF-κB, Ras, and C/EBP. These protein–protein interactions control the regulation of pro-inflammatory target genes. A number of in vitro and in vivo studies using mouse models of inflammatory diseases demonstrate an anti-inflammatory role for GILZ. Here, authors summarize the studies that make GILZ eligible as an anti-inflammatory protein through which GCs can act. These findings permit the future development of pharmacological tools that mimic the therapeutic effects of GCs while avoiding the detrimental ones.
Introduction
Glucocorticoid-induced leucine zipper (GILZ) is a dexamethasone-inducible gene belonging to the TSC-22 family of proteins, which are characterized by the presence of TSC box and leucine zipper domains (1). GILZ expression is rapidly and ubiquitously induced by glucocorticoids (GCs) in various types of cells, including lymphoid cells, in which it regulates activation and apoptosis. An emerging body of literature has established GILZ as an important mediator of GC anti-inflammatory effects in cell lineages and in inflammatory mouse models, by inhibiting expression of pro-inflammatory genes (2–4). This effect resides in the ability of GILZ to homo- and hetero-dimerize with known transcription factors, thereby influencing gene transcription. Some of these partner proteins are NF-κB, Raf-1, TORC2, AP-1, Ras, and C/EBPs; all of these transcription factors are pivotal players in pro-inflammatory signaling pathways. Some of them, such as NF-κB and Raf-1/Ras, are even involved in oncogenesis signaling pathways. TORC2 is specifically bound by GILZ in BCR–ABL cells, leading to apoptosis (5). GILZ has also been found to be involved in allergy, being downregulated in DCs of respiratory allergic patients or in the airways of allergen-challenged asthmatic subjects and upregulated after GC treatment (6, 7). Thus, GILZ plays a pivotal role into the inflammatory processes of various sources.
Glucocorticoids are the most used anti-inflammatory drugs by virtue of their powerful effects on the cells of the immune system. However, these drugs also exhibit detrimental side effects, which can limit their usefulness. It is commonly assumed that transactivation accounts for the onset of side effects as a consequence of the metabolic activity of GCs, and transrepression for the therapeutic effects via inhibition of pro-inflammatory proteins. Nonetheless, GILZ is one of the earliest and most GC inducible genes by transactivation, as well as other genes with anti-inflammatory properties (e.g., Iκ-B). Therefore, the transactivation process is responsible also for some anti-inflammatory effects of GCs, through the use of GILZ as a mediator (4, 8, 9).
Glucocorticoid-induced leucine zipper is currently considered an important molecular player in the multiple mechanisms of GC-influenced gene regulation of inflammation and a new focus for anti-inflammatory strategies.
GILZ Interaction with Transcription Factors
Inhibition of the transcription factor NF-κB represents one of the mechanisms by which GCs repress gene activation without binding DNA, through an indirect pathway, in a process called transrepression (10, 11). GILZ is postulated to be a mediator of GC-induced transrepression because it can bind NF-κB and inhibit nuclear translocation of the transcription factor, in T cells and TCR-triggered thymocytes (12–14). As a consequence of this interaction, expression of some immune and inflammatory response genes is repressed. In T cells, overexpression of GILZ counteracts the NF-κB-induced up-regulation of the Fas/FasL system, thus preventing activation-induced apoptosis (1). In thymocytes, the same mechanism seems to be responsible for the inhibition of cell death, controlled by some NF-κB-regulated genes (12, 15). Very recently, in B cells, lack of GILZ has been found to cause increased NF-κB transcriptional activity with consequent contribution to the development of non-lethal B lymphocytosis (16). In bone marrow mesenchymal stem cells, GILZ inhibits COX-2 expression through inhibition of NF-κB. Furthermore, GILZ knockdown reduces GC inhibitory effect on cytokine-induced COX-2 expression, further demonstrating that GILZ can be a mediator of GC effects (17).
