
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 06 March 2015
Sec. Cancer Endocrinology
Volume 6 - 2015 | https://doi.org/10.3389/fendo.2015.00030
This article is part of the Research TopicThe role of the Insulin/IGFs system in cancer: critical points in dysregulation, targeting problems and new blocking strategiesView all 17 articles
 Paola De Marco1Francesca Cirillo1Adele Vivacqua1
Paola De Marco1Francesca Cirillo1Adele Vivacqua1 Roberta Malaguarnera2
Roberta Malaguarnera2 Antonino Belfiore2*
Antonino Belfiore2* Marcello Maggiolini1*
Marcello Maggiolini1*The insulin/IGF system plays an important role in cancer progression. Accordingly, elevated levels of circulating insulin have been associated with an increased cancer risk as well as with aggressive and metastatic cancer phenotypes. Numerous studies have documented that estrogens cooperate with the insulin/IGF system in multiple pathophysiological conditions. The biological responses to estrogens are mainly mediated by the estrogen receptors (ER)α and ERβ, which act as transcription factors; however, several studies have recently demonstrated that a member of the G protein-coupled receptors, named GPR30/G-protein estrogen receptor (GPER), is also involved in the estrogen signaling in normal and malignant cells as well as in cancer-associated fibroblasts (CAFs). In this regard, novel mechanisms linking the action of estrogens through GPER with the insulin/IGF system have been recently demonstrated. This review recapitulates the relevant aspects of this functional cross-talk between the insulin/IGF and the estrogenic GPER transduction pathways, which occurs in various cell types and may account for cancer progression.
IGF-1 and IGF-2 (IGFs) as well as insulin and cognate receptors (the type I IGF-1 receptor, IGF-1R, and the insulin receptor, IR) belong to a complex system, the insulin/IGF system, which is essential for normal development and growth of cells, organs, and whole animals (1). Insulin, which secretion is modulated by nutrients, namely glucose, is exclusively produced by the endocrine pancreas and is the main regulator of glucose homeostasis. The insulin pro-hormone, proinsulin, is partially also secreted in the bloodstream and its biological role, if any, is unknown (2). IGF-1, which is regulated by the hypothalamus/pituitary axis through pituitary GH, is mainly produced by the liver and plays a major role in linear growth. IGF-2 is produced in the liver and many other tissues, and is scarcely responsive to GH. It is mainly involved in trophic, survival, and differentiation effects (3). IGFs, but not insulin or proinsulin, associate with six different binding proteins (IGFBPs) that regulate their bioactivity. IGFs act on most cells and tissues and regulate survival, mitogenesis, cell migration, and differentiation (3).
Insulin and IGFs all activate a very well-conserved signaling pathway, the PI3K/Akt/FoxO pathway, which has a crucial role in the regulation of metabolism, growth, and apoptosis processes (4). In particular, upon ligand binding, the tyrosine kinase domains of both IR and IGF-1R catalyze the phosphorylation of specific substrates, such as the insulin receptor substrate (IRS)-1, IRS-2, IRS-3, and IRS-4, Gab-1, Cbl, and Shc (5). Activated IRS proteins interact with the growth factor receptor binding protein 2 (GRB2) and the p85 regulatory subunit of phosphoinositide 3-kinase (PI3K), which then activates the Akt pathway that regulates metabolic enzymes and mediates cell growth, proliferation, and survival (4). Moreover, IR and IGF-1R activate a second major signaling pathway, the Ras/Raf/MEK/ERK transduction cascade, which is involved in important biological responses like gene expression, cell motility, proliferation, survival, differentiation, and death (6). Another pathway, which integrates signals from both the PI3K–Akt activation and the Ras/Raf/MEK/ERK, as well as signals coming from other growth factors and from nutrients, is the mammalian target of rapamycin (mTOR) pathway, which is also a central regulator of cell growth and metabolism through the control of mRNA translation (7). Moreover, insulin/IGFs also cooperate with other receptor and signaling pathways, thus exerting a crucial role in normal development and homeostasis and tissue repair.
It is now well established that the insulin/IGF system is frequently dysregulated in cancer, thus contributing to cancer progression, metastases, and resistance to cancer therapies (8, 9). Common alterations include overexpression of IR and IGF-1R by the malignant cells, increased IR/IGF-1R hybrid formation, deregulated autocrine secretion of IGFs, and increased IGFs secretion by the tumor stroma. IGFBPs production in the tumor microenvironment may also be dysregulated (8, 10–12). Indeed, an overexpression of IR/IGF-1R has been detected in breast malignancy (13, 14), and a high expression of these receptors was shown to be tumorigenic in mouse tumor models (15). Moreover, epidemiological studies have shown that elevated IGF-1 plasma concentrations are associated with a higher risk of developing various malignancies, like breast, colon, prostate, and lung carcinomas (16–18). Notably, the human IR exists in two isoforms characterized by the inclusion (IR-B) or the exclusion (IR-A) of exon 11, which encodes a 12 amino acid residue located at the carboxyl terminus of the IR alpha-subunit (5). In adult life, the metabolic effects of insulin in target organs (liver, muscle, and fat) are predominantly mediated by the IR-B. In contrast, IR-A plays a role in growth and development in prenatal life, while its role in adult life is unknown. Recently, IR-A has been shown to be almost invariably overexpressed in both epithelial and non-epithelial malignancies (19–21). Interestingly, while IR-B is a highly specific receptor for insulin, IR-A is a more promiscuous receptor, exhibiting high affinity for insulin, intermediate affinity for IGF-2 and proinsulin, and low affinity for IGF-1 (19, 22, 23). IR-A overexpression and enhanced IGF-1/IGF-2 autocrine/paracrine production often coexist in several malignancies and is associated with an aggressive and dedifferentiated tumor phenotype (20, 24, 25). In this scenario, it is most likely, that IR-A functions as the main IGF-2 receptor, as the IGF-1R is often saturated by the high levels of IGF-1 of tumor microenvironment. Further corroborating these findings, phosphorylated IR was found in different breast-cancer subtypes and correlated with a poor survival (26). Noteworthy, IGF-1R and IR function as transcriptional regulators of the IGF-1R promoter activity in cells with reduced estrogen receptor (ER) levels (27). In particular, in ER depleted C4.12.5 breast-cancer cells but not in ER-positive MCF-7 cells, IGF-1R and IR were able to bind to the promoter of IGF-1R after translocation into the nucleus. IGF-1R enhanced the activity of its own promoter, whereas IR acted as a negative regulator of the IGF-1R promoter activity (27).
IR-A overexpression in cancer has helped us in explaining recent findings indicating that high levels of circulating insulin (hyperinsulinemia) may be related to cancer. Indeed, various lines of evidence indicate that compensatory hyperinsulinemia associated with insulin resistance is significantly associated with increased risk for various cancer histotypes (28) and are also associated to poor cancer prognosis (29). The “Women’s Health Initiative Study” showed that women exhibiting insulin levels in the upper tertile were more than twice as likely to develop breast cancer compared with those presenting insulin levels in the lowest tertile (30). It has been also shown that increased insulin levels are associated with an augmented risk for benign proliferative breast disease (BPBD) (31). Likewise, the relationship between hyperinsulinemia and breast cancer was demonstrated using an insulin-resistant and hyperinsulinemic transgenic mouse model (32). Insulin resistance and hyperinsulinemia are common in metabolic disorders, such obesity and type 2 diabetes mellitus (T2DM), which are both associated with up to two- to threefold increased risk for various malignancies (33–35). The rising occurrence of obesity and T2DM worldwide is causally associated with changing diet and lifestyle, which cause excessive adiposity, especially in the visceral region. Interestingly, in insulin-resistant states, the increased levels of circulating insulin produce biased biological effects whereby glucose metabolism is impaired whereas proliferative effects are unimpaired or enhanced (36, 37). In fact, obesity and T2DM should be considered low-grade inflammatory disorders, owing to macrophage infiltration of hypoxic visceral adipose tissue, and it is well known that inflammatory cytokines impair the IRS/PI3K/Akt pathway (so called metabolic pathway) but do not affect the Ras/Raf/MEK/ERK pathway, which may produce unbalanced stimulation of cancer cells (38, 39). The most recent World Health Organization (WHO) World Cancer report (2014) acknowledge these findings and states that the association of waist size and BMI with cancer risk follows a dose–response relationship, and that overall cancer mortality also increases in a linear fashion with increasing BMI. There is also evidence for a direct association between T2DM and cancer mortality (35, 38). Therefore, insulin resistance, obesity, and T2DM should be considered major preventable cancer risk factors (40, 41). This increased awareness may have important implications in T2DM patient’s management. Diabetic patients are exposed to endogenous hyperinsulinemia both in the pre-diabetic state and at the early stages of the disease, but also when treated with insulin or insulin analogs at later stages of the disease. Moreover, there is concern that insulin analogs may increase cancer because of a biased effect on IR signaling pathways (42). Because of the recognized role of the insulin/IGF system in cancer, in the last decade, this system has been seriously considered as a therapeutic target (43, 44). Indeed, several blocking strategies have been developed in order to selectively target the IGF-1R, with the aim to block protumoral effects without causing deterioration of glucose metabolism. Although pre-clinical studies and some phase I and II clinical trials have been very promising (45), results from phase III trials were disappointing showing clinical benefits only in small subsets of patients (44). A number of factors account for these disappointing results (46). Major factors include insulin resistance and compensatory hyperinsulinemia and increased IR-A in malignant cells (47). The cross-talk of IR/IGF-1R with matrix receptors and with other signaling pathways involved in induction and maintenance of cell stemness features may also have a role (48). As we will summarize below, previously unappreciated modalities of cross-talk with estrogen transduction pathways may also partially account for resistance to selective IGF-1R inhibitors.
The insulin/IGF system and estrogens act synergistically as potent mitogens in normal breast as well as in breast tumor cells (49). Originally, it was considered that these agents display their actions through separate pathways, but a growing body of evidence has suggested that the insulin/IGF system and estrogen-mediated signaling pathways are strictly interconnected (49). Both classical and non-classical ERs have been shown to concur to this extensive cross-talk.
Classical ERs include two subtypes, ERα and ERβ, which belong to the nuclear receptor family of transcription factors. Both receptor subtypes exert a role in cancer as suggested by the observation that ERα is overexpressed in breast-cancer cells while ERβ in prostate cancer metastases (50, 51). Both ERα and ERβ may act through ligand-dependent and ligand-independent mechanisms. The first pathway includes the well-known genomic actions and the membrane-initiated rapid effects. The ligand-independent pathway comprises the activation of other signaling effectors, like growth factors, which, after binding to kinase receptors, induce ERs phosphorylation and, thereby, activate them to dimerize, bind DNA, and regulate genes (52). The cross-talk between classical ERs and the IGF system may occur through both ligand-dependent and -independent activation. However, the ligand-independent signaling are the mechanisms especially evident in cancer, where they may contribute to tumor progression and endocrine resistance (53).
Early studies describing a cross-talk between classical ERs and the IGFs pathway were conducted in breast and prostate tumors but may be recapitulated in all tumors where both signaling are simultaneously active in inducing a positive feedback cycle of cell survival and proliferation stimuli. For example, a role of ERα in mediating insulin and IGF-I growth effects, also in absence of estrogens, was found in a pituitary tumor cell line (54) as well as in SK-ER3 neuroblastoma cells (55). In human breast-cancer cells, ligand-dependent stimulation of ERs has been shown to enhance IGF signaling at multiple levels (56). For instance, 17β-Estradiol (E2) upregulates the expression of several IGF family members including IGF-1, IGF-2, IGF-BP2, IGF-1R, and IRS-1, whereas the expression of IGFBP3 decreases upon estrogen exposure (57–61). In addition, activated ERα by E2 induces the phosphorylation of IGF-1R, which triggers downstream transduction pathways (62). IRS-1 upregulation by E2 was associated with a direct positive regulatory role on the IRS-1 promoter (60), while IGF-IR upregulation by E2 appears to involve, at least in part, the transcription factor Sp1 (57). In turn, IGF-1 stimulation may induce ligand-independent ER activation by inducing ER phosphorylation. Akt activation appears to be required and a constitutively active Akt was able to mimic IGF-1 effects (63, 64). Other studies indicated that the main molecular mechanism responsible is the activation of the PI3K/mTOR/S6K1 pathway, which phosphorylates ERα at S167 in a mitogen-activated protein kinase (MAPK)-independent manner (65). Phosphorylated ERαS167 may bind and stimulate ERE sequences, and promote gene transcription, growth, and proliferation (65). Interestingly, this response was abrogated by the mTOR1 inhibitor rapamycin (65). However, it has also been shown that E2 and IGF-1 differentially regulates ER-dependent transcription both at ERE and AP-1 sites, indicating that the effects of ligand-dependent and ligand-independent ER activation are not identical (66). At least some of these functional interactions between ERs and the IGF system may be recapitulated in other tissues and tumors (67, 68). To reinforce the relevance of the ER–IGF-1 cross-talk in cancer, microarray data have suggested that a gene signature co-regulated by IGF-1 and estrogens associates with poor prognosis in breast cancer, indicating that the inhibition of both IGF-1R and ER may be necessary in certain subtypes of breast cancer (69). Nonetheless, tamoxifen-resistant (TamR) breast-cancer cells may exhibit reduced levels of IGF-1R (70). Thus, in breast malignancies characterized by a tamoxifen resistance, IGF-1R has been proposed as a poor therapeutic target (70).
