 Nathalie Castanon
Nathalie Castanon Julie Lasselin
Julie Lasselin Lucile Capuron
Lucile Capuron- 1UMR 1286, Laboratory of Nutrition and Integrative Neurobiology (NutriNeuro), French National Institute for Agricultural Research (INRA), Bordeaux, France
- 2UMR 1286, Laboratory of Nutrition and Integrative Neurobiology (NutriNeuro), University of Bordeaux, Bordeaux, France
- 3Stress Research Institute (Stressforskningsinstitutet), Stockholm University, Stockholm, Sweden
Neuropsychiatric symptoms are frequent in obesity. In addition to their substantial economic and health impact, these symptoms significantly interfere with the quality of life and social function of obese individuals. While the pathophysiological mechanisms underlying obesity-related neuropsychiatric symptoms are still under investigation and remain to be clearly identified, there is increasing evidence for a role of inflammatory processes. Obesity is characterized by a chronic low-grade inflammatory state that is likely to influence neuropsychiatric status given the well-known and highly documented effects of inflammation on brain activity/function and behavior. This hypothesis is supported by recent findings emanating from clinical investigations in obese subjects and from experimentations conducted in animal models of obesity. These studies converge to show that obesity-related inflammatory processes, originating either from the adipose tissue or gut microbiota environment, spread to the brain where they lead to substantial changes in neurocircuitry, neuroendocrine activity, neurotransmitter metabolism and activity, and neurogenesis. Together, these alterations contribute to shape the propitious bases for the development of obesity-related neuropsychiatric comorbidities.
Introduction
The pandemic of obesity represents a major public health concern, as this disorder is associated with an increased risk of medical comorbidities contributing to a significant rise in mortality. Among those comorbidities related to obesity, neuropsychiatric disorders are particularly preoccupying. Not only neuropsychiatric symptoms affect the quality of life of obese subjects and contribute to their social impairment, but also they represent potent risk factors for aggravation of obesity. Given the growing prevalence of both obesity and neuropsychiatric disorders worldwide, the identification of the mechanisms underlying their comorbid association is urgently needed.
While different mechanisms are likely to be involved in the development of neuropsychiatric comorbidity in obesity, there is increasing evidence for a role of inflammatory processes. Chronic low-grade inflammation is an important characteristic of obesity and inflammatory processes are notorious for modulating brain functions and causing behavioral alterations. Recent clinical findings indicate that the increased systemic expression of inflammatory markers (e.g., cytokines) in obesity correlates with neuropsychiatric status, notably as it relates to mood and cognitive function. Moreover, studies in experimental animal models of obesity contribute to show that obesity-related inflammation manifests not only at the periphery but also within the brain where it modulates neurocircuitry, neurochemistry, and behavior. These findings that provide strong support to the notion that obesity-related inflammation plays an important role in the pathophysiology of neuropsychiatric symptoms will be presented and discussed in the present review.
Neuropsychiatric Comorbidity in Obesity
Neuropsychiatric comorbidity, including mood and anxiety disorders, binge eating, and mild cognitive impairment, is frequent in obesity and is associated with a significant reduction in the quality of life and social functioning of obese individuals. Among those disorders often seen in obese subjects, depressive symptoms are particularly frequent with a prevalence rate significantly higher (up to 30%) compared to the general age-matched population (1–5). Similarly, reports of cognitive disturbances in obesity are multiple. Those alterations concern primarily planning function, problem solving, mental flexibility, and inhibitory processes, suggestive of frontal lobe alterations (6–9). Impairment in memory, regardless of age, has been also reported (10, 11). Associations between obesity and cognitive impairment have been more often reported in cross-sectional studies comparing performance from overweight/obese subjects to performance from lean participants. Nevertheless, a predictive longitudinal association of obesity with the development of age-related cognitive deficit has also been documented in several reports (12–14).
The directionality of the relationship between obesity and neuropsychiatric symptoms is usually difficult to determine from clinical studies. The significant improvement in mood and cognitive function reported after weight loss induced by bariatric surgery or diet restriction in obese subjects supports the hypothesis that obesity significantly impacts neuropsychiatric status and contributes to the development of neuropsychiatric comorbidity (15–22). Nevertheless, other reports indicate that preexisting mood and cognitive alterations can promote and/or predict the development of later obesity (23–26), attesting of the bidirectional link between obesity and neuropsychiatric status. To further address this issue, animal models of obesity represent certainly a useful and unique opportunity. Several models of obesity resulting either from genetic manipulations or diet modifications have been developed over the last decades (27). Among those models, diet-induced obesity (DIO) is probably the closest to human obesity with respect to etiological aspects. Moreover, because of its longitudinal characteristic, DIO allows the investigation of the mechanisms and pathophysiological changes preceding the development of obesity-related comorbidities, including neuropsychiatric alterations. Genetic models of obesity, in particular severe obesity, are also of great interest to explore the genetic–metabolic–brain interactions associated with obesity-related comorbidities. In that context, ob/ob (deficient for leptin) and db/db (deficient for functional leptin receptor) mice are particularly relevant as, in addition to metabolic disorders, these mice also display brain alterations (28–30). Overall, behavioral changes reported in experimental models of obesity include alterations in emotional reactivity and impairment in learning and memory [for review, see Ref. (31)]. Relevant to neural function, significant decreases in hippocampal-dependent learning together with impaired hippocampal neurogenesis and neuronal plasticity have been documented in animal models of DIO, notably in young mice (32–35). Similar results were described in db/db mice (28–30), supporting the notion that the hippocampus plays a major role in mediating obesity-associated cognitive impairment. Interestingly, db/db mice also display anxiety-like behavior (36). In contrast, depressive-like behavior appears to be mostly unchanged in animal models of obesity (36, 37), except in challenging conditions including stimulation of the immune system (38–40). Altogether, these data comfort the notion that neuropsychiatric comorbidities in obesity rely on interactions involving multiple systems, including metabolic characteristics, environmental influences, and immune-related processes.
Obesity and Inflammation
Basal systemic low-grade inflammation is a fundamental characteristic of obesity, which is now considered not only as a metabolic disorder but also as an inflammatory condition affecting both the innate and acquired immune systems (41, 42). Obesity is characterized by increased levels of circulating proinflammatory cytokines [including interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and IL-6], accumulation of leukocytes within the adipose tissue and other organs, activation of macrophages in the liver and fat, and activation of proinflammatory signaling pathways in multiple organs (43, 44). Inflammatory markers in obesity correlate more with measures of central adiposity, such as waist circumference and waist-to-hip ratio, rather than with the general measure of body mass index (BMI) (45–48). Interestingly, significant improvement in inflammatory profile is obtained after weight loss induced by low-caloric diet or bariatric surgery in obese individuals (49–54).
Different mechanisms have been identified as playing a major role in the instauration of the chronic low-grade inflammatory state that characterizes obesity (Figure 1). One major player is the adipose tissue that has the ability to secrete adipokines and in which macrophages accumulate and potently secrete inflammatory factors (55–59). Moreover, an additional role for T cells in the development of adiposity-related inflammation is supported by several recent studies (57, 60–63). Obesity-related inflammation can also be triggered by pathogens, as there is now evidence of gut microbiota alterations associated with inflammatory processes in obesity (64, 65). In obese animals, gut microbial population is altered independently of diet characteristics (66), notably in the form of decreased Bacteroidetes and Bifidobacterium populations together with increases in the number of Firmicutes (64, 66, 67). High-fat diet is also associated with alterations in gut permeability leading to the instauration of a state of chronic low-grade endotoxemia (presence of lipopolysaccharide, LPS, in the blood) that is believed to contribute to obesity-related inflammation by the activation of systemic macrophages through the binding of LPS on TLR4 (68, 69). Similarly, recent clinical data in obese individuals indicate significant associations between gut microbiota modifications, reflected by the reduction in Bacteroidetes/Firmicutes ratio, and markers of local and systemic inflammation (67) and document improvement in the intestinal microbiota profile and in serum levels of an endotoxemia marker (LPS binding protein) following weight loss (70–72).