The interaction between GILZ and NF-κB has many other consequences. A particular, and still unexplained, interaction with NF-κB comes from studies in endothelial cells. IL-10 was found to upregulate GILZ in human umbilical vein endothelial cells (HUVEC), thus contributing to the failure to endothelium-dependent costimulation of CD4 T cells (18). In another study with the same cell type, overexpression of GILZ decreased TNF-induced transmigration of leukocytes by inhibition of NF-κB binding to DNA (19). These results would suggest that GILZ is involved in leukocyte rolling and adhesion. Interestingly, neither physical interaction between GILZ and the p65 NF-κB subunit nor inhibition of NF-κB translocation to the nucleus was observed. Thus, the exact mechanism through which GILZ inhibits NF-κB is still unclear. Similarly, another study demonstrated that endogenous expression of GILZ in HUVEC prevents vascular inflammation via inhibition of NF-κB nuclear translocation (20). Thus, NF-κB represents a key target for GILZ-mediated GC anti-inflammatory activity, and, by consequence, GILZ represents a potential target for treatment of inflammation in the endothelium.
Glucocorticoid-induced leucine zipper was also found to homo- and hetero-dimerize with AP-1 pathway components, such as c-Jun and c-Fos. GILZ can bind c-Jun and c-Fos through its N-terminal region, thereby inhibiting AP-1-driven activation. Such an interaction contributes to the regulation of AP-1 activity by GCs through a mechanism different from GR direct binding to transcription factors (21).
Although GILZ has been found to antagonize the activity of transcription factors, such as NF-κB and AP-1, it can also act as a chaperone protein. GILZ expression is induced by aldosterone in cortical collecting tubules in the kidney. Once induced, GILZ contributes to the stimulation of epithelial sodium channel (ENaC)-mediated Na+ transport via mineralocorticoids through the inhibition of the ERK cascade (22, 23). Through a second GILZ-dependent mechanism, ion transport can be controlled through physical interaction between GILZ and serum, and glucocorticoid-induced kinase 1 (SGK1). GILZ stabilizes SGK1 by recruiting it to the ER. This event inhibits SGK1 ubiquitination and, as a result, ENaC is activated (24). Therefore, GILZ represents a key regulator of transepithelial ion transport in the kidneys.
Pharmacological treatment with GCs is known to promote osteoporosis; however, osteoporosis can be induced by chronic inflammation as well (25). GILZ, despite being a GC-induced gene, is involved in the regulation of osteogenesis. In mesenchymal stem cells, GILZ acts as a transcriptional repressor by inhibiting C/EBP-δ-induced PPAR-γ2 expression, a key regulator of adipogenesis. GILZ binds specifically to tandem C/EBP sites in the promoter, forming complexes with C/EBP-δ and other still unknown transcription factors, thus inhibiting expression of the target genes (26, 27). Therefore, GILZ can drive the differentiation of mesenchymal stem cells toward the osteogenic pathway. These findings suggest that GILZ expression is important for osteoblast differentiation, whereas lack of GILZ can trigger the maturation of adipocytes. Overall, GILZ expression tips the balance of mesenchymal stem cell commitment in the bone marrow, playing an opposing role to GCs, which induce the differentiation toward adipocytes (28). Most likely, GILZ can temper the damage caused to bones by chronic inflammation because of its anti-inflammatory properties.
GILZ and T Lymphocytes
Control of lymphocyte function during an immune response by means of GCs implies regulation of activation and apoptosis of T cells. GILZ was found to modulate T lymphocyte response in the murine cell line 3DO; overexpression of GILZ inhibited anti-CD3-induced apoptosis through the regulation of the Fas/FasL system (1). Thereafter, our laboratory reported that GILZ inhibits TCR-induced IL-2 and IL-2 receptor expression, which can explain the aforementioned effect on the inhibition of Fas/FasL expression and consequent T cell survival (12). These results indicate GILZ counters TCR-induced T cell activation, thus contributing to T cell anergy (Figure 1). Since then, additional studies have been published, supporting a role of GILZ as a mediator of GC action on T lymphocytes, through its function in regulating the homeostasis of these cells (3). One mechanism by which GILZ regulates T lymphocyte is through IL-2, the main survival interleukin in activated T lymphocytes. IL-2 is downregulated by GILZ, and it also inhibits GILZ expression through the repression of the transcription factor FoxO3, which directly acts on GILZ promoter (12, 29). Therefore, FoxO3 seems to be the mediator of GC action and IL-2 antagonism for GILZ regulation in T lymphocytes. In turn, GILZ negatively regulates FoxO factors, such as FoxO1, FoxO3, and FoxO4 (30).
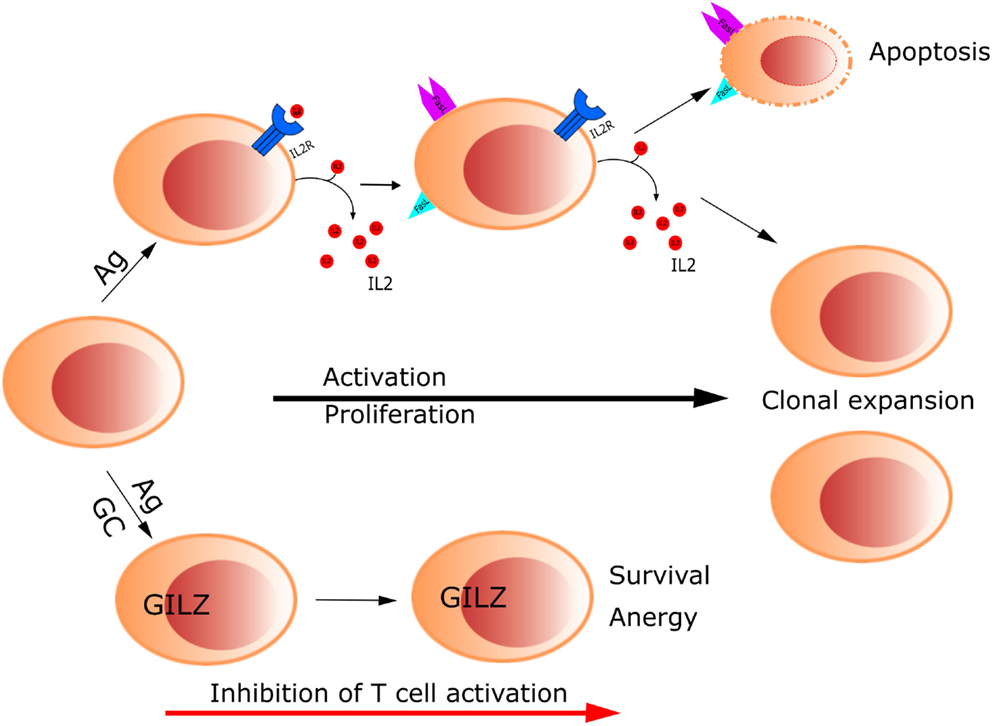
Figure 1. Glucocorticoids control T cell activation through up-regulation of GILZ. GILZ inhibits TCR-induced IL-2 and IL-2 receptor expression, which in turn inhibits Fas/FasL expression, promoting T cell survival and anergy.
Like GCs, GILZ can modulate a switch from Th1 to Th2 immune phenotypes, as demonstrated by the up-regulation and down-regulation of Th2 and Th1 cytokines, respectively. This conclusion was demonstrated in CD4+ cells of GILZ transgenic mice (TG), where an increase of IL-4 and a decrease of IFN-γ were observed. This shift in cytokine expression causes GILZ TG mice to be less susceptible to the induction of colitis. Much like what has been observed for the other transcription factors, GILZ can regulate the transcription of factors that control Th1- and Th2-specific cytokine expression. GILZ up-regulates GATA 3 and STAT6 to modulate the Th2 phenotype, and downregulates T-bet to modulate the Th1 phenotype (9).
GILZ plays another important role by contributing to the TGF-β-mediated generation of peripheral Treg (pTreg). GCs synergize with TGF-β signaling to increase Treg formation in upregulating Foxp3, the master regulator transcription factor of Tregs (31). GILZ-knock-out (KO) mice show a reduction of pTreg cells due to a diminished TGF-β signaling. As a consequence, an increased susceptibility to DNBS-induced Th1-type colitis is associated with GILZ absence. Thus, GILZ represents the means by which stress hormones encounter TGF-β signaling in the control of pTreg production (32).