Another intriguing cross-talk with the IGF system is elicited by the small fraction of classical ER located at the level of the cell membrane and acting via MISS (membrane-initiated steroid signaling). We have recently described a novel mechanism of cross-talk between estrogens and IGF system in prostate cancer cells, involving the upregulation of the IGF-1R through the classical ERs acting via MISS (71). Both ER isoforms behave similarly in activating this pathway that requires the activation of Src, ERK, and PI3K, and results in the phosphorylation of CREB transcription factor. These findings are in close agreement with previous studies indicating that E2 activates a Src-dependent pathway by inducing an interaction between the ER phosphotyrosine 537 and the SH2 domain of Src (72, 73). These authors have also shown that ER, Src, and p85 form a ternary complex, whose assembly is stimulated by E2 (72). In turn, this complex activates both the Src and the PI3K/Akt pathways and will eventually affect gene expression by affecting multiple transcription factors, including Elk-1, c-fos, and down-regulation of C/EBPbeta and c-Jun (74). We found that CREB responsive elements are present in the 5′UTR region of IGF-IR promoter. IGF-1R upregulation by this mechanism is able to enhance IGFs effects in prostate cancer cells (71). Moreover, IGF-IR itself may phosphorylate CREB and induce CREB-dependent genes (75, 76), therefore regulating its own gene expression. Notably, this pathway is only partially blocked by classical anti-androgens or anti-estrogens, which preferentially block the genomic pathway, but it may be sensitive to inhibitors of the Scr/ERK/PI3K/CREB pathway (71) and to the antidiabetic drug metformin, which blocks MISS at multiple levels (48).
The G-protein estrogen receptor (GPER), formerly known as G protein receptor 30 (GPR30), mediates rapid responses to estrogens in several types of normal and tumor cells as well as cancer-associated fibroblasts (CAFs) (77–80). Ligand-activated GPER leads to EGFR transactivation and rapid phosphorylation of MAPKs ERK1/2 as well as phosphatidylinositol 3-kinase (PI3K) (81, 82). In addition, GPER signaling stimulates adenylyl cyclase, PKA and PKC activation (83), cAMP accumulation (81, 84), and calcium mobilization (85, 86). The identification of GPER-selective ligands has allowed a better evaluation of GPER-mediated signaling and has further supported the association of GPER with biological responses like gene expression changes, proliferation, migration, and invasion (87–92). In this regard, it has been demonstrated that GPER agonists upregulate the expression of genes associated with tumor progression like c-fos (93–96), cyclins A, D1, and E (97, 98), the connective tissue growth factor (CTGF) (99), and the early growth response-1 (Egr-1) (100). Additionally, GPER has been shown to contribute to the HIF1α-dependent expression of VEGF, which mainly supports angiogenesis and tumor progression (101). It has been also suggested that the localization of GPER at the nuclear level in CAFs occurs via an importin-dependent mechanism and is involved in its transcriptional activity (79, 102). In addition, the potential of GPER in mediating the production of E2 in breast CAFs has been recently highlighted (103) together with the observation that hydroxy tamoxifen induces through GPER the aromatase expression in both the SKBR3 breast-cancer cells and CAFs (104). GPER has been implicated not only in cancer but also in cardiovascular, immunological, and neurological functions as well as diabetes (105–107). Accordingly, GPER has been detected in pancreatic β-cells and GPER-ligands have shown insulinotropic effects by mediating pancreatic β-cell survival and stimulating insulin release (108, 109). Pharmacological manipulations and gene deletion of GPER in mice (GPR30−/−) have been associated with altered insulin release upon estrogen exposure (106, 108). Likewise, GPER deficiency resulted in insulin resistance, dyslipidemia, obesity, and increased circulating pro-inflammatory cytokines, suggesting a role of GPER in metabolism and inflammatory state (110). With regard to the clinical effects mediated by GPER, previous views might be reassessed regarding, for instance, the action of raloxifene and Fulvestrant or Faslodex. Instead of acting solely as ER-modulating agents, these drugs have the potential to act also as agonists for GPER in vitro and in vivo (111). In this regard, the fact that the activation of GPER causes vasodilation may be consistent with the hypotensive side effects observed in some patients receiving Faslodex (112). Anyway, a better understanding of the Faslodex action is challenging as this compound may act as an agonist of mutated ERα in the activation function-2 (AF-2) (113, 114).
It has been previously shown that GPER is regulated by EGF and TGFα as well as by hypoxia, one of the main factors involved in tumor aggressiveness (115, 116). Notably, an elevated expression of GPER has been associated with a high risk of metastatic diseases and poor survival rates in breast, endometrial, and ovarian tumors (112). Increased levels of GPER have been also identified in inflammatory breast cancer (IBC), an aggressive hormone-independent form of this malignancy (117). Recently, the overexpression of GPER and its plasma membrane localization were shown to be critical events in breast-cancer progression, whereas the lack of GPER in the plasma membrane was associated with an excellent long-term prognosis in ER-positive tamoxifen-treated breast tumors (118). Therefore, the expression of GPER may characterize not only the estrogen sensitivity and the response to endocrine pharmacological intervention in the above-mentioned tumors but could also be predictive of biologically aggressive phenotypes consistent with adverse outcomes and low survival rates. A cross-talk between the insulin/IGF system and the G protein-coupled receptors (GPCRs) plays a critical role in the regulation of multiple physiological functions and a variety of pathophysiological processes like cardiovascular and renal diseases, obesity, metabolic syndrome, and type II diabetes (78, 112, 119). At the cellular level, insulin as well as IGF-1 dramatically synergizes with GPCR agonists in inducing mitogenic signaling in multiple solid tumors including pancreas, colon, prostate, and breast tumors (119). In addition, recent findings have identified a new cross-talk between the insulin/IGF-1 system and GPER signaling. In particular, IGF-1 has been shown to transactivate the promoter sequence of GPER and to upregulate the expression of GPER at both the mRNA and protein levels in ERα-positive breast (MCF-7) and endometrial (Ishikawa) cancer cells (120). The aforementioned stimulatory action was exhibited by insulin in leiomyosarcoma SKUT-1 cells and in breast CAFs (80). The induction of GPER by both insulin and IGF-1 was mediated by the rapid activation of PKCδ and ERK1/2 transduction pathways and the stimulation of c-fos, which was recruited to the AP-1 site located within the promoter sequence of GPER (Figure 1). The functional role exerted by AP-1 was demonstrated to be essential for the transactivation of the GPER promoter sequence and the GPER upregulation, as the transfection of a construct encoding a dominant-negative form of c-fos abrogated these responses in cell models used. Noteworthy, GPER and one of its main target genes, named CTGF, were required for cell migration induced by IGF-1 and insulin (80, 121). Previous studies have indicated that estrogens increase insulin sensitivity and stimulate glucose uptake in target tissues and breast-cancer cells (122, 123). Of note, GPER has been involved in insulin-regulated metabolic functions in mice and humans (106, 110) as well as in the glucose uptake induced by estrogens (80). In this regard, the insulin-induced expression of GPER was found to boost the glucose uptake stimulated by estrogens and cell-cycle progression (80).