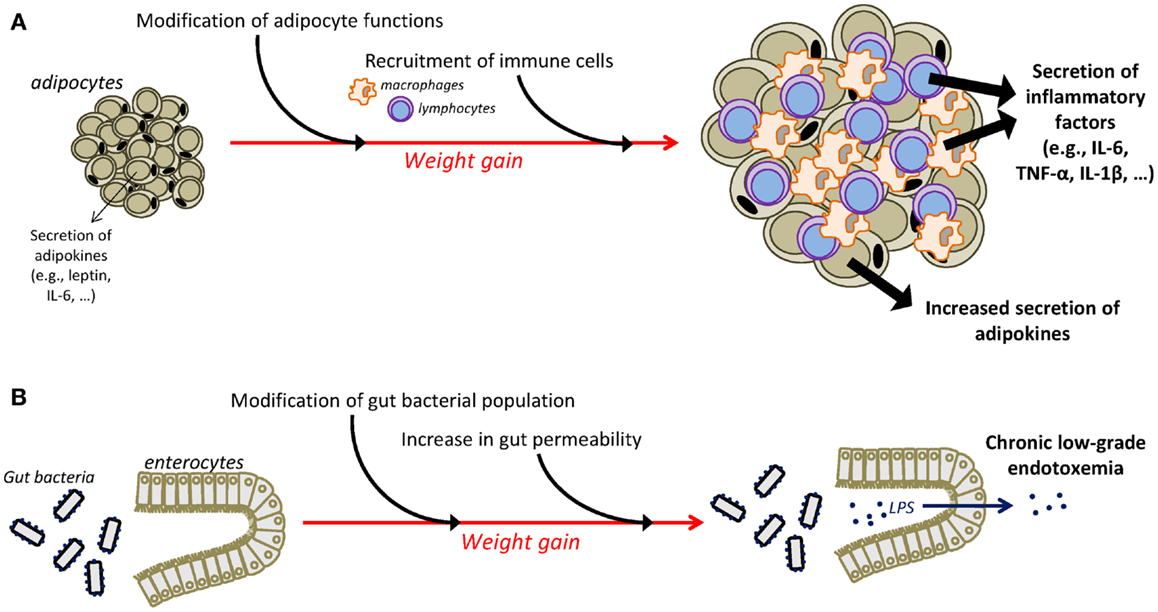
Figure 1. Mechanisms underlying chronic low-grade inflammatory state in obesity. The adipose tissue is an important contributor of chronic low-grade inflammation in obesity (A). Weight gain is associated with substantial changes in the function of adipocytes that increase their secretion of adipokines, including inflammatory factors. Moreover, infiltration of immune cells, i.e., macrophages and T cells, in the adipose tissue is also responsible for the adipose secretion of inflammatory cytokines. Additional mechanisms, including alterations in the gut microbiota, contribute also to the instauration of obesity-related inflammation (B). Obesity is associated with modifications in gut microbial population and with an increased permeability of the intestinal wall that promotes the passage of LPS in the circulation, leading thus to the development of chronic low-grade endotoxemia and the increased production of inflammatory factors. IL-1β, interleukin-1β; IL-6, interleukin-6; LPS, lipopolysaccharide; TNF-α, tumor necrosis factor-α.
It is now clear that systemic inflammation in obesity contributes to both increased central inflammatory processes (notably in the hippocampus and hypothalamus) and metabolic dysregulations, including insulin-resistance. While these mechanisms are believed to play a major role in the development and maintenance of obesity (68, 73), their specific contribution to the development of the disorder is still under investigation. With respect to central inflammation, db/db mice that exhibit immune defects such as increased systemic inflammation and reduced immune competence (64, 74, 75) also show increased hippocampus cytokine expression (36, 37), and these effects are associated. Moreover, there is mounting evidence for enhanced cytokine expression and microglial activation in the hypothalamus in animal models of obesity (76–78). Consistent with these experimental findings, clinical indication of gliosis was recently reported in the mediobasal hypothalamus of obese individuals (79). While it is likely that central inflammation in obesity results from adiposity-related systemic inflammatory processes, the increases in circulating levels of TLR4 ligands and free saturated fatty acids following impaired gut permeability in high-fat diet mice (68, 80) may alternatively contribute to central inflammation, through activation of TLR4/MyD88 signaling (81, 82). Further investigation is needed to determine whether central activation of inflammatory processes occurs as a result of peripheral inflammation or whether it represents an early event promoting the development of obesity following a high-fat diet.
With regard to inflammation-related metabolic dysregulations, the inflammatory cytokine, TNF-α, was found to play an important role in the pathogenesis of insulin-resistance and type 2 diabetes (83–85). Inflammatory factors are also strong modulators of energy balance mainly due to their effects on the brain, in particular the hypothalamus (76), a mechanism that may promote weight gain through impairment of local peptidergic neuronal networks involved in food intake (73, 86). Interestingly, obesity-associated inflammation, notably as it relates to the visceral adipose tissue, was found to impact obesity treatment outcomes, with increased adipose expression of immune cells and inflammatory markers being associated with lower BMI reduction after bariatric surgery in severely obese patients (57).
Central Effects of Inflammation: Evidence and Mechanisms
There is clear evidence for a role of proinflammatory cytokines in the development of neuropsychiatric symptoms (87). Proinflammatory cytokines released locally by activated innate immune cells have access to the brain, through different mechanisms that have been reviewed elsewhere (87). These pathways, which include humoral, neural, and cellular routes, ultimately lead to the production of cytokines by activated glial cells, in particular microglia, within the central nervous system (CNS). While microglia activation normally exerts a protective action on the CNS, its unregulated and chronic activation may in contrast become deleterious. Within the brain, proinflammatory cytokines activate the neuroendocrine system, impair neurotransmitter metabolism and function, and alter neural plasticity and brain circuitry (87). These biological alterations are associated with a large number of behavioral changes that have been referred to as sickness behavior (88). Necessary for the recovery of the host to the infection, sickness behavior usually resolves within few days. However, in cases of chronic and unregulated activation of the immune system, sickness behavior may evolve into clinically relevant neuropsychiatric symptoms, including depression and cognitive symptoms (88). At the clinical level, strong evidence for a role of cytokines in the development of neuropsychiatric comorbidity emanates from the model of cytokine therapy, based in particular on interferon (IFN)-α administration. Using this model, we and others have shown that IFN-α induces major depression in up to 45% of treated patients (89, 90) and that this effect relates on inflammation-induced alterations in the hypothalamic–pituitary–adrenal (HPA) axis, neurotransmitter function, and enzymatic pathways involved in the metabolism of monoamines (87, 91). Consistent with this last point, inflammatory factors are able to induce the synthesis of the enzymes indoleamine 2,3-dioxygenase (IDO) and GTP-cyclohydrolase 1 (GTP-CH1) in monocytes/macrophages and dendritic cells, which results in significant alterations in the biosynthesis of key monoamines (e.g., serotonin, dopamine) known to play a major role in mood regulation and cognitive function. Moreover, IDO is the first and rate-limiting enzyme that catabolizes tryptophan along the kynurenine pathway, a pathway leading ultimately to the production of neuroactive metabolites that have been associated with depressive symptoms in IFN-α-treated patients (92). In particular, IDO activation results in an increased production of the glutamatergic metabolites, 3-hydroxykynurenine, and quinolinic acid, which are well-known to induce neuronal death and to whom brain or cerebrospinal fluid concentrations were found to be increased in several neuropsychiatric or neurodegenerative diseases (93–96). Consistent with these data, inflammation-induced depressive- and anxiety-like behaviors in mice can be prevented by pharmacological or genetic inhibition of brain IDO activation (97–101). Moreover, NMDA receptor blockade abrogates cytokine-induced depressive-like behavior in mice (102). Interestingly, the hippocampus was found to play an important role in cytokine and IDO activation (103–106) and dysregulated activation of hippocampal microglia was associated with sustained IDO activity and protracted depressive-like behavior (104). Moreover, emotional alterations linked to inflammation-induced hippocampus IDO activation in mice was associated with reduced hippocampal expression of the brain-derived neurotrophic factor (BDNF) (107) that contributes to mood regulation and memory function. Altogether, these results point to a pivotal role of IDO activation, particularly in the hippocampus, in mediating cytokine-induced mood and cognitive alterations.