Most recently, GILZ was found to regulate Th17 cells. GILZ controls the secretion of IL-17A by CD4 T cells by regulating the expression of cytokines favoring the Th17 phenotype, such as IL-1α, IL-23, and IL-6. In the mouse imiquimod model of psoriasis, in which Th17 lymphocytes play a pathogenic role, GILZ-KO mice developed excessive inflammation and exhibited enhanced pro-inflammatory cytokine expression. These results suggest that Th17 cells are another T cell subtype controlled by GILZ (33).
Glucocorticoid-induced leucine zipper is also pivotal in controlling T lymphocyte functions prior to their maturation in the thymus. Constitutive GILZ overexpression in GILZ-TG mice causes an increase in apoptosis, which is associated with an augmented activation of the caspase-8/caspase-3 pathway. As a consequence, adult GILZ-TG mice show a reduced number of CD4 and CD8 double positive thymocytes (34). In addition, GILZ prevents apoptosis induced by stimulation of TCR in thymocytes through the inhibition of NF-κB (15). Because GCs induce apoptosis in thymocytes and rapidly upregulate GILZ, the latter represents the mediator of GC action in the thymus.
GILZ Function in Mouse Models of Inflammation
Because GILZ is involved in the inflammatory process, it has been studied in many of the mouse models for inflammatory diseases. Inflammatory bowel diseases (IBDs) are autoimmune/inflammatory diseases of the gastrointestinal tract that can be easily reproduced in mice. IBDs are usually treated with GCs; thus, given the relationship between GILZ and GCs, the role of GILZ has been studied in rodent models of IBDs. In TG mice overexpressing GILZ in CD4+ cells, the severity of colonic inflammation is diminished compared to the control mice. This result suggests that GILZ inhibits Th1 activation in favor of the Th2 commitment of CD4+ cells exerted. Furthermore, treatment of IL-10-KO mice with a recombinant TAT–GILZ protein reduces the severity of spontaneously developed colitis (8). The Th2 phenotype of GILZ-TG mice was shown previously in another Th2-driven disease and model of inflammation: bleomycin-induced lung injury. GILZ-TG mice exhibit a more severe bleomycin-induced lung injury, but they are protected under a Th1-dependent delayed-type hypersensitivity response (9). From these data, one might hypothesize that the effect of GILZ on Th2-driven differentiation overlaps with the action of GC.
Overexpression of GILZ in the TG mice has proven to be a helpful tool in the study of GILZ in another model of inflammation, i.e., the post-traumatic disease that develops after spinal cord injury (SCI). GILZ prevents the development of SCI mainly by inhibiting T cell activation and the release of pro-inflammatory cytokines, such as TNF-α and IL-1β (35). Similar effects on activated T cells were found in a mouse model of experimental autoimmune encephalomyelitis, in which a GILZ peptide (GILZ-P) interacting with NF-κB was administered on the day of disease onset. Treated mice were protected against the disease, demonstrating the powerful control GILZ can exert on T cell activity (36). Using the same inflammatory model, another study demonstrated that the induction of GILZ in dendritic cells mirrors the immunomodulation by GC in the control of autoaggressive T cell responses (37).
Anti-inflammatory actions of GILZ are evident even in mouse models of acute inflammation, such as endotoxemia. GILZ expression led to the resistance of SPRET/Ei mice to LPS-induced endotoxemia through an altered cytokine production. Furthermore, mice that received in vivo administration of a pES34–TAT–GILZ expression vector exhibited increased resistance to lethal effects of LPS (38). In a second example, GILZ was studied in the rodent model of LPS-induced pleurisy. Administration of recombinant TAT–GILZ protein accelerated the resolution phase of acute inflammation, reducing the magnitude of PMN infiltration by inducing apoptosis (39). Lastly, GILZ modulates acute inflammation in other cells of the innate system, the macrophages. In a mouse model for LPS tolerance, GILZ was upregulated in alveolar macrophages, whereas in endotoxin-tolerized GILZ-KO macrophages, cytokine induction and MAPK activation were rescued (40).
Glucocorticoid-induced leucine zipper plays a role in arthritis as well. In the CIA model of arthritis, GILZ expression mimicked the therapeutic effect of GCs, and deletion of GILZ increased the severity of the disease together with the expression of TNF and IL-1. Importantly, GILZ was found in the sinovium of patients with active RA, suggesting GILZ is crucial for the regulation of the local inflammatory response functioning as an endogenous inhibitor of chronic inflammation (41). Overall, these examples of GILZ involvement in inflammatory diseases clearly illustrate that GILZ acts as a mediator for the GC therapeutic effect, both in T lymphocytes and other cells of the innate immune system.
