 Anastasia Hodes
Anastasia Hodes David Lichtstein
David Lichtstein
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 28 November 2014
Sec. Neuroendocrine Science
Volume 5 - 2014 | https://doi.org/10.3389/fendo.2014.00201
This article is part of the Research Topic The Natriuretic Hormones View all 10 articles
Natriuretic hormones (NH) include three groups of compounds: the natriuretic peptides (ANP, BNP and CNP), the gastrointestinal peptides (guanylin and uroguanylin), and endogenous cardiac steroids. These substances induce the kidney to excrete sodium and therefore participate in the regulation of sodium and water homeostasis, blood volume, and blood pressure (BP). In addition to their peripheral functions, these hormones act as neurotransmitters or neuromodulators in the brain. In this review, the established information on the biosynthesis, release and function of NH is discussed, with particular focus on their role in brain function. The available literature on the expression patterns of each of the NH and their receptors in the brain is summarized, followed by the evidence for their roles in modulating brain function. Although numerous open questions exist regarding this issue, the available data support the notion that NH participate in the central regulation of BP, neuroprotection, satiety, and various psychiatric conditions, including anxiety, addiction, and depressive disorders. In addition, the interactions between the different NH in the periphery and the brain are discussed.
Natriuretic hormones (NH) are compounds that act in an endocrine or paracrine fashion to regulate extracellular fluid volume and blood pressure (BP) through the stimulation of sodium excretion by the kidney. Three groups of compounds fall into this broad definition: the natriuretic peptides (NP: ANP, BNP, and CNP), the guanylin peptides (GP), and the endogenous cardiac steroids (CS: ouabain, digoxin, and marinobufagenin). A large body of evidence supports the notion that in addition to their natriuretic effects, these hormones participate in numerous brain functions. Our goal is to review the established information on the biosynthesis, release, and physiological roles of NH, with particular focus on the brain. The available literature on the interactions between the different NH families in the periphery and in the brain is also addressed.
The first demonstration of an endocrine link between the heart and kidneys came from the pioneering experiments of De Bold, which led to the discovery of atrial NP (ANP), the founding member of the family of NP. De Bold and his colleagues found that injecting rats with an atrial homogenate caused significant natriuresis and diuresis (1). Additional members of this family of peptides were purified over the course of the following years: B-type NP (BNP) (2) and C-type NP (CNP) (3). ANP, BNP, and CNP are expressed as pre-pro-hormones and are proteolytically processed to form the mature peptides. The three peptides share a similar structure consisting of two cysteine residues flanking a 17-residue disulfide-linked ring that is essential for biological activity (3). The main inducer of ANP release is atrial wall stretch (4). BNP is released from the atrium, as is ANP, but its main sources are the ventricles, where BNP is transcriptionally regulated by cardiac wall stretch resulting from volume overload (5). There are three known NP receptors: NP receptor-A (NPR-A), or guanylyl cyclase A (GC-A), which binds ANP and BNP (6); NPR-B (GC-B), which is highly specific for CNP (7); and NPR-C. NPR-A and NPR-B are membrane-bound receptors consisting of an extracellular ligand binding domain, a single transmembrane region and an intracellular GC domain that rapidly releases cyclic guanosine monophosphate (cGMP) in response to the NP binding (8). The cGMP then acts as a second messenger that activates protein kinase-G (PKG) and subsequent cellular signaling cascades (9). A third receptor, NPR-C, contains only a short intracellular fragment and has no GC activity. The main function of NPR-C is to clear NP through receptor-mediated internalization and degradation (10).
Atrial natriuretic peptide has a major role in the regulation of BP. In the kidney, ANP induces natriuresis and diuresis by increasing the glomerular filtration rate (GFR) and inhibiting sodium and water reabsorption (11). ANP acts as a functional antagonist of the renin–angiotensin–aldosteron system by inhibiting renin secretion from the kidney and aldosterone production in the adrenal glands (12). It stimulates smooth muscle cell relaxation in blood vessels, causing vasorelaxation (13). It also regulates the intravascular volume by increasing endothelium permeability (14). In accordance with these effects, it was found that ANP knockout mice developed salt-sensitive hypertension (15). ANP also directly affects the heart by inhibiting cardiac hypertrophy (16). BNP activates the same receptor as ANP but its precise functional significance is not well understood. Studies on mice with targeted disruption of BNP (17) and on cultured cardiac fibroblasts (18) established BNP as an antifibrotic factor that plays a role in ventricular remodeling. Indeed, high concentrations of BNP were found in the ventricles following congestive heart failure or myocardial infarction, rendering it an important biomarker for these conditions (19). Unlike the other two family members, CNP acts in an autocrine/paracrine fashion. Although NPR-B is present in the kidney, CNP has little natriuretic or diuretic effect, and it is a much more potent cardiovascular effector (20). CNP is produced by the endothelium and induces vasorelaxation (21). It also participates in vascular remodeling following injury (22). In addition to their cardiovascular and renal effects, NP show a wide-spread effect throughout the body (8): They act as bronchodilators and vasorelaxants in the lungs (23), elicit anti-inflammatory effects in the immune system (24), and have metabolic effects on the adipose tissue (25) and on long bones (26).
The ANP, BNP, and CNP and their receptors are expressed in the brain, which implies a possible role for these peptides in brain function. CNP is the most abundantly present NP in the brain (27), suggesting that it acts as a neurotransmitter or neuromodulator rather than a cardiac hormone (28). Accordingly, the CNP-specific receptor – NPR-B is widely spread throughout the brain: NPR-B mRNA was detected in the cerebral cortex, the limbic area, preoptic-hypothalamic regions, motor nuclei, and the brainstem (29). ANP and BNP are also present in the brain and have interesting neuromodulatory functions. ANP expression was first found in the hypothalamus (30), which is the main source of NP in the brain (31, 32). ANP is present in neurons and glia in the cerebral cortex (33) and in the cerebellum (34). ANP was also described in neurons and fibers in the limbic area, olfactory bulb, thalamus, and striatum (31, 35–37). BNP was found in the hypothalamus (38) and cerebral cortex (33). Unlike ANP and CNP, no BNP mRNA was detected in the brain (39), suggesting the peripheral origin of this peptide. Interestingly, ANP and BNP were described in some of the circumventricular organs in the brain – the highly vasculated structures in the hypothalamus that allow endocrine communication between the periphery and CNS (40). Considerable ANP-like immunoreactivity was found in nerve fibers of the vascular organ of the lamina terminalis and the subfornical organ in rat brain (31). Neurons in the subfornical organ were shown to respond to ANP by increased cGMP production (41). Neurons of the circumventricular organs express receptors for the majority of the cardiovascular hormones (42), including NP receptors: NPR-A and NPR-B were found in the vascular organ of lamina terminalis, the subfornical organ, area postrema, and the choroid plexus (43).
The presence of NP and their receptors in the brain, and in the circumventricular organs in particular, led to the postulation that NP, either locally produced in the brain or arriving via the peripheral circulation, might affect neuronal pathways that centrally regulate BP. However, the results are inconsistent. Intracerebroventricular (i.c.v.) administration of ANP was reported to cause a decrease in BP in normal and spontaneously hypertensive conscious rats, but only at concentrations 10 times higher than the physiological level (44). Low concentrations of ANP were shown to have a depressor effect in anesthetized rats with sinoartic denervation, leading to a decrease in BP and sympathetic outflow (45, 46). A study performed on conscious sheep showed that CNP, but not ANP, decreased BP upon i.c.v. administration (47). Numerous studies found no change in BP upon central administration of ANP (48–52) or BNP (53). However, there are reports describing a decrease in vasopressin secretion following central ANP infusion, suggesting that ANP and vasopressin may interact to attenuate the central pressor effects of vasopressin (49, 51–54). Pretreatment of rats with i.c.v. BNP was also shown to suppress vasopressin secretion (53). In several studies it was postulated that ANP acts in the brain by partially inhibiting the angiotensin II (ANG II) pathway. ANP injection prevented the pressor effect of centrally administered ANG II (46, 51). On the behavioral level, centrally administered ANP was shown to inhibit water intake induced by ANG II or dehydration in rats (55). It was also found to attenuate salt appetite in spontaneously hypertensive rats (SHR) (48). These results suggest that ANP may not be directly involved in central regulation of BP, but rather act as a secondary modulator of other mechanisms, perhaps, similar to its peripheral effect, by counteracting to the effects of vasopressin and ANG II.
Natriuretic peptides were shown to exert a neuroprotective effect in cultured cells and in vivo. Cortical spreading depression (CSD) is a wave of depolarization followed by transient suppression of electrical activity in the brain (56). Rats preconditioned with an evoked episode of CSD were protected from neuronal damage following cerebral ischemia (57). Wiggins and his colleagues found that an acute episode of CSD caused an elevation in ANP mRNA and peptide levels in the rat cortex. The elevation was prolonged, overlapped the time window for CSD-induced neuroprotection and accompanied by ANP-dependent activation of cGMP signaling cascades (58). Increased cGMP levels were previously implicated in the neuroprotective mechanism of CSD (59). This notion is supported by studies showing that ANP and BNP caused an elevation in cGMP levels and inhibited apoptosis of PC12 cells (60). However, there is no direct evidence of this effect in the brain. A neuroprotective effect was also demonstrated in rat retinal neurons, where ANP was shown to ameliorate NMDA-induced neurotoxicity, presumably in a dopamine-dependent manner (61). It was postulated that the ANP neuroprotective effect is mediated via the cerebral blood flow. Indeed, an increased number of ANP-immunoreactive astrocytes and other glial cells were found in the white matter surrounding an infarction area in rats (62). This neuroprotective effect may be modulated by cGMP signaling, since cGMP-phosphodiesterase inhibitor was found to have a protective effect in a focal brain injury model in rats (63). In ischemic brain edema induced in rats, intravenous (i.v.) administration of ANP proved to have a beneficial effect. At pharmacological doses, the peptide significantly suppressed the elevation of the brain’s water and sodium content and reduced the area of edema, as revealed by magnetic resonance imaging (MRI) (64). ANP was beneficial even after delayed administration, and reduced brain edema when injected i.c.v. 4 h after induction of hemorrhagic brain injury in rats (65). BNP too was implicated in neuroprotection following brain injury. James and colleagues demonstrated that i.v. administration of BNP improved cerebral blood flow and reduced inflammation in brain injury models in mice, as manifested by reduced neurodegeneration and improved functional outcome (66). Although these experiments were performed using high doses of exogenous human recombinant BNP (nesiritide), endogenous BNP may play a role in recovery from brain injury, as elevated BNP levels have been associated with this condition. Elevated plasma BNP levels were described in patients with traumatic brain injury (67, 68), stroke (69), and other brain injuries (70, 71). Elevated BNP levels were also reported in the cerebrospinal fluid (CSF) of brain trauma patients (67). These changes, however, correlated with a poor clinical outcome in trauma and stroke patients (72, 73). This may indicate an insufficiency of the endogenous neuroprotective mechanism. The mechanism of NP neuroprotection could be mediated through immunomodulation, as was demonstrated in the periphery (74). All these clinical and pre-clinical observations lead to the premise that ANP and BNP are part of an endogenous protective mechanism in the brain against injury or damage.
Natriuretic peptides modulate the function of the hypothalamic–pituitary–adrenal (HPA) axis and influence anxiety and addictive behavior. NP regulate the HPA-axis at several levels: ANP inhibits the release of corticotrophin (ACTH) and corticortrophin releasing hormone (CRH) (75, 76), which, in turn, stimulate ANP release, acting in a feedback loop (76). ANP also directly inhibits cortisol secretion, whereas CNP exerts the opposite effect (77). Central or peripheral administration of ANP decreased anxiety-associated behavior in rats (78). In humans, lower levels of ANP were described in patients with anxiety-related disorders, including panic disorder (79) and posttraumatic stress disorder (80), and high ANP levels were associated with lower anxiety levels in patients recovering from cardiac failure (81). Experimentally induced panic attacks were followed by an increase in plasma ANP levels, which was faster and more pronounced in panic disorder patients (79, 82). These observations suggest a therapeutic potential for ANP agonists in the treatment of anxiety-related disorders (83). Indeed, pretreatment with i.v. ANP significantly reduced the number of experimentally induced panic attacks in panic disorder patients and in healthy individuals (84, 85). The effects of ANP on anxiety are presumed to be mediated through inhibition of the HPA-axis. In healthy individuals, pretreatment with ANP was able to partially block the sympathetic activation induced by a bolus injection of CRH (86). However, further investigation is needed to fully understand the interplay between ANP and the HPA-axis.