Role of Inflammatory Processes in Obesity-Related Neuropsychiatric Symptoms: Clinical and Experimental Findings
Recent clinical findings support the hypothesis that inflammatory processes contribute to neuropsychiatric comorbidity in obesity, notably as it relates to mood status and cognitive function [see for review, Ref. (108)]. In support of this notion, concentrations of the inflammatory markers, C reactive protein (CRP) and IL-6, have been associated with depressive symptoms in obese subjects or in patients afflicted with the metabolic syndrome (109–111). Similar associations have been reported with leptin (112). Moreover, it was recently shown that CRP levels explained approximately 20% of the increase in depression scores over time in obese subjects (113). Consistent with the role of adiposity in these associations, reductions in inflammatory markers following weight loss induced by bariatric surgery were found to correlate with significant improvement in the emotional status and depression scores of severely obese individuals (114, 115). Given the bidirectional link reported between obesity and depressive symptoms, it is highly probable that depressive symptoms occurring in the context of obesity-related inflammation may in turn contribute to obesity maintenance, promoting thus the instauration of a vicious circle. Regarding cognitive function, a significant relationship was reported between CRP levels and decreased performance on cognitive tests targeting frontal lobe function in obese and overweight women (116). Moreover, in patients with the metabolic syndrome, higher levels of CRP and IL-6 were found to increase the risk of age-related cognitive decline (117). While these data support a role for obesity-related inflammation in the development of neuropsychiatric symptoms, the literature is still sparse regarding the causality of the events and the mechanisms that specifically underlie these effects in the context of obesity. Result from animal models of obesity may help to start to address this issue. In genetically or diet-induced obese rodents, increased cytokine expression in the hippocampus and cortex is associated with emotional and cognitive alterations (30, 31, 36, 37, 118). Interestingly, hippocampal IL-1β expression in db/db mice is related to adiposity and its blockade normalizes hippocampal dendritic spine density and prevents synaptic dysfunction and cognitive impairment (30). Associations have also been found between hippocampal microgliosis and obesity-related elevation in plasma glucocorticoids in the same mice (119). Consistent with the role of inflammatory processes, a direct relationship has been recently reported by our group between inflammation-related brain IDO activation and the development of depressive-like behavior in db/db mice (37). Similarly, we showed that DIO exacerbates both hippocampal induction of cytokines and IDO in response to an immune challenge and related behavioral changes (40). Interestingly, exacerbated depressive-like behavior is also associated in DIO mice with increased hypothalamic inflammation (39, 40). Beyond its impact on energy homeostasis, hypothalamic inflammation might also influence obesity-related emotional alterations. In addition to inflammatory processes, metabolic factors associated with obesity, including insulin or leptin, may also be able to act within the brain and lead to behavioral alterations (120). Nevertheless, several studies suggest that these factors per se are not sufficient to explain neuropsychiatric symptoms occurring in contexts of obesity. In support of this, increased emotional behaviors and cognitive impairment have been reported in animal models of obesity in the absence of any significant hyperinsulinemia (32, 36, 40). Reciprocally, the normalization of hyperglycemia in db/db mice was not effective in reversing spatial cognitive impairment or anxiety-like behavior (28, 29). Moreover, no difference in brain concentrations of glucose and insulin was measured in both db/db and db/+ mice and these concentrations remained the same when peripheral hyperinsulinemia was normalized (28). Altogether, these data point to brain inflammation as a major player in the development of obesity-related neuropsychiatric symptoms, although the pathways linking inflammation to these symptoms still need to be thoroughly studied.
Inflammation-Driven Neuropsychiatric Comorbidity in Obesity: Potential Underlying Pathways
There are several pathways by which inflammation may promote the development of neuropsychiatric comorbidity in the context of obesity. Some of these mechanisms may be common to various situations of chronic inflammation and some may be more specific to the condition of obesity. Non-specific mechanisms include diffusion of inflammatory markers from the adipose tissue to the circulation and activation of relevant immune-to-brain pathways including humoral, neural, and cellular routes leading ultimately to the production de novo of inflammatory cytokines within the CNS and subsequent alterations in CNS functions (e.g., changes in neuroendocrine function, neurocircuitry, enzymatic pathways, and neurotransmitter metabolism/function) as described above. In particular, similar to other inflammatory conditions, obesity has been often associated with alterations in basal ganglia/reward circuitry and dopamine function (121, 122). Accordingly, several studies have indicated that obesity is associated with reduced striatal dopamine D2 receptor availability together with alterations in the fronto-striatal network (123–125). Moreover, investigations in rodents with DIO have shown significant associations between alterations in striatal circuitry and depressive-like behavior, suggesting a role for dopamine-related disruptions in obesity-associated depressive symptoms (38). Relevant to the contribution of inflammatory processes to these effects, the basal ganglia and dopamine system are highly targeted by inflammatory factors (126). In addition, inflammation-induced neuropsychiatric symptoms, in particular fatigue, anhedonia, psychomotor slowing, decreased motivation, and depressed mood, have been found to relate to alterations in basal ganglia/dopamine function and striatal circuitry in subjects treated with immune agents (127–129).
Alterations in neuroendocrine function – a mechanism highly described in the neurobiology of mood disorders – represent another common feature of inflammatory conditions, including obesity. In particular, obese subjects have been shown to exhibit an impaired feedback response to cortisol, similar to what is observed in depression (130). The immune system and neuroendocrine system are in constant communication and immune alterations are notorious for causing significant changes in neuroendocrine activity and vice versa. While the association between low-grade inflammation and alterations in the neuroendocrine system remains to be determined in obese subjects, it is highly possible that obesity-related neuroendocrine dysfunction contributes to neuropsychiatric comorbidity in obese individuals.
Converging findings have highlighted the consequences of deregulated hippocampal cytokines and neurotrophins expression on mood, learning, and memory (88, 131, 132), and the negative impact of cytokines on neurotrophins and synaptic plasticity (107, 133). Alterations in these mechanisms have been fully documented in models of chronic stress exposure, a well-admitted contributor of mood disorders (134, 135). Several clinical and experimental data strongly suggest that these deregulations may similarly participate to obesity-related neuropsychiatric alterations. Indeed, body weight loss induced by lifestyle intervention program in young obese patients normalizes plasma levels of BDNF (136). Moreover, cognitive impairment and emotional alterations reported in DIO and genetic models of obesity are linked to increased inflammation and reduced BDNF levels in the cortex (118) and the hippocampus (36, 37). Reciprocally, anti-inflammatory interventions in DIO mice reduce body weight, normalize hippocampal levels of BDNF, and prevent hippocampus-mediated cognitive impairments (137).