Beyond Inflammation
The ability of GILZ to hetero-dimerize even with “non-LZ” proteins extends its functions beyond inflammation to a number of additional pathways. GILZ can form heterodimers with Ras and Raf from the MAP-kinase/ERK-kinase pathway, as well as ternary complex with both binding partners, thereby controlling growth of the cell (42). Signaling through the Ras family of small GTPases is pivotal in the transmission of mitogenic stimuli to the cell cycle machinery. The fact that GILZ binds predominantly to activated Ras, but not to Raf, suggests a fine tuning of the regulation of this pathway. Interestingly, GILZ binds to Raf when Ras in not activated. Thus, in the case of activated Ras, both Akt/Pkb serine (threonine kinase) and ERK pathways are inhibited by GILZ. When Ras is not activated, GILZ can influence only the ERK pathway via interaction with Raf (Figure 2).
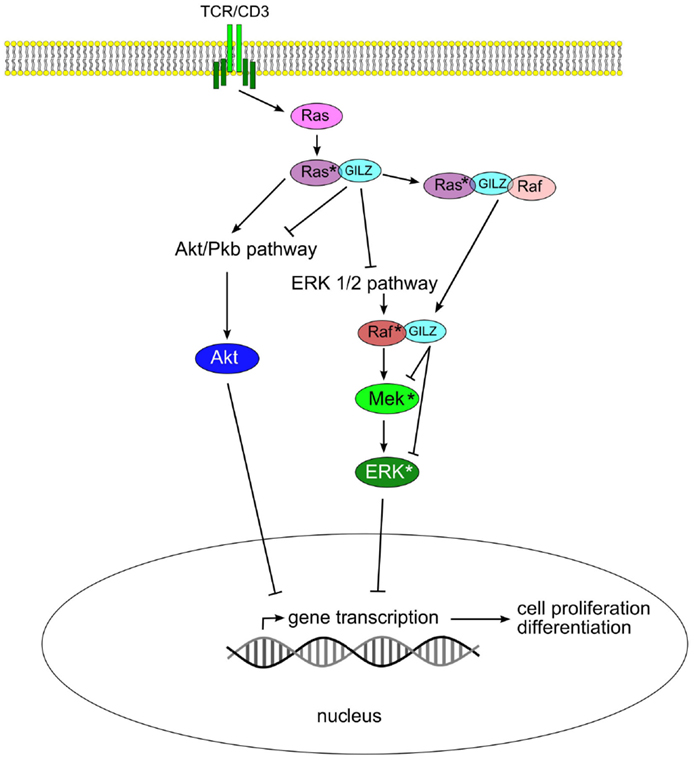
Figure 2. Glucocorticoid-induced leucine zipper inhibits Akt/Pkb and ERK 1/2 pathways by binding to Ras and Raf, thus blocking cell proliferation and transformation.
Conclusion
In recent years, GILZ has emerged as a pivotal mediator of GC action in the inflammatory process, as demonstrated by many in vitro and in vivo studies. This GILZ function opens the possibility to develop new or repurposed pharmaceutical tools for the treatment of inflammation-based diseases. These new therapies could achieve obtaining the same effect as GC treatment, but circumvent the undesirable side effects caused by GCs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by a grant from the Associazione Italiana per la Ricerca sul Cancro (AIRC), Milan, Italy (IG14291).
