 Antje M. Richter
Antje M. Richter Tobias Zimmermann
Tobias Zimmermann- Institute for Genetics, University of Giessen, Giessen, Germany
Pheochromocytomas (PCCs) are rare neuroendocrine tumors that arise from the medulla of the adrenal gland or the sympathetic ganglia and are characterized by the secretion of catecholamines. In 30–40% of patients, PCCs are genetically determined by susceptibility genes as various as RET, VHL, and NF1. We have analyzed the Ras-association domain family members (RASSFs) in PCCs regarding their inactivating promoter hypermethylation status. Previously, we reported a promoter methylation in PCC for the first family member RASSF1A. Promoter hypermethylation of CpG islands leads to the silencing of the according transcript and is a common mechanism for inactivation of tumor suppressors. In this study, we observed inactivating DNA modifications for the RASSF members RASSF2, RASSF5A, RASSF9, and RASSF10, but not for the members RASSF3, RASSF4, RASSF5C, RASSF6, RASSF7, and RASSF8. The degree of promoter methylation was 19% for RASSF2, 67% for RASSF5A, 18% for RASSF9, and 74% for RASSF10. Interestingly, the degree of hypermethylation for RASSF10 in hereditary PCCs was 89 vs. 60% in sporadic PCCs. A similar but less dramatic effect was observed in RASSF5A and RASSF9. Including all RASSF members, we found that of 25 PCCs, 92% show promoter methylation in at least in one RASSF member. In 75% of the hereditary PCC samples, we found two or more methylated RASSF promoters, whereas in sporadic PCCs only 46% were observed. In summary, we could show that in PCC several RASSF members are strongly hypermethylated in their promoter regions and methylation of more than one RASSF member occurs in the majority of PCCs. This adds the inactivation of genes of the RASSF tumor suppressor family to the already known deregulated genes of PCC.
Introduction
Pheochromocytomas (PCCs) are rare catecholamine-secreting tumors derived from chromaffin cells and 10% of PCCs are malignant. Tumors are malign when the capsule is invaded or metastases occur. The hormone imbalance affects the cardiovascular system leading to hypertension and other metabolic processes. PCC treatment involves treatment of hypertension and surgical removal of the tumor. (1, 2) The occurrence peaks between the ages of 20–40 and up to 30–40% occur due to germline mutations of 11 known susceptibility genes like HIF2A (hypoxia-inducible factor 2α) or SDH (succinate dehydrogenase) (3). The syndromes familial multiple endocrine neoplasia (MEN) type 2A/2B, von Hippel-Lindau disease, and neurofibromatosis are also associated with PCC development (4). Regarding RASSF1A, which is frequently inactivated in different tumor entities (5, 6), we have reported its inactivation in the tumor entity of PCC (7). Tumor suppressors act antiproliferatively and are known to be negatively regulated by their CpG island promoter hypermethylation, leading to the inactivation of the according transcript in neoplasia (8). Our focus lies on the tumor suppressors of the Ras-association domain family (RASSF) containing 10 members (RASSF1A to RASSF10), which are frequently inactivated via the methylation of their CpG island promoter (9). The RASSFs were first described in 2000, by the characterization of the first family member RASSF1A (10). The family members are characterized by the presence of the Ras-association domain (C-terminus or N-terminus) and additional protein–protein interaction domains [SARAH (11) or coiled-coil domains]. Their functions range from microtubule network interaction, RAS interaction, Hippo pathway involvement, cell cycle regulation to apoptosis induction (9, 12). Interestingly, mice studies exist for several RASSFs and suggest an involvement in tissue or organ size control (13–17). Due to our earlier studies of RASSF1A inactivation in PCC (7) and the known contribution of tumor suppressor inactivation to cancer development (8), we were interested in the possibility that inactivation of more than one RASSF member occurs in PCC. Their inactivation could contribute to tumor formation of PCC and we therefore used the combined bisulfite restriction analysis (COBRA) methylation analysis to study the promoter methylation status of the RASSF members RASSF2, RASSF3, RASSF4, RASSF5A, RASSF5C, RASSF6, RASSF7, RASSF8, RASSF9, and RASSF10.
Experimental
CpG Island Prediction, PCR Product Size, and Digestion Products
Promoter regions of RASSF2, RASSF3, RASSF4, RASSF5A, RASSF5C, RASSF6, RASSF7, RASSF8, RASSF9, and RASSF10 were analyzed for their CpG islands by CpG plot http://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/ and ucsc genome browser https://genome.ucsc.edu/. Except for RASSF6 and RASSF9, all members show prominent CpG islands. Primers for COBRA methylation analysis were designed to frame at least one restriction site for TaqI enzyme on bisulfite treated and therefore fully converted DNA (unmethylated cytosine to uracil). The resulting PCR products lengths, digestion products, and PCR conditions are listed in Table S1 in Supplementary Material.
Pheochromocytoma and Controls
Primary tissues were previously published (7). All patients signed informed consent at initial clinical investigation. The study was approved by a local ethic committee (Martin-Luther University, Halle, Germany).
Methylation Analysis by COBRA
DNA was isolated by phenol-chloroform extraction. Two micrograms of genomic DNA from PCC was bisulfite treated (12 μl of 0.1 M hydroquinone, 208 μl of 1.9 M sodium metabisulfite, and pH 5.5 with NaOH) and incubated over night at 50°C. Then DNA was purified using MSB Spin PCRapace (STRATEC Molecular, Berlin, Germany), eluted in 50 μl H2O and followed by 10 min incubation with 5 μl of 3 M NaOH at 37°C. DNA was then precipitated with 100% ethanol and 7.5 M ammonium acetate and resolved in 1× TE buffer. Two hundred nanograms were subsequently used for 25 μl PCR reaction with COBRA primers. The PCR product was either mock digested or digested with 0.5 μl of TaqI (Fermentas GmbH, St.Leon-Rot, Germany) 1 h at 65°C and resolved on 2% TBE gel together with mock control (18, 19). Digestion of the PCR product indicates an underlying CpG methylation at the analyzed restriction site. We used an in vitro methylated (ivm) DNA as a positive control for COBRA. For in vitro methylation of genomic DNA, we used M.SssI methylase (NEB, Frankfurt, Germany) (20). Human fibroblasts (HF) were used as a normal unmethylated control cell line.
Results
In the current study, we have analyzed a total of 25 PCC patient samples of which 13 were sporadic and 12 hereditary (Table 1). We have studied the promoter methylation status of RASSF2, RASSF3, RASSF4, RASSF5A, RASSF5C, RASSF6, RASSF7, RASSF8, RASSF9, and RASSF10. DNA promoter methylation is an indicator of epigenetic inactivation of tumor suppressor genes (TSGs) (8). The CpG island sizes are 1.1 kb for RASSF2, 1.1 kb for RASSF3, 0.5 kb for RASSF4, 1.2 kb for RASSF5A, 0.5 kb for RASSF5C, 0.2 kb for RASSF6, 1 kb for RASSF7, 1.2 kb for RASSF8, not predicted for RASSF9, and 2.3 kb for RASSF10. We used the COBRA method for promoter methylation analysis, which is based on bisulfite treatment of genomic DNA for conversion of unmethylated cytosines to uracils. The following PCR amplifies the defined region and is followed by digestion with TaqI restriction enzyme (Figure 1). PCR fragments still containing restriction sites (methylated and therefore unchanged CpG) are digested and fragments no longer containing restriction sites (earlier unmethylated CpG) remain undigested. Therefore, a digestion of the PCR product indicates a methylation of the according genomic CpGs that were analyzed. Localization of COBRA primers within the analyzed CpG promoter region, transcription start site, CpG enrichment, restriction site in PCR product, and PCR product size are shown for each analyzed family member (Figure 1). Apparently, RASSF6 and RASSF9 show little CpG island presence, but were analyzed as well.
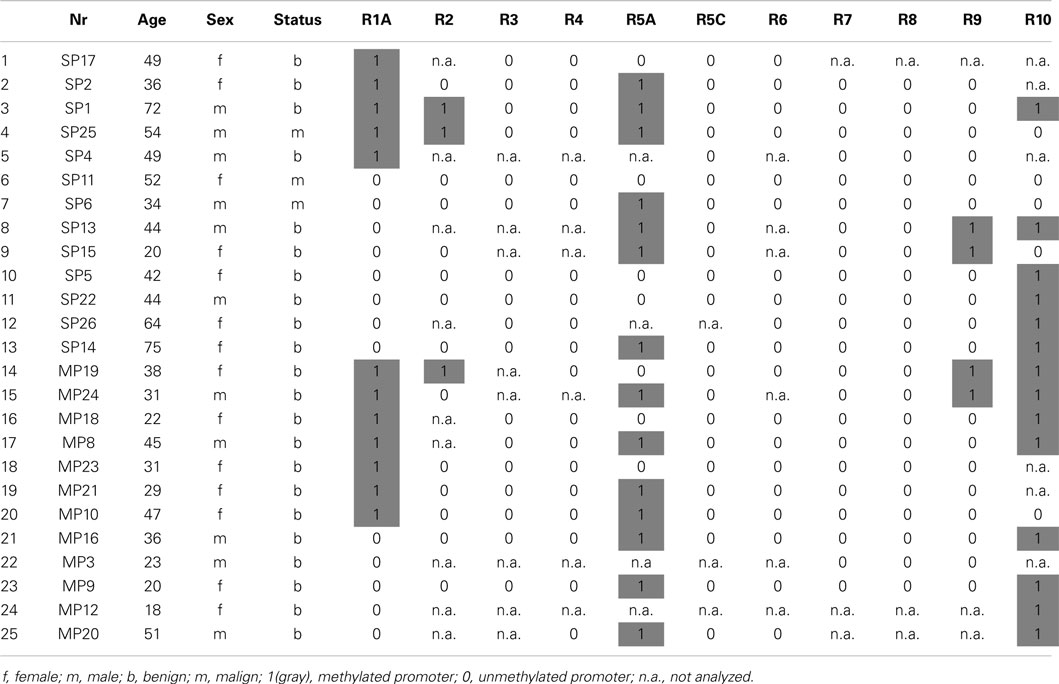
Table 1. RASSF promoter methylation in sporadic (SP) and hereditary (MP) pheochromocytoma.

Figure 1. Genomic RASSF promoter regions. RASSF family members RASSF2, RASSF3, RASSF4, RASSF5A, RASSF5C, RASSF6, RASSF7, RASSF8, RASSF9, and RASSF10 are shown with their promoter region. Single CpGs are represented as vertical black lines, restriction enzyme TaqI recognition sites are marked with T, and bent arrows indicate transcriptional start sites. Horizontal arrows mark the COBRA PCR products obtained after bisulfite treatment of DNA and according PCR fragment size. Hundred base pair standard is shown below each promoter region. Graphics were generated with the python.vs.cobra program (https://launchpad.net/python.vs.cobra).
Our results show promoter methylation for RASSF2, RASSF5A, RASSF9, and RASSF10, but not for any of the other analyzed members of the family (Figure 2). RASSF2 shows in 3 of 16 samples (19%), a methylation, RASSF5A in 14 of 21 samples (67%), RASSF9 in 4 out of 22 samples (18%), and RASSF10 in 14 out of 19 samples (74%). For RASSF3 0/17, RASSF4 0/19, RASSF5C 0/22, RASSF6 0/19, RASSF7 0/22, and RASSF8 0/22 and therefore no samples showed methylation of their promoter region (Tables 1 and 2). For COBRA analysis, we used ivm DNA as a positive control for methylation detection after bisulfite treatment. In this study, HF serve as a negative control for promoter methylation analysis. This non-cancerous cell line reaches senescence after few passages like normal tissues (21).
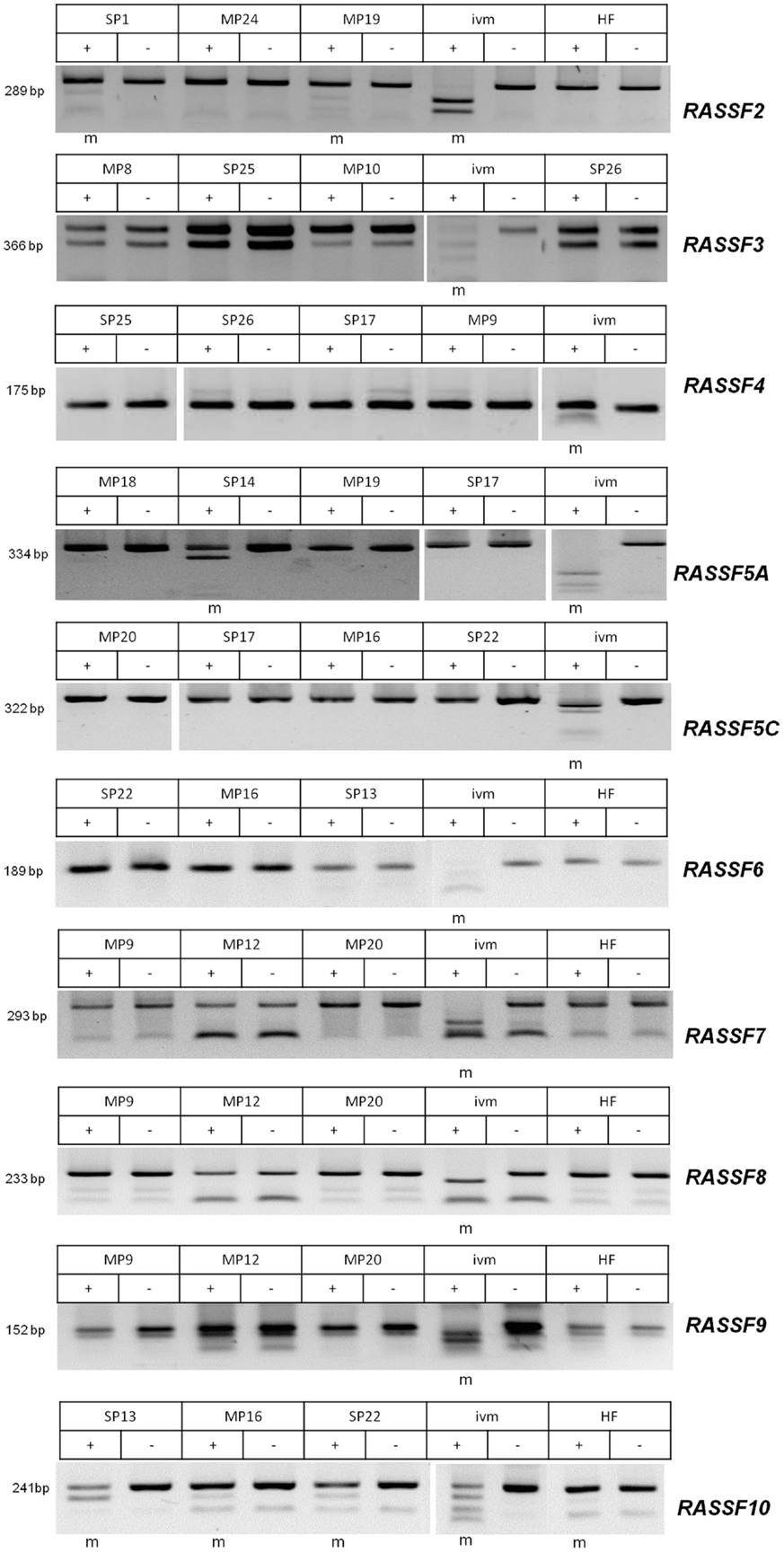
Figure 2. Methylation analyses by COBRA for RASSF2, −3, −4, −5A, −5C, −6, −7, −8, −9, and −10. Methylation analysis by COBRA method for RASSF2, −3, −4, −5A, −5C, −6, −7, −8, −9, and −10 was performed and PCR product sizes are shown. PCR products were digested with TaqI enzyme (+) or mock digested (−). Sporadic (SP) and hereditary (MP) PCCs are exemplarily shown for each RASSF together with in vitro methylated (ivm) positive control sample and human fibroblasts (HF) as normal cell control. Methylated samples are marked (m) below each gel.
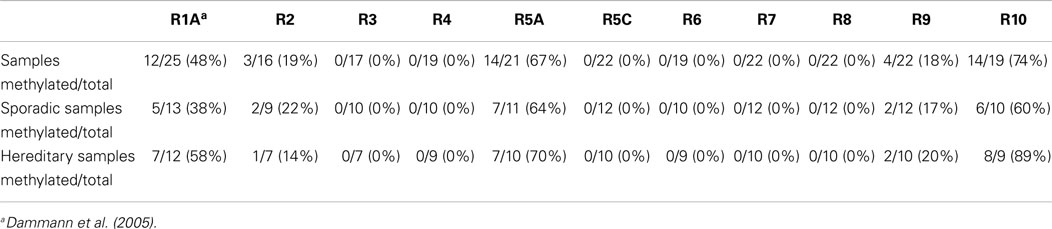
Table 2. Summary of RASSF promoter methylation.
We have analyzed sporadic as well as hereditary PCC samples and found promoter hypermethylation in both subsets of PCC for RASSF2, RASSF5A, RASSF9, and RASSF10 (Table 1). Interestingly, the promoter of RASSF10 showed an increase in methylation from 60% of sporadic samples to 89% of hereditary PCCs. A similar but less dramatic effect was observed in RASSF5A and RASSF9. Furthermore, correlation analysis neither showed an association of methylation status of RASSF2, RASSF5A, RASSF9, and RASSF10 with age, sex, bilateral localization, noradrenalin/adrenalin expression nor the presence of p16, VHL, MSH2, MLH1 promoter methylation, which we studied earlier (7). We compared the number of co-methylated promoters from all RASSFs in the 25 PCCs, which included RASSF1A (7). We found that only two samples remained unmethylated for all RASSF promoters (Figure 3). The other 23 PCC samples showed one up to four methylated RASSF promoters per sample. In all PCCs, we observed a distribution of eight samples with one promoter methylation, nine samples with two methylated promoters, and six samples with three or four methylated promoters (Figure 3). Six sporadic PCC samples showed one promoter methylation and six samples had two or more promoters being methylated. In hereditary PCCs, we found however only two samples with one promoter methylation vs. nine samples with two or more methylated promoters (Figure 3).
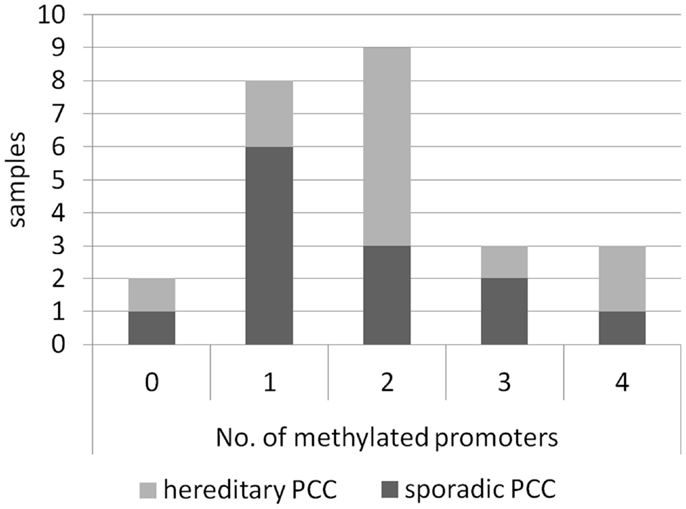
Figure 3. Occurrence of co-methylated RASSF promoters in PCCs. Samples of sporadic (n = 13) and hereditary (n = 12) PCCs are shown regarding their number of methylated RASSF promoters for all RASSFs. For details, see Table 1.
Discussion
The aim of our study was the comparative analysis of RASSFs regarding their promoter methylation status in PCC. The tumor suppressor family RASSF consists of 10 members, with the first member RASSF1 being the best studied one. Tumor suppressors are known to be inactivated by CpG island promoter hypermethylation (8) and we and others have already shown the RASSF1A promoter methylation in PCC (7, 22). A later study even correlated RASSF1A promoter methylation with PCC malignancy (23). In this study, we analyzed the family members RASSF2, RASSF3, RASSF4, RASSF5A, RASSF5C, RASSF6, RASSF7, RASSF8, RASSF9, and RASSF10, of which some have been characterized as tumor suppressors (9, 12). So far, no other study analyzed PCC and the promoter methylation of our studied RASSFs except for one publication showing no promoter methylation for RASSF5A in primary PCCs (23). However, a significant downregulation of RASSF5A has been reported in PCC (19), although the relevant mechanism was not identified. We have studied sporadic and hereditary PCCs regarding their promoter methylation status of RASSFs. We observed a strong hypermethylation in PCCs for RASSF2 (19%), RASSF5A (67%), RASSF9 (18%), and RASSF10 (74%), but not in the other family members (Table 2). Interestingly, the degree of methylation was higher in hereditary PCCs when compared to sporadic PCCs for RASSF5A (70 vs. 64%) and RASSF10 (89 vs. 60%; Table 1), which is in accordance with the RASSF1A (58 vs. 38%) methylation status (7).
Regarding all analyzed PCCs, we found that 92% show promoter methylation of at least one RASSF member. Additionally, we observed the co-methylation of several RASSF promoters in the same PCC sample. The comparison of sporadic to hereditary PCCs shows an increase in the number of samples with two or more methylated RASSF promoters from 46% of sporadic to 75% of hereditary PCCs (Figure 3). In a hereditary PCC background, an additional RASSF inactivation seems a driver of tumor formation and underlines the importance of expression regulation of RASSFs. It would be interesting to analyze this positive association in a larger cohort of PCCs. We found an inverse correlation of RASSF10 methylation with malignancy and infiltration of the capsule, which very likely must be attributed to the rather small sample number. Interestingly, we observed a promoter methylation for RASSF9 despite the absence of a predictable CpG island. It can be presumed that even the RASSF9 promoter methylation plays a role in its inactivation.
The family members that showed no methylation in our PCC analysis were studied earlier in different cancer types with differing results. Regarding RASSF3 and RASSF4, very little data exist, but it was reported that RASSF3 is epigenetically inactivated in somatotroph adenomas (24) and RASSF4 in human tumor cells (25). RASSF5C is the 3′ transcript of the RASSF5 locus (26), similar to the isoforms RASSF1A and C, containing a CpG island, but hardly ever being methylated and thereby inactivated (18, 26–28). Due to the small size of the RASSF6 CpG island, an epigenetic regulation is rather unlikely. However, in neuroblastoma, RASSF6 and RASSF7 showed a promoter methylation and RASSF6 promoter methylation was correlated with an unfavorable outcome (29). RASSF8 methylation was reported in a small subset of leukemia (30). In further studies, we would like to address a larger set of PCCs regarding promoter methylation of RASSFs and if possible include normal adrenal gland tissue as well as the PCC cell line PC12. It will be fascinating to further study the functional contribution of inactivated RASSFs for PCC development or progression. It will be especially interesting to dissect why several RASSF members are inactivated in the same samples and therefore presumably missing. This co-inactivation in PCC suggests distinct and separate pathways through which RASSF1A, RASSF2, RASSF5A, RASSF9, and RASSF10 function in the normal adrenal gland. It seems plausible that inactivation of several members of the RASSF family contributes to tumor formation and progression. It could also underline that RASSFs are not all redundant and interchangeable. Knockdown experiments of each of the members will be of valuable use and studies using the available knockout mice could indicate the contribution of co-inactivation of the RASSFs.
Conclusion
In our study, we are the first to show the promoter methylation status of all RASSFs in sporadic and hereditary PCC. We have found promoter hypermethylation for the family members RASSF1A, RASSF2, RASSF5A, RASSF9, and RASSF10, but not for RASSF3, RASSF4, RASSF5C, RASSF6, RASSF7, or RASSF8. Our observations show co-appearance of CpG promoter hypermethylation of several RASSF members in PCC, especially in the hereditary PCCs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Michelle Woods for correcting the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fendo.2015.00021/abstract
Abbreviations
COBRA, combined bisulfite restriction analysis; PCC, pheochromocytoma; RASSF, Ras-association domain family; TSG, tumor suppressor gene.
References
1. Manger WM. An overview of pheochromocytoma: history, current concepts, vagaries, and diagnostic challenges. Ann N Y Acad Sci (2006) 1073:1–20. doi: 10.1196/annals.1353.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Harari A, Inabnet WB III. Malignant pheochromocytoma: a review. Am J Surg (2011) 201:700–8. doi:10.1016/j.amjsurg.2010.04.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Gruber M, Darr R, Eisenhofer G. [Pheochromocytoma: update on diagnosis and therapy]. Dtsch Med Wochenschr (2014) 139:486–90. doi:10.1055/s-0033-1360082
4. Karagiannis A, Mikhailidis DP, Athyros VG, Harsoulis F. Pheochromocytoma: an update on genetics and management. Endocr Relat Cancer (2007) 14:935–56. doi:10.1677/ERC-07-0142
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Agathanggelou A, Cooper WN, Latif F. Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res (2005) 65:3497–508. doi:10.1158/0008-5472.CAN-04-4088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Dammann R, Schagdarsurengin U, Seidel C, Strunnikova M, Rastetter M, Baier K, et al. The tumor suppressor RASSF1A in human carcinogenesis: an update. Histol Histopathol (2005) 20:645–63.
7. Dammann R, Schagdarsurengin U, Seidel C, Trümpler C, Hoang-Vu C, Gimm O, et al. Frequent promoter methylation of tumor-related genes in sporadic and men2-associated pheochromocytomas. Exp Clin Endocrinol Diabetes (2005) 113:1–7. doi:10.1055/s-2004-830522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Jones PA. The DNA methylation paradox. Trends Genet (1999) 15:34–7. doi:10.1016/S0168-9525(98)01636-9
9. Richter AM, Pfeifer GP, Dammann RH. The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim Biophys Acta (2009) 1796:114–28. doi:10.1016/j.bbcan.2009.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet (2000) 25:315–9. doi:10.1038/77083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Scheel H, Hofmann K. A novel interaction motif, SARAH, connects three classes of tumor suppressor. Curr Biol (2003) 13:R899–900. doi:10.1016/j.cub.2003.11.007
12. Sherwood V, Recino A, Jeffries A, Ward A, Chalmers AD. The N-terminal RASSF family: a new group of Ras-association-domain-containing proteins, with emerging links to cancer formation. Biochem J (2010) 425:303–11. doi:10.1042/BJ20091318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Tommasi S, Dammann R, Zhang Z, Wang Y, Liu L, Tsark WM, et al. Tumor susceptibility of Rassf1a knockout mice. Cancer Res (2005) 65:92–8.
14. Del Re DP, Matsuda T, Zhai P, Gao S, Clark GJ, Van Der Weyden L, et al. Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. J Clin Invest (2010) 120:3555–67. doi:10.1172/JCI43569
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Song H, Kim H, Lee K, Lee DH, Kim TS, Song JY, et al. Ablation of Rassf2 induces bone defects and subsequent haematopoietic anomalies in mice. EMBO J (2012) 31:1147–59. doi:10.1038/emboj.2011.480
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Lee CM, Yang P, Chen LC, Chen CC, Wu SC, Cheng HY, et al. A novel role of RASSF9 in maintaining epidermal homeostasis. PLoS One (2011) 6:e17867. doi:10.1371/journal.pone.0017867
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Park J, Kang SI, Lee SY, Zhang XF, Kim MS, Beers LF, et al. Tumor suppressor Ras association domain family 5 (RASSF5/NORE1) mediates death receptor ligand-induced apoptosis. J Biol Chem (2010) 285:35029–38. doi:10.1074/jbc.M110.165506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Richter AM, Haag T, Walesch S, Herrmann-Trost P, Marsch WC, Kutzner H, et al. Aberrant promoter hypermethylation of RASSF family members in Merkel cell carcinoma. Cancers (Basel) (2013) 5:1566–76. doi:10.3390/cancers5041566
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Kiehl S, Herkt SC, Richter AM, Fuhrmann L, El-Nikhely N, Seeger W, et al. ABCB4 is frequently epigenetically silenced in human cancers and inhibits tumor growth. Sci Rep (2014) 4:6899. doi:10.1038/srep06899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Haag T, Herkt CE, Walesch SK, Richter AM, Dammann RH. The apoptosis associated tyrosine kinase gene is frequently hypermethylated in human cancer and is regulated by epigenetic mechanisms. Genes Cancer (2014) 5:365–74.
21. Goldstein S. Replicative senescence: the human fibroblast comes of age. Science (1990) 249:1129–33. doi:10.1126/science.2204114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Astuti D, Agathanggelou A, Honorio S, Dallol A, Martinsson T, Kogner P, et al. RASSF1A promoter region CpG island hypermethylation in pheochromocytomas and neuroblastoma tumours. Oncogene (2001) 20:7573–7. doi:10.1038/sj.onc.1204968
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Geli J, Kiss N, Lanner F, Foukakis T, Natalishvili N, Larsson O, et al. The Ras effectors NORE1A and RASSF1A are frequently inactivated in pheochromocytoma and abdominal paraganglioma. Endocr Relat Cancer (2007) 14:125–34. doi:10.1677/ERC-06-0031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Peng H, Liu H, Zhao S, Wu J, Fan J, Liao J. Silencing of RASSF3 by DNA hypermethylation is associated with tumorigenesis in somatotroph adenomas. PLoS One (2013) 8:e59024. doi:10.1371/journal.pone.0059024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Eckfeld K, Hesson L, Vos MD, Bieche I, Latif F, Clark GJ. RASSF4/AD037 is a potential Ras effector/tumor suppressor of the RASSF family. Cancer Res (2004) 64:8688–93. doi:10.1158/0008-5472.CAN-04-2065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Tommasi S, Dammann R, Jin SG, Zhang XF, Avruch J, Pfeifer GP. RASSF3 and NORE1: identification and cloning of two human homologues of the putative tumor suppressor gene RASSF1. Oncogene (2002) 21:2713–20. doi:10.1038/sj.onc.1205365
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Hesson L, Bièche I, Krex D, Criniere E, Hoang-Xuan K, Maher ER, et al. Frequent epigenetic inactivation of RASSF1A and BLU genes located within the critical 3p21.3 region in gliomas. Oncogene (2004) 23:2408–19. doi:10.1038/sj.onc.1207407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Schagdarsurengin U, Richter AM, Hornung J, Lange C, Steinmann K, Dammann RH. Frequent epigenetic inactivation of RASSF2 in thyroid cancer and functional consequences. Mol Cancer (2010) 9:264. doi:10.1186/1476-4598-9-264
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Djos A, Martinsson T, Kogner P, Caren H. The RASSF gene family members RASSF5, RASSF6 and RASSF7 show frequent DNA methylation in neuroblastoma. Mol Cancer (2012) 11:40. doi:10.1186/1476-4598-11-40
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Hesson LB, Dunwell TL, Cooper WN, Catchpoole D, Brini AT, Chiaramonte R, et al. The novel RASSF6 and RASSF10 candidate tumour suppressor genes are frequently epigenetically inactivated in childhood leukaemias. Mol Cancer (2009) 8:42. doi:10.1186/1476-4598-8-42
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: pheochromocytoma, tumor suppressor, DNA methylation, epigenetics, RASSF
Citation: Richter AM, Zimmermann T, Haag T, Walesch SK and Dammann RH (2015) Promoter methylation status of Ras-association domain family members in pheochromocytoma. Front. Endocrinol. 6:21. doi: 10.3389/fendo.2015.00021
Received: 19 December 2014; Accepted: 05 February 2015;
Published online: 19 February 2015.
Edited by:
Fabio Coppedè, University of Pisa, ItalyReviewed by:
Daniela Pasquali, Seconda Università degli Studi Napoli, ItalyFrancesca Peruzzi, LSU Health Sciences Center, USA
Copyright: © 2015 Richter, Zimmermann, Haag, Walesch and Dammann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antje M. Richter, Heinrich-Buff Ring 58, Giessen, Hessen, Germany e-mail: antje.m.richter@gen.bio.uni-giessen.de