 Qun Lin
Qun Lin Zhong Yun*
Zhong Yun*- Department of Therapeutic Radiology, Yale School of Medicine, New Haven, CT, USA
Under obese conditions, adipose tissue can become oxygen-deficient or hypoxic. Extensive work has been done using various diet-induced obesity models to demonstrate an important role of hypoxia-induced signaling in adipose tissue and its impact on adipose functions related to adipogenesis, insulin sensitivity, and inflammation. We have recently identified a new mechanism connecting activation of the hypoxia-sensing pathway manifested by hypoxia-inducible factor (HIF) 2α to adipose tissue inflammation and hypertrophic cardiomyopathy. Interestingly, this observation is consistent with the clinical evidence showing that obesity is often associated with ventricular hypertrophy and dysfunction as well as congestive heart failure independent of other well-established risk factors including diabetes, hypertension, and coronary artery disease. This brief review will discuss the currently published genetic mouse models to determine the role of the HIF pathway in adipose tissue-associated diseases with a focus on the newly identified role of adipocyte HIF-2 in the development of hypertrophic cardiomyopathy.
Introduction
Molecular oxygen (O2) is essential for maintaining normal tissue functions from cellular energy production to regulation of a multitude of intracellular signal transduction pathways. Under obese conditions, O2 concentrations in adipose tissue can be negatively affected by a number of factors. Pathological obesity is often associated with adipocyte hypertrophy (1–3). Enlarged adipocytes can reduce the effective range of O2 diffusion in adipose tissue. On the other hand, O2 supply to adipose tissue is reduced because of decreased capillary density as well as subdued blood flow in obese subjects compared to lean subjects [see reviews by Ye (4) and Trayhurn (5)]. As shown by a recent study, obesity and high fat diet (HFD) can also increase O2 consumption in adipocytes likely due to uncoupled respiration induced by free fatty acids (6). As a result of these and other changes, insufficient oxygenation, i.e., hypoxia occurs in adipose tissue under pathologically obese conditions.
Using a needle-type pO2 microelectrode to directly measured tissue pO2 concentrations, Ye et al. have found that the average pO2 is low at 15.2 mmHg in adipose tissue of the genetically obese ob/ob mice compared to the average of 47.9 mmHg in that of lean mice (7). Decreased adipose pO2 has further been independently confirmed in HFD-induced obese mice and ob/ob mice (8, 9). Adipose tissue hypoxia is also found in humans. The mean adipose pO2 in overweight or obese patients is approximately 15% lower than that of lean subjects; adipose pO2 decreases even further with increasing body fat percentage (10). Such direct evidence indicates that adipose tissue hypoxia is a common pathological feature of obese subjects. In contrast, other studies have found no evidence of obesity-associated hypoxia in human abdominal subcutaneous fat (11, 12). These discrepancies in adipose tissue oxygenation are likely due to the use of different pO2 measurement techniques [see review by Hodson (13)] as well as fat depot-dependent differences in O2 supply and consumption.
Increasing amounts of evidence nonetheless suggest that hypoxia can exert profound impact on adipose tissue function. It has been shown that hypoxia inhibits adipogenic differentiation (14, 15), which may further enhance adipocyte hypertrophy due to inadequate de novo adipogenesis (16). Hypoxia affects glucose homeostasis, lipid metabolism, and production of adipokines and pro-inflammatory cytokines in adipose tissue (7, 10, 17). It has also been shown that macrophages are preferentially localized in hypoxic regions of adipose tissue with obesity (8). These observations strongly suggest adipose tissue hypoxia is an important etiological entity closely involved in onset and/or progression of obesity-associated diseases.
The Hypoxia-Inducible Factor Pathway
Mammalian cells respond to pO2 variations via the canonical multi-step O2-sensing pathway leading to the eventual activation of the hypoxia-inducible factors (HIF), a class of heterodimeric transcription factors containing the basic helix-loop-helix and PER/SIM/aryl hydrocarbon receptor nuclear translocator (ARNT) (bHLH-PAS) domains (Figure 1). Each heterodimer consists of an O2-sensitive HIF-α (HIF-1α or HIF-2α) and the O2-insensitive HIF-1β subunit (18). The quintessential aspect of this pathway is the O2-dependent regulation of HIF-α protein stability. Changes in pO2 are first “sensed” by HIF prolyl hydroxylases (PHDs), a family of O2-binding dioxygenases (19–22). In mammals, HIF-α proteins are regulated primarily by three PHD isoforms (PHD1, 2, and 3) among which PHD2 is the most abundant and widely expressed hydroxylase (23, 24). Under normoxic conditions, PHDs catalyze hydroxylation of the two conserved proline residues within the O2-dependent degradation domain (ODD) of HIF-1α or HIF-2α subunit. The hydroxylated HIF-α proteins interact with the von Hippel–Lindau tumor suppressor protein pVHL, undergo polyubiquitination, and are finally degraded by proteasomes (25). Under hypoxic conditions (e.g., <2% O2), PHDs are rendered inactive and HIF-α is no longer hydroxylated. The stabilized HIF-α forms a dimer with the constitutively expressed HIF-1β to activate transcription of a wide range of genes including those involved in the regulation of angiogenesis, metabolism, and inflammation (18, 26).
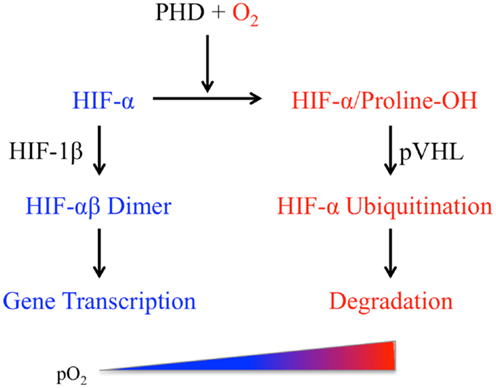
Figure 1. The hypoxia-inducible factor (HIF) pathway (see text for details).
Murine Hif1a mRNA is expressed in both mature adipocytes and progenitor cells (14, 15), whereas both Hif2a mRNA and HIF-2α protein are found in differentiated adipocytes even under normoxic conditions (14), suggesting that HIF-2α might have a unique role in mature adipocytes. Animal model studies have shown that HIF-1α protein levels (7), as well as HIF DNA-binding activities (27), are elevated in adipose tissue of obese mice. In addition, HIF-2α protein levels are also increased in adipose tissue of mice fed with HFD for 4 weeks (28). However, activation of HIF-1 leads to suppression of adipogenesis (14, 15). Collectively, these observations suggest that the HIF pathway can play a potentially significant role in obesity, diabetes, and metabolic syndrome (29).
The efforts to study the whole-body effects of the HIF-dependent O2-sensing pathway have been hampered by the embryonic lethality of homozygous deletion of the key genes of this pathway including Hif1a, Hif2a, Hif1b, Phd2, and Vhl. Several genetic mouse models have nonetheless demonstrated a global impact of the HIF pathway on body weight and metabolism. Hif1a+/− mice are viable but gain slightly less weight under normoxia but lost significantly more weight when maintained at 10% ambient O2 compared to wild-type littermates (30). Zhang et al. (31) have found that glucose increases HIF-2α protein synthesis and stability in proopiomelanocortin (POMC) neurons. Genetic deletion of Hif1b in POMC neurons facilitates obesity development via impaired hypothalamic glucose sensing (31). On the other hand, the PHD2 hypomorphic (Hif-p4h-2gt/gt) mice have less adipose tissue, smaller adipocytes, but improved glucose tolerance and insulin sensitivity, as well as less adipose tissue inflammation compared to their wild-type littermates (32). In this review, we will focus on the adipocyte-specific knockout mouse models.
Genetic Mouse Models to Investigate the Role of the HIF Pathway in Adipocytes
A number of genetic approaches have been used to either inactivate or activate the HIF pathway in adipocytes. The tissue-specific gene deletion or knockout is achieved by adipocyte-targeted expression of the cre recombinase under the transcriptional control by the gene promoter of fatty acid binding protein 4 (Fabp4) or aP2. The majority of currently published work uses either of the two aP2-cre mouse strains: one is developed by Dr. Ronald M. Evans’ group of the Salk Institute (33) and commercially available from the Jackson Laboratory (#005069), and the other is created by Dr. Barbara B. Kahn’s group of the Beth Israel Deaconess Medical Center and Harvard Medical School (34). Although the same 5.4-kbp aP2 gene promoter/enhancer region was used to generate the aP2-cre construct, the two strains of aP2-cre transgenic mice appear to show differences in tissue-specific expression of cre, which is likely due to different sites of integration of the aP2-cre transgene in the moue genome. Two reports have shown that the Evans/aP2-cre strain has leaky expression in brain and liver during embryonic development (35, 36). As an alternative to the aP2 promoter, the promoter of adiponectin (Apn), another adipocyte-specific gene, has been used to generate transgenic mice with adipocyte-targeted gene expression (9). Although Apn expression appears to be more restricted to adipocytes than aP2, both genes can be expressed albeit at lower levels in other tissues. Transcription of Apn and aP2 also appears to be differentially regulated in adipocytes (37–39). Therefore, it is highly possible that the phenotypes of mice derived from the Apn-transgenic mice would not be identical to those derived from the aP2-transgenic mice. It is also worth noting that the promoter/enhancer fragment used for controlling transgene expression contains limited numbers of enhancer elements only and thus is not likely to fully recapitulate the expression profile of the corresponding endogenous gene.
HIF Inactivation in Adipocytes
The direct role of the HIF pathway in adipocytes in live animals has been examined using two transgenic mouse approaches: adipocyte-targeted expression of a dominant-negative HIF (dnHIF) and cre-mediated deletion of individual HIF genes in adipocytes. Two groups generated mouse models with targeted deletion of HIF-1β, a common binding partner for both HIF-1α and HIF-2α, in adipocytes by crossing mice carrying floxed Hif1b alleles (Hif1bf/f) with the Kahn/aP2-cre mice (40, 41). Both groups found that mice with adipocyte-targeted Hif1b deletion were lean with reduced fat formation and were protected from HFD-induced glucose intolerance compared to wild-type littermates under the same conditions (40, 41). Adipose expression of glucose transporter 1 (Glut1) was decreased (40, 41) and glucose uptake by adipocytes was also reduced (41) in the adipocyte-targeted Hif1b knockout mice. Furthermore, both expression of vascular endothelial growth factor (Vegf) and vascular permeability were decreased in fat tissues of the adipocyte-targeted Hif1b knockout mice although vascular density was unchanged in fat (41). These independent results demonstrate an important role of HIF-1β in the regulation of formation, development, and function of adipocytes. However, it should bear in mind that HIF-1β, also known as ARNT, can also interact with a number of other proteins (42, 43) and therefore, some of the HIF-1β-associated phenotypes may be unrelated to the HIF pathway.
As discussed above, functions of the HIF pathway are manifested primarily by HIF-1 and/or HIF-2. A recently published work (6) examined the respective and collective roles of HIF-1α and HIF-2α in adipocytes using mouse models with adipocyte-targeted deletion of either Hif1a (HAKO) or Hif2a (H2AKO) as well as double knockout of both Hif1a and Hif2a (DHKO) that were generated by crossing with the Evans/aP2-cre transgenic mice. The main phenotypes of HAKO mice are in good agreement with an independent study using a similar HIF-1α knockout mouse model generated from the Evans/aP2-cre transgenic mice (44). The body weights of HAKO and H2AKO mice were, by and large, comparable to that of wild-type littermates fed with regular chow or HFD, which was somewhat inconsistent with the lean phenotype of the adipocyte-targeted Hif1b knockout mice (40, 41). Nonetheless, the vascular density in adipose tissue was little changed in HAKO mice, just like that of the Hif1b knockout mice (41). Interestingly, HAKO mice were refractory to HFD-induced insulin resistance, which was also found in mice with adipocyte-targeted deletion of HIF-1α generated by cross-breeding with the Kahn/aP2-cre mice (40). Furthermore, HAKO mice resisted the development of adipose inflammation induced by HFD as compared to their wild-type littermates (6). In contrast to adipocyte-targeted deletion of HIF-1α, loss of HIF-2α in adipocytes resulted in more pronounced HFD-induced inflammation, glucose intolerance, and insulin resistance compared to wild-type littermates (6). On the other hand, DHKO mice with adipocyte-targeted deletion of both Hif1a and Hif2a exhibited the same phenotype as that of the HAKO mice. These results suggest that HIF-1α and HIF-2α have opposing functions in adipocytes with HIF-2α playing a protective role whereas HIF-1α facilitating adipose tissue pathology under HFD conditions.
Alternatively, Zhang et al. (45) created a transgenic C57Bl/6xCBA mouse model in which a dominant-negative HIF-1α (dnHIF-1α) protein lacking amino acids 30–389 was selectively expressed in adipocytes under the transcriptional control by the 5.4-kpb aP2 promoter/enhancer region. The dnHIF-1α transgenic mice gained weight more quickly and had more body fat mass than the wild-type littermates did on either regular chow or HFD (45). These mice also developed glucose intolerance and insulin resistance especially when fed with HFD. Furthermore, the dnHIF-1α transgenic mice exhibited increased sizes of lipid droplets but decreased mitochondrial biogenesis and angiogenesis in interscapular brown adipose tissue (BAT) (45). These findings are somewhat opposite to those from the Hif1a knockout models (6, 40, 41). It is possible that dnHIF-1α retains some of the HIF-1α functions and thus cannot fully recapitulate the genetic loss of Hif1a.
Sun et al. generated a double transgenic mouse (C57BL/6) model with doxycycline (DOX)-inducible overexpression of the dnHIF-1α protein lacking amino acids 30–389 (TRE-dnHIF-1α) under the control by the adiponectin promoter-driven-rtTA transgene (Apn-rtTA) (9). In contrast to the aP2 promoter-driven dnHIF-1α (45), Apn promoter-driven overexpression of dnHIF-1α in adipose tissue results in decreased body weight gain and adipocyte size but increased energy expenditure and improved insulin sensitivity in the transgenic mice on HFD (9). These inconsistencies between these two transgenic studies are likely due to differences in promoters driving the same dnHIF-1α and modes of gene regulation, i.e., constitutive versus DOX-induced expression. Nonetheless, the observations made by Sun et al. are consistent with those found in mice with adipocyte-targeted Hif1b deletion (40, 41). Also in agreement with the Hif1a knockout study (6), DOX-induced overexpression of dn-HIF-1α reduced local adipose tissue inflammation in HFD-fed transgenic mice (9).
HIF Activation in Adipocytes
Halberg et al. generated a transgenic FVB mouse model with adipocyte-targeted expression of a constitutively active HIF-1α mutant lacking amino acids 401–603 (ODD) under the control of the 5.4-kbp aP2 promoter/enhancer (27). Expression of the HIF-1α-ΔODD mutant varied significantly in a depot-dependent manner with, relatively speaking, the highest level of expression in subcutaneous fat and the lowest in epididymal fat (27). Consistent with high transgene expression, there was a significant increase in the sizes of subcutaneous adipocytes. The hemizygous transgenic mice gained more weight than their wild-type littermates did on either regular chow diet or HFD. Plasma glucose concentrations of the HIF-1α-ΔODD transgenic mice were moderately higher than those of their wild-type littermates during the oral glucose tolerance test. Furthermore, overexpression of HIF-1α-ΔODD in adipocytes resulted in local inflammation and fibrosis in adipose tissue (27). It is quite surprising that some of the phenotypes exhibited by transgenic mice expressing the constitutively active HIF-1α lacking amino acids 401–603 are rather similar to those displayed by transgenic mice expressing the dnHIF-1α without amino acids 30–389 (9, 45). It remains to be seen whether the unexpectedly similar results from these two opposite approaches could be due to the shared C-terminal domain of HIF-1α in addition to other genetic differences in these two transgenic strains.
In contrast to the transgenic approach, the endogenous HIF pathway can be activated as a result of genetic deletion of a HIF prolyl 4-hydroxylase or the VHL gene, which leads to stabilization of both HIF-1α and HIF-2α proteins. Being the most commonly expressed member of the PHD family, Phd2 has been targeted for genetic deletion. Matsuura et al. generated a mouse model (Phd2f/f/ap2-Cre) with adipocyte-targeted deletion of Phd2 using the Evans/aP2-cre transgenic mice (46). The epididymal white fat pads of the Phd2f/f/ap2-Cre mice weighed less and the adipocytes were smaller in size compared to wild-type littermates. The Phd2f/f/ap2-Cre mice were resistant to HFD-induced obesity and exhibited improved glucose homeostasis. Expression of pro-inflammatory cytokines in epididymal white fat was comparable between Phd2f/f/ap2-Cre mice and their wild-type littermates under the same HFD conditions, although macrophage infiltration appeared to be reduced in epididymal white fat Phd2f/f/ap2-Cre mice (46).
Using the same genetic approach, Michailidou et al. have made quite different observations (47). Mice with deletion of Phd2 in adipocytes (aP2-Phd2KO) gained more weight mainly with larger white adipose tissue mass and bigger adipocytes than their wild-type littermates fed with the same chow diet. The increased adiposity correlated with enhanced angiogenesis in the aP2-Phd2KO adipose tissue. Nonetheless, the aP2-Phd2KO mice showed no significant differences in glucose homeostasis but basal lipolysis was significantly lower compared to that of wild-type controls (47).
In addition to PHD2, PHD1 and PHD3 can also catalyze proline hydroxylation of HIF-1α and HIF-2α proteins. Therefore, possibilities exist that loss of PHD2 might, at least, be partially compensated by PHD1 and/or PHD3 in adipocytes. As reported by Matsuura et al., Phd3 mRNA levels increased by several fold whereas the increase of Phd1 mRNA levels was not significant in white fat of Phd2f/f/aP2-Cre mice (46). Furthermore, expression of Hif1a and Hif2a mRNA was increased more than twofold in adipose tissue of the Phd2f/f/aP2-Cre mice (46). In contrast, Michailidou et al. found that expression of Phd1 and Phd3 mRNA in adipose tissue was not affected in their aP2-Phd2KO mice (47). Using the murine 3T3-L1-derived adipocytes, Floyd et al. found that pharmacological inhibition of PHD activity did not consistently affect HIF-1α protein levels, suggesting that other pathways might also be involved in HIF activation (48). These differences, among others, may partially explain why there are so many discrepancies between these two studies despite their nearly identical genetic approach using the same Evans/aP2-cre transgenic mice from the Jackson Laboratory and the same congenic C57Bl6 genetic background.
In contrast to the multiple isoforms of PHDs, there is only one VHL gene in mammals. Therefore, genetic deletion of the VHL gene would be a more efficient approach to activate the entire HIF pathway without compensation by other isoforms. We created a genetic mouse model (fatVHLko) with adipocyte-targeted deletion of Vhl by crossing the Vhlf/f mice with the Kahn/aP2-cre transgenic mice (17). Consistent with reports from other groups who used the Kahn/aP2-cre transgenic mice (34, 36, 49, 50), genetic deletion of Vhl was highly restricted to adipocytes. Both brown and white adipocytes of the homozygous fatVHLko mice were enlarged in size, consistent with the findings from the aP2-Phd2KO mice (47). The fatVHLko mice showed normal fetal development and no apparent defects at birth. Interscapular brown fat developed normally although intracellular fat contents were significantly increased. In contrast, visceral white fat was generally reduced, suggesting a depot-dependent effect of HIF activation or Vhl deletion on adipose development. It is worth noting that reduced white fat development of fatVHLko mice (17) is similar to that found in Phd2f/f/ap2-Cre mice (46). Because hypoxia or HIF activation can suppress adipogenic differentiation of preadipocytes (14, 15), it is possible that white fat tissue development may be more sensitive than brown fat tissue to HIF-dependent inhibition of adipogenesis. This concept is further supported by the finding that levels of Hif1a and Hif2a mRNA were significantly increased in white fat tissue of Phd2f/f/aP2-Cre mice (46), suggesting that further elevated HIF activation in adipocytes of Phd2f/f/ap2-Cre mice potentially contributes to reduction of white adipose tissue.
HIF-2, Adipose Inflammation, and Hypertrophic Cardiomyopathy
Some 50 years ago, it was reported (51) that increase in heart weight above the predicted normal value was proportional to the increase of body weight over the normal range and grossly obese patients presented with ventricular hypertrophy as the predominant and most specific alteration in the heart. Ensuing studies further found that obese patients often developed cardiomyopathy with increased wall thickness and cavity volume of the left ventricle (52). As discussed below, activation of the hypoxia-sensing pathway in adipocytes could potentially be a leading cause of obesity-associated cardiomyopathy.
As our study has shown, all fatVHLko mice developed cardiomegaly within 7 days after birth, while all other major organs appeared to develop normally (17). Incidentally, this is also the time frame when rapid adipose tissue development occurs postnatally (53). The heart of fatVHLko mice showed enlarged atria, ventricles, and wall thicknesses. Cardiomyocyte hypertrophy and increased numbers of Ki67+ cells, along with increased cardiac angiogenesis, were apparent in the heart. There was also fibrosis in the heart of fatVHLko mice. Functions of the fatVHLko heart were also compromised with reduced ejection fraction, increased end diastolic pressure, and decreased dynamic regulation of blood pressure, especially −dP/dt. The fatVHLko mice died in early adulthood with a median survival time of 10 weeks, likely due to sudden heart failure (17). This phenotype occurred in the homozygous fatVHLko mice only as Vhl heterozygous mice were developmentally indistinguishable from wild-type littermates. In contrast to this study, the Phd2f/f/aP2-Cre mice appeared to have normal cardiac development (46), whereas the heart phenotype was not examined in the aP2-Phd2KO mice (47). The differences between Vhl knockout and Phd2 knockout could be attributed, in part, to the potential compensation by other PHD isoforms and the different strains of the aP2-cre mice, as discussed above.
As primary effectors of the hypoxia-signaling pathway, both HIF-1α and HIF-2α proteins are stabilized with Vhl deletion, but they have non-overlapping functions (54). We further created mouse models with different combinations of adipocyte-specific double or triple knockout to determine the differential roles of HIF-1α and HIF-2α in the pathological progression of fatVHLko mice (17). Genetic deletion of both Vhl and Hif2a completely normalized the development of the fatVHLko mice with no apparent signs of any developmental or behavioral defects. In stark contrast, deletion of Hif1a together with Vhl (fatVHL1Ako) significantly exacerbated hypertrophic cardiomyopathy and resulted in much earlier onset of death with a median survival time of only 4 weeks for fatVHL1Ako mice (17). These results suggest that chronic activation of HIF-2α, but not HIF-1α, in adipocytes is the main cause of adipose tissue pathology and hypertrophic cardiomyopathy. Consistent with these findings, cardiac abnormalities were not found in transgenic mice with adipocyte-targeted expression of the constitutively active HIF-1α-ΔODD (27) although the study by Lee et al. suggested otherwise (6).
The basal plasma glucose levels of fatVHLko mice were lower than those of their wild-type littermates. The fatVHLko mice showed by and large normal responses to intraperitoneal injection of glucose or insulin and showed moderate abnormalities in lipid metabolism. These findings suggest that the development of hypertrophic heart disease in pathologically obese subjects is likely independent of insulin resistance and/or hyperlipidemia, two commonly regarded key risk factors for cardiovascular diseases. Nevertheless, adipocyte-targeted deletion of Vhl led to both systemic and local adipose tissue inflammation. Expression of immunoglobulin genes, chemokines (Ccl2, Ccl3, Ccl7, and Ccl8), and cytokines (Il1b, Il6, and Tnf) was robustly increased in Vhl-deleted adipose tissue. Serum levels of IL-1β, IL-6, MCP-1/CCL2, and TNFα among others were also significantly increased in fatVHLko mice. Consistent with the systemic inflammatory phenotype, the NFκB pathway is strongly activated in the heart of fatVHLko mice (17). Furthermore, the NFAT pathway, a key player in development of cardiac hypertrophy (55), was also activated in the heart of fatVHLko mice (17). Consistent with these observations, it has been shown that NFκB can activate NFAT via protein–protein interaction to induce cardiac hypertrophy (56). Collectively, these findings strongly support the emerging concept that chronic inflammation is a leading cause of heart diseases (57–61).
Mechanistically, the inflammation phenotype was mainly dependent on activation of HIF-2α because double knockout of Vhl with Hif2a, but not with Hif1a, normalized expression of pro-inflammatory genes in adipose tissue and reduced activation of NFκB and the NFAT pathways (17). These results clearly demonstrate that HIF-1α and HIF-2α play fundamentally different roles in adipocytes. Importantly, chronic activation of HIF-2α in adipocytes elicits local inflammation in adipose tissue and increases production of circulating pro-inflammatory cytokines and chemokines, which then induces hypertrophic cardiomyopathy by activating NFκB and NFAT pathways.
Several similarities can be drawn between the fatVHLko models and clinical obesity with regard to the heart phenotype. Both clinical and epidemiological studies have shown that obesity is associated with left ventricle dysfunction and heart failure independent of diabetes, hypertension, and coronary artery disease (52). Some obese patients develop ventricular hypertrophy and congestive heart failure without other comorbidities in the heart (51, 62) while others present with diastolic dysfunctions but without hypertension (63). Similarly, the fatVHLko and fatVHL1Ako mice develop lethal cardiomegaly without presenting with severely compromised heart functions except reduced diastolic −dP/dt or significant metabolic abnormalities including insulin resistance and hyperlipidemia (17). In light of adipose tissue hypoxia associated with severe obesity (7, 10, 27), our study has led to a new disease mechanism underlying the etiology of obesity-associated cardiomyopathy. As this model suggests, obesity-induced pathological changes adipose tissue, such as adipocyte hypertrophy, decreased O2 supply, and increased O2 consumption, lead to the development of adipose tissue hypoxia that, in turn, induces sustained activation of HIF-2α in adipocytes, which then results in both local and systemic inflammation, and contributes to the development of hypertrophic cardiomyopathy.
Summary
The various genetic mouse model studies discussed above have illustrated an important role of the HIF pathway in adipose tissue under both physiological and pathological conditions, despite a lack of consistent observations in some of the studies. New genetic approaches would be required to further clarify these inconsistent results. Nonetheless, the current studies have presented strong evidence showing that HIF pathway can play a critical role in the development of adipose tissue inflammation and other pathological abnormalities reminiscent of obesity-induced diseases including obesity-associated hypertrophic cardiomyopathy. Pharmacological intervention of the HIF pathway will likely have the potential to control the progression of obesity and obesity-related symptoms.
Conflict of Interest Statement
Qun Lin and Zhong Yun declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is in part supported by a Grant-in-Aid from the American Heart Association (11GRNT7970029) awarded to ZY.
References
1. Cotillard A, Poitou C, Torcivia A, Bouillot JL, Dietrich A, Kloting N, et al. Adipocyte size threshold matters: link with risk of type 2 diabetes and improved insulin resistance after gastric bypass. J Clin Endocrinol Metab (2014) 99:E1466–70. doi: 10.1210/jc.2014-1074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Salans LB, Knittle JL, Hirsch J. The role of adipose cell size and adipose tissue insulin sensitivity in the carbohydrate intolerance of human obesity. J Clin Invest (1968) 47:153–65. doi:10.1172/JCI105705
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Stern JS, Batchelor BR, Hollander N, Cohn CK, Hirsch J. Adipose-cell size and immunoreactive insulin levels in obese and normal-weight adults. Lancet (1972) 2:948–51. doi:10.1016/S0140-6736(72)92474-9
4. Ye J. Adipose tissue vascularization: its role in chronic inflammation. Curr Diab Rep (2011) 11:203–10. doi:10.1007/s11892-011-0183-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Trayhurn P. Hypoxia and adipocyte physiology: implications for adipose tissue dysfunction in obesity. Annu Rev Nutr (2014) 34:207–36. doi:10.1146/annurev-nutr-071812-161156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Lee YS, Kim JW, Osborne O, Oh da Y, Sasik R, Schenk S, et al. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell (2014) 157:1339–52. doi:10.1016/j.cell.2014.05.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab (2007) 293:E1118–28. doi:10.1152/ajpendo.00435.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Rausch ME, Weisberg S, Vardhana P, Tortoriello DV. Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. Int J Obes (Lond) (2008) 32:451–63. doi:10.1038/sj.ijo.0803744
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Sun K, Halberg N, Khan M, Magalang UJ, Scherer PE. Selective inhibition of hypoxia-inducible factor 1alpha ameliorates adipose tissue dysfunction. Mol Cell Biol (2013) 33:904–17. doi:10.1128/MCB.00951-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Pasarica M, Sereda OR, Redman LM, Albarado DC, Hymel DT, Roan LE, et al. Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes (2009) 58:718–25. doi:10.2337/db08-1098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Goossens GH, Bizzarri A, Venteclef N, Essers Y, Cleutjens JP, Konings E, et al. Increased adipose tissue oxygen tension in obese compared with lean men is accompanied by insulin resistance, impaired adipose tissue capillarization, and inflammation. Circulation (2011) 124:67–76. doi:10.1161/CIRCULATIONAHA.111.027813
12. Hodson L, Humphreys SM, Karpe F, Frayn KN. Metabolic signatures of human adipose tissue hypoxia in obesity. Diabetes (2013) 62:1417–25. doi:10.2337/db12-1032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Hodson L. Adipose tissue oxygenation: effects on metabolic function. Adipocyte (2014) 3:75–80. doi:10.4161/adip.27114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Lin Q, Lee YJ, Yun Z. Differentiation arrest by hypoxia. J Biol Chem (2006) 281:30678–83. doi:10.1074/jbc.C600120200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Yun Z, Maecker HL, Johnson RS, Giaccia AJ. Inhibition of PPARγ2 gene expression by the HIF-1-regulated gene DEC1/Stra13: a mechanism for regulation of adipogenesis by hypoxia. Dev Cell (2002) 2:331–41. doi:10.1016/S1534-5807(02)00131-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Arner E, Westermark PO, Spalding KL, Britton T, Ryden M, Frisen J, et al. Adipocyte turnover: relevance to human adipose tissue morphology. Diabetes (2010) 59:105–9. doi:10.2337/db09-0942
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Lin Q, Huang Y, Booth CJ, Haase VH, Johnson RS, Celeste Simon M, et al. Activation of hypoxia-inducible factor-2 in adipocytes results in pathological cardiac hypertrophy. J Am Heart Assoc (2013) 2:e000548. doi:10.1161/JAHA.113.000548
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol (1999) 15:551–78. doi:10.1146/annurev.cellbio.15.1.551
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Bruick RK. Oxygen sensing in the hypoxic response pathway: regulation of the hypoxia-inducible transcription factor. Genes Dev (2003) 17:2614–23. doi:10.1101/gad.1145503
20. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell (2001) 107:43–54. doi:10.1016/S0092-8674(01)00507-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science (2001) 292:464–8. doi:10.1126/science.1059817
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science (2001) 292:468–72. doi:10.1126/science.1059796
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem (2004) 279:38458–65. doi:10.1074/jbc.M406026200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J (2003) 22:4082–90. doi:10.1093/emboj/cdg392
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Kim W, Kaelin WG Jr. The von Hippel-Lindau tumor suppressor protein: new insights into oxygen sensing and cancer. Curr Opin Genet Dev (2003) 13:55–60. doi:10.1016/S0959-437X(02)00010-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Aragones J, Fraisl P, Baes M, Carmeliet P. Oxygen sensors at the crossroad of metabolism. Cell Metab (2009) 9:11–22. doi:10.1016/j.cmet.2008.10.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, et al. Hypoxia-inducible factor 1α induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol (2009) 29:4467–83. doi:10.1128/MCB.00192-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Shimba S, Wada T, Hara S, Tezuka M. EPAS1 promotes adipose differentiation in 3T3-L1 cells. J Biol Chem (2004) 279:40946–53. doi:10.1074/jbc.M400840200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Girgis CM, Cheng K, Scott CH, Gunton JE. Novel links between HIFs, type 2 diabetes, and metabolic syndrome. Trends Endocrinol Metab (2012) 23:372–80. doi:10.1016/j.tem.2012.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α<ATΛ>. J Clin Invest (1999) 103:691–6. doi:10.1172/JCI5912
31. Zhang H, Zhang G, Gonzalez FJ, Park SM, Cai D. Hypoxia-inducible factor directs POMC gene to mediate hypothalamic glucose sensing and energy balance regulation. PLoS Biol (2011) 9:e1001112. doi:10.1371/journal.pbio.1001112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Rahtu-Korpela L, Karsikas S, Horkko S, Sequeiros RB, Lammentausta E, Makela KA, et al. HIF prolyl 4-hydroxylase-2 inhibition improves glucose and lipid metabolism and protects against obesity and metabolic dysfunction. Diabetes (2014) 63(10):3324–33. doi:10.2337/db14-0472
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. He W, Barak Y, Hevener A, Olson P, Liao D, Le J, et al. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A (2003) 100:15712–7. doi:10.1073/pnas.2536828100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, et al. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature (2001) 409:729–33. doi:10.1038/35055575
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Zhang J, Wang Y, Gao Z, Yun Z, Ye J. Hypoxia-inducible factor 1 activation from adipose protein 2-cre mediated knockout of von Hippel-Lindau gene leads to embryonic lethality. Clin Exp Pharmacol Physiol (2012) 39:145–50. doi:10.1111/j.1440-1681.2011.05656.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Martens K, Bottelbergs A, Baes M. Ectopic recombination in the central and peripheral nervous system by aP2/FABP4-Cre mice: implications for metabolism research. FEBS Lett (2010) 584:1054–8. doi:10.1016/j.febslet.2010.01.061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Liu J, Farmer SR. Regulating the balance between peroxisome proliferator-activated receptor gamma and beta-catenin signaling during adipogenesis. A glycogen synthase kinase 3beta phosphorylation-defective mutant of beta-catenin inhibits expression of a subset of adipogenic genes. J Biol Chem (2004) 279:45020–7. doi:10.1074/jbc.M407050200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Gustafson B, Jack MM, Cushman SW, Smith U. Adiponectin gene activation by thiazolidinediones requires PPAR gamma 2, but not C/EBP alpha-evidence for differential regulation of the aP2 and adiponectin genes. Biochem Biophys Res Commun (2003) 308:933–9. doi:10.1016/S0006-291X(03)01518-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Korner A, Wabitsch M, Seidel B, Fischer-Posovszky P, Berthold A, Stumvoll M, et al. Adiponectin expression in humans is dependent on differentiation of adipocytes and down-regulated by humoral serum components of high molecular weight. Biochem Biophys Res Commun (2005) 337:540–50. doi:10.1016/j.bbrc.2005.09.064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Jiang C, Qu A, Matsubara T, Chanturiya T, Jou W, Gavrilova O, et al. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes (2011) 60(10):2484–95. doi:10.2337/db11-0174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Lee KY, Gesta S, Boucher J, Wang XL, Kahn CR. The differential role of Hif1β/Arnt and the hypoxic response in adipose function, fibrosis, and inflammation. Cell Metab (2011) 14:491–503. doi:10.1016/j.cmet.2011.08.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Beischlag TV, Luis Morales J, Hollingshead BD, Perdew GH. The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr (2008) 18:207–50. doi:10.1615/CritRevEukarGeneExpr.v18.i3.20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Bersten DC, Sullivan AE, Peet DJ, Whitelaw ML. bHLH-PAS proteins in cancer. Nat Rev Cancer (2013) 13:827–41. doi:10.1038/nrc3621
44. Kihira Y, Miyake M, Hirata M, Hoshina Y, Kato K, Shirakawa H, et al. Deletion of hypoxia-inducible factor-1alpha in adipocytes enhances glucagon-like peptide-1 secretion and reduces adipose tissue inflammation. PLoS One (2014) 9:e93856. doi:10.1371/journal.pone.0093856
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Zhang X, Lam KS, Ye H, Chung SK, Zhou M, Wang Y, et al. Adipose tissue-specific inhibition of hypoxia-inducible factor 1α induces obesity and glucose intolerance by impeding energy expenditure in mice. J Biol Chem (2010) 285:32869–77. doi:10.1074/jbc.M110.135509
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Matsuura H, Ichiki T, Inoue E, Nomura M, Miyazaki R, Hashimoto T, et al. Prolyl hydroxylase domain protein 2 plays a critical role in diet-induced obesity and glucose intolerance. Circulation (2013) 127:2078–87. doi:10.1161/CIRCULATIONAHA.113.001742
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Michailidou Z, Morton NM, Moreno Navarrete JM, West CC, Stewart KJ, Fernandez-Real JM, et al. Adipocyte pseudohypoxia suppresses lipolysis and facilitates benign adipose tissue expansion. Diabetes (2014) 64(3):733–45. doi:10.2337/db14-0233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Floyd ZE, Kilroy G, Wu X, Gimble JM. Effects of prolyl hydroxylase inhibitors on adipogenesis and hypoxia inducible factor 1 alpha levels under normoxic conditions. J Cell Biochem (2007) 101(6):1545–57. doi:10.1002/jcb.21266
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Jones JR, Barrick C, Kim KA, Lindner J, Blondeau B, Fujimoto Y, et al. Deletion of PPARγ in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc Natl Acad Sci U S A (2005) 102:6207–12. doi:10.1073/pnas.0306743102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Morroni M, Giordano A, Zingaretti MC, Boiani R, De Matteis R, Kahn BB, et al. Reversible transdifferentiation of secretory epithelial cells into adipocytes in the mammary gland. Proc Natl Acad Sci U S A (2004) 101:16801–6. doi:10.1073/pnas.0407647101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Amad KH, Brennan JC, Alexander JK. The cardiac pathology of chronic exogenous obesity. Circulation (1965) 32:740–5. doi:10.1161/01.CIR.32.5.740
52. Wong C, Marwick TH. Obesity cardiomyopathy: pathogenesis and pathophysiology. Nat Clin Pract Cardiovasc Med (2007) 4:436–43. doi:10.1038/ncpcardio0943
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Greenwood MR, Hirsch J. Postnatal development of adipocyte cellularity in the normal rat. J Lipid Res (1974) 15:474–83.
54. Pawlus MR, Hu CJ. Enhanceosomes as integrators of hypoxia inducible factor (HIF) and other transcription factors in the hypoxic transcriptional response. Cell Signal (2013) 25:1895–903. doi:10.1016/j.cellsig.2013.05.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Barry SP, Townsend PA. What causes a broken heart – molecular insights into heart failure. Int Rev Cell Mol Biol (2010) 284:113–79. doi:10.1016/S1937-6448(10)84003-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Liu Q, Chen Y, Auger-Messier M, Molkentin JD. Interaction between NFkappaB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ Res (2012) 110:1077–86. doi:10.1161/CIRCRESAHA.111.260729
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Hotamisligil GS. Inflammation and metabolic disorders. Nature (2006) 444:860–7. doi:10.1038/nature05485
58. Lavie CJ, Milani RV, Ventura HO. Obesity and cardiovascular disease: risk factor, paradox, and impact of weight loss. J Am Coll Cardiol (2009) 53:1925–32. doi:10.1016/j.jacc.2008.12.068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Lazar MA. How obesity causes diabetes: not a tall tale. Science (2005) 307:373–5. doi:10.1126/science.1104342
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Litwin SE. The growing problem of obesity and the heart: the plot “thickens”. J Am Coll Cardiol (2006) 47:617–9. doi:10.1016/j.jacc.2005.11.014
61. Walsh K. Adipokines, myokines and cardiovascular disease. Circ J (2009) 73:13–8. doi:10.1253/circj.CJ-08-0961
62. Smith HL, Willius FA. Adiposity of the heart: a clinical and pathologic study of one hundred and thirty-six obese patients. Arch Intern Med (Chic) (1933) 52:911–31. doi:10.1001/archinte.1933.00160060085007
Keywords: adipocyte, cardiomyopathy, heart, hypertrophy, hypoxia, HIF-2, obesity, von Hippel–Lindau
Citation: Lin Q and Yun Z (2015) The hypoxia-inducible factor pathway in adipocytes: the role of HIF-2 in adipose inflammation and hypertrophic cardiomyopathy. Front. Endocrinol. 6:39. doi: 10.3389/fendo.2015.00039
Received: 14 January 2015; Accepted: 07 March 2015;
Published online: 23 March 2015.
Edited by:
Daniela Patrizia Foti, University “Magna Græcia” of Catanzaro, ItalyReviewed by:
Sinan Tanyolac, Istanbul Science University, TurkeyZoi Michailidou, University of Edinburgh, UK
Copyright: © 2015 Lin and Yun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhong Yun, Department of Therapeutic Radiology, Yale School of Medicine, P. O. Box 208040, New Haven, CT 06520-8040, USA e-mail:emhvbmcueXVuQHlhbGUuZWR1