 Vincenzo De Tata
Vincenzo De Tata- Department of Translational Research and New Technologies in Medicine and Surgery, University of Pisa, Pisa, Italy
The incidence of type 2 diabetes significantly increases with age. The relevance of this association is dramatically magnified by the concomitant global aging of the population, but the underlying mechanisms remain to be fully elucidated. Here, some recent advances in this field are reviewed at the level of both the pathophysiology of glucose homeostasis and the cellular senescence of pancreatic islets. Overall, recent results highlight the crucial role of beta-cell dysfunction in the age-related impairment of pancreatic endocrine function and delineate the possibility of new original therapeutic interventions.
Type 2 Diabetes: An Age-Related Disease
Diabetes mellitus, a disease characterized by high blood glucose levels resulting from a combination of genetic and acquired factors, represents the most prevalent metabolic disorders. Indeed, the prevalence of the most common form, type 2 diabetes or non-insulin-dependent diabetes mellitus (NIDDM), exploded over the last several decades. Data from the World Health Organization (WHO) and the International Diabetes Federation (IDF) show that the prevalence of type 2 diabetes increased from 100–135 million affected adults worldwide in 1994–1995 to approximately 336 million in 2011, and it is expected to rise to 439 million by 2030 (1–3). These data are even more dramatic considered in the light of the concomitant global aging of the population. Elderly people (by definition, person of over 65 years of age) represented 12–15% of the population in 2008, whereas it has been estimated that the same age group would account for 26% of the population in 2026 and will reach 2 billion people in 2050 (4, 5). Population aging is rapidly becoming a global issue with a major impact on health policies and programs. Such a remarkable improvement in life expectancy considerably contributed to a shift in the leading causes of diseases and death from infectious and parasitic diseases to non-communicable diseases (such as heart disease, cancer, and diabetes) that more commonly affect adults and older adults (6, 7). In particular, aging is an important risk factor for metabolic disorders, including obesity, impaired glucose tolerance, and type-2 diabetes (8, 9). The prevalence of type 2 diabetes increases with age (in older adults it is more than twice that of middle-aged adults) and peaks at 60–74 years of age (10–12). In consideration of the already mentioned nearly doubling of the numbers of elderly persons by the year 2030, it is easy to see why diabetes in older adults is considered as a growing public health concern.
Normal aging is usually associated with a progressive deterioration in most endocrine functions that may be responsible for serious disturbances of metabolic homeostasis (13–16). Actually, an impairment of glucose tolerance has been recognized for a long time as a well-known feature of aging in both humans and experimental animals (17, 18). Nevertheless, the underlying biological mechanism(s) is still not clearly understood.
Aging and Insulin Resistance
Type 2 diabetes mellitus is a metabolic disorder characterized by high blood glucose levels as a result of the complex interplay of multiple genetic and environmental factors that cause both impaired insulin action on target tissues and defective pancreatic beta-cell insulin secretion in response to glucose (19).
Traditional views of type 2 diabetes pathophysiology indicated peripheral insulin resistance, i.e., the inability of insulin-target tissues to respond properly to the hormone, as the main driver of altered glucose homeostasis (20). Clinically, the term insulin resistance is utilized to indicate that higher-than-normal circulating levels of insulin are required to maintain normoglycemia. At the cellular level, insulin action, initiated by the binding to its cell surface receptor, involves a series of signaling cascades that can be schematically summarized as follows: receptor autophosphorylation and activation of receptor tyrosine kinase; tyrosine phosphorylation of insulin receptor substrates (IRSs) 1 and 2; activation of phosphatidylinositol 3-kinase (PI3K); activation of Akt and its downstream mediator, AS160, which stimulates the translocation of insulin-mediated GLUT4 from intracellular vesicles to the plasma membrane (21, 22).
It is well documented that aging is associated with a decline of insulin action. Studies utilizing the euglycemic hyperinsulinemic clamp technique to assess insulin effectiveness in regulating glucose transport usually stress the relevance of the diminished insulin sensitivity on target tissues in the development of age-related glucose intolerance (17, 20, 23, 24). Insulin resistance could increase with age in relation to several well-known age-related changes, such as: (i) increased adiposity (with particular reference to abdominal fat mass) (5); (ii) decreased lean muscle mass (sarcopenia) (25–27); (iii) mitochondrial dysfunctions (28–32); (iv) hormonal changes (33, 34); (v) increased oxidative stress and inflammation (35–39); (vi) changes in dietary habits (40–42); (vii) reduced physical activity (43, 44). However, it has also been claimed that these factors alone cannot fully account for the age-related glucose-intolerance (11), and other studies seem to indicate that age per se could be not responsible for the increased insulin resistance (45–47).
Aging and Insulin Secretion
On the other hand, several observations clearly show that insulin resistance alone is not sufficient to lead to type 2 diabetes in the absence of a beta-cell defect associated with abnormal insulin secretion. Consequently, beta-cell dysfunction is increasingly recognized to play a fundamental role in type 2 diabetes pathophysiology (48, 49) and could represent another significant contributing factor to abnormal glucose metabolism with age (9, 50). Indeed, it has been repeatedly reported that the ability of pancreatic beta cells to maintain an insulin secretory function adequate for metabolic demand is impaired with increasing age in both experimental animals (51–55) and humans (11, 56–65), although some of these studies (especially in humans) were characterized by a significant degree of variability (66).
This age-related impairment of beta-cell secretory capabilities has been variously attributed to several factors, including: (i) mitochondrial dysfunction (34, 67–69); (ii) reduced GLUT2 levels (54, 70); (iii) accumulation of advanced glycation end products (AGEs) (71, 72); (iv) telomerase deficiency and reduced telomere length (73, 74): (v) reduced expression of β2-adrenergic receptors (75); (vi) impaired Ca++ handling (76, 77); (vii) reduced response to GLP-1 stimulation (62, 65, 78–83); (vii) increased autophagy (84); (viii) reduced expression of beta-cell-specific genes and transcription factors such as PDX-1 (54).
Among the above mentioned factors, mitochondrial dysfunction may deserve a particular discussion because mitochondria play a crucial role in the physiological stimulus-secretion coupling in beta cells. In these cells, mitochondria serve as nutrient sensors and signal generators for insulin secretion. In particular, the mitochondrial metabolism of pyruvate, glycolitically derived from glucose, generates ATP, which in turn promotes the closure of ATP-sensitive K+ channels and the consequent cell depolarization, inducing Ca2+ influx through voltage-gated Ca2+ channels, increased cytosolic [Ca2+], and finally triggering insulin exocytosis (85). On the other hand, due to the central role played in the generation of reactive oxygen species (ROS) at the level of the electron transport chain and ATP production, it has been proposed that mitochondria could represent a primary target of ROS damage (mitochondrial free radical theory of aging) (86). Indeed, increasing evidence suggests that abnormal mitochondrial ROS production and detoxification contribute to mitochondrial dysfunction in old age (87). Thus, age-related impairment of mitochondrial function could easily result in decreased beta-cell function and insulin secretion (88).