B-type natriuretic peptide, like ANP, was found to have an anxiolytic effect (87). On the other hand, CNP enhances cortisol secretion (77) and has an anxiogenic effect in rodents and humans (88, 89). However, it is worth mentioning that high doses of CNP (up to 5 μg), were used in these experiments; at low doses (100 ng), CNP reduced anxiety-like behavior in rats (87). At doses similar to those used for ANP, CNP increased the levels of anxiety-related behavior when administered i.c.v. in rats (88). This effect was abolished by a CRH antagonist, pointing toward an HPA-axis related mechanism (89). In humans, pretreatment with CNP enhanced the emotional effect of the anxiogenic agent CCK-4, and increased the release of ACTH following this treatment (90). CNP was also shown to stimulate cortisol and prolactin release (91). All these finding indicate that CNP is a potent anxiogenic substance that acts by stimulating the HPA-axis. It is therefore that CNP antagonists were considered in anti-anxiety therapy (83).
Atrial natriuretic peptide may modulate alcohol withdrawal-related anxiety. In alcohol-dependant patients, abrupt cessation of alcohol consumption is accompanied by an array of symptoms known as alcohol withdrawal (92). ANP is involved in some of the neurobehavioral aspects of alcohol withdrawal, including anxiety and alcohol craving (93). In mice, i.p. ANP administration attenuated anxiety-like behavior following alcohol withdrawal (94). Handling-induced convulsions resulting from withdrawal were reduced by i.c.v. infusion of ANP, whereas anti-ANP antibodies had the opposite effect (95). Consistently, NPR-A knockout mice showed increased stress-related alcohol consumption and aggravated withdrawal symptoms (96). In humans, acute alcohol consumption elevated plasma ANP levels in healthy individuals (97). In patients with alcohol-dependence, plasma ANP levels were lower during detoxification compared with those in non-drinking individuals (93). The lower levels correlated with alterations in promoter DNA methylation, which was significantly reduced as compared with that in healthy controls (98). On the emotional level, lower ANP levels were associated with increased anxiety and alcohol craving during withdrawal (93, 99). It was postulated that the mechanism of ANP involvement in withdrawal-related stress is mediated through the HPA-axis (100). However, although the HPA-axis stress response affects the patient’s recovery from alcohol addiction (101) as well as relapse rate (102), cortisol and ACTH levels do not correlate with those factors affected by ANP, such as alcohol craving (102) and perceived stress (99). ANP involvement in alcohol dependence is supported by recent genetic studies. A genome-wide association study (GWAS) revealed alcohol dependence to be associated with a single-nucleotide polymorphism located in gene GATA4, which encodes a transcription factor regulating ANP (103, 104). This finding was confirmed by a candidate association study, which found GATA4 to be linked to alcohol dependence at the gene level (105). This genetic variation in GATA4 was also shown to be associated with an increased relapse rate in patients (106), and greater reactivity in the amygdala to alcohol-related images, as shown by functional MRI (107). These results suggest that NP, possibly by modifying the stress response of the HPA-axis, are involved in the pathological states of anxiety disorders and alcohol dependence.
As described above, there is evidence for the involvement of NP in several brain functions. These observations open exciting new venues for research and drug development. However, many open questions remain to be clarified. In all the cases described above, it appears that NP do not regulate brain functions directly, but rather modulate other endocrine pathways, such as ANG II in BP regulation, or the HPA-axis in anxiety-related disorders. As for their neuroprotective qualities, those could be mediated via other mechanisms activated by brain injury, such as the immune system. The intricate interactions between NP and other cellular systems need to be studied in depth. On the more basic level, although the control of NP biosynthesis and release in the periphery is well established, not much is known about locally produced NP in non-cardiac tissues, the brain in particular. Information is lacking as to the specific cell types in the brain that produce NP, the factors regulating NP production and release, the modes of their elimination, and the neuronal signaling pathways that they affect. Electrophysiological studies are necessary to establish the effects of NP on neuronal excitation and channel activation. It is possible that NP do not directly regulate neuronal activity, but rather modulate it via their effect on glial cells. Studies on the NP effect on calcium release and neurotransmitters uptake in glial cells may help elucidate this point. Also, the interactions between NP and known neurotransmitters and their receptors may be of importance, and should be addressed.
Dietary sodium leads to increased natriuresis in an aldosterone-independent manner. This observation led to the postulation that an additional NH is released from the intestine in response to salt intake (108). Such a hormone was discovered in 1992 – the previously unknown endogenous ligand of the intestinal receptor GC-C, activated by bacterial enterotoxins (109), and termed guanylin. Guanylin was purified from rat jejunum, and it was shown to activate GC-C in T84 human intestinal cells (109). One year later, a second endogenous ligand of GC-C was purified from the urine and intestinal mucosa of opossums, and named uroguanylin (110). More recently, additional members of the family, such as lymphoguanylin and renoguanylin were identified (111, 112). GP are expressed as pre-pro-hormones and are proteolytically cleaved to produce the biologically active peptides (113). They share a similar structure two pairs of cysteine residues forming disulfide bonds in conserved positions that are essential for their biological function (114, 115). Like NP, guanylin and uroguanylin bind to a single-membrane-spanning receptor GC (116). GC-C has a similar GC-C domain architecture to GC-A and GC-B and elicit an increase in cellular cGMP (8), which may account for the similar function of the two peptide families. GC-C is mainly expressed in the intestine, but GC-C transcripts were also found in the adrenal gland, kidney, lung, reproductive system, lymphatic organs, and brain (117, 118).
Guanylin peptides are produced in the intestine after oral sodium intake and are secreted into the intestinal lumen (119). The resulting increase in enterocyte cGMP stimulates chloride and bicarbonate secretion and inhibits sodium absorption, causing greater secretion of fluids into the intestine (113). Guanylin and uroguanylin also exert long-term effects on the intestine by regulating intestinal cell proliferation (120). In the kidney, these peptides cause increased natriuresis, kaliuresis, and diuresis without changes in GFR or renal blood flow (121). The renal effects of GP are maintained in GC-C null mice (122), suggesting the existence of an additional receptor whose identity is yet to be discovered. Indeed, novel members of the receptor GC family were described in specific cell types in the olfactory system (123, 124).
There are few reports on the expression of GP in the brain (118). Their main effect in the CNS is likely endocrine: guanylin and uroguanylin are secreted from the gut and enter the circulation, mainly as prohormones (125, 126), and subsequently affect extra-intestinal tissues such as the kidney (127) and the brain (128). GC-C, the main intestinal receptor for GP, was found in the midbrain (129) and the hypothalamus (128).
The intestine is an important endocrine organ, secreting hormones that centrally regulate satiety and food intake (130). The intestinal hormones are vigorously studied as possible therapeutic targets for the growing public health concern regarding obesity and metabolic diseases (131). Valentino and colleagues identified uroguanylin as a novel satiety hormone and showed that food intake causes increased intestinal prouroguanylin secretion in fasting individuals and mice (128). Administration of bacterial enterotoxins (a GC-C agonist) i.v. or i.c.v. (but not orally) reduced food intake in fasting mice, and i.v. administration of anti-prouroguanylin antibodies blocked this response (128). The receptor GC-C is expressed in the mouse hypothalamus. However, uroguanylin expression was not found in this region, suggesting an endocrine rather than a paracrine mode of regulation (128). To strengthen this postulation, Valentino showed an increase in cGMP in the hypothalamus in response to treatment with prouroguanylin, suggesting that this prohormone is cleaved in the hypothalamus by an unknown enzyme to produce the active peptide form (128). Mice lacking GC-C exhibited impaired satiety that resulted in increased food intake, obesity, and metabolic syndrome. In these animals, as opposed to the normal controls, i.v. administration of bacterial enterotoxins did not reduce food intake (128). Although further validation of this new endocrine pathway is necessary, the study provides strong evidence that uroguanylin is a central mediator of food intake, and it may provide a new therapeutic target for obesity and metabolic diseases (132).
Guanylate cyclase C is expressed in dopaminergic neurons in the midbrain, and GC-C knockout was associated with behavioral changes in mice (129). Gong and collogues showed the expression of the GC-C protein in the ventral tegmental area and substantia nigra compacta in mice (129). Voltage clamp recordings from mouse dopaminergic neurons revealed that guanylin and uroguanylin significantly increased the neuronal activation evoked by metabotropic and muscarinic receptor agonists. This effect was abolished by blocking PKG signaling downstream from GC-C, and it was absent in GC-C knockout mice (129). The knockout mice exhibited increased locomotor activity, high levels of novelty seeking behavior and impulsivity. Such behavior was attenuated by low doses (1 mg/kg) of amphetamine, used to treat attention deficit hyperactivity disorder (ADHD) in humans. It is widely accepted that the dopaminergic system in the midbrain is involved in the etiology of ADHD (133). The GC-C knockout mice were described by the authors as a new animal model for ADHD, as they exhibit some of the symptoms related to this condition (129, 134). However, as the behavioral changes described could mimic several human conditions, further validation of the model is needed, and evidence of the involvement of GC-C in ADHD in humans is required. This pathway can provide new therapeutic targets for diseases involving the dopamine system, such as ADHD and schizophrenia.
Unlike NP and CS, it appears that GP are not synthesized in the brain, but rather arrive via the circulation from the intestine. However, the link between the intestine and the brain is not clear. Which brain-derived factors, if any, induce GP release from the intestine, and what signaling pathways they regulate require further investigation. The humoral or neuronal pathways that mediate the differential endocrine and paracrine effects of GP on remote organs such as the brain and kidney need to be established. Additionally, new members of the GC receptor family have been recently described in specific sensory neurons (135). There is a strong possibility that there are additional receptors which mediate GP functions in specific brain areas.
Cardiac steroids are a group of compounds that bind to and inhibit Na+, K+-ATPase. These compounds, originally discovered in plants and toad skin, have been used for centuries in Eastern and Western medicine to treat cardiac failure (136). Investigation into these substances started with the search for a missing “third factor” in the regulation of sodium homeostasis, as described in the classic work by de Wardener et al. (137). Although the interest in endogenous CS as the “third factor” has subsided with the finding of the NH, these studies paved the way for the recognition of CS in mammalian tissues and circulation. Rat brain extracts were shown to inhibit Na+, K+-ATPase activity and ouabain binding (138–140). Consequently, ouabain (141, 142), digoxin (143), and several bufadienolides (144–148) were identified in mammalian tissues, urine and plasma. CS are considered to be produced in the adrenal cortex and hypothalamus (149, 150), although their complete synthetic pathway is unknown. CS are subdivided into cardenolides, such as ouabain and digoxin, and bufadienolides, including bufalin and marinobufagenin. All CS have a steroid nucleus with a lactone ring at position C-17, and a hydroxyl group at C-14. The 5-member- and 6-member lactone rings are essential for the biological function of the cardenolides and bufadienolides, respectively (151). The only established receptor for all CS is the catalytic α subunit of the plasma membrane Na+, K+-ATPase. In addition to the inhibition of the Na+, K+-ATPase pumping function (152), the binding of CS results in the activation of signaling transduction cascades, including the Src-kinase, the MAP-kinase, and the PKC signaling pathways (153, 154).
Na+, K+-ATPase is an essential enzyme expressed in all mammalian cells. CS have widespread effects in different types of cells, including cardiac myocytes, smooth muscle cells, epithelial cells, and neurons (153, 154). CS play important roles in many physiological and pathological processes, among them sodium homeostasis (155), cardiac function (156), BP (157), cell growth (158), and behavior (159). CS form the link between dietary sodium intake and salt-sensitive hypertension (155). As described below, CS regulate BP and hypertension by their effects in the periphery and in the CNS. Given their presence in the brain and CSF, these substances were postulated to act as neurotransmitters or neuromodulators, and they were shown to be involved in psychiatric conditions such as depressive disorders (159). On the cellular level, CS were found to function in cell growth and proliferation (158) as well as in cell migration (160) and they may be associated with the development of cancer (161).