References
1. D’Adamio F, Zollo O, Moraca R, Ayroldi E, Bruscoli S, Bartoli A, et al. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity (1997) 7(6):803–12. doi: 10.1016/S1074-7613(00)80398-2
2. Ayroldi E, Macchiarulo A, Riccardi C. Targeting glucocorticoid side effects: selective glucocorticoid receptor modulator or glucocorticoid-induced leucine zipper? A perspective. FASEB J (2014) 28(12):5055–70. doi:10.1096/fj.14-254755
3. Ayroldi E, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J (2009) 23(11):3649–58. doi:10.1096/fj.09-134684
4. Cheng Q, Morand E, Yang YH. Development of novel treatment strategies for inflammatory diseases-similarities and divergence between glucocorticoids and GILZ. Front Pharmacol (2014) 5:169. doi:10.3389/fphar.2014.00169
5. Joha S, Nugues AL, Hetuin D, Berthon C, Dezitter X, Dauphin V, et al. GILZ inhibits the mTORC2/AKT pathway in BCR-ABL(+) cells. Oncogene (2012) 31(11):1419–30. doi:10.1038/onc.2011.328
6. Karaki S, Garcia G, Tcherakian C, Capel F, Tran T, Pallardy M, et al. Enhanced glucocorticoid-induced leucine zipper in dendritic cells induces allergen-specific regulatory CD4(+) T-cells in respiratory allergies. Allergy (2014) 69(5):624–31. doi:10.1111/all.12379
7. Kelly MM, King EM, Rider CF, Gwozd C, Holden NS, Eddleston J, et al. Corticosteroid-induced gene expression in allergen-challenged asthmatic subjects taking inhaled budesonide. Br J Pharmacol (2012) 165(6):1737–47. doi:10.1111/j.1476-5381.2011.01620.x
8. Cannarile L, Cuzzocrea S, Santucci L, Agostini M, Mazzon E, Esposito E, et al. Glucocorticoid-induced leucine zipper is protective in Th1-mediated models of colitis. Gastroenterology (2009) 136(2):530–41. doi:10.1053/j.gastro.2008.09.024
9. Cannarile L, Fallarino F, Agostini M, Cuzzocrea S, Mazzon E, Vacca C, et al. Increased GILZ expression in transgenic mice up-regulates Th-2 lymphokines. Blood (2006) 107(3):1039–47. doi:10.1182/blood-2005-05-2183
10. Almawi WY, Melemedjian OK. Negative regulation of nuclear factor-kappaB activation and function by glucocorticoids. J Mol Endocrinol (2002) 28(2):69–78. doi:10.1677/jme.0.0280069
11. Strehl C, Buttgereit F. Optimized glucocorticoid therapy: teaching old drugs new tricks. Mol Cell Endocrinol (2013) 380(1–2):32–40. doi:10.1016/j.mce.2013.01.026
12. Ayroldi E, Migliorati G, Bruscoli S, Marchetti C, Zollo O, Cannarile L, et al. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood (2001) 98(3):743–53. doi:10.1182/blood.V98.3.743
13. Di Marco B, Massetti M, Bruscoli S, Macchiarulo A, Di Virgilio R, Velardi E, et al. Glucocorticoid-induced leucine zipper (GILZ)/NF-kappaB interaction: role of GILZ homo-dimerization and C-terminal domain. Nucleic Acids Res (2007) 35(2):517–28. doi:10.1093/nar/gkl1080
14. Riccardi C, Bruscoli S, Ayroldi E, Agostini M, Migliorati G. GILZ, a glucocorticoid hormone induced gene, modulates T lymphocytes activation and death through interaction with NF-kB. Adv Exp Med Biol (2001) 495:31–9. doi:10.1007/978-1-4615-0685-0_5
15. Delfino DV, Agostini M, Spinicelli S, Vacca C, Riccardi C. Inhibited cell death, NF-kappaB activity and increased IL-10 in TCR-triggered thymocytes of transgenic mice overexpressing the glucocorticoid-induced protein GILZ. Int Immunopharmacol (2006) 6(7):1126–34. doi:10.1016/j.intimp.2006.02.001
16. Bruscoli S, Biagioli M, Sorcini D, Frammartino T, Cimino M, Sportoletti P, et al. Lack of glucocorticoid-induced leucine zipper (GILZ) deregulates B cell survival and results in B cell lymphocytosis in mice. Blood (2015) 126(15):1790–801. doi:10.1182/blood-2015-03-631580
17. Yang N, Zhang W, Shi XM. Glucocorticoid-induced leucine zipper (GILZ) mediates glucocorticoid action and inhibits inflammatory cytokine-induced COX-2 expression. J Cell Biochem (2008) 103(6):1760–71. doi:10.1002/jcb.21562
18. Gleissner CA, Zastrow A, Klingenberg R, Kluger MS, Konstandin M, Celik S, et al. IL-10 inhibits endothelium-dependent T cell costimulation by up-regulation of ILT3/4 in human vascular endothelial cells. Eur J Immunol (2007) 37(1):177–92. doi:10.1002/eji.200636498
19. Cheng Q, Fan H, Ngo D, Beaulieu E, Leung P, Lo CY, et al. GILZ overexpression inhibits endothelial cell adhesive function through regulation of NF-kappaB and MAPK activity. J Immunol (2013) 191(1):424–33. doi:10.4049/jimmunol.1202662
20. Hahn RT, Hoppstadter J, Hirschfelder K, Hachenthal N, Diesel B, Kessler SM, et al. Downregulation of the glucocorticoid-induced leucine zipper (GILZ) promotes vascular inflammation. Atherosclerosis (2014) 234(2):391–400. doi:10.1016/j.atherosclerosis.2014.03.028
21. Mittelstadt PR, Ashwell JD. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J Biol Chem (2001) 276(31):29603–10. doi:10.1074/jbc.M101522200
22. Muller OG, Parnova RG, Centeno G, Rossier BC, Firsov D, Horisberger JD. Mineralocorticoid effects in the kidney: correlation between alphaENaC, GILZ, and Sgk-1 mRNA expression and urinary excretion of Na+ and K+. J Am Soc Nephrol (2003) 14(5):1107–15. doi:10.1097/01.ASN.0000061777.67332.77
23. Soundararajan R, Zhang TT, Wang J, Vandewalle A, Pearce D. A novel role for glucocorticoid-induced leucine zipper protein in epithelial sodium channel-mediated sodium transport. J Biol Chem (2005) 280(48):39970–81. doi:10.1074/jbc.M508658200
24. Soundararajan R, Wang J, Melters D, Pearce D. Glucocorticoid-induced Leucine zipper 1 stimulates the epithelial sodium channel by regulating serum- and glucocorticoid-induced kinase 1 stability and subcellular localization. J Biol Chem (2010) 285(51):39905–13. doi:10.1074/jbc.M110.161133
25. Armour KJ, Armour KE, van’t Hof RJ, Reid DM, Wei XQ, Liew FY, et al. Activation of the inducible nitric oxide synthase pathway contributes to inflammation-induced osteoporosis by suppressing bone formation and causing osteoblast apoptosis. Arthritis Rheum (2001) 44(12):2790–6. doi:10.1002/1529-0131(200112)44:12<2790::AID-ART466>3.0.CO;2-X
26. Pan G, Cao J, Yang N, Ding K, Fan C, Xiong WC, et al. Role of glucocorticoid-induced leucine zipper (GILZ) in bone acquisition. J Biol Chem (2014) 289(28):19373–82. doi:10.1074/jbc.M113.535237
27. Shi X, Shi W, Li Q, Song B, Wan M, Bai S, et al. A glucocorticoid-induced leucine-zipper protein, GILZ, inhibits adipogenesis of mesenchymal cells. EMBO Rep (2003) 4(4):374–80. doi:10.1038/sj.embor.embor805
28. Zhang W, Yang N, Shi XM. Regulation of mesenchymal stem cell osteogenic differentiation by glucocorticoid-induced leucine zipper (GILZ). J Biol Chem (2008) 283(8):4723–9. doi:10.1074/jbc.M704147200
29. Asselin-Labat ML, Biola-Vidamment A, Kerbrat S, Lombes M, Bertoglio J, Pallardy M. FoxO3 mediates antagonistic effects of glucocorticoids and interleukin-2 on glucocorticoid-induced leucine zipper expression. Mol Endocrinol (2005) 19(7):1752–64. doi:10.1210/me.2004-0206
30. Latre de Late P, Pepin A, Assaf-Vandecasteele H, Espinasse C, Nicolas V, Asselin-Labat ML, et al. Glucocorticoid-induced leucine zipper (GILZ) promotes the nuclear exclusion of FOXO3 in a Crm1-dependent manner. J Biol Chem (2010) 285(8):5594–605. doi:10.1074/jbc.M109.068346
31. Karagiannidis C, Akdis M, Holopainen P, Woolley NJ, Hense G, Ruckert B, et al. Glucocorticoids upregulate FOXP3 expression and regulatory T cells in asthma. J Allergy Clin Immunol (2004) 114(6):1425–33. doi:10.1016/j.jaci.2004.07.014
32. Bereshchenko O, Coppo M, Bruscoli S, Biagioli M, Cimino M, Frammartino T, et al. GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF-beta signaling. Cell Rep (2014) 7(2):464–75. doi:10.1016/j.celrep.2014.03.004
33. Jones SA, Perera DN, Fan H, Russ BE, Harris J, Morand EF. GILZ regulates Th17 responses and restrains IL-17-mediated skin inflammation. J Autoimmun (2015) 61:73–80. doi:10.1016/j.jaut.2015.05.010
34. Delfino DV, Agostini M, Spinicelli S, Vito P, Riccardi C. Decrease of Bcl-xL and augmentation of thymocyte apoptosis in GILZ overexpressing transgenic mice. Blood (2004) 104(13):4134–41. doi:10.1182/blood-2004-03-0920
35. Esposito E, Bruscoli S, Mazzon E, Paterniti I, Coppo M, Velardi E, et al. Glucocorticoid-induced leucine zipper (GILZ) over-expression in T lymphocytes inhibits inflammation and tissue damage in spinal cord injury. Neurotherapeutics (2012) 9(1):210–25. doi:10.1007/s13311-011-0084-7
36. Srinivasan M, Janardhanam S. Novel p65 binding glucocorticoid-induced leucine zipper peptide suppresses experimental autoimmune encephalomyelitis. J Biol Chem (2011) 286(52):44799–810. doi:10.1074/jbc.M111.279257
37. Benkhoucha M, Molnarfi N, Dunand-Sauthier I, Merkler D, Schneiter G, Bruscoli S, et al. Hepatocyte growth factor limits autoimmune neuroinflammation via glucocorticoid-induced leucine zipper expression in dendritic cells. J Immunol (2014) 193(6):2743–52. doi:10.4049/jimmunol.1302338
38. Pinheiro I, Dejager L, Petta I, Vandevyver S, Puimege L, Mahieu T, et al. LPS resistance of SPRET/Ei mice is mediated by Gilz, encoded by the Tsc22d3 gene on the X chromosome. EMBO Mol Med (2013) 5(3):456–70. doi:10.1002/emmm.201201683
39. Vago JP, Tavares LP, Garcia CC, Lima KM, Perucci LO, Vieira EL, et al. The role and effects of glucocorticoid-induced leucine zipper in the context of inflammation resolution. J Immunol (2015) 194(10):4940–50. doi:10.4049/jimmunol.1401722
40. Hoppstadter J, Kessler SM, Bruscoli S, Huwer H, Riccardi C, Kiemer AK. Glucocorticoid-induced leucine zipper: a critical factor in macrophage endotoxin tolerance. J Immunol (2015) 194(12):6057–67. doi:10.4049/jimmunol.1403207
41. Beaulieu E, Ngo D, Santos L, Yang YH, Smith M, Jorgensen C, et al. Glucocorticoid-induced leucine zipper is an endogenous antiinflammatory mediator in arthritis. Arthritis Rheum (2010) 62(9):2651–61. doi:10.1002/art.27566
Keywords: glucocorticoids, GILZ, inflammation, immune cells, transcription factor
Citation: Ronchetti S, Migliorati G and Riccardi C (2015) GILZ as a Mediator of the Anti-Inflammatory Effects of Glucocorticoids. Front. Endocrinol. 6:170. doi: 10.3389/fendo.2015.00170
Received: 31 August 2015; Accepted: 23 October 2015;
Published: 09 November 2015
Edited by:
Isaias Glezer, Universidade Federal de São Paulo, BrazilReviewed by:
John Anthony Cidlowski, National Institutes of Health, USAOmar Tliba, Thomas Jefferson University, USA
Copyright: © 2015 Ronchetti, Migliorati and Riccardi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlo Riccardi, Y2FybG8ucmljY2FyZGlAdW5pcGcuaXQ=