We can tentatively conclude this brief survey of the pathophysiology of glucose homeostasis by observing that several risk factors for diabetes associated with aging likely contribute to the development of age-related glucose intolerance and insulin resistance. Adaptation to insulin resistance normally requires compensatory hyperinsulinemia to maintain normal glucose metabolism. On the average, many studies show that, when considered in light of the degree of insulin resistance, all the indexes of insulin secretion appear to be decreased with age, indicating decreased beta-cell secretory reserve. Thus, the main homeostatic defect could be ascribed to age-dependent failure of the endocrine pancreas to provide enough insulin to overcome the state of increased peripheral insulin resistance.
Beta-Cell Senescence
Studies on the age-related glucose intolerance at the pathophysiological level may be difficult to interpret because the development of this condition could depend on a combination of many different factors whose independent influence is not easily controlled, thus making their relative importance a matter of debate. Therefore, more recently several researchers shifted the focus of their interest on the effect of aging on islet biology, with particular reference to the proliferative and regenerative capacity of beta cells. This paradigmatic change arises mainly from the consideration that aging represents a major risk factor for many generally chronic diseases (including cancer, neurodegeneration, and diabetes) and from the related possibility that these pathologies could be linked by a common biology. In the last few decades, a growing consensus has been reached and now it is considered likely that one or more basic aging processes underlie most, if not all, age-related pathologies (89). One basic process that may contribute to age-related dysfunction, including decreased secretory function (90), is cellular senescence. Cellular senescence was firstly described more than 50 years ago by Hayflick and Moorhead (91) as a process limiting the proliferation of normal human fibroblasts in culture, and this term is now generally used to indicate the essentially irreversible growth arrest that occurs when cells that can divide are challenged by a potentially oncogenic stress (92, 93). Senescent cells have clearly been shown to disrupt normal tissue structures and differentiated functions in complex cell culture models (89).
The growing interest in the cellular mechanisms responsible for the age-related decline in beta-cell proliferation originated from two distinct considerations with either fundamental or clinical implications. (A) Since insulin secretion by pancreatic beta cells represents the key point of the endocrine axis regulating glucose homeostasis, it is obvious that maintenance of beta-cell number and islet mass must be considered crucial in order to sustain normoglycemia. (B) Beta-cell replication represents a major goal of the cellular therapy of diabetes. Indeed, the promising attempt to develop a therapy based on pancreatic islets transplantation is still seriously hampered by the scarcity of cadaver-derived islets. The possibility to enhance replication of islet cells in vitro has been proposed as a solution to overcome the limited supply. Similarly, the expansion of potentially reduced functional beta-cell mass in vivo might represent another therapeutic strategy in type 1 and type 2 diabetes.
In normal healthy conditions, beta cells have a long lifespan with a low proliferation rate (94). However, it has been shown that in particular conditions, such as in response to increased metabolic demand or after injury, the adult pancreas could be able to produce new cells, particularly beta cells. Recent experimental evidences indicate that beta-cell mass, like many other tissues, could be dynamically regulated with ongoing beta-cell regeneration throughout life to replace lost or damaged beta cells (95).
Molecular Mechanism of Age-Related Beta-Cell Growth Arrest
Beta-cell cycling is driven by cyclin D1/D2-Cdk activity and is repressed by the Cdk-inhibitor p16INK4a (Figure 1) (96). In mice, it has been shown that beta-cell proliferation is an age-related process and that the expansion of beta-cell mass after pancreatic injury is more robust in young than in old animals (97). However, several pieces of experimental evidence indicate that aging mouse beta cells maintain a partially preserved ability to proliferate when specifically stimulated, both after pancreas injury (such as partial pancreatectomy or beta-cell-specific cell ablation) (98–102) and after islet transplantation in hyperglycemic recipients (103, 104). On the other hand, in recent years it became increasingly apparent that many of the mechanisms identified in these rodent models cannot be transferred easily to human islet cells. Human studies generally consist of observations made from pancreases obtained at autopsy, pancreas donation, and surgical resection, and are mainly based on immunohistochemical markers of proliferation (such as the nuclear Ki-67). As a consequence, data obtained in humans are often less conclusive than those obtained in rodent experimental models (105). It has been shown that human beta-cell mass can increase in obesity, although to a lesser degree than in rodents (30–40% estimated increase in humans with respect to a 30-fold increase observed in mice) (106–108). On the contrary, recent studies failed to detect an increased rate of beta-cell proliferation in pregnant individuals and in type 2 diabetes patients (109).
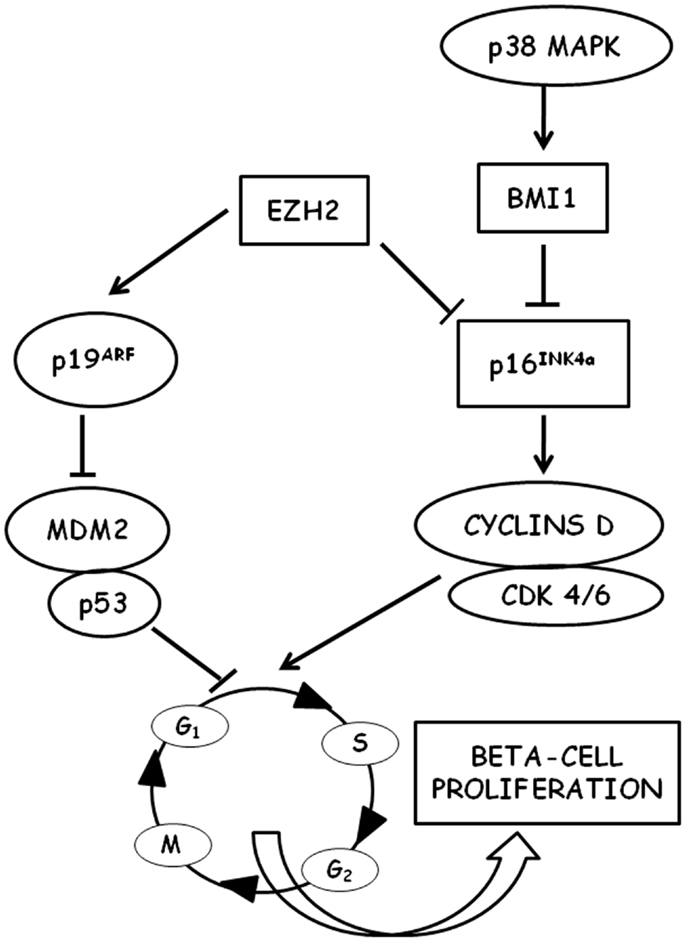
Figure 1. Schematic representation of the molecular pathways involved in the regulation of beta-cell proliferation is shown. P16INK4a is a key regulator of cell cycle entry in aged beta cells through D-type cyclins and cyclin-dependent kinases (CDK). P16INK4a is negatively regulated by the polycomb proteins EHZ3 (enhancer of zeste homolog 2) and BMI1 (B lymphoma Mo-MLV insertion region 1 homolog). BMI1 is stimulated by p38 MAPK.