Based on their ability to inhibit ouabain binding and Na+, K+-ATPase activity, CS were identified in bovine hypothalamus (140), rat brain (138), and CSF from humans (162, 163). Immunohistochemical studies of mammalian brains revealed high concentrations of CS in the paraventricular nucleus and the supraoptic nucleus (164). Cultured rat hypothalamic neurons were shown to secrete CS in vitro (164, 165), supporting the premise that the hypothalamus is the source of endogenous brain CS. The only established CS receptor, Na+, K+-ATPase, is expressed throughout the brain. Three isoforms of this enzyme are expressed in the brain: α1, α2, and α3. They display a complex expression pattern: neurons are the principal source of the α3 isoform (166) [although some express α2, especially in the neonate (167)], whereas glial cells predominantly express α2 (168). The α1 isoform is expressed in all cell types, and considered a house keeping protein. The different subunit isoforms vary in their sensitivity to CS and may mediate differential functions of these substances.
It is widely accepted that excess dietary sodium is an extremely important factor in essential hypertension (169), although the mechanism by which sodium elevates BP is not clear. A large body of evidence links endogenous CS to the regulation of BP and hypertension. In patients with essential hypertension, plasma levels of ouabain and marinobufagenin were increased in about 40%, with a high correlation with BP (170–175). The plasma levels of these substances in hypertensive patients and in rats increased with sodium intake (176–178). Several animal models for hypertension showed increased circulating levels of CS (178–180). Furthermore, prolonged infusion of ouabain produced hypertension in animals (181–183), but had no effect in genetically modified ouabain-insensitive mice (183, 184). In transgenic mice, a greater natriuretic response to sodium loading was demonstrated in animals expressing a highly CS-sensitive Na+, K+-ATPase α1 subunit (185). Studies on mice carrying mutations in the gene encoding α2 showed that ouabain-induced elevation of BP in rodents was mediated via this isoform: reduction of the expression level of α2 was associated with increased BP (186). In contrast, animals overexpressing α2 were hypotensive (187). Treatment of hypertensive rats with anti-digoxin antibodies (185, 188) or anti-marinobufogenin antibodies (178) administered to rats on a high sodium intake, resulted in a marked reduction in BP. Endogenous ouabain was put forward as a putative target for the treatment of hypertension; the ouabain inhibitor rostafuroxin showed promising results in hypertensive rats (189). Studies by Leenen and colleagues indicated that CS involvement in BP regulation is partially mediated by their effect in the CNS. The first indication of brain involvement came from experiments in SHR, in which adrenalectomy did not prevent the increase in CS levels following high sodium intake (177). Lesions in the most anteroventral part of the third ventricle (AV3V) showed that this region is essential in mediating the pressor effects of increased CSF sodium concentration via endogenous ouabain (190, 191). The effects of both acute and prolonged ouabain infusion in sodium-loaded rodents were abolished by administration of ANG II type 1 receptor blockers such as losartan (192, 193), as well as in transgenic rats with reduced brain renin-angiotensin pathway activity (194). These results pointed to the involvement of this pathway in the effect of ouabain. All of these finding led to a unifying hypothesis regarding the role of CS in sodium-induced hypertension: sodium loading increases the levels of ouabain in salt-sensitive individuals (195, 196). In addition to induction of vasoconstriction in the periphery, ouabain also acts in the brain, where it activates the renin-angiotensin pathway, causing sympathetic activation, vasoconstriction and consequently, an elevation in BP.
Mood disorders include major depression and dysthymia, characterized by depressive episodes, and bipolar disorder (BD) marked by both depressive and manic episodes. These conditions pose a growing public health concern in the Western world. The etiology of these diseases is not completely understood. Early reports of the psychiatric effects of CS came from doctors describing a syndrome termed “foxglove frenzy” or “digitalis delirium” in patients with digitalis intoxication (197). More recently, a comprehensive hypothesis was put forward, linking brain CS levels and Na+, K+-ATPase activity with BD (198, 199). BD has consistently been associated with abnormalities in Na+, K+-ATPase activity in erythrocytes (200, 201). A significant mood-related decrease in the enzyme’s activity was found in manic BD patients (202). Furthermore, Na+, K+-ATPase density was significantly lower in BD patients than in major depressed and schizophrenic patients (159). The plasma levels of endogenous CS were found to be significantly decreased in manic individuals as compared with those in normal controls (203, 204). Conversely, the levels of these compounds were higher in the parietal cortex of BD patients (159). More recently, it was found that there is a reduction in brain Na+, K+-ATPase α1 isoform expression in mice treated with the mood stabilizer lithium (205). An allelic association between BD and a Na+, K+-ATPase α subunit gene (ATP1A3) was reported (206). We have demonstrated the prominent linkage to BD of six single-nucleotide polymorphisms (SNPs) in the three genes of the Na+, K+-ATPase α isoforms. Haplotype analysis of the α2 isoform showed the significant association of two loci haplotypes with BD (207). A genetic knockdown of the neuron-specific Na+, K+-ATPase α3 isoform induced manic-like behavior in mice (208). Numerous studies have demonstrated that i.c.v. injection of ouabain induces hyperactive behavior in rats (159, 209), which could be ameliorated by administration of mood stabilizing drugs such as lithium (210). Reduction of the endogenous brain CS level by i.c.v. injection of anti-ouabain antibodies had anti-depressive effects in rats (159, 211). This was reflected by significant changes in catecholamine metabolism in the hippocampus and ventral tegmentum, two regions known to be associated with mood disorders (211). The molecular pathway underlying the CS behavioral effect is unknown. Ouabain injected i.c.v. elicited the activation of the ERK and Akt signaling pathways in the brain (212, 213), which are known to be activated via Na+, K+-ATPase. Other effects of ouabain include a reduction in brain-derived neurotrophic factor (BDNF) (214), activation of mammalian target of rapamycin (mTOR) signaling (213) and an increased level of oxidative stress (215). These findings strongly link the Na+, K+-ATPase and CS system to the etiology of depressive disorders, and BD in particular, and suggest their potential application in future drug development.
Despite the identification of cardenolides and bufadienolides in mammalian tissue in many independent studies (see Cardiac steroids), some still question the validity of these findings. Recently, it was claimed that ouabain, the most studied cardenolide, could not be detected in human plasma (216). This issue must be clarified. An additional major missing piece of information for the establishment of CS as neurotransmitters or neuromodulators is the elucidation of their biosynthetic pathway in the adrenal gland and brain. Although the available literature supports the notion that these steroids are synthesized in mammals, the key enzymes involved have not been identified. This issue was recently reviewed in Ref. (217). Several CS were identified in the human body. It was postulated that the different α isoforms of the Na+, K+-ATPase serve as receptors for the different CS. Which of the CS are involved in brain functions, and which isoform combinations they activate are topics for future research.
Mutual interactions exist between CS and NP in the periphery and in the brain. Ouabain and digoxin were shown to cause increased ANP expression and secretion in rat and rabbit atria (218–221), and in anesthetized dogs (222). In patients with congestive heart failure, i.v. administration of digitalis increased the plasma levels of ANP and BNP (223). Indeed, ANP regulates the secretion of CS in the brain (224–226). ANP decreased the release of CS from rat brain extract when added to the tissue, or when administered i.v. to the animals prior to their sacrifice (224). On the other hand, another study showed that i.c.v. injection of synthetic ANP increased blood CS levels, whereas i.v. administration or incubation with this peptide had no effect (225). The effect of ANP on CS release was abolished by lesions in the AV3V region (226), the area in the hypothalamus that is thought to mediate CS central regulation of BP (191). In addition to secretion regulation, NP and CS interact at the functional level. ANP differentially modulates the effect of marinobufogenin in the rat heart and kidney (227). Ouabain was shown to antagonize the effect of ANP on vasorelaxation in rabbit aorta and in dogs (228, 229), whereas ANP abolished an ouabain-induced increase in aldosteron secretion (230). Administration of anti-ouabain antibodies caused increased sensitivity to ANP-induced vasodilation in rat aorta (231). In rat heart muscle preparations, ANP was shown to attenuate several effects of ouabain, including ouabain-induced increase in contractility, Na+, K+-ATPase and ERK phosphorylation (232). ANP also interacts with GP. Santos-Neto and colleagues showed synergism between ANP, guanylin, and uroguanylin in the kidney (233). They demonstrated in an isolated perfused rat kidney that pretreatment with ANP significantly enhanced the natriuretic, kaliuretic, and chloriuretic responses to low doses of guanylin and uroguanylin (233). Low doses of ANP enhanced GP induced diuresis, and vice versa (233). Since GP and NP activate GC receptors, their interaction may be mediated through the shared second messenger, cGMP (6, 116, 234). These initial studies suggest the physiological crosstalk between ANP and CS, particularly in the cardiovascular system and in the brain, and between ANP and GP in the kidney. More studies are needed to deepen our understanding of the nature of these interactions, which may be of significance in the regulation of peripheral and central functions of the NH.
This review summarizes the available data implicating NH in brain function. There is a vast amount of data supporting the assessment that the three families of NH, NP, Guanylins, and endogenous CS are present in the brain and participate in high brain functions (see Figure 1). These include central regulation of BP, satiety, neuroprotection, and behavior. In depth research of these effects will not only increase our thorough understanding of brain function but may also lead to new treatments for brain-related diseases.
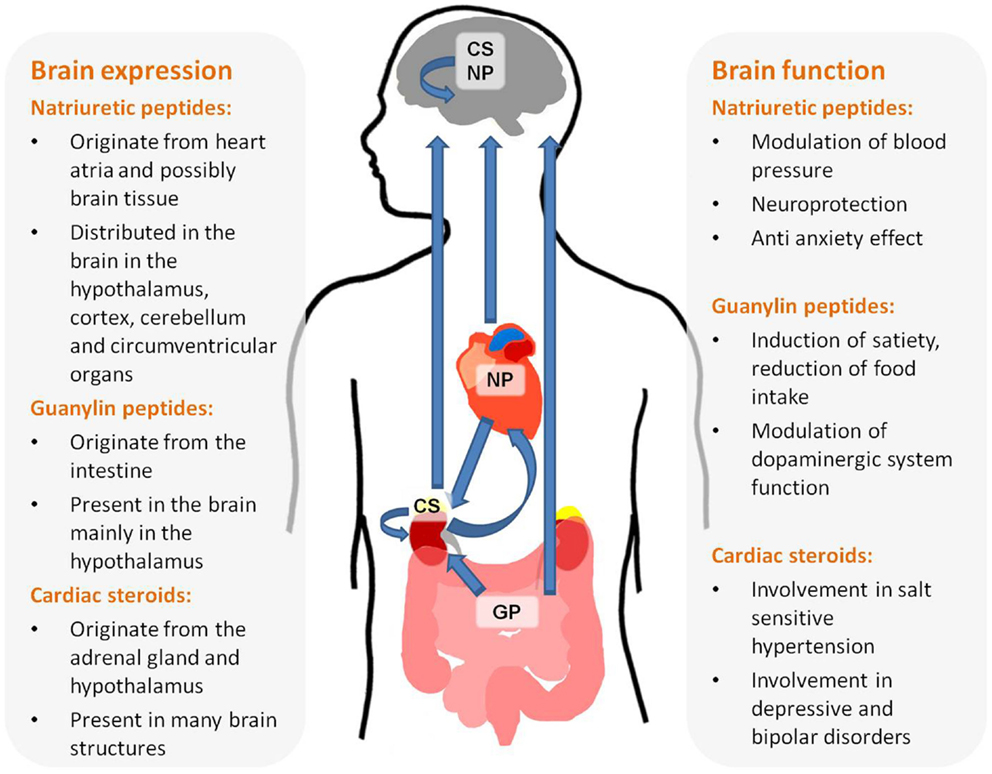
Figure 1. Origin, brain distribution, and function of natriuretic hormones.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. de Bold AJ, Borenstein HB, Veress AT, Sonnenberg H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci (1981) 28:89–94. doi: 10.1016/0024-3205(81)90370-2
2. Sudoh T, Kangawa K, Minamino N, Matsuo H. A new natriuretic peptide in porcine brain. Nature (1988) 332:78–81. doi:10.1038/332078a0
3. Sudoh T, Minamino N, Kangawa K, Matsuo H. C-type natriuretic peptide (CNP): a new member of natriuretic peptide family identified in porcine brain. Biochem Biophys Res Commun (1990) 168:863–70. doi:10.1016/0006-291X(90)92401-K
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Edwards BS, Zimmerman RS, Schwab TR, Heublein DM, Burnett JC Jr. Atrial stretch, not pressure, is the principal determinant controlling the acute release of atrial natriuretic factor. Circ Res (1988) 62:191–5. doi:10.1161/01.RES.62.2.191
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Thuerauf DJ, Hanford DS, Glembotski CC. Regulation of rat brain natriuretic peptide transcription. A potential role for GATA-related transcription factors in myocardial cell gene expression. J Biol Chem (1994) 269:17772–5.