Alterations in the gut–brain axis represent one mechanism of inflammatory-driven neuropsychiatric comorbidity that may be more relevant to the condition of obesity. As already mentioned, obesity is associated with alterations in the gut microbiota in the form of modifications in microbiota populations, increased gut permeability, and activated inflammatory processes. A rich and complex communication network exists between the gut and the brain that involves endocrine, immune, and neural pathways (138), and there are now multiple evidences that impairment or dysregulation in the gut–brain axis impacts on mood and cognitive function (139). These data suggest that gut microbiota alterations found in obesity may modulate gut-to-brain communication pathways, leading thus to the development of neuropsychiatric comorbidity.
Altogether, these data are in favor of the involvement of inflammation-related complex non-exclusive pathophysiological processes in the development of neuropsychiatric symptoms in obesity. In that context, strategies to reduce inflammation either by pharmacological or non-pharmacological interventions (e.g., diet, surgical weight reduction strategies, exercise) may help in the prevention and management of obesity-related neuropsychiatric comorbidity. In particular, nutritional factors with immunomodulatory properties (i.e., omega-3 fatty acids, antioxidants) may worth being considered.
Conclusion
Data presented in this review strongly support the notion that inflammatory processes represent key players in the development of neuropsychiatric comorbidities in obesity. In addition to clinical investigations that clearly highlight the relationship between adiposity-related inflammation and neuropsychiatric symptoms in obese individuals, animal studies provide strong evidence of the direct effects of obesity-related neuroinflammatory processes on brain function and neurocircuitry and on the development of behavioral symptoms. The mechanisms and pathways leading to neuropsychiatric comorbidities in obesity are also discussed, starting from a general aspect to a viewpoint more specific to the condition of obesity. The effects rely on complex communication networks including the immune system, the gut, the neuroendocrine system, and key brain areas, including the hypothalamus, the hippocampus, and the basal ganglia (Figure 2). Alterations in monoamine metabolism and function, impaired neurotransmitter activity together with the occurrence of neurotoxic effects likely to promote neuronal death and decreased neurogenesis appear to represent major pathophysiological pathways to neuropsychiatric morbidity in obese individuals.
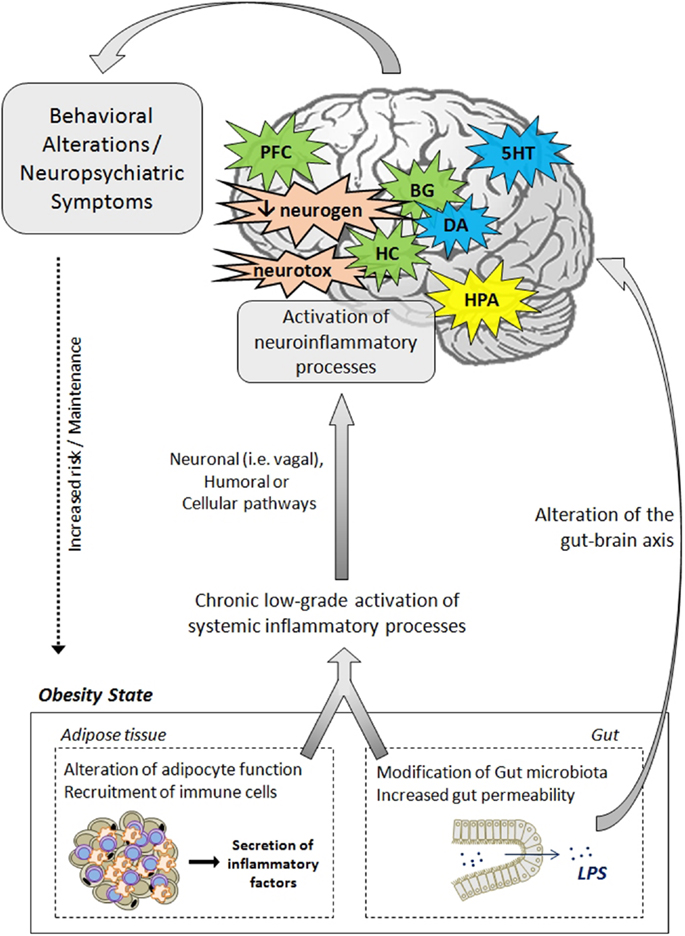
Figure 2. Pathophysiological mechanisms likely to underlie neuropsychiatric comorbidities associated with obesity. The activation of systemic inflammatory processes, originating from alterations in adipose tissue and gut functions, can contribute to the development of obesity-associated neuropsychiatric comorbidities. Proinflammatory cytokines released at the periphery can access the brain via several pathways (e.g., neural, humoral, and cellular routes) and induce the activation of neuroinflammatory processes, primarily by activating microglia. In the brain, proinflammatory cytokines impair neuroendocrine activity, neurotransmitter function (e.g., 5HT, DA, glutamate), and neurocircuitry, involving notably the hippocampus, the hypothalamus, the basal ganglia, and the prefrontal cortex. Cytokines can also disturb neurogenesis and induce neurotoxic effects through induction of IDO-derived neuroactive/neurotoxic metabolites. Altogether, these brain alterations lead ultimately to the development of behavioral/neuropsychiatric symptoms. Deregulations of the gut–brain axis, originating from changes in gut microbiota and permeability, may also contribute to mood and cognitive symptoms. These behavioral/neuropsychiatric symptoms can in turn promote the development or maintenance of obesity through risky or unadjusted eating behaviors. 5HT, serotonin; BG, basal ganglia; CNS, central nervous system; DA, dopamine; IDO, indoleamine 2,3-dioxygenase; HC, hippocampus; HPA, hypothalamic–pituitary–adrenal axis; LPS, lipopolysaccharide; PFC, prefrontal cortex; neurogen, neurogenesis; neurotox, neurotoxicity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Roberts RO, Geda YE, Knopman DS, Cha RH, Boeve BF, Ivnik RJ, et al. Metabolic syndrome, inflammation, and nonamnestic mild cognitive impairment in older persons: a population-based study. Alzheimer Dis Assoc Disord (2010) 24:11–8. doi:10.1097/WAD.0b013e3181a4485c
2. Goldbacher EM, Bromberger J, Matthews KA. Lifetime history of major depression predicts the development of the metabolic syndrome in middle-aged women. Psychosom Med (2009) 71:266–72. doi:10.1097/PSY.0b013e318197a4d5
3. Lasselin J, Capuron L. Chronic low-grade inflammation in metabolic disorders: relevance for behavioral symptoms. Neuroimmunomodulation (2014) 21:95–101. doi:10.1159/000356535
4. Lin HY, Huang CK, Tai CM, Kao YH, Tsai CC, Hsuan CF, et al. Psychiatric disorders of patients seeking obesity treatment. BMC Psychiatry (2013) 13:1. doi:10.1186/1471-244X-13-1
5. Evans DL, Charney DS, Lewis L, Golden RN, Gorman JM, Krishnan KR, et al. Mood disorders in the medically ill: scientific review and recommendations. Biol Psychiatry (2005) 58:175–89. doi:10.1016/j.biopsych.2005.05.001
6. Boeka AG, Lokken KL. Neuropsychological performance of a clinical sample of extremely obese individuals. Arch Clin Neuropsychol (2008) 23:467–74. doi:10.1016/j.acn.2008.03.003
7. Fagundo AB, de la Torre R, Jimenez-Murcia S, Aguera Z, Granero R, Tarrega S, et al. Executive functions profile in extreme eating/weight conditions: from anorexia nervosa to obesity. PLoS One (2012) 7:e43382. doi:10.1371/journal.pone.0043382
8. Fergenbaum JH, Bruce S, Lou W, Hanley AJ, Greenwood C, Young TK. Obesity and lowered cognitive performance in a Canadian First Nations population. Obesity (Silver Spring) (2009) 17:1957–63. doi:10.1038/oby.2009.161
9. Lokken KL, Boeka AG, Yellumahanthi K, Wesley M, Clements RH. Cognitive performance of morbidly obese patients seeking bariatric surgery. Am Surg (2010) 76:55–9.
10. Elias MF, Elias PK, Sullivan LM, Wolf PA, D’Agostino RB. Lower cognitive function in the presence of obesity and hypertension: the Framingham heart study. Int J Obes Relat Metab Disord (2003) 27:260–8. doi:10.1038/sj.ijo.802225
11. Gunstad J, Paul RH, Cohen RA, Tate DF, Gordon E. Obesity is associated with memory deficits in young and middle-aged adults. Eat Weight Disord (2006) 11:e15–9. doi:10.1007/BF03327747
12. Cournot M, Marquie JC, Ansiau D, Martinaud C, Fonds H, Ferrieres J, et al. Relation between body mass index and cognitive function in healthy middle-aged men and women. Neurology (2006) 67:1208–14. doi:10.1212/01.wnl.0000238082.13860.50
13. Dahl AK, Hassing LB, Fransson EI, Gatz M, Reynolds CA, Pedersen NL. Body mass index across midlife and cognitive change in late life. Int J Obes (Lond) (2013) 37:296–302. doi:10.1038/ijo.2012.37
14. Sabia S, Kivimaki M, Shipley MJ, Marmot MG, Singh-Manoux A. Body mass index over the adult life course and cognition in late midlife: the Whitehall II Cohort Study. Am J Clin Nutr (2009) 89:601–7. doi:10.3945/ajcn.2008.26482
15. Siervo M, Arnold R, Wells JC, Tagliabue A, Colantuoni A, Albanese E, et al. Intentional weight loss in overweight and obese individuals and cognitive function: a systematic review and meta-analysis. Obes Rev (2011) 12:968–83. doi:10.1111/j.1467-789X.2011.00903.x
16. Alosco ML, Spitznagel MB, Strain G, Devlin M, Cohen R, Paul R, et al. Improved memory function two years after bariatric surgery. Obesity (Silver Spring) (2014) 22:32–8. doi:10.1002/oby.20494
17. Miller LA, Crosby RD, Galioto R, Strain G, Devlin MJ, Wing R, et al. Bariatric surgery patients exhibit improved memory function 12 months postoperatively. Obes Surg (2013) 23:1527–35. doi:10.1007/s11695-013-0970-7
18. Brinkworth GD, Buckley JD, Noakes M, Clifton PM, Wilson CJ. Long-term effects of a very low-carbohydrate diet and a low-fat diet on mood and cognitive function. Arch Intern Med (2009) 169:1873–80. doi:10.1001/archinternmed.2009.329
19. Halyburton AK, Brinkworth GD, Wilson CJ, Noakes M, Buckley JD, Keogh JB, et al. Low- and high-carbohydrate weight-loss diets have similar effects on mood but not cognitive performance. Am J Clin Nutr (2007) 86:580–7.
20. Andersen JR, Aasprang A, Bergsholm P, Sletteskog N, Vage V, Natvig GK. Anxiety and depression in association with morbid obesity: changes with improved physical health after duodenal switch. Health Qual Life Outcomes (2010) 8:52. doi:10.1186/1477-7525-8-52
21. Dixon JB, Dixon ME, O’Brien PE. Depression in association with severe obesity: changes with weight loss. Arch Intern Med (2003) 163:2058–65. doi:10.1001/archinte.163.17.2058
22. Thonney B, Pataky Z, Badel S, Bobbioni-Harsch E, Golay A. The relationship between weight loss and psychosocial functioning among bariatric surgery patients. Am J Surg (2010) 199:183–8. doi:10.1016/j.amjsurg.2008.12.028
23. Luppino FS, de Wit LM, Bouvy PF, Stijnen T, Cuijpers P, Penninx BW, et al. Overweight, obesity, and depression: a systematic review and meta-analysis of longitudinal studies. Arch Gen Psychiatry (2010) 67:220–9. doi:10.1001/archgenpsychiatry.2010.2
24. Guxens M, Mendez MA, Julvez J, Plana E, Forns J, Basagana X, et al. Cognitive function and overweight in preschool children. Am J Epidemiol (2009) 170:438–46. doi:10.1093/aje/kwp140
25. Osika W, Montgomery SM, Longitudinal Birth Cohort Study. Physical control and coordination in childhood and adult obesity: Longitudinal Birth Cohort Study. BMJ (2008) 337:a699. doi:10.1136/bmj.a699
26. Davis C, Levitan RD, Muglia P, Bewell C, Kennedy JL. Decision-making deficits and overeating: a risk model for obesity. Obes Res (2004) 12:929–35. doi:10.1038/oby.2004.113
27. Kanasaki K, Koya D. Biology of obesity: lessons from animal models of obesity. J Biomed Biotechnol (2011) 2011:197636. doi:10.1155/2011/197636
28. Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci (2008) 11:309–17. doi:10.1038/nn2055
29. Stranahan AM, Lee K, Martin B, Maudsley S, Golden E, Cutler RG, et al. Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus (2009) 19:951–61. doi:10.1002/hipo.20577
30. Erion JR, Wosiski-Kuhn M, Dey A, Hao S, Davis CL, Pollock NK, et al. Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J Neurosci (2014) 34:2618–31. doi:10.1523/JNEUROSCI.4200-13.2014
31. Kanoski SE, Davidson TL. Western diet consumption and cognitive impairment: links to hippocampal dysfunction and obesity. Physiol Behav (2011) 103:59–68. doi:10.1016/j.physbeh.2010.12.003
32. Boitard C, Cavaroc A, Sauvant J, Aubert A, Castanon N, Layé S, et al. Impairment of hippocampal-dependent memory induced by juvenile high-fat diet intake is associated with enhanced hippocampal inflammation in rats. Brain Behav Immun (2014). doi:10.1016/j.bbi.2014.03.005
33. Boitard C, Etchamendy N, Sauvant J, Aubert A, Tronel S, Marighetto A, et al. Juvenile, but not adult exposure to high-fat diet impairs relational memory and hippocampal neurogenesis in mice. Hippocampus (2012) 22:2095–100. doi:10.1002/hipo.22032
34. Valladolid-Acebes I, Merino B, Principato A, Fole A, Barbas C, Lorenzo MP, et al. High-fat diets induce changes in hippocampal glutamate metabolism and neurotransmission. Am J Physiol Endocrinol Metab (2012) 302:E396–402. doi:10.1152/ajpendo.00343.2011
35. Molteni R, Barnard RJ, Ying Z, Roberts CK, Gomez-Pinilla F. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience (2002) 112:803–14. doi:10.1016/S0306-4522(02)00123-9
36. Dinel AL, Andre C, Aubert A, Ferreira G, Laye S, Castanon N. Cognitive and emotional alterations are related to hippocampal inflammation in a mouse model of metabolic syndrome. PLoS One (2011) 6:e24325. doi:10.1371/journal.pone.0024325
37. Dinel AL, André C, Aubert A, Ferreira G, Layé S, Castanon N. Lipopolysaccharide-induced brain activation of the indoleamine 2,3-dioxygenase and depressive-like behavior are impaired in a mouse model of metabolic syndrome. Psychoneuroendocrinology (2014) 40:48–59. doi:10.1016/j.psyneuen.2013.10.014
38. Sharma S, Fulton S. Diet-induced obesity promotes depressive-like behaviour that is associated with neural adaptations in brain reward circuitry. Int J Obes (Lond) (2013) 37:382–9. doi:10.1038/ijo.2012.48
39. Aguliar-Valles A, Kim J, Jung S, Woodside B, Luheshi GN. Role of brain transmigrating neutrophils in depression-like behavior during systemic infection. Mol Psychiatry (2014) 19:599–606. doi:10.1038/mp.2013.154
40. André C, Dinel AL, Ferreira G, Layé S, Castanon N. Diet-induced obesity progressively alters cognition, anxiety-like behavior and lipopolysaccharide-induced depressive-like behavior: focus on brain indoleamine 2,3-dioxygenase activation. Brain Behav Immun (2014). doi:10.1016/j.bbi.2014.03.012
41. Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology (2007) 132:2169–80. doi:10.1053/j.gastro.2007.03.059
42. Nieman DC, Henson DA, Nehlsen-Cannarella SL, Ekkens M, Utter AC, Butterworth DE, et al. Influence of obesity on immune function. J Am Diet Assoc (1999) 99:294–9. doi:10.1016/S0002-8223(99)00077-2
43. Cancello R, Clement K. Is obesity an inflammatory illness? Role of low-grade inflammation and macrophage infiltration in human white adipose tissue. BJOG (2006) 113:1141–7. doi:10.1111/j.1471-0528.2006.01004.x
44. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol (2011) 29:415–45. doi:10.1146/annurev-immunol-031210-101322
45. Brooks GC, Blaha MJ, Blumenthal RS. Relation of C-reactive protein to abdominal adiposity. Am J Cardiol (2010) 106:56–61. doi:10.1016/j.amjcard.2010.02.017
46. Hermsdorff HH, Zulet MA, Puchau B, Martinez JA. Central adiposity rather than total adiposity measurements are specifically involved in the inflammatory status from healthy young adults. Inflammation (2011) 34:161–70. doi:10.1007/s10753-010-9219-y
47. Thorand B, Baumert J, Doring A, Herder C, Kolb H, Rathmann W, et al. Sex differences in the relation of body composition to markers of inflammation. Atherosclerosis (2006) 184:216–24. doi:10.1016/j.atherosclerosis.2005.04.011
48. Thewissen MM, Damoiseaux JG, Duijvestijn AM, van Greevenbroek MM, van der Kallen CJ, Feskens EJ, et al. Abdominal fat mass is associated with adaptive immune activation: the CODAM Study. Obesity (Silver Spring) (2011) 19:1690–8. doi:10.1038/oby.2010.337
49. Belza A, Toubro S, Stender S, Astrup A. Effect of diet-induced energy deficit and body fat reduction on high-sensitive CRP and other inflammatory markers in obese subjects. Int J Obes (Lond) (2009) 33:456–64. doi:10.1038/ijo.2009.27
50. Dalmas E, Rouault C, Abdennour M, Rovere C, Rizkalla S, Bar-Hen A, et al. Variations in circulating inflammatory factors are related to changes in calorie and carbohydrate intakes early in the course of surgery-induced weight reduction. Am J Clin Nutr (2011) 94:450–8. doi:10.3945/ajcn.111.013771
51. Esposito K, Pontillo A, Di Palo C, Giugliano G, Masella M, Marfella R, et al. Effect of weight loss and lifestyle changes on vascular inflammatory markers in obese women: a randomized trial. JAMA (2003) 289:1799–804. doi:10.1001/jama.289.14.1799
52. Hakeam HA, O’Regan PJ, Salem AM, Bamehriz FY, Jomaa LF. Inhibition of C-reactive protein in morbidly obese patients after laparoscopic sleeve gastrectomy. Obes Surg (2009) 19:456–60. doi:10.1007/s11695-008-9729-y
53. Manco M, Fernandez-Real JM, Equitani F, Vendrell J, Valera Mora ME, Nanni G, et al. Effect of massive weight loss on inflammatory adipocytokines and the innate immune system in morbidly obese women. J Clin Endocrinol Metab (2007) 92:483–90. doi:10.1210/jc.2006-0960
54. Viana EC, Araujo-Dasilio KL, Miguel GP, Bressan J, Lemos EM, Moyses MR, et al. Gastric bypass and sleeve gastrectomy: the same impact on IL-6 and TNF-alpha. Prospective clinical trial. Obes Surg (2013) 23:1252–61. doi:10.1007/s11695-013-0894-2
55. Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr (2004) 92:347–55. doi:10.1079/BJN20041213
56. Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest (2003) 112:1785–8. doi:10.1172/JCI200320514
57. Lasselin J, Magne E, Beau C, Ledaguenel P, Dexpert S, Aubert A, et al. Adipose inflammation in obesity: relationship with circulating levels of inflammatory markers and association with surgery-induced weight loss. J Clin Endocrinol Metab (2014) 99:E53–61. doi:10.1210/jc.2013-2673
58. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112:1796–808. doi:10.1172/JCI19246
59. Cancello R, Henegar C, Viguerie N, Taleb S, Poitou C, Rouault C, et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes (2005) 54:2277–86. doi:10.2337/diabetes.54.8.2277
60. Kintscher U, Hartge M, Hess K, Foryst-Ludwig A, Clemenz M, Wabitsch M, et al. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol (2008) 28:1304–10. doi:10.1161/ATVBAHA.108.165100
61. Deiuliis J, Shah Z, Shah N, Needleman B, Mikami D, Narula V, et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PLoS One (2011) 6:e16376. doi:10.1371/journal.pone.0016376
62. Zeyda M, Huber J, Prager G, Stulnig TM. Inflammation correlates with markers of T-cell subsets including regulatory T cells in adipose tissue from obese patients. Obesity (Silver Spring) (2011) 19:743–8. doi:10.1038/oby.2010.123
63. Goossens GH, Blaak EE, Theunissen R, Duijvestijn AM, Clement K, Tervaert JW, et al. Expression of NLRP3 inflammasome and T cell population markers in adipose tissue are associated with insulin resistance and impaired glucose metabolism in humans. Mol Immunol (2012) 50:142–9. doi:10.1016/j.molimm.2012.01.005
64. Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut (2009) 58:1091–103. doi:10.1136/gut.2008.165886
65. Flint HJ. Obesity and the gut microbiota. J Clin Gastroenterol (2011) 45(Suppl):S128–32. doi:10.1097/MCG.0b013e31821f44c4
66. Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A (2005) 102:11070–5. doi:10.1073/pnas.0504978102
67. Verdam FJ, Fuentes S, de Jonge C, Zoetendal EG, Erbil R, Greve JW, et al. Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity (Silver Spring) (2013) 21:E607–15. doi:10.1002/oby.20466
68. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes (2007) 56:1761–72. doi:10.2337/db06-1491
69. Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes (2008) 57:1470–81. doi:10.2337/db07-1403
70. Furet JP, Kong LC, Tap J, Poitou C, Basdevant A, Bouillot JL, et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: links with metabolic and low-grade inflammation markers. Diabetes (2010) 59:3049–57. doi:10.2337/db10-0253
71. Aron-Wisnewsky J, Dore J, Clement K. The importance of the gut microbiota after bariatric surgery. Nat Rev Gastroenterol Hepatol (2012) 9:590–8. doi:10.1038/nrgastro.2012.161
72. Yang PJ, Lee WJ, Tseng PH, Lee PH, Lin MT, Yang WS. Bariatric surgery decreased the serum level of an endotoxin-associated marker: lipopolysaccharide-binding protein. Surg Obes Relat Dis (2014). doi:10.1016/j.soard.2014.02.022
73. Velloso LA, Araújo EP, de Souza CT. Diet-induced inflammation of the hypothalamus in obesity. Neuroimmunomodulation (2008) 15:189–93. doi:10.1159/000153423
74. Lawrence CB, Brough D, Knight EM. Obese mice exhibit an altered behavioural and inflammatory response to lipopolysaccharide. Dis Model Mech (2012) 5:649–59. doi:10.1242/dmm.009068
75. Rummel C, Inoue W, Poole S, Luheshi GN. Leptin regulates leukocyte recruitment into the brain following systemic LPS-induced inflammation. Mol Psychiatry (2010) 15:523–34. doi:10.1038/mp.2009.98
76. Cai D, Liu T. Inflammatory cause of metabolic syndrome via brain stress and NF-kappaB. Aging (Albany NY) (2012) 4:98–115.
77. De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology (2005) 146:4192–9. doi:10.1210/en.2004-1520
78. Gao Y, Ottaway N, Schriever SC, Legutko B, García-Cáceres C, de la Fuente E, et al. Hormones and diet, but not body weight, control hypothalamic microglial activity. Glia (2014) 62:17–25. doi:10.1002/glia.22580
79. Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest (2012) 122:153–62. doi:10.1172/JCI59660
80. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest (2006) 116:3015–25. doi:10.1172/JCI28898
81. Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci (2009) 29:359–70. doi:10.1523/JNEUROSCI.2760-08.2009
82. Kleinridders A, Schenten D, Könner AC, Belgardt BF, Mauer J, Okamura T, et al. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab (2009) 10:249–59. doi:10.1016/j.cmet.2009.08.013
83. Lann D, LeRoith D. Insulin resistance as the underlying cause for the metabolic syndrome. Med Clin North Am (2007) 91:1063–77, viii. doi:10.1016/j.mcna.2007.06.012
84. Hotamisligil GS. Inflammation and metabolic disorders. Nature (2006) 444:860–7. doi:10.1038/nature05485
85. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest (2003) 112:1821–30. doi:10.1172/JCI19451
86. Thaler JP, Choi SJ, Schwartz MW, Wisse BE. Hypothalamic inflammation and energy homeostasis: resolving the paradox. Front Neuroendocrinol (2010) 31:79–84. doi:10.1016/j.yfrne.2009.10.002
87. Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther (2011) 130:226–38. doi:10.1016/j.pharmthera.2011.01.014
88. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci (2008) 9:46–56. doi:10.1038/nrn2297
89. Musselman DL, Lawson DH, Gumnick JF, Manatunga AK, Penna S, Goodkin RS, et al. Paroxetine for the prevention of depression induced by high-dose interferon alfa. N Engl J Med (2001) 344:961–6. doi:10.1056/NEJM200103293441303
90. Capuron L, Gumnick JF, Musselman DL, Lawson DH, Reemsnyder A, Nemeroff CB, et al. Neurobehavioral effects of interferon-alpha in cancer patients: phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology (2002) 26:643–52. doi:10.1016/S0893-133X(01)00407-9
91. Capuron L, Schroecksnadel S, Feart C, Aubert A, Higueret D, Barberger-Gateau P, et al. Chronic low-grade inflammation in elderly persons is associated with altered tryptophan and tyrosine metabolism: role in neuropsychiatric symptoms. Biol Psychiatry (2011) 70:175–82. doi:10.1016/j.biopsych.2010.12.006
92. Raison CL, Dantzer R, Kelley KW, Lawson MA, Woolwine BJ, Vogt G, et al. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol Psychiatry (2010) 15:393–403. doi:10.1038/mp.2009.116
93. Myint AM, Kim YK, Verkerk R, Scharpe S, Steinbusch H, Leonard B. Kynurenine pathway in major depression: evidence of impaired neuroprotection. J Affect Disord (2007) 98:143–51. doi:10.1016/j.jad.2006.07.013
94. Stone TW, Forrest CM, Stoy N, Darlington LG. Involvement of kynurenines in Huntington’s disease and stroke-induced brain damage. J Neural Transm (2012) 119:261–74. doi:10.1007/s00702-011-0676-8
95. Schwarcz R, Rassoulpour A, Wu HQ, Medoff D, Tamminga CA, Roberts RC. Increased cortical kynurenate content in schizophrenia. Biol Psychiatry (2001) 50:521–30. doi:10.1016/S0006-3223(01)01078-2
96. Gulaj E, Pawlak K, Bien B, Pawlak D. Kynurenine and its metabolites in Alzheimer’s disease patients. Adv Med Sci (2010) 55:204–11. doi:10.2478/v10039-010-0023-6
97. Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun (2009) 23:309–17. doi:10.1016/j.bbi.2008.09.002
98. O’Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, et al. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry (2009) 14:511–22. doi:10.1038/sj.mp.4002148
99. O’Connor JC, Lawson MA, Andre C, Briley EM, Szegedi SS, Lestage J, et al. Induction of IDO by bacille Calmette-Guerin is responsible for development of murine depressive-like behavior. J Immunol (2009) 182:3202–12. doi:10.4049/jimmunol.0802722
100. O’Connor JC, Andre C, Wang Y, Lawson MA, Szegedi SS, Lestage J, et al. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J Neurosci (2009) 29:4200–9. doi:10.1523/JNEUROSCI.5032-08.2009
101. Salazar A, Gonzalez-Rivera BL, Redus L, Parrott JM, O’Connor JC. Indoleamine 2,3-dioxygenase mediates anhedonia and anxiety-like behaviors caused by peripheral lipopolysaccharide immune challenge. Horm Behav (2012) 62:202–9. doi:10.1016/j.yhbeh.2012.03.010
102. Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B, et al. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology (2013) 38:1609–16. doi:10.1038/npp.2013.71
103. Andre C, O’Connor JC, Kelley KW, Lestage J, Dantzer R, Castanon N. Spatio-temporal differences in the profile of murine brain expression of proinflammatory cytokines and indoleamine 2,3-dioxygenase in response to peripheral lipopolysaccharide administration. J Neuroimmunol (2008) 200:90–9. doi:10.1016/j.jneuroim.2008.06.011
104. Corona AW, Huang Y, O’Connor JC, Dantzer R, Kelley KW, Popovich PG, et al. Fractalkine receptor (CX3CR1) deficiency sensitizes mice to the behavioral changes induced by lipopolysaccharide. J Neuroinflammation (2010) 7:93. doi:10.1186/1742-2094-7-93
105. Frenois F, Moreau M, O’Connor J, Lawson M, Micon C, Lestage J, et al. Lipopolysaccharide induces delayed FosB/DeltaFosB immunostaining within the mouse extended amygdala, hippocampus and hypothalamus, that parallel the expression of depressive-like behavior. Psychoneuroendocrinology (2007) 32:516–31. doi:10.1016/j.psyneuen.2007.03.005
106. Fu X, Zunich SM, O’Connor JC, Kavelaars A, Dantzer R, Kelley KW. Central administration of lipopolysaccharide induces depressive-like behavior in vivo and activates brain indoleamine 2,3 dioxygenase in murine organotypic hippocampal slice cultures. J Neuroinflammation (2010) 7:43. doi:10.1186/1742-2094-7-43
107. Gibney SM, McGuinness B, Prendergast C, Harkin A, Connor TJ. Poly I:C-induced activation of the immune response is accompanied by depression and anxiety-like behaviours, kynurenine pathway activation and reduced BDNF expression. Brain Behav Immun (2013) 28:170–81. doi:10.1016/j.bbi.2012.11.010
108. Shelton RC, Miller AH. Eating ourselves to death (and despair): the contribution of adiposity and inflammation to depression. Prog Neurobiol (2010) 91:275–99. doi:10.1016/j.pneurobio.2010.04.004
109. Ladwig KH, Marten-Mittag B, Lowel H, Doring A, Koenig W. Influence of depressive mood on the association of CRP and obesity in 3205 middle aged healthy men. Brain Behav Immun (2003) 17:268–75. doi:10.1016/S0889-1591(03)00056-4
110. Dixon JB, Hayden MJ, Lambert GW, Dawood T, Anderson ML, Dixon ME, et al. Raised CRP levels in obese patients: symptoms of depression have an independent positive association. Obesity (Silver Spring) (2008) 16:2010–5. doi:10.1038/oby.2008.271
111. Capuron L, Su S, Miller AH, Bremner JD, Goldberg J, Vogt GJ, et al. Depressive symptoms and metabolic syndrome: is inflammation the underlying link? Biol Psychiatry (2008) 64:896–900. doi:10.1016/j.biopsych.2008.05.019
112. Chirinos DA, Goldberg R, Gellman M, Mendez AJ, Gutt M, McCalla JR, et al. Leptin and its association with somatic depressive symptoms in patients with the metabolic syndrome. Ann Behav Med (2013) 46:31–9. doi:10.1007/s12160-013-9479-5
113. Daly M. The relationship of C-reactive protein to obesity-related depressive symptoms: a longitudinal study. Obesity (Silver Spring) (2013) 21:248–50. doi:10.1002/oby.20051
114. Capuron L, Poitou C, Machaux-Tholliez D, Frochot V, Bouillot JL, Basdevant A, et al. Relationship between adiposity, emotional status and eating behaviour in obese women: role of inflammation. Psychol Med (2011) 41:1517–28. doi:10.1017/S0033291710001984
115. Emery CF, Fondow MD, Schneider CM, Christofi FL, Hunt C, Busby AK, et al. Gastric bypass surgery is associated with reduced inflammation and less depression: a preliminary investigation. Obes Surg (2007) 17:759–63. doi:10.1007/s11695-007-9140-0
116. Sweat V, Starr V, Bruehl H, Arentoft A, Tirsi A, Javier E, et al. C-reactive protein is linked to lower cognitive performance in overweight and obese women. Inflammation (2008) 31:198–207. doi:10.1007/s10753-008-9065-3
117. Yaffe K, Kanaya A, Lindquist K, Simonsick EM, Harris T, Shorr RI, et al. The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA (2004) 292:2237–42. doi:10.1001/jama.292.18.2237
118. Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK, et al. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J Neuroimmunol (2010) 219:25–32. doi:10.1016/j.jneuroim.2009.11.010
119. Dey A, Hao S, Erion JR, Wosiski-Kuhn M, Stranahan AM. Glucocorticoid sensitization of microglia in a genetic mouse model of obesity and diabetes. J Neuroimmunol (2014) 269:20–7. doi:10.1016/j.jneuroim.2014.01.013
120. Yates KF, Sweat V, Yau PL, Turchiano MM, Convit A. Impact of metabolic syndrome on cognition and brain: a selected review of the literature. Arterioscler Thromb Vasc Biol (2012) 32:2060–7. doi:10.1161/ATVBAHA.112.252759
121. Volkow ND, Wang GJ, Baler RD. Reward, dopamine and the control of food intake: implications for obesity. Trends Cogn Sci (2011) 15:37–46. doi:10.1016/j.tics.2010.11.001
122. Volkow ND, Wang GJ, Tomasi D, Baler RD. The addictive dimensionality of obesity. Biol Psychiatry (2013) 73:811–8. doi:10.1016/j.biopsych.2012.12.020
123. de Weijer BA, van de Giessen E, van Amelsvoort TA, Boot E, Braak B, Janssen IM, et al. Lower striatal dopamine D2/3 receptor availability in obese compared with non-obese subjects. EJNMMI Res (2011) 1:37. doi:10.1186/2191-219X-1-37
124. Wang GJ, Volkow ND, Logan J, Pappas NR, Wong CT, Zhu W, et al. Brain dopamine and obesity. Lancet (2001) 357:354–7. doi:10.1016/S0140-6736(00)03643-6
125. Tomasi D, Volkow ND. Brain activation and neurochemistry. Proc Natl Acad Sci U S A (2013) 110:10888–9. doi:10.1073/pnas.1309471110
126. Felger JC, Miller AH. Cytokine effects on the basal ganglia and dopamine function: the subcortical source of inflammatory malaise. Front Neuroendocrinol (2012) 33:315–27. doi:10.1016/j.yfrne.2012.09.003
127. Capuron L, Pagnoni G, Drake DF, Woolwine BJ, Spivey JR, Crowe RJ, et al. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch Gen Psychiatry (2012) 69:1044–53. doi:10.1001/archgenpsychiatry.2011.2094
128. Capuron L, Pagnoni G, Demetrashvili MF, Lawson DH, Fornwalt FB, Woolwine B, et al. Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology (2007) 32:2384–92. doi:10.1038/sj.npp.1301362
129. Brydon L, Harrison NA, Walker C, Steptoe A, Critchley HD. Peripheral inflammation is associated with altered substantia nigra activity and psychomotor slowing in humans. Biol Psychiatry (2008) 63:1022–9. doi:10.1016/j.biopsych.2007.12.007
130. Mattsson C, Reynolds RM, Simonyte K, Olsson T, Walker BR. Combined receptor antagonist stimulation of the hypothalamic-pituitary-adrenal axis test identifies impaired negative feedback sensitivity to cortisol in obese men. J Clin Endocrinol Metab (2009) 94:1347–52. doi:10.1210/jc.2008-2054
131. Martinowich K, Manji H, Lu B. New insights into BDNF function in depression and anxiety. Nat Neurosci (2007) 10:1089–93. doi:10.1038/nn1971
132. Yamada K, Nabeshima T. Brain-derived neurotrophic factor/TrkB signaling in memory processes. J Pharmacol Sci (2003) 91:267–70. doi:10.1254/jphs.91.267
133. Barrientos RM, Sprunger DB, Campeau S, Watkins LR, Rudy JW, Maier SF. BDNF mRNA expression in rat hippocampus following contextual learning is blocked by intrahippocampal IL-1beta administration. J Neuroimmunol (2004) 155:119–26. doi:10.1016/j.jneuroim.2004.06.009
134. Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry (2006) 59:1116–27. doi:10.1016/j.biopsych.2006.02.013
135. Goshen I, Kreisel T, Ben-Menachem-Zidon O, Licht T, Weidenfeld J, Ben-Hur T, et al. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry (2008) 13:717–28. doi:10.1038/sj.mp.4002055
136. Corripio R, Gonzalez-Clemente JM, Jacobo PS, Silvia N, Lluis G, Joan V, et al. Plasma brain-derived neurotrophic factor in prepubertal obese children: results from a 2-year lifestyle intervention programme. Clin Endocrinol (Oxf) (2012) 77:715–20. doi:10.1111/j.1365-2265.2012.04431.x
137. Moy GA, McNay EC. Caffeine prevents weight gain and cognitive impairment caused by a high-fat diet while elevating hippocampal BDNF. Physiol Behav (2013) 109:69–74. doi:10.1016/j.physbeh.2012.11.008
138. Grenham S, Clarke G, Cryan JF, Dinan TG. Brain-gut-microbe communication in health and disease. Front Physiol (2011) 2:94. doi:10.3389/fphys.2011.00094
Keywords: obesity, inflammation, neuroinflammation, cytokines, gut-brain axis, mood, cognition, neuropsychiatric symptoms
Citation: Castanon N, Lasselin J and Capuron L (2014) Neuropsychiatric comorbidity in obesity: role of inflammatory processes. Front. Endocrinol. 5:74. doi: 10.3389/fendo.2014.00074
Received: 22 March 2014; Accepted: 02 May 2014;
Published online: 15 May 2014.
Edited by:
Jacques Epelbaum, INSERM, FranceReviewed by:
Jacques Epelbaum, INSERM, FranceCarole Rovere-Jovene, CNRS-University of Nice Sophia Antipolis, France
Copyright: © 2014 Castanon, Lasselin and Capuron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucile Capuron, UMR 1286, Laboratory of Nutrition and Integrative Neurobiology (NutriNeuro), INRA, University of Bordeaux, 146 rue Léo Saignat, F-33076 Bordeaux, France e-mail:bHVjaWxlLmNhcHVyb25AYm9yZGVhdXguaW5yYS5mcg==