A major difference between mice and humans is telomere shortening that limits proliferation and leads to cellular senescence in humans (110, 111), whereas in mice that have long telomeres no impairment of replication has been detected for several generations after ablation of telomerase (112, 113). This difference may account for the differential response observed between mice and humans (proliferation vs. differentiation from non-beta-cell progenitors) in beta-cell compensation (114). Human beta cells in adults appear to be largely postmitotic with very low rates of cell proliferation after the age of 20–30 years, as determined by Ki-67 content (115–117), thymidine analog incorporation (118), and increased in vivo lipofuscin accumulation (119, 120). Growth arrest of adult human beta cells cannot be reversed by procedures inducing proliferation in vitro (121, 122). This decline in the proliferative capacity of aging beta cells is directly associated with a decreased expression of the pancreatic and duodenal homeobox 1 (Pdx1) (121, 123), a transcription factor that plays a crucial role in beta-cell replication (124). Several experimental pieces of evidence demonstrated a decreased expression of cell cycle activators (such as, e.g., the transcription factor FoxM1) in aging beta cells with a simultaneous decrease in the expression of cell cycle inhibitors [for a review see Ref. (10)]. p16INK4a tumor suppressor protein has emerged from these studies as a key control point for cell cycle entry of beta cells. p16INK4a is a cyclin-dependent kinase inhibitor (CDKI) encoded by the Cdkn2a locus, which sequesters cdk4 and cdk6, thus preventing their interaction with the D cyclins. It has been shown that p16INK4a expression increases with age in several mouse tissues, including islets (125), and that proliferation of beta cells in young mice was reduced to levels observed in older mice when the transgenic overexpression of p16INK4a was induced (125, 126). On the other hand, in p16INK4a knockout mice, beta-cell proliferation was significantly increased (126). In this context, it could be very intriguing to mention that genome-wide association studies revealed an association between SNPs near Cdk2a (the locus encoding p16INK4a) and increased risk of type 2 diabetes (113, 127, 128). It has also been shown that free fatty acids, whose levels were typically increased in type 2 diabetes and that could be responsible for beta-cell damage (129), can induce p16INK4a expression in islets (130). Thus, p16INK4a could represent a potential link between aging, metabolic derangements, and beta-cell failure in type 2 diabets (131). More recently, it has been shown that the age-associated decrease in p16INK4a expression in pancreatic islets could be related to the decreased expression of BMI1 (132) and EZH2 (133), two chromatin-regulating polycomb group proteins, indicating the crucial role that epigenetic regulation could play in the control of cell cycle progression of beta cells in both aging and type 2 diabetes (134). Indeed, mice with conditional gene inactivation of EZH2 in beta cells exhibited a premature increase in p16INK4a and p19arf expression and a reduced beta-cell proliferation, whereas no changes were observed in the levels of other CDK inhibitors, suggesting a specific effect of EZH2 on the INK4a/arf locus in beta cells (133). However, the transgenic expression of EZH2 was unable to repress INK4a in mice older than 8 months, unless EZH2 was expressed in conjunction with knockdown of trithorax group (TrxG) protein complex components (135).
Overall, these results indicate that cellular senescence could be responsible for the observed decline in the proliferative capacity of pancreatic beta cells. It has been reported that Akita mice with short telomeres are characterized by slower proliferation of beta cells and accumulation of p16INK4a (74). More recently, Zeng et al. (136) showed that in mice the beta-cell-specific genetic deletion of Pten (phosphatase and tensin homolog), encoding a tumor suppressor protein involved in the regulation of the cell cycle (137), prevents the age-related decline in beta-cell proliferation and restores the ability of beta cells to respond to injury-mediated regeneration. Interestingly, the ability of Pten deletion to remove the block in cell cycle re-entry seems to be mediated by a decrease in p16INK4a expression.
The decline in beta-cell proliferation with age may also be the result of an age-related impairment of mitotic signal transduction pathways. It has been shown that p38 MAPK signals are able to influence CDKI expression in aged islets: the destruction of p38 MAPK signals in aged mutant mice has as a consequence a reduced expression of p16INK4a, p19arf, and other CDKI with a related increase of beta-cell proliferation (138). This effect seems to be counterbalanced by the p53-induced phosphatase 1 (WIP1), whose overexpression in middle-aged transgenic mice causes a reduced p16INK4a expression as well as an improved capacity of beta-cell regeneration after selective beta-cell destruction by streptozotocin (138). A further important component linking growth signals to beta-cell expansion could likely be represented by Akt activation and its downstream mTORC1 signaling (137). It is well known that alterations in the nutrient-sensing pathways (such as the insulin/IGF-1 and the TOR pathways) have been proposed to underlie the aging process and modulate longevity (139). mTOR is an evolutionarily conserved nutrient-sensing cytoplasmic protein kinase that regulates cell growth and metabolism in response to mitogens, nutrients, and hormones in all eukaryotic cells (140). However, later in life, when growth has been completed, mTOR can drive cellular and organismal aging (141) and can be involved in age-related diseases (138). Indeed, the most well-known TOR inhibitor, rapamycin, is able to extend lifespan in yeast, flies, worms, and rodents (142). Glucose, amino acids, and fatty acids activate mTOR in beta cells, and the consequent increase in beta-cell mass and function may help to compensate the age-related development of insulin resistance (143). However, it has been proposed that, during aging, the chronic hyperstimulation of mTOR could contribute to the development of beta-cell failure (143). Interestingly, metformin, the most widely used anti-diabetic drug, has been shown to be an inhibitor of mTORC1 and to decrease the phosphorylation of its substrates S6K1 and 4E-BP1 (144). Metformin was also shown to increase longevity in species ranging from yeast to mice (145). The underlying mechanism of this action of metformin is not fully understood. However, it is known that metformin inhibits the activity of mitochondrial complex I and increases the activity of AMPK, which in turn inhibits mTORC1 complex activity, thus suggesting a possible link between rapamycin and metformin actions on longevity.
Little is known about the upstream signals that could be responsible for the regulation of beta-cell proliferation and its decline with age. It has been reported that PDGF treatment increased beta-cell proliferation in cultured human islets from young donors but not in islets from adults. Interestingly, PDGF receptor signals seem to act in part via EZH2 (146). Treatment with the glucagon-like peptide 1 (GLP-1) analog, exendin-4 is able to increase beta-cell mass and markedly decrease p16INK4a expression in young but not in middle-aged mice (147). Recently, it has been shown with parabiosis experiments that a systemic factor (whose exact nature is still unknown) found in the circulation of young mice seems to be able to increase the proliferation rate of old pancreatic beta cells (148).
Conclusion
Alterations of glucose homeostasis increase with age and represent leading causes of morbidity and mortality, mainly linked to both the complications associated with type 2 diabetes and the increased risk for several other age-related diseases (149). The classical pathophysiological factors responsible for this age-related failure of glucose homeostasis (insulin resistance and decreased secretory capability of beta cells) are quite well characterized, but new mechanisms have recently been revealed (Figure 2). Central to this new development is the key concept that loss or dysfunction of pancreatic beta cells plays a crucial role in the pathogenesis of type 2 diabetes. Since the predominant mechanism of beta-cell generation seems to be self-renewal, the senescence-associated cell cycle dysregulation and the consequent proliferative arrest assume a particular relevance. In recent years, some of the cellular and molecular mechanisms associated with the decreased proliferation capability of senescent beta cells have been explored, but some others remain to be fully elucidated, and a further effort will be requested in order to efficiently translate this new insight into successful new therapeutic strategies.