6. Suga S, Nakao K, Hosoda K, Mukoyama M, Ogawa Y, Shirakami G, et al. Receptor selectivity of natriuretic peptide family, atrial natriuretic peptide, brain natriuretic peptide, and C-type natriuretic peptide. Endocrinology (1992) 130:229–39. doi:10.1210/en.130.1.229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Koller KJ, Lowe DG, Bennett GL, Minamino N, Kangawa K, Matsuo H, et al. Selective activation of the B natriuretic peptide receptor by C-type natriuretic peptide (CNP). Science (1991) 252:120–3. doi:10.1126/science.1672777
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Potter LR, Yoder AR, Flora DR, Antos LK, Dickey DM. Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. Handb Exp Pharmacol (2009) 191:341–66. doi:10.1007/978-3-540-68964-5_15
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Tremblay J, Desjardins R, Hum D, Gutkowska J, Hamet P. Biochemistry and physiology of the natriuretic peptide receptor guanylyl cyclases. Mol Cell Biochem (2002) 230:31–47. doi:10.1023/A:1014260204524
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Matsukawa N, Grzesik WJ, Takahashi N, Pandey KN, Pang S, Yamauchi M, et al. The natriuretic peptide clearance receptor locally modulates the physiological effects of the natriuretic peptide system. Proc Natl Acad Sci U S A (1999) 96:7403–8. doi:10.1073/pnas.96.13.7403
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Marin-Grez M, Fleming JT, Steinhausen M. Atrial natriuretic peptide causes pre-glomerular vasodilatation and post-glomerular vasoconstriction in rat kidney. Nature (1986) 324:473–6. doi:10.1038/324473a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Richards AM, Mcdonald D, Fitzpatrick MA, Nicholls MG, Espiner EA, Ikram H, et al. Atrial natriuretic hormone has biological effects in man at physiological plasma concentrations. J Clin Endocrinol Metab (1988) 67:1134–9. doi:10.1210/jcem-67-6-1134
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Currie MG, Geller DM, Cole BR, Boylan JG, Yusheng W, Holmberg SW, et al. Bioactive cardiac substances: potent vasorelaxant activity in mammalian atria. Science (1983) 221:71–3. doi:10.1126/science.6857267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Baron DA, Lofton CE, Newman WH, Currie MG. Atriopeptin inhibition of thrombin-mediated changes in the morphology and permeability of endothelial monolayers. Proc Natl Acad Sci U S A (1989) 86:3394–8. doi:10.1073/pnas.86.9.3394
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. John SW, Krege JH, Oliver PM, Hagaman JR, Hodgin JB, Pang SC, et al. Genetic decreases in atrial natriuretic peptide and salt-sensitive hypertension. Science (1995) 267:679–81. doi:10.1126/science.7839143
16. Holtwick R, Van Eickels M, Skryabin BV, Baba HA, Bubikat A, Begrow F, et al. Pressure-independent cardiac hypertrophy in mice with cardiomyocyte-restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase-A. J Clin Invest (2003) 111:1399–407. doi:10.1172/JCI17061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Tamura N, Ogawa Y, Chusho H, Nakamura K, Nakao K, Suda M, et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc Natl Acad Sci U S A (2000) 97:4239–44. doi:10.1073/pnas.070371497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Cao L, Gardner DG. Natriuretic peptides inhibit DNA synthesis in cardiac fibroblasts. Hypertension (1995) 25:227–34. doi:10.1161/01.HYP.25.2.227
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Nagaya N, Nishikimi T, Uematsu M, Satoh T, Kyotani S, Sakamaki F, et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation (2000) 102:865–70. doi:10.1161/01.CIR.102.8.865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Schulz S. C-type natriuretic peptide and guanylyl cyclase B receptor. Peptides (2005) 26:1024–34. doi:10.1016/j.peptides.2004.08.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Drewett JG, Fendly BM, Garbers DL, Lowe DG. Natriuretic peptide receptor-B (guanylyl cyclase-B) mediates C-type natriuretic peptide relaxation of precontracted rat aorta. J Biol Chem (1995) 270:4668–74. doi:10.1074/jbc.270.9.4668
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Matsuo H, Furuya M. C-type natriuretic protein inhibits intimal thickening after vascular injury. Ann N Y Acad Sci (1997) 811:45–7. doi:10.1111/j.1749-6632.1997.tb51987.x
23. Hamad AM, Clayton A, Islam B, Knox AJ. Guanylyl cyclases, nitric oxide, natriuretic peptides, and airway smooth muscle function. Am J Physiol Lung Cell Mol Physiol (2003) 285:L973–83. doi:10.1152/ajplung.00033.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Morita R, Ukyo N, Furuya M, Uchiyama T, Hori T. Atrial natriuretic peptide polarizes human dendritic cells toward a Th2-promoting phenotype through its receptor guanylyl cyclase-coupled receptor A. J Immunol (2003) 170:5869–75. doi:10.4049/jimmunol.170.12.5869
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Sengenes C, Berlan M, De Glisezinski I, Lafontan M, Galitzky J. Natriuretic peptides: a new lipolytic pathway in human adipocytes. FASEB J (2000) 14:1345–51. doi:10.1096/fj.14.10.1345
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Chusho H, Tamura N, Ogawa Y, Yasoda A, Suda M, Miyazawa T, et al. Dwarfism and early death in mice lacking C-type natriuretic peptide. Proc Natl Acad Sci U S A (2001) 98:4016–21. doi:10.1073/pnas.071389098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Kaneko T, Shirakami G, Nakao K, Nagata I, Nakagawa O, Hama N, et al. C-type natriuretic peptide (CNP) is the major natriuretic peptide in human cerebrospinal fluid. Brain Res (1993) 612:104–9. doi:10.1016/0006-8993(93)91649-D
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Komatsu Y, Nakao K, Suga S, Ogawa Y, Mukoyama M, Arai H, et al. C-type natriuretic peptide (CNP) in rats and humans. Endocrinology (1991) 129:1104–6. doi:10.1210/endo-129-2-1104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Herman JP, Dolgas CM, Rucker D, Langub MC Jr. Localization of natriuretic peptide-activated guanylate cyclase mRNAs in the rat brain. J Comp Neurol (1996) 369:165–87. doi:10.1002/(SICI)1096-9861(19960527)369:2<165::AID-CNE1>3.0.CO;2-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Tanaka I, Misono KS, Inagami T. Atrial natriuretic factor in rat hypothalamus, atria and plasma: determination by specific radioimmunoassay. Biochem Biophys Res Commun (1984) 124:663–8. doi:10.1016/0006-291X(84)91606-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Kawata M, Nakao K, Morii N, Kiso Y, Yamashita H, Imura H, et al. Atrial natriuretic polypeptide: topographical distribution in the rat brain by radioimmunoassay and immunohistochemistry. Neuroscience (1985) 16:521–46. doi:10.1016/0306-4522(85)90190-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Marei HE. Fine structural and immunohistochemical localization of cardiac hormones (ANP) in the right atrium and hypothalamus of the white rat. Eur J Morphol (2002) 40:37–41. doi:10.1076/ejom.40.1.0037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Mckenzie JC, Berman NE, Thomas CR, Young JK, Compton LY, Cothran LN, et al. Atrial natriuretic peptide-like (ANP-LIR) and ANP prohormone immunoreactive astrocytes and neurons of human cerebral cortex. Glia (1994) 12:228–43. doi:10.1002/glia.440120308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Mckenzie JC, Juan YW, Thomas CR, Berman NE, Klein RM. Atrial natriuretic peptide-like immunoreactivity in neurons and astrocytes of human cerebellum and inferior olivary complex. J Histochem Cytochem (2001) 49:1453–67. doi:10.1177/002215540104901113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Standaert DG, Needleman P, Saper CB. Organization of atriopeptin-like immunoreactive neurons in the central nervous system of the rat. J Comp Neurol (1986) 253:315–41. doi:10.1002/cne.902530304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Brown J, Czarnecki A. Autoradiographic localization of atrial and brain natriuretic peptide receptors in rat brain. Am J Physiol (1990) 258:R57–63.
37. Ryan MC, Gundlach AL. Anatomical localisation of preproatrial natriuretic peptide mRNA in the rat brain by in situ hybridisation histochemistry: novel identification in olfactory regions. J Comp Neurol (1995) 356:168–82. doi:10.1002/cne.903560204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Abdelalim EM, Takada T, Torii R, Tooyama I. Molecular cloning of BNP from heart and its immunohistochemical localization in the hypothalamus of monkey. Peptides (2006) 27:1886–93. doi:10.1016/j.peptides.2006.01.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Langub MC Jr, Watson RE Jr, Herman JP. Distribution of natriuretic peptide precursor mRNAs in the rat brain. J Comp Neurol (1995) 356:183–99. doi:10.1002/cne.903560205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Ganong WF. Circumventricular organs: definition and role in the regulation of endocrine and autonomic function. Clin Exp Pharmacol Physiol (2000) 27:422–7. doi:10.1046/j.1440-1681.2000.03259.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. de Vente J, Bol JG, Steinbusch HW. cGMP-producing, atrial natriuretic factor-responding cells in the rat brain. Eur J Neurosci (1989) 1:436–60. doi:10.1111/j.1460-9568.1989.tb00351.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Smith PM, Ferguson AV. Circulating signals as critical regulators of autonomic state – central roles for the subfornical organ. Am J Physiol Regul Integr Comp Physiol (2010) 299:R405–15. doi:10.1152/ajpregu.00103.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Saavedra JM, Kurihara M. Autoradiography of atrial natriuretic peptide (ANP) receptors in the rat brain. Can J Physiol Pharmacol (1991) 69:1567–75. doi:10.1139/y91-233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Levin ER, Weber MA, Mills S. Atrial natriuretic factor-induced vasodepression occurs through central nervous system. Am J Physiol (1988) 255:H616–22.
45. Schultz HD, Steele MK, Gardner DG. Central administration of atrial peptide decreases sympathetic outflow in rats. Am J Physiol (1990) 258:R1250–6.
46. Steele MK, Gardner DG, Xie PL, Schultz HD. Interactions between ANP and ANG II in regulating blood pressure and sympathetic outflow. Am J Physiol (1991) 260:R1145–51.
47. Charles CJ, Richards AM, Espiner EA. Central C-type natriuretic peptide but not atrial natriuretic factor lowers blood pressure and adrenocortical secretion in normal conscious sheep. Endocrinology (1992) 131:1721–6. doi:10.1210/endo.131.4.1396317
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Itoh H, Nakao K, Katsuura G, Morii N, Shiono S, Sakamoto M, et al. Centrally infused atrial natriuretic polypeptide attenuates exaggerated salt appetite in spontaneously hypertensive rats. Circ Res (1986) 59:342–7. doi:10.1161/01.RES.59.3.342
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Lee J, Malvin RL, Claybaugh JR, Huang BS. Atrial natriuretic factor inhibits vasopressin secretion in conscious sheep. Proc Soc Exp Biol Med (1987) 185:272–6. doi:10.3181/00379727-185-42544
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Shoji M, Kimura T, Matsui K, Ota K, Iitake K, Inoue M, et al. Effects of centrally administered atrial natriuretic peptide on renal functions. Acta Endocrinol (Copenh) (1987) 115:433–40.
51. Al-Barazanji KA, Balment RJ. The renal and vascular effects of central angiotensin II and atrial natriuretic factor in the anaesthetized rat. J Physiol (1990) 423:485–93.
