 Xiaoxia Zhu
Xiaoxia Zhu Wen Zhou
Wen Zhou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 09 October 2015
Sec. Cancer Endocrinology
Volume 6 - 2015 | https://doi.org/10.3389/fendo.2015.00159
Vascular endothelial growth factor-A (VEGF) signals vascular development and angiogenesis mainly by binding to VEGF receptor family member 2 (VEGFR-2). Adaptor proteins mediate many VEGFR-2’s functions in the development of blood vessels. Cancer cells secrete VEGF to activate VEGFR-2 pathway in their neighboring endothelial cells in the process of cancer-related angiogenesis. Interestingly, activation of VEGFR-2 signaling is found in breast cancer cells, but its role and regulation are not clear. We highlighted research advances of VEGFR-2, with a focus on VEGFR-2’s regulation by mutant p53 in breast cancer. In addition, we reviewed recent Food and Drug Administration-approved tyrosine kinase inhibitor drugs that can inhibit the function of VEGFR-2. Ongoing preclinical and clinical studies might prove that pharmaceutically targeting VEGFR-2 could be an effective therapeutic strategy in treating triple-negative breast cancer.
Vascular endothelial growth factor-A (VEGF-A, also known as vascular permeability factor) is a major factor in regulating functions of endothelial cells in vasculogenesis and angiogenesis (1, 2). VEGF family consists of five members, VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placenta growth factor (3). This review focuses exclusively on VEGF-A since this isoform of VEGF is the most extensively studied, and hereafter, VEGF refers only to VEGF-A. The importance of VEGF in endothelial cells has been demonstrated by mouse models in which both VEGF-A−/− and VEGF-A+/− are embryonic lethal, and the mouse embryos died at embryonic day E9.5 and E11, respectively (4, 5). Solid tumors secrete VEGF to induce endothelial cells forming blood vessels in order to gain adequate blood supply for tumors (6). Blood vessel formations further stimulate tumor proliferation and metastasis (7–10). In breast cancer, the expression of VEGF correlates well with decreased overall survival and disease-free survival (8).
Current data reveal many facets of VEGF function diversity in both normal and cancer cells, and some of these VEGF functions in promoting breast cancer are depicted in Figure 1. Apart from its well-known role in angiogenesis, VEGF plays a critical role in stem cell maintenance. VEGF has been shown to be important for stem cells in hemopoietic, endothelial, muscle, cardiac, neuronal, and adipose tissues (3, 11–17). Recently, VEGF was found to regulate cancer stem cells (CSCs) self-renewal in brain, lung, and breast tumors (18, 19).
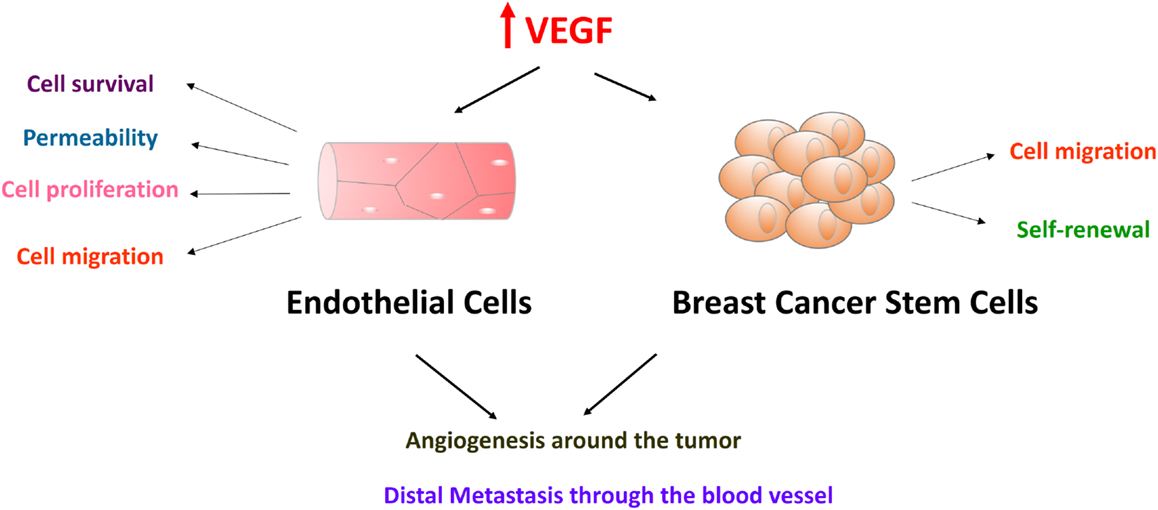
Figure 1. Vascular endothelial growth factor-A in endothelial cells and breast cancer stem cells promote breast cancer progression and metastasis. VEGF receptors are expressed in both endothelial cells and breast cancer stem cells. Endothelial cells form endothelium with tight cell–cell junction. Effects of VEGF on endothelial cells include cell survival, permeability, cell proliferation, and cell migration. VEGF also plays an important role in breast cancer cell migration and cancer stem cell self-renewal. These functions converge on promoting breast cancer progression and metastasis. Endothelial cells will be attracted and activated by high levels of VEGF proteins in the tumor-stromal niche. After endothelial cells form new blood vessels surrounding tumor cells, tumor cells will be stimulated by the cytokines in the blood supply. Tumor cells go through epithelial-to-mesenchymal transition, intravasation, circulation, extravasation, and finally form distal metastases.
Vascular endothelial growth factor, as a ligand, executes its functions through VEGF receptors. In humans, there are at least three VEGF receptors, VEGFR-1 (20, 21), VEGFR-2 (22), and VEGFR-3 (6, 23). VEGFR-2 is the principal VEGFR in humans (24). It is abundantly expressed in vascular endothelial cells and lymphatic endothelial cells (25). VEGFR-2 is also expressed in neuronal cells, megakaryocytes, hematopoietic stem cells, and different cancer cells (26–30). This review discusses the relevance of VEGFR-2 in breast cancer, particularly in breast cancer CSCs. We further discuss the mechanism through which mutant p53 activates VEGFR-2 gene expression in breast cancer. The therapeutic implications of these findings for breast cancer are also discussed.
Vascular endothelial growth factor receptor-2 is a receptor tyrosine kinase and a master node in VEGF signaling. VEGFR-2 has an extracellular portion consisting of seven immunoglobulin-like domains, a transmembrane domain, and an intracellular portion containing two tyrosine kinase domains (31). A kinase-insert domain splits its two tyrosine kinase domains. VEGF binds to and triggers two VEGFR-2 monomers to dimerize and to be autophosphorylated (3, 32, 33). A major phosphorylation site Y951 is in the kinase-insert domain (34). Four other major sites are Y1054/Y1059 in the tyrosine kinase domain and Y1175/Y1214 in the C-terminus (35, 36). Properly phosphorylated tyrosine residues serve as a binding surface for SRC homology 2 (SH2)-domain-containing adaptor proteins, as discussed below and depicted in Figure 2.
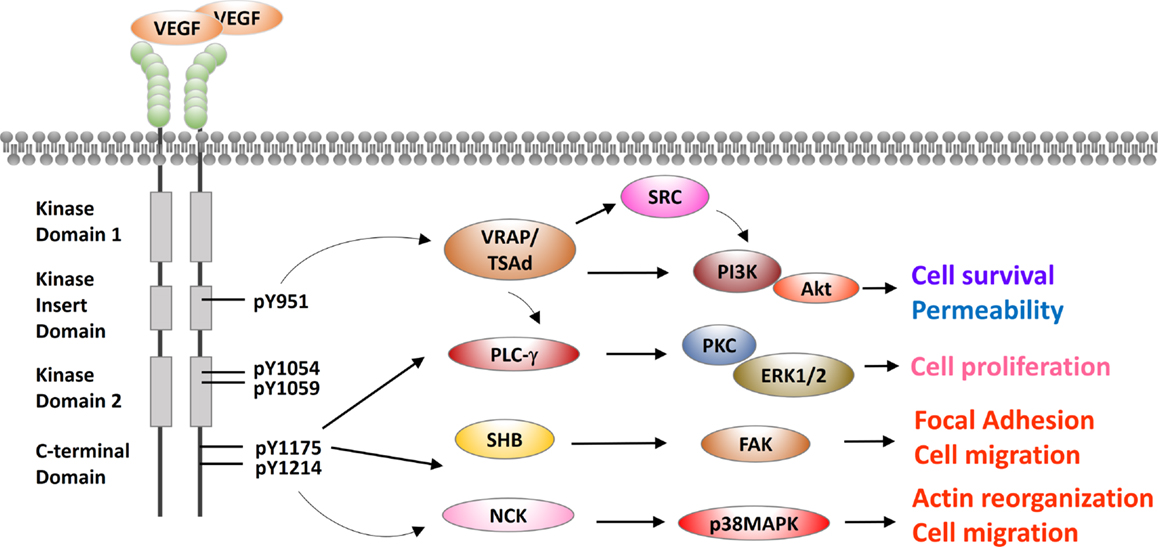
Figure 2. Cross talks between VEGFR-2 and other signaling pathways in endothelial cells. VEGFR-2 is presented in a typical receptor tyrosine kinase scheme with an extracellular domain, a juxtaposed transmembrane domain and intracellular kinase domains. Extracellular domain of VEGFR-2 is composed of seven IgG-like domains to bind to its cognate ligand VEGF. Intracellular domain has two tyrosine kinase domains, which are split by a kinase-insert domain of 70 amino acids. Five major phosphorylation residues Y951, Y1054, Y1059, Y1175, and Y1214 are labeled. SH2 domain-containing adaptor proteins are recruited by these phosphorylated tyrosine residues, including VRAP/TSAd, PLC-γ, SHB, and NCK. These adaptors mediate the downstream effects of VEGFR-2, including cell proliferation, permeability, cell survival, and cell migration.
In human umbilical vein endothelial cell models, VEGF stimulation triggers VEGFR-2 phosphorylation at Y951 and subsequent recruitment of VEGF-receptor-associated proteins (VRAPs; also known as T-cell-specific adapter molecule, TSAd) (37). VRAP contains an SH2 domain and a C-terminal proline-rich motif. VRAP is constitutively associated with SRC and phosphatidylinositol 3-kinase (PI3K) (Figure 2). SRC is a non-receptor tyrosine kinase regulating cytoskeleton reorganization, metastasis, and proliferation (38). VRAP recruits and activates PI3K either directly or through SRC. PI3K then phosphorylates membrane-bound phosphatidylinositol-3,4-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3). The binding of PIP3 to the pleckstrin homology domain of Akt leads to Akt activation. Akt has numerous and diverse biological effects by phosphorylating a variety of substrates. These effects include involvement in metabolism, protein synthesis, apoptosis pathways, transcription factor regulation, and cell cycle regulation (39–41). The overall effect of Akt activation is antiapoptosis or cell survival. In conclusion, VEGFR-2 cross talks with SRC or PI3K/Akt are mediated by VRAP/TSAd, and these cross talks are important to VEGF-induced cytoskeletal reorganization, migration, cell survival, and proliferation (34).
In porcine aortic endothelial cell models and human umbilical vein endothelial cell models, VEGFR-2 pY1175 recruits and activates phospholipase C-γ (PLC-γ), which is essential for generating inositol phosphates (35, 42). PLC-γ hydrolyzes PIP2 to release second messengers 1,2-diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). DAG is an activator of protein kinase C (PKC). PKC activates extracellular signal regulated kinases 1/2 (ERK1/2), which result in cell survival (Figure 2). IP3 binds to its receptor IP3R in endoplasmic reticulum to release Ca2+ from endoplasmic reticulum to cytoplasm. Ca2+ activates calmodulin, which further activates calcineurin. Calcineurin facilitates calcium-sensitive nuclear factor of activated T-cells (NFAT) to promote cell proliferation. Aforementioned VEGFR-2 adaptor VRAP can also activate PLC-γ. Moreover, the essential in vivo role of PLC-γ in vasculogenesis has been verified in a mutant Vegfr-2 Y1173F knock-in mouse model (murine Y1173 corresponding to Y1175 in human VEGFR-2). Vegfr-2 Y1173F mice died between embryonic days 8.5 and 9.5 without any organized blood vessels or yolk sac blood islands, and hematopoietic progenitors were severely reduced, phenotypically mimicking Vegf2−/− mice (43).
In pig aortic endothelial cells expressing human VEGFR-2 molecules, VEGF stimulation induces VEGFR-2 phosphorylation at Y1175, and pY1175 recruits SH2 domain-containing adaptor protein B (SHB) (44). SHB activates focal adhesion kinase (FAK), which is a highly conserved tyrosine kinase regulating focal adhesions (Figure 2). FAK activates small Rho GTPase RAC1, which drives actin polymerization, forms lamellipodia, and promotes cell migration (45, 46). Furthermore, Shb−/− mouse model has revealed the essential in vivo role of SHB in vasculogenesis. Shb−/− mice have abnormal endothelial ultrastructures in liver sinusoids and heart capillaries (47).
In porcine aortic endothelial cell models and human umbilical vein endothelial models, VEGFR-2 pY1214 recruits SH2/SH3 adaptor protein neuronal CDK (NCK) (48, 49). NCK activates SRC family kinase FYN (36). FYN activates p21-activated protein kinase-2 (PAK-2), and PAK-2 activates CDC42 (36). CDC42 subsequently activates p38 mitogen-activated protein kinase (MAPK) (50). p38 MAPK is a stress-activated protein kinase, and its activation is known to promote VEGF-triggered stress fiber formation and endothelial cell migration in human umbilical vein endothelial cells (51). To conclude, VEGFR2 recruits NCK/FYN to activate p38, which promotes stress fiber formation and cell migration (Figure 2).
The knowledge of VEGFR-2 signaling gained from endothelial cell model may be applied to VEGFR-2 signaling in cancer cells. For example, PLC-γ activating mutant R707Q is observed in human primary cardiac angiosarcoma. PLC-γ R707Q leads to a hyperactive VEGFR-2 signaling and increases apoptotic resistance in cancer cells (52). Furthermore, Shb−/− mice have impaired tumor growth (47).
Vascular endothelial growth factor receptor-2’s regulatory role for cancer development is largely unknown. Pfister et al. recently identified that mutant p53 activates VEGFR-2 gene expression (53). Mutant p53 and histone remodeling complex switch/sucrose non-fermentable (SWI/SNF) colocalize to the VEGFR-2 promoter. SWI/SNF remodels the VEGFR-2 promoter and keeps the promoter at an “open” configuration (Figure 3, bottom left). Pfister et al. also revealed an interesting aspect of the relationship between SWI/SNF and mutant p53 from RNA-seq analyses. The results showed that more than 40% of mutant p53-regulated genes are also under the regulation of SWI/SNF (53). It is inconclusive whether SWI/SNF coactivates all types of mutant p53s due to limited cell lines and mutation types assayed in this study. This study is important because it identifies SWI/SNF as a general cofactor of p53 mutants. Disrupting the mutant p53-SWI/SNF interaction would be an effective strategy in treating triple-negative breast cancer.
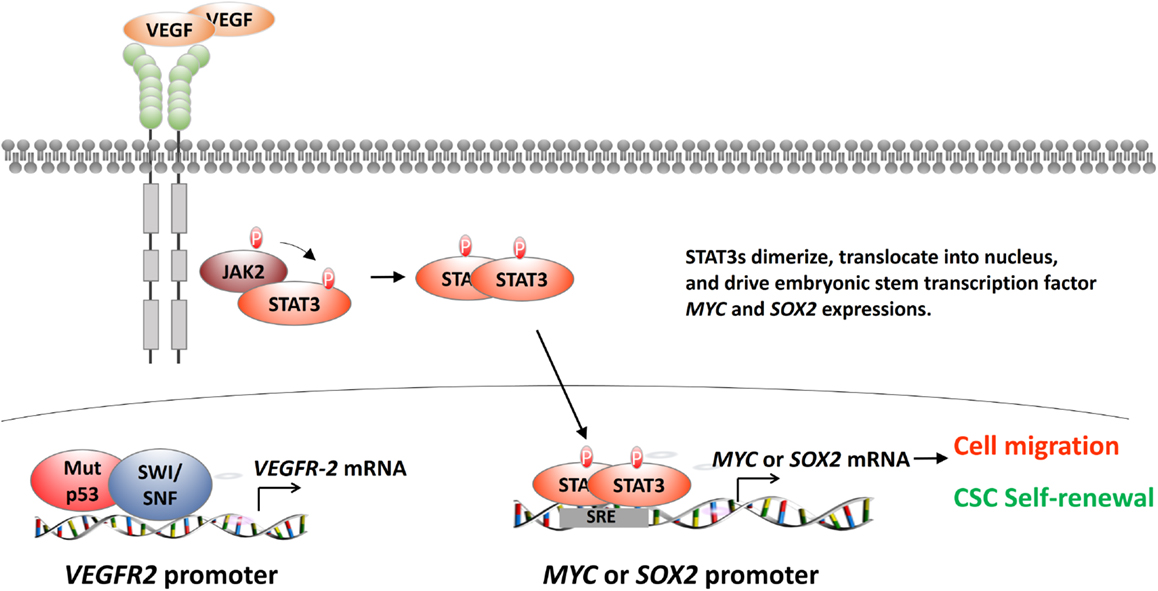
Figure 3. The regulation of VEGFR-2 in breast cancer cells. After the binding of VEGF to its cognate receptor VEGFR-2, VEGFR-2 activates STAT3 dimer formation. STAT3 dimer activates STAT3-response element (SRE)-containing genes, including MYC and SOX2 (top and lower right). MYC and SOX2 are embryonic stem cell transcription factors (ES-TF). They will affect many downstream EMT-related genes, such as up-regulating SNAIL, SLUG, ZEB1, and ZEB2 and down-regulating CDH1. These genes will endow the breast cancer cells the capability of cell motility and cancer stem cell self-renewal. Another exciting advance found that mutant p53, together with SWI/SNF histone remodeling complex, will be recruited to the VEGFR-2 promoter to activate VEGFR-2 in triple-negative breast cancer cell lines (lower left).
More importantly, Pfister et al. showed that VEGFR-2’s expression is critical for mutant p53-containing breast cancer cell growth and migration (53). The results should be appreciated because Pfister et al. extensively used mammosphere assay and other three-dimensional (3D) culture techniques (53). These 3D techniques are commonly accepted as a good indicator for clinical response. These in vitro data support that targeting VEGFR-2 might be beneficial as a cancer stem cell therapy.
Interestingly, Zhao et al. explored the role of VEGFR-2 in CSCs using established triple-negative breast cancer cell lines, dissociated primary breast tumor cells, and mice xenograft models (19). Zhao et al. found that VEGF-triggered VEGFR-2 activation increases mammospheres and aldehyde dehydrogenase activity in triple-negative breast cancer lines and dissociated primary cancers in vitro. EMT generates cancer cells with stem cell properties (54). Next, Zhao et al. focused on the role of VEGFR-2 in CSC populations and found that VEGFR-2 increases breast cancer CSCs, orthotopic tumors, and metastasis in vivo (19). More importantly, Zhao et al. delineated the downstream signaling of VEGFR-2 in CSCs (19). Zhao et al. found that VEGFR-2 recruits Janus-family tyrosine kinase 2/signal transducers and activators of transcription 3 (JAK2/STAT3) and STAT3 dimer induces MYC and SOX2 expression (Figure 3, top and bottom right). Meta-analysis of over a thousand primary breast cancers showed that high VEGF expression is strongly associated with STAT3 and MYC expression, supporting the link between VEGFR-2 and CSC self-renewal (19).
The regulation of breast cancer CSCs by VEGFR-2 is an important finding, which further supports preclinical investigation of anti-VEGFR-2 in breast cancer treatments. Targeting CSC-addicted signaling pathways is attractive. Increasing evidence suggests that cells within a tumor can exhibit heterogeneity and cancer originates from CSCs (55, 56). CSCs are thought to be responsible for many attributes of cancer, including radiation resistance/chemoresistance, metastasis, and relapse of disease (57, 58). Targeting VEGFR-2 and/or downstream JAK2 or STAT3 might overcome the radiation resistance and chemoresistance in triple-negative breast cancer by eliminating CSCs.
Breast cancer is the most frequent cancer and the second-most common cause of death from cancer in women worldwide (59). Two thirds of new breast cancers express estrogen receptor α (ER) protein, and the growth of these primary tumors is predominantly depend on estrogen (60). Till date, Food and Drug Administration (FDA) has proved three selective ER modulators (SERMs) – raloxifene, toremifene, and tamoxifen – and three aromatase inhibitors (AIs) – anastrozole, letrozole, and exemestane (61). Although patients with ER-positive breast cancers can be treated with these drugs successfully, it is in the treatment of triple-negative (ER−, PR−, Her2−) breast cancer where there is a clear demand for the development of new therapies (62). Triple-negative breast cancers commonly cause mortality when these tumors metastasize to distant organs including lung and brain. Bone metastasis can cause significant morbidity.
Anti-VEGF therapy in metastatic breast cancer was initially embraced with great enthusiasm. Two commonly used reagents are bevacizumab (Avastin) (27), an anti-VEGF monoclonal antibody, and its antibody derivative ranibizumab (Lucentis) (63). However, anti-VEGF therapy for breast cancer has been a “veritable roller coaster of results” (64). Bevacizumab, initially on FDA “fast track” for metastatic breast cancer, was revoked of approval in breast cancer in 2011 (65). The efficacy of bevacizumab in breast cancer is unclear. Bevacizumab delayed metastatic breast progression in early trials with paclitaxel, whereas subsequent trials showed no increase in overall survival (66). Moreover, other preclinical studies suggested that bevacizumab promotes more aggressive metastatic behavior in surviving cells (67, 68). Causes of resistance to bevacizumab are that bevacizumab reduces tumor vessel supply, decreases drug penetration, and increases hypoxia to stimulate even greater VEGF production to overcome drug effects (69).
To overcome drug resistance to bevacizumab, chemical inhibitor drugs against VEGFR-2 may be proven effective. Indeed, FDA has proved several small compound drugs inhibiting VEGFR-2, including sunitinib (Sutent) (70–72), sorafenib (Nexavar) (73–75), axitinib (Inlyta) (76), and pazopanib (Votrient) (77, 78). The development of these VEGFR-2 inhibitors supports the further investigation of their clinical benefits for a selective subset of breast cancer patients whose mutant p53 activates VEGFR-2. At present, the benefit of sunitinib in breast cancer is undefined. Sunitinib had shown single-agent activity in the treatment of metastatic breast cancer (79). In other trials, sunitinib has failed to demonstrate therapeutic benefit in either first-line or refractory breast cancer (80, 81). The clinical experience with sorafenib in breast cancer is limited, with only a recently completed phase I/II trial of combination of sorafenib and anastrozole (NCT00217399). Axitinib has significant benefits only in patients who have previously received paclitaxel (NCT00076024), which suggested anti-VEGFR-2 therapy might best work on patients receiving prior paclitaxel (82). Similar to axitinib, pazopanib showed additional benefits to paclitaxel treatment in breast cancer from a recent clinical trial (NCT01644825). Pazopanib plus paclitaxel group has a significant longer progression-free survival than the paclitaxel only group [median, 6.35 months (95% CI, 5.36–11.02) versus 3.49 months (2.01–5.66); hazard ratio, 0.42 (95% CI, 0.25–0.69); p = 0.0002] (83). To reduce the complexity of drug response, it is of particular interest to identify the subgroups of breast cancer patients who will preferentially benefit from anti-VEGFR-2 therapy with combinations of biomarkers. Mutant p53 could be one of the predictive biomarkers.
Much progress has been made in understanding the biology of VEGFR-2 in breast cancer. One breakthrough is that mutant p53 recruits SWI/SNF to activate VEGFR-2 expression (53). This finding suggests compounds disrupting mutant p53–SWI/SNF interaction might be effective in treating breast cancer. Another interesting study found that VEGFR-2 recruits JAK2/STAT3 to activate embryonic stem cell transcription factors MYC and SOX2 in breast cancer CSCs (19). This finding offers another possibility that VEGFR-2 inhibitor and/or JAK2/STAT3 inhibitors could be used as a cancer stem cell-targeted therapy in breast cancer.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors have been unable to discuss many important publications in the VEGF research field and apologize to those authors whose work has not been included. We thank Kirkwood Adams for carefully reading and editing the manuscript.
1. Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science (1983) 219:983–5. doi:10.1126/science.6823562
2. Thomas JL, Eichmann A. The power of VEGF (vascular endothelial growth factor) family molecules. Cell Mol Life Sci (2013) 70:1673–4. doi:10.1007/s00018-013-1276-6
3. Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal (2007) 19:2003–12. doi:10.1016/j.cellsig.2007.05.013
4. Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature (1996) 380:435–9. doi:10.1038/380435a0
5. Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature (1996) 380:439–42. doi:10.1038/380439a0
6. Chung AS, Lee J, Ferrara N. Targeting the tumour vasculature: insights from physiological angiogenesis. Nat Rev Cancer (2010) 10:505–14. doi:10.1038/nrc2868
7. Ria R, Roccaro AM, Merchionne F, Vacca A, Dammacco F, Ribatti D. Vascular endothelial growth factor and its receptors in multiple myeloma. Leukemia (2003) 17:1961–6. doi:10.1038/sj.leu.2403076
8. Ghosh S, Sullivan CA, Zerkowski MP, Molinaro AM, Rimm DL, Camp RL, et al. High levels of vascular endothelial growth factor and its receptors (VEGFR-1, VEGFR-2, neuropilin-1) are associated with worse outcome in breast cancer. Hum Pathol (2008) 39:1835–43. doi:10.1016/j.humpath.2008.06.004
9. Barquet LA. [Role of VEGF in diseases of the retina]. Arch Soc Esp Oftalmol (2015) 90(Suppl 1):3–5. doi:10.1016/S0365-6691(15)30002-2
10. De Brot S, Ntekim A, Cardenas R, James V, Allegrucci C, Heery DM, et al. Regulation of vascular endothelial growth factor in prostate cancer. Endocr Relat Cancer (2015) 22:R107–23. doi:10.1530/ERC-15-0123
11. Gerber HP, Malik AK, Solar GP, Sherman D, Liang XH, Meng G, et al. VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature (2002) 417:954–8. doi:10.1038/nature00821
12. Brusselmans K, Bono F, Collen D, Herbert JM, Carmeliet P, Dewerchin M. A novel role for vascular endothelial growth factor as an autocrine survival factor for embryonic stem cells during hypoxia. J Biol Chem (2005) 280:3493–9. doi:10.1074/jbc.M406613200
13. Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM. Self-renewal and expansion of single transplanted muscle stem cells. Nature (2008) 456:502–6. doi:10.1038/nature07384
14. Deasy BM, Feduska JM, Payne TR, Li Y, Ambrosio F, Huard J. Effect of VEGF on the regenerative capacity of muscle stem cells in dystrophic skeletal muscle. Mol Ther (2009) 17:1788–98. doi:10.1038/mt.2009.136
15. He S, Nakada D, Morrison SJ. Mechanisms of stem cell self-renewal. Annu Rev Cell Dev Biol (2009) 25:377–406. doi:10.1146/annurev.cellbio.042308.113248
16. Tang JM, Wang JN, Zhang L, Zheng F, Yang JY, Kong X, et al. VEGF/SDF-1 promotes cardiac stem cell mobilization and myocardial repair in the infarcted heart. Cardiovasc Res (2011) 91:402–11. doi:10.1093/cvr/cvr053
17. Fuchs E, Chen T. A matter of life and death: self-renewal in stem cells. EMBO Rep (2013) 14:39–48. doi:10.1038/embor.2012.197
18. Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res (2006) 66:7843–8. doi:10.1158/0008-5472.CAN-06-1010
19. Zhao D, Pan C, Sun J, Gilbert C, Drews-Elger K, Azzam DJ, et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene (2015) 34:3107–19. doi:10.1038/onc.2014.257
20. De Vries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science (1992) 255:989–91. doi:10.1126/science.1312256
21. Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature (1995) 376:66–70. doi:10.1038/376066a0
22. Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature (1995) 376:62–6. doi:10.1038/376062a0
23. Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling – in control of vascular function. Nat Rev Mol Cell Biol (2006) 7:359–71. doi:10.1038/nrm1911
24. Terman BI, Dougher-Vermazen M, Carrion ME, Dimitrov D, Armellino DC, Gospodarowicz D, et al. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem Biophys Res Commun (1992) 187:1579–86. doi:10.1016/0006-291X(92)90483-2
25. Karkkainen MJ, Petrova TV. Vascular endothelial growth factor receptors in the regulation of angiogenesis and lymphangiogenesis. Oncogene (2000) 19:5598–605. doi:10.1038/sj.onc.1203855
26. Ryden L, Linderholm B, Nielsen NH, Emdin S, Jonsson PE, Landberg G. Tumor specific VEGF-A and VEGFR2/KDR protein are co-expressed in breast cancer. Breast Cancer Res Treat (2003) 82:147–54. doi:10.1023/B:BREA.0000004357.92232.cb
27. Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev (2004) 25:581–611. doi:10.1210/er.2003-0027
28. Tanno S, Ohsaki Y, Nakanishi K, Toyoshima E, Kikuchi K. Human small cell lung cancer cells express functional VEGF receptors, VEGFR-2 and VEGFR-3. Lung Cancer (2004) 46:11–9. doi:10.1016/j.lungcan.2004.03.006
29. Guo S, Colbert LS, Fuller M, Zhang Y, Gonzalez-Perez RR. Vascular endothelial growth factor receptor-2 in breast cancer. Biochim Biophys Acta (2010) 1806:108–21. doi:10.1016/j.bbcan.2010.04.004
30. Hamerlik P, Lathia JD, Rasmussen R, Wu Q, Bartkova J, Lee M, et al. Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med (2012) 209:507–20. doi:10.1084/jem.20111424
31. Matsumoto T, Claesson-Welsh L. VEGF receptor signal transduction. Sci STKE (2001) 2001:re21. doi:10.1126/stke.2001.112.re21
32. Loureiro RM, D’Amore PA. Transcriptional regulation of vascular endothelial growth factor in cancer. Cytokine Growth Factor Rev (2005) 16:77–89. doi:10.1016/j.cytogfr.2005.01.005
33. Rini BI, Rathmell WK. Biological aspects and binding strategies of vascular endothelial growth factor in renal cell carcinoma. Clin Cancer Res (2007) 13:741s–6s. doi:10.1158/1078-0432.CCR-06-2110
34. Matsumoto T, Bohman S, Dixelius J, Berge T, Dimberg A, Magnusson P, et al. VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. EMBO J (2005) 24:2342–53. doi:10.1038/sj.emboj.7600709
35. Takahashi T, Yamaguchi S, Chida K, Shibuya M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J (2001) 20:2768–78. doi:10.1093/emboj/20.11.2768
36. Lamalice L, Houle F, Huot J. Phosphorylation of Tyr1214 within VEGFR-2 triggers the recruitment of Nck and activation of Fyn leading to SAPK2/p38 activation and endothelial cell migration in response to VEGF. J Biol Chem (2006) 281:34009–20. doi:10.1074/jbc.M603928200
37. Wu LW, Mayo LD, Dunbar JD, Kessler KM, Ozes ON, Warren RS, et al. VRAP is an adaptor protein that binds KDR, a receptor for vascular endothelial cell growth factor. J Biol Chem (2000) 275:6059–62. doi:10.1074/jbc.275.9.6059
38. Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol (1997) 13:513–609. doi:10.1146/annurev.cellbio.13.1.513
39. Li Y, Dowbenko D, Lasky LA. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J Biol Chem (2002) 277:11352–61. doi:10.1074/jbc.M109062200
40. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer (2003) 3:11–22. doi:10.1038/nrc969
41. Min YH, Cheong JW, Kim JY, Eom JI, Lee ST, Hahn JS, et al. Cytoplasmic mislocalization of p27Kip1 protein is associated with constitutive phosphorylation of Akt or protein kinase B and poor prognosis in acute myelogenous leukemia. Cancer Res (2004) 64:5225–31. doi:10.1158/0008-5472.CAN-04-0174
42. Knight EL, Warner AJ, Maxwell A, Prigent SA. Chimeric VEGFRs are activated by a small-molecule dimerizer and mediate downstream signalling cascades in endothelial cells. Oncogene (2000) 19:5398–405. doi:10.1038/sj.onc.1203915
43. Sakurai Y, Ohgimoto K, Kataoka Y, Yoshida N, Shibuya M. Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc Natl Acad Sci U S A (2005) 102:1076–81. doi:10.1073/pnas.0404984102
44. Holmqvist K, Cross MJ, Rolny C, Hagerkvist R, Rahimi N, Matsumoto T, et al. The adaptor protein shb binds to tyrosine 1175 in vascular endothelial growth factor (VEGF) receptor-2 and regulates VEGF-dependent cellular migration. J Biol Chem (2004) 279:22267–75. doi:10.1074/jbc.M312729200
45. Le Boeuf F, Houle F, Huot J. Regulation of vascular endothelial growth factor receptor 2-mediated phosphorylation of focal adhesion kinase by heat shock protein 90 and Src kinase activities. J Biol Chem (2004) 279:39175–85. doi:10.1074/jbc.M405493200
46. Le Boeuf F, Houle F, Sussman M, Huot J. Phosphorylation of focal adhesion kinase (FAK) on Ser732 is induced by rho-dependent kinase and is essential for proline-rich tyrosine kinase-2-mediated phosphorylation of FAK on Tyr407 in response to vascular endothelial growth factor. Mol Biol Cell (2006) 17:3508–20. doi:10.1091/mbc.E05-12-1158
47. Funa NS, Kriz V, Zang G, Calounova G, Akerblom B, Mares J, et al. Dysfunctional microvasculature as a consequence of shb gene inactivation causes impaired tumor growth. Cancer Res (2009) 69:2141–8. doi:10.1158/0008-5472.CAN-08-3797
48. Kroll J, Waltenberger J. The vascular endothelial growth factor receptor KDR activates multiple signal transduction pathways in porcine aortic endothelial cells. J Biol Chem (1997) 272:32521–7. doi:10.1074/jbc.272.51.32521
49. Stoletov KV, Ratcliffe KE, Spring SC, Terman BI. NCK and PAK participate in the signaling pathway by which vascular endothelial growth factor stimulates the assembly of focal adhesions. J Biol Chem (2001) 276:22748–55. doi:10.1074/jbc.M009720200
50. Rousseau S, Houle F, Kotanides H, Witte L, Waltenberger J, Landry J, et al. Vascular endothelial growth factor (VEGF)-driven actin-based motility is mediated by VEGFR2 and requires concerted activation of stress-activated protein kinase 2 (SAPK2/p38) and geldanamycin-sensitive phosphorylation of focal adhesion kinase. J Biol Chem (2000) 275:10661–72. doi:10.1074/jbc.275.14.10661
51. McMullen M, Keller R, Sussman M, Pumiglia K. Vascular endothelial growth factor-mediated activation of p38 is dependent upon Src and RAFTK/Pyk2. Oncogene (2004) 23:1275–82. doi:10.1038/sj.onc.1207243
52. Kunze K, Spieker T, Gamerdinger U, Nau K, Berger J, Dreyer T, et al. A recurrent activating PLCG1 mutation in cardiac angiosarcomas increases apoptosis resistance and invasiveness of endothelial cells. Cancer Res (2014) 74:6173–83. doi:10.1158/0008-5472.CAN-14-1162
53. Pfister NT, Fomin V, Regunath K, Zhou JY, Zhou W, Silwal-Pandit L, et al. Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes Dev (2015) 29:1298–315. doi:10.1101/gad.263202.115
54. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell (2008) 133:704–15. doi:10.1016/j.cell.2008.03.027
55. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer (2005) 5:275–84. doi:10.1038/nrc1590
56. Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell (2007) 131:1109–23. doi:10.1016/j.cell.2007.10.054
57. Kucia M, Reca R, Miekus K, Wanzeck J, Wojakowski W, Janowska-Wieczorek A, et al. Trafficking of normal stem cells and metastasis of cancer stem cells involve similar mechanisms: pivotal role of the SDF-1-CXCR4 axis. Stem Cells (2005) 23:879–94. doi:10.1634/stemcells.2004-0342
58. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell (2007) 1:313–23. doi:10.1016/j.stem.2007.06.002
59. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer (2015) 136(5):E359–86. doi:10.1002/ijc.29210
60. McClelland RA, Berger U, Miller LS, Powles TJ, Coombes RC. Immunocytochemical assay for estrogen receptor in patients with breast cancer: relationship to a biochemical assay and to outcome of therapy. J Clin Oncol (1986) 4:1171–6.
61. Liang J, Shang Y. Estrogen and cancer. Annu Rev Physiol (2013) 75:225–40. doi:10.1146/annurev-physiol-030212-183708
62. Zhou W, Slingerland JM. Links between oestrogen receptor activation and proteolysis: relevance to hormone-regulated cancer therapy. Nat Rev Cancer (2014) 14:26–38. doi:10.1038/nrc3622
63. Chakravarthy U, Harding SP, Rogers CA, Downes SM, Lotery AJ, Culliford LA, et al. Alternative treatments to inhibit VEGF in age-related choroidal neovascularisation: 2-year findings of the IVAN randomised controlled trial. Lancet (2013) 382:1258–67. doi:10.1016/S0140-6736(13)61501-9
64. Schneider BP, Sledge GW Jr. Anti-vascular endothelial growth factor therapy for breast cancer: can we pick the winners? J Clin Oncol (2011) 29:2444–7. doi:10.1200/JCO.2011.34.9266
65. Couzin-Frankel J, Ogale Y. FDA. Once on ‘fast track,’ avastin now derailed. Science (2011) 333:143–4. doi:10.1126/science.333.6039.143
66. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer (2008) 8:592–603. doi:10.1038/nrc2442
67. Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell (2009) 15:232–9. doi:10.1016/j.ccr.2009.01.021
68. Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell (2009) 15:220–31. doi:10.1016/j.ccr.2009.01.027
69. Ebos JM, Lee CR, Kerbel RS. Tumor and host-mediated pathways of resistance and disease progression in response to antiangiogenic therapy. Clin Cancer Res (2009) 15:5020–5. doi:10.1158/1078-0432.CCR-09-0095
70. Demetri GD, Baker LH, Beech D, Benjamin R, Casper ES, Conrad EU III, et al. Soft tissue sarcoma clinical practice guidelines in oncology. J Natl Compr Canc Netw (2005) 3:158–94.
71. Motzer RJ, Basch E. Targeted drugs for metastatic renal cell carcinoma. Lancet (2007) 370:2071–3. doi:10.1016/S0140-6736(07)61874-1
72. Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med (2007) 356:115–24. doi:10.1056/NEJMoa065044
73. Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med (2007) 356:125–34. doi:10.1056/NEJMoa060655
74. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med (2008) 359:378–90. doi:10.1056/NEJMoa0708857
75. Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther (2008) 7:3129–40. doi:10.1158/1535-7163.MCT-08-0013
76. Escudier B, Gore M. Axitinib for the management of metastatic renal cell carcinoma. Drugs R D (2011) 11:113–26. doi:10.2165/11591240-000000000-00000
77. Zivi A, Cerbone L, Recine F, Sternberg CN. Safety and tolerability of pazopanib in the treatment of renal cell carcinoma. Expert Opin Drug Saf (2012) 11:851–9. doi:10.1517/14740338.2012.712108
78. Verweij J, Sleijfer S. Pazopanib, a new therapy for metastatic soft tissue sarcoma. Expert Opin Pharmacother (2013) 14:929–35. doi:10.1517/14656566.2013.780030
79. Burstein HJ, Elias AD, Rugo HS, Cobleigh MA, Wolff AC, Eisenberg PD, et al. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol (2008) 26:1810–6. doi:10.1200/JCO.2007.14.5375
80. Bergh J, Greil R, Voytko N, Makhson A, Cortes J, Lortholary A, et al. Sunitinib (SU) in combination with docetaxel (D) versus D alone for the first-line treatment of advanced breast cancer (ABC). J Clin Oncol (2010) 30(9):921–9. doi:10.1200/JCO.2011.35.7376
81. Crown J, Dieras V, Staroslawska E, Yardley DA, Davidson N, Bachelot TD, et al. Phase III trial of sunitinib (SU) in combination with capecitabine (C) versus C in previously treated advanced breast cancer (ABC). J Clin Oncol (2010) 28:LBA1011.
82. Rugo HS, Stopeck AT, Joy AA, Chan S, Verma S, Lluch A, et al. Randomized, placebo-controlled, double-blind, phase II study of axitinib plus docetaxel versus docetaxel plus placebo in patients with metastatic breast cancer. J Clin Oncol (2011) 29:2459–65. doi:10.1200/JCO.2010.31.2975
83. Pignata S, Lorusso D, Scambia G, Sambataro D, Tamberi S, Cinieri S, et al. Pazopanib plus weekly paclitaxel versus weekly paclitaxel alone for platinum-resistant or platinum-refractory advanced ovarian cancer (MITO 11): a randomised, open-label, phase 2 trial. Lancet Oncol (2015) 16:561–8. doi:10.1016/S1470-2045(15)70115-4
Keywords: VEGF, VEGFR-2, VEGFR inhibitor, JAK2, STAT3, mutant p53, breast cancer
Citation: Zhu X and Zhou W (2015) The emerging regulation of VEGFR-2 in triple-negative breast cancer. Front. Endocrinol. 6:159. doi: 10.3389/fendo.2015.00159
Received: 10 August 2015; Accepted: 25 September 2015;
Published: 09 October 2015
Edited by:
Gabriella Castoria, Second University of Naples, ItalyReviewed by:
Silvio Naviglio, Second University of Naples, ItalyCopyright: © 2015 Zhu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen Zhou, d3oyMzEzQGNvbHVtYmlhLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.