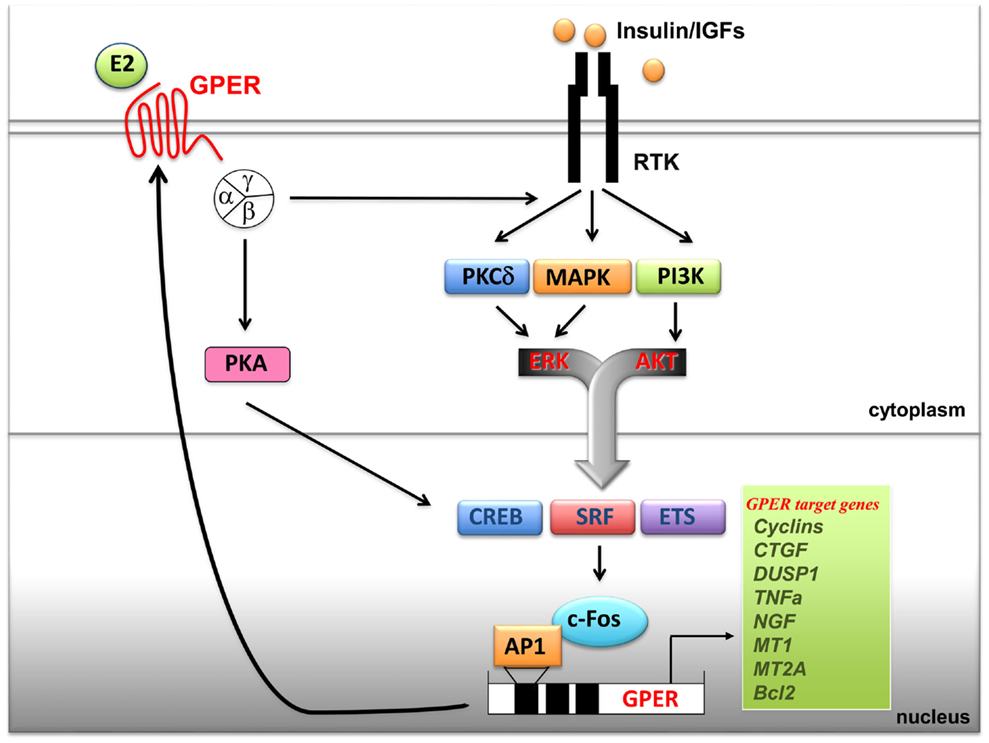
Figure 1. Cross-talk between GPER and the IGF system. Upon binding to their specific tyrosine kinase receptors (RTKs), insulin and IGF-I stimulate rapid signals converging on the activation of PI3K, MAPK and PKCδ networks. These pathways, in turn, trigger the activation of transcription factors including CREB, SRF and ETS, which favor c-fos induction and its recruitment to the AP-1 site located next to the GPER 5’ flanking region. Transactivation of GPER promoter sequences induces GPER upregulation at both mRNA and protein levels and, as a consequence, enhanced transcription of GPER target genes. In turn, GPER, upon estrogens binding, activates heterotrimeric G proteins, which trigger multiple effectors including PKA, and also PKCδ, MAPK and PI3K, converging on c-fos induction and GPER gene activation. The resulting effects of these signaling and transcriptional events lead to enhanced mitogenic signals. Abbreviations: PKA, protein kinase A; PKCδ, protein kinase C, δ isoform; MAPK, mitogen activated protein kinases; PI3K, phosphatidyl-inositol-3-kinases; ERK, extracellular signal-regulated kinases; AKT, protein kinase B; CREB, cAMP-response element-binding protein; ETS, E26 transformation specific; SRF, serum response factor; c-fos, FBJ murine osteosarcoma virus; AP-1, activator protein-1; CTGF, connective tissue growth factor; DUSP1, dual specificity protein phosphatase 1; TNFα, tumor necrosis factor α; NGF, nerve growth factor; MT1, metallothionein 1; MT2A, metallothionein 2A; Bcl2, B-cell lymphoma 2.
Many tumors are characterized not only by profound dysregulation of the insulin/IGF axis involving overexpression of receptors, ligands, and intracellular mediators, but also by deregulated expression and trafficking of classical and non-classical ERs and related adaptors/mediators. These conditions greatly enhance the complexity of the cross-talk between the insulin/IGF system and estrogens, which has been largely reported. In this respect, the upregulation of GPER triggered by ligand-activated IGF-1R and IR further contributes to the potentiation of the biological effects induced by estrogens and the insulin/IGF system in cancer. A better understanding of the mechanisms involved in the cooperation of these signaling pathways would provide further opportunities toward innovative anticancer treatments.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part by grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC) (grant no. 14066/14 to AB and 12849/2012 to MM), AIRC project Calabria 2013 and Fondazione Cassa di Risparmio di Calabria e Lucania (to AB and MM), PON01_01078 (to AB and MM), and from Ministero della Salute, 67/GR-2010-2319511 (to RM and MM).
1. Stewart CE, Rotwein P. Growth, differentiation, and survival: multiple physiological functions for insulin-like growth factors. Physiol Rev (1996) 76:1005–26.
2. Hernandez-Sanchez C, Mansilla A, de la Rosa EJ, de Pablo F. Proinsulin in development: new roles for an ancient prohormone. Diabetologia (2006) 49:1142–50. doi: 10.1007/s00125-006-0232-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Holly JMP, Perks CM. Insulin-like growth factor physiology: what we have learned from human studies. Endocrinol Metab Clin North Am (2012) 41:249–63. doi:10.1016/j.ecl.2012.04.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Hers I, Vincent EE, Tavaré JM. Akt signalling in health and disease. Cell Signal (2011) 23:1515–27. doi:10.1016/j.cellsig.2011.05.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev (2009) 30:586–623. doi:10.1210/er.2008-0047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Ahmad T, Farnie G, Bundred NJ, Anderson NG. The mitogenic action of insulin-like growth factor I in normal human mammary epithelial cells requires the epidermal growth factor receptor tyrosine kinase. J Biol Chem (2004) 279:1713–9. doi:10.1074/jbc.M306156200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev (2004) 18:1926–45. doi:10.1101/gad.1212704
8. Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev (2007) 28:20–47. doi:10.1210/er.2006-0001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Belfiore A. The role of insulin receptor isoforms and hybrid insulin/IGF-1 receptors in human cancer. Curr Pharm Des (2007) 13:671–86. doi:10.2174/138161207780249173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Aiello A, Pandini G, Sarfstein R, Werner H, Manfioletti G, Vigneri R, et al. HMGA1 protein is a positive regulator of the insulin-like growth factor-I receptor gene. Eur J Cancer (2010) 46:1919–26. doi:10.1016/j.ejca.2010.02.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Pandini G, Medico E, Conte E, Sciacca L, Vigneri R, Belfiore A. Differential gene expression induced by insulin and insulin-like growth factor-II through the insulin receptor isoform A. J Biol Chem (2003) 278:42178–89. doi:10.1074/jbc.M304980200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Belfiore A, Pandini G, Vella V, Squatrito S, Vigneri R. Insulin/IGF-I hybrid receptors play a major role in IGF-I signaling in thyroid cancer. Biochimie (1999) 81:403–7. doi:10.1016/S0300-9084(99)80088-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Papa V, Pezzino V, Costantino A, Belfiore A, Giuffrida D, Frittitta L, et al. Elevated insulin receptor content in human breast cancer. J Clin Invest (1990) 86:1503–10. doi:10.1172/JCI114868
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Pandini G, Vigneri R, Costantino A, Frasca F, Ippolito A, Fujita-Yamaguchi Y, et al. Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: evidence for a second mechanism of IGF-I signaling. Clin Cancer Res (1999) 5:1935–44.