52. Stepniakowski K, Budzikowski A, Lon S, Szczepanska-Sadowska E. Central ANP attenuates pressor responses to central AVP in WKY and SHR. Brain Res Bull (1991) 27:247–9. doi:10.1016/0361-9230(91)90076-V
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Yamada T, Nakao K, Itoh H, Shirakami G, Kangawa K, Minamino N, et al. Intracerebroventricular injection of brain natriuretic peptide inhibits vasopressin secretion in conscious rats. Neurosci Lett (1988) 95:223–8. doi:10.1016/0304-3940(88)90661-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Samson WK, Aguila MC, Martinovic J, Antunes-Rodrigues J, Norris M. Hypothalamic action of atrial natriuretic factor to inhibit vasopressin secretion. Peptides (1987) 8:449–54. doi:10.1016/0196-9781(87)90008-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Antunes-Rodrigues J, Mccann SM, Rogers LC, Samson WK. Atrial natriuretic factor inhibits dehydration- and angiotensin II-induced water intake in the conscious, unrestrained rat. Proc Natl Acad Sci U S A (1985) 82:8720–3. doi:10.1073/pnas.82.24.8720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Leao AAP. Spreading depression of activity in the cerebral cortex. J Neurophysiol (1944) 7:359–90.
57. Kawahara N, Ruetzler CA, Klatzo I. Protective effect of spreading depression against neuronal damage following cardiac arrest cerebral ischaemia. Neurol Res (1995) 17:9–16.
58. Wiggins AK, Shen PJ, Gundlach AL. Atrial natriuretic peptide expression is increased in rat cerebral cortex following spreading depression: possible contribution to sd-induced neuroprotection. Neuroscience (2003) 118:715–26. doi:10.1016/S0306-4522(03)00006-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Read SJ, Hirst WD, Upton N, Parsons AA. Cortical spreading depression produces increased cGMP levels in cortex and brain stem that is inhibited by tonabersat (SB-220453) but not sumatriptan. Brain Res (2001) 891:69–77. doi:10.1016/S0006-8993(00)03191-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Fiscus RR, Tu AW, Chew SB. Natriuretic peptides inhibit apoptosis and prolong the survival of serum-deprived PC12 cells. Neuroreport (2001) 12:185–9. doi:10.1097/00001756-200102120-00003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Kuribayashi K, Kitaoka Y, Kumai T, Munemasa Y, Isenoumi K, Motoki M, et al. Neuroprotective effect of atrial natriuretic peptide against NMDA-induced neurotoxicity in the rat retina. Brain Res (2006) 1071:34–41. doi:10.1016/j.brainres.2005.11.068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Nogami M, Shiga J, Takatsu A, Endo N, Ishiyama I. Immunohistochemistry of atrial natriuretic peptide in brain infarction. Histochem J (2001) 33:87–90. doi:10.1023/A:1017996113871
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Pifarre P, Prado J, Giralt M, Molinero A, Hidalgo J, Garcia A. Cyclic GMP phosphodiesterase inhibition alters the glial inflammatory response, reduces oxidative stress and cell death and increases angiogenesis following focal brain injury. J Neurochem (2010) 112:807–17. doi:10.1111/j.1471-4159.2009.06518.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Naruse S, Aoki Y, Takei R, Horikawa Y, Ueda S. Effects of atrial natriuretic peptide on ischemic brain edema in rats evaluated by proton magnetic resonance method. Stroke (1991) 22:61–5. doi:10.1161/01.STR.22.1.61
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Rosenberg GA, Estrada EY. Atrial natriuretic peptide blocks hemorrhagic brain edema after 4-hour delay in rats. Stroke (1995) 26:874–7. doi:10.1161/01.STR.26.5.874
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. James ML, Wang H, Venkatraman T, Song P, Lascola CD, Laskowitz DT. Brain natriuretic peptide improves long-term functional recovery after acute CNS injury in mice. J Neurotrauma (2010) 27:217–28. doi:10.1089/neu.2009.1022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Kirchhoff C, Stegmaier J, Bogner V, Buhmann S, Mussack T, Kreimeier U, et al. Intrathecal and systemic concentration of NT-proBNP in patients with severe traumatic brain injury. J Neurotrauma (2006) 23:943–9. doi:10.1089/neu.2006.23.943
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Powner DJ, Hergenroeder GW, Awili M, Atik MA, Robertson C. Hyponatremia and comparison of NT-pro-BNP concentrations in blood samples from jugular bulb and arterial sites after traumatic brain injury in adults: a pilot study. Neurocrit Care (2007) 7:119–23. doi:10.1007/s12028-007-0079-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Koenig MA, Puttgen HA, Prabhakaran V, Reich D, Stevens RD. B-type natriuretic peptide as a marker for heart failure in patients with acute stroke. Intensive Care Med (2007) 33:1587–93. doi:10.1007/s00134-007-0704-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Mcgirt MJ, Blessing R, Nimjee SM, Friedman AH, Alexander MJ, Laskowitz DT, et al. Correlation of serum brain natriuretic peptide with hyponatremia and delayed ischemic neurological deficits after subarachnoid hemorrhage. Neurosurgery (2004) 54:1369–73. doi:10.1227/01.NEU.0000125016.37332.50
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. James ML, Blessing R, Phillips-Bute BG, Bennett E, Laskowitz DT. S100B and brain natriuretic peptide predict functional neurological outcome after intracerebral haemorrhage. Biomarkers (2009) 14:388–94. doi:10.1080/13547500903015784
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Sviri GE, Soustiel JF, Zaaroor M. Alteration in brain natriuretic peptide (BNP) plasma concentration following severe traumatic brain injury. Acta Neurochir (Wien) (2006) 148:529–33. doi:10.1007/s00701-005-0666-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Montaner J, Garcia-Berrocoso T, Mendioroz M, Palacios M, Perea-Gainza M, Delgado P, et al. Brain natriuretic peptide is associated with worsening and mortality in acute stroke patients but adds no prognostic value to clinical predictors of outcome. Cerebrovasc Dis (2012) 34:240–5. doi:10.1159/000341858
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. De Vito P. Atrial natriuretic peptide: an old hormone or a new cytokine? Peptides (2014) 58:108–16. doi:10.1016/j.peptides.2014.06.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Fink G, Dow RC, Casley D, Johnston CI, Lim AT, Copolov DL, et al. Atrial natriuretic peptide is a physiological inhibitor of ACTH release: evidence from immunoneutralization in vivo. J Endocrinol (1991) 131:R9–12. doi:10.1677/joe.0.131R009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Vesely DL, San Miguel GI, Hassan I, Schocken DD. Atrial natriuretic hormone, vessel dilator, long-acting natriuretic hormone, and kaliuretic hormone decrease the circulating concentrations of CRH, corticotropin, and cortisol. J Clin Endocrinol Metab (2001) 86:4244–9. doi:10.1210/jcem.86.9.7829
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Charles CJ, Espiner EA, Richards AM, Donald RA. Central C-type natriuretic peptide augments the hormone response to hemorrhage in conscious sheep. Peptides (1995) 16:129–32. doi:10.1016/0196-9781(94)00160-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Strohle A, Jahn H, Montkowski A, Liebsch G, Boll E, Landgraf R, et al. Central and peripheral administration of atriopeptin is anxiolytic in rats. Neuroendocrinology (1997) 65:210–5. doi:10.1159/000127274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Kellner M, Knaudt K, Jahn H, Holsboer F, Wiedemann K. Atrial natriuretic hormone in lactate-induced panic attacks: mode of release and endocrine and pathophysiological consequences. J Psychiatr Res (1998) 32:37–48. doi:10.1016/S0022-3956(97)00034-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Levinson DF, Zubenko GS, Crowe RR, Depaulo RJ, Scheftner WS, Weissman MM, et al. Genetics of recurrent early-onset depression (GenRED): design and preliminary clinical characteristics of a repository sample for genetic linkage studies. Am J Med Genet B Neuropsychiatr Genet (2003) 119B:118–30. doi:10.1002/ajmg.b.20009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Herrmann-Lingen C, Binder L, Klinge M, Sander J, Schenker W, Beyermann B, et al. High plasma levels of N-terminal pro-atrial natriuretic peptide associated with low anxiety in severe heart failure. Psychosom Med (2003) 65:517–22. doi:10.1097/01.PSY.0000073870.93003.C4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Kellner M, Herzog L, Yassouridis A, Holsboer F, Wiedemann K. Possible role of atrial natriuretic hormone in pituitary-adrenocortical unresponsiveness in lactate-induced panic. Am J Psychiatry (1995) 152:1365–7.
83. Kellner M, Jahn H, Wiedemann K. Natriuretic peptides and panic disorder: therapeutic prospects. Expert Rev Neurother (2003) 3:381–6. doi:10.1586/14737175.3.3.381
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Strohle A, Kellner M, Holsboer F, Wiedemann K. Anxiolytic activity of atrial natriuretic peptide in patients with panic disorder. Am J Psychiatry (2001) 158:1514–6. doi:10.1176/appi.ajp.158.9.1514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Wiedemann K, Jahn H, Yassouridis A, Kellner M. Anxiolyticlike effects of atrial natriuretic peptide on cholecystokinin tetrapeptide-induced panic attacks: preliminary findings. Arch Gen Psychiatry (2001) 58:371–7. doi:10.1001/archpsyc.58.4.371
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Arlt J, Jahn H, Kellner M, Strohle A, Yassouridis A, Wiedemann K. Modulation of sympathetic activity by corticotropin-releasing hormone and atrial natriuretic peptide. Neuropeptides (2003) 37:362–8. doi:10.1016/j.npep.2003.09.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Biro E, Toth G, Telegdy G. Effect of receptor blockers on brain natriuretic peptide and C-type natriuretic peptide caused anxiolytic state in rats. Neuropeptides (1996) 30:59–65. doi:10.1016/S0143-4179(96)90056-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Montkowski A, Jahn H, Strohle A, Poettig M, Holsboer F, Wiedemann K. C-type natriuretic peptide exerts effects opposing those of atrial natriuretic peptide on anxiety-related behaviour in rats. Brain Res (1998) 792:358–60. doi:10.1016/S0006-8993(98)00274-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Jahn H, Montkowski A, Knaudt K, Strohle A, Kiefer F, Schick M, et al. Alpha-helical-corticotropin-releasing hormone reverses anxiogenic effects of C-type natriuretic peptide in rats. Brain Res (2001) 893:21–8. doi:10.1016/S0006-8993(00)03275-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Kellner M, Yassouridis A, Hua Y, Wendrich M, Jahn H, Wiedemann K. Intravenous C-type natriuretic peptide augments behavioral and endocrine effects of cholecystokinin tetrapeptide in healthy men. J Psychiatr Res (2002) 36:1–6. doi:10.1016/S0022-3956(01)00042-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Kellner M, Diehl I, Knaudt K, Schule C, Jahn H, Wiedemann K. C-type natriuretic peptide exerts stimulatory effects on the corticotropin-releasing hormone-induced secretion of hormones in normal man. Eur J Endocrinol (1997) 136:388–93. doi:10.1530/eje.0.1360388
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Noble JM, Weimer LH. Neurologic complications of alcoholism. Continuum (Minneap Minn) (2014) 20:624–41. doi:10.1212/01.CON.0000450970.99322.84
93. Kiefer F, Andersohn F, Jahn H, Wolf K, Raedler TJ, Wiedemann K. Involvement of plasma atrial natriuretic peptide in protracted alcohol withdrawal. Acta Psychiatr Scand (2002) 105:65–70. doi:10.1034/j.1600-0447.2002.0_011.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. von der Goltz C, Jahn H, Mutschler J, Wiedemann K, Kiefer F. Intraperitoneal atrial natriuretic peptide attenuates anxiety-related behaviour during alcohol withdrawal in mice. Pharmacopsychiatry (2014) 47:97–100. doi:10.1055/s-0034-1372645
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Kovacs GL. Alpha-atrial natriuretic peptide attenuates ethanol withdrawal symptoms. Eur J Pharmacol (1993) 238:417–9. doi:10.1016/0014-2999(93)90878-L
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Mutschler J, Bilbao A, von der Goltz C, Demiralay C, Jahn H, Wiedemann K, et al. Augmented stress-induced alcohol drinking and withdrawal in mice lacking functional natriuretic peptide-A receptors. Alcohol Alcohol (2010) 45:13–6. doi:10.1093/alcalc/agp065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Gianoulakis C, Guillaume P, Thavundayil J, Gutkowska J. Increased plasma atrial natriuretic peptide after ingestion of low doses of ethanol in humans. Alcohol Clin Exp Res (1997) 21:162–70. doi:10.1111/j.1530-0277.1997.tb03744.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Glahn A, Riera Knorrenschild R, Rhein M, Haschemi Nassab M, Groschl M, Heberlein A, et al. Alcohol-induced changes in methylation status of individual CpG sites, and serum levels of vasopressin and atrial natriuretic peptide in alcohol-dependent patients during detoxification treatment. Eur Addict Res (2014) 20:143–50. doi:10.1159/000357473
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Koopmann A, Lemenager T, Wolf ND, Reinhard I, Hermann D, Koch J, et al. The impact of atrial natriuretic peptide on anxiety, stress and craving in patients with alcohol dependence. Alcohol Alcohol (2014) 49:282–6. doi:10.1093/alcalc/agt160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Kiefer F, Jahn H, Schick M, Wiedemann K. Alcohol self-administration, craving and HPA-axis activity: an intriguing relationship. Psychopharmacology (Berl) (2002) 164:239–40. doi:10.1007/s00213-002-1255-3
101. Adinoff B, Junghanns K, Kiefer F, Krishnan-Sarin S. Suppression of the HPA axis stress-response: implications for relapse. Alcohol Clin Exp Res (2005) 29:1351–5. doi:10.1097/01.ALC.0000176356.97620.84
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Kiefer F, Jahn H, Tarnaske T, Helwig H, Briken P, Holzbach R, et al. Comparing and combining naltrexone and acamprosate in relapse prevention of alcoholism: a double-blind, placebo-controlled study. Arch Gen Psychiatry (2003) 60:92–9. doi:10.1001/archpsyc.60.1.92
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Treutlein J, Cichon S, Ridinger M, Wodarz N, Soyka M, Zill P, et al. Genome-wide association study of alcohol dependence. Arch Gen Psychiatry (2009) 66:773–84. doi:10.1001/archgenpsychiatry.2009.83
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Edenberg HJ, Koller DL, Xuei X, Wetherill L, Mcclintick JN, Almasy L, et al. Genome-wide association study of alcohol dependence implicates a region on chromosome 11. Alcohol Clin Exp Res (2010) 34:840–52. doi:10.1111/j.1530-0277.2010.01156.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Karpyak VM, Winham SJ, Biernacka JM, Cunningham JM, Lewis KA, Geske JR, et al. Association of GATA4 sequence variation with alcohol dependence. Addict Biol (2014) 19:312–5. doi:10.1111/j.1369-1600.2012.00482.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Kiefer F, Witt SH, Frank J, Richter A, Treutlein J, Lemenager T, et al. Involvement of the atrial natriuretic peptide transcription factor GATA4 in alcohol dependence, relapse risk and treatment response to acamprosate. Pharmacogenomics J (2011) 11:368–74. doi:10.1038/tpj.2010.51
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Jorde A, Bach P, Witt SH, Becker K, Reinhard I, Vollstadt-Klein S, et al. Genetic variation in the atrial natriuretic peptide transcription factor GATA4 modulates amygdala responsiveness in alcohol dependence. Biol Psychiatry (2014) 75:790–7. doi:10.1016/j.biopsych.2013.10.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Carey RM. Evidence for a splanchnic sodium input monitor regulating renal sodium excretion in man. Lack of dependence upon aldosterone. Circ Res (1978) 43:19–23. doi:10.1161/01.RES.43.1.19
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Currie MG, Fok KF, Kato J, Moore RJ, Hamra FK, Duffin KL, et al. Guanylin: an endogenous activator of intestinal guanylate cyclase. Proc Natl Acad Sci U S A (1992) 89:947–51. doi:10.1073/pnas.89.3.947
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Hamra FK, Forte LR, Eber SL, Pidhorodeckyj NV, Krause WJ, Freeman RH, et al. Uroguanylin: structure and activity of a second endogenous peptide that stimulates intestinal guanylate cyclase. Proc Natl Acad Sci U S A (1993) 90:10464–8. doi:10.1073/pnas.90.22.10464
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Forte LR, Eber SL, Fan X, London RM, Wang Y, Rowland LM, et al. Lymphoguanylin: cloning and characterization of a unique member of the guanylin peptide family. Endocrinology (1999) 140:1800–6. doi:10.1210/endo.140.4.6630
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Yuge S, Inoue K, Hyodo S, Takei Y. A novel guanylin family (guanylin, uroguanylin, and renoguanylin) in eels: possible osmoregulatory hormones in intestine and kidney. J Biol Chem (2003) 278:22726–33. doi:10.1074/jbc.M303111200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Hamra FK, Fan X, Krause WJ, Freeman RH, Chin DT, Smith CE, et al. Prouroguanylin and proguanylin: purification from colon, structure, and modulation of bioactivity by proteases. Endocrinology (1996) 137:257–65. doi:10.1210/endo.137.1.8536621
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. de Sauvage FJ, Keshav S, Kuang WJ, Gillett N, Henzel W, Goeddel DV. Precursor structure, expression, and tissue distribution of human guanylin. Proc Natl Acad Sci U S A (1992) 89:9089–93. doi:10.1073/pnas.89.19.9089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Kita T, Smith CE, Fok KF, Duffin KL, Moore WM, Karabatsos PJ, et al. Characterization of human uroguanylin: a member of the guanylin peptide family. Am J Physiol (1994) 266:F342–8.
116. Forte LR, Eber SL, Turner JT, Freeman RH, Fok KF, Currie MG. Guanylin stimulation of Cl-secretion in human intestinal T84 cells via cyclic guanosine monophosphate. J Clin Invest (1993) 91:2423–8. doi:10.1172/JCI116476
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Schulz S, Chrisman TD, Garbers DL. Cloning and expression of guanylin. Its existence in various mammalian tissues. J Biol Chem (1992) 267:16019–21.
118. Fan X, Wang Y, London RM, Eber SL, Krause WJ, Freeman RH, et al. Signaling pathways for guanylin and uroguanylin in the digestive, renal, central nervous, reproductive, and lymphoid systems. Endocrinology (1997) 138:4636–48. doi:10.1210/endo.138.11.5539
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Kita T, Kitamura K, Sakata J, Eto T. Marked increase of guanylin secretion in response to salt loading in the rat small intestine. Am J Physiol (1999) 277:G960–6.
120. Pitari GM, Di Guglielmo MD, Park J, Schulz S, Waldman SA. Guanylyl cyclase C agonists regulate progression through the cell cycle of human colon carcinoma cells. Proc Natl Acad Sci U S A (2001) 98:7846–51. doi:10.1073/pnas.141124698
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Fonteles MC, Greenberg RN, Monteiro HS, Currie MG, Forte LR. Natriuretic and kaliuretic activities of guanylin and uroguanylin in the isolated perfused rat kidney. Am J Physiol (1998) 275:F191–7.
122. Carrithers SL, Ott CE, Hill MJ, Johnson BR, Cai W, Chang JJ, et al. Guanylin and uroguanylin induce natriuresis in mice lacking guanylyl cyclase-C receptor. Kidney Int (2004) 65:40–53. doi:10.1111/j.1523-1755.2004.00375.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Fulle HJ, Vassar R, Foster DC, Yang RB, Axel R, Garbers DL. A receptor guanylyl cyclase expressed specifically in olfactory sensory neurons. Proc Natl Acad Sci U S A (1995) 92:3571–5. doi:10.1073/pnas.92.8.3571
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Chao YC, Cheng CJ, Hsieh HT, Lin CC, Chen CC, Yang RB. Guanylate cyclase-G, expressed in the Grueneberg ganglion olfactory subsystem, is activated by bicarbonate. Biochem J (2010) 432:267–73. doi:10.1042/BJ20100617
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Nakazato M, Yamaguchi H, Shiomi K, Date Y, Fujimoto S, Kangawa K, et al. Identification of 10-kDa proguanylin as a major guanylin molecule in human intestine and plasma and its increase in renal insufficiency. Biochem Biophys Res Commun (1994) 205:1966–75. doi:10.1006/bbrc.1994.2901
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Moss NG, Fellner RC, Qian X, Yu SJ, Li Z, Nakazato M, et al. Uroguanylin, an intestinal natriuretic peptide, is delivered to the kidney as an unprocessed propeptide. Endocrinology (2008) 149:4486–98. doi:10.1210/en.2007-1725
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Kinoshita H, Fujimoto S, Nakazato M, Yokota N, Date Y, Yamaguchi H, et al. Urine and plasma levels of uroguanylin and its molecular forms in renal diseases. Kidney Int (1997) 52:1028–34. doi:10.1038/ki.1997.424
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Valentino MA, Lin JE, Snook AE, Li P, Kim GW, Marszalowicz G, et al. A uroguanylin-GUCY2C endocrine axis regulates feeding in mice. J Clin Invest (2011) 121:3578–88. doi:10.1172/JCI57925
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Gong R, Ding C, Hu J, Lu Y, Liu F, Mann E, et al. Role for the membrane receptor guanylyl cyclase-C in attention deficiency and hyperactive behavior. Science (2011) 333:1642–6. doi:10.1126/science.1207675
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Strader AD, Woods SC. Gastrointestinal hormones and food intake. Gastroenterology (2005) 128:175–91. doi:10.1053/j.gastro.2004.10.043
131. Badman MK, Flier JS. The gut and energy balance: visceral allies in the obesity wars. Science (2005) 307:1909–14. doi:10.1126/science.1109951
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Seeley RJ, Tschop MH. Uroguanylin: how the gut got another satiety hormone. J Clin Invest (2011) 121:3384–6. doi:10.1172/JCI58297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Volkow ND, Wang GJ, Kollins SH, Wigal TL, Newcorn JH, Telang F, et al. Evaluating dopamine reward pathway in ADHD: clinical implications. JAMA (2009) 302:1084–91. doi:10.1001/jama.2009.1308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Sagvolden T, Russell VA, Aase H, Johansen EB, Farshbaf M. Rodent models of attention-deficit/hyperactivity disorder. Biol Psychiatry (2005) 57:1239–47. doi:10.1016/j.biopsych.2005.02.002
135. Zufall F, Munger SD. Receptor guanylyl cyclases in mammalian olfactory function. Mol Cell Biochem (2010) 334:191–7. doi:10.1007/s11010-009-0325-9
136. Chen KK, Kovarikova A. Pharmacology and toxicology of toad venom. J Pharm Sci (1967) 56:1535–41. doi:10.1002/jps.2600561202
137. de Wardener HE, Clarkson EM, Nutbourne DM, Schrier RW, Talner LB, Ventom MG, et al. Evidence for a hormone other than aldosterone which controls urinary sodium excretion. Adv Nephrol Necker Hosp (1971) 1:97–111.
138. Lichtstein D, Samuelov S. Endogenous ‘ouabain like’ activity in rat brain. Biochem Biophys Res Commun (1980) 96:1518–23. doi:10.1016/0006-291X(80)91346-7
139. Fishman MC. Endogenous digitalis-like activity in mammalian brain. Proc Natl Acad Sci U S A (1979) 76:4661–3. doi:10.1073/pnas.76.9.4661
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Haupert GT Jr, Sancho JM. Sodium transport inhibitor from bovine hypothalamus. Proc Natl Acad Sci U S A (1979) 76:4658–60. doi:10.1073/pnas.76.9.4658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Hamlyn JM, Blaustein MP, Bova S, Ducharme DW, Harris DW, Mandel F, et al. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci U S A (1991) 88:6259–63. doi:10.1073/pnas.88.21.9907-d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Ludens JH, Clark MA, Ducharme DW, Harris DW, Lutzke BS, Mandel F, et al. Purification of an endogenous digitalis like factor from human plasma for structural analysis. Hypertension (1991) 17:923–9. doi:10.1161/01.HYP.17.6.923
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Goto A, Ishiguro T, Yamada K, Ishii M, Yoshioka M, Eguchi C, et al. Isolation of a urinary digitalis-like factor indistinguishable from digoxin. Biochem Biophys Res Commun (1990) 173:1093–101. doi:10.1016/S0006-291X(05)80898-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Lichtstein D, Gati I, Samuelov S, Berson D, Rozenman Y, Landau L, et al. Identification of digitalis-like compounds in human cataractous lenses. Eur J Biochem (1993) 216:261–8. doi:10.1111/j.1432-1033.1993.tb18141.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Hilton PJ, White RW, Lord GA, Garner GV, Gordon DB, Hilton MJ, et al. An inhibitor of the sodium pump obtained from human placenta. Lancet (1996) 348:303–5. doi:10.1016/S0140-6736(96)02257-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Bagrov AY, Fedorova OV, Dmitrieva RI, Howald WN, Hunter AP, Kuznetsova EA, et al. Characterization of a urinary bufodienolide Na+,K+-ATPase inhibitor in patients after acute myocardial infarction. Hypertension (1998) 31:1097–103. doi:10.1161/01.HYP.31.5.1097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Schneider R, Antolovic R, Kost H, Sich B, Kirch U, Tepel M, et al. Proscillaridin A immunoreactivity: its purification, transport in blood by a specific binding protein and its correlation with blood pressure. Clin Exp Hypertens (1998) 20:593–9. doi:10.3109/10641969809053237
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Komiyama Y, Dong XH, Nishimura N, Masaki H, Yoshika M, Masuda M, et al. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin Biochem (2005) 38:36–45. doi:10.1016/j.clinbiochem.2004.08.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Lichtstein D, Steinitz M, Gati I, Samuelov S, Deutsch J, Orly J. Biosynthesis of digitalis-like compounds in rat adrenal cells: hydroxycholesterol as possible precursor. Life Sci (1998) 62:2109–26. doi:10.1016/S0024-3205(98)00186-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Murrell JR, Randall JD, Rosoff J, Zhao JL, Jensen RV, Gullans SR, et al. Endogenous ouabain: upregulation of steroidogenic genes in hypertensive hypothalamus but not adrenal. Circulation (2005) 112:1301–8. doi:10.1161/CIRCULATIONAHA.105.554071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Greeff K, Gãntert TW, Linde HHA. Chemistry and Structure-Activity Relationships of Cardioactive Steroids,” in Cardiac Glycosides. Berlin Heidelberg: Springer (1981). p. 13–24.
152. Klein M, Nejad NS, Lown B, Hagemeijer F, Barr I. Correlation of the electrical and mechanical changes in the dog heart during progressive digitalization. Circ Res (1971) 29:635–45.
153. Xie Z, Cai T. Na+-K+ – ATPase-mediated signal transduction: from protein interaction to cellular function. Mol Interv (2003) 3:157–68. doi:10.1124/mi.3.3.157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides and their mechanisms of action. Am J Cardiovasc Drugs (2007) 7:173–89. doi:10.2165/00129784-200707030-00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Huang BS, Amin MS, Leenen FH. The central role of the brain in salt-sensitive hypertension. Curr Opin Cardiol (2006) 21:295–304. doi:10.1097/01.hco.0000231398.64362.94
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Lichtstein D. Na+, K(+)-ATPase and heart excitability. Adv Exp Med Biol (1995) 382:23–30. doi:10.1007/978-1-4615-1893-8_3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Blaustein MP, Zhang J, Chen L, Hamilton BP. How does salt retention raise blood pressure? Am J Physiol Regul Integr Comp Physiol (2006) 290:R514–23. doi:10.1152/ajpregu.00819.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Dvela M, Rosen H, Ben-Ami HC, Lichtstein D. Endogenous ouabain regulates cell viability. Am J Physiol Cell Physiol (2012) 302:C442–52. doi:10.1152/ajpcell.00336.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Goldstein I, Levy T, Galili D, Ovadia H, Yirmiya R, Rosen H, et al. Involvement of Na(+), K(+)-ATPase and endogenous digitalis-like compounds in depressive disorders. Biol Psychiatry (2006) 60:491–9. doi:10.1016/j.biopsych.2005.12.021
160. Pongrakhananon V, Chunhacha P, Chanvorachote P. Ouabain suppresses the migratory behavior of lung cancer cells. PLoS One (2013) 8:e68623. doi:10.1371/journal.pone.0068623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Newman RA, Yang P, Pawlus AD, Block KI. Cardiac glycosides as novel cancer therapeutic agents. Mol Interv (2008) 8:36–49. doi:10.1124/mi.8.1.8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Halperin J, Schaeffer R, Galvez L, Malave S. Ouabain-like activity in human cerebrospinal fluid. Proc Natl Acad Sci U S A (1983) 80:6101–4. doi:10.1073/pnas.80.19.6101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Lichtstein D, Minc D, Bourrit A, Deutsch J, Karlish SJ, Belmaker H, et al. Evidence for the presence of ‘ouabain like’ compound in human cerebrospinal fluid. Brain Res (1985) 325:13–9. doi:10.1016/0006-8993(85)90297-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Yamada H, Ihara N, Takahashi H, Yoshimura M, Sano Y. Distribution of the endogenous digitalis-like substance (EDLS)-containing neurons labeled by digoxin antibody in hypothalamus and three circumventricular organs of dog and macaque. Brain Res (1992) 584:237–43. doi:10.1016/0006-8993(92)90900-T
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Morgan K, Lewis MD, Spurlock G, Collins PA, Foord SM, Southgate K, et al. Characterization and partial purification of the sodium-potassium-ATPase inhibitor released from cultured rat hypothalamic cells. J Biol Chem (1985) 260:13595–600.
166. Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol (1998) 275:F633–50.
167. Moseley AE, Lieske SP, Wetzel RK, James PF, He S, Shelly DA, et al. The Na,K-ATPase alpha 2 isoform is expressed in neurons, and its absence disrupts neuronal activity in newborn mice. J Biol Chem (2003) 278:5317–24. doi:10.1074/jbc.M211315200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Mcgrail KM, Phillips JM, Sweadner KJ. Immunofluorescent localization of three Na,K-ATPase isozymes in the rat central nervous system: both neurons and glia can express more than one Na,K-ATPase. J Neurosci (1991) 11:381–91.
169. Meneton P, Jeunemaitre X, De Wardener HE, Macgregor GA. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol Rev (2005) 85:679–715. doi:10.1152/physrev.00056.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Goto A, Yamada K, Ishii M, Sugimoto T. Digitalis-like activity in human plasma: relation to blood pressure and sodium balance. Am J Med (1990) 89:420–6. doi:10.1016/0002-9343(90)90369-O
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Rossi G, Manunta P, Hamlyn JM, Pavan E, De Toni R, Semplicini A, et al. Immunoreactive endogenous ouabain in primary aldosteronism and essential hypertension: relationship with plasma renin, aldosterone and blood pressure levels. J Hypertens (1995) 13:1181–91. doi:10.1097/00004872-199510000-00013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Gonick HC, Ding Y, Vaziri ND, Bagrov AY, Fedorova OV. Simultaneous measurement of marinobufagenin, ouabain, and hypertension-associated protein in various disease states. Clin Exp Hypertens (1998) 20:617–27. doi:10.3109/10641969809053240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Manunta P, Messaggio E, Ballabeni C, Sciarrone MT, Lanzani C, Ferrandi M, et al. Plasma ouabain-like factor during acute and chronic changes in sodium balance in essential hypertension. Hypertension (2001) 38:198–203. doi:10.1161/01.HYP.38.2.198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Pierdomenico SD, Bucci A, Manunta P, Rivera R, Ferrandi M, Hamlyn JM, et al. Endogenous ouabain and hemodynamic and left ventricular geometric patterns in essential hypertension. Am J Hypertens (2001) 14:44–50. doi:10.1016/S0895-7061(00)01225-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Fridman AI, Matveev SA, Agalakova NI, Fedorova OV, Lakatta EG, Bagrov AY. Marinobufagenin, an endogenous ligand of alpha-1 sodium pump, is a marker of congestive heart failure severity. J Hypertens (2002) 20:1189–94. doi:10.1097/00004872-200206000-00032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Hasegawa T, Masugi F, Ogihara T, Kumahara Y. Increase in plasma ouabainlike inhibitor of Na+, K+-ATPase with high sodium intake in patients with essential hypertension. J Clin Hypertens (1987) 3:419–29.
177. Leenen FH, Harmsen E, Yu H, Yuan B. Dietary sodium stimulates ouabainlike activity in adrenalectomized spontaneously hypertensive rats. Am J Physiol (1993) 265:H421–4.
178. Fedorova OV, Talan MI, Agalakova NI, Lakatta EG, Bagrov AY. Endogenous ligand of alpha(1) sodium pump, marinobufagenin, is a novel mediator of sodium chloride – dependent hypertension. Circulation (2002) 105:1122–7. doi:10.1161/hc0902.104710
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Manunta P, Rogowski AC, Hamilton BP, Hamlyn JM. Ouabain-induced hypertension in the rat: relationships among plasma and tissue ouabain and blood pressure. J Hypertens (1994) 12:549–60. doi:10.1097/00004872-199405000-00008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Yamada K, Goto A, Omata M. Adrenocorticotropin-induced hypertension in rats: role of ouabain-like compound. Am J Hypertens (1997) 10:403–8. doi:10.1016/S0895-7061(97)90523-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Yuan CM, Manunta P, Hamlyn JM, Chen S, Bohen E, Yeun J, et al. Long-term ouabain administration produces hypertension in rats. Hypertension (1993) 22:178–87. doi:10.1161/01.HYP.22.2.178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Manunta P, Hamilton J, Rogowski AC, Hamilton BP, Hamlyn JM. Chronic hypertension induced by ouabain but not digoxin in the rat: antihypertensive effect of digoxin and digitoxin. Hypertens Res (2000) 23(Suppl):S77–85. doi:10.1291/hypres.23.Supplement_S77
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Dostanic I, Paul RJ, Lorenz JN, Theriault S, Van Huysse JW, Lingrel JB. The alpha2-isoform of Na-K-ATPase mediates ouabain-induced hypertension in mice and increased vascular contractility in vitro. Am J Physiol Heart Circ Physiol (2005) 288:H477–85. doi:10.1152/ajpheart.00083.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Dostanic-Larson I, Lorenz JN, Van Huysse JW, Neumann JC, Moseley AE, Lingrel JB. Physiological role of the alpha1- and alpha2-isoforms of the Na+-K+-ATPase and biological significance of their cardiac glycoside binding site. Am J Physiol Regul Integr Comp Physiol (2006) 290:R524–8. doi:10.1152/ajpregu.00838.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Loreaux EL, Kaul B, Lorenz JN, Lingrel JB. Ouabain-sensitive alpha1 Na,K-ATPase enhances natriuretic response to saline load. J Am Soc Nephrol (2008) 19:1947–54. doi:10.1681/ASN.2008020174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Zhang J, Lee MY, Cavalli M, Chen L, Berra-Romani R, Balke CW, et al. Sodium pump alpha2 subunits control myogenic tone and blood pressure in mice. J Physiol (2005) 569:243–56. doi:10.1113/jphysiol.2005.091801
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Poburko D, Liao CH, Lemos VS, Lin E, Maruyama Y, Cole WC, et al. Transient receptor potential channel 6-mediated, localized cytosolic [Na+] transients drive Na+/Ca2+ exchanger-mediated Ca2+ entry in purinergically stimulated aorta smooth muscle cells. Circ Res (2007) 101:1030–8. doi:10.1161/CIRCRESAHA.107.155531
188. Kaide J, Ura N, Torii T, Nakagawa M, Takada T, Shimamoto K. Effects of digoxin-specific antibody Fab fragment (Digibind) on blood pressure and renal water-sodium metabolism in 5/6 reduced renal mass hypertensive rats. Am J Hypertens (1999) 12:611–9. doi:10.1016/S0895-7061(99)00029-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Wenceslau CF, Rossoni LV. Rostafuroxin ameliorates endothelial dysfunction and oxidative stress in resistance arteries from deoxycorticosterone acetate-salt hypertensive rats: the role of Na+K+-ATPase/cSRC pathway. J Hypertens (2014) 32:542–54. doi:10.1097/HJH.0000000000000059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Veerasingham SJ, Leenen FH. Excitotoxic lesions of the ventral anteroventral third ventricle and pressor responses to central sodium, ouabain and angiotensin II. Brain Res (1997) 749:157–60. doi:10.1016/S0006-8993(96)01381-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Veerasingham SJ, Leenen FH. Ouabain- and central sodium-induced hypertension depend on the ventral anteroventral third ventricle region. Am J Physiol (1999) 276:H63–70.
192. Huang BS, Leenen FH. Sympathoexcitatory and pressor responses to increased brain sodium and ouabain are mediated via brain ANG II. Am J Physiol (1996) 270:H275–80.
193. Huang BS, Leenen FH. Brain renin-angiotensin system and ouabain-induced sympathetic hyperactivity and hypertension in Wistar rats. Hypertension (1999) 34:107–12. doi:10.1161/01.HYP.34.1.107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
194. Huang BS, Ganten D, Leenen FH. Responses to central Na(+) and ouabain are attenuated in transgenic rats deficient in brain angiotensinogen. Hypertension (2001) 37:683–6. doi:10.1161/01.HYP.37.2.683
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Kawano Y, Yoshida K, Kawamura M, Yoshimi H, Ashida T, Abe H, et al. Sodium and noradrenaline in cerebrospinal fluid and blood in salt-sensitive and non-salt-sensitive essential hypertension. Clin Exp Pharmacol Physiol (1992) 19:235–41. doi:10.1111/j.1440-1681.1992.tb00444.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Huang BS, Van Vliet BN, Leenen FH. Increases in CSF [Na+] precede the increases in blood pressure in Dahl S rats and SHR on a high-salt diet. Am J Physiol Heart Circ Physiol (2004) 287:H1160–6. doi:10.1152/ajpheart.00126.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
198. el-Mallakh RS, Wyatt RJ. The Na,K-ATPase hypothesis for bipolar illness. Biol Psychiatry (1995) 37:235–44. doi:10.1016/0006-3223(94)00201-D
199. Traub N, Lichtstein D. The mood cycle hypothesis: possible involvement of steroid hormones in mood regulation by means of Na+, K+-ATPase inhibition. J Basic Clin Physiol Pharmacol (2000) 11:375–94. doi:10.1515/JBCPP.2000.11.4.375
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
200. Naylor GJ, Mcnamee HB, Moody JP. Changes in erythrocyte sodium and potassium on recovery from a depressive illness. Br J Psychiatry (1971) 118:219–23. doi:10.1192/bjp.118.543.219
201. Nurnberger J Jr, Jimerson DC, Allen JR, Simmons S, Gershon E. Red cell ouabain-sensitive Na+-K+-adenosine triphosphatase: a state marker in affective disorder inversely related to plasma cortisol. Biol Psychiatry (1982) 17:981–92.
202. Looney SW, el-Mallakh RS. Meta-analysis of erythrocyte Na,K-ATPase activity in bipolar illness. Depress Anxiety (1997) 5:53–65. doi:10.1002/(SICI)1520-6394(1997)5:2<53::AID-DA1>3.0.CO;2-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
203. Grider G, El-Mallakh RS, Huff MO, Buss TJ, Miller J, Valdes R Jr. Endogenous digoxin-like immunoreactive factor (DLIF) serum concentrations are decreased in manic bipolar patients compared to normal controls. J Affect Disord (1999) 54:261–7. doi:10.1016/S0165-0327(98)00208-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
204. El-Mallakh RS, Stoddard M, Jortani SA, El-Masri MA, Sephton S, Valdes R Jr. Aberrant regulation of endogenous ouabain-like factor in bipolar subjects. Psychiatry Res (2010) 178:116–20. doi:10.1016/j.psychres.2009.03.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
205. Chetcuti A, Adams LJ, Mitchell PB, Schofield PR. Microarray gene expression profiling of mouse brain mRNA in a model of lithium treatment. Psychiatr Genet (2008) 18:64–72. doi:10.1097/YPG.0b013e3282fb0051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
206. Mynett-Johnson L, Murphy V, Mccormack J, Shields DC, Claffey E, Manley P, et al. ). Evidence for an allelic association between bipolar disorder and a Na+, K+ adenosine triphosphatase alpha subunit gene (ATP1A3). Biol Psychiatry (1998) 44:47–51. doi:10.1016/S0006-3223(97)00343-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
207. Goldstein I, Lerer E, Laiba E, Mallet J, Mujaheed M, Laurent C, et al. Association between sodium- and potassium-activated adenosine triphosphatase alpha isoforms and bipolar disorders. Biol Psychiatry (2009) 65:985–91. doi:10.1016/j.biopsych.2008.10.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
208. Kirshenbaum GS, Clapcote SJ, Duffy S, Burgess CR, Petersen J, Jarowek KJ, et al. Mania-like behavior induced by genetic dysfunction of the neuron-specific Na+,K+-ATPase alpha3 sodium pump. Proc Natl Acad Sci U S A (2011) 108:18144–9. doi:10.1073/pnas.1108416108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
209. El-Mallakh RS, El-Masri MA, Huff MO, Li XP, Decker S, Levy RS. Intracerebroventricular administration of ouabain as a model of mania in rats. Bipolar Disord (2003) 5:362–5. doi:10.1034/j.1399-5618.2003.00053.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
210. Brocardo PS, Budni J, Pavesi E, Franco JL, Uliano-Silva M, Trevisan R, et al. Folic acid administration prevents ouabain-induced hyperlocomotion and alterations in oxidative stress markers in the rat brain. Bipolar Disord (2010) 12:414–24. doi:10.1111/j.1399-5618.2010.00827.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
211. Goldstein I, Lax E, Gispan-Herman I, Ovadia H, Rosen H, Yadid G, et al. Neutralization of endogenous digitalis-like compounds alters catecholamines metabolism in the brain and elicits anti-depressive behavior. Eur Neuropsychopharmacol (2012) 22:72–9. doi:10.1016/j.euroneuro.2011.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
212. Yu HS, Kim SH, Park HG, Kim YS, Ahn YM. Activation of Akt signaling in rat brain by intracerebroventricular injection of ouabain: a rat model for mania. Prog Neuropsychopharmacol Biol Psychiatry (2010) 34:888–94. doi:10.1016/j.pnpbp.2010.04.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
213. Kim SH, Yu HS, Park HG, Ha K, Kim YS, Shin SY, et al. Intracerebroventricular administration of ouabain, a Na/K-ATPase inhibitor, activates mTOR signal pathways and protein translation in the rat frontal cortex. Prog Neuropsychopharmacol Biol Psychiatry (2013) 45:73–82. doi:10.1016/j.pnpbp.2013.04.018
214. Freitas TP, Rezin GT, Goncalves CL, Jeremias GC, Gomes LM, Scaini G, et al. Evaluation of citrate synthase activity in brain of rats submitted to an animal model of mania induced by ouabain. Mol Cell Biochem (2010) 341:245–9. doi:10.1007/s11010-010-0455-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
215. Riegel RE, Valvassori SS, Moretti M, Ferreira CL, Steckert AV, De Souza B, et al. Intracerebroventricular ouabain administration induces oxidative stress in the rat brain. Int J Dev Neurosci (2010) 28:233–7. doi:10.1016/j.ijdevneu.2010.02.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
216. Lewis LK, Yandle TG, Hilton PJ, Jensen BP, Begg EJ, Nicholls MG. Endogenous ouabain is not ouabain. Hypertension (2014) 64:680–3. doi:10.1161/HYPERTENSIONAHA.114.03919
217. Lichtstein D, Rosen H, Dvela M. Cardenolides and bufadienolides as hormones: what is missing? Am J Physiol Renal Physiol (2012) 302:F957–8. doi:10.1152/ajprenal.00042.2012
218. Bloch KD, Zamir N, Lichtstein D, Seidman CE, Seidman JG. Ouabain induces secretion of proatrial natriuretic factor by rat atrial cardiocytes. Am J Physiol (1988) 255:E383–7.
219. Morise T, Takeuchi Y, Okamoto S, Takeda R. Stimulation of atrial natriuretic peptide secretion and synthesis by Na-K-ATPase inhibitors. Biochem Biophys Res Commun (1991) 176:875–81. doi:10.1016/S0006-291X(05)80267-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
220. Schiebinger RJ, Cragoe EJ Jr. Ouabain. A stimulator of atrial natriuretic peptide secretion and its mechanism of action. Circ Res (1993) 72:1035–43. doi:10.1161/01.RES.72.5.1035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
221. Liu LP, Hong L, Yu L, Li HY, Ding DZ, Jin SJ, et al. Ouabain stimulates atrial natriuretic peptide secretion via the endothelin-1/ET(B) receptor-mediated pathway in beating rabbit atria. Life Sci (2012) 90:793–8. doi:10.1016/j.lfs.2012.04.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
222. Yamamoto A, Shouji T, Kimura S, Aki Y, Nakamura A, Fukui K, et al. Effects of hypercalcemia and ouabain on plasma atrial natriuretic polypeptide in anesthetized dogs. Am J Physiol (1988) 255:E437–41.
223. Tsutamoto T, Wada A, Maeda K, Hisanaga T, Fukai D, Maeda Y, et al. Digitalis increases brain natriuretic peptide in patients with severe congestive heart failure. Am Heart J (1997) 134:910–6. doi:10.1016/S0002-8703(97)80014-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
224. Crabos M, Ausiello DA, Haupert GT Jr, Cantiello HF. Atrial natriuretic peptide regulates release of Na+-K+-ATPase inhibitor from rat brain. Am J Physiol (1988) 254:F912–7.
225. Songu-Mize E, Bealer SL, Hassid AI. Centrally administered ANF promotes appearance of a circulating sodium pump inhibitor. Am J Physiol (1990) 258:H1655–9.
226. Songu-Mize E, Bealer SL. Effect of hypothalamic lesions on interaction of centrally administered ANF and the circulating sodium-pump inhibitor. J Cardiovasc Pharmacol (1993) 22(Suppl 2):S4–6. doi:10.1097/00005344-199322002-00003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
227. Fedorova OV, Agalakova NI, Morrell CH, Lakatta EG, Bagrov AY. ANP differentially modulates marinobufagenin-induced sodium pump inhibition in kidney and aorta. Hypertension (2006) 48:1160–8. doi:10.1161/01.HYP.0000248129.20524.d0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
228. Sybertz EJ, Desiderio DM. The role of Na+–K+-ATPase in the vasorelaxant actions of synthetic atrial natriuretic factor. Arch Int Pharmacodyn Ther (1985) 278:142–9.
229. Chintala MS, Jandhyala BS. Interaction between atrial natriuretic factor and ouabain: vascular reactivity to noradrenaline in pentobarbital anaesthetized dogs. Clin Exp Pharmacol Physiol (1988) 15:591–9. doi:10.1111/j.1440-1681.1988.tb01118.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
230. Szalay KS, Beck M, Toth M, De Chatel R. Interactions between ouabain, atrial natriuretic peptide, angiotensin-II and potassium: effects on rat zona glomerulosa aldosterone production. Life Sci (1998) 62:1845–52. doi:10.1016/S0024-3205(98)00150-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
231. Nesher M, Dvela M, Igbokwe VU, Rosen H, Lichtstein D. Physiological roles of endogenous ouabain in normal rats. Am J Physiol Heart Circ Physiol (2009) 297:H2026–34. doi:10.1152/ajpheart.00734.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
232. Nesher M, Bai Y, Li D, Rosen H, Lichtstein D, Liu L. Interaction of atrial natriuretic peptide and ouabain in the myocardium. Can J Physiol Pharmacol (2012) 90:1386–93. doi:10.1139/y2012-112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
233. Santos-Neto MS, Carvalho AF, Monteiro HS, Forte LR, Fonteles MC. Interaction of atrial natriuretic peptide, urodilatin, guanylin and uroguanylin in the isolated perfused rat kidney. Regul Pept (2006) 136:14–22. doi:10.1016/j.regpep.2006.04.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
234. Potter LR. Regulation and therapeutic targeting of peptide-activated receptor guanylyl cyclases. Pharmacol Ther (2011) 130:71–82. doi:10.1016/j.pharmthera.2010.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: atrial natriuretic peptide, cardiac steroids, ouabain, guanylin, brain function
Citation: Hodes A and Lichtstein D (2014) Natriuretic hormones in brain function. Front. Endocrinol. 5:201. doi: 10.3389/fendo.2014.00201
Received: 09 September 2014; Accepted: 12 November 2014;
Published online: 28 November 2014.
Edited by:
Harvey Craig Gonick, University of California Berkeley, USAReviewed by:
Joao Carlos Dos Reis Cardoso, University of Algarve, PortugalCopyright: © 2014 Hodes and Lichtstein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Lichtstein, Faculty of Medicine, Department of Medical Neurobiology, Institute for Medical Research Israel-Canada, The Hebrew University of Jerusalem, Ein-Kerem, Jerusalem 91120, Israel e-mail:ZGF2aWRsaUBla21kLmh1amkuYWMuaWw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.