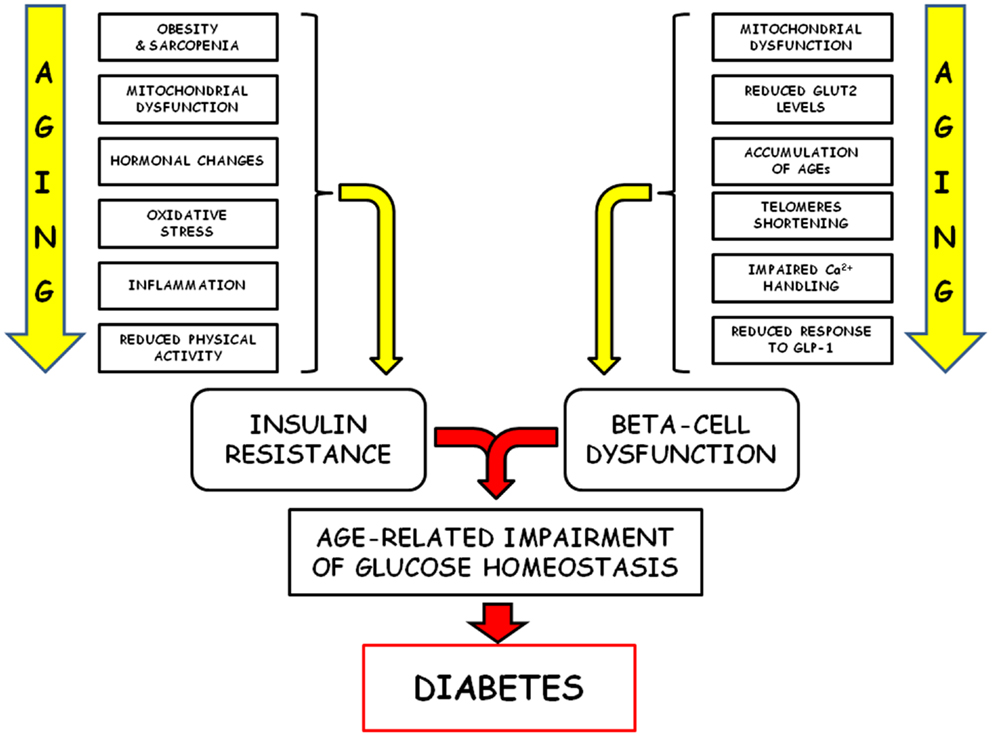
Figure 2. Schematic representation of the pathophysiological factors responsible of the age-related failure of glucose homeostasis is shown.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care (2004) 27:1047–53. doi: 10.2337/diacare.27.10.2569-a
2. Kolb H, Mandrup-Poulsen T. The global diabetes epidemics as a consequence of lifestyle-induced low-grade inflammation. Diabetologia (2010) 53:10–20. doi:10.1007/s00125-009-1573-7
3. Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract (2010) 87:4–14. doi:10.1016/j.diabres.2009.10.007
4. Kowal P, Chatterji S, Naidoo N, Biritwum R, Fan W, Lopez Ridaura R, et al. Data resource profile: the World Health Organization Study on global AGEing and adult health (SAGE). Int J Epidemiol (2012) 41:1639–49. doi:10.1093/ije/dys210
5. Michalakis K, Goulis DG, Vazaiou A, Mintziori G, Polymeris A, Abrahamian-Michalakis A. Obesity in the ageing man. Metabolism (2013) 62:1341–9. doi:10.1016/j.metabol.2013.05.019
6. Christensen K, Doblhammer G, Rau R, Vaupel JW. Ageing populations: the challenges ahead. Lancet (2009) 374:1196–208. doi:10.1016/S0140-6736(09)61460-4
7. National Institutes of Health (NIH). Global Health and Aging. NIH Publication no. 11-7737. Washington, DC: Department of Health and Uman Services (2011).
8. Kalyani RR, Egan JM. Diabetes and altered glucose metabolism with aging. Endocrinol Metab Clin North Am (2013) 42:333–47. doi:10.1016/j.ecl.2013.02.010
9. Gong Z, Muzumdar RH. Pancreatic function, type 2 diabetes, and metabolism in aging. Int J Endocrinol (2012) 2012:320482. doi:10.1155/2012/320482
10. Gunasekaran U, Gannon M. Type 2 diabetes and the aging pancreatic beta cell. Aging (Albany NY) (2011) 3:565–75.
11. Iozzo P, Beck-Nielsen H, Laakso M, Smith U, Yki-Järvinen H, Ferrannini E. Independent influence of age on basal insulin secretion in nondiabetic humans. European Group for the Study of Insulin Resistance. J Clin Endocrinol Metab (1999) 84:863–8. doi:10.1210/jcem.84.3.5542
12. Cowie CC, Rust KF, Byrd-Holt DD, Eberhardt MS, Flegal KM, Engelgau MM, et al. Prevalence of diabetes and impaired fasting glucose in adults in the U.S. population: National Health And Nutrition Examination Survey 1999-2002. Diabetes Care (2006) 29:1263–8. doi:10.2337/dc06-0062
13. Yeap BB. Hormones and health outcomes in aging men. Exp Gerontol (2013) 48:677–81. doi:10.1016/j.exger.2012.07.012
14. Batrinos ML. The aging of the endocrine hypothalamus and its dependent endocrine glands. Hormones (Athens) (2012) 11:241–53. doi:10.14310/horm.2002.1354
15. Clegg A, Young J, Iliffe S, Rikkert MO, Rockwood K. Frailty in elderly people. Lancet (2013) 381:752–62. doi:10.1016/S0140-6736(12)62167-9
16. Vitale G, Salvioli S, Franceschi C. Oxidative stress and the ageing endocrine system. Nat Rev Endocrinol (2013) 9:228–40. doi:10.1038/nrendo.2013.29
17. DeFronzo RA. Glucose intolerance and aging. Diabetes Care (1981) 4:493–501. doi:10.2337/diacare.4.4.493
18. Jackson RA. Mechanisms of age-related glucose intolerance. Diabetes Care (1990) 13(Suppl 2):9–19.
19. Nolan CJ, Damm P, Prentki M. Type 2 diabetes across generations: from pathophysiology to prevention and management. Lancet (2011) 378:169–81. doi:10.1016/S0140-6736(11)60614-4
20. Fink RI, Kolterman OG, Griffin J, Olefsky JM. Mechanisms of insulin resistance in aging. J Clin Invest (1983) 71:1523–35. doi:10.1172/JCI110908
21. Choi K, Kim YB. Molecular mechanism of insulin resistance in obesity and type 2 diabetes. Korean J Intern Med (2010) 25:119–29. doi:10.3904/kjim.2010.25.2.119
22. Saini V. Molecular mechanisms of insulin resistance in type 2 diabetes mellitus. World J Diabetes (2010) 1:68–75. doi:10.4239/wjd.v1.i3.68
23. Defronzo RA. Glucose intolerance and aging: evidence for tissue insensitivity to insulin. Diabetes (1979) 28:1095–101. doi:10.2337/diab.28.12.1095
24. Rowe JW, Minaker KL, Pallotta JA, Flier JS. Characterization of the insulin resistance of aging. J Clin Invest (1983) 71:1581–7. doi:10.1172/JCI110914
25. Kim JE, Lee YH, Huh JH, Kang DR, Rhee Y, Lim SK. Early-stage chronic kidney disease, insulin resistance, and osteoporosis as risk factors of sarcopenia in aged population: the Fourth Korea National Health and Nutrition Examination Survey (KNHANES IV), 2008-2009. Osteoporos Int (2014) 25:2189–98. doi:10.1007/s00198-014-2745-y
26. Ostler JE, Maurya SK, Dials J, Roof SR, Devor ST, Ziolo MT, et al. Effects of insulin resistance on skeletal muscle growth and exercise capacity in type 2 diabetic mouse models. Am J Physiol Endocrinol Metab (2014) 306:E592–605. doi:10.1152/ajpendo.00277.2013
27. Atkins JL, Whincup PH, Morris RW, Wannamethee SG. Low muscle mass in older men: the role of lifestyle, diet and cardiovascular risk factors. J Nutr Health Aging (2014) 18:26–33. doi:10.1007/s12603-013-0336-9
28. Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science (2003) 300:1140–2. doi:10.1126/science.1082889
29. Toledo FG, Goodpaster BH. The role of weight loss and exercise in correcting skeletal muscle mitochondrial abnormalities in obesity, diabetes and aging. Mol Cell Endocrinol (2013) 379:30–4. doi:10.1016/j.mce.2013.06.018
30. Phielix E, Szendroedi J, Roden M. Mitochondrial function and insulin resistance during aging: a mini-review. Gerontology (2011) 57:387–96. doi:10.1159/000317691
31. Dela F, Helge JW. Insulin resistance and mitochondrial function in skeletal muscle. Int J Biochem Cell Biol (2013) 45:11–5. doi:10.1016/j.biocel.2012.09.019
32. Bertrand C, Blanchet E, Pessemesse L, Annicotte JS, Feillet-Coudray C, Chabi B, et al. Mice lacking the p43 mitochondrial T3 receptor become glucose intolerant and insulin resistant during aging. PLoS One (2013) 8:e75111. doi:10.1371/journal.pone.0075111
33. Maggio M, Lauretani F, Ceda GP, Bandinelli S, Basaria S, Paolisso G, et al. Association of hormonal dysregulation with metabolic syndrome in older women: data from the InCHIANTI study. Am J Physiol Endocrinol Metab (2007) 292:E353–8. doi:10.1152/ajpendo.00339.2006
34. Barzilai N, Huffman DM, Muzumdar RH, Bartke A. The critical role of metabolic pathways in aging. Diabetes (2012) 61:1315–22. doi:10.2337/db11-1300
35. Junnila RK, List EO, Berryman DE, Murrey JW, Kopchick JJ. The GH/IGF-1 axis in ageing and longevity. Nat Rev Endocrinol (2013) 9:366–76. doi:10.1038/nrendo.2013.67
36. Rains JL, Jain SK. Oxidative stress, insulin signaling, and diabetes. Free Radic Biol Med (2011) 50:567–75. doi:10.1016/j.freeradbiomed.2010.12.006
37. Solas M, Aisa B, Tordera RM, Mugueta MC, Ramírez MJ. Stress contributes to the development of central insulin resistance during aging: implications for Alzheimer’s disease. Biochim Biophys Acta (2013) 1832:2332–9. doi:10.1016/j.bbadis.2013.09.013
38. Kim JH, Park HY, Bae S, Lim YH, Hong YC. Diethylhexyl phthalates is associated with insulin resistance via oxidative stress in the elderly: a panel study. PLoS One (2013) 8:e71392. doi:10.1371/journal.pone.0071392
39. Singh T, Newman AB. Inflammatory markers in population studies of aging. Ageing Res Rev (2011) 10:319–29. doi:10.1016/j.arr.2010.11.002
40. Morley JE. Nutrition and the aging male. Clin Geriatr Med (2010) 26:287–99. doi:10.1016/j.cger.2010.02.008
41. Anderson AL, Harris TB, Tylavsky FA, Perry SE, Houston DK, Lee JS, et al. Dietary patterns, insulin sensitivity and inflammation in older adults. Eur J Clin Nutr (2012) 66:18–24. doi:10.1038/ejcn.2011.162
42. Ruiz-Núñez B, Pruimboom L, Dijck-Brouwer DA, Muskiet FA. Lifestyle and nutritional imbalances associated with Western diseases: causes and consequences of chronic systemic low-grade inflammation in an evolutionary context. J Nutr Biochem (2013) 24:1183–201. doi:10.1016/j.jnutbio.2013.02.009
43. Bunprajun T, Henriksen TI, Scheele C, Pedersen BK, Green CJ. Lifelong physical activity prevents aging-associated insulin resistance in human skeletal muscle myotubes via increased glucose transporter expression. PLoS One (2013) 8:e66628. doi:10.1371/journal.pone.0066628
44. León-Latre M, Moreno-Franco B, Andrés-Esteban EM, Ledesma M, Laclaustra M, Alcalde V, et al. Sedentary lifestyle and its relation to cardiovascular risk factors, insulin resistance and inflammatory profile. Rev Esp Cardiol (2014) 67:449–55. doi:10.1016/j.rec.2013.10.015
45. Coon PJ, Rogus EM, Drinkwater D, Muller DC, Goldberg AP. Role of body fat distribution in the decline in insulin sensitivity and glucose tolerance with age. J Clin Endocrinol Metab (1992) 75:1125–32. doi:10.1210/jc.75.4.1125
46. Goulet ED, Hassaine A, Dionne IJ, Gaudreau P, Khalil A, Fulop T, et al. Frailty in the elderly is associated with insulin resistance of glucose metabolism in the postabsorptive state only in the presence of increased abdominal fat. Exp Gerontol (2009) 44:740–4. doi:10.1016/j.exger.2009.08.008
47. Soriguer F, Colomo N, Valdés S, Goday A, Rubio-Martín E, Esteva I, et al. Modifications of the homeostasis model assessment of insulin resistance index with age. Acta Diabetol (2014). doi:10.1007/s00592-013-0523-5
48. Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest (2006) 116:1802–12. doi:10.1172/JCI29103
49. Ashcroft FM, Rorsman P. Diabetes mellitus and the β cell: the last ten years. Cell (2012) 148:1160–71. doi:10.1016/j.cell.2012.02.010
50. Scheen AJ. Diabetes mellitus in the elderly: insulin resistance and/or impaired insulin secretion? Diabetes Metab (2005) 31:S27–34. doi:10.1016/S1262-3636(05)73649-1
51. Bergamini E, Bombara M, Fierabracci V, Masiello P, Novelli M. Effects of different regimens of dietary restriction on the age-related decline in insulin secretory response of isolated rat pancreatic islets. Ann N Y Acad Sci (1991) 621:327–36. doi:10.1111/j.1749-6632.1991.tb16989.x
52. Novelli M, De Tata V, Bombara M, Lorenzini A, Masini M, Pollera M, et al. Insufficient adaptive capability of pancreatic endocrine function in dexamethasone-treated ageing rats. J Endocrinol (1999) 162:425–32. doi:10.1677/joe.0.1620425
53. Barbera M, Fierabracci V, Novelli M, Bombara M, Masiello P, Bergamini E, et al. Dexamethasone-induced insulin resistance and pancreatic adaptive response in aging rats are not modified by oral vanadyl sulfate treatment. Eur J Endocrinol (2001) 145:799–806. doi:10.1530/eje.0.1450799
54. Ihm SH, Moon HJ, Kang JG, Park CY, Oh KW, Jeong IK, et al. Effect of aging on insulin secretory function and expression of beta cell function-related genes of islets. Diabetes Res Clin Pract (2007) 77(Suppl 1):S150–4. doi:10.1016/j.diabres.2007.01.049
55. Wu D, Yue F, Zou C, Chan P, Alex Zhang Y. Analysis of glucose metabolism in cynomolgus monkeys during aging. Biogerontology (2012) 13:147–55. doi:10.1007/s10522-011-9364-1
56. Ihm SH, Matsumoto I, Sawada T, Nakano M, Zhang HJ, Ansite JD, et al. Effect of donor age on function of isolated human islets. Diabetes (2006) 55:1361–8. doi:10.2337/db05-1333
57. Chang AM, Smith MJ, Galecki AT, Bloem CJ, Halter JB. Impaired beta-cell function in human aging: response to nicotinic acid-induced insulin resistance. J Clin Endocrinol Metab (2006) 91:3303–9. doi:10.1210/jc.2006-0913
58. Cobelli C, Toffolo GM, Dalla Man C, Campioni M, Denti P, Caumo A, et al. Assessment of beta-cell function in humans, simultaneously with insulin sensitivity and hepatic extraction, from intravenous and oral glucose tests. Am J Physiol Endocrinol Metab (2007) 293:E1–15. doi:10.1152/ajpendo.00421.2006
59. Szoke E, Shrayyef MZ, Messing S, Woerle HJ, van Haeften TW, Meyer C, et al. Effect of aging on glucose homeostasis: accelerated deterioration of beta-cell function in individuals with impaired glucose tolerance. Diabetes Care (2008) 31:539–43. doi:10.2337/dc07-1443
60. Haupt A, Staiger H, Schäfer SA, Kirchhoff K, Guthoff M, Machicao F, et al. The risk allele load accelerates the age-dependent decline in beta cell function. Diabetologia (2009) 52:457–62. doi:10.1007/s00125-008-1250-2
61. Ma X, Becker D, Arena VC, Vicini P, Greenbaum C. The effect of age on insulin sensitivity and insulin secretion in first-degree relatives of type 1 diabetic patients: a population analysis. J Clin Endocrinol Metab (2009) 94:2446–51. doi:10.1210/jc.2008-2687
62. Trahair LG, Horowitz M, Rayner CK, Gentilcore D, Lange K, Wishart JM, et al. Comparative effects of variations in duodenal glucose load on glycemic, insulinemic, and incretin responses in healthy young and older subjects. J Clin Endocrinol Metab (2012) 97:844–51. doi:10.1210/jc.2011-2583
63. Xiao J, Weng J, Ji L, Jia W, Lu J, Shan Z, et al. China National Diabetes and Metabolic Disorders Study Group. Worse pancreatic β-cell function and better insulin sensitivity in older Chinese without diabetes. J Gerontol A Biol Sci Med Sci (2014) 69:463–70. doi:10.1093/gerona/glt104
64. Oya J, Nakagami T, Yamamoto Y, Fukushima S, Takeda M, Endo Y, et al. Effects of age on insulin resistance and secretion in subjects without diabetes. Intern Med (2014) 53:941–7. doi:10.2169/internalmedicine.53.1580
65. Geloneze B, de Oliveira MD, Vasques AC, Novaes FS, Pareja JC, Tambascia MA. Impaired incretin secretion and pancreatic dysfunction with older age and diabetes. Metabolism (2014) 63:922–9. doi:10.1016/j.metabol.2014.04.004
66. Chang AM, Halter JB. Aging and insulin secretion. Am J Physiol Endocrinol Metab (2003) 284:E7–12.
67. Cree LM, Patel SK, Pyle A, Lynn S, Turnbull DM, Chinnery PF, et al. Age-related decline in mitochondrial DNA copy number in isolated human pancreatic islets. Diabetologia (2008) 51:1440–3. doi:10.1007/s00125-008-1054-4
68. Supale S, Thorel F, Merkwirth C, Gjinovci A, Herrera PL, Scorrano L, et al. Loss of prohibitin induces mitochondrial damages altering β-cell function and survival and is responsible for gradual diabetes development. Diabetes (2013) 62:3488–99. doi:10.2337/db13-0152
69. Jain D, Jain R, Eberhard D, Eglinger J, Bugliani M, Piemonti L, et al. Age- and diet-dependent requirement of DJ-1 for glucose homeostasis in mice with implications for human type 2 diabetes. J Mol Cell Biol (2012) 4:221–30. doi:10.1093/jmcb/mjs025
70. Novelli M, De Tata V, Bombara M, Bergamini E, Masiello P. Age-dependent reduction in GLUT-2 levels is correlated with the impairment of the insulin secretory response in isolated islets of Sprague-Dawley rats. Exp Gerontol (0000) 200035:641–51.
71. Coughlan MT, Yap FY, Tong DC, Andrikopoulos S, Gasser A, Thallas-Bonke V, et al. Advanced glycation end products are direct modulators of β-cell function. Diabetes (2011) 60:2523–32. doi:10.2337/db10-1033
72. Zhao Z, Zhao C, Zhang XH, Zheng F, Cai W, Vlassara H, et al. Advanced glycation end products inhibit glucose-stimulated insulin secretion through nitric oxide-dependent inhibition of cytochrome c oxidase and adenosine triphosphate synthesis. Endocrinology (2009) 150:2569–76. doi:10.1210/en.2008-1342
73. Kuhlow D, Florian S, von Figura G, Weimer S, Schulz N, Petzke KJ, et al. Telomerase deficiency impairs glucose metabolism and insulin secretion. Aging (Albany NY) (2010) 2:650–8.
74. Guo N, Parry EM, Li LS, Kembou F, Lauder N, Hussain MA, et al. Short telomeres compromise β-cell signaling and survival. PLoS One (2011) 6:e17858. doi:10.1371/journal.pone.0017858
75. Santulli G, Lombardi A, Sorriento D, Anastasio A, Del Giudice C, Formisano P, et al. Age-related impairment in insulin release: the essential role of β(2)-adrenergic receptor. Diabetes (2012) 61:692–701. doi:10.2337/db11-1027
76. Ribeiro RA, Batista TM, Coelho FM, Boschero AC, Lopes GS, Carneiro EM. Decreased β-cell insulin secretory function in aged rats due to impaired Ca(2+) handling. Exp Physiol (2012) 97:1065–73. doi:10.1113/expphysiol.2012.064790
77. Lin Y, Sun Z. Antiaging gene Klotho enhances glucose-induced insulin secretion by up-regulating plasma membrane levels of TRPV2 in MIN6 β-cells. Endocrinology (2012) 153:3029–39. doi:10.1210/en.2012-1091
78. Elahi D, Andersen DK, Muller DC, Tobin JD, Brown JC, Andres R. The enteric enhancement of glucose-stimulated insulin release. The role of GIP in aging, obesity, and non-insulin-dependent diabetes mellitus. Diabetes (1984) 33:950–7. doi:10.2337/diab.33.10.950
79. Meneilly GS, Ryan AS, Minaker KL, Elahi D. The effect of age and glycemic level on the response of the beta-cell to glucose-dependent insulinotropic polypeptide and peripheral tissue sensitivity to endogenously released insulin. J Clin Endocrinol Metab (1998) 83:2925–32. doi:10.1210/jc.83.8.2925
80. Wang Y, Perfetti R, Greig NH, Holloway HW, Deore KA, Montrose-Rafizadeh C, et al. Glucagon-like peptide-1 can reverse the age-related decline in glucose tolerance in rats. J Clin Invest (1997) 99:2883–9. doi:10.1172/JCI119482
81. Fan R, Kang Z, He L, Chan J, Xu G. Exendin-4 improves blood glucose control in both young and aging normal non-diabetic mice, possible contribution of beta cell independent effects. PLoS One (2011) 6:e20443. doi:10.1371/journal.pone.0020443
82. Irwin N, McClean PL, Harriott P, Flatt PR. Beneficial effects of sub-chronic activation of glucagon-like peptide-1 (GLP-1) receptors on deterioration of glucose homeostasis and insulin secretion in aging mice. Exp Gerontol (2007) 42:296–300. doi:10.1016/j.exger.2006.10.017
83. Meneilly GS, Veldhuis JD, Elahi D. Deconvolution analysis of rapid insulin pulses before and after six weeks of continuous subcutaneous administration of glucagon-like peptide-1 in elderly patients with type 2 diabetes. J Clin Endocrinol Metab (2005) 90:6251–6. doi:10.1210/jc.2004-2100
84. Wang S, Sun QQ, Xiang B, Li XJ. Pancreatic islet cell autophagy during aging in rats. Clin Invest Med (2013) 36:E72–80.
85. Maechler P, Carobbio S, Rubi B. In beta-cells, mitochondria integrate and generate metabolic signals controlling insulin secretion. Int J Biochem Cell Biol (2006) 38:696–709.
87. Dai DF, Chiao YA, Marcinek DJ, Szeto HH, Rabinovitch PS. Mitochondrial oxidative stress in aging and healthspan. Longev Healthspan (2014) 3:6. doi:10.1186/2046-2395-3-6
88. Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab (2013) 17:491–506. doi:10.1016/j.cmet.2013.03.002
89. Campisi J, Andersen JK, Kapahi P, Melov S. Cellular senescence: a link between cancer and age-related degenerative disease? Semin Cancer Biol (2011) 21:354–9. doi:10.1016/j.semcancer.2011.09.001
90. Basu R, Breda E, Oberg AL, Powell CC, Dalla Man C, Basu A, et al. Mechanisms of the age-associated deterioration in glucose tolerance: contribution of alterations in insulin secretion, action, and clearance. Diabetes (2003) 52:1738–48.
91. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res (1961) 25:585–621. doi:10.1016/0014-4827(61)90192-6
92. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol (2011) 192:547–56. doi:10.1083/jcb.201009094
93. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest (2013) 123:966–72. doi:10.1172/JCI64098
94. Desgraz R, Bonal C, Herrera PL. β-cell regeneration: the pancreatic intrinsic faculty. Trends Endocrinol Metab (2010) 22:34–43. doi:10.1016/j.tem.2010.09.004
95. Ding L, Gysemans C, Mathieu C. β-cell differentiation and regeneration in type 1 diabetes. Diabetes Obes Metab (2013) 15(Suppl 3):98–104. doi:10.1111/dom.12164
96. Tavana O, Zhu C. Too many breaks (brakes): pancreatic β-cell senescence leads to diabetes. Cell Cycle (2011) 10:2471–84. doi:10.4161/cc.10.15.16741
97. Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes (2005) 54:2557–67. doi:10.2337/diabetes.54.9.2557
98. Herrera PL, Huarte J, Zufferey R, Nichols A, Mermillod B, Philippe J, et al. Ablation of islet endocrine cells by targeted expression of hormone-promoter-driven toxigenes. Proc Natl Acad Sci U S A (1994) 91:12999–3003. doi:10.1073/pnas.91.26.12999
99. Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. J Clin Invest (2007) 117:2553–61. doi:10.1172/JCI32959
100. Thorel F, Népote V, Avril I, Kohno K, Desgraz R, Chera S, et al. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature (2010) 464:1149–54. doi:10.1038/nature08894
101. Xu X, D’Hoker J, Stangé G, Bonné S, De Leu N, Xiao X, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell (2008) 132:197–207. doi:10.1016/j.cell.2007.12.015
102. Stolovich-Rain M, Hija A, Grimsby J, Glaser B, Dor Y. Pancreatic beta cells in very old mice retain capacity for compensatory proliferation. J Biol Chem (2012) 287:27407–14. doi:10.1074/jbc.M112.350736
103. Chen X, Zhang X, Chen F, Larson CS, Wang LJ, Kaufman DB. Comparative study of regenerative potential of beta cells from young and aged donor mice using a novel islet transplantation model. Transplantation (2009) 88:496–503. doi:10.1097/TP.0b013e3181b0d2ee
104. Tian L, Gao J, Weng G, Yi H, Tian B, O’Brien TD, et al. Comparison of exendin-4 on beta-cell replication in mouse and human islet grafts. Transpl Int (2011) 24:856–64. doi:10.1111/j.1432-2277.2011.01275.x
105. Gianani R. Beta cell regeneration in human pancreas. Semin Immunopathol (2011) 33:23–7. doi:10.1007/s00281-010-0235-7
106. Brüning JC, Winnay J, Bonner-Weir S, Taylor SI, Accili D, Kahn CR. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell (1997) 88:561–72. doi:10.1016/S0092-8674(00)81896-6
107. Klöppel G, Löhr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res (1985) 4:110–25.
108. Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab (2008) 10(Suppl 4):32–42. doi:10.1111/j.1463-1326.2008.00969.x
109. Rieck S, Kaestner KH. Expansion of beta-cell mass in response to pregnancy. Trends Endocrinol Metab (2010) 21:151–8. doi:10.1016/j.tem.2009.11.001
110. Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev (1998) 12:3008–19. doi:10.1101/gad.12.19.3008
111. Zhu J, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev (1998) 12:2997–3007. doi:10.1101/gad.12.19.2997
112. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes (2003) 52:102–10. doi:10.2337/diabetes.52.1.102
113. Reers C, Erbel S, Esposito I, Schmied B, Büchler MW, Nawroth PP, et al. Impaired islet turnover in human donor pancreata with aging. Eur J Endocrinol (2009) 160:185–91. doi:10.1530/EJE-08-0596
114. Forsyth NR, Wright WE, Shay JW. Telomerase and differentiation in multicellular organisms: turn it off, turn it on, and turn it off again. Differentiation (2002) 69:188–97. doi:10.1046/j.1432-0436.2002.690412.x
115. Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes (2008) 57:1584–94. doi:10.2337/db07-1369
116. Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care (2013) 36:111–7. doi:10.2337/dc12-0421
117. Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, et al. Formation of a human β-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab (2012) 97:3197–206. doi:10.1210/jc.2012-1206
118. Perl S, Kushner JA, Buchholz BA, Meeker AK, Stein GM, Hsieh M, et al. Significant human beta-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J Clin Endocrinol Metab (2010) 95:E234–9. doi:10.1210/jc.2010-0932
119. Cnop M, Hughes SJ, Igoillo-Esteve M, Hoppa MB, Sayyed F, van de Laar L, et al. The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia (2010) 53:321–30. doi:10.1007/s00125-009-1562-x
120. Cnop M, Igoillo-Esteve M, Hughes SJ, Walker JN, Cnop I, Clark A. Longevity of human islet α- and β-cells. Diabetes Obes Metab (2011) 13(Suppl 1):39–46. doi:10.1111/j.1463-1326.2011.01443.x
121. Maedler K, Schumann DM, Schulthess F, Oberholzer J, Bosco D, Berney T, et al. Aging correlates with decreased beta-cell proliferative capacity and enhanced sensitivity to apoptosis: a potential role for Fas and pancreatic duodenal homeobox-1. Diabetes (2006) 55:2455–62. doi:10.2337/db05-1586
122. Scharfmann R. Expanding human beta cells. Diabetologia (2008) 51:692–3. doi:10.1007/s00125-007-0909-4
123. Gannon M, Ables ET, Crawford L, Lowe D, Offield MF, Magnuson MA, et al. Pdx-1 function is specifically required in embryonic beta cells to generate appropriate numbers of endocrine cell types and maintain glucose homeostasis. Dev Biol (2008) 314:406–17. doi:10.1016/j.ydbio.2007.10.038
124. Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature (2006) 443:453–7. doi:10.1038/nature05092
125. Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Invest (2004) 114:1299–307. doi:10.1172/JCI200422475
126. Diabetes Genetics Initiative of Broad Institute of Harvard and MITL and University and Novartis Institutes of BioMedical Research, Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science (2007) 316:1331–6. doi:10.1126/science.1142358
127. Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science (2007) 316:1341–5. doi:10.1126/science.1142382
128. Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science (2007) 316:1336–41. doi:10.1126/science.1142364
129. Martino L, Masini M, Novelli M, Beffy P, Bugliani M, Marselli L, et al. Palmitate activates autophagy in INS-1E β-cells and in isolated rat and human pancreatic islets. PLoS One (2012) 7:e36188. doi:10.1371/journal.pone.0036188
130. Pascoe J, Hollern D, Stamateris R, Abbasi M, Romano LC, Zou B, et al. Free fatty acids block glucose-induced β-cell proliferation in mice by inducing cell cycle inhibitors p16 and p18. Diabetes (2012) 61:632–41. doi:10.2337/db11-0991
131. Kushner JA. The role of aging upon β cell turnover. J Clin Invest (2013) 123:990–5. doi:10.1172/JCI64095
132. Dhawan S, Tschen SI, Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev (2009) 23:906–11. doi:10.1101/gad.1742609
133. Chen H, Gu X, Su IH, Bottino R, Contreras JL, Tarakhovsky A, et al. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev (2009) 23:975–85. doi:10.1101/gad.1742509
134. Gilbert ER, Liu D. Epigenetics: the missing link to understanding β-cell dysfunction in the pathogenesis of type 2 diabetes. Epigenetics (2012) 7:841–52. doi:10.4161/epi.21238
135. Zhou JX, Dhawan S, Fu H, Snyder E, Bottino R, Kundu S, et al. Combined modulation of polycomb and trithorax genes rejuvenates β cell replication. J Clin Invest (2013) 123:4849–58. doi:10.1172/JCI69468
136. Zeng N, Yang KT, Bayan JA, He L, Aggarwal R, Stiles JW, et al. PTEN controls β-cell regeneration in aged mice by regulating cell cycle inhibitor p16ink4a. Aging Cell (2013) 12:1000–11. doi:10.1111/acel.12132
137. Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature (2005) 436:725–30. doi:10.1038/nature03918
138. Wong ES, Le Guezennec X, Demidov ON, Marshall NT, Wang ST, Krishnamurthy J, et al. p38MAPK controls expression of multiple cell cycle inhibitors and islet proliferation with advancing age. Dev Cell (2009) 17:142–9. doi:10.1016/j.devcel.2009.05.009
139. Newgard CB, Sharpless NE. Coming of age: molecular drivers of aging and therapeutic opportunities. J Clin Invest (2013) 123:946–50. doi:10.1172/JCI68833
140. Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, et al. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab (2010) 11:453–65. doi:10.1016/j.cmet.2010.05.001
141. Blandino-Rosano M, Chen AY, Scheys JO, Alejandro EU, Gould AP, Taranukha T, et al. mTORC1 signaling and regulation of pancreatic β-cell mass. Cell Cycle (2012) 11:1892–902. doi:10.4161/cc.20036
142. Galluzzi L, Kepp O, Kroemer G. TP53 and MTOR crosstalk to regulate cellular senescence. Aging (Albany NY) (2010) 2:535–7.
143. Blagosklonny MV. TOR-centric view on insulin resistance and diabetic complications: perspective for endocrinologists and gerontologists. Cell Death Dis (2013) 4:e964. doi:10.1038/cddis.2013.506
144. Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res (2007) 67:10804–12. doi:10.1158/0008-5472.CAN-07-2310
145. Lamming DW, Ye L, Sabatini DM, Baur JA. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest (2013) 123:980–9. doi:10.1172/JCI64099
146. Chen H, Gu X, Liu Y, Wang J, Wirt SE, Bottino R, et al. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature (2011) 478:349–55. doi:10.1038/nature10502
147. Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes (2009) 58:1312–20. doi:10.2337/db08-1651
148. Salpeter SJ, Khalaileh A, Weinberg-Corem N, Ziv O, Glaser B, Dor Y. Systemic regulation of the age-related decline of pancreatic β-cell replication. Diabetes (2013) 62:2843–8. doi:10.2337/db13-0160
Keywords: aging, pancreatic beta cells, insulin secretion, insulin resistance, cellular senescence
Citation: De Tata V (2014) Age-related impairment of pancreatic beta-cell function: pathophysiological and cellular mechanisms. Front. Endocrinol. 5:138. doi: 10.3389/fendo.2014.00138
Received: 26 June 2014; Accepted: 07 August 2014;
Published online: 03 September 2014.
Edited by:
Andrzej Bartke, Southern Illinois University School of Medicine, USAReviewed by:
Rong Yuan, Southern Illinois University School of Medicine, USACiro Isidoro, Università del Piemonte Orientale, Italy
Copyright: © 2014 De Tata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincenzo De Tata, Department of Translational Research and New Technologies in Medicine and Surgery, University of Pisa, Via Roma, 55 Scuola Medica, Pisa 56126, Italy e-mail:dmluY2Vuem8uZGV0YXRhQG1lZC51bmlwaS5pdA==