15. Buck E, Gokhale PC, Koujak S, Brown E, Eyzaguirre A, Tao N, et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol Cancer Ther (2010) 9:2652–64. doi:10.1158/1535-7163.MCT-10-0318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Giovannucci E, Pollak M, Liu Y, Platz EA, Majeed N, Rimm EB, et al. Nutritional predictors of insulin-like growth factor I and their relationships to cancer in men. Cancer Epidemiol Biomarkers Prev (2003) 12:84–9. Available from: http://cebp.aacrjournals.org/content/12/2/84.long
17. Stattin P, Rinaldi S, Biessy C, Stenman UH, Hallmans G, Kaaks R. High levels of circulating insulin-like growth factor-I increase prostate cancer risk: a prospective study in a population-based nonscreened cohort. J Clin Oncol (2004) 22:3104–12. doi:10.1200/JCO.2004.10.105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, et al. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet (1998) 351:1393–6. doi:10.1016/S0140-6736(97)10384-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol (1999) 19:3278–88.
20. Sciacca L, Costantino A, Pandini G, Mineo R, Frasca F, Scalia P, et al. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene (1999) 18:2471–9. doi:10.1038/sj.onc.1202600
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Vella V, Sciacca L, Pandini G, Mineo R, Squatrito S, Vigneri R, et al. The IGF system in thyroid cancer: new concepts. Mol Pathol (2001) 54:121–4. doi:10.1136/mp.54.3.121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Sacco A, Morcavallo A, Pandini G, Vigneri R, Belfiore A. Differential signaling activation by insulin and insulin-like growth factors I and II upon binding to insulin receptor isoform A. Endocrinology (2009) 150:3594–602. doi:10.1210/en.2009-0377
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Malaguarnera R, Sacco A, Voci C, Pandini G, Vigneri R, Belfiore A. Proinsulin binds with high affinity the insulin receptor isoform A and predominantly activates the mitogenic pathway. Endocrinology (2012) 153(5):2152–63. doi:10.1210/en.2011-1843
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Vella V, Pandini G, Sciacca L, Mineo R, Vigneri R, Pezzino V, et al. A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J Clin Endocrinol Metab (2002) 87:245–54. doi:10.1210/jcem.87.1.8142
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Sciacca L, Prisco M, Wu A, Belfiore A, Vigneri R, Baserga R. Signaling differences from the A and B isoforms of the insulin receptor (IR) in 32D cells in the presence or absence of IR substrate-1. Endocrinology (2003) 144:2650–8. doi:10.1210/en.2002-0136
26. Law JH, Habibi G, Hu K, Masoudi H, Wang MY, Stratford AL, et al. Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res (2008) 68:10238–46. doi:10.1158/0008-5472.CAN-08-2755
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Sarfstein R, Pasmanik-Chor M, Yeheskel A, Edry L, Shomron N, Warman N, et al. Insulin-like growth factor-I receptor (IGF-IR) translocates to nucleus and autoregulates IGF-IR gene expression in breast cancer cells. J Biol Chem (2012) 287:2766–76. doi:10.1074/jbc.M111.281782
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Pisani P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch Physiol Biochem (2008) 114:63–70. doi:10.1080/13813450801954451
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Rose DP, Vona-Davis L. The cellular and molecular mechanisms by which insulin influences breast cancer risk and progression. Endocr Relat Cancer (2012) 19:R225–41. doi:10.1530/ERC-12-0203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Kabat GC, Kim M, Caan BJ, Chlebowski RT, Gunter MJ, Ho GY, et al. Repeated measures of serum glucose and insulin in relation to postmenopausal breast cancer. Int J Cancer (2009) 125:2704–10. doi:10.1002/ijc.24609
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Catsburg C, Gunter MJ, Chen C, Cote ML, Kabat GC, Nassir R, et al. Insulin, estrogen, inflammatory markers, and risk of benign proliferative breast disease. Cancer Res (2014) 74:3248–58. doi:10.1158/0008-5472.CAN-13-3514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Novosyadlyy R, Lann DE, Vijayakumar A, Rowzee A, Lazzarino DA, Fierz Y, et al. Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Res (2010) 70:741–51. doi:10.1158/0008-5472.CAN-09-2141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer (2004) 4:579–91. doi:10.1038/nrc1408
34. Vigneri P, Frasca F, Sciacca L, Frittitta L, Vigneri R. Obesity and cancer. Nutr Metab Cardiovasc Dis (2006) 16:1–7. doi:10.1016/j.numecd.2005.10.013
35. Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R. Diabetes and cancer. Endocr Relat Cancer (2009) 16:1103–23. doi:10.1677/ERC-09-0087
36. Bruning PF, Bonfrer JM, van Noord PA, Hart AA, de Jong-Bakker M, Nooijen WJ. Insulin resistance and breast-cancer risk. Int J Cancer (1992) 52:511–6. doi:10.1002/ijc.2910520402
37. Belfiore A, Frasca F. IGF and insulin receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia (2008) 13:381–406. doi:10.1007/s10911-008-9099-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Gallagher EJ, LeRoith D. Epidemiology and molecular mechanisms tying obesity, diabetes, and the metabolic syndrome with cancer. Diabetes Care (2013) 36(Suppl 2):S233–9. doi:10.2337/dcS13-2001
39. Willemse PP, van der Meer RW, Burggraaf J, van Elderen SG, de Kam ML, de Roos A, et al. Abdominal visceral and subcutaneous fat increase, insulin resistance and hyperlipidemia in testicular cancer patients treated with cisplatin-based chemotherapy. Acta Oncol (2014) 53:351–60. doi:10.3109/0284186X.2013.819116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet (2008) 371:569–78. doi:10.1016/S0140-6736(08)60269-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Belfiore A, Malaguarnera R. Insulin receptor and cancer. Endocr Relat Cancer (2011) 18:R125–47. doi:10.1530/ERC-11-0074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Sciacca L, Cassarino MF, Genua M, Pandini G, Le Moli R, Squatrito S, et al. Insulin analogues differently activate insulin receptor isoforms and post-receptor signalling. Diabetologia (2010) 53:1743–53. doi:10.1007/s00125-010-1760-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Gualberto A, Pollak M. Clinical development of inhibitors of the insulin-like growth factor receptor in oncology. Curr Drug Targets (2009) 10:923–36. doi:10.2174/138945009789577945
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Singh P, Alex JM, Bast F. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: novel treatment strategies for cancer. Med Oncol (2014) 31:805. doi:10.1007/s12032-013-0805-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer (2008) 8:915–28. doi:10.1038/nrc2536
46. King H, Aleksic T, Haluska P, Macaulay VM. Can we unlock the potential of IGF-1R inhibition in cancer therapy? Cancer Treat Rev (2014) 40:1096–105. doi:10.1016/j.ctrv.2014.07.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Garofalo C, Manara MC, Nicoletti G, Marino MT, Lollini PL, Astolfi A, et al. Efficacy of and resistance to anti-IGF-1R therapies in Ewing’s sarcoma is dependent on insulin receptor signaling. Oncogene (2011) 30(24):2730–40. doi:10.1038/onc.2010.640
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Malaguarnera R, Belfiore A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front Endocrinol (Lausanne) (2014) 5:10. doi:10.3389/fendo.2014.00010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Bradley LM, Gierthy JF, Pentecost BT. Role of insulin-like growth factor system on an estrogen-dependent cancer phenotype in the MCF-7 human breast cancer cell line. J Steroid Biochem Mol Biol (2008) 109:185–96. doi:10.1016/j.jsbmb.2007.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Lau KM, LaSpina M, Long J, Ho SM. Expression of estrogen receptor (ER)-alpha and ER-beta in normal and malignant prostatic epithelial cells: regulation by methylation and involvement in growth regulation. Cancer Res (2000) 60:3175–82. Available from: http://escholarship.umassmed.edu/cgi/viewcontent.cgi?article=1363&context=oapubs
51. Lai JS, Brown LG, True LD, Hawley SJ, Etzioni RB, Higano CS, et al. Metastases of prostate cancer express estrogen receptor-beta. Urology (2004) 64:814–20. doi:10.1016/j.urology.2004.05.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Bennesch MA, Picard D. Minireview: tipping the balance: ligand-independent activation of steroid receptors. Mol Endocrinol (2015) 29:349–63. doi:10.1210/me.2014-1315
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Bartella V, De Marco P, Malaguarnera R, Belfiore A, Maggiolini M. New advances on the functional cross-talk between insulin-like growth factor-I and estrogen signaling in cancer. Cell Signal (2012) 24:1515–21. doi:10.1016/j.cellsig.2012.03.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Newton CJ, Buric R, Trapp T, Brockmeier S, Pagotto U, Stalla GK. The unliganded estrogen receptor (ER) transduces growth factor signals. J Steroid Biochem Mol Biol (1994) 48(5–6):481–6. doi:10.1016/0960-0760(94)90197-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Ma ZQ, Santagati S, Patrone C, Pollio G, Vegeto E, Maggi A. Insulin-like growth factors activate estrogen receptor to control the growth and differentiation of the human neuroblastoma cell line SK-ER3. Mol Endocrinol (1994) 8:910–8. doi:10.1210/me.8.7.910
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Lanzino M, Morelli C, Garofalo C, Panno ML, Mauro L, Andò S, et al. Interaction between estrogen receptor alpha and insulin/IGF signaling in breast cancer. Curr Cancer Drug Targets (2008) 8:597–610. doi:10.2174/156800908786241104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Maor S, Mayer D, Yarden RI, Lee AV, Sarfstein R, Werner H, et al. Estrogen receptor regulates insulin-like growth factor-I receptor gene expression in breast tumor cells: involvement of transcription factor Sp1. J Endocrinol (2006) 191:605–12. doi:10.1677/joe.1.07016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Sisci D, Morelli C, Cascio S, Lanzino M, Garofalo C, Reiss K, et al. The estrogen receptor a: insulin receptor substrate 1 complex in breast cancer: structure-function relationships. Ann Oncol (2007) 18:vi81–6. doi:10.1093/annonc/mdm232
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Lee AV, Weng CN, Jackson JG, Yee D. Activation of estrogen receptor-mediated gene transcription by IGF-I in human breast cancer cells. J Endocrinol (1997) 1:39–47. doi:10.1677/joe.0.1520039
60. Mauro L, Salerno M, Panno ML, Bellizzi D, Sisci D, Miglietta A, et al. Estradiol increases IRS-1 gene expression and insulin signaling in breast cancer cells. Biochem Biophys Res Commun (2001) 288:685–9. doi:10.1006/bbrc.2001.5815
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Huynh H, Yang X, Pollak M. Estradiol and antiestrogens regulate a growth inhibitory insulin-like growth factor binding protein 3 autocrine loop in human breast cancer cells. J Biol Chem (1996) 271:1016–21. doi:10.1074/jbc.271.2.1016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Kahlert S, Nuedling S, van Eickels M, Vetter H, Meyer R, Grohe C. Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway. J Biol Chem (2000) 275:18447–53. doi:10.1074/jbc.M910345199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Gaben AM, Sabbah M, Redeuilh G, Bedin M, Mester J. Ligand-free estrogen receptor activity complements IGF1R to induce the proliferation of the MCF-7 breast cancer cells. BMC Cancer (2012) 12:291. doi:10.1186/1471-2407-12-291
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Martin MB, Franke TF, Stoica GE, Chambon P, Katzenellenbogen BS, Stoica BA, et al. A role for Akt in mediating the estrogenic functions of epidermal growth factor and insulin-like growth factor I. Endocrinology (2000) 141(12):4503–11. doi:10.1210/endo.141.12.7836
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Becker MA, Ibrahim YH, Cui X, Lee AV, Yee D. The IGF pathway regulates ERalpha through a S6K1-dependent mechanism in breast cancer cells. Mol Endocrinol (2011) 25:51628. doi:10.1210/me.2010-0373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Cascio S, Bartella V, Garofalo C, Russo A, Giordano A, Surmacz E. Insulin-like growth factor 1 differentially regulates estrogen receptor-dependent transcription at estrogen response element and AP-1 sites in breast cancer cells. J Biol Chem (2007) 282(6):3498–506. doi:10.1074/jbc.M606244200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Takeo C, Ikeda K, Horie-Inoue K, Inoue S. Identification of Igf2, Igfbp2 and Enpp2 as estrogen-responsive genes in rat hippocampus. Endocr J (2009) 56(1):113–20. doi:10.1507/endocrj.K08E-220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Ramachandran C, Khatib Z, Petkarou A, Fort J, Fonseca HB, Melnick SJ, et al. Tamoxifen modulation of etoposide cytotoxicity involves inhibition of protein kinase C activity and insulin-like growth factor II expression in brain tumor cells. J Neurooncol (2004) 67(1–2):19–28. doi:10.1023/B:NEON.0000021738.77612.1b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Casa AJ, Potter AS, Malik S, Lazard Z, Kuiatse I, Kim HT, et al. Estrogen and insulin-like growth factor-I (IGF-I) independently down-regulate critical repressors of breast cancer growth. Breast Cancer Res Treat (2012) 132:61–73. doi:10.1007/s10549-011-1540-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Fagan DH, Uselman RR, Sachdev D, Yee D. Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor (IGF1R): implications for breast cancer treatment. Cancer Res (2012) 72:3372–80. doi:10.1158/0008-5472.CAN-12-0684
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Genua M, Pandini G, Sisci D, Castoria G, Maggiolini M, Vigneri R, et al. Role of cyclic AMP response element-binding protein in insulin-like growth factor-i receptor up-regulation by sex steroids in prostate cancer cells. Cancer Res (2009) 69:7270–7.
72. Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Lombardi M, et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J (2000) 19(20):5406–17. doi:10.1093/emboj/19.20.5406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Castoria G, Migliaccio A, Bilancio A, Di Domenico M, de Falco A, Lombardi M, et al. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. EMBO J (2001) 21:6050–9. doi:10.1093/emboj/20.21.6050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Kousteni S, Bellido T, Plotkin LI, O’Brien CA, Bodenner DL, Han L, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell (2001) 104(5):719–30. doi:10.1016/S0092-8674(02)08100-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Monnier D, Boutillier AL, Giraud P, Chiu R, Aunis D, Feltz P, et al. Insulin-like growth factor-I stimulates c-fos and c-jun transcription in PC12 cells. Mol Cell Endocrinol (1994) 104(2):139–45. doi:10.1016/0303-7207(94)90116-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Linnerth NM, Baldwin M, Campbell C, Brown M, McGowan H, Moorehead RA. IGF-II induces CREB phosphorylation and cell survival in human lung cancer cells. Oncogene (2005) 24(49):7310–9. doi:10.1038/sj.onc.1208882
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Maggiolini M, Picard D. The unfolding stories of GPR30, a new membrane bound estrogen receptor. J Endocrinol (2010) 204:105–14. doi:10.1677/JOE-09-0242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Lappano R, Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov (2011) 10:47–60. doi:10.1038/nrd3320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Pupo M, Vivacqua A, Perrotta I, Pisano A, Aquila S, Abonante S, et al. The nuclear localization signalis required for nuclear GPER translocation and function in breast cancer-associated fibroblasts (CAFs). Mol Cell Endocrinol (2013) 376:23–32. doi:10.1016/j.mce.2013.05.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. De Marco P, Romeo E, Vivacqua A, Malaguarnera R, Abonante S, Romeo F, et al. GPER1 is regulated by insulin in cancer cells and cancer-associated fibroblasts. Endocr Relat Cancer (2014) 21:739–53. doi:10.1530/ERC-14-0245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen-induced activation of Erk-1and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol (2002) 14:1649–60. doi:10.1210/mend.14.10.0532
82. Petrie WK, Dennis MK, Hu C, Dai D, Arterburn JB, Smith HO, et al. G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth. Obstet Gynecol Int (2013) 2013:472720. doi:10.1155/2013/472720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Goswami C, Kuhn J, Dina OA, Fernandez-Ballester G, Levine JD, FerrerMontiel A, et al. Estrogen destabilizes microtubules through an ion conductivity-independent TRPV1 pathway. J Neurochem (2011) 117:995–1008. doi:10.1111/j.1471-4159.2011.07270.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Zucchetti AE, Barosso IR, Boaglio AC, Basiglio CL, Misczuk G, Larocca MC, et al. G protein coupled receptor30-adenylyl cyclase-protein kinase A pathway is involved in estradiol 17ss-d-glucuronide-induced cholestasis. Hepatology (2013) 59:1016–29. doi:10.1002/hep.26752
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science (2005) 11(307):1625–30. doi:10.1126/science.1106943
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Tica AA, Dun EC, Tica OS, Gao X, Arterburn JB, Brailoiu GC, et al. G protein-coupled estrogen receptor 1-mediated effects in the rat myometrium. Am J Physiol Cell Physiol (2011) 301(5):C1262–9. doi:10.1152/ajpcell.00501.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Prossnitz ER, Oprea TI, Sklar LA, Arterburn JB. The ins and outs of GPR30: a transmembrane estrogen receptor. J Steroid Biochem Mol Biol (2008) 109:350–3. doi:10.1016/j.jsbmb.2008.03.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Lappano R, Rosano C, De Marco P, De Francesco EM, Pezzi V, Maggiolini M. Estriol acts as a GPR30 antagonist in estrogen receptor-negative breast cancer cells. Mol Cell Endocrinol (2010) 320:162–70. doi:10.1016/j.mce.2010.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counter selectivity. J Steroid Biochem Mol Biol (2011) 127:358–66. doi:10.1016/j.jsbmb.2011.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Lappano R, Santolla MF, Pupo M, Sinicropi MS, Caruso A, Rosano C, et al. MIBE acts as antagonist ligand of both estrogen receptor a and GPER in breast cancer cells. Breast Cancer Res (2012) 14:R12. doi:10.1186/bcr3096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Rosano C, Lappano R, Santolla MF, Ponassi M, Donadini A, Maggiolini M. Recent advances in the rationale design of GPER ligands. Curr Med Chem (2012) 19:6199–206. doi:10.2174/092986712804485755
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Santolla MF, De Francesco EM, Lappano R, Rosano C, Abonante S, Maggiolini M. Niacin activates the G protein estrogen receptor (GPER)-mediated signalling. Cell Signal (2014) 26:1466–75. doi:10.1016/j.cellsig.2014.03.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. O’Brien JE, Peterson TJ, Tong MH, Lee EJ, Pfaff LE, Hewitt SC, et al. Estrogen-induced proliferation of uterine epithelial cells is independent of estrogen receptor alpha binding to classical estrogen response elements. J Biol Chem (2006) 281:26683–92. doi:10.1074/jbc.M601522200
94. Vivacqua A, Bonofiglio D, Recchia AG, Musti AM, Picard D, Andò S, et al. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17beta-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol Endocrinol (2006) 20(3):631–46. doi:10.1210/me.2005-0280
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Albanito L, Sisci D, Aquila S, Brunelli E, Vivacqua A, Madeo A, et al. Epidermal growth factor induces G protein-coupled receptor 30 expression in estrogen receptor-negative breast cancer cells. Endocrinology (2008) 8:3799–808. doi:10.1210/en.2008-0117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Prossnitz ER, Maggiolini M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol Cell Endocrinol (2009) 308:32–8. doi:10.1016/j.mce.2009.03.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17β-estradiol and selective GPR30 ligand G1 in ovarian cancer cells. Cancer Res (2007) 67:1859–66. doi:10.1158/0008-5472.CAN-06-2909
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Li Y, Chen Y, Zhu ZX, Liu XH, Yang L, Wan L, et al. 4-Hydroxytamoxifen-stimulated processing of cyclin E is mediated via G protein-coupled receptor 30 (GPR30) and accompanied by enhanced migration in MCF-7 breast cancer cells. Toxicology (2013) 5(309):61–5. doi:10.1016/j.tox.2013.04.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Pandey DP, Lappano R, Albanito L, Madeo A, Maggiolini M, Picard D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J (2009) 28:523–32. doi:10.1038/emboj.2008.304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Vivacqua A, Romeo E, De Marco P, De Francesco EM, Abonante S, Maggiolini M. GPER mediates the Egr-1 expression induced by 17β-estradiol and 4-hydroxitamoxifen in breast and endometrial cancer cells. Breast Cancer Res Treat (2012) 133:1025–35. doi:10.1007/s10549-011-1901-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. De Francesco EM, Pellegrino M, Santolla MF, Lappano R, Ricchio E, Abonante S, et al. GPER mediates activation of HIF-1a/VEGF signaling by estrogens. Cancer Res (2014) 74:1–12. doi:10.1158/0008-5472.CAN-13-3590
102. Madeo A, Maggiolini M. Nuclear alternate estrogen receptor GPR30 mediates 17beta-estradiol-induced gene expression and migration in breast cancer-associated fibroblasts. Cancer Res (2010) 70:6036–46. doi:10.1158/0008-5472.CAN-10-0408
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Luo H, Yang G, Yu T, Luo S, Wu C, Sun Y, et al. GPER-mediated proliferation and estradiol production in breast cancer associated fibroblasts. Endocr Relat Cancer (2014) 21:355–69. doi:10.1530/ERC-13-0237
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Catalano S, Giordano C, Panza S, Chemi F, Bonofiglio D, Lanzino M, et al. Tamoxifen through GPER upregulates aromatase expression: a novel mechanism sustaining tamoxifen-resistant breast cancer cell growth. Breast Cancer Res Treat (2014) 146:273–85. doi:10.1007/s10549-014-3017-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Mizukami Y. In vivo functions of GPR30/GPER-1, a membrane receptor for estrogen: from discovery to functions in vivo. Endocr J (2010) 57:101–7. doi:10.1507/endocrj.K09E-332
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Mårtensson UE, Salehi SA, Windahl S, Gomez MF, Swärd K, Daszkiewicz-Nilsson J, et al. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology (2009) 150:687–98. doi:10.1210/en.2008-0623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Liu S, Kilic G, Meyers MS, Navarro G, Wang Y, Oberholzer J, et al. Oestrogens improve human pancreatic islet transplantation in a mouse model of insulin deficient diabetes. Diabetologia (2013) 56:370–81. doi:10.1007/s00125-012-2764-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Balhuizen A, Kumar R, Amisten S, Lundquist I, Salehi A. Activation of G protein-coupled receptor 30 modulates hormone secretion and counteracts cytokine-induced apoptosis in pancreatic islets of female mice. Mol Cell Endocrinol (2010) 320:16–24. doi:10.1016/j.mce.2010.01.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Liu S, Le May C, Wong WP, Ward RD, Clegg DJ, Marcelli M, et al. Importance of extranuclear estrogen receptor-alpha and membrane G protein-coupled estrogen receptor in pancreatic islet survival. Diabetes (2009) 58:2292–302. doi:10.2337/db09-0257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Sharma G, Prossnitz ER. Mechanisms of estradiol-induced insulin secretion by the G protein-coupled estrogen receptor GPR30/GPER in pancreatic β-Cells. Endocrinology (2013) 152:3030–9. doi:10.1210/en.2011-0091
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Barton M. Position paper: the membrane estrogen receptor GPER-clues and questions. Steroids (2012) 10:935–42. doi:10.1016/j.steroids.2012.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Prossnitz ER, Barton M. Estrogen biology: new insights into GPER function and clinical opportunities. Mol Cell Endocrinol (2014) 389:71–83. doi:10.1016/j.mce.2014.02.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Borjesson AE, Windahl SH, Lagerquist MK, Engdahl C, Frenkel B, Moverare-Skrtic S, et al. Roles of transactivating functions1and 2 of estrogen receptor-alpha in bone. Proc Natl Acad Sci U S A (2011) 108:6288–93. doi:10.1073/pnas.1100454108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Moverare-Skrtic S, Borjesson AE, Farman HH, Sjogren K, Windahl SH, Lagerquist MK, et al. The estrogen receptor antagonist ICI182,780 can act both as an agonist and an inverse agonist when estrogen receptor alpha AF-2 is modified. Proc Natl Acad Sci U S A (2014) 111:1180–5. doi:10.1073/pnas.1322910111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Vivacqua A, Lappano R, De Marco P, Sisci D, Aquila S, De Amicis F, et al. G protein coupled receptor 30 expression is up-regulated by EGF and TGF a in estrogen receptor alpha-positive cancer cells. Mol Endocrinol (2009) 23:1815–26. doi:10.1210/me.2009-0120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Recchia AG, De Francesco EM, Vivacqua A, Sisci D, Panno ML, Ando S, et al. The G protein-coupled receptor 30 is up-regulated by hypoxia inducible factor-1a (HIF-1a) in breast cancer cells and cardiomyocytes. J Biol Chem (2011) 286:10773–82. doi:10.1074/jbc.M110.172247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Arias-Pulido H, Royce M, Gong Y, Joste N, Lomo L, Lee SJ, et al. GPR30 and estrogen receptor expression: new insights into hormone dependence of inflammatory breast cancer. Breast Cancer Res Treat (2010) 123:51–8. doi:10.1007/s10549-009-0631-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Sjöström M, Hartman L, Grabau D, Fornander T, Malmström P, Nordenskjöld B, et al. Lack of G protein-coupled estrogen receptor (GPER) in the plasma membrane is associated with excellent long-term prognosis in breast cancer. Breast Cancer Res Treat (2014) 145:61–71. doi:10.1007/s10549-014-2936-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Rozengurt E, Sinnett-Smith J, Kisfalvi K. Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein-coupled receptor signaling systems: a novel target for the antidiabetic drug metformin in pancreatic cancer. Clin Cancer Res (2010) 16:2505–11. doi:10.1158/1078-0432.CCR-09-2229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Rozengurt E. Mitogenic signaling pathways induced by G protein coupled receptors. J Cell Physiol (2007) 213:589–602. doi:10.1002/jcp.21246
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. De Marco P, Bartella V, Vivacqua A, Lappano R, Santolla MF, Morcavallo A, et al. Insulin-like growth factor-I regulates GPER expression and function in cancer cells. Oncogene (2013) 6:678–88. doi:10.1038/onc.2012.97
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Garrido P, Moran J, Alonso A, Gonzàlez S, Gonzàlez C. 17b-estradiol activates glucose uptake via GLUT4 translocation and PI3K/Akt signaling pathway in MCF-7 cells. Endocrinology (2013) 154:1979–89. doi:10.1210/en.2012-1558
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Alonso A, Fernàndez R, Moreno M, Ordònêz P, Gonzalez-Pardo H, Conejo NM, et al. Positive effects of 17b-estradiol on insulin sensitivity in aged ovariectomized female rats. J Gerontol A Biol Sci Med Sci (2006) 61:419–26. doi:10.1093/gerona/61.5.419
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: insulin/IGF system, GPR30/GPER, estrogen receptor, cancer cells, cancer-associated fibroblasts, signal transduction
Citation: De Marco P, Cirillo F, Vivacqua A, Malaguarnera R, Belfiore A and Maggiolini M (2015) Novel aspects concerning the functional cross-talk between the insulin/IGF-I system and estrogen signaling in cancer cells. Front. Endocrinol. 6:30. doi: 10.3389/fendo.2015.00030
Received: 13 January 2015; Paper pending published: 04 February 2015;
Accepted: 19 February 2015; Published online: 06 March 2015.
Edited by:
Briony Forbes, The University of Adelaide, AustraliaReviewed by:
Antimo Migliaccio, Seconda Università degli Studi di Napoli, ItalyCopyright: © 2015 De Marco, Cirillo, Vivacqua, Malaguarnera, Belfiore and Maggiolini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonino Belfiore, Università degli Studi Magna Graecia di Catanzaro, Viale Europa, Loc. Germaneto, Catanzaro 88100, Italy e-mail:YmVsZmlvcmVAdW5pY3ouaXQ=;
Marcello Maggiolini, Università della Calabria, via P. Bucci, Rende 87036, Italy e-mail:bWFyY2VsbG9tYWdnaW9saW5pQHlhaG9vLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.