 Ana I. Pérez-Hernández1
Ana I. Pérez-Hernández1 Victoria Catalán1,2
Victoria Catalán1,2 Javier Gómez-Ambrosi1,2
Javier Gómez-Ambrosi1,2 Amaia Rodríguez1,2
Amaia Rodríguez1,2 Gema Frühbeck1,2,3*
Gema Frühbeck1,2,3*- 1Metabolic Research Laboratory, Clínica Universidad de Navarra, Pamplona, Spain
- 2Centro de Investigación Biomédica en Red de Fisiopatología de la Obesidad y Nutrición, Instituto de Salud Carlos III, Madrid, Spain
- 3Department of Endocrinology and Nutrition, Clínica Universidad de Navarra, Pamplona, Spain
Obesity constitutes one of the most important metabolic diseases being associated to insulin resistance development and increased cardiovascular risk. Association between obesity and cancer has also been well established for several tumor types, such as breast cancer in post-menopausal women, colorectal, and prostate cancer. Cancer is the first death cause in developed countries and the second one in developing countries, with high incidence rates around the world. Furthermore, it has been estimated that 15–20% of all cancer deaths may be attributable to obesity. Tumor growth is regulated by interactions between tumor cells and their tissue microenvironment. In this sense, obesity may lead to cancer development through dysfunctional adipose tissue and altered signaling pathways. In this review, three main pathways relating obesity and cancer development are examined: (i) inflammatory changes leading to macrophage polarization and altered adipokine profile; (ii) insulin resistance development; and (iii) adipose tissue hypoxia. Since obesity and cancer present a high prevalence, the association between these conditions is of great public health significance and studies showing mechanisms by which obesity lead to cancer development and progression are needed to improve prevention and management of these diseases.
Introduction
Obesity has reached epidemic proportions constituting one of the most important diseases of this century (1). Despite body mass index (BMI) being used for a long time as the best way to diagnose and define obesity, recent studies have proposed that BMI misclassified subjects with increased cardiovascular risk factors (2). This finding highlights that BMI underestimates the real prevalence of overweight and obesity, since it only considers weight and height of the patients and not total body fat, which really defines the obesity-associated comorbidity risk. In this regard, other indicators to determine the prevalence of obesity have been proposed with body fat percentage being one of the most useful due to its superior ability to stratify patients according to their metabolic and cardiovascular risks (3, 4).
Health care expenditure in severe obesity (BMI ≥ 40 or ≥35 kg/m2 with manifest serious comorbidities) is greater than the investment in any other medical condition (5). This is not only due to direct consequences of obesity, but also due to their associated comorbidities. There are numerous evidences of the relationship between obesity and type 2 diabetes mellitus (T2D) (6), non-alcoholic fatty liver disease (NAFLD) (7, 8) and other chronic liver diseases such as cirrhosis (9), obstructive sleep apnea (OSA) syndrome (10), other respiratory alterations (11) as well as cardiovascular diseases (CVD) including cerebrovascular disorders and hypertension (12–14). Furthermore, evidence about the relationship between obesity and several types of cancer is increasing (15–19). These tumors include colon, breast, endometrium, prostate, thyroid, renal, esophagus, gastric cardia, pancreas, gallbladder, and liver cancer as well as non-Hodgkin lymphoma and multiple myeloma (15, 20–23). Nowadays, cancer is probably one of the most important diseases due to its high prevalence and mortality rates (24). According to the National Cancer Institute, a total of 1,660,290 new cancer cases and 580,350 cancer deaths would take place in the United States in 2013 (25). Increasing trends in incidence of the most common cancers in Europe are also of concern, in particular for colorectal cancer, where incidence is high and still continues to increase (26, 27). A large proportion of the studied cancers are potentially avoidable (28), and several programs to prevent or detect early stages of cancer have been proposed, such as gynecologic or colon cancer population screenings (29–31). Importantly, it has been estimated that 15–20% of all cancer deaths in the United States can be attributed to overweight and obesity (32–34). Obesity and cancer affect millions of people with important consequences; thereby a comprehensive approximation to the mechanisms underlying their relationship is needed to ensure better prevention and treatment strategies.
Higher BMIs have been associated with increased risk of disease while at the same time they have been related with a lower mortality and a better outcome in several chronic diseases (35). This paradoxical finding is shared over a variety of cardiovascular, pulmonary, and renal diseases and is known as the obesity paradox (36). There is increasing evidence that patients, especially elderly, with elevated BMI and several chronic diseases exhibit lower all-cause mortality compared with normal-weight patients (37). However, it should be highlighted that the BMI is an anthropometric marker that does not consider total body fat mass and distribution, nutritional status, or other factors influencing health risks (2, 3, 38).
Relationship between Obesity and Cancer
Large prospective studies show a significant association between excess adiposity and several cancers. There is sufficient evidence relating obesity and tumor development of the colon, breast (post-menopausal women), and prostate among others (18).
Breast cancer is the most frequently diagnosed cancer globally and the second cause of cancer death in women. It has been estimated that breast cancer constituted 30% of all new cancer cases in women in 2013 (25). Overweight and obesity are important risk factors for breast cancer development and, in contrast to other risk factors, such as menstruation age, familiar antecedents or aging, both overweight and obesity are preventable. It has been shown that post-menopausal obese women present a threefold increase in the risk of breast cancer compared to the non-obese control group (39). Moreover, other studies have revealed that higher BMI is associated with worse response to neoadjuvant chemotherapy and worse overall survival (40). Other anthropometric measurements, such as the waist-to-hip ratio, also correlate with breast cancer death risk (41). Worse prognosis or survival rates in obese women with breast cancer have been established in different studies (42–44), showing that obesity is an important risk factor for breast cancer and, therefore, therapeutic measures must be adopted to reduce obesity incidence. It is important to take into account that hormone replacement therapy (45, 46) and mammographic density (47) could be additional confounders in these association studies. The obesity-associated increased risk of post-menopausal breast cancer might be explained by higher rates of peripheral conversion of androgenic precursors to estradiol due to an increased aromatase enzyme activity in adipose tissue (48).
Globally, colorectal cancer remains the third most commonly diagnosed in males and the second in females with a cumulative life risk of developing colorectal cancer of 5% in the general population (49). Although both, the incidence and mortality have been slowly but steadily decreasing, colorectal cancer is the third leading cause of cancer-related deaths (25). The relationship between obesity and colon cancer is well established (50–53). In this regard, it has been estimated that about 30% of all colon cancer cases could be attributable to a BMI higher than 22.5 kg/m2 (54). BMI appears to be consistently associated with an increased risk of colorectal cancer in men, but only a weak association in women has been described (48). This gender difference might be explained by sex differences in prevalence and age of the onset of metabolic syndrome, or by a protective effect of estrogens, inducing apoptosis and inhibition of cell proliferation (55, 56). However, a recent meta-analysis showed that BMI is positively related with colon cancer in both men and women, and no gender differences were found (57). In this sense, a positive association between BMI and prevalence of colonic adenoma and advanced polyps has been demonstrated in pre-menopausal women according to hormonal status (58). Obesity has also been associated with worse cancer outcomes, such as recurrence of the primary cancer or increased mortality. Several factors, including reduced sensitivity to antiangiogenic–therapeutic regimens, might explain these differences. The underlying mechanisms linking obesity to colorectal cancer are not completely elucidated, but inflammation, insulin resistance, and a dysregulated adipokine profile are proposed as important factors. Other biological factors such as the gut microbiota (59) or bile acid concentrations are emerging as novel influential factors in obesity-associated tumors (55).
Prostate cancer is the second diagnosed cancer and the sixth cause of cancer-related mortality among men worldwide (60). Because obesity and prostate cancer affect substantial proportions of the male population, the association between these conditions is of great public health significance. In the last decade, multiple epidemiologic studies have suggested that obesity is associated with an increased risk of death from numerous cancer types including prostate cancer (32, 48). Three meta-analyses reported a positive association between obesity and prostate cancer incidence and, although modest, the relative risks were consistent (48, 61, 62). Obesity has also been associated with worse prognostic and malignant transformation of epithelial cells (63, 64). However, there are contradictory studies about the link between obesity and prostate cancer (65) reporting no association (64, 66), and even a protective effect of obesity (67–71). Proposed obesity-associated alterations related to prostate tumor development include lower levels of sex hormone-binding globulin that increase the fraction of biologically available testosterone (15).
Lung cancer is the main cause of cancer-related death in both genders, with an estimated incidence of more than 200,000 cases in 2013 in the United States (25). To date, the relationship between obesity and the incidence of lung cancer remains unclear (72). Controversy still focuses on the effect of smoking in studies investigating the relationship between BMI and lung cancer (73). In case–control studies, odds ratios for lung cancer by levels of BMI showed an increasing linear trend with a lower threshold BMI for current smokers and ex-smokers of both sexes (74, 75). Moreover, an inverse association between BMI and lung cancer has been shown in other studies after adjusting for smoking or waist circumference (76–78). In this sense, a recent meta-analysis indicates that overweight and obesity are protective factors against lung cancer, especially in current and former smokers (72). Recently, the involvement of adipokines in lung cancer is under study due to their emerging carcinogenic and immunomodulatory properties, making them potential mediators of the complex and still unclear multistep carcinogenetic process (79).
Association with obesity has also been proposed for other cancer types, as thyroid, kidney, esophagus, gastric cardia, pancreas, gallbladder, or liver cancer, but further studies are needed to confirm the obesity-attributable risk of tumor development.
Dysfunctional Adipose Tissue in Obesity and Tumor Development
Obesity is described as an excess of adiposity due to a prolonged status of positive energy balance, which leads not only to changes in adipose tissue distribution but also to metabolic alterations as well as altered cytokine and lipid secretion profiles. Energy balance and fuel homeostasis require a tight equilibrium between energy intake (chemical energy from diet-derived macronutrients) and energy expenditure [basal metabolic rate plus mechanical energy by muscle contractions due to physical activity (PA)] that are regulated by both central and peripheral mechanisms. In this sense, substantial changes have occurred in the patterns of foods consumed with special concern about excess of sugar intake in the diet in many industrialized countries mainly as sugar-sweetened drinks (80, 81).
There are two main types of adipose tissue, white and brown, with different origins and functions (82). The major role of white adipose tissue (WAT) is related to maintaining energy homeostasis by storing triglycerides and releasing fatty acids for energy synthesis. It has been well established that WAT also controls a wide variety of functions including immune and inflammatory regulation, glucose and lipid homeostasis, food intake control, or metabolism by secreting a great number of adipokines (83, 84). WAT is a heterogeneous tissue, consisting of a peripheral subcutaneous component (SAT) and a central intra-abdominal component [visceral adipose tissue (VAT)] (85). Abdominal obesity seems to be of greater pathophysiological concern than subcutaneous fat with an increase in VAT being strongly correlated with the metabolic syndrome and tumor development (86–88). Brown adipose tissue (BAT) was initially described to play a physiological role in animals and infants. In human neonates, BAT is located in specific depots, the interscapular and axillary region, and to a lesser extent, near to the thymus and in the dorsal midline region of the thorax and abdomen (89). However, recent studies using positron emission tomography imaging techniques documented the presence of functional BAT in adult humans (90–92). BAT is specialized in thermogenesis, the production of heat mainly mediated by the uncoupling protein-1 (UCP-1), which produces heat by uncoupling mitochondrial respiration for ATP synthesis (93, 94). Cancer has been associated with cachexia, a complex syndrome that involves profound metabolic imbalances (95). The colorectal tumor-induced cachexia on BAT in mice has been described, finding smaller brown adipocytes with profound delipidation in cachectic tumor-bearing mice (96). Recently, the existence of a third kind of adipose tissue has been proposed, the “beige” or “brite” (brown-like adipocytes) adipose tissue (97). Beige adipose tissue is also a thermogenic adipose tissue influenced by cold-induced signals via the sympathetic nervous system.
Adipose tissue from obese subjects experiments a wide range of modifications due to its expansion and the excessive amount of lipids stored. The limitation in adipose tissue expandability leads to fat accumulation in other parts of the body with lipotoxic consequences (98). Expanded fat depots in obesity are less efficient in storing dietary fatty acids, so that obese subjects exhibit an increase in plasma free fatty acids (FFA) and ectopic fat depots. The higher levels of FFA are susceptible to be substrates for oxidative processes and also may function as mediators of insulin resistance in muscle (99) or liver (100, 101). An analysis of cancer-induced modifications in the lipid profile reveals important clues linking obesity and cancer. Changes in lipid metabolism may promote cancer development by an FFA increase, due to its function as oncogenic signals (18). Reportedly, increased fatty acid synthase (FASN) activity has been shown in breast cancer cell lines (102), ovarian tumors (103), or cancer precursor lesions in different locations (colon, stomach, esophagus, and oral cavity) (104). The inhibition of the FASN activity reduces the cancer cells’ proliferative capacity, suggesting that FFA act as an energy source for cancer cells. In addition, some studies show a relationship between mutations in the FASN enzyme and cancer incidence (105). Furthermore, FASN has been proposed as an important biomarker of overnutrition-induced insulin resistance (106). Elevated FFA levels derived either from the cancer cell or exogenous fat sources may promote a more aggressive tumorigenic phenotype with the implication of monoacylglycerol lipase (107). Finally, different studies suggest that cancer cells may use lipids directly from adipocytes as energy source, promoting tumor growth (108).
Pathophysiological Mechanisms Relating Visceral Adipose Tissue Excess and Cancer
Several mechanisms whereby obesity can favor cancer development and progression have been proposed, including the obesity-associated low-grade inflammation, endocrine alterations, insulin resistance, and hypoxia–angiogenesis processes.
Adipose Tissue Inflammation, a Microenvironment Favoring Tumorigenesis
Obesity is characterized by a chronic state of low-grade inflammation (109). Adipose tissue is composed of mature adipocytes and stromal cells, which include preadipocytes, fibroblasts as well as vascular and immune cells. The interaction between immune cells and adipocytes leads to inflammation and subsequent adipose tissue dysfunction. Chronic inflammation is characterized by a sustained response to diverse stress signals, which leads to adipose tissue remodeling (110–112). This process includes adipocyte hypertrophy, immune cell infiltration, angiogenesis, and fibrosis during the progression of inflammation (113–116) accompanied with changes in the adipokine production profile (117). The link between chronic inflammation and cancer development was first noticed over 100 years ago by Virchow, when he observed an abundance of leukocytes in neoplastic tissue (118). Since then, the role of chronic inflammation as a precursor of cancer development has been observed in multiple cancer types (119). In animal models of human cancers, inflammation has been shown to influence tumor promotion and progression (120–122). Like adipose tissue, tumor microenvironment is composed of multiple cell types including epithelial cells, fibroblasts, mast cells, and cells of the innate and adaptive immune system that favors a pro-inflammatory and pro-tumorigenic environment (123–127).
Macrophages, obesity, and cancer
Growing evidence reveals that adipose tissue from obese subjects is markedly infiltrated by macrophages that participate in inflammatory pathways with important roles in obesity-associated comorbidities (128). Adipose tissue macrophages (ATM) enhance the levels of inflammatory markers dysregulated in cancer, suggesting that macrophages from peritumoral adipose tissue are locally involved in promoting carcinogenesis (129–131). The presence of tumor-associated macrophages (TAM) also contributes to the pro-inflammatory tumor environment. The recruitment of TAM to the tumor microenvironment is largely dependent on the monocyte chemoattractant protein-1 (MCP-1). In this regard, levels of MCP-1 in tumor tissue have been highly correlated with the accumulation of TAM in ovarian, breast, and pancreatic cancer (123, 132).
Local environment factors are able to determine polarization of macrophages through two general states: M1 (pro-inflammatory) and M2 (anti-inflammatory) macrophages (Figure 1). Activation of M1 macrophages is mediated mainly through the nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) and Jun N-terminal kinase (JNK)/activator protein-1 (AP-1) system. M1 macrophage-derived tumor necrosis factor (TNF)-α and interleukin (IL)-18, 12, and 23 have been identified as important mediators in several chronic inflammatory diseases including obesity and cancer (133–136). Excess of M1 macrophage polarization is related to insulin resistance in obese subjects (137) with M1 macrophages being also differentiated in the tumor initiation stages. Obesity could lead to tumor initiation by the denominated extrinsic pathway, in which tumorigenesis is attributable to a chronic inflammatory condition through the NF-κB pathway (138). NF-κB activation in tumor cells promotes carcinogenesis by increasing their aggressive potential by triggering autocrine growth factor cascades and by inhibiting proliferation control mechanisms, including apoptotic signals (139). NF-κB involvement in tumorigenesis has been shown in various tumor models (140). M1 macrophages also contribute to tumor development by TNF-α and IL-6 signaling, enhancing carcinogenesis by increasing cell proliferation and neoangiogenic cell properties (141, 142). In addition, TNF-α production by macrophages activates the Wnt/β-catenin pathway, which is also associated with tumor development by a non-resulting anti-inflammatory response (143, 144). M2 macrophage activation is mainly mediated by the IL-4-induced activation of Janus kinase (JAK)/signal transducer and activator of transcription (STAT) transduction signaling. It has been shown that IL-10 secreted by M2 macrophages improves insulin signaling, with a protective role in obesity-induced insulin resistance (145). Moreover, several factors are involved in M2 activation, such as peroxisome proliferator-activated receptor (PPAR)-γ, Krüppel-like factor 4, AMP-activated protein kinase (AMPK), or sirtuin-1 (SIRT1) (146).
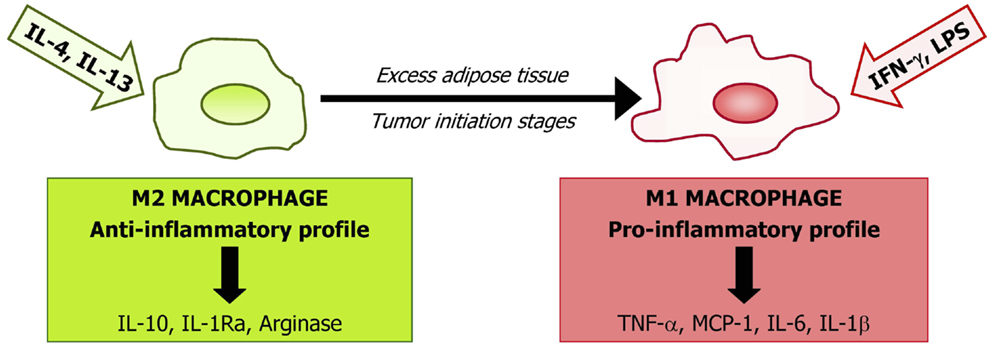
Figure 1. Macrophages are representative of the innate immune system and represent a large proportion of the stromovascular cell fraction in adipose tissue. The phenotype of macrophages depends on the subset of monocytes upon arrival at target tissues being probably determined by the local microenvironment. Based on their cytokine profile secretion and cell surface markers, macrophages are classified into two main types: the “classical” macrophages named M1 in contrast to the “alternatively activated” M2. M1 macrophages are the first line of defense against intracellular pathogens and are classically stimulated by interferon (IFN)-γ or by lipopolysaccharide (LPS). M1 induce the secretion of inflammatory cytokines [interleukin (IL)-1, IL-6, tumor necrosis factor (TNF)-α, or monocyte chemoattractant protein (MCP)-1]. Alternative activation, resulting from induction by the Th2 cytokines interleukin IL-4 and IL-13, is associated with tissue repair and humoral immunity producing immunosuppressive factors, including IL-10, IL-1Ra, and arginase. Obesity and initial tumor stages induce a phenotypic switch from an anti-inflammatory M2-polarized state to a pro-inflammatory M1 state.
In adipose tissue from obese subjects, there is a phenotype switch from M2 to M1 macrophages leading to an increase in general inflammatory markers (147). Moreover, during tumor progression, an M2 to M1 transition of ATM has been shown. However, in later tumor stages, a trophic and immunomodulatory M2-like adipose tissue macrophage phenotype is recruited to the tumor environment. It has been proposed that M2 differentiation could act as a compensatory mechanism, trying to re-establish a homeostatic environment in a pathological condition (144). Mechanisms involved in this M1–M2 switch are not well understood, but molecular pathways related to cyclooxygenase (COX)-2, toll-like receptor (TLR), or Notch signaling have been proposed to play an important role (148–150). Macrophage switch from M1 to M2 in tumor setting occurs via many different receptors, signaling pathways, and transcription factors as a result of the coexistence of pro- and anti-inflammatory signals present during tumor progression in the tumor environment.
Dysregulated adipokine profile in obesity and cancer
The progression of tumors toward malignancy requires the interaction of various cytokines, growth factors, and transcription factors. Adipose tissue acts as an important endocrine organ via the synthesis of several adipokines, which regulate insulin sensitivity, lipolysis, control of energy intake, or inflammatory processes (151–153). Obesity affects the secretory profile of adipokines leading to alteration in multiple physiological processes (154–164). Adipokines are linked to tumor development and progression due to their plentiful actions on different cell types, mainly by exerting their effects through inflammatory pathways. Adiponectin and leptin are two of the most important adipocyte-specific adipokines.
Adiponectin. Adiponectin, also known as Acrp30, ADIPOQ, apM1, or GBP28, is a 30-kDa protein mainly secreted by adipocytes (165). Adiponectin presents a carboxy-terminal globular domain and an amino-terminal collagen domain that forms characteristic multimers (166). Adiponectin exists as full-length adiponectin or as a globular fragment. Globular adiponectin is generated by proteolytic cleavage of full-length adiponectin, but it circulates in very small amounts in the bloodstream (167). Globular adiponectin exists as a trimer, whereas full-length adiponectin can be found as a low-molecular weight (trimer), middle-molecular weight (hexamers), and high-molecular weight (12–18mers) with different biological functions (168, 169). Both globular and full-length adiponectin promote an increased fatty acid oxidation and glucose uptake in the liver and skeletal muscle as well as a decreased hepatic gluconeogenesis. These biological effects are mediated by two transmembrane adiponectin receptors, AdipoR1 and AdipoR2, which are expressed in many tissues and activate the downstream targets AMPK, PPAR-α, and p38 MAPK (167).
Several studies suggest that hypoadiponectinemia or reduced concentrations of adiponectin and PPAR-α in obesity may be one of the mechanisms linking obesity and cancer development as well as progression. Importantly, adiponectin levels are decreased in obesity-associated insulin resistance (170) and cancer (171). A negative correlation between adiponectin levels and the risk of developing colorectal (52, 172, 173), endometrium (174), breast (175), prostate (176), or pancreas cancer (177) has been shown. In animal models, a protective role for adiponectin by suppressing colorectal cancer development has been suggested (178). Adipoq-knockout mice showed a significant higher polyp formation compared with wild-type mice under a high-fat diet. Furthermore, it was also demonstrated that Adipor1, but not Adipor2-deficient mice show an increase in colonic epithelial cell proliferation, suggesting that adiponectin suppresses this biological process under a high-fat diet (178). Reduced adiponectin levels in the obese state lead to the development of insulin resistance and compensatory chronic hyperinsulinemia. Increased insulin levels lead to reduced liver synthesis and secretion of insulin-like growth factor-binding protein (IGFBP)-1 and -2, resulting in increased levels of bioavailable insulin-like growth factor (IGF)-1. Insulin and IGF-1 promote cellular proliferation and inhibit apoptosis in many tissue types, leading to carcinogenesis (15, 171). Low adiponectin levels are potentially associated with carcinogenesis, indirectly through its effects on TNF-α and tumor cell proliferation and directly by its effects on the regulation of hematopoiesis and system, selective binding to several mitogenic growth factors, and inhibition of NF-κB (171, 179). The adiponectin protective effects in tumors also include the inhibition of leptin proliferative signaling and inducing cell apoptosis (180).
Leptin. Leptin, the OB gene product, acts as a key mediator in body weight regulation (181–184). In adipose tissue, the secretion levels of leptin are strictly controlled and maintain a balance to ensure adequate regulation of food intake and energy expenditure under physiological conditions. Contrary to adiponectin, leptin levels are increased in obese individuals (185). Reportedly, treatment with leptin promotes cell growth, inhibits apoptosis, and modulates migration of cancer cells (186). In this regard, an overexpression of leptin receptors in various cancers like breast (187) or colon (188) has been also shown. Tumor cells from papillary thyroid cancer also show increased expression levels of the leptin receptor being associated with a more aggressive phenotype (189). Mature adipocytes secrete both adiponectin and leptin with preadipocytes showing a primarily secretion of high leptin levels (190). An increase of the preadipocyte pool in obese subjects is related to an increase in leptin levels, with proangiogenic and promitogenic properties (191). At the same time, high leptin levels attract more inflammatory cells and promote monocyte to macrophage differentiation, maintaining the obesity-associated state of chronic inflammation. In summary, leptin has an important role in the development of a large variety of malignancies, predominantly acting through the JAK/STAT pathway, which modulates phosphatidylinositol 3-kinase (PI3K)/Akt and extracellular signal-regulated kinase (ERK) 1/2 signaling pathways increasing the expression of anti-apoptotic proteins (XIAP), inflammatory markers (TNF-α, IL-6), angiogenic factors [vascular endothelial growth factor (VEGF)], and also the hypoxia-inducible factor-1α (HIF-1α). These processes promote cancer cell survival, proliferation, and migration.
Tumor necrosis factor-α. TNF-α is a pro-inflammatory cytokine secreted by adipocytes with increased secretion levels in obese subjects (192). TNF-α was first identified as a macrophage-derived factor that induces the necrosis of tumor cells, but when its antitumoral activity was tested on cancer patients, a paradoxical tumor-promoting role became apparent (193–195). At present, the pro-inflammatory role of TNF-α has been linked to all steps involved in tumorigenesis, including cellular transformation, survival, proliferation, invasion, angiogenesis, and metastasis (195, 196). Animal models have shown a positive relationship between TNF-α and tumor development and progression in liver (197) and colorectal cancer (195) with elevated circulating concentrations in different tumoral types (198, 199). In addition, TNF-α is not only produced by a wide variety of tumor cells (200) but also by adipocytes. Levels of TNF-α are increased in obesity, indicating a role for this cytokine in the obesity-associated inflammation and particularly in insulin resistance and diabetes.
Interleukin-6. Interleukin-6 is another major pro-inflammatory cytokine secreted by adipose tissue, which shows increased levels in obese subjects. It has been implicated in inflammation-associated carcinogenesis (201, 202). IL-6 modulates the expression of genes involved in proliferation, survival, and angiogenesis via the JAK/STAT signaling pathway (203). A relationship between IL-6 and carcinogenesis has been shown for renal cell carcinoma (204), gastric cancer (205), or colorectal cancer, among others (200, 206). Moreover, elevated levels of IL-6 in cancer patients correlate with disease aggressiveness and poor prognosis (207, 208). Other studies have revealed the role of IL-6 as an anti-inflammatory cytokine acting via the classic signaling through the activation of the IL-6 receptor on the cellular membrane of specific cell types such as macrophages, neutrophils, some T-cells, and hepatocytes (209, 210).
C-reactive protein. C-reactive protein (CRP) is an acute-phase reactant protein induced as a response to inflammatory conditions and different cytokines including IL-6. Elevated levels of CRP in adipose tissue and serum of obese subjects have been shown (211) as well as in patients with a variety of malignancies (212). Prospective studies have shown a higher risk of developing cancer in those with elevated serum CRP (213, 214). Elevated CRP is a significant predictor of lower survival rates in patients with several cancers, including esophageal, colorectal, hepatocellular, pancreatic, urinary bladder, renal, ovarian, and cervical cancer, after surgical resection. CRP can serve as an additional prognostic predictor for survival and post-treatment monitoring in cancer patients (215).
Resistin. Resistin, also known as Fizz3, is a 12-kDa cysteine-rich protein that plays a role in the development of insulin resistance and obesity in rodents (216). While adipose tissue constitutes the main source of resistin in mice, this molecule is secreted mainly by monocytes and macrophages in humans (217). Human resistin is implicated in the pathogenesis of atherosclerosis by promoting smooth muscle cell proliferation and migration, ICAM-1 and VCAM-1 expression, as well as via regulating patterns of adhesion and inflammation in atherosclerotic plaques (218, 219). Several colorectal cancer studies have shown a positive correlation between resistin and tumor size based on T-staging and tumor grading (172, 220). Resistin has also been postulated as a target molecule, which associates with clinicopathological features and prognosis of pancreatic ductal adenocarcinoma (221). Resistin has been postulated as a pro-inflammatory adipokine (222), exhibiting significantly higher serum levels in patients affected by breast cancer in comparison to controls. Additionally, resistin has been proposed as a regulator of the human choriocarcinoma cell invasive behavior and endothelial cell angiogenic processes (223).
Monocyte chemoattractant protein-1. MCP-1 is a chemokine implicated in the infiltration of macrophages in adipose tissue. Higher levels of MCP-1 can be found in obese subjects compared with lean subjects (224). The presence of infiltrating macrophages as a consequence of increased MCP-1 levels has been shown to correlate with cancer metastasis and poor prognosis in a variety of human carcinomas (172, 225). MCP-1 overexpression has been reported in both ovarian cancer (226, 227) and colorectal cancer (228).
Visfatin. Visfatin also known as pre-B cell colony enhancing factor (PBEF) or nicotinamide phosphoribosyltransferase (NAMPT) is a 52-kDa protein with apparently insulin-mimetic actions (229) and increased plasma concentrations in patients with obesity and T2D (230). Since several authors could not replicate the insulin-mimetic activities of visfatin as well as its ability to bind the insulin receptor, Fukuhara and colleagues were forced to retract some of their original findings (231). However, visfatin stimulates glucose-induced insulin secretion in β-pancreatic cells (232, 233). Visfatin has been further identified in inflammatory cells with its levels being reportedly increased in various inflammatory conditions (229). Visfatin has been related with carcinogenesis and tumor progression, as well as with chemotherapy response (234) in colorectal (235, 236) and breast cancer (237).
Osteopontin. Osteopontin (OPN), also known as secreted phosphoprotein-1 (SPP1) and bone sialoprotein-1, among others, is a phosphoprotein expressed by a wide variety of cell types, including adipocytes (238). The expression levels of OPN are increased in obesity (160, 239) as well as in other pathophysiological processes including neoplastic transformation, progression of metastases, and promotion of cell survival (240). OPN is also expressed in many cancers, with elevated circulating levels and tissular tumor expression being associated with poor prognosis in gastric and liver cancers (241, 242).
Chitinase-3-like protein-1. Chitinase-3-like protein-1 (CHI3L1), also known as YKL-40 or human cartilage glucoprotein-39, is a member of mammalian chitinase-like proteins with increased gene expression levels in obesity and T2D (155, 243). Recently, YKL-40 has been proposed as a new inflammatory marker related to insulin resistance (244). YKL-40 is also increased in several types of solid tumors contributing not only to the development of an inflammatory state but also to cell proliferation, inhibition of apoptosis, stimulation of angiogenesis, and regulation of extracellular tissue remodeling (245). Increased YKL-40 expression has been detected in glioblastoma multiforme (246), papillary thyroid carcinoma (247), extracellular myxoid chondrosarcoma (248), colon cancer (131), and diverse cancer cell lines, being suggested as a useful cancer prognostic biomarker (249).
Consequences of oxidative stress
Reactive oxygen species (ROS) comprise hydrogen peroxide (H2O2), superoxide anion (·O2), and hydroxyl radical (·HO) and induce DNA mutations contributing to cancer development and progression. Potential sources for ROS production in mitochondria include xanthine oxidase, cytochrome P450 oxidases, uncoupled nitric oxide synthases, and NADPH oxidases (250).
Reactive oxygen species exert a wide range of effects in cancer cells depending on their concentrations. In general, low levels of ROS are mitogenic and promote cell proliferation and survival, while intermediate levels cause transient or permanent cell cycle arrest and induce cell differentiation (251). At high levels, ROS can easily react with membrane lipids (causing membrane permeability alteration), DNA (inducing damage and genomic instability), and proteins (promoting oxidative modifications that result in less active enzymes or proteins more susceptible to proteolytic degradation). These facts suggest a possible protective effect of high local ROS levels in cancer progression (252). However, although ROS production does not irreversibly alter cell viability, they can act as a primary messenger, modulating several intracellular signaling cascades leading to cancer progression. Indeed, it has been demonstrated that ROS activate the MAPK, PI3K/Akt, phospholipase C-γ1 (PLCγ1), protein kinase C, NF-κB, and JAK/STAT pathways (253). The increase of fat mass in obesity has been correlated with the increase of markers of systemic oxidative stress in both human and mice (254, 255). In this regard, an increase in the NADPH oxidase subunits in obesity has been described (254), suggesting that adipose tissue may constitute a source of ROS, releasing them into the peripheral blood, and affecting the function of remote organs (256). Adipose tissue from lean subjects expresses antioxidant enzymes (catalase, superoxide dismutase-1, and glutathione peroxidase) for managing ROS production, with the expression of these antioxidant enzymes downregulated in adipose tissue from obese individuals (254, 255, 257, 258).
Therapeutic approaches to cancer involving ROS metabolism have been proposed in recent years. Experimental results have proposed that the ROS increase is involved in apoptosis induction by chemotherapeutic anticancer agents (259). Cancer cells with increased oxidative stress are likely to be more vulnerable to damage by further ROS insults induced by exogenous agents (260). Therefore, manipulating ROS levels by redox modulation constitutes a way to selectively induce cancer cell death without causing significant toxicity to normal cells (261).
Hormonal Links in Obesity-Associated Cancers
A substantial number of cancers are linked to a hormonal etiology such as tumors of the breast, endometrium, ovary, prostate, testis, and thyroid. Adipose tissue excess is thought to be related with these types of tumors due to the abnormal levels of several hormones encountered in obesity, most notably increased estrogen levels (262, 263). Estrogen is a known growth factor with different biosynthesis between pre- and post-menopausal women (264). Pre-menopausal women mainly synthesize estrogens in the ovary, however, after the menopause peripheral sites including adipose tissue, are the primary source of estrogens. In this regard, for post-menopausal women, significant increases in estrone, estradiol, and free estradiol are associated with increasing BMI (262). Estrogen binds to at least three major classes of receptors, ER-α, ER-β, and GPR30 (265) activating numerous growth-promoting genes, including growth factors and growth-enhancing protooncogenes such as c-fos and c-myc (266–268). The primary mediator of post-menopausal estrogen biosynthesis is aromatase, which is found in adipose tissue as well as tumor tissue itself (269). In the expanding adipose tissue, this mechanism of production leads to a local increase in estrogen levels favoring tumor development (15). Different meta-analysis indicated a higher risk of breast cancer in post-menopausal women with high serum estrogen levels (270, 271). However, the role of blood levels of estrogen in obesity-associated colon cancer is controversial. Evidence showed a worse marked effect of obesity in men than women in the carcinogenesis of colon cancer, pointing to a protective effect of estrogens, which are increased in obesity (56).
In addition to excess estrogen, hyperplasia and carcinogenesis may also stem from a decrease in circulating progesterone levels. In obese women, lack of progesterone due to anovulation, similar to that observed in the polycystic ovarian syndrome, may contribute to endometrial cancer risk (272).
Obesity-Associated Insulin Resistance
Insulin is a key hormone involved in the control of glucose and lipid homeostasis with insulin actions being mediated by a transmembrane insulin receptor, a heterotetrameric glycoprotein consisting of two α-subunits and two β-subunits linked by disulfide bonds (273). Insulin promotes cellular response through many pathways, including PI3K/Akt signaling leading to AMPK activation. AMPK has been implicated in regulating cellular functions including energy state, fuel metabolism, mitochondrial biogenesis, protein and ceramide synthesis, as well as cell growth and proliferation (274). In addition, its activation was initially shown to inhibit TNF-α-induced inflammation, insulin resistance, apoptosis, and oxidative stress. AMPK activity is diminished in adipose tissue from severely obese subjects who are insulin resistant compared to equally obese individuals who are insulin sensitive (275). Studies evaluating insulin secretion, as reflected by C-peptide levels, have pointed out a correlation between hyperinsulinemia and poor clinical outcome and death in prostate cancer (276). A recent study has shown that aldo–keto reductase 1B10, which plays a critical role in tumor development and progression through promoting lipogenesis and eliminating cytotoxic carbonyls, is induced by insulin through the activator protein-1 (AP-1) signaling pathway in human hepatocellular carcinoma cells (277). Most recent reports have also suggested that insulin has mitogenic and anti-apoptotic effects in endometrial cancer and that the activation of insulin receptors and Akt is associated with more aggressive features (278, 279).
Since insulin also acts as a growth factor, there are some molecules, called insulin-like peptides (ILPs), which can bind to the insulin receptor and trigger intracellular responses similar to those triggered by insulin. In humans, the most important ILPs are IGF-1 and IGF-2. The mitogenic and anti-apoptotic environment caused by elevated levels of insulin and IGF-1 in obesity accelerates the stepwise accumulation of mutations and, hence, favor carcinogenesis (280). Studies evaluating IGF-1 circulating concentrations and cancer risk show important effects on cancer development and progression. Circulating IGF-1 binds mainly to the major IGF-binding protein, IGFBP-3 (281). Results of early studies evaluating the risk of prostate (282), breast (283), colorectal (284), and lung cancer (285) suggest that high circulating IGF-1 concentrations are associated with an increased risk, whereas high IGFBP-3 concentrations are associated with a lower risk (286, 287). Associations between IGF-1, IGFBP-3, and cancer risk vary by cancer site. Circulating concentrations of IGF-1 increase the risk of non-smoking-related malignant diseases (288). Other ILPs, like IGF-2, have also been proposed to have a relationship with carcinogenesis (279).
Hypoxia, Angiogenesis, and Cancer Development
Adipose tissue presents an adequate exposure to nutrients and oxygen due to the extensive capillary network surrounding each adipocyte. However, there is substantial evidence that hypoxia develops in adipose tissue as the tissue mass expands, and the reduction in O2 concentrations is considered to underlie the obesity-associated inflammatory response (289). Cancer may be promoted by the hypoxic and angiogenic environment of obese adipose tissue, in conjunction with the described elevated circulating levels of cytokines existing in obese subjects (290).
Studies in hepatocellular carcinoma have shown a higher accumulation of ATM in tumor regions poorly vascularized (291, 292). Under low oxygen conditions, both tumor cells and macrophages establish a proangiogenic program mediated by HIF-1, which is a transcriptional activator complex constituted with two types of subunits, an inducible α subunit (HIF-1α, HIF-2α, or HIF-3α), and the constitutively expressed HIF-1β subunit. Hypoxia stabilizes HIF-1α, preventing its post-translational hydroxylation and consequently proteasome-mediated degradation. In addition, hypoxia promotes HIF-1α association with HIF-1β, as well as cofactor recruitment (293). HIF-1α is also transcriptionally regulated by NF-κB (294). ATM adaptation to hypoxia is mediated by the induction of HIF-1 and HIF-2-regulated genes, including VEGF, fibroblast growth factor (FGF)-β, and IL-8, as well as glycolytic enzymes (295). Furthermore, the HIF-1 pathway has been demonstrated to play a role in macrophage recruitment and activation. As suggested for NF-κB and STAT-3, HIF-1 might be a potential target in hepatocellular carcinoma therapy (296). HIF-1 has been also proposed as a key molecule implicated in tumor metastasis (297, 298) and a possible metabolic target in cancer therapy (299–301). Inflammatory molecules like TNF-α are also secreted by adipose tissue in response to hypoxia and macrophage–monocyte infiltration (280).
Therapeutic Strategies
Since obesity is a result of a prolonged positive energy balance, caloric restriction and PA are key strategies to reduce BMI, fat mass, and improve metabolic abnormalities. Although pharmaceutical approaches for obesity and T2D are used, the main pillar of treatment is lifestyle changes. In this regard, it has been recently reported that behaviors concordant with the Nutrition and Physical Activity Cancer Prevention Guidelines are associated with lower risk of total, breast, and colorectal cancers and lower cancer-specific mortality in post-menopausal women (302).
Physical Activity
Sedentary behavior and insufficient PA are strongly related with obesity and metabolic syndrome development. Animal studies have shown metabolic beneficial effects of aerobic exercise training independent of dietary changes (303). However, it has been demonstrated that the combination of PA and dietary intervention is more effective than only an increase in PA (304). There are evidences of benefits of physical exercise to normalize obesity-altered lipid patterns (305, 306), improve glucose metabolism (307), change the growth hormone/IGF-1 axis (308), and consequently reduce the risk of obesity-associated comorbidities. Studies in humans have also shown a direct relationship between BMI, PA, and mortality with cancer (309). In a large cohort study of middle-age women, both the excess of weight (considered as a BMI ≥ 25 kg/m2) and sedentary habits were significantly associated with an increased mortality. It has been estimated that excess weight and physical inactivity could account for 31% of all premature deaths and 21% of deaths from cancer among non-smoking women (310). A great variety of studies has reported the advantages of PA in several types of cancer, such as breast (311, 312), colon (313, 314), or prostate cancer (315).
Diet
Both epidemiological studies and experiments in animals suggest that alterations in caloric intake or in the quality of the diet may significantly influence the risk of cancer development and progression. A higher risk of cancer development related to diets rich in calories, overabundant in alcohol and animal fats, and/or deficient in vegetal products has been reported. By contrast, cancer risk was lower in diets with a higher intake of fruit and vegetables. High-fiber cereal consumption has also been linked to a reduced risk of colorectal cancer (16, 316). Recently, the role of ω-3 polyunsaturated fatty acids in cancer prevention has been highlighted (317, 318). These fatty acids exert their effects by targeting different stages of cancer development, including cell proliferation and survival, inflammation, or angiogenesis (319, 320).
A cell-cycle regulation by nutrients has been shown in studies on Saccharomyces cerevisiae. Growth experiments under glucose-limited conditions suggested that coupling of circadian, metabolic, and cell division cycles is essential for genome integrity (321). However, the coupling of these cycles is highly dependent on the experimental conditions and can be uncoupled under other nutrient-limiting conditions (322). The relationship between nutrient availability and genome alterations could play a role in obesity-related cancer due to an abnormal energy state in obese subjects. Therefore, aerobic glycolysis or the Warburg effect links the high rate of glucose fermentation to cancer (323). Moreover, sugar intake and consumption of sweet cakes and cookies have been associated with increased risk of endometrial cancer (324). Biological mechanisms whereby high-sugar foods consumption might increase risk of cancer development are related to the development of hyperglycemia, stimulation of insulin production, and insulin resistance (325, 326).
Caloric restriction could be a strategy to prevent cancer by reducing changes induced by obesity and general inflammatory profile (327). In this regard, studies in prostate cancer and murine prostate cancer models suggest that caloric restriction and weight loss may reduce the risk of prostate cancer-specific mortality (71). Positive effects of weight loss and PA have also been demonstrated in colorectal cancer (55).
Bariatric Surgery
When PA and dietary changes are not enough to reduce BMI, bariatric surgery is a useful strategy to reduce excess of fat mass and prevent weight gain in morbid obesity. There are different surgical techniques that combine variable degrees of restriction and malabsorption. Beneficial effects of bariatric surgery in reducing obesity-associated alterations have been largely described. A clinical improvement in patients with T2D after bariatric surgery procedures has been shown (328, 329). Noteworthy, there are studies that suggest a preventive effect of bariatric surgery on diabetes development (330). Positive effects over other comorbidities include OSA (331), dyslipidemia (332), hepatic fibrosis (333), and cardiovascular risk (334, 335). Furthermore, there is evidence showing that bariatric surgery-induced weight loss reduces cancer risk in some retrospective cohort studies (336, 337). The mechanisms that may lead to a reduced cancer incidence in subjects under bariatric surgery include reduction in sex steroid plasma levels; decreased circulating fatty acid, adipokines, and pro-inflammatory cytokines levels; improvement in insulin sensitivity and reduction of fat mass leading to a lower ectopic fat accumulation (338). The Swedish Obese Subjects (SOS) study, a prospective and controlled intervention trial involving 2,010 obese patients who underwent bariatric surgery and 1,037 contemporaneously matched obese subjects, found a significantly lower number of first-time cancers after inclusion in the surgery group (gastric banding, vertical banded gastroplasty, and gastric bypass) than in the control group, but only in women. It has been postulated that since gastric bypass reduces hyperinsulinemia, this could also be a biological mechanism by which bariatric surgery exerts protective effects on cancer incidence (339). Future studies with larger patient numbers and longer follow-up periods are required to firmly establish whether this might also have beneficial effects for men in the long term (340).
Conclusion
Obesity is a potentially avoidable high prevalent risk factor for developing different health problems including tumor development. There are increasing evidences relating obesity and cancer, with a well-established relationship between obesity and breast, colorectal, prostate, or lung cancer among others. It has been proposed that an important percentage of all cancer deaths may be attributable to obesity. Due to the high prevalence and repercussion of these conditions, the understanding of mechanisms of obesity-associated tumorigenesis is an important objective in the prevention and treatment of these pathologies. Future interventions to reduce the prevalence of obesity are needed to prevent cancer development in these patients. In this regard, it has been shown that increased PA, healthy dietary habits, and bariatric surgery may be appropriate to reduce cancer risk in obese patients. In addition, a comprehensive approximation to the underlying molecular mechanisms involved in obesity-related carcinogenesis may provide specific targets implicated in pathways by which obesity leads to tumor progression. Reduction of inflammatory signaling, improvement of insulin sensitivity, or counteracting the hypoxia-inducible factors appears to be relevant pathways in prevention of cancer development and reducing its progression (Figure 2).
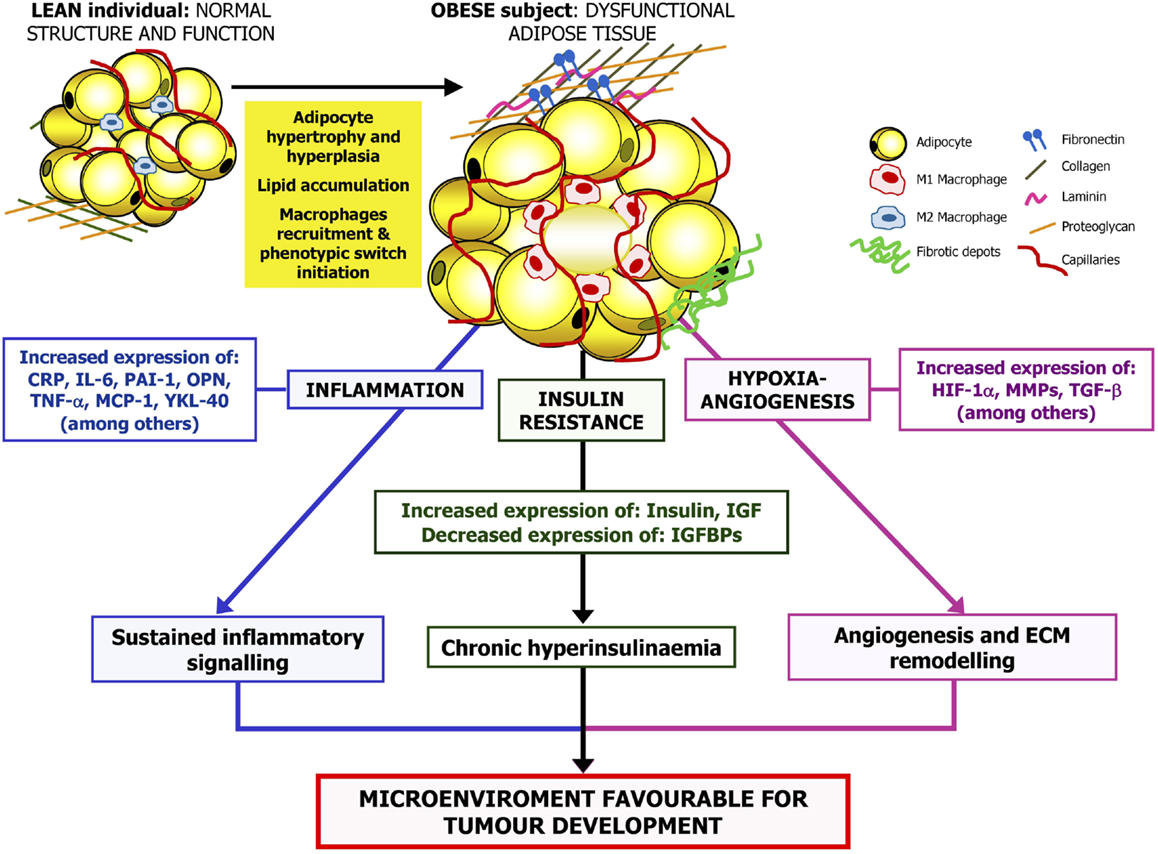
Figure 2. Proposed mechanisms linking obesity and cancer. The excess of adipose tissue, especially abdominal obesity, is related to changes in circulating lipid concentrations, reactive oxygen species levels as well as adipokine and hormone secretion profile. Obesity is also linked to adipocyte hypertrophy and hypoxia, aggravating the inflammatory state. Therefore, the adipose tissue-derived inflammatory cytokines, the production of angiogenic factors by adipocytes, or infiltrating M1 macrophages that take place in obesity together with the obesity-associated insulin resistance may promote the stimulation of a microenvironment favorable for tumorigenesis. CRP, C-reactive protein; HIF-1α, hypoxia-inducible factor-1α; IGF, insulin growth factor; IGFBP, insulin-like growth factor-binding protein; IL, interleukin; MCP-1, monocyte chemoattractant protein 1; MMP, matrix metalloproteinase; OPN, osteopontin; PAI-1, plasminogen activator inhibitor-1; TGF-β, transforming growth factor β; TNF-α, tumor necrosis factor α; YKL-40, chitinase-3-like protein.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Fondo de Investigación Sanitaria-FEDER (PI11/02681, PI12/00515, PI13/00460) from the Spanish Instituto de Salud Carlos III and by the Department of Health (48/2011 and 58/2011) of the Gobierno de Navarra of Spain. Centro de Investigación Biomédica en Red de Fisiopatología de la Obesidad y Nutrición is an initiative of the Instituto de Salud Carlos III (Spain).
References
1. Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999-2010. JAMA (2012) 307:491–7. doi: 10.1001/jama.2012.39
2. Gómez-Ambrosi J, Silva C, Galofré JC, Escalada J, Santos S, Millán D, et al. Body mass index classification misses subjects with increased cardiometabolic risk factors related to elevated adiposity. Int J Obes (2012) 36:286–94. doi:10.1038/ijo.2011.100
3. Frühbeck G. Obesity: screening for the evident in obesity. Nat Rev Endocrinol (2012) 8:570–2. doi:10.1038/nrendo.2012.165
4. Frühbeck G, Toplak H, Woodward E, Yumuk V, Maislos M, Oppert JM. Obesity: the gateway to ill health – an EASO position statement on a rising public health, clinical and scientific challenge in Europe. Obes Facts (2013) 6:117–20. doi:10.1159/000350627
5. Kral JG, Kava RA, Catalano PM, Moore BJ. Severe obesity: the neglected epidemic. Obes Facts (2012) 5:254–69. doi:10.1159/000338566
6. Mokdad AH, Ford ES, Bowman BA, Dietz WH, Vinicor F, Bales VS, et al. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA (2003) 289:76–9. doi:10.1001/jama.289.1.76
7. Angulo P. NAFLD, obesity, and bariatric surgery. Gastroenterology (2006) 130:1848–52. doi:10.1053/j.gastro.2006.03.041
8. Moore JB. Non-alcoholic fatty liver disease: the hepatic consequence of obesity and the metabolic syndrome. Proc Nutr Soc (2010) 69:211–20. doi:10.1017/S0029665110000030
9. Diehl AM. Hepatic complications of obesity. Gastroenterol Clin North Am (2010) 39:57–68. doi:10.1016/j.gtc.2009.12.001
10. de Sousa AG, Cercato C, Mancini MC, Halpern A. Obesity and obstructive sleep apnea-hypopnea syndrome. Obes Rev (2008) 9:340–54. doi:10.1111/j.1467-789X.2008.00478.x
11. Campo A, Frühbeck G, Zulueta JJ, Iriarte J, Seijo LM, Alcaide AB, et al. Hyperleptinaemia, respiratory drive and hypercapnic response in obese patients. Eur Respir J (2007) 30:223–31. doi:10.1183/09031936.00115006
12. Gómez-Ambrosi J, Salvador J, Paramo JA, Orbe J, de Irala J, Diez-Caballero A, et al. Involvement of leptin in the association between percentage of body fat and cardiovascular risk factors. Clin Biochem (2002) 35:315–20. doi:10.1016/S0009-9120(02)00320-X
13. Landsberg L, Aronne LJ, Beilin LJ, Burke V, Igel LI, Lloyd-Jones D, et al. Obesity-related hypertension: pathogenesis, cardiovascular risk, and treatment – a position paper of the obesity society and the American society of hypertension. Obesity (2013) 21:8–24. doi:10.1002/oby.20181
14. Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature (2006) 444:875–80. doi:10.1038/nature05487
15. Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer (2004) 4:579–91. doi:10.1038/nrc1408
16. De Pergola G, Silvestris F. Obesity as a major risk factor for cancer. J Obes (2013) 2013:291546. doi:10.1155/2013/291546
17. Hursting SD. Minireview: the year in obesity and cancer. Mol Endocrinol (2012) 26:1961–6. doi:10.1210/me.2012-1283
18. Louie SM, Roberts LS, Nomura DK. Mechanisms linking obesity and cancer. Biochim Biophys Acta (2013) 1831:1499–508. doi:10.1016/j.bbalip.2013.02.008
19. Sundaram S, Johnson AR, Makowski L. Obesity, metabolism and the microenvironment: links to cancer. J Carcinog (2013) 12:19. doi:10.4103/1477-3163.119606
20. Aleman JO, Eusebi LH, Ricciardiello L, Patidar K, Sanyal AJ, Holt PR. Mechanisms of obesity-induced gastrointestinal neoplasia. Gastroenterology (2013) 146:357–73. doi:10.1053/j.gastro.2013.11.051
22. Dyson J, Jaques B, Chattopadyhay D, Lochan R, Graham J, Das D, et al. Hepatocellular cancer: the impact of obesity, type 2 diabetes and a multidisciplinary team. J Hepatol (2014) 60:110–7. doi:10.1016/j.jhep.2013.08.011
23. Kitahara CM, Berndt SI, de Gonzalez AB, Coleman HG, Schoen RE, Hayes RB, et al. Prospective investigation of body mass index, colorectal adenoma, and colorectal cancer in the prostate, lung, colorectal, and ovarian cancer screening trial. J Clin Oncol (2013) 31:2450–9. doi:10.1200/JCO.2012.48.4691
24. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin (2011) 61:69–90. doi:10.3322/caac.20107
25. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin (2013) 63:11–30. doi:10.3322/caac.21166
26. Arnold M, Karim-Kos HE, Coebergh JW, Byrnes G, Antilla A, Ferlay J, et al. Recent trends in incidence of five common cancers in 26 European countries since 1988: analysis of the European cancer observatory. Eur J Cancer (2013). doi:10.1016/j.ejca.2013.09.002
27. Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer (2013) 49:1374–403. doi:10.1016/j.ejca.2012.12.027
28. de Vries E, Soerjomataram I, Lemmens VE, Coebergh JW, Barendregt JJ, Oenema A, et al. Lifestyle changes and reduction of colon cancer incidence in Europe: a scenario study of physical activity promotion and weight reduction. Eur J Cancer (2010) 46:2605–16. doi:10.1016/j.ejca.2010.07.040
29. Lieberman D. Colorectal cancer screening: practice guidelines. Dig Dis (2012) 30(Suppl 2):34–8. doi:10.1159/000341891
30. Roobol MJ, Carlsson SV. Risk stratification in prostate cancer screening. Nat Rev Urol (2013) 10:38–48. doi:10.1038/nrurol.2013.84
31. Smith RA, Brooks D, Cokkinides V, Saslow D, Brawley OW. Cancer screening in the United States, 2013: a review of current American cancer society guidelines, current issues in cancer screening, and new guidance on cervical cancer screening and lung cancer screening. CA Cancer J Clin (2013) 63:88–105. doi:10.3322/caac.21174
32. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med (2003) 348:1625–38. doi:10.1056/NEJMoa021423
33. Chang SH, Pollack LM, Colditz GA. Obesity, mortality, and life years lost associated with breast cancer in nonsmoking US women, national health interview survey, 1997-2000. Prev Chronic Dis (2013) 10:E186. doi:10.5888/pcd10.130112
34. Ghaem Maralani H, Tai BC, Wong TY, Tai ES, Li J, Wang JJ, et al. The prognostic role of body mass index on mortality amongst the middle-aged and elderly: a competing risk analysis. Diabetes Res Clin Pract (2013) 103(1):42–50. doi:10.1016/j.diabres.2013.11.025
35. Hainer V, Aldhoon-Hainerova I. Obesity paradox does exist. Diabetes Care (2013) 36(Suppl 2):S276–81. doi:10.2337/dcS13-2023
36. Gruberg L, Weissman NJ, Waksman R, Fuchs S, Deible R, Pinnow EE, et al. The impact of obesity on the short-term and long-term outcomes after percutaneous coronary intervention: the obesity paradox? J Am Coll Cardiol (2002) 39:578–84. doi:10.1016/S0735-1097(01)01802-2
37. Shah R, Gayat E, Januzzi JL Jr, Sato N, Cohen-Solal A, Disomma S, et al. Body mass index and mortality in acutely decompensated heart failure across the world: a global obesity paradox. J Am Coll Cardiol (2014) 63:778–85. doi:10.1016/j.jacc.2013.09.072
38. Heymsfield SB, Cefalu WT. Does body mass index adequately convey a patient’s mortality risk? JAMA (2013) 309:87–8. doi:10.1001/jama.2012.185445
39. Montazeri A, Sadighi J, Farzadi F, Maftoon F, Vahdaninia M, Ansari M, et al. Weight, height, body mass index and risk of breast cancer in postmenopausal women: a case-control study. BMC Cancer (2008) 8:278. doi:10.1186/1471-2407-8-278
40. Litton JK, Gonzalez-Angulo AM, Warneke CL, Buzdar AU, Kau SW, Bondy M, et al. Relationship between obesity and pathologic response to neoadjuvant chemotherapy among women with operable breast cancer. J Clin Oncol (2008) 26:4072–7. doi:10.1200/JCO.2007.14.4527
41. Dal Maso L, Zucchetto A, Talamini R, Serraino D, Stocco CF, Vercelli M, et al. Effect of obesity and other lifestyle factors on mortality in women with breast cancer. Int J Cancer (2008) 123:2188–94. doi:10.1002/ijc.23747
42. Dawood S, Broglio K, Gonzalez-Angulo AM, Kau SW, Islam R, Hortobagyi GN, et al. Prognostic value of body mass index in locally advanced breast cancer. Clin Cancer Res (2008) 14:1718–25. doi:10.1158/1078-0432.CCR-07-1479
43. Majed B, Moreau T, Senouci K, Salmon RJ, Fourquet A, Asselain B. Is obesity an independent prognosis factor in woman breast cancer? Breast Cancer Res Treat (2008) 111:329–42. doi:10.1007/s10549-007-9785-3
44. Cleveland RJ, Eng SM, Abrahamson PE, Britton JA, Teitelbaum SL, Neugut AI, et al. Weight gain prior to diagnosis and survival from breast cancer. Cancer Epidemiol Biomarkers Prev (2007) 16:1803–11. doi:10.1158/1055-9965.EPI-06-0889
45. Lahmann PH, Hoffmann K, Allen N, van Gils CH, Khaw KT, Tehard B, et al. Body size and breast cancer risk: findings from the European prospective investigation into cancer and nutrition (EPIC). Int J Cancer (2004) 111:762–71. doi:10.1002/ijc.20315
46. Mellemkjaer L, Bigaard J, Tjonneland A, Christensen J, Thomsen B, Johansen C, et al. Body composition and breast cancer in postmenopausal women: a Danish prospective cohort study. Obesity (2006) 14:1854–62. doi:10.1038/oby.2006.214
47. Boyd NF, Martin LJ, Sun L, Guo H, Chiarelli A, Hislop G, et al. Body size, mammographic density, and breast cancer risk. Cancer Epidemiol Biomarkers Prev (2006) 15:2086–92. doi:10.1158/1055-9965.EPI-05-0762
48. Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet (2008) 371:569–78. doi:10.1016/S0140-6736(08)60269-X
49. Andrieu N, Launoy G, Guillois R, Ory-Paoletti C, Gignoux M. Familial relative risk of colorectal cancer: a population-based study. Eur J Cancer (2003) 39:1904–11. doi:10.1016/S0959-8049(03)00420-9
50. Kim Y, Lee S. An association between colonic adenoma and abdominal obesity: a cross-sectional study. BMC Gastroenterol (2009) 9:4. doi:10.1186/1471-230X-9-4
51. Thygesen LC, Gronbaek M, Johansen C, Fuchs CS, Willett WC, Giovannucci E. Prospective weight change and colon cancer risk in male US health professionals. Int J Cancer (2008) 123:1160–5. doi:10.1002/ijc.23612
52. Guffey CR, Fan D, Singh UP, Murphy EA. Linking obesity to colorectal cancer: recent insights into plausible biological mechanisms. Curr Opin Clin Nutr Metab Care (2013) 16:595–600. doi:10.1097/MCO.0b013e328362d10b
53. Ma Y, Yang Y, Wang F, Zhang P, Shi C, Zou Y, et al. Obesity and risk of colorectal cancer: a systematic review of prospective studies. PLoS One (2013) 8:e53916. doi:10.1371/journal.pone.0053916
54. Larsson SC, Bergkvist L, Wolk A. High-fat dairy food and conjugated linoleic acid intakes in relation to colorectal cancer incidence in the Swedish mammography cohort. Am J Clin Nutr (2005) 82:894–900.
55. Bardou M, Barkun AN, Martel M. Obesity and colorectal cancer. Gut (2013) 62:933–47. doi:10.1136/gutjnl-2013-304701
56. Chen J, Iverson D. Estrogen in obesity-associated colon cancer: friend or foe? Protecting postmenopausal women but promoting late-stage colon cancer. Cancer Causes Control (2012) 23:1767–73. doi:10.1007/s10552-012-0066-z
57. Ben Q, An W, Jiang Y, Zhan X, Du Y, Cai QC, et al. Body mass index increases risk for colorectal adenomas based on meta-analysis. Gastroenterology (2012) 142:762–72. doi:10.1053/j.gastro.2011.12.050
58. Lin J, Zhang SM, Cook NR, Rexrode KM, Lee IM, Buring JE. Body mass index and risk of colorectal cancer in women (United States). Cancer Causes Control (2004) 15:581–9. doi:10.1023/B:CACO.0000036168.23351.f1
59. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature (2013) 499:97–101. doi:10.1038/nature12347
60. Center MM, Jemal A, Lortet-Tieulent J, Ward E, Ferlay J, Brawley O, et al. International variation in prostate cancer incidence and mortality rates. Eur Urol (2012) 61:1079–92. doi:10.1016/j.eururo.2012.02.054
61. Bergström A, Pisani P, Tenet V, Wolk A, Adami HO. Overweight as an avoidable cause of cancer in Europe. Int J Cancer (2001) 91:421–30. doi:10.1002/1097-0215(200002)9999:9999<::AID-IJC1053>3.0.CO;2-T
62. MacInnis RJ, English DR. Body size and composition and prostate cancer risk: systematic review and meta-regression analysis. Cancer Causes Control (2006) 17:989–1003. doi:10.1007/s10552-006-0074-y
63. Spangler E, Zeigler-Johnson CM, Coomes M, Malkowicz SB, Wein A, Rebbeck TR. Association of obesity with tumor characteristics and treatment failure of prostate cancer in African-American and European American men. J Urol (2007) 178:1939–44. doi:10.1016/j.juro.2007.07.021
64. Andersson SO, Wolk A, Bergstrom R, Adami HO, Engholm G, Englund A, et al. Body size and prostate cancer: a 20-year follow-up study among 135006 Swedish construction workers. J Natl Cancer Inst (1997) 89:385–9. doi:10.1093/jnci/89.5.385
65. Baillargeon J, Platz EA, Rose DP, Pollock BH, Ankerst DP, Haffner S, et al. Obesity, adipokines, and prostate cancer in a prospective population-based study. Cancer Epidemiol Biomarkers Prev (2006) 15:1331–5. doi:10.1158/1055-9965.EPI-06-0082
66. Engeland A, Tretli S, Bjorge T. Height, body mass index, and prostate cancer: a follow-up of 950000 Norwegian men. Br J Cancer (2003) 89:1237–42. doi:10.1038/sj.bjc.6601206
67. Porter MP, Stanford JL. Obesity and the risk of prostate cancer. Prostate (2005) 62:316–21. doi:10.1002/pros.20121
68. Gong Z, Neuhouser ML, Goodman PJ, Albanes D, Chi C, Hsing AW, et al. Obesity, diabetes, and risk of prostate cancer: results from the prostate cancer prevention trial. Cancer Epidemiol Biomarkers Prev (2006) 15:1977–83. doi:10.1158/1055-9965.EPI-06-0477
69. Rodriguez C, Freedland SJ, Deka A, Jacobs EJ, McCullough ML, Patel AV, et al. Body mass index, weight change, and risk of prostate cancer in the cancer prevention study II nutrition cohort. Cancer Epidemiol Biomarkers Prev (2007) 16:63–9. doi:10.1158/1055-9965.EPI-06-0754
70. Wright ME, Chang SC, Schatzkin A, Albanes D, Kipnis V, Mouw T, et al. Prospective study of adiposity and weight change in relation to prostate cancer incidence and mortality. Cancer (2007) 109:675–84. doi:10.1002/cncr.22443
71. Allott EH, Masko EM, Freedland SJ. Obesity and prostate cancer: weighing the evidence. Eur Urol (2013) 63:800–9. doi:10.1016/j.eururo.2012.11.013
72. Yang Y, Dong J, Sun K, Zhao L, Zhao F, Wang L, et al. Obesity and incidence of lung cancer: a meta-analysis. Int J Cancer (2013) 132:1162–9. doi:10.1002/ijc.27719
73. El-Zein M, Parent ME, Rousseau MC. Comments on a recent meta-analysis: obesity and lung cancer. Int J Cancer (2013) 132:1962–3. doi:10.1002/ijc.27854
75. Khan N, Afaq F, Mukhtar H. Lifestyle as risk factor for cancer: evidence from human studies. Cancer Lett (2010) 293:133–43. doi:10.1016/j.canlet.2009.12.013
76. Kabat GC, Kim M, Hunt JR, Chlebowski RT, Rohan TE. Body mass index and waist circumference in relation to lung cancer risk in the women’s health initiative. Am J Epidemiol (2008) 168:158–69. doi:10.1093/aje/kwn109
77. Kanashiki M, Sairenchi T, Saito Y, Ishikawa H, Satoh H, Sekizawa K. Body mass index and lung cancer: a case-control study of subjects participating in a mass-screening program. Chest (2005) 128:1490–6. doi:10.1378/chest.128.3.1490
78. Kabat GC, Miller AB, Rohan TE. Body mass index and lung cancer risk in women. Epidemiology (2007) 18:607–12. doi:10.1097/EDE.0b013e31812713d1
79. Ntikoudi E, Kiagia M, Boura P, Syrigos KN. Hormones of adipose tissue and their biologic role in lung cancer. Cancer Treat Rev (2014) 40:22–30. doi:10.1016/j.ctrv.2013.06.005
80. Bray GA, Popkin BM. Dietary sugar and body weight: have we reached a crisis in the epidemic of obesity and diabetes? Health be damned! Pour on the sugar. Diabetes Care (2014) 37:950–6. doi:10.2337/dc13-2085
81. Ludwig DS, Peterson KE, Gortmaker SL. Relation between consumption of sugar-sweetened drinks and childhood obesity: a prospective, observational analysis. Lancet (2001) 357:505–8. doi:10.1016/S0140-6736(00)04041-1
82. Frühbeck G, Becerril S, Sáinz N, Garrastachu P, García-Velloso MJ. BAT: a new target for human obesity? Trends Pharmacol Sci (2009) 30:387–96. doi:10.1016/j.tips.2009.05.003
83. Catalán V, Gómez-Ambrosi J, Rodríguez A, Salvador J, Frühbeck G. Adipokines in the treatment of diabetes mellitus and obesity. Expert Opin Pharmacother (2009) 10:239–54. doi:10.1517/14656560802618811
84. Falcao-Pires I, Castro-Chaves P, Miranda-Silva D, Lourenco AP, Leite-Moreira AF. Physiological, pathological and potential therapeutic roles of adipokines. Drug Discov Today (2012) 17:880–9. doi:10.1016/j.drudis.2012.04.007
85. Rodríguez A, Catalán V, Gómez-Ambrosi J, Frühbeck G. Visceral and subcutaneous adiposity: are both potential therapeutic targets for tackling the metabolic syndrome? Curr Pharm Des (2007) 13:2169–75. doi:10.2174/138161207781039599
86. Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, Liu CY, et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation (2007) 116:39–48. doi:10.1161/CIRCULATIONAHA.106.675355
87. Koster A, Stenholm S, Alley DE, Kim LJ, Simonsick EM, Kanaya AM, et al. Body fat distribution and inflammation among obese older adults with and without metabolic syndrome. Obesity (2010) 18:2354–61. doi:10.1038/oby.2010.86
88. Bjorntorp P. The associations between obesity, adipose tissue distribution and disease. Acta Med Scand Suppl (1988) 723:121–34.
89. Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell (2007) 131:242–56. doi:10.1016/j.cell.2007.10.004
90. Frühbeck G, Sesma P, Burrell MA. PRDM16: the interconvertible adipo-myocyte switch. Trends Cell Biol (2009) 19:141–6. doi:10.1016/j.tcb.2009.01.007
91. Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, et al. Functional brown adipose tissue in healthy adults. N Engl J Med (2009) 360:1518–25. doi:10.1056/NEJMoa0808949
92. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med (2009) 360:1509–17. doi:10.1056/NEJMoa0810780
93. Feng B, Zhang T, Xu H. Human adipose dynamics and metabolic health. Ann N Y Acad Sci (2013) 1281:160–77. doi:10.1111/nyas.12009
94. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev (2004) 84:277–359. doi:10.1152/physrev.00015.2003
95. Bruera E. ABC of palliative care. Anorexia, cachexia, and nutrition. BMJ (1997) 315:1219–22. doi:10.1136/bmj.315.7117.1219
96. Tsoli M, Moore M, Burg D, Painter A, Taylor R, Lockie SH, et al. Activation of thermogenesis in brown adipose tissue and dysregulated lipid metabolism associated with cancer cachexia in mice. Cancer Res (2012) 72:4372–82. doi:10.1158/0008-5472.CAN-11-3536
97. Wu J, Cohen P, Spiegelman BM. Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev (2013) 27:234–50. doi:10.1101/gad.211649.112
98. Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the metabolic syndrome – an allostatic perspective. Biochim Biophys Acta (2010) 1801:338–49. doi:10.1016/j.bbalip.2009.12.006
99. Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, et al. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest (1996) 97:2859–65. doi:10.1172/JCI118742
100. Rebrin K, Steil GM, Getty L, Bergman RN. Free fatty acid as a link in the regulation of hepatic glucose output by peripheral insulin. Diabetes (1995) 44:1038–45. doi:10.2337/diabetes.44.9.1038
101. Hennes MM, Shrago E, Kissebah AH. Receptor and postreceptor effects of free fatty acids (FFA) on hepatocyte insulin dynamics. Int J Obes (1990) 14:831–41.
102. Hunt DA, Lane HM, Zygmont ME, Dervan PA, Hennigar RA. MRNA stability and overexpression of fatty acid synthase in human breast cancer cell lines. Anticancer Res (2007) 27:27–34.
103. Gansler TS, Hardman W III, Hunt DA, Schaffel S, Hennigar RA. Increased expression of fatty acid synthase (OA-519) in ovarian neoplasms predicts shorter survival. Hum Pathol (1997) 28:686–92. doi:10.1016/S0046-8177(97)90177-5
104. Alli PM, Pinn ML, Jaffee EM, McFadden JM, Kuhajda FP. Fatty acid synthase inhibitors are chemopreventive for mammary cancer in neu-N transgenic mice. Oncogene (2005) 24:39–46. doi:10.1038/sj.onc.1208174
105. Nguyen PL, Ma J, Chavarro JE, Freedman ML, Lis R, Fedele G, et al. Fatty acid synthase polymorphisms, tumor expression, body mass index, prostate cancer risk, and survival. J Clin Oncol (2010) 28:3958–64. doi:10.1200/JCO.2009.27.0793
106. Fernández-Real JM, Menéndez JA, Moreno-Navarrete JM, Bluher M, Vazquez-Martín A, Vazquez MJ, et al. Extracellular fatty acid synthase: a possible surrogate biomarker of insulin resistance. Diabetes (2010) 59:1506–11. doi:10.2337/db09-1756
107. Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell (2010) 140:49–61. doi:10.1016/j.cell.2009.11.027
108. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med (2011) 17:1498–503. doi:10.1038/nm.2492
109. Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest (2003) 112:1785–8. doi:10.1172/JCI200320514
110. Catalán V, Gómez-Ambrosi J, Rodríguez A, Frühbeck G. Role of extracellular matrix remodelling in adipose tissue pathophysiology: relevance in the development of obesity. Histol Histopathol (2012) 27:1515–28.
111. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454:428–35. doi:10.1038/nature07201
112. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol (2005) 6:1191–7. doi:10.1038/ni1276
113. Divoux A, Tordjman J, Lacasa D, Veyrie N, Hugol D, Aissat A, et al. Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes (2010) 59:2817–25. doi:10.2337/db10-0585
114. Hotamisligil GS. Inflammation and metabolic disorders. Nature (2006) 444:860–7. doi:10.1038/nature05485
115. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med (2009) 15:914–20. doi:10.1038/nm.1964
116. Nishimura S, Manabe I, Nagasaki M, Hosoya Y, Yamashita H, Fujita H, et al. Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes (2007) 56:1517–26. doi:10.2337/db06-1749
117. Suganami T, Ogawa Y. Adipose tissue macrophages: their role in adipose tissue remodeling. J Leukoc Biol (2010) 88:33–9. doi:10.1189/jlb.0210072
118. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet (2001) 357:539–45. doi:10.1016/S0140-6736(00)04046-0
120. Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell (2005) 7:211–7. doi:10.1016/j.ccr.2005.02.013
121. Karin M. Inflammation and cancer: the long reach of Ras. Nat Med (2005) 11:20–1. doi:10.1038/nm0105-20
122. Karin M. Nuclear factor-κB in cancer development and progression. Nature (2006) 441:431–6. doi:10.1038/nature04870
123. Harvey AE, Lashinger LM, Hursting SD. The growing challenge of obesity and cancer: an inflammatory issue. Ann N Y Acad Sci (2011) 1229:45–52. doi:10.1111/j.1749-6632.2011.06096.x
124. Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, et al. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer (2000) 88:577–83. doi:10.1002/(SICI)1097-0142(20000201)88:3<577::AID-CNCR13>3.0.CO;2-V
125. Ribatti D, Ennas MG, Vacca A, Ferreli F, Nico B, Orru S, et al. Tumor vascularity and tryptase-positive mast cells correlate with a poor prognosis in melanoma. Eur J Clin Invest (2003) 33:420–5. doi:10.1046/j.1365-2362.2003.01152.x
126. Leek RD, Landers RJ, Harris AL, Lewis CE. Necrosis correlates with high vascular density and focal macrophage infiltration in invasive carcinoma of the breast. Br J Cancer (1999) 79:991–5. doi:10.1038/sj.bjc.6690158
127. Catalán V, Gómez-Ambrosi J, Rodríguez A, Frühbeck G. Adipose tissue immunity and cancer. Front Physiol (2013) 4:275. doi:10.3389/fphys.2013.00275
128. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112:1796–808. doi:10.1172/JCI19246
129. Mayi TH, Daoudi M, Derudas B, Gross B, Bories G, Wouters K, et al. Human adipose tissue macrophages display activation of cancer-related pathways. J Biol Chem (2012) 287:21904–13. doi:10.1074/jbc.M111.315200
130. Ribeiro R, Monteiro C, Catalán V, Hu P, Cunha V, Rodríguez A, et al. Obesity and prostate cancer: gene expression signature of human periprostatic adipose tissue. BMC Med (2012) 10:108. doi:10.1186/1741-7015-10-108
131. Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Silva C, Rotellar F, et al. Up-regulation of the novel proinflammatory adipokines lipocalin-2, chitinase-3 like-1 and osteopontin as well as angiogenic-related factors in visceral adipose tissue of patients with colon cancer. J Nutr Biochem (2011) 22:634–41. doi:10.1016/j.jnutbio.2010.04.015
132. Allavena P, Sica A, Garlanda C, Mantovani A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol Rev (2008) 222:155–61. doi:10.1111/j.1600-065X.2008.00607.x
133. Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, et al. Divergent pro- and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med (2003) 198:1951–7. doi:10.1084/jem.20030896
134. Smith AM, Rahman FZ, Hayee B, Graham SJ, Marks DJ, Sewell GW, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med (2009) 206:1883–97. doi:10.1084/jem.20091233
135. Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature (2006) 443:998–1002. doi:10.1038/nature05245
136. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol (2011) 11:723–37. doi:10.1038/nri3073
137. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2010) 72:219–46. doi:10.1146/annurev-physiol-021909-135846
138. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454:436–44. doi:10.1038/nature07205
139. Karin M, Cao Y, Greten FR, Li ZW. NF-κB in cancer: from innocent bystander to major culprit. Nat Rev Cancer (2002) 2:301–10. doi:10.1038/nrc780
140. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol (2011) 12:715–23. doi:10.1038/ni.2060
141. Kulbe H, Thompson R, Wilson JL, Robinson S, Hagemann T, Fatah R, et al. The inflammatory cytokine tumor necrosis factor-α generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res (2007) 67:585–92. doi:10.1158/0008-5472.CAN-06-2941
142. Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science (2007) 317:121–4. doi:10.1126/science.1140485
143. Oguma K, Oshima H, Aoki M, Uchio R, Naka K, Nakamura S, et al. Activated macrophages promote Wnt signalling through tumour necrosis factor-α in gastric tumour cells. EMBO J (2008) 27:1671–81. doi:10.1038/emboj.2008.105
144. Schmieder A, Michel J, Schonhaar K, Goerdt S, Schledzewski K. Differentiation and gene expression profile of tumor-associated macrophages. Semin Cancer Biol (2012) 22:289–97. doi:10.1016/j.semcancer.2012.02.002
145. Cintra DE, Pauli JR, Araujo EP, Moraes JC, de Souza CT, Milanski M, et al. Interleukin-10 is a protective factor against diet-induced insulin resistance in liver. J Hepatol (2008) 48:628–37. doi:10.1016/j.jhep.2007.12.017
146. Tateya S, Kim F, Tamori Y. Recent advances in obesity-induced inflammation and insulin resistance. Front Endocrinol (Lausanne) (2013) 4:93. doi:10.3389/fendo.2013.00093
147. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest (2007) 117:175–84. doi:10.1172/JCI29881
148. Nakanishi Y, Nakatsuji M, Seno H, Ishizu S, Akitake-Kawano R, Kanda K, et al. COX-2 inhibition alters the phenotype of tumor-associated macrophages from M2 to M1 in ApcMin/+ mouse polyps. Carcinogenesis (2011) 32:1333–9. doi:10.1093/carcin/bgr128
149. Poulain-Godefroy O, Lecoeur C, Pattou F, Frühbeck G, Froguel P. Inflammation is associated with a decrease of lipogenic factors in omental fat in women. Am J Physiol Regul Integr Comp Physiol (2008) 295:R1–7. doi:10.1152/ajpregu.00926.2007
150. Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C, et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal (2013) 26:192–7. doi:10.1016/j.cellsig.2013.11.004
151. Frühbeck G. The Sir David Cuthbertson Medal Lecture. Hunting for new pieces to the complex puzzle of obesity. Proc Nutr Soc (2006) 65:329–47. doi:10.1079/PNS2006510
152. Frühbeck G, Gómez-Ambrosi J. Rationale for the existence of additional adipostatic hormones. FASEB J (2001) 15:1996–2006. doi:10.1096/fj.00-0829hyp
153. Frühbeck G, Gómez-Ambrosi J, Salvador J. Leptin-induced lipolysis opposes the tonic inhibition of endogenous adenosine in white adipocytes. FASEB J (2001) 15:333–40. doi:10.1096/fj.00-0249com
154. Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Rotellar F, Valenti V, et al. Increased levels of calprotectin in obesity are related to macrophage content: impact on inflammation and effect of weight loss. Mol Med (2011) 17:1157–67. doi:10.2119/molmed.2011.00144
155. Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Rotellar F, Valenti V, et al. Increased circulating and visceral adipose tissue expression levels of YKL-40 in obesity-associated type 2 diabetes are related to inflammation: impact of conventional weight loss and gastric bypass. J Clin Endocrinol Metab (2011) 96:200–9. doi:10.1210/jc.2010-0994
156. Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Rotellar F, Valentí V, et al. Increased tenascin C and toll-like receptor 4 levels in visceral adipose tissue as a link between inflammation and extracellular matrix remodeling in obesity. J Clin Endocrinol Metab (2012) 97:E1880–9. doi:10.1210/jc.2012-1670
157. Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Rotellar F, Valentí V, et al. Increased levels of chemerin and its receptor, chemokine-like receptor-1, in obesity are related to inflammation: tumor necrosis factor-α stimulates mRNA levels of chemerin in visceral adipocytes from obese patients. Surg Obes Relat Dis (2013) 9:306–14. doi:10.1016/j.soard.2011.11.001
158. Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Rotellar F, Valentí V, et al. Six-transmembrane epithelial antigen of prostate 4 and neutrophil gelatinase-associated lipocalin expression in visceral adipose tissue is related to iron status and inflammation in human obesity. Eur J Nutr (2013) 52:1587–95. doi:10.1007/s00394-012-0464-8
159. Frühbeck G. Obesity: aquaporin enters the picture. Nature (2005) 438:436–7. doi:10.1038/438436b
160. Gómez-Ambrosi J, Catalán V, Ramírez B, Rodríguez A, Colina I, Silva C, et al. Plasma osteopontin levels and expression in adipose tissue are increased in obesity. J Clin Endocrinol Metab (2007) 92:3719–27. doi:10.1210/jc.2007-0349
161. Gómez-Ambrosi J, Frühbeck G. Do resistin and resistin-like molecules also link obesity to inflammatory diseases? Ann Intern Med (2001) 135:306–7. doi:10.7326/0003-4819-135-4-200108210-00030
162. Lancha A, Frühbeck G, Gómez-Ambrosi J. Peripheral signalling involved in energy homeostasis control. Nutr Res Rev (2012) 25:223–48. doi:10.1017/S0954422412000145
163. Moreno-Navarrete JM, Catalán V, Whyte L, Diaz-Arteaga A, Vazquez-Martinez R, Rotellar F, et al. The l-α-lysophosphatidylinositol/GPR55 system and its potential role in human obesity. Diabetes (2012) 61:281–91. doi:10.2337/db11-0649
164. Muruzábal FJ, Frühbeck G, Gómez-Ambrosi J, Archanco M, Burrell MA. Immunocytochemical detection of leptin in non-mammalian vertebrate stomach. Gen Comp Endocrinol (2002) 128:149–52. doi:10.1016/S0016-6480(02)00072-2
165. Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem (1995) 270:26746–9. doi:10.1074/jbc.270.45.26746
166. Waki H, Yamauchi T, Kamon J, Ito Y, Uchida S, Kita S, et al. Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J Biol Chem (2003) 278:40352–63. doi:10.1074/jbc.M300365200
167. Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev (2005) 26:439–51. doi:10.1210/er.2005-0005
168. Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest (2006) 116:1784–92. doi:10.1172/JCI29126
169. Pajvani UB, Du X, Combs TP, Berg AH, Rajala MW, Schulthess T, et al. Structure-function studies of the adipocyte-secreted hormone Acrp30/adiponectin. Implications fpr metabolic regulation and bioactivity. J Biol Chem (2003) 278:9073–85. doi:10.1074/jbc.M207198200
170. Kern PA, Di Gregorio GB, Lu T, Rassouli N, Ranganathan G. Adiponectin expression from human adipose tissue: relation to obesity, insulin resistance, and tumor necrosis factor-α expression. Diabetes (2003) 52:1779–85. doi:10.2337/diabetes.52.7.1779
171. Kelesidis I, Kelesidis T, Mantzoros CS. Adiponectin and cancer: a systematic review. Br J Cancer (2006) 94:1221–5. doi:10.1038/sj.bjc.6603051
172. Hillenbrand A, Fassler J, Huber N, Xu P, Henne-Bruns D, Templin M, et al. Changed adipocytokine concentrations in colorectal tumor patients and morbidly obese patients compared to healthy controls. BMC Cancer (2012) 12:545. doi:10.1186/1471-2407-12-545
173. Wei EK, Giovannucci E, Fuchs CS, Willett WC, Mantzoros CS. Low plasma adiponectin levels and risk of colorectal cancer in men: a prospective study. J Natl Cancer Inst (2005) 97:1688–94. doi:10.1093/jnci/dji376
174. Moon HS, Chamberland JP, Aronis K, Tseleni-Balafouta S, Mantzoros CS. Direct role of adiponectin and adiponectin receptors in endometrial cancer: in vitro and ex vivo studies in humans. Mol Cancer Ther (2011) 10:2234–43. doi:10.1158/1535-7163.MCT-11-0545
175. Liu LY, Wang M, Ma ZB, Yu LX, Zhang Q, Gao DZ, et al. The role of adiponectin in breast cancer: a meta-analysis. PLoS One (2013) 8:e73183. doi:10.1371/journal.pone.0073183
176. Perrier S, Jarde T. Adiponectin, an anti-carcinogenic hormone? A systematic review on breast, colorectal, liver and prostate cancer. Curr Med Chem (2012) 19:5501–12. doi:10.2174/092986712803833137
177. Zhang J, Hochwald SN. Plasma adiponectin: a possible link between fat metabolism and pancreatic cancer risk. J Natl Cancer Inst (2013) 105:79–80. doi:10.1093/jnci/djs522
178. Fujisawa T, Endo H, Tomimoto A, Sugiyama M, Takahashi H, Saito S, et al. Adiponectin suppresses colorectal carcinogenesis under the high-fat diet condition. Gut (2008) 57:1531–8. doi:10.1136/gut.2008.159293
179. Dalamaga M, Diakopoulos KN, Mantzoros CS. The role of adiponectin in cancer: a review of current evidence. Endocr Rev (2012) 33:547–94. doi:10.1210/er.2011-1015
180. Clemons N, Phillips W, Lord RV. Signaling pathways in the molecular pathogenesis of adenocarcinomas of the esophagus and gastresophageal junction. Cancer Biol Ther (2013) 14:782–95. doi:10.4161/cbt.25362
181. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372:425–32. doi:10.1038/372425a0
182. Frühbeck G. Intracellular signalling pathways activated by leptin. Biochem J (2006) 393:7–20. doi:10.1042/BJ20051578
183. Frühbeck G. Peripheral actions of leptin and its involvement in disease. Nutr Rev (2002) 60:S47–55. doi:10.1301/002966402320634931
184. Frühbeck G. A heliocentric view of leptin. Proc Nutr Soc (2001) 60:301–18. doi:10.1079/PNS200196
185. Ryan AS, Berman DM, Nicklas BJ, Sinha M, Gingerich RL, Meneilly GS, et al. Plasma adiponectin and leptin levels, body composition, and glucose utilization in adult women with wide ranges of age and obesity. Diabetes Care (2003) 26:2383–8. doi:10.2337/diacare.26.8.2383
186. Cheng SP, Yin PH, Chang YC, Lee CH, Huang SY, Chi CW. Differential roles of leptin in regulating cell migration in thyroid cancer cells. Oncol Rep (2010) 23:1721–7. doi:10.3892/or_00000817
187. Zheng Q, Banaszak L, Fracci S, Basali D, Dunlap SM, Hursting SD, et al. Leptin receptor maintains cancer stem-like properties in triple negative breast cancer cells. Endocr Relat Cancer (2013) 20:797–808. doi:10.1530/ERC-13-0329
188. Howard JM, Pidgeon GP, Reynolds JV. Leptin and gastro-intestinal malignancies. Obes Rev (2010) 11:863–74. doi:10.1111/j.1467-789X.2010.00718.x
189. Uddin S, Bu R, Ahmed M, Hussain AR, Ajarim D, Al-Dayel F, et al. Leptin receptor expression and its association with PI3K/AKT signaling pathway in diffuse large B-cell lymphoma. Leuk Lymphoma (2010) 51:1305–14. doi:10.3109/10428191003802365
190. Simons PJ, van den Pangaart PS, van Roomen CP, Aerts JM, Boon L. Cytokine-mediated modulation of leptin and adiponectin secretion during in vitro adipogenesis: evidence that tumor necrosis factor-α- and interleukin-1β-treated human preadipocytes are potent leptin producers. Cytokine (2005) 32:94–103. doi:10.1016/j.cyto.2005.08.003
191. Cirillo D, Rachiglio AM, la Montagna R, Giordano A, Normanno N. Leptin signaling in breast cancer: an overview. J Cell Biochem (2008) 105:956–64. doi:10.1002/jcb.21911
192. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science (1993) 259:87–91. doi:10.1126/science.7678183
193. Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol (2003) 3:745–56. doi:10.1038/nri1184
194. Sethi G, Sung B, Aggarwal BB. TNF: a master switch for inflammation to cancer. Front Biosci (2008) 13:5094–107. doi:10.2741/3066
195. Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer (2009) 9:361–71. doi:10.1038/nrc2628
196. Balkwill F. TNF-α in promotion and progression of cancer. Cancer Metastasis Rev (2006) 25:409–16. doi:10.1007/s10555-006-9005-3
197. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature (2004) 431:461–6. doi:10.1038/nature02924
198. Naylor MS, Stamp GW, Foulkes WD, Eccles D, Balkwill FR. Tumor necrosis factor and its receptors in human ovarian cancer. Potential role in disease progression. J Clin Invest (1993) 91:2194–206. doi:10.1172/JCI116446
199. Ferrajoli A, Keating MJ, Manshouri T, Giles FJ, Dey A, Estrov Z, et al. The clinical significance of tumor necrosis factor-α plasma level in patients having chronic lymphocytic leukemia. Blood (2002) 100:1215–9.
200. Sethi G, Shanmugam MK, Ramachandran L, Kumar AP, Tergaonkar V. Multifaceted link between cancer and inflammation. Biosci Rep (2012) 32:1–15. doi:10.1042/BSR20100136
201. Hong DS, Angelo LS, Kurzrock R. Interleukin-6 and its receptor in cancer: implications for translational therapeutics. Cancer (2007) 110:1911–28. doi:10.1002/cncr.22999
202. Naugler WE, Karin M. The wolf in sheep’s clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med (2008) 14:109–19. doi:10.1016/j.molmed.2007.12.007
203. Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest (2007) 117:1175–83. doi:10.1172/JCI31537
204. Angelo LS, Talpaz M, Kurzrock R. Autocrine interleukin-6 production in renal cell carcinoma: evidence for the involvement of p53. Cancer Res (2002) 62:932–40.
205. Kai H, Kitadai Y, Kodama M, Cho S, Kuroda T, Ito M, et al. Involvement of proinflammatory cytokines IL-1β and IL-6 in progression of human gastric carcinoma. Anticancer Res (2005) 25:709–13.
206. Chung YC, Chang YF. Serum interleukin-6 levels reflect the disease status of colorectal cancer. J Surg Oncol (2003) 83:222–6. doi:10.1002/jso.10269
207. Klein B, Bataille R. Cytokine network in human multiple myeloma. Hematol Oncol Clin North Am (1992) 6:273–84.
208. Ghosh S, Ashcraft K. An IL-6 link between obesity and cancer. Front Biosci (2013) 5:461–78. doi:10.2741/E628
209. Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol (2014) 15:423–30. doi:10.1038/ni.2865
210. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta (2011) 1813:878–88. doi:10.1016/j.bbamcr.2011.01.034
211. Choi J, Joseph L, Pilote L. Obesity and C-reactive protein in various populations: a systematic review and meta-analysis. Obes Rev (2013) 14:232–44. doi:10.1111/obr.12003
212. Heikkila K, Ebrahim S, Lawlor DA. A systematic review of the association between circulating concentrations of C reactive protein and cancer. J Epidemiol Community Health (2007) 61:824–33. doi:10.1136/jech.2006.051292
213. Helzlsouer KJ, Erlinger TP, Platz EA. C-reactive protein levels and subsequent cancer outcomes: results from a prospective cohort study. Eur J Cancer (2006) 42:704–7. doi:10.1016/j.ejca.2006.01.008
214. Trichopoulos D, Psaltopoulou T, Orfanos P, Trichopoulou A, Boffetta P. Plasma C-reactive protein and risk of cancer: a prospective study from Greece. Cancer Epidemiol Biomarkers Prev (2006) 15:381–4. doi:10.1158/1055-9965.EPI-05-0626
215. Baba H, Kuwabara K, Ishiguro T, Hatano S, Matsuzawa T, Fukuchi M, et al. C-reactive protein as a significant prognostic factor for stage IV gastric cancer patients. Anticancer Res (2013) 33:5591–5.
216. Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature (2001) 409:307–12. doi:10.1038/35053000
217. Dupont J, Reverchon M, Cloix L, Froment P, Rame C. Involvement of adipokines, AMPK, PI3K and the PPAR signaling pathways in ovarian follicle development and cancer. Int J Dev Biol (2012) 56:959–67. doi:10.1387/ijdb.120134jd
218. Filkova M, Haluzik M, Gay S, Senolt L. The role of resistin as a regulator of inflammation: implications for various human pathologies. Clin Immunol (2009) 133:157–70. doi:10.1016/j.clim.2009.07.013
219. Hsu WY, Chao YW, Tsai YL, Lien CC, Chang CF, Deng MC, et al. Resistin induces monocyte-endothelial cell adhesion by increasing ICAM-1 and VCAM-1 expression in endothelial cells via p38MAPK-dependent pathway. J Cell Physiol (2011) 226:2181–8. doi:10.1002/jcp.22555
220. Nakajima TE, Yamada Y, Hamano T, Furuta K, Matsuda T, Fujita S, et al. Adipocytokines as new promising markers of colorectal tumors: adiponectin for colorectal adenoma, and resistin and visfatin for colorectal cancer. Cancer Sci (2010) 101:1286–91. doi:10.1111/j.1349-7006.2010.01518.x
221. Jiang CY, Wang W, Yin YL, Yuan ZR, Wang LB. Expression of the adipocytokine resistin and its association with the clinicopathological features and prognosis of pancreatic ductal adenocarcinoma. Oncol Lett (2012) 4:960–4. doi:10.3892/ol.2012.865
222. Ho GY, Wang T, Gunter MJ, Strickler HD, Cushman M, Kaplan RC, et al. Adipokines linking obesity with colorectal cancer risk in postmenopausal women. Cancer Res (2012) 72:3029–37. doi:10.1158/0008-5472.CAN-11-2771
223. Di Simone N, Di Nicuolo F, Sanguinetti M, Castellani R, D’Asta M, Caforio L, et al. Resistin regulates human choriocarcinoma cell invasive behaviour and endothelial cell angiogenic processes. J Endocrinol (2006) 189:691–9. doi:10.1677/joe.1.06610
224. Catalán V, Gómez-Ambrosi J, Ramírez B, Rotellar F, Pastor C, Silva C, et al. Proinflammatory cytokines in obesity: impact of type 2 diabetes mellitus and gastric bypass. Obes Surg (2007) 17:1464–74. doi:10.1007/s11695-008-9424-z
225. Siveen KS, Kuttan G. Role of macrophages in tumour progression. Immunol Lett (2009) 123:97–102. doi:10.1016/j.imlet.2009.02.011
226. Hong S, Zhang P, Zhang H, Jia L, Qu X, Yang Q, et al. Enforced effect of tk-MCP-1 fusion gene in ovarian cancer. J Exp Clin Cancer Res (2012) 31:74. doi:10.1186/1756-9966-31-74
227. Matte I, Lane D, Laplante C, Rancourt C, Piche A. Profiling of cytokines in human epithelial ovarian cancer ascites. Am J Cancer Res (2012) 2:566–80.
228. Baier PK, Eggstein S, Wolff-Vorbeck G, Baumgartner U, Hopt UT. Chemokines in human colorectal carcinoma. Anticancer Res (2005) 25:3581–4.
229. Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, et al. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science (2005) 307:426–30. doi:10.1126/science.1097243
230. Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Silva C, Rotellar F, et al. Association of increased visfatin/PBEF/NAMPT circulating concentrations and gene expression levels in peripheral blood cells with lipid metabolism and fatty liver in human morbid obesity. Nutr Metab Cardiovasc Dis (2011) 21:245–53. doi:10.1016/j.numecd.2009.09.008
231. Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, et al. Retraction. Science (2007) 318:565. doi:10.1126/science.318.5850.565b
232. Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, et al. Nampt/PBEF/visfatin regulates insulin secretion in β cells as a systemic NAD biosynthetic enzyme. Cell Metab (2007) 6:363–75. doi:10.1016/j.cmet.2007.09.003
233. Revollo JR, Grimm AA, Imai S. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol (2007) 23:164–70. doi:10.1097/MOG.0b013e32801b3c8f
234. Bi TQ, Che XM. Nampt/PBEF/visfatin and cancer. Cancer Biol Ther (2010) 10:119–25. doi:10.4161/cbt.10.2.12581
235. Hufton SE, Moerkerk PT, Brandwijk R, de Bruine AP, Arends JW, Hoogenboom HR. A profile of differentially expressed genes in primary colorectal cancer using suppression subtractive hybridization. FEBS Lett (1999) 463:77–82. doi:10.1016/S0014-5793(99)01578-1
236. Van Beijnum JR, Moerkerk PT, Gerbers AJ, De Bruine AP, Arends JW, Hoogenboom HR, et al. Target validation for genomics using peptide-specific phage antibodies: a study of five gene products overexpressed in colorectal cancer. Int J Cancer (2002) 101:118–27. doi:10.1002/ijc.10584
237. Folgueira MA, Carraro DM, Brentani H, Patrao DF, Barbosa EM, Netto MM, et al. Gene expression profile associated with response to doxorubicin-based therapy in breast cancer. Clin Cancer Res (2005) 11:7434–43. doi:10.1158/1078-0432.CCR-04-0548
238. Gómez-Ambrosi J, Rodríguez A, Catalán V, Frühbeck G. The bone-adipose axis in obesity and weight loss. Obes Surg (2008) 18:1134–43. doi:10.1007/s11695-008-9548-1
239. Nomiyama T, Perez-Tilve D, Ogawa D, Gizard F, Zhao Y, Heywood EB, et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Invest (2007) 117:2877–88. doi:10.1172/JCI31986
240. Rangaswami H, Bulbule A, Kundu GC. Osteopontin: role in cell signaling and cancer progression. Trends Cell Biol (2006) 16:79–87. doi:10.1016/j.tcb.2005.12.005
241. Anborgh PH, Mutrie JC, Tuck AB, Chambers AF. Role of the metastasis-promoting protein osteopontin in the tumour microenvironment. J Cell Mol Med (2010) 14:2037–44. doi:10.1111/j.1582-4934.2010.01115.x
242. Cao DX, Li ZJ, Jiang XO, Lum YL, Khin E, Lee NP, et al. Osteopontin as potential biomarker and therapeutic target in gastric and liver cancers. World J Gastroenterol (2012) 18:3923–30. doi:10.3748/wjg.v18.i30.3923
243. El-Mesallamy HO, Mostafa AM, Amin AI, El Demerdash E. The interplay of YKL-40 and leptin in type 2 diabetic obese patients. Diabetes Res Clin Pract (2011) 93:e113–6. doi:10.1016/j.diabres.2011.05.040
244. Rathcke CN, Vestergaard H. YKL-40, a new inflammatory marker with relation to insulin resistance and with a role in endothelial dysfunction and atherosclerosis. Inflamm Res (2006) 55:221–7. doi:10.1007/s00011-006-0076-y
245. Johansen JS. Studies on serum YKL-40 as a biomarker in diseases with inflammation, tissue remodelling, fibroses and cancer. Dan Med Bull (2006) 53:172–209.
246. Nigro JM, Misra A, Zhang L, Smirnov I, Colman H, Griffin C, et al. Integrated array-comparative genomic hybridization and expression array profiles identify clinically relevant molecular subtypes of glioblastoma. Cancer Res (2005) 65:1678–86. doi:10.1158/0008-5472.CAN-04-2921
247. Huang Y, Prasad M, Lemon WJ, Hampel H, Wright FA, Kornacker K, et al. Gene expression in papillary thyroid carcinoma reveals highly consistent profiles. Proc Natl Acad Sci U S A (2001) 98:15044–9. doi:10.1073/pnas.251547398
248. Sjogren H, Meis-Kindblom JM, Orndal C, Bergh P, Ptaszynski K, Aman P, et al. Studies on the molecular pathogenesis of extraskeletal myxoid chondrosarcoma-cytogenetic, molecular genetic, and cDNA microarray analyses. Am J Pathol (2003) 162:781–92. doi:10.1016/S0002-9440(10)63875-8
249. Johansen JS, Jensen BV, Roslind A, Nielsen D, Price PA. Serum YKL-40, a new prognostic biomarker in cancer patients? Cancer Epidemiol Biomarkers Prev (2006) 15:194–202. doi:10.1158/1055-9965.EPI-05-0011
250. Finkel T. Signal transduction by reactive oxygen species. J Cell Biol (2011) 194:7–15. doi:10.1083/jcb.201102095
251. Parola M, Bellomo G, Robino G, Barrera G, Dianzani MU. 4-Hydroxynonenal as a biological signal: molecular basis and pathophysiological implications. Antioxid Redox Signal (1999) 1:255–84. doi:10.1089/ars.1999.1.3-255
252. Wang J, Yi J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol Ther (2008) 7:1875–84. doi:10.4161/cbt.7.12.7067
253. Son Y, Kim S, Chung HT, Pae HO. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol (2013) 528:27–48. doi:10.1016/B978-0-12-405881-1.00002-1
254. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest (2004) 114:1752–61. doi:10.1172/JCI21625
255. Matsuda M, Shimomura I. Increased oxidative stress in obesity: implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes Res Clin Pract (2013) 7:e330–41. doi:10.1016/j.orcp.2013.05.004
256. Keaney JF Jr, Larson MG, Vasan RS, Wilson PW, Lipinska I, Corey D, et al. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol (2003) 23:434–9. doi:10.1161/01.ATV.0000058402.34138.11
257. Okuno Y, Matsuda M, Kobayashi H, Morita K, Suzuki E, Fukuhara A, et al. Adipose expression of catalase is regulated via a novel remote PPARγ-responsive region. Biochem Biophys Res Commun (2008) 366:698–704. doi:10.1016/j.bbrc.2007.12.001
258. Kobayashi H, Matsuda M, Fukuhara A, Komuro R, Shimomura I. Dysregulated glutathione metabolism links to impaired insulin action in adipocytes. Am J Physiol Endocrinol Metab (2009) 296:E1326–34. doi:10.1152/ajpendo.90921.2008
259. Ozben T. Oxidative stress and apoptosis: impact on cancer therapy. J Pharm Sci (2007) 96:2181–96. doi:10.1002/jps.20874
260. Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat (2004) 7:97–110. doi:10.1016/j.drup.2004.01.004
261. Halliwell B, Chirico S. Lipid peroxidation: its mechanism, measurement, and significance. Am J Clin Nutr (1993) 57:715S–24S.
262. Lukanova A, Lundin E, Zeleniuch-Jacquotte A, Muti P, Mure A, Rinaldi S, et al. Body mass index, circulating levels of sex-steroid hormones, IGF-I and IGF-binding protein-3: a cross-sectional study in healthy women. Eur J Endocrinol (2004) 150:161–71. doi:10.1530/eje.0.1500161
263. Warner M, Gustafsson JA. On estrogen, cholesterol metabolism, and breast cancer. N Engl J Med (2014) 370:572–3. doi:10.1056/NEJMcibr1315176
264. Cleary MP, Grossmann ME. Minireview: obesity and breast cancer: the estrogen connection. Endocrinology (2009) 150:2537–42. doi:10.1210/en.2009-0070
265. Leslie KK, Thiel KW, Reyes HD, Yang S, Zhang Y, Carlson MJ, et al. The estrogen receptor joins other cancer biomarkers as a predictor of outcome. Obstet Gynecol Int (2013) 2013:479541. doi:10.1155/2013/479541
266. Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet (2006) 38:1289–97. doi:10.1038/ng1901
267. Charpentier AH, Bednarek AK, Daniel RL, Hawkins KA, Laflin KJ, Gaddis S, et al. Effects of estrogen on global gene expression: identification of novel targets of estrogen action. Cancer Res (2000) 60:5977–83.
268. Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology (2003) 144:4562–74. doi:10.1210/en.2003-0567
269. Stocco C. Tissue physiology and pathology of aromatase. Steroids (2012) 77:27–35. doi:10.1016/j.steroids.2011.10.013
270. Key T, Appleby P, Barnes I, Reeves G. Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J Natl Cancer Inst (2002) 94:606–16.
271. Thomas HV, Reeves GK, Key TJ. Endogenous estrogen and postmenopausal breast cancer: a quantitative review. Cancer Causes Control (1997) 8:922–8. doi:10.1023/A:1018476631561
272. Carlson MJ, Thiel KW, Yang S, Leslie KK. Catch it before it kills: progesterone, obesity, and the prevention of endometrial cancer. Discov Med (2012) 14:215–22.
273. Kahn CR, White MF. The insulin receptor and the molecular mechanism of insulin action. J Clin Invest (1988) 82:1151–6. doi:10.1172/JCI113711
274. Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res (2007) 100:328–41. doi:10.1161/01.RES.0000256090.42690.05
275. Xu XJ, Pories WJ, Dohm LG, Ruderman NB. What distinguishes adipose tissue of severely obese humans who are insulin sensitive and resistant? Curr Opin Lipidol (2013) 24:49–56. doi:10.1097/MOL.0b013e32835b465b
276. Ma J, Li H, Giovannucci E, Mucci L, Qiu W, Nguyen PL, et al. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: a long-term survival analysis. Lancet Oncol (2008) 9:1039–47. doi:10.1016/S1470-2045(08)70235-3
277. Liu Z, Yan R, Al-Salman A, Shen Y, Bu Y, Ma J, et al. Epidermal growth factor induces tumour marker AKR1B10 expression through activator protein-1 signalling in hepatocellular carcinoma cells. Biochem J (2012) 442:273–82. doi:10.1042/BJ20111322
278. Wang Y, Hua S, Tian W, Zhang L, Zhao J, Zhang H, et al. Mitogenic and anti-apoptotic effects of insulin in endometrial cancer are phosphatidylinositol 3-kinase/Akt dependent. Gynecol Oncol (2012) 125:734–41. doi:10.1016/j.ygyno.2012.03.012
279. Djiogue S, Nwabo Kamdje AH, Vecchio L, Kipanyula MJ, Farahna M, Aldebasi Y, et al. Insulin resistance and cancer: the role of insulin and IGFs. Endocr Relat Cancer (2013) 20:R1–17. doi:10.1530/ERC-12-0324
280. van Kruijsdijk RC, van der Wall E, Visseren FL. Obesity and cancer: the role of dysfunctional adipose tissue. Cancer Epidemiol Biomarkers Prev (2009) 18:2569–78. doi:10.1158/1055-9965.EPI-09-0372
281. Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev (1995) 16:3–34. doi:10.1210/edrv-16-1-3
282. Chan JM, Stampfer MJ, Giovannucci E, Gann PH, Ma J, Wilkinson P, et al. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science (1998) 279:563–6. doi:10.1126/science.279.5350.563
283. Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, et al. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet (1998) 351:1393–6. doi:10.1016/S0140-6736(97)10384-1
284. Ma J, Pollak MN, Giovannucