 Sara Salehi Hammerstad
Sara Salehi Hammerstad Shira Frankel Grock1
Shira Frankel Grock1 Hanna J. Lee
Hanna J. Lee Alia Hasham
Alia Hasham Nina Sundaram
Nina Sundaram Yaron Tomer
Yaron Tomer- 1Department of Medicine, Division of Endocrinology, Icahn School of Medicine at Mount Sinai, New York, NY, USA
- 2Department of Pediatrics, Oslo University Hospital Ullevål, Oslo, Norway
- 3James J. Peters VA Medical Center, Bronx, NY, USA
Diabetes and hepatitis C infection are both prevalent diseases worldwide, and are associated with increased morbidity and mortality. Most studies, but not all, have shown that patients with chronic hepatitis C are more prone to develop type 2 diabetes (T2D) compared to healthy controls, as well as when compared to patients with other liver diseases, including hepatitis B. Furthermore, epidemiological studies have revealed that patients with T2D may also be at higher risk for worse outcomes of their hepatitis C infection, including reduced rate of sustained virological response, progression to fibrosis and cirrhosis, and higher risk for development of hepatocellular carcinoma. Moreover, hepatitis C infection and mainly its treatment, interferon α, can trigger the development of type 1 diabetes. In this review, we discuss the existing data on this two-way association between diabetes and hepatitis C infection with emphasis on possible mechanisms. It remains to be determined whether the new curative therapies for chronic hepatitis C will improve outcomes in diabetic hepatitis C patients, and conversely whether treatment with Metformin will reduce complications from hepatitis C virus infection. We propose an algorithm for diabetes screening and follow-up in hepatitis C patients.
Introduction
Type 2 diabetes (T2D) and hepatitis C are prevalent diseases worldwide. The World Health Organization (WHO) reported that 170 million people are chronically infected with hepatitis C virus (HCV) globally (1), and 347 million have diabetes mellitus (DM). Despite decreasing prevalence of hepatitis C infection in the United States, the disease burden continues to grow due to hepatitis C-related diseases (2). Studies have shown that more than one-third of patients with chronic HCV infection will develop at least one extrahepatic manifestation (3, 4).
A large number of studies report an increased risk for T2D in patients with chronic HCV infection (5). While a few studies could not confirm this association in the absence of liver dysfunction (6), the majority of studies strongly support this association. This association holds even when comparing patients with chronic HCV to patients with other progressive viral liver diseases (7, 8). There are also studies reporting T2D as a predisposing factor for HCV infection (9–11).
While insulin resistance (IR) and T2D are more frequently reported as complications of HCV infection, HCV is also known to be associated with several autoimmune manifestations, including type 1 diabetes (T1D) (12, 13). Therapy for chronic HCV infection, in particular interferon alpha (IFNα), can also trigger diabetes. The immunomodulatory effect of IFNα can induce or exacerbate autoimmune diseases. While interferon-induced thyroiditis (IIT) is the most frequently reported autoimmune complication of IFNα therapy (14, 15), other autoimmune conditions, such as T1D (16), systemic lupus erythematosus (17), rheumatoid arthritis (18), pernicious anemia (19), optic neuritis (20), vitiligo (21), and autoimmune hemolytic anemia (22) are also associated with HCV infection [for review, see Ref. (23)].
Recently, new non-interferon-based therapeutic regimens for hepatitis C have been introduced; however, chronic HCV infection and its sequelae remain a serious public health concern especially cirrhosis and hepatocellular carcinoma (HCC) which are the leading causes of death in patients with chronic HCV (24). Numerous studies illustrate that IR and T2D have a negative impact on clinical outcomes for patients with chronic HCV infection and it is possible that insulin-sensitizing therapy with metformin might be beneficial for reducing HCV complications. In this review, we analyze the literature linking HCV infection and its therapy – IFNα – with diabetes. We focus on the mechanisms by which both HCV infection and IFNα treatment may contribute to the development of T2D and T1D (in this review, we refer to all forms of autoimmune diabetes as T1D), and the mechanisms by which diabetes can worsen outcomes in HCV infection.
We systematically searched PubMed databases from 1990 to 2015.The search was limited to studies reported in English. The reference lists from identified studies were further searched for primary sources.
Type 2 Diabetes and Insulin Resistance in HCV Infection
Hepatitis C Infection, Insulin Resistance, and Type 2 Diabetes
An association between cirrhosis and glucose intolerance has long been known. Indeed, cirrhosis itself is “diabetogenic” with various studies describing the majority of cirrhotics as having impaired glucose tolerance (25–27). Beginning with Allison’s group in 1994 (26), epidemiological studies have since specifically linked HCV and HCV cirrhosis with DM (25, 26). The prevalence of DM, the majority assumed to be T2D, was significantly higher among patients with HCV cirrhosis than in patients with cirrhosis due to other etiologies (26). HCV infection precedes the diagnosis of T2D in as many as 73% of cases, further suggesting HCV’s pathogenic role in the development of T2D (28–30).
It is estimated that up to 33% of chronic hepatitis C patients have T2D (31). One meta-analysis of 34 studies showed a significantly higher risk for T2D in HCV patients are compared to hepatitis B virus (HBV) patients [odds ratio (OR) 1.8], matched controls, and patients with other forms of chronic liver disease infection (5). Perhaps due to subtle variations in HCV genotype, ethnicity, severity of liver disease, and other variables, a few studies have found no association (32–34). Mangia et al. prospectively followed 385 non-cirrhotic hospitalized patients and concluded that the prevalence of DM was not significantly different than that in the general Italian population. A recent NHANES study also had conflicting results and showed that HCV infection was only associated with ALT and GGT elevations, not diabetes. However, there was still a clear trend for higher homeostatic model assessment-estimated insulin resistance (HOMA-IR) scores in both the HCV antibody and RNA-positive groups compared to controls (35). Regardless, the vast majority of studies have noted a 2- to 10-fold increase of T2D in chronic HCV infection compared to other liver diseases (31, 36–38). The cumulative data suggest that T2D is approximately two to threefold more prevalent in HCV than in HBV infection. We therefore believe that the risk for T2D among HCV patients is significant and is more than just coincidental or cirrhosis-dependent (5, 39, 40).
Epidemiology
A positive association between chronic HCV infection and T2D has been consistently demonstrated across different ethnicities and geographic regions, in both developed and developing countries. The overall prevalence of DM among chronic HCV-seropositive populations in North America, Europe, the Middle East, and Asia ranges from 13 to 33% (39, 41, 42). It is estimated that 20% of chronic HCV patients will develop cirrhosis and as many as ~50% of these patients will have T2D (26, 40). This translates into 47 million people worldwide and 750,000 people in the USA alone who are expected to develop HCV-associated T2D (31).
The landmark cross-sectional NHANES III study that surveyed 9,841 US adults concluded that HCV-positive U.S. patients older than 40 years old had a threefold increased risk for T2D, compared to those without HCV (OR 3.77) (43). Other studies from the U.S. and abroad have corroborated this association between hepatitis C and T2D (7). The highest rate of HCV infection in the world is found in Egypt where 12% of its general population and up to 40% of its older, rural population is HCV seropositive (41). The prevalence of DM is 25.4% among the HCV population in Egypt which indicates that chronic HCV patients are three times more likely to develop DM than the HCV seronegative population (38). In Italy, the frequency of HCV infection varies depending on certain geographic regions but ranges from 3 to 15% in the general population (44). One retrospective study showed that the relative risk for T2D in non-cirrhotic HCV patients was as high as 2.71 compared to that of non-cirrhotic HBV patients and 1.81 compared to controls without hepatitis (45). Likewise, the prevalence of DM (presuming that the majority are type 2 DM) in South Korea is significantly increased in the chronic HCV patients (23.5%) compared to chronic HBV patients (8.2%) (46). In a Japanese retrospective study, the prevalence of DM (also presumed to be mostly T2D) was 20.9% among the non-cirrhotic HCV patients versus 11.9% among the non-cirrhotic HBV patients (47). Among the cirrhotic population, the prevalence rates for diabetes showed analogous findings; DM was found in 30.8% of the HCV cirrhotics versus 11.9% of the HBV cirrhotics (47). The general trend, regardless of country of origin, suggests that T2D is approximately two to threefold more prevalent in HCV than in HBV infection.
The association between hepatitis C and T2D is also seen in the reverse direction, namely that diabetics are more prone to acquiring HCV. Although two small cohorts from Turkey and Nigeria failed to demonstrate an increased prevalence of hepatitis C among T2D patients, the vast majority of studies do. For instance, one US study showed the prevalence of hepatitis C to be 4.2% among diabetics compared to only 1.2% among the non-diabetic controls (7). By contrast, the prevalence of HBV in diabetics (0.3%) was not significantly increased compared to non-diabetics. A study in Taiwan that included 820 type 2 diabetics showed that HCV seropositivity was 2.8 times greater among T2D patients than in non-diabetic controls (48). Similarly, a study from Italy showed that the prevalence of HCV infection among 1,516 T2D patients was 7.6% compared to 2.3% in the non-diabetic controls (49). Comparable trends were also observed in Pakistan with an OR for HCV infection of 3.03 among T2D patients (50).
Risk Factors for Type 2 Diabetes in HCV Patients
Age and gender. Age has been shown to be a risk factor and an independent predictor for the development of diabetes in chronic HCV patients, although not consistently in every study. When separated into quintiles by age, one study noted an increased frequency of diabetes in all HCV patients except in the youngest quintile that included those less than 38 years old (7). In the NHANES III cohort of almost 10,000 U.S. adults, the likelihood for T2D among HCV patients older than 40 years old was twofold compared to those who were younger than 40 years old (43). When further evaluated for confounders, including ethnicity, sex, body mass index (BMI), and socioeconomic factors, the OR for T2D increased to 3.77 (43). Moreover, in Egypt it was also noted that the diabetic HCV patients were older than the non-diabetics (mean age 48.1 versus 40.7 years old) (41). Longer duration of hepatitis C infection has also been noted to increase the risk for diabetes.
Hepatitis C is more common among men than women (51), and male gender is also associated with more hepatitis C disease progression to fibrosis and cirrhosis. It is difficult, however, to precisely tease out the relative contribution of gender itself from confounders like fibrosis and hepatosteatosis, which are themselves markers of more severe liver disease and are associated with higher BMI, larger waist circumference, and a higher HOMA index (34).
Family history. Family history of diabetes has been demonstrated to be a risk factor for the development of T2D in HCV patients as in the general population (52). One case-control study showed that, among 45 non-cirrhotic HCV-seropositive patients, 67% of those with T2D had a family history for diabetes versus only 7% of those without T2D (31). Another study showed a positive family history for diabetes in 29% of the HCV diabetic group while only 1.8% had pertinent family history in the non-diabetic group (40). This risk is comparable to the two to sixfold increased risk for T2D in the general non-HCV population who have a positive family history for diabetes (52). Across various ethnic groups (Pima Indians and populations in the US, Norway, South Africa, and the UK), the risk of developing diabetes is consistently increased among individuals with a positive family history and is up to 4.4-fold higher for individuals with a single diabetic parent (52).
Genotype. T2D in chronic HCV infection has also been correlated with specific HCV genotypes. In North America, ~70% of HCV patients are infected with genotype 1a and 1b while a mere 4% are infected with genotypes 2a (53). Despite these numbers, genotype 1 is relatively less associated with diabetes in the HCV population. For instance, one US case-control analysis showed that HCV genotype 2 was present in as many as 29% of 594 HCV-positive diabetic patients versus only 3% of the local HCV control patients (7). In Japan, HCV genotype 2 patients also have a higher rate of DM than genotype 1 (47). Non-genotype 3 HCV (particularly genotypes 1, 2, and 4) has been more closely associated with the development of T2D among the hepatitis C population, and interestingly, genotype 2a in particular has been preferentially associated with extrahepatic manifestations of HCV. Fewer cases of IR were consistently demonstrated among HCV genotype 3 patients, even after adjusting for confounders like BMI and stage of fibrosis (52, 54). However, this has not been consistently demonstrated in all studies (40). These discrepancies may be due to the differences in the number of patients with available genotype data.
Cirrhosis. Chronic HCV infection in the absence of cirrhosis or antiviral therapy is associated with T2D (7, 31, 40, 55). This indicates that it is not the stage of liver disease alone that influences the development of T2D. The presence of fibrosis and cirrhosis, however, in some studies has been shown to be an independent risk factor that contributes to the progression to T2D (31, 33, 40, 42). Various studies have shown that the rate of T2D is higher among HCV cirrhotics than in other forms of cirrhosis, including cirrhosis caused by HBV, alcohol, or cholestatic disease (5, 40). Seventy to eighty percent of HCV cirrhotics have been reported to have glucose intolerance (31), and 50% of these cirrhotics have developed DM compared to only 9% of the non-HCV cirrhotics (26). This suggests a direct diabetogenic effect of the HCV beyond the damage to the liver. Furthermore, the rates of DM are even more increased on the background of higher grades of hepatic fibrosis, steatosis, or cirrhosis (31, 33, 40, 42, 56). For example, the prevalence of T2D was shown to increase with every rise in the fibrosis score of HCV patients with an OR of 3.83 (51). One prospective case series with 361 HCV patients from Pakistan showed an OR for T2D of 2.0 in HCV cirrhotics compared to that of HCV non-cirrhotics. Conversely, DM itself has recently been demonstrated to be hasten the progression of fibrosis and cirrhosis underscoring the bidirectionality of the association between chronic HCV infection and IR in chronic HCV infection (57) (see Hepatic Fibrosis and Cirrhosis).
Therapeutic regimen and virological response. Studies have shown that non-responders to HCV treatment have a greater incidence of DM and IR (28, 58). In a study of 234 patients with chronic HCV infection who were treated with IFNα ± ribavirin (RBV) and were followed for at least 3 years, impaired fasting glucose was found in 34.1% of the non-sustained responders versus 14.6% of the patients with sustained virological response (SVR) (28). SVR is defined as undetectable viral load after 24 weeks of treatment. Furthermore, none of the patients with SVR developed DM while nine of the non-responders eventually developed T2D. In another study, treatment failure for HCV was associated with an OR of 2.81 for the development of new-onset IR (59) while other groups have demonstrated improvements in IR after viral clearance, strongly suggesting causality (i.e., that HCV is responsible for the IR) (28, 60). It is unclear, however, if IR is simply a consequence of poor response to IFN–RBV therapy or if it, on the contrary, mechanistically contributes to treatment resistance. Conversely, there is very strong evidence that hepatic disease progression and response to antiviral therapy are worse with concomitant DM (28, 54, 61) (see Virological Response to Antiviral Therapy). This two-way association between hepatitis C and diabetes further supports a potential direct role of HCV in IR and T2D.
Mechanisms
Hepatic fibrosis and cirrhosis are not the only mechanisms leading to glucose intolerance and frank DM in HCV patients (27, 62). It may be assumed that HCV infection could primarily predispose to T2D through the induction of hepatic steatosis. Chronic HCV infection after all carries a higher prevalence of steatosis (50%) compared to other liver diseases, including chronic HBV infection (18%) and autoimmune liver disease (16–21%) (63). Viral steatosis, however, is most frequently present in chronic infections with HCV genotype 3 and yet genotype 3 is the least associated with IR. Studies also suggest that viral steatosis may behave differently from metabolic steatosis and that the steatosis seen in non-genotype 3 HCV infections is more a marker and consequence of the underlying IR (27). There is growing evidence demonstrating that specific and direct effects of the HCV itself are responsible for triggering the development of T2D.
Increase in reactive oxygen species. The HCV genome comprises 10 mature proteins, including structural proteins (core, E1, E2, p7) and non-structural proteins (NS2–NS5). Various in vitro studies have demonstrated chronic background inflammation and an increase in mitochondrial reactive oxygen species (ROS) in HCV infection. NS3 and NS5, in particular, were shown to trigger oxidative stress responses. Human monocytes incubated with various HCV proteins demonstrated that NS3 selectively generated ROS by activation of NADPH oxidase, Nox2 (64). Human hepatoma Huh-7 cells transfected with an NS5A vector have shown an elevation of ROS with consequent activation of STAT-3 and NF-KB pathways (65) that then led to the release of an array of cytokines, including TNFα, TGFβ, IL-6, and IL-8. The structural core protein has also been demonstrated to induce an increase in ROS, mitochondrial dysfunction, and ER stress by possibly overwhelming glutathione stores and ER chaperones during viral replication (66, 67).
TNFα and other inflammatory cytokines. The intense inflammatory response to HCV is deemed central to the development of peripheral and hepatic IR in chronic HCV infection, primarily through disruptions in the insulin signaling pathway. Several studies have reported that TNFα can directly interfere with insulin signaling in HCV patients (68–70). Knobler and colleagues noted significantly more detectable serum TNFα (measured as soluble TNFR1 and 2) in diabetic HCV+ patients than in non-diabetic HCV+ patients (respectively, 74 versus 64%; p-value <0.0001), independent of prior IFNα treatment or cirrhosis (71). CRP was higher in both diabetic groups (regardless of HCV seropositivity) while levels of IL-6 and IL-8 remained consistently low. Similar upregulation of TNFα and downregulation of IL-6 were observed in hepatic tissues of chronically infected HCV patients (72). Numerous animal and human studies with T2D patients have positively correlated TNFα with obesity and IR (73–76), and have implicated a role for TNFα in the pathogenesis of IR primarily through its postreceptor effects (71, 77). Animal pancreatic clamp studies have linked TNFα to increased IR and decreased glucose uptake (75). Similarly, in vitro studies using cultured adipocytes stimulated with insulin showed that chronic TNFα exposure reduced tyrosine kinase activities and decreased autophosphorylation of the insulin receptor and tyrosine-phosphorylation of insulin receptor substrate 1 (IRS-1) (71, 77, 78). Moreover, incubation of Huh-7 hepatocytes with TNFα increased serine-phosphorylation of IRS-1 (79), also resulting in inhibition of the insulin signaling cascade. TNFα also downregulated GLUT4 mRNA expression in muscle and adipose tissues and has been implicated in reduced expression of IRS-1 and PPARs (80, 81). A few studies have questioned the role of TNFα in IR and have shown no significant change in insulin signaling in skeletal muscle exposed to TNFα (82).
Direct alterations in insulin signaling by HCV. There is increasing evidence that HCV has direct effects on insulin signaling (Figure 1). One study found that, compared to control livers, the livers of the non-obese HCV patients showed a twofold decrease in insulin-stimulated tyrosine-phosphorylation of IRS-1 and a significantly blunted activation of two downstream targets that are critical for most of the metabolic effects of insulin: phosphoinositide 3-kinase (PI3-kinase) and Akt (Protein Kinase B, a downstream target of PI3-kinase) (37). An imbalance in the levels of activating tyrosine-phosphorylated IRS-1, the inhibitory serine-phosphorylated IRS-1, and threonine-phosphorylated Akt seems to play an integral part in the development of IR in hepatitis C (79, 83, 84).
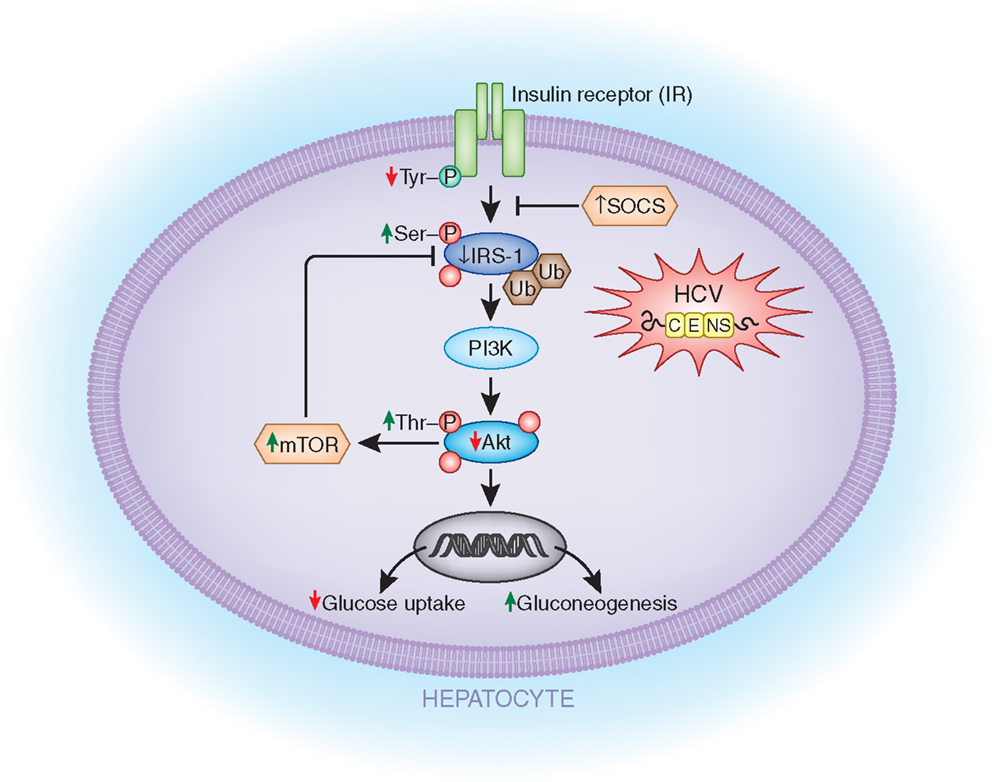
Figure 1. Potential mechanisms by which HCV directly affects the insulin signaling cascade. HCV infection of liver cells can lead to (1) decreased insulin receptor (IR in the figure) auto phosphorylation; (2) decreased IRS-1 activation due to increased serine-phosphorylation of IRS-1; (3) decreased IRS-1 levels due to increased ubiquitin-mediated proteasomal degradation induced by SOCS 3/7 and mTOR upregulation; (4) reduced Akt activity due to increased threonine-phosphorylation of Akt; (5) decreased GLUT4 expression; and (6) increased gluconeogenic enzymes (GC6P and PCK2). IR, insulin receptor; IRS-1, insulin receptor substrate-1; SOCS 3/7, suppressor of cytokine signaling; PI3K, phosphoinositide 3-kinase; mTOR, mammalian target of rapamycin; GC6P, glucose-6-phosphatase; PCK2, phosphoenolpyruvate carboxykinase 2.
Another group demonstrated that mice transgenic for the HCV core protein showed decreased expression of IRS-1 and IRS-2 (85). This group further showed that there was an accumulation of ubiquitin-conjugated IRS-1 and IRS-2 in HepG2 human hepatic cells transfected with the HCV core protein. Proteasome-mediated degradation was further suggested by the increased IRS-1 and IRS-2 expression levels after treatment with MG 132, a proteasome inhibitor (84, 85). The inhibition of IRS-1 may be due to mTOR activation by the HCV 2a core protein which then ultimately leads to IRS-1 degradation (84).
These findings were replicated in another in vitro study using Huh-7 cells transfected with the HCV 1b or 3a core proteins (86). Ubiquitin-mediated proteasomal degradation of IRS-1 via SOCS-7, a negative regulator of IRS expression, was found only in the cells transfected with the HCV 3a core protein. These HCV 3a transfected cells also had significantly reduced mRNA levels of PPARγ, an insulin-sensitizing nuclear receptor, and showed an inhibition in insulin-induced Akt phosphorylation. When subsequently treated with a PPARγ agonist, not only were these effects reversed but also IRS-1 levels were noted to be increased (86). These results suggest that there may be genotype-specific mechanisms, although other groups have shown HCV 1b-mediated downregulation of IRS-1 by SOCS1 and SOCS3 (85).
Beta cell dysfunction. Histological evaluation of the pancreases of HCV-seropositive patients has shown evidence for pancreatic β-cells that are infected with HCV. These HCV-infected beta cells have been noted to have both morphological and functional defects, including a blunted insulin response to glucose (62). Narita’s group examined 131 chronic HCV patients with normal fasting serum glucose levels who were later diagnosed as either glucose tolerant, glucose intolerant, or overt diabetic after a 75 g oral glucose tolerance test (OGTT). These patients were further evaluated for various parameters of IR and of beta cell function (HOMA-β, C-peptide and insulin levels, ΔC-peptide 30, insulinogenic index). Significant decreases in the ΔC-peptide 30 and the insulinogenic index, both markers of early phase pancreatic insulin secretion, were consistently noted in the diabetic group suggesting a direct effect of HCV on beta cells (87). Others have suggested that proinflammatory cytokines secreted in chronically infected HCV patients, such as TNFα, may also affect beta cell function by disrupting insulin signaling/secretion and by sensitizing the beta cell to the toxic effects of free radicals (76, 88). These findings support the notion that HCV infection induces significant beta cell dysfunction either directly or through cytokine release.
IFNα Therapy – Insulin Resistance and Type 2 Diabetes
The literature is mixed on the effects of IFNα on IR. Some studies have shown an improvement in glucose levels during treatment of HCV infection. Huang et al. observed that 63 out of 180 hepatitis C patients with pre-diabetes (34.8%) became normoglycemic after PegIFNα–RBV therapy, while 10 (5.5%) developed DM (89).
It has also been documented that SVR may be an important factor in the reduction of IR after IFNα therapy (89, 90). The results from a cohort that included patients without diabetes, who were treated with IFNα for hepatitis C, showed that 143 out of 2,842 patients developed T2D during a mean observation time of 6.4 years. Predictive factors for development of diabetes in this study were older age, histological changes (cirrhosis), and non-SVR (60).
Also, injection of IFNα to healthy persons impaired glucose tolerance and insulin sensitivity, stimulated counter regulatory hormone secretion, and stimulated insulin clearance (91). In contrary, Ito et al. could not reveal any effect on insulin sensitivity and glucose tolerance at 3-month follow-up after IFNα treatment for hepatitis C (92).
Oral administration of low-dose IFNα (5,000 U) to patients with newly diagnosed T1D was shown to maintain more beta cell function after 1 year compared to high-dose (30,000 U) groups (93, 94). However, these studies had methodological limitations and there were no statistically significant differences in HbA1c or insulin dose between the two groups (93, 94).
Pancreatic Islets Autoimmunity – Type 1 Diabetes and Hepatitis C Virus Infection of the Islets
Even though HCV is a liver-tropic pathogen, the antigen and viral sequences have been detected in other organs, such as the thyroid (95) and the pancreas (62, 96). Using RT-PCR on pancreatic tissue removed at autopsy from HCV-infected individuals, Yan et al. showed the presence of HCV RNA in all nine pancreata tested (96). However, the viral load was lower than in the liver. These results were confirmed in 5/9 pancreata by in situ hybridization (ISH), and by immunohistochemistry (IHC) in 6/9 pancreata (96). The authors suggested that the infected pancreas might act as a reservoir for HCV and may play a role in persistence of infection. Conversely, it is plausible that the infection may cause pancreatic islet dysfunction and/or trigger islet autoimmunity. Interestingly, epidemiological studies have shown a possible relationship between hepatitis C and pancreatic carcinoma (97), also suggesting direct infection of the pancreas by HCV and induction of local inflammation (98). If indeed direct infection of islets or acinar cells by HCV is confirmed, it is possible that it can trigger autoimmunity in susceptible individuals by inducing local inflammation with cytokine and chemokine secretion (also called “bystander mechanism”).
HCV Infection and Type 1 Diabetes
Epidemiological studies have suggested but not confirmed an association between HCV infection and islet autoimmunity. In several studies, the frequency of T1D autoantibodies was not significantly higher in chronic HCV-infected patients selected for IFNα therapy than that in controls (99, 100). Hieronimus et al. reported the presence of glutamic-acid decarboxylase antibodies (GAD-antibodies) in 1 out of 47 untreated HCV patients with known T1D (99). Piquer and colleagues showed the presence of GAD-antibodies in 4 out of 277 (1.4%) non-diabetic HCV patients, and 1 out of 273 (0.4%) in control population. However, this difference did not reach statistical significance (101). Nevertheless, there are several cases of T1D reported in the literature that were associated with acute hepatitis C infection. Chen reported a case of a male patient who developed acute hepatitis C after blood transfusion. The patient presented GAD-antibodies and islet cell antibodies (ICA Abs) 4 weeks after transfusion, and developed T1D 1 year later (12). The patient had HLA DR3 and DR7, which are not associated with T1D in China. Another case, a 22-year-old female with type 1 diabetes and acute hepatitis C, was reported by Masuda (13). Therefore, it seems that the majority of the risk for T1D in hepatitis C patients is due to IFNα therapy (see below); however, HCV infection itself may also contribute synergistically to the association, possible by local production of IFNα in the islets if HCV indeed is proven to infect the islets (Figure 2).
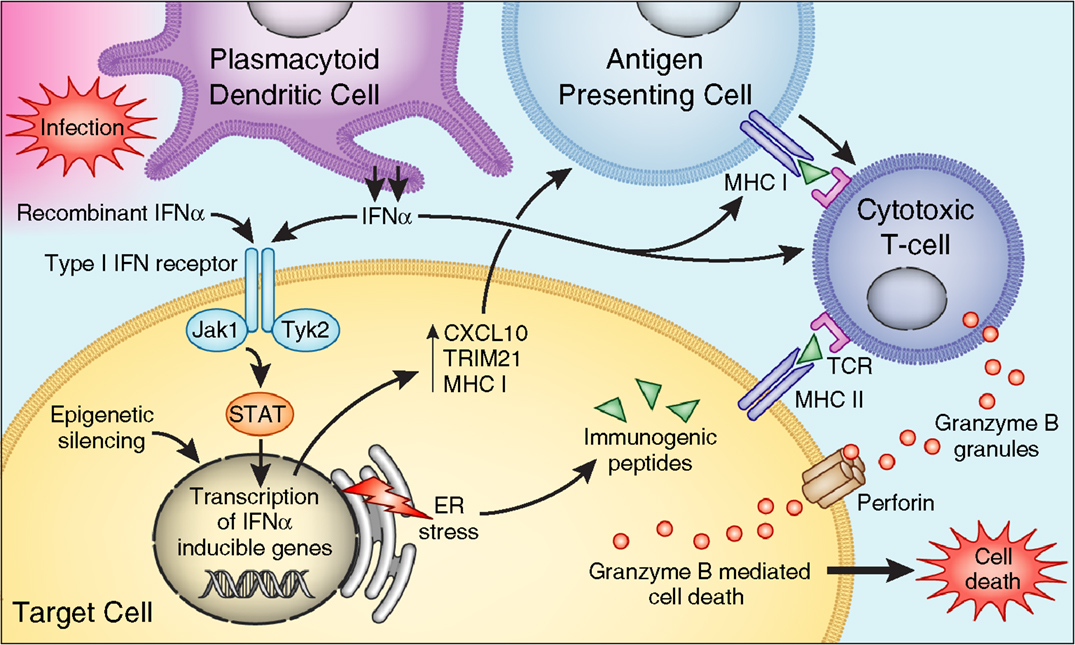
Figure 2. Mechanisms by which IFNα affects beta cells to trigger islet autoimmunity. PDCs produce IFNα in response to infection. Binding to IFN type I receptor, IFNα or therapeutic IFNα, activates Jak/STAT pathways – inducing transcription of IFN-inducible genes leading to (1) production of proinflammatory cytokines, (2) increase expression of HLA class I, (3) stimulation of cytotoxic T cells. IFNα also induces ER stress, and epigenetic modification of genes involved in the pathogenesis of type 1 diabetes.
IFNα Therapy in HCV Infection and Type 1 Diabetes
Interferon alpha was discovered more than 50 years ago (102), and is now recognized as a key component of the innate immune response and the first line of defense against viral infections. IFNα therapy was shown to be particularly effective in HCV infection. Indeed, hepatitis C is the most frequent indication for IFNα therapy (103), even though recently non-interferon-based therapies for HCV infection have been replaced interferon-based therapies. IFNα treatment has considerably improved the outcomes in patients with chronic HCV infection, however, with its potent immunomodulatory effect it may contribute to development of autoimmunity.
Epidemiology
The first case of IFNα-induced T1D was reported by Fabris et al. (104). Although the exact incidence of IFNα-induced diabetes is unknown, several large retrospective studies investigating adverse effects of IFNα in patients with chronic viral hepatitis, mainly hepatitis C, reported incidences of 0.08% (10/11.241) (105) in Italy, and 0.7 (5/667)–0.96% (12/1.250) in Japan (106, 107). Nakamura et al., in a nationwide study in Japan, identified 91 patients with IFNα-induced T1D (108). Furthermore, a Dutch study reported development of T1D in 5 out of 189 patients (2.6%) treated with Peg-IFN + RBV (109). In a study from Spain, including 46 patients, no evidence of increased prevalence of beta cell autoimmunity was found 6 and 12 months after IFNα therapy (101).
In most reported cases, the onset of T1D occurred during or shortly after treatment with IFNα [reviewed in Ref. (110)]. To date, there are more than 45 case reports of T1D developing after treatment with IFNα. The clinical presentation is usually of an acute onset as is typical for T1D (108). The levels of GAD-antibodies were reported to be significantly higher in IFN-induced T1D compared to classical T1D (106). It is important to suspect T1D in HCV patients experiencing polyuria and polydypsia, or other T1D symptoms after initiation of IFNα therapy. Of note, treatment with IFNα is associated with many side effects such as flu-like symptoms, gastrointestinal (GI) symptoms, and headaches (111) that can mask the symptoms of diabetes, resulting in delayed diagnosis and complications.
Risk Factors for IFN-Induced Type 1 Diabetes
Age and gender. Whether age is a risk factor for IFN-induced T1D is difficult to answer since the age of onset of T1D in HCV patients likely reflects the age at which IFNα was initiated. The mean age at the onset of IFN-induced T1D was 56 years in a nationwide survey in Japan that included 91 patients who developed interferon-induced T1D (most received IFNα for HCV infection) (106).
The reported female to male (F/M) ratio of IFN-induced T1D in the same survey in Japan was 0.90, which is lower than the 1.4–1.5 F/M ratio for T1D in the Japanese population (106) and likely reflects the male preponderance of HCV. Another study also found no significant difference in the male and female frequency in children or adults who developed IFN-induced T1D (112). Similarly, the F/M ratio for IFN-induced T1D in Europe (~0.5) was also lower than that for classical T1D (~1) (105, 109). This ratio may reflect the gender distribution of hepatitis C as numerous large cohorts reported that overall two-third of reported cases of chronic HCV infection were males [for review, see Ref. (113)].
Preexisting islet autoantibodies. Autoantibodies are often reported in both adults and children with HCV infection. Wesche et al. reported development of islet autoantibodies in 9.7% (6/62) of patients after initiation of IFNα therapy (114). In another cohort, 2.5% (5/186) non-diabetic patients who received Peg-IFN + RBV developed T1D, and 1.6% (2/186) developed T2D (109). Two out of the five patients with IFN-induced T1D were tested positive for islet antibodies at the baseline prior to initiation of interferon therapy. Results from a recent cohort reported development of T1D in 1 out of 114 children (age 4–17 years) with chronic hepatitis C treated with Peg-IFN, while 9.3% had positive diabetes-related antibodies at baseline (4% with GAD-antibodies) (112).
Even though islets autoantibodies, diagnostic of T1D, have not been commonly measured pre- and post-treatment with IFNα, seroconversion has been observed following IFNα therapy. Fabris reported clinical features of 31 cases of IFN-induced T1D of whom 23 were treated for chronic hepatitis C (1992–2002) (16). For those who were tested for antibodies, markers for pancreatic islet autoimmunity were present prior to IFNα therapy in 50% of cases with IFN-induced T1D. These data are consistent with the well-known fact that islet autoantibodies are the best biomarkers of genetic risk factors for T1D. These findings are similar to those observed for IFNα-induced thyroiditis where thyroid Abs were shown to be the strongest risk factor for IFN-induced thyroiditis (115).
Family history. One study that examined family history in patients that developed T1D during or following IFNα therapy did not find history of T1D in first degree relatives of patients with IFNα-induced T1D (108). It is, however, interesting that 15% of patients with chronic HCV infection had T2D, and 23% had first degree family history of T2D (108). Among 31 cases of IFN-induced T1D reported by Fabris, family history of T1D was reported in 3/31 (9%) patients who developed IFN-induced T1D and family history of T2D was reported in 6/31 (19%) (16). Koivisto postulated that the impairment in insulin sensitivity (91), and the resulting stimulation of beta cells may make islets more vulnerable to destructive process of IFNα therapy (116).
Viral genotype. The HCV genome was determined by Choo et al. (117). Based on the identification of genomic differences, HCV has been classified into distinct subtypes. Genotype 1 (1a and 1b) is the most common form in the USA and Europe (118), while genotype 1b is common in Japan. Genotype 2 is the second commonest strain in North America, Europe, and Japan (119, 120). Genetic heterogeneity of HCV may account for some of the differences in disease outcome and response to treatment observed in HCV-infected individuals (121).
In the Japanese nationwide survey, genotype 1b was most frequently detected (81%) in patients that developed IFN-induced T1D (106), but this likely reflected the fact that genotype 1b is the most prevalent HCV subtype in the Japanese (121) and Chinese populations (122). Patients with genotype 1 that is more resistant to therapy, usually receive higher doses of IFNα and for longer duration contributing to adverse effects (113). In a recent study, Nakanishi and co-workers could not find an association between genotype 1b and the development of T1D; however, the study was limited as the control group included very few patients (106). Similarly, Hasham et al. reported that 74% of patients with IFN-induced thyroiditis were infected with HCV genotype 1, however, 76% of hepatitis C controls also had genotype 1 (123). Thus, viral genotype does not contribute to the association of interferon therapy in HCV patients with T1D.
Therapeutic regimen and virological response to therapy. Data regarding the impact of therapeutic regimen and SVR on development of IFN-induced diabetes are difficult to compare as different forms, dosages, and durations of IFN therapy were used in different studies. Until 2011, the standard IFNα regimen for HCV infection included pegylated IFNα (peg-IFN) 2a in combination with RBV, with genotype-dependent dosage and duration (124). Genotype 1 has the lowest SVR to this regimen compared to genotypes 2 and 3, and it requires higher doses of IFNα. This might contribute to more side effects (113, 125). RBV also has immunomodulatory effects, but RBV is rarely used as monotherapy. However, there is a case report describing a patient who developed T1D after RBV was added to Peg-IFN (126).
The median time from initiation of IFN therapy to the onset of T1D was 8 months in a Japanese cohort, and was significantly shorter in those who were treated with the Peg-IFNα in combination with RBV compared to those who were treated with non-Peg-IFN in mono therapy (108). Interestingly, almost 60% of patients who developed T1D were negative for HCV RNA at the onset of T1D. Overall, the mean latency period in reported cases of IFN-induced T1D was 5 months and ranged from 10 days to 4 years [reviewed in Ref. (16)]. However, no correlation was found between the rate of virological response and the latency of development of T1D (16, 108). Supporting this observation is an Italian cohort that did not show a significant difference in the incidence of glucose abnormalities between long-term responders and non-responders (58).
The current guidelines for treatment of chronic HCV infection recommend using protease inhibitors in combination with Peg-IFN with or without RBV for genotype 1 (127). However, protease inhibitors have side effects too, and recently a case of T1D induced shortly after triple therapy was reported (128). Further studies are needed to determine the impact of protease inhibitors in combination with IFN, or in monotherapy, on the development of autoimmunity. In addition, as the therapy for chronic HCV infection switches to non-interferon-based therapies, it is possible that T1D will no longer be triggered by therapy for hepatitis C.
Genetic factors. Genetic susceptibility plays a major role in the etiology of T1D (129). Therefore, genetic predisposition may also play a role in the etiology of IFNα-induced T1D. In a Japanese cohort, HLA-A*2402 [associated with T1D in Japan (130)] was shown to be associated with the development of T1D in patients treated with IFNα (106). It is intriguing that HLA-A24 has also been shown to be associated with the development of IFNα-induced thyroiditis in Japan (131), suggesting a general role for HLA-A24 in IFNα-induced autoimmunity. The frequency of HLA-DR4 and DR9 (also susceptibility genes for T1D in Japan), as well as DR13, was also significantly higher in patients with IFNα-induced T1D when compared to healthy controls (108). Moreover, HLA-DR13 was also significantly more frequent in patients with IFNα-induced T1D compared to patients with classical T1D, suggesting a direct role in IFNα-induced autoimmunity through an environmental–genetic interaction (108). Studies and results from reported cases, including patients with different ethnicity, showed association between IFN-induced T1D and classical T1D HLA haplotypes (DR3, DR4, DQ8) (16, 109, 110).
Interferon alpha may also trigger or accelerate disease development in genetically predisposed individuals. We previously showed that injecting IFNα to NOD H2h4 mice, a strain genetically susceptible to spontaneous autoimmune thyroiditis, caused a higher frequency of autoimmune thyroiditis. These findings support the notion that IFNα triggers or accelerates autoimmunity in genetically susceptible individuals (132). However, similar studies have not been performed in mice genetically predisposed to autoimmune diabetes.
Taken together, these data support two potential mechanisms by which genetic susceptibility can contribute to IFN-induced T1D: (1) some individuals inherit classical T1D susceptibility genes and IFNα simply triggers or accelerates the progression of T1D in them; alternatively, (2) other individuals inherit genetic variants that uniquely predispose to IFNα-induced autoimmunity.
Mechanisms
The mechanisms by which IFNα can induce T1D have not been thoroughly studied (Figure 2). IFNα has diverse effects on the immune system that can be implicated in the development of autoimmunity. By binding to its receptor, IFNα activates the JAK–STAT pathway (133–135), leading to expression of interferon-stimulated genes (ISGs), including cytokines and adhesion molecules genes. These proinflammatory cytokines can trigger autoimmunity in genetically susceptible individuals (Figure 2).
A direct effect of IFNα on the pancreas might also trigger T1D. Indeed, several studies have shown elevated levels of IFNα in the pancreas of patients with a recent onset of T1D (136–138). Transgenic mice over-expressing IFNα in their beta cells developed hypoinsulinemic diabetes associated with islets inflammation, demonstrating that IFNα can trigger T1D locally by direct effects on the islets. This inflammatory disease process was prevented through the use of an IFNα neutralizing antibody (139). Our group has previously shown similar results in transgenic mice over-producing IFNα in the thyroid; these transgenic mice developed severe inflammatory thyroiditis (140). In our thyroidal model, IFNα triggered the recruitment of inflammatory cells through the induction of various cytokines and chemokines, and it activated cytotoxic T cells causing thyroid cell necrosis (Figure 2). Moreover, we were also able to show direct toxic effects of IFNα on thyroid cells (140). If a similar mechanism occurs in the islets, it is possible that IFNα can also induce toxic effects in islet cells. Li et al. demonstrated that blockade of IFNα signaling in 2- to 3-week-old NOD mice by anti-IFNAR1 mAb resulted in delayed onset and decreased incidence of diabetes (141). This was accompanied by an increase in the number of immature dendritic cells in draining popliteal lymph nodes and enhanced production of the cytokines IL-4 and IL-10. Furthermore, interferon regulatory factor 1-deficient NOD mice were protected from T1D (142).
Interferon alpha modulates the ability of immune effector cells to interact with infected cells, partly through upregulation of MHC class I molecules on target infected cells (143). Moreover, overexpression of MHC class I antigens is associated with activation of cytotoxic T cells. Upregulation of MHC class I molecule expression on islets cells is a prominent, early feature during the development of T1D (144), and it is possible that this is mediated by IFNα. In one study, 33 of 34 pancreata removed at necropsy from patients with T1D revealed beta cells that were positive for immunoreactive IFNα and showed hyperexpression of MHC class I antigens (136). Moreover, type I interferons decrease regulatory T cell activation, another effect that may play a role in development of autoimmunity (145, 146). In contrast to these data, a recent study showed an increase in the number of regulatory T cells and a decrease in CD8+ T cells in the livers of mice treated with IFNα (147).
Effects of Insulin Resistance and Type 2 Diabetes on Outcome of Chronic HCV Infection
While HCV infection has been demonstrated to be a risk factor for T2D (see Type 2 Diabetes and Insulin Resistance in HCV Infection), patients with HCV and IR have also been shown to have significantly worse clinical outcomes. This two-way association further exemplifies the integral role of IR in HCV infection. IR is associated with increased liver fibrosis, reduced virological response to antiviral therapy, and higher rates of HCC. There is also some evidence that IR increases the risk for liver-related death and is associated with shorter time to transplantation (148).
Association of Diabetes/Insulin Resistance with Worse Outcomes in Hepatitis C
Hepatic Fibrosis and Cirrhosis
A number of factors are known to increase the risk of liver fibrosis and subsequent cirrhosis. These include longer duration of infection, older age at time of exposure, male gender, co-infection with other viruses, and alcohol consumption (149, 150). In addition to these factors, IR is increasingly recognized as a separate risk factor for fibrosis progression and cirrhosis.
Two studies by Hui et al. demonstrated that IR is an independent predictor of fibrosis in chronic HCV infection (54, 151). Similarly, Hsu et al. found a strong association between diabetes and severity of fibrosis (152). A recent, large cohort study from Taiwan also concluded that patients with chronic HCV infection who developed diabetes had a two to threefold increased risk for development of cirrhosis and hepatic decompensation (57). However, these findings are not uniform and a few studies failed to show this correlation (153–155). While Svegliati-Baroni et al. did not find an association between HOMA-IR and liver fibrosis, they did find post-load IR to be a predictor of fibrosis progression correlation (153). Overall, the majority of studies support the notion that IR and diabetes predispose to liver fibrosis and cirrhosis.
Virological Response to Antiviral Therapy
While there is significant evidence to suggest that DM worsens response to antiviral therapy, a few studies could not confirm this relationship. A large study of 330 patients found that reduced HOMA-IR correlated with lower rates of SVR, particularly in patients with “difficult to treat” chronic HCV populations, including cirrhotics, the overweight, and HIV-coinfected individuals (156). Other studies suggested that HOMA-IR only predicts treatment response in patients with genotype 1 HCV (157). An Italian study of 412 patients showed a trend for association (albeit not statistically significant) between higher HOMA-IR and reduced SVR (p = 0.06) (158).
To resolve these conflicting data, a recent meta-analysis of 14 studies showed that patients with IR treated with pegIFN–RBV had a 20% lower rate of SVR compared to patients without IR (95% CI: −29.9 to −9.4%, p < 0.001). In addition, responders had a lower HOMA-IR compared to non-responders (mean difference: −0.92, 95% CI: −1.53 to −0.32, p < 0.001) (159).
Hepatocellular Carcinoma
Hepatocellular carcinoma is the third leading cause of cancer-related mortality worldwide (160). Chronic HCV infection is an established risk factor for HCC. Recent studies suggest that diabetes also increases the risk of malignancy, including HCC (148, 161, 162). Additionally, diabetes is a risk factor for HCC-related mortality (163).
A recent Taiwanese study found that HCV patients with diabetes had a hazard ratio of 1.73 for HCC compared to non-diabetic HCV patients (161). Of note, patients with hepatitis B did not demonstrate a similar increase in HCC incidence (161). Another study of HCV patients with Ishak fibrosis score 4–6 found HCC incidence to be 11.4% in diabetics, and 5.0% in non-diabetics (162). The relationship persisted even when controlling for albumin, bilirubin, and platelet count. The degree of IR also appears to correlate with HCC risk. A study from France in patients with HCV cirrhosis found the 5-year incidence of HCC to be 7, 18, and 27% for the lowest, middle, and highest tertile of HOMA-IR, respectively. In a multivariate analysis, HOMA-IR was still predictive of HCC incidence, albeit with a hazard ratio of 1.1 (148).
While the majority of published studies support an association between diabetes and HCC in HCV patients, there are a few reports showing otherwise. However, a 2006 literature review found an association between diabetes and HCV-related HCC in 9 of 13 case-control studies and 7 of 13 cohort studies (164). Despite some inconsistencies, the pooled risk ratio and pooled OR were both significant at 2.5.
Mechanisms of Accelerated Disease Progression in HCV Patients with Diabetes and Insulin Resistance
Hepatic Fibrosis and Cirrhosis
Liver fibrosis is a reversible wound-healing response to hepatic cell injury. Parenchymal cell regeneration in response to acute injury generally results in minimal deposition of extracellular matrix (ECM). However, chronic hepatic injury disrupts the balance between liver repair and scar formation. Hepatocytes are replaced with excessive ECM produced primarily by hepatic stellate cells (HSC) (165).
Excessive HSC activation is one proposed mechanism by which IR can induce fibrosis. Supporting this hypothesis, in vitro studies showed that hyperglycemia and hyperinsulinemia upregulate the secretion of connective tissue growth factor mRNA, a growth factor involved in HSC activation and subsequent fibrogenesis (166). In addition, insulin can directly stimulate HSC proliferation in a dose-dependent fashion (167).
Hepatitis C virus is associated with accelerated steatosis that is mediated through increased production of lipogenic substrate, upregulation of lipogenesis, and disruptions of fatty acid metabolism (168). The magnitude of HCV-related steatosis is compounded by IR. IR activates the lipid biosynthetic pathway in the liver resulting in dyslipidemia and increased steatosis, which can accelerate liver fibrosis (169). Decreased insulin sensitivity also increases release of free fatty acids from adipose leading to increased hepatic lipid deposition (170). Enhanced hepatic lipid deposition and steatosis are independently associated with increased fibrosis in patients with genotype 1 HCV (171).
Lastly, a reciprocal regulation between HCV and SNF1/AMP kinase-related kinase (SNARK) promotes TGF-β signaling, a major driver of hepatic fibrogenesis. In vitro studies have shown that metformin suppressed both HCV replication and SNARK-mediated enhancement of TGF-β signaling (172).
Virological Response to Antiviral Therapy
IR is associated with increased levels of protein tyrosine phosphatases (PTP). PTPs inhibit phosphorylation of signal transducers and activators of transcription (STATs), thus preventing activation of a group of key transcription factors in the interferon signaling pathway that can lead to decreased effectiveness of IFNα therapy and lower rates of SVR (173, 174). This has been corroborated by an in vitro study that demonstrated an insulin-driven dose-dependent decreased expression of IFN-stimulated genes, including PKR, MxA, and OAS-1 (175).
In vitro studies have shown that metformin downregulates PTP-1B and improves response to IFNα in liver cell lines (174), and that metformin inhibited HCV replication by activating AKT (176). However, clinical trials examining the role of metformin in antiviral treatment regimens provided mixed results (157). The TRIC-1 study group compared metformin versus placebo in HCV patients treated with pegINF–RBV. While intention to treat analysis showed no effect of metformin, per protocol analysis displayed a 10% absolute increase in treatment success with metformin (157). Moreover, subgroup analysis of females showed an SVR of 57.7% with metformin versus 28.6% with placebo (p = 0.03). A recent open-label study by Yu et al. found that addition of metformin to pegIFN–RBV resulted in 59.2% SVR compared to 38.8% SVR without metformin (177). In contrast, a placebo-controlled trial by Sharifi and colleagues found no correlation (178).
Additionally, HCV replication is thought to be downregulated by p21-activated kinase 1 (PAK1). IR may reduce PAK1 activity leading to upregulation of HCV replication (179, 180). IR is also associated with increased viral load (181) and progression of fibrosis (54, 152), which independently influence response to interferon-based therapy (182). It remains to be determined the extent to which SVR will continue to be affected by IR when using novel anti-HCV regimens.
Hepatocellular Carcinoma
Patients with diabetes are at increased risk for various types of malignancies (183–185). HCC appears to be one of the malignancies most strongly linked to diabetes with a relative risk of 2.5 (184, 185). Chronic HCV infection thus promotes HCC both directly and indirectly through the virus’s diabetogenic effects (184). While the exact mechanisms have yet to be elucidated, insulin, insulin-like growth factor (IGF), and chronic inflammation have all been implicated in tumorigenesis (184, 186). Additionally, hyperglycemia is thought to promote tumorigenesis by providing the metabolic substrate necessary for cancer cell survival (184).
The insulin receptor is regulated by alternative splicing mechanisms to yield two isoforms, IR-A and IR-B. IR-A expression is increased in cancer cells, including HCC, and primarily promotes the mitogenic role of insulin, while IR-B primarily controls insulin’s metabolic effects (187, 188). High insulin levels in the setting of IR likely activate the IR-A pathway driving tumorigenesis, while the IR-B pathway remains downregulated. This phenomenon would be amplified in the setting of IR-A upregulation, which occurs in cancer cells including HCC (187). Chettouh et al. demonstrated that HCC cell lines with enhanced IR-A expression proliferated in response to insulin while normal human hepatocytes did not (187). Insulin binds to its receptor, likely IR-A, in the setting of tumorigenesis, and activates downstream signaling cascades via insulin receptor substrate (IRS) proteins (189). Indeed, levels of IRS proteins are elevated in human cancers, including HCC (181). IRS proteins can activate mitogen-activated protein kinase (MAPK) and the phosphoinositol-3-kinase (PI3K)/AKT pathway (186, 188, 189). These pathways are involved in cell growth and proliferation and inhibition of apoptosis as shown in Figure 3 (186, 188, 189).
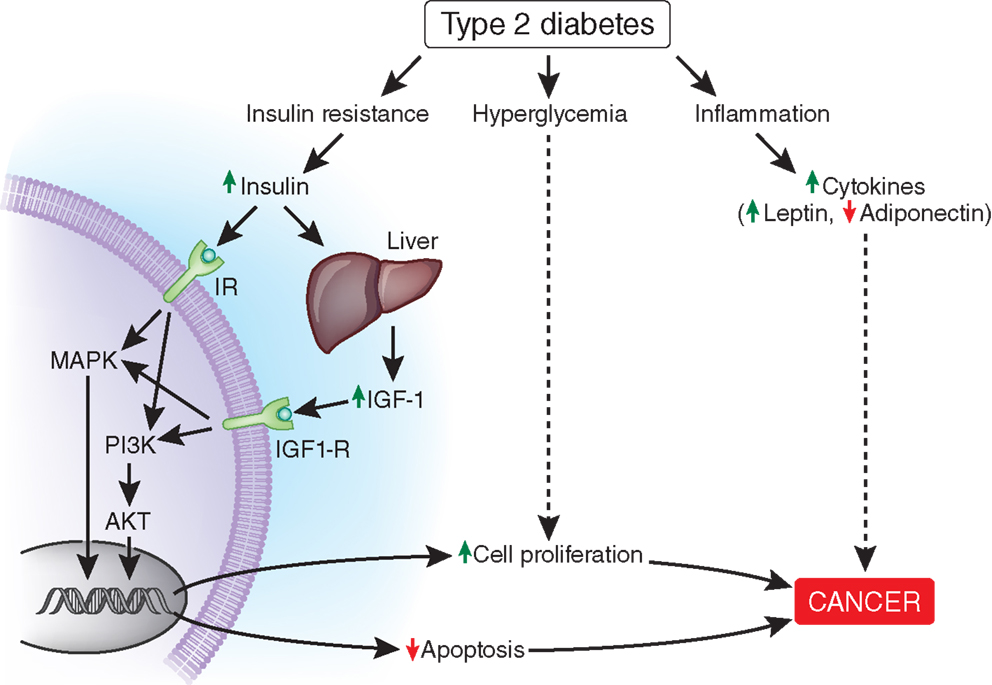
Figure 3. Possible mechanisms linking diabetes to accelerated cancer development. Binding of insulin to the insulin receptor (IR in the figure), likely IR-A and/or binding of IGF-1 to the IGF-1 receptor activate MAPK and PI3K/AKT signaling pathways leading to increased cell proliferation and decreased apoptosis. Hyperglycemia and chronic inflammation also contribute to cancer progression through other mechanisms. The mechanisms by which HCV promotes the development of HCC were not included in this figure. IGF-1, insulin-like growth factor 1; IGF-1R, insulin-like growth factor 1 receptor; IR-A, insulin receptor A; MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B.
Hyperinsulinemia also augments IGF-1 receptor (IGF-1R) signaling through increased hepatic production of IGF-1 and decreased hepatic production of IGF-binding proteins (IGF-BPs), thereby increasing levels of free IGF-1 (184, 190). Similar to activation of IR-A, IGF-1R activation leads to stimulation of the MAPK and PI3K/AKT pathways (Figure 3) (184, 186, 189). The LeRoith group demonstrated that administration of IGF-1 to liver-specific IGF-1-deficient (LID) mice with orthotopically implanted colon adenocarcinoma resulted in increased liver metastasis compared with saline controls (191). Therefore, it is possible that elevated levels of IGF-1 induced by hyperinsulinemia could also promote HCC growth. Notably, IGF-1 and IGF-BPs are primarily produced in the liver. Greater than 90% of patients with HCC suffer from cirrhosis (192), which may impair IGF-1 and IGF-BP synthesis.
Diabetes and obesity are states of chronic inflammation associated with elevated levels of leptin and low levels of adiponectin (193). Leptin is a proinflammatory adipokine that is linked to cell proliferation and invasion of cancer cells, while adiponectin is an anti-inflammatory adipokine that has been linked to reduced tumor growth (184, 188, 193). While the precise relationship between leptin, adiponectin, and HCC is still poorly understood (194), the chronic inflammation associated with diabetes may also promote HCC.
Further supporting the association between HCC and diabetes is recent evidence that metformin decreases the risk of HCC in patients with chronic HCV infection. A 2009 study of patients with HCV cirrhosis and diabetes found the 5-year incidence of HCC to be 9.5% for patients treated with metformin versus 31.2% without metformin therapy (p = 0.001) (182). Several additional studies, including a recent meta-analysis, show similar findings (183, 184). While clinical data are suggestive that metformin has anti-neoplastic properties, supporting laboratory models have been criticized for using concentrations of metformin that greatly exceed clinical dosing (195). Further studies are needed to determine whether metformin’s anti-neoplastic effects are secondary to direct effects of metformin on hepatocytes or due to the insulin-sensitizing effect of metformin.
Screening for Diabetes in HCV Patients and Therapeutic Considerations
Screening
Who?
As summarized above, epidemiological data clearly show that patients with chronic HCV infection are more susceptible to develop T2D. Moreover, there is negative impact of IR and diabetes on virological response and long-term outcomes of HCV infection. Therefore, we recommend screening for diabetes in all HCV-infected patients regardless of age or medical history. There is also epidemiological evidence for increased prevalence of hepatitis C in patients with diabetes, and some authors find it reasonable to screen patients with diabetes and persistently elevated ALT for HCV (196). T2D patients frequently present with abnormal liver enzymes (197); however, in most cases, the elevated ALT levels may represent non-alcoholic fatty liver disease (NAFLD). Still testing for hepatitis C could be considered in diabetic patients with elevated liver enzymes, especially in cases with no evidence of NAFLD. We recommend that each case be evaluated individually.
Whether screening for islet autoantibodies is necessary prior to IFN therapy, which has been shown to trigger islet autoimmunity, is unclear. Fabris reported the presence of autoantibodies in 50% of patients with IFN-induced T1D (16). In our opinion, screening for islet antibodies should be considered in patients with increased risk for T1D, such as patients with first degree relatives with T1D, patients with known autoimmune conditions, and HCV patients receiving IFNα (Figure 4). Pretreatment screening for diabetes (prior to IFNα therapy) and periodic monitoring of blood glucose (BG) should be performed during and after IFNα treatment.
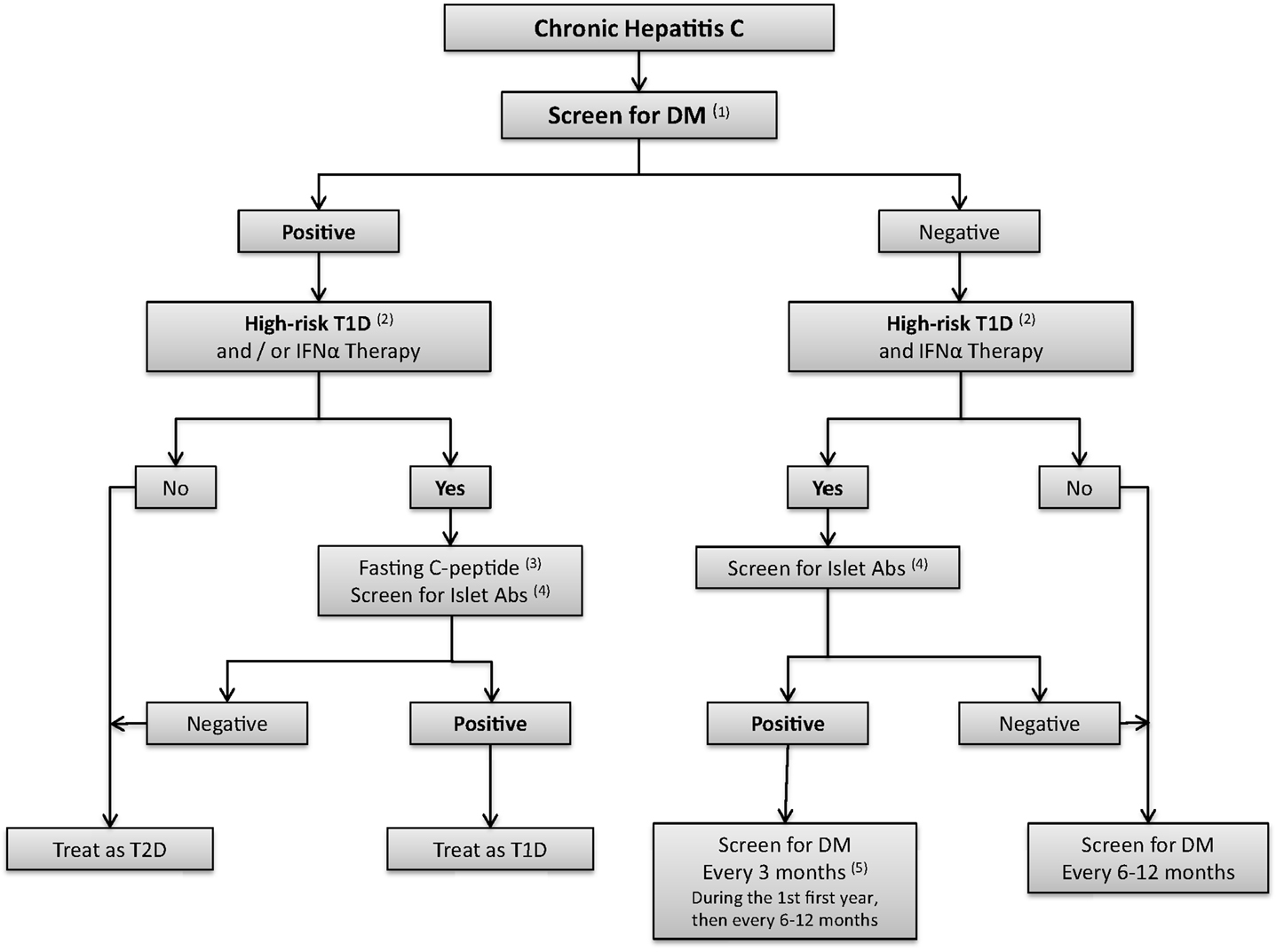
Figure 4. Algorithm for screening for diabetes in hepatitis C patients. (1) Screen for DM: fasting blood sugar (FBS) and/or oral glucose tolerance test (OGTT). HbA 1c might be considered if the measurement methods are well established. (2) High risk Type 1 diabetes patients are defined as individuals with first degree relatives with type 1 diabetes (T1D); patients with other autoimmune conditions; or patients might be considered as high risk, if IFNα therapy is considered. (3) Fasting C-peptide levels. (4) Islet antibodies include autoantibodies to glutamic-acid decarboxylase (GAD65), autoantibodies to insulin (IAA2), autoantibodies to the tyrosine phosphatase IA-2 and IA2B, autoantibodies to zinc transporter 8 (ZnT8), (5) IFNα may induce autoimmunity and therefore measurement of islet antibodies should be considered before and after IFNα therapy in high risk patients, and in patients with suspicion of T1D after initiation of IFNα.
How?
According to ADA guidelines, the standard screening test for diabetes is fasting plasma Glucose (FPG) (198). The 75-gr oral glucose test (OGT) should be considered in patients with pre-diabetes (fasting blood sugar (FBS) 100–125 mg/dL or 6–6.9 mmol/L) or high risk of diabetes.
HbA1c is an important tool for follow-up of diabetic patients; however, recent prospective studies have shown a continuous association between HbA1c at baseline and subsequent diabetes. Therefore, HbA1c levels in the range of 5.7–6.4 can also be considered as pre-diabetes (198). Treatment with IFNα and RBV may induce hemolytic anemia, thereby falsely decreasing HbA1c levels (199). Testing for fructosamine may be valuable in these cases as well as in cases with known hemoglobinopathies.
How Often?
Screening for T2D is recommended every 3 years in the general population of individuals older than 45 years (198). In our opinion, screening for diabetes should be performed every 6–12 months in patients with hepatitis C in view of the strong association between the two diseases. We recommend testing for T1D if the patient develops diabetes after initiation of IFNα therapy (Figure 4).
Therapeutic Considerations
Diabetes in HCV patients should be treated according to the ADA guidelines (198). Management of diabetes in patients with liver diseases is, however, complicated by the effects of drug metabolism by the liver, drug interactions, and potential liver toxicity of some medications used to treat diabetes [For review, see Ref. (196)].
IFNα-Induced Type 1 Diabetes
To date, no consensus exists regarding continuation or withdrawal of IFNα–RBV in IFN-induced T1D. However, reported cases show that IFNα–RBV are frequently stopped after diagnosis of IFN-induced T1D, presumably in the hope of reversing the diabetes. Permanent insulin treatment has been required in most cases of IFN-induced T1D (16); however, improvement has been observed in a few cases (110).
Insulin Resistance and Type 2 Diabetes
Metformin was not reported to cause hepatotoxicity and therefore should be considered as the drug of choice in HCV patients with IR or T2D. Although there is no clear evidence that treatment with metformin will increase the sustained viral response in HCV patients, there is increasing evidence that metformin is independently associated with reduced risk for HCC and liver-related death/transplantation (157, 177, 178, 200). The use of metformin, however, is not recommended in advanced hepatic disease because of increased risk for lactic acidosis (201).
Use of Metformin for other liver pathologies associated with IR, such as NAFLD, did not improve hepatic histopathology (202). By contrast, recent studies on another class of insulin sensitizers – thiazolidinediones (TZDs) – in NAFLD showed improvement in ALT and liver histology [for review, see Ref. (203)]. Sumie et al. assessed the effect of pioglitazone in patients with HCV-induced HCC and found no effect on recurrence free survival (204). Nevertheless, the treatment improved the IR (204). Yet, due to reported cases of acute cholestatic hepatitis (205, 206), TZD use is not recommended in advanced liver cirrhosis.
Glucosidase inhibitors act directly on the GI tract and in a recent placebo-controlled cross over study in patients with grade 1–2 hepatic encephalopathy, acarbose decreased both fasting and postprandial glucose (207). Acarbose increases the risk of hyperammonemia (208), and can induce mild transient elevation of ALT. Rare reversible cases of severe liver disease have been reported [for review, see Ref. (209)], and therefore, it should be used with caution.
Glucagon-like peptide-1 (GLP-1) is a gut-derived incretin hormone that stimulates insulin secretion. Dipeptidyl peptidase-4 (DPP-4) inhibitors increase incretin hormone levels, such as GLP-1. Itou et al. demonstrated that the serum GLP-1 levels were significantly decreased in HCV patients compared to control group and HBV group; and that DPP-4 expression was significantly increased in the ileum, liver, and serum in the HCV group. The authors concluded that altered expression of GLP-1 may be involved in the development of HCV-associated glucose intolerance (210). Recent studies on GLP-1 have shown a slowing of the progression of NAFLD by direct effects on lipid metabolism in hepatocytes, and on inflammation in the liver. DPP-4 inhibitors may also affect hepatic pathways of fat elimination [for review, see Ref. (211, 212)]. A case-control study assessed the efficacy and safety of DPP-4 inhibitors in HCV patients (16 patients with T2D, included 3 patients with hepatic cirrhosis) and reported a reduction in HbA1C without side effects (213). Further larger studies are needed to support their use in patients with advanced hepatic diseases.
There are no trials examining the safety of sulfonylureas (SUs) in patients with chronic liver diseases. The use of SU with short half-life is generally considered safe in patients with liver disease (196). However, close monitoring of BG is necessary since there might be increased risk of hypoglycemia. SUs are metabolized by the liver and excreted by the kidneys, and their use is contraindicated in advanced liver diseases.
Insulin has been considered as the drug of choice in patients with diabetes and decompensated liver disease due to short half-life to prevent hypoglycemia and hyperglycemia by frequent adjustment of the doses. However, a nested case-control study conducted in Japan found that use of insulin and second-generation SU were independent variables associated with incidence of HCC (OR = 2.96 95% confidence interval 1.29–6.82) (214). Other studies showed that use of metformin was associated with reduced risk of HCC compared with SUs and insulin (215, 216). A meta-analysis of observational studies reported that insulin and SU increased the risk for HCC, while metformin reduced the risk, and TDZs did not change it (217). Prospective studies are required to assess the link between exogenous insulin therapy and SU, and the risk for HCC.
Conclusion
Hepatitis C and DM are chronic diseases with high global prevalence of epidemic proportions. The association between hepatitis C and DM has been substantiated by numerous studies in the past two decades and represents a two-way association. On the one hand, HCV infection triggers diabetes, mostly type 2 but occasionally (especially in patients treated with IFNα) type 1 diabetes; and, on the other hand, diabetes worsens hepatitis C outcomes, including increasing the risk for cirrhosis and HCC. Mechanistic studies support the hypothesis that HCV infection leads to defects in insulin signaling pathways, and that direct effects of HCV on the beta cells are possible. Metformin is the drug of choice in chronic HCV infection as data provided show a significant reduction in the risk for HCC. Further studies are needed to assess the long-term effect of TDZs and DPP-4 inhibitors on glycemic control in HCV patients, and the effects of insulin and SU on cancer progression.
In the light of these data, it is essential that physicians caring for HCV patients are aware of the high risk for T2D and T1D and that they screen HCV patients for diabetes. In addition, the presence of diabetes in an HCV patient should alert the clinician to the possibility of worse outcomes. It remains to be determined whether the new non-interferon-based therapeutic regimens for chronic HCV infection (such as protease inhibitors) will reduce the frequency of diabetes in HCV patients or improve glycemic control if diabetes has already developed.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by DK061659, DK067555, and DK073681 from NIDDK and Helse Sør-Øst. We wish to thank Dr. Emily Gallagher for helpful discussions.
References
1. Hepatitis C. World Health Organisation Media Center (2013). Available from: http://www.who.int/mediacentre/factsheets/fs164/en/
2. Razavi H, Elkhoury AC, Elbasha E, Estes C, Pasini K, Poynard T, et al. Chronic hepatitis C virus (HCV) disease burden and cost in the United States. Hepatology (2013) 57(6):2164–70. doi: 10.1002/hep.26218
3. Mayo MJ. Extrahepatic manifestations of hepatitis C infection. Am J Med Sci (2003) 325(3):135–48. doi:10.1097/00000441-200303000-00006
4. Jacobson IM, Cacoub P, Dal ML, Harrison SA, Younossi ZM. Manifestations of chronic hepatitis C virus infection beyond the liver. Clin Gastroenterol Hepatol (2010) 8(12):1017–29. doi:10.1016/j.cgh.2010.08.026
5. White DL, Ratziu V, El-Serag HB. Hepatitis C infection and risk of diabetes: a systematic review and meta-analysis. J Hepatol (2008) 49(5):831–44. doi:10.1016/j.jhep.2008.08.006
6. Montenegro L, De MA, Misciagna G, Guerra V, Di LA. Virus C hepatitis and type 2 diabetes: a cohort study in southern Italy. Am J Gastroenterol (2013) 108(7):1108–11. doi:10.1038/ajg.2013.90
7. Mason AL, Lau JY, Hoang N, Qian K, Alexander GJ, Xu L, et al. Association of diabetes mellitus and chronic hepatitis C virus infection. Hepatology (1999) 29(2):328–33. doi:10.1002/hep.510290235
8. Montano-Loza AJ, Sultan A, Falanga D, Loss G, Mason AL. Immunogenetic susceptibility to diabetes mellitus in patients with liver disease. Liver Int (2009) 29(10):1543–51. doi:10.1111/j.1478-3231.2009.02095.x
9. Simo R, Hernandez C, Genesca J, Jardi R, Mesa J. High prevalence of hepatitis C virus infection in diabetic patients. Diabetes Care (1996) 19(9):998–1000. doi:10.2337/diacare.19.9.998
10. Soma J, Saito T, Taguma Y, Chiba S, Sato H, Sugimura K, et al. High prevalence and adverse effect of hepatitis C virus infection in type II diabetic-related nephropathy. J Am Soc Nephrol (2000) 11(4):690–9.
11. Guo X, Jin M, Yang M, Liu K. Type 2 diabetes mellitus and the risk of hepatitis C virus infection: a systematic review. Sci Rep (2013) 18(3):2981. doi:10.1038/srep02981
12. Chen LK, Chou YC, Tsai ST, Hwang SJ, Lee SD. Hepatitis C virus infection-related type 1 diabetes mellitus. Diabet Med (2005) 22(3):340–3. doi:10.1111/j.1464-5491.2005.01412.x
13. Masuda H, Atsumi T, Fujisaku A, Shimizu C, Yoshioka N, Koike T. Acute onset of type 1 diabetes accompanied by acute hepatitis C: the potential role of proinflammatory cytokine in the pathogenesis of autoimmune diabetes. Diabetes Res Clin Pract (2007) 75(3):357–61. doi:10.1016/j.diabres.2006.07.016
14. Tomer Y. Hepatitis C and interferon induced thyroiditis. J Autoimmun (2010) 34(3):J322–6. doi:10.1016/j.jaut.2009.11.008
15. Hasham A, Zhang W, Lotay V, Haggerty S, Stefan M, Concepcion E, et al. Genetic analysis of interferon induced thyroiditis (IIT): evidence for a key role for MHC and apoptosis related genes and pathways. J Autoimmun (2013) 44:61–70. doi:10.1016/j.jaut.2013.04.002
16. Fabris P, Floreani A, Tositti G, Vergani D, De LF, Betterle C. Type 1 diabetes mellitus in patients with chronic hepatitis C before and after interferon therapy. Aliment Pharmacol Ther (2003) 18(6):549–58. doi:10.1046/j.1365-2036.2003.01681.x
17. Niewold TB, Swedler WI. Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin Rheumatol (2005) 24(2):178–81. doi:10.1007/s10067-004-1024-2
18. Cacopardo B, Benanti F, Pinzone MR, Nunnari G. Rheumatoid arthritis following PEG-interferon-alfa-2a plus ribavirin treatment for chronic hepatitis C: a case report and review of the literature. BMC Res Notes (2013) 6:437. doi:10.1186/1756-0500-6-437
19. Borgia G, Reynaud L, Gentile I, Borrelli F, Cerini R, Ciampi R, et al. Pernicious anemia during IFN-alpha treatment for chronic hepatitis C. J Interferon Cytokine Res (2003) 23(1):11–2. doi:10.1089/10799900360520405
20. Kawazoe T, Araki M, Lin Y, Ogawa M, Okamoto T, Yamamura T, et al. New-onset type 1 diabetes mellitus and anti-aquaporin-4 antibody positive optic neuritis associated with type 1 interferon therapy for chronic hepatitis C. Intern Med (2012) 51(18):2625–9. doi:10.2169/internalmedicine.51.7771
21. Tomasiewicz K, Modrzewska R, Semczuk G. Vitiligo associated with pegylated interferon and ribavirin treatment of patients with chronic hepatitis C: a case report. Adv Ther (2006) 23(1):139–42. doi:10.1007/BF02850354
22. Sykia A, Gigi E, Sinakos E, Bibashi E, Bellou A, Raptopoulou-Gigi M. Severe autoimmune hemolytic anemia complicated with liver decompensation and invasive aspergillosis in a patient with chronic hepatitis C during treatment with peg-interferon-a and ribavirin. J Gastrointestin Liver Dis (2009) 18(1):118–9.
23. Burdick LM, Somani N, Somani AK. Type I IFNs and their role in the development of autoimmune diseases. Expert Opin Drug Saf (2009) 8(4):459–72. doi:10.1517/14740330903066726
24. Lee MH, Yang HI, Lu SN, Jen CL, You SL, Wang LY, et al. Chronic hepatitis C virus infection increases mortality from hepatic and extrahepatic diseases: a community-based long-term prospective study. J Infect Dis (2012) 206(4):469–77. doi:10.1093/infdis/jis385
25. Kruszynska YT, NcIntyre N. Carbohydrate metabolism. In: McIntyre N, Benhamou JP, Bircher J, Rizzetto M, Rodes J, editors. Oxford Textbook of Clinical Hepatology. Oxford, England: Oxford University press (1991). p. 129–34.
26. Allison ME, Wreghitt T, Palmer CR, Alexander GJ. Evidence for a link between hepatitis C virus infection and diabetes mellitus in a cirrhotic population. J Hepatol (1994) 21(6):1135–9. doi:10.1016/S0168-8278(05)80631-2
27. Lonardo A, Adinolfi LE, Petta S, Craxi A, Loria P. Hepatitis C and diabetes: the inevitable coincidence? Expert Rev Anti Infect Ther (2009) 7(3):293–308. doi:10.1586/eri.09.3
28. Romero-Gomez M, Del Mar Viloria M, Andrade RJ, Salmeron J, Diago M, Fernandez-Rodriguez CM, et al. Insulin resistance impairs sustained response rate to peginterferon plus ribavirin in chronic hepatitis C patients. Gastroenterology (2005) 128(3):636–41. doi:10.1053/j.gastro.2004.12.049
29. Grimbert S, Valensi P, Levy-Marchal C, Perret G, Richardet JP, Raffoux C, et al. High prevalence of diabetes mellitus in patients with chronic hepatitis C. A case-control study. Gastroenterol Clin Biol (1996) 20(6–7):544–8.
30. Mehta SH, Brancati FL, Strathdee SA, Pankow JS, Netski D, Coresh J, et al. Hepatitis C virus infection and incident type 2 diabetes. Hepatology (2003) 38(1):50–6. doi:10.1053/jhep.2003.50291
31. Knobler H, Schihmanter R, Zifroni A, Fenakel G, Schattner A. Increased risk of type 2 diabetes in noncirrhotic patients with chronic hepatitis C virus infection. Mayo Clin Proc (2000) 75(4):355–9. doi:10.4065/75.4.355
32. Mangia A, Schiavone G, Lezzi G, Marmo R, Bruno F, Villani MR, et al. HCV and diabetes mellitus: evidence for a negative association. Am J Gastroenterol (1998) 93(12):2363–7. doi:10.1111/j.1572-0241.1998.00688.x
33. Papatheodoridis GV, Chrysanthos N, Savvas S, Sevastianos V, Kafiri G, Petraki K, et al. Diabetes mellitus in chronic hepatitis B and C: prevalence and potential association with the extent of liver fibrosis. J Viral Hepat (2006) 13(5):303–10. doi:10.1111/j.1365-2893.2005.00677.x
34. Missiha SB, Ostrowski M, Heathcote EJ. Disease progression in chronic hepatitis C: modifiable and nonmodifiable factors. Gastroenterology (2008) 134(6):1699–714. doi:10.1053/j.gastro.2008.02.069
35. Ruhl CE, Menke A, Cowie CC, Everhart JE. Relationship of hepatitis C virus infection with diabetes in the U.S. population. Hepatology (2014) 60(4):1139–49. doi:10.1002/hep.27047
36. Fraser GM, Harman I, Meller N, Niv Y, Porath A. Diabetes mellitus is associated with chronic hepatitis C but not chronic hepatitis B infection. Isr J Med Sci (1996) 32(7):526–30.
37. Aytug S, Reich D, Sapiro LE, Bernstein D, Begum N. Impaired IRS-1/PI3-kinase signaling in patients with HCV: a mechanism for increased prevalence of type 2 diabetes. Hepatology (2003) 38(6):1384–92. doi:10.1053/jhep.2003.09012
38. el-Zayadi AR, Selim OE, Hamdy H, Dabbous H, Ahdy A, Moniem SA. Association of chronic hepatitis C infection and diabetes mellitus. Trop Gastroenterol (1998) 19(4):141–4.
39. Ozyilkan E, Arslan M. Increased prevalence of diabetes mellitus in patients with chronic hepatitis C virus infection. Am J Gastroenterol (1996) 91(7):1480–1.
40. Zein NN, Abdulkarim AS, Wiesner RH, Egan KS, Persing DH. Prevalence of diabetes mellitus in patients with end-stage liver cirrhosis due to hepatitis C, alcohol, or cholestatic disease. J Hepatol (2000) 32(2):209–17. doi:10.1016/S0168-8278(00)80065-3
41. Elhawary EI, Mahmoud GF, El-Daly MA, Mekky FA, Esmat GG, Abdel-Hamid M. Association of HCV with diabetes mellitus: an Egyptian case-control study. Virol J (2011) 8:367. doi:10.1186/1743-422X-8-367
42. Memon MS, Arain ZI, Naz F, Zaki M, Kumar S, Burney AA. Prevalence of type 2 diabetes mellitus in hepatitis C virus infected population: a Southeast Asian study. J Diabetes Res (2013) 2013:539361. doi:10.1155/2013/539361
43. Mehta SH, Brancati FL, Sulkowski MS, Strathdee SA, Szklo M, Thomas DL. Prevalence of type 2 diabetes mellitus among persons with hepatitis C virus infection in the United States. Ann Intern Med (2000) 133(8):592–9. doi:10.7326/0003-4819-133-8-200010170-00009
44. Soverini V, Persico M, Bugianesi E, Forlani G, Salamone F, Massarone M, et al. HBV and HCV infection in type 2 diabetes mellitus: a survey in three diabetes units in different Italian areas. Acta Diabetol (2011) 48(4):337–43. doi:10.1007/s00592-011-0293-x
45. Antonelli A, Ferri C, Fallahi P, Pampana A, Ferrari SM, Goglia F, et al. Hepatitis C virus infection: evidence for an association with type 2 diabetes. Diabetes Care (2005) 28(10):2548–50. doi:10.2337/diacare.28.10.2548
46. Ryu JK, Lee SB, Hong SJ, Lee S. Association of chronic hepatitis C virus infection and diabetes mellitus in Korean patients. Korean J Intern Med (2001) 16(1):18–23. doi:10.3904/kjim.2001.16.1.18
47. Arao M, Murase K, Kusakabe A, Yoshioka K, Fukuzawa Y, Ishikawa T, et al. Prevalence of diabetes mellitus in Japanese patients infected chronically with hepatitis C virus. J Gastroenterol (2003) 38(4):355–60. doi:10.1007/s005350300063
48. Chen HF, Li CY, Chen P, See TT, Lee HY. Seroprevalence of hepatitis B and C in type 2 diabetic patients. J Chin Med Assoc (2006) 69(4):146–52. doi:10.1016/S1726-4901(09)70195-9
49. Sangiorgio L, Attardo T, Gangemi R, Rubino C, Barone M, Lunetta M. Increased frequency of HCV and HBV infection in type 2 diabetic patients. Diabetes Res Clin Pract (2000) 48(2):147–51. doi:10.1016/S0168-8227(99)00135-7
50. Jadoon NA, Shahzad MA, Yaqoob R, Hussain M, Ali N. Seroprevalence of hepatitis C in type 2 diabetes: evidence for a positive association. Virol J (2010) 7:304. doi:10.1186/1743-422X-7-304
51. Caronia S, Taylor K, Pagliaro L, Carr C, Palazzo U, Petrik J, et al. Further evidence for an association between non-insulin-dependent diabetes mellitus and chronic hepatitis C virus infection. Hepatology (1999) 30(4):1059–63. doi:10.1002/hep.510300416
52. Harrison TA, Hindorff LA, Kim H, Wines RC, Bowen DJ, McGrath BB, et al. Family history of diabetes as a potential public health tool. Am J Prev Med (2003) 24(2):152–9. doi:10.1016/S0749-3797(02)00588-3
53. Lau JY, Davis GL, Prescott LE, Maertens G, Lindsay KL, Qian K, et al. Distribution of hepatitis C virus genotypes determined by line probe assay in patients with chronic hepatitis C seen at tertiary referral centers in the United States. Hepatitis interventional therapy group. Ann Intern Med (1996) 124(10):868–76. doi:10.7326/0003-4819-124-10-199605150-00002
54. Hui JM, Sud A, Farrell GC, Bandara P, Byth K, Kench JG, et al. Insulin resistance is associated with chronic hepatitis C virus infection and fibrosis progression [corrected]. Gastroenterology (2003) 125(6):1695–704. doi:10.1053/j.gastro.2003.08.032
55. Pazhanivel M, Jayanthi V. Diabetes mellitus and cirrhosis liver. Minerva Gastroenterol Dietol (2010) 56(1):7–11.
56. Custro N, Carroccio A, Ganci A, Scafidi V, Campagna P, Di PL, et al. Glycemic homeostasis in chronic viral hepatitis and liver cirrhosis. Diabetes Metab (2001) 27(4 Pt 1):476–81. doi:09.2001/27-4-1262-101019
57. Huang YW, Yang SS, Fu SC, Wang TC, Hsu CK, Chen DS, et al. Increased risk of cirrhosis and its decompensation in chronic hepatitis C patients with new-onset diabetes: a nationwide cohort study. Hepatology (2014) 60(3):807–14. doi:10.1002/hep.27212
58. Giordanino C, Bugianesi E, Smedile A, Ciancio A, Abate ML, Olivero A, et al. Incidence of type 2 diabetes mellitus and glucose abnormalities in patients with chronic hepatitis C infection by response to treatment: results of a cohort study. Am J Gastroenterol (2008) 103(10):2481–7. doi:10.1111/j.1572-0241.2008.02002.x
59. Aghemo A, Prati GM, Rumi MG, Soffredini R, D’Ambrosio R, Orsi E, et al. Sustained virological response prevents the development of insulin resistance in patients with chronic hepatitis C. Hepatology (2012) 56(5):1681–7. doi:10.1002/hep.25867
60. Arase Y, Suzuki F, Suzuki Y, Akuta N, Kobayashi M, Kawamura Y, et al. Sustained virological response reduces incidence of onset of type 2 diabetes in chronic hepatitis C. Hepatology (2009) 49(3):739–44. doi:10.1002/hep.22703
61. Cimino L, Oriani G, D’Arienzo A, Manguso F, Loguercio C, Ascione A, et al. Interactions between metabolic disorders (diabetes, gallstones, and dyslipidaemia) and the progression of chronic hepatitis C virus infection to cirrhosis and hepatocellular carcinoma. A cross-sectional multicentre survey. Dig Liver Dis (2001) 33(3):240–6. doi:10.1016/S1590-8658(01)80714-3
62. Masini M, Campani D, Boggi U, Menicagli M, Funel N, Pollera M, et al. Hepatitis C virus infection and human pancreatic beta-cell dysfunction. Diabetes Care (2005) 28(4):940–1. doi:10.2337/diacare.28.4.940
63. Mihm S. Hepatitis C virus, diabetes and steatosis: clinical evidence in favor of a linkage and role of genotypes. Dig Dis (2010) 28(1):280–4. doi:10.1159/000282103
64. Bureau C, Bernad J, Chaouche N, Orfila C, Beraud M, Gonindard C, et al. Nonstructural 3 protein of hepatitis C virus triggers an oxidative burst in human monocytes via activation of NADPH oxidase. J Biol Chem (2001) 276(25):23077–83. doi:10.1074/jbc.M100698200
65. Gong G, Waris G, Tanveer R, Siddiqui A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc Natl Acad Sci U S A (2001) 98(17):9599–604. doi:10.1073/pnas.171311298
66. Korenaga M, Wang T, Li Y, Showalter LA, Chan T, Sun J, et al. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J Biol Chem (2005) 280(45):37481–8. doi:10.1074/jbc.M506412200
67. Tardif KD, Mori K, Siddiqui A. Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J Virol (2002) 76(15):7453–9. doi:10.1128/JVI.76.15.7453-7459.2002
68. Durante-Mangoni E, Zampino R, Marrone A, Tripodi MF, Rinaldi L, Restivo L, et al. Hepatic steatosis and insulin resistance are associated with serum imbalance of adiponectin/tumour necrosis factor-alpha in chronic hepatitis C patients. Aliment Pharmacol Ther (2006) 24(9):1349–57. doi:10.1111/j.1365-2036.2006.03114.x
69. Elsammak M, Refai W, Elsawaf A, Abdel-Fattah I, Abd EE, Ghazal A. Elevated serum tumor necrosis factor alpha and ferritin may contribute to the insulin resistance found in HCV positive Egyptian patients. Curr Med Res Opin (2005) 21(4):527–34. doi:10.1185/030079905X38141
70. Shintani Y, Fujie H, Miyoshi H, Tsutsumi T, Tsukamoto K, Kimura S, et al. Hepatitis C virus infection and diabetes: direct involvement of the virus in the development of insulin resistance. Gastroenterology (2004) 126(3):840–8. doi:10.1053/j.gastro.2003.11.056
71. Knobler H, Zhornicky T, Sandler A, Haran N, Ashur Y, Schattner A. Tumor necrosis factor-alpha-induced insulin resistance may mediate the hepatitis C virus-diabetes association. Am J Gastroenterol (2003) 98(12):2751–6. doi:10.1016/j.amjgastroenterol.2003.06.002
72. McGuinness PH, Painter D, Davies S, McCaughan GW. Increases in intrahepatic CD68 positive cells, MAC387 positive cells, and proinflammatory cytokines (particularly interleukin 18) in chronic hepatitis C infection. Gut (2000) 46(2):260–9. doi:10.1136/gut.46.2.260
73. Hotamisligil GS, Budavari A, Murray D, Spiegelman BM. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-alpha. J Clin Invest (1994) 94(4):1543–9. doi:10.1172/JCI117495
74. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science (1993) 259(5091):87–91. doi:10.1126/science.7678183
75. Lang CH, Dobrescu C, Bagby GJ. Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology (1992) 130(1):43–52. doi:10.1210/en.130.1.43
76. Greenberg AS, McDaniel ML. Identifying the links between obesity, insulin resistance and beta-cell function: potential role of adipocyte-derived cytokines in the pathogenesis of type 2 diabetes. Eur J Clin Invest (2002) 32(Suppl 3):24–34. doi:10.1046/j.1365-2362.32.s3.4.x
77. Hotamisligil GS. The role of TNFalpha and TNF receptors in obesity and insulin resistance. J Intern Med (1999) 245(6):621–5. doi:10.1046/j.1365-2796.1999.00490.x
78. Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci U S A (1994) 91(11):4854–8. doi:10.1073/pnas.91.11.4854
79. Banerjee S, Saito K, Ait-Goughoulte M, Meyer K, Ray RB, Ray R. Hepatitis C virus core protein upregulates serine phosphorylation of insulin receptor substrate-1 and impairs the downstream akt/protein kinase B signaling pathway for insulin resistance. J Virol (2008) 82(6):2606–12. doi:10.1128/JVI.01672-07
80. Peltier HJ, Latham GJ. Normalization of microRNA expression levels in quantitative RT-PCR assays: identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA (2008) 14(5):844–52. doi:10.1261/rna.939908
81. Im SS, Kwon SK, Kim TH, Kim HI, Ahn YH. Regulation of glucose transporter type 4 isoform gene expression in muscle and adipocytes. IUBMB Life (2007) 59(3):134–45. doi:10.1080/15216540701313788
82. Nolte LA, Hansen PA, Chen MM, Schluter JM, Gulve EA, Holloszy JO. Short-term exposure to tumor necrosis factor-alpha does not affect insulin-stimulated glucose uptake in skeletal muscle. Diabetes (1998) 47(5):721–6. doi:10.2337/diabetes.47.5.721
83. Bose SK, Ray R. Hepatitis C virus infection and insulin resistance. World J Diabetes (2014) 5(1):52–8. doi:10.4239/wjd.v5.i1.52
84. Bose SK, Shrivastava S, Meyer K, Ray RB, Ray R. Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. J Virol (2012) 86(11):6315–22. doi:10.1128/JVI.00050-12
85. Kawaguchi T, Yoshida T, Harada M, Hisamoto T, Nagao Y, Ide T, et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am J Pathol (2004) 165(5):1499–508. doi:10.1016/S0002-9440(10)63408-6
86. Pazienza V, Clement S, Pugnale P, Conzelman S, Foti M, Mangia A, et al. The hepatitis C virus core protein of genotypes 3a and 1b downregulates insulin receptor substrate 1 through genotype-specific mechanisms. Hepatology (2007) 45(5):1164–71. doi:10.1002/hep.21634
87. Narita R, Abe S, Kihara Y, Akiyama T, Tabaru A, Otsuki M. Insulin resistance and insulin secretion in chronic hepatitis C virus infection. J Hepatol (2004) 41(1):132–8. doi:10.1016/j.jhep.2004.03.020
88. Knobler H, Schattner A. TNF-{alpha}, chronic hepatitis C and diabetes: a novel triad. QJM (2005) 98(1):1–6. doi:10.1093/qjmed/hci001
89. Huang JF, Dai CY, Yu ML, Huang CF, Huang CI, Yeh ML, et al. Pegylated interferon plus ribavirin therapy improves pancreatic beta-cell function in chronic hepatitis C patients. Liver Int (2011) 31(8):1155–62. doi:10.1111/j.1478-3231.2011.02545.x
90. Kawaguchi T, Ide T, Taniguchi E, Hirano E, Itou M, Sumie S, et al. Clearance of HCV improves insulin resistance, beta-cell function, and hepatic expression of insulin receptor substrate 1 and 2. Am J Gastroenterol (2007) 102(3):570–6. doi:10.1111/j.1572-0241.2006.01038.x
91. Koivisto VA, Pelkonen R, Cantell K. Effect of interferon on glucose tolerance and insulin sensitivity. Diabetes (1989) 38(5):641–7. doi:10.2337/diabetes.38.5.641
92. Ito Y, Takeda N, Ishimori M, Akai A, Miura K, Yasuda K. Effects of long-term interferon-alpha treatment on glucose tolerance in patients with chronic hepatitis C. J Hepatol (1999) 31(2):215–20. doi:10.1016/S0168-8278(99)80216-5
93. Rother KI, Brown RJ, Morales MM, Wright E, Duan Z, Campbell C, et al. Effect of ingested interferon-alpha on beta-cell function in children with new-onset type 1 diabetes. Diabetes Care (2009) 32(7):1250–5. doi:10.2337/dc08-2029
94. Brod SA, Atkinson M, Lavis VR, Brosnan PG, Hardin DS, Orlander PR, et al. Ingested IFN-alpha preserves residual beta cell function in type 1 diabetes. J Interferon Cytokine Res (2001) 21(12):1021–30. doi:10.1089/107999001317205141
95. Bartolome J, Rodriguez-Inigo E, Quadros P, Vidal S, Pascual-Miguelanez I, Rodriguez-Montes JA, et al. Detection of hepatitis C virus in thyroid tissue from patients with chronic HCV infection. J Med Virol (2008) 80(9):1588–94. doi:10.1002/jmv.21269
96. Yan FM, Chen AS, Hao F, Zhao XP, Gu CH, Zhao LB, et al. Hepatitis C virus may infect extrahepatic tissues in patients with hepatitis C. World J Gastroenterol (2000) 6(6):805–11.
97. Fiorino S, Cuppini A, Castellani G, Bacchi-Reggiani ML, Jovine E. HBV- and HCV-related infections and risk of pancreatic cancer. JOP (2013) 14(6):603–9. doi:10.6092/1590-8577/1948
98. Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene (2007) 26(15):2166–76. doi:10.1038/sj.onc.1210279
99. Hieronimus S, Fredenrich A, Tran A, Benzaken S, Fenichel P. Antibodies to GAD in chronic hepatitis C patients. Diabetes Care (1997) 20(6):1044. doi:10.2337/diacare.20.6.1044a
100. Ando H, Nagai Y, Yokoyama M, Takamura T, Kobayashi K. Antibodies to GAD in diabetic patients with chronic hepatitis C. Diabet Med (1998) 15(9):797. doi:10.1002/(SICI)1096-9136(199809)15:9<797::AID-DIA675>3.3.CO;2-D
101. Piquer S, Hernandez C, Enriquez J, Ross A, Esteban JI, Genesca J, et al. Islet cell and thyroid antibody prevalence in patients with hepatitis C virus infection: effect of treatment with interferon. J Lab Clin Med (2001) 137(1):38–42. doi:10.1067/mlc.2001.111515
102. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci (1957) 147(927):258–67. doi:10.1098/rspb.1957.0048
103. Davis GL, Balart LA, Schiff ER, Lindsay K, Bodenheimer HC Jr, Perrillo RP, et al. Treatment of chronic hepatitis C with recombinant interferon alfa. A multicenter randomized, controlled trial. Hepatitis interventional therapy group. N Engl J Med (1989) 321(22):1501–6. doi:10.1056/NEJM198911303212203
104. Fabris P, Betterle C, Floreani A, Greggio NA, de LF, Naccarato R, et al. Development of type 1 diabetes mellitus during interferon alfa therapy for chronic HCV hepatitis. Lancet (1992) 340(8818):548. doi:10.1016/0140-6736(92)91744-S
105. Fattovich G, Giustina G, Favarato S, Ruol A. A survey of adverse events in 11,241 patients with chronic viral hepatitis treated with alfa interferon. J Hepatol (1996) 24(1):38–47. doi:10.1016/S0168-8278(96)80184-X
106. Nakanishi K, Saitoh S. Clinical and genetic characteristics of patients with type 1 diabetes associated with interferon therapy. Diabetes Care (2011) 34(2):471–3. doi:10.2337/dc10-1237
107. Okanoue T, Sakamoto S, Itoh Y, Minami M, Yasui K, Sakamoto M, et al. Side effects of high-dose interferon therapy for chronic hepatitis C. J Hepatol (1996) 25(3):283–91. doi:10.1016/S0168-8278(96)80113-9
108. Nakamura K, Kawasaki E, Imagawa A, Awata T, Ikegami H, Uchigata Y, et al. Type 1 diabetes and interferon therapy: a nationwide survey in Japan. Diabetes Care (2011) 34(9):2084–9. doi:10.2337/dc10-2274
109. Schreuder TC, Gelderblom HC, Weegink CJ, Hamann D, Reesink HW, Devries JH, et al. High incidence of type 1 diabetes mellitus during or shortly after treatment with pegylated interferon alpha for chronic hepatitis C virus infection. Liver Int (2008) 28(1):39–46. doi:10.1111/j.1478-3231.2007.01610.x
110. Oka R, Hiroi N, Shigemitsu R, Sue M, Oshima Y, Yoshida-Hiroi M. Type 1 diabetes mellitus associated with pegylated interferon-alpha plus ribavirin treatment for chronic hepatitis C: case report and literature review. Clin Med Insights Endocrinol Diabetes (2011) 4:39–45. doi:10.4137/CMED.S7815
111. Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL Jr, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med (2002) 347(13):975–82. doi:10.1056/NEJMoa020047
112. Molleston JP, Mellman W, Narkewicz MR, Balistreri WF, Gonzalez-Peralta RP, Jonas MM, et al. Autoantibodies and autoimmune disease during treatment of children with chronic hepatitis C. J Pediatr Gastroenterol Nutr (2013) 56(3):304–10. doi:10.1097/MPG.0b013e3182774cae
113. Minami T, Kishikawa T, Sato M, Tateishi R, Yoshida H, Koike K. Meta-analysis: mortality and serious adverse events of peginterferon plus ribavirin therapy for chronic hepatitis C. J Gastroenterol (2013) 48(2):254–68. doi:10.1007/s00535-012-0631-y
114. Wesche B, Jaeckel E, Trautwein C, Wedemeyer H, Falorni A, Frank H, et al. Induction of autoantibodies to the adrenal cortex and pancreatic islet cells by interferon alpha therapy for chronic hepatitis C. Gut (2001) 48(3):378–83. doi:10.1136/gut.48.3.378
115. Tomer Y, Blackard JT, Akeno N. Interferon alpha treatment and thyroid dysfunction. Endocrinol Metab Clin North Am (2007) 36(4):1051–66. doi:10.1016/j.ecl.2007.07.001
116. Koivisto VA. Interferon alfa and development of type 1 diabetes mellitus. Lancet (1992) 340(8829):1236. doi:10.1016/0140-6736(92)92944-B
117. Choo QL, Richman KH, Han JH, Berger K, Lee C, Dong C, et al. Genetic organization and diversity of the hepatitis C virus. Proc Natl Acad Sci U S A (1991) 88(6):2451–5. doi:10.1073/pnas.88.6.2451
118. Zein NN, Rakela J, Krawitt EL, Reddy KR, Tominaga T, Persing DH. Hepatitis C virus genotypes in the United States: epidemiology, pathogenicity, and response to interferon therapy. Collaborative study group. Ann Intern Med (1996) 125(8):634–9. doi:10.7326/0003-4819-125-8-199610150-00002
119. Takada N, Takase S, Takada A, Date T. Differences in the hepatitis C virus genotypes in different countries. J Hepatol (1993) 17(3):277–83. doi:10.1016/S0168-8278(05)80205-3
120. Takada A, Tsutsumi M, Okanoue T, Matsushima T, Komatsu M, Fujiyama S. Distribution of the different subtypes of hepatitis C virus in Japan and the effects of interferon: a nationwide survey. J Gastroenterol Hepatol (1996) 11(3):201–7. doi:10.1111/j.1440-1746.1996.tb00063.x
121. Zein NN. Clinical significance of hepatitis C virus genotypes. Clin Microbiol Rev (2000) 13(2):223–35. doi:10.1128/CMR.13.2.223-235.2000
122. Dong ZX, Zhou HJ, Wang JH, Xiang XG, Zhuang Y, Guo SM, et al. Distribution of hepatitis C virus genotypes in Chinese patients with chronic hepatitis C: correlation with patients’ characteristics and clinical parameters. J Dig Dis (2012) 13(11):564–70. doi:10.1111/j.1751-2980.2012.00636.x
123. Hasham A, Tomer Y. Genetic and epigenetic mechanisms in thyroid autoimmunity. Immunol Res (2012) 54(1–3):204–13. doi:10.1007/s12026-012-8302-x
124. European Association for the Study of the Liver. EASL clinical practice guidelines: management of hepatitis C virus infection. J Hepatol (2011) 55(2):245–64. doi:10.1016/j.jhep.2011.02.023
125. Antaki N, Craxi A, Kamal S, Moucari R, Van der Merwe S, Haffar S, et al. The neglected hepatitis C virus genotypes 4, 5 and 6: an international consensus report. Liver Int (2010) 30(3):342–55. doi:10.1111/j.1478-3231.2009.02188.x
126. Ogihara T, Katagiri H, Yamada T, Kudo H, Imai J, Ishigaki Y, et al. Peginterferon (PEG-IFN) plus ribavirin combination therapy, but neither interferon nor PGE-IFN alone, induced type 1 diabetes in a patient with chronic hepatitis C. Intern Med (2009) 48(16):1387–90. doi:10.2169/internalmedicine.48.2220
127. European Association for Study of Liver. EASL clinical practice guidelines: management of hepatitis C virus infection. J Hepatol (2014) 60(2):392–420. doi:10.1016/j.jhep.2013.11.003
128. Alsabbagh ME, Eisa N, Alraiyes AH, Alraies MC. Chronic hepatitis C therapy: a rare complication revisited. BMJ Case Rep (2013) 2013:bcr2013200514. doi:10.1136/bcr-2013-200514
129. Huber A, Menconi F, Corathers S, Jacobson EM, Tomer Y. Joint genetic susceptibility to type 1 diabetes and autoimmune thyroiditis: from epidemiology to mechanisms. Endocr Rev (2008) 29(6):697–725. doi:10.1210/er.2008-0015
130. Nakanishi K, Kobayashi T, Murase T, Naruse T, Nose Y, Inoko H. Human leukocyte antigen-A24 and -DQA1*0301 in Japanese insulin-dependent diabetes mellitus: independent contributions to susceptibility to the disease and additive contributions to acceleration of beta-cell destruction. J Clin Endocrinol Metab (1999) 84(10):3721–5. doi:10.1210/jcem.84.10.6045
131. Kakizaki S, Takagi H, Murakami M, Takayama H, Mori M. HLA antigens in patients with interferon-alpha-induced autoimmune thyroid disorders in chronic hepatitis C. J Hepatol (1999) 30(5):794–800. doi:10.1016/S0168-8278(99)80131-7
132. Oppenheim Y, Kim G, Ban Y, Unger P, Concepcion E, Ando T, et al. The effects of alpha interferon on the development of autoimmune thyroiditis in the NOD H2h4 mouse. Clin Dev Immunol (2003) 10(2–4):161–5. doi:10.1080/10446670310001642177
133. Guo D, Dunbar JD, Yang CH, Pfeffer LM, Donner DB. Induction of Jak/STAT signaling by activation of the type 1 TNF receptor. J Immunol (1998) 160(6):2742–50.
134. Haque SJ, Williams BR. Signal transduction in the interferon system. Semin Oncol (1998) 25(1 Suppl 1):14–22.
135. Nguyen KB, Watford WT, Salomon R, Hofmann SR, Pien GC, Morinobu A, et al. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science (2002) 297(5589):2063–6. doi:10.1126/science.1074900
136. Foulis AK, Farquharson MA, Meager A. Immunoreactive alpha-interferon in insulin-secreting beta cells in type 1 diabetes mellitus. Lancet (1987) 2(8573):1423–7. doi:10.1016/S0140-6736(87)91128-7
137. Somoza N, Vargas F, Roura-Mir C, Vives-Pi M, Fernandez-Figueras MT, Ariza A, et al. Pancreas in recent onset insulin-dependent diabetes mellitus. Changes in HLA, adhesion molecules and autoantigens, restricted T cell receptor V beta usage, and cytokine profile. J Immunol (1994) 153(3):1360–77.
138. Huang X, Yuang J, Goddard A, Foulis A, James RF, Lernmark A, et al. Interferon expression in the pancreases of patients with type I diabetes. Diabetes (1995) 44(6):658–64. doi:10.2337/diab.44.6.658
139. Stewart TA, Hultgren B, Huang X, Pitts-Meek S, Hully J, MacLachlan NJ. Induction of type I diabetes by interferon-alpha in transgenic mice. Science (1993) 260(5116):1942–6. doi:10.1126/science.8100367
140. Akeno N, Smith EP, Stefan M, Huber AK, Zhang W, Keddache M, et al. IFN-alpha mediates the development of autoimmunity both by direct tissue toxicity and through immune cell recruitment mechanisms. J Immunol (2011) 186(8):4693–706. doi:10.4049/jimmunol.1002631
141. Li Q, Xu B, Michie SA, Rubins KH, Schreriber RD, McDevitt HO. Interferon-alpha initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci U S A (2008) 105(34):12439–44. doi:10.1073/pnas.0806439105
142. Nakazawa T, Satoh J, Takahashi K, Sakata Y, Ikehata F, Takizawa Y, et al. Complete suppression of insulitis and diabetes in NOD mice lacking interferon regulatory factor-1. J Autoimmun (2001) 17(2):119–25. doi:10.1006/jaut.2001.0531
143. Pfeffer LM, Dinarello CA, Herberman RB, Williams BR, Borden EC, Bordens R, et al. Biological properties of recombinant alpha-interferons: 40th anniversary of the discovery of interferons. Cancer Res (1998) 58(12):2489–99.
144. Harrison LC, Campbell IL, Allison J, Miller JF. MHC molecules and beta-cell destruction. Immune and nonimmune mechanisms. Diabetes (1989) 38(7):815–8. doi:10.2337/diab.38.7.815
145. Srivastava S, Koch MA, Pepper M, Campbell DJ. Type I interferons directly inhibit regulatory T cells to allow optimal antiviral T cell responses during acute LCMV infection. J Exp Med (2014) 211(5):961–74. doi:10.1084/jem.20131556
146. Mozzillo N, Ascierto P. Reduction of circulating regulatory T cells by intravenous high-dose interferon alfa-2b treatment in melanoma patients. Clin Exp Metastasis (2012) 29(7):801–5. doi:10.1007/s10585-012-9504-2
147. Feng L, Kang H, Liu LN, Cao YM. CD4+CD25+Foxp3+ regulatory T cells contribute in liver fibrosis improvement with interferon alpha. Inflammation (2013) 36(6):1374–82. doi:10.1007/s10753-013-9677-0
148. Nkontchou G, Bastard JP, Ziol M, Aout M, Cosson E, Ganne-Carrie N, et al. Insulin resistance, serum leptin, and adiponectin levels and outcomes of viral hepatitis C cirrhosis. J Hepatol (2010) 53(5):827–33. doi:10.1016/j.jhep.2010.04.035
149. Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet (1997) 349(9055):825–32. doi:10.1016/S0140-6736(96)07642-8
150. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology (2008) 134(6):1655–69. doi:10.1053/j.gastro.2008.03.003
151. Hui JM, Kench J, Farrell GC, Lin R, Samarasinghe D, Liddle C, et al. Genotype-specific mechanisms for hepatic steatosis in chronic hepatitis C infection. J Gastroenterol Hepatol (2002) 17(8):873–81. doi:10.1046/j.1440-1746.2002.02813.x
152. Hsu CS, Liu CH, Liu CJ, Hsu SJ, Chen CL, Hwang JJ, et al. Association of metabolic profiles with hepatic fibrosis in chronic hepatitis C patients with genotype 1 or 2 infection. J Gastroenterol Hepatol (2010) 25(5):970–7. doi:10.1111/j.1440-1746.2009.06186.x
153. Svegliati-Baroni G, Bugianesi E, Bouserhal T, Marini F, Ridolfi F, Tarsetti F, et al. Post-load insulin resistance is an independent predictor of hepatic fibrosis in virus C chronic hepatitis and in non-alcoholic fatty liver disease. Gut (2007) 56(9):1296–301. doi:10.1136/gut.2006.107946
154. Merchante N, Rivero A, de LS-G I, Merino D, Marquez M, Lopez-Ruz MA, et al. Insulin resistance is associated with liver stiffness in HIV/HCV co-infected patients. Gut (2009) 58(12):1654–60. doi:10.1136/gut.2009.179085
155. Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci (2009) 54(12):2699–705. doi:10.1007/s10620-008-0683-2
156. Dai CY, Huang JF, Hsieh MY, Hou NJ, Lin ZY, Chen SC, et al. Insulin resistance predicts response to peginterferon-alpha/ribavirin combination therapy in chronic hepatitis C patients. J Hepatol (2009) 50(4):712–8. doi:10.1016/j.jhep.2008.12.017
157. Romero-Gomez M, Diago M, Andrade RJ, Calleja JL, Salmeron J, Fernandez-Rodriguez CM, et al. Treatment of insulin resistance with metformin in naive genotype 1 chronic hepatitis C patients receiving peginterferon alfa-2a plus ribavirin. Hepatology (2009) 50(6):1702–8. doi:10.1002/hep.23206
158. Fattovich G, Covolo L, Pasino M, Perini E, Rossi L, Brocco G, et al. The homeostasis model assessment of the insulin resistance score is not predictive of a sustained virological response in chronic hepatitis C patients. Liver Int (2011) 31(1):66–74. doi:10.1111/j.1478-3231.2010.02343.x
159. Deltenre P, Louvet A, Lemoine M, Mourad A, Fartoux L, Moreno C, et al. Impact of insulin resistance on sustained response in HCV patients treated with pegylated interferon and ribavirin: a meta-analysis. J Hepatol (2011) 55(6):1187–94. doi:10.1016/j.jhep.2011.03.010
160. In: Boyle P, Bernard L, editors. World Cancer Report 2008 [Article Online]. World Health Organization, International Agency for Research on Cancer (2008). Available from: http://www.iarc.fr/en/publications/pdfs-online/wcr/index.php
161. Lai SW, Chen PC, Liao KF, Muo CH, Lin CC, Sung FC. Risk of hepatocellular carcinoma in diabetic patients and risk reduction associated with anti-diabetic therapy: a population-based cohort study. Am J Gastroenterol (2012) 107(1):46–52. doi:10.1038/ajg.2011.384
162. Veldt BJ, Chen W, Heathcote EJ, Wedemeyer H, Reichen J, Hofmann WP, et al. Increased risk of hepatocellular carcinoma among patients with hepatitis C cirrhosis and diabetes mellitus. Hepatology (2008) 47(6):1856–62. doi:10.1002/hep.22251
163. Seshasai SR, Kaptoge S, Thompson A, Di AE, Gao P, Sarwar N, et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N Engl J Med (2011) 364(9):829–41. doi:10.1056/NEJMoa1008862
164. El-Serag HB, Hampel H, Javadi F. The association between diabetes and hepatocellular carcinoma: a systematic review of epidemiologic evidence. Clin Gastroenterol Hepatol (2006) 4(3):369–80. doi:10.1016/j.cgh.2005.12.007
165. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest (2005) 115(2):209–18. doi:10.1172/JCI24282
166. Paradis V, Perlemuter G, Bonvoust F, Dargere D, Parfait B, Vidaud M, et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: a potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology (2001) 34(4 Pt 1):738–44. doi:10.1053/jhep.2001.28055
167. Garcia-Monzon C, Lo IO, Mayoral R, Gonzalez-Rodriguez A, Miquilena-Colina ME, Lozano-Rodriguez T, et al. Hepatic insulin resistance is associated with increased apoptosis and fibrogenesis in nonalcoholic steatohepatitis and chronic hepatitis C. J Hepatol (2011) 54(1):142–52. doi:10.1016/j.jhep.2010.06.021
168. Lonardo A, Adinolfi LE, Restivo L, Ballestri S, Romagnoli D, Baldelli E, et al. Pathogenesis and significance of hepatitis C virus steatosis: an update on survival strategy of a successful pathogen. World J Gastroenterol (2014) 20(23):7089–103. doi:10.3748/wjg.v20.i23.7089
169. Capeau J. Insulin resistance and steatosis in humans. Diabetes Metab (2008) 34(6 Pt 2):649–57. doi:10.1016/S1262-3636(08)74600-7
170. Frohnert BI, Jacobs DR Jr, Steinberger J, Moran A, Steffen LM, Sinaiko AR. Relation between serum free fatty acids and adiposity, insulin resistance, and cardiovascular risk factors from adolescence to adulthood. Diabetes (2013) 62(9):3163–9. doi:10.2337/db12-1122
171. Fartoux L, Poujol-Robert A, Guechot J, Wendum D, Poupon R, Serfaty L. Insulin resistance is a cause of steatosis and fibrosis progression in chronic hepatitis C. Gut (2005) 54(7):1003–8. doi:10.1136/gut.2004.050302
172. Goto K, Lin W, Zhang L, Jilg N, Shao RX, Schaefer EA, et al. The AMPK-related kinase SNARK regulates hepatitis C virus replication and pathogenesis through enhancement of TGF-beta signaling. J Hepatol (2013) 59(5):942–8. doi:10.1016/j.jhep.2013.06.025
173. del Campo JA, Lopez RA, Romero-Gomez M. Insulin resistance and response to antiviral therapy in chronic hepatitis C: mechanisms and management. Dig Dis (2010) 28(1):285–93. doi:10.1159/000282104
174. Garcia-Ruiz I, Solis-Munoz P, Gomez-Izquierdo E, Munoz-Yague MT, Valverde AM, Solis-Herruzo JA. Protein-tyrosine phosphatases are involved in interferon resistance associated with insulin resistance in HepG2 cells and obese mice. J Biol Chem (2012) 287(23):19564–73. doi:10.1074/jbc.M112.342709
175. Franceschini L, Realdon S, Marcolongo M, Mirandola S, Bortoletto G, Alberti A. Reciprocal interference between insulin and interferon-alpha signaling in hepatic cells: a vicious circle of clinical significance? Hepatology (2011) 54(2):484–94. doi:10.1002/hep.24394
176. Huang H, Kang R, Wang J, Luo G, Yang W, Zhao Z. Hepatitis C virus inhibits AKT-tuberous sclerosis complex (TSC), the mechanistic target of rapamycin (MTOR) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy (2013) 9(2):175–95. doi:10.4161/auto.22791
177. Yu JW, Sun LJ, Zhao YH, Kang P, Yan BZ. The effect of metformin on the efficacy of antiviral therapy in patients with genotype 1 chronic hepatitis C and insulin resistance. Int J Infect Dis (2012) 16(6):e436–41. doi:10.1016/j.ijid.2012.02.004
178. Sharifi AH, Mohammadi M, Fakharzadeh E, Zamini H, Zaer-Rezaee H, Jabbari H, et al. Efficacy of adding metformin to pegylated interferon and ribavirin in treatment naive patients with chronic hepatitis C: a randomized double-blind controlled trial. Middle East J Dig Dis (2014) 6(1):13–7.
179. Ishida H, Li K, Yi M, Lemon SM. p21-activated kinase 1 is activated through the mammalian target of rapamycin/p70 S6 kinase pathway and regulates the replication of hepatitis C virus in human hepatoma cells. J Biol Chem (2007) 282(16):11836–48. doi:10.1074/jbc.M610106200
180. Douglas MW, George J. Molecular mechanisms of insulin resistance in chronic hepatitis C. World J Gastroenterol (2009) 15(35):4356–64. doi:10.3748/wjg.15.4356
181. Moucari R, Asselah T, Cazals-Hatem D, Voitot H, Boyer N, Ripault MP, et al. Insulin resistance in chronic hepatitis C: association with genotypes 1 and 4, serum HCV RNA level, and liver fibrosis. Gastroenterology (2008) 134(2):416–23. doi:10.1053/j.gastro.2007.11.010
182. Myers RP, Patel K, Pianko S, Poynard T, McHutchison JG. The rate of fibrosis progression is an independent predictor of the response to antiviral therapy in chronic hepatitis C. J Viral Hepat (2003) 10(1):16–22. doi:10.1046/j.1365-2893.2003.00387.x
183. Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, et al. Diabetes and cancer: a consensus report. Diabetes Care (2010) 33(7):1674–85. doi:10.2337/dc10-0666
184. Zelenko Z, Gallagher EJ. Diabetes and cancer. Endocrinol Metab Clin North Am (2014) 43(1):167–85. doi:10.1016/j.ecl.2013.09.008
185. Noto H, Osame K, Sasazuki T, Noda M. Substantially increased risk of cancer in patients with diabetes mellitus: a systematic review and meta-analysis of epidemiologic evidence in Japan. J Diabetes Complications (2010) 24(5):345–53. doi:10.1016/j.jdiacomp.2010.06.004
186. Taniguchi CM, Ronald Kahn C. Chapter 2 insulin/IGF-1 signaling nodes and their role in carcinogenesis. In: George Fantus editor. Energy Balance and Cancer. Springer (2011). p. 53–76.
187. Chettouh H, Fartoux L, Aoudjehane L, Wendum D, Claperon A, Chretien Y, et al. Mitogenic insulin receptor-A is overexpressed in human hepatocellular carcinoma due to EGFR-mediated dysregulation of RNA splicing factors. Cancer Res (2013) 73(13):3974–86. doi:10.1158/0008-5472.CAN-12-3824
188. Gallagher EJ, LeRoith D. Diabetes, antihyperglycemic medications and cancer risk: smoke or fire? Curr Opin Endocrinol Diabetes Obes (2013) 20(5):485–94. doi:10.1097/01.med.0000433065.16918.83
189. Ferguson RD, Gallagher EJ, Scheinman EJ, Damouni R, LeRoith D. The epidemiology and molecular mechanisms linking obesity, diabetes, and cancer. Vitam Horm (2013) 93:51–98. doi:10.1016/B978-0-12-416673-8.00010-1
190. Brismar K, Fernqvist-Forbes E, Wahren J, Hall K. Effect of insulin on the hepatic production of insulin-like growth factor-binding protein-1 (IGFBP-1), IGFBP-3, and IGF-I in insulin-dependent diabetes. J Clin Endocrinol Metab (1994) 79(3):872–8. doi:10.1210/jcem.79.3.7521354
191. Wu Y, Yakar S, Zhao L, Hennighausen L, LeRoith D. Circulating insulin-like growth factor-I levels regulate colon cancer growth and metastasis. Cancer Res (2002) 62(4):1030–5.
192. Flemming JA, Yang JD, Vittinghoff E, Kim WR, Terrault NA. Risk prediction of hepatocellular carcinoma in patients with cirrhosis: the ADRESS-HCC risk model. Cancer (2014) 120(22):3485–93. doi:10.1002/cncr.28832
193. Sharma D, Wang J, Fu PP, Sharma S, Nagalingam A, Mells J, et al. Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology (2010) 52(5):1713–22. doi:10.1002/hep.23892
194. Wang SN, Lee KT, Ker CG. Leptin in hepatocellular carcinoma. World J Gastroenterol (2010) 16(46):5801–9. doi:10.3748/wjg.v16.i46.5801
195. Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov (2012) 2(9):778–90. doi:10.1158/2159-8290.CD-12-0263
196. Tolman KG, Fonseca V, Dalpiaz A, Tan MH. Spectrum of liver disease in type 2 diabetes and management of patients with diabetes and liver disease. Diabetes Care (2007) 30(3):734–43. doi:10.2337/dc06-1539
197. Armstrong MJ, Houlihan DD, Rowe IA, Clausen WH, Elbrond B, Gough SC, et al. Safety and efficacy of liraglutide in patients with type 2 diabetes and elevated liver enzymes: individual patient data meta-analysis of the LEAD program. Aliment Pharmacol Ther (2013) 37(2):234–42. doi:10.1111/apt.12149
198. American Diabetes Association. Standards of medical care in diabetes – 2014. Diabetes Care (2014) 37(Suppl 1):S14–80. doi:10.2337/dc14-S014
199. Tai YK, Wu CS, Tsai CY. Combination effect of ribavirin and erythropoietin treatment on hemoglobin A1c in a diabetic patient with chronic hepatitis C. J Chin Med Assoc (2012) 75(10):539–42. doi:10.1016/j.jcma.2012.07.008
200. Nkontchou G, Cosson E, Aout M, Mahmoudi A, Bourcier V, Charif I, et al. Impact of metformin on the prognosis of cirrhosis induced by viral hepatitis C in diabetic patients. J Clin Endocrinol Metab (2011) 96(8):2601–8. doi:10.1210/jc.2010-2415
201. Brackett CC. Clarifying metformin’s role and risks in liver dysfunction. J Am Pharm Assoc (2003) (2010) 50(3):407–10. doi:10.1331/JAPhA.2010.08090
202. Musso G, Gambino R, Cassader M, Pagano G. A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology (2010) 52(1):79–104. doi:10.1002/hep.23623
203. Van Wagner LB, Rinella ME. The role of insulin-sensitizing agents in the treatment of nonalcoholic steatohepatitis. Therap Adv Gastroenterol (2011) 4(4):249–63. doi:10.1177/1756283X11403809
204. Sumie S, Kawaguchi T, Kawaguchi A, Kuromatsu R, Nakano M, Satani M, et al. Effect of pioglitazone on outcome following curative treatment for hepatocellular carcinoma in patients with hepatitis C virus infection: a prospective study. Mol Clin Oncol (2015) 3(1):115–20. doi:10.3892/mco.2014.435
205. Al-Salman J, Arjomand H, Kemp DG, Mittal M. Hepatocellular injury in a patient receiving rosiglitazone. A case report. Ann Intern Med (2000) 132(2):121–4. doi:10.7326/0003-4819-132-2-200001180-00006
206. Bonkovsky HL, Azar R, Bird S, Szabo G, Banner B. Severe cholestatic hepatitis caused by thiazolidinediones: risks associated with substituting rosiglitazone for troglitazone. Dig Dis Sci (2002) 47(7):1632–7. doi:10.1023/A:1015895925374
207. Gentile S, Guarino G, Romano M, Alagia IA, Fierro M, Annunziata S, et al. A randomized controlled trial of acarbose in hepatic encephalopathy. Clin Gastroenterol Hepatol (2005) 3(2):184–91. doi:10.1016/S1542-3565(04)00667-6
208. Kihara Y, Ogami Y, Tabaru A, Unoki H, Otsuki M. Safe and effective treatment of diabetes mellitus associated with chronic liver diseases with an alpha-glucosidase inhibitor, acarbose. J Gastroenterol (1997) 32(6):777–82. doi:10.1007/BF02936954
209. Chitturi S, George J. Hepatotoxicity of commonly used drugs: nonsteroidal anti-inflammatory drugs, antihypertensives, antidiabetic agents, anticonvulsants, lipid-lowering agents, psychotropic drugs. Semin Liver Dis (2002) 22(2):169–83. doi:10.1055/s-2002-30102
210. Itou M, Kawaguchi T, Taniguchi E, Sumie S, Oriishi T, Mitsuyama K, et al. Altered expression of glucagon-like peptide-1 and dipeptidyl peptidase IV in patients with HCV-related glucose intolerance. J Gastroenterol Hepatol (2008) 23(2):244–51. doi:10.1111/j.1440-1746.2007.05183.x
211. Liu J, Wang G, Jia Y, Xu Y. GLP-1 receptor agonists: effects on the progression of non-alcoholic fatty liver disease. Diabetes Metab Res Rev (2014) 31(4):329–35. doi:10.1002/dmrr.2580
212. Blaslov K, Bulum T, Zibar K, Duvnjak L. Incretin based therapies: a novel treatment approach for non-alcoholic fatty liver disease. World J Gastroenterol (2014) 20(23):7356–65. doi:10.3748/wjg.v20.i23.7356
213. Arase Y, Suzuki F, Kobayashi M, Suzuki Y, Kawamura Y, Matsumoto N, et al. Efficacy and safety in sitagliptin therapy for diabetes complicated by chronic liver disease caused by hepatitis C virus. Hepatol Res (2011) 41(6):524–9. doi:10.1111/j.1872-034X.2011.00798.x
214. Kawaguchi T, Taniguchi E, Morita Y, Shirachi M, Tateishi I, Nagata E, et al. Association of exogenous insulin or sulphonylurea treatment with an increased incidence of hepatoma in patients with hepatitis C virus infection. Liver Int (2010) 30(3):479–86. doi:10.1111/j.1478-3231.2009.02191.x
215. Donadon V, Balbi M, Mas MD, Casarin P, Zanette G. Metformin and reduced risk of hepatocellular carcinoma in diabetic patients with chronic liver disease. Liver Int (2010) 30(5):750–8. doi:10.1111/j.1478-3231.2010.02223.x
216. Donadon V, Balbi M, Ghersetti M, Grazioli S, Perciaccante A, Della VG, et al. Antidiabetic therapy and increased risk of hepatocellular carcinoma in chronic liver disease. World J Gastroenterol (2009) 15(20):2506–11. doi:10.3748/wjg.15.2506
Keywords: type 1 diabetes, type 2 diabetes, hepatitis C virus, hepatitis C, insulin resistance, interferon alpha, hepatocellular carcinoma, outcomes, treatment
Citation: Hammerstad SS, Grock SF, Lee HJ, Hasham A, Sundaram N and Tomer Y (2015) Diabetes and hepatitis C: a two-way association. Front. Endocrinol. 6:134. doi: 10.3389/fendo.2015.00134
Received: 15 June 2015; Accepted: 17 August 2015;
Published: 14 September 2015
Edited by:
Anca Dana Dobrian, Eastern Virginia Medical School, USAReviewed by:
Hiroki Mizukami, Hirosaki University Graduate School of Medicine, JapanRosana De Morais Borges Marques, Universidade Federal de Goias, Brazil
Copyright: © 2015 Hammerstad, Grock, Lee, Hasham, Sundaram and Tomer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaron Tomer, Division of Endocrinology, Icahn School of Medicine at Mount Sinai, Box 1055, One Gustave L. Levy Place, New York, NY 10029, USA,eWFyb24udG9tZXJAbXNzbS5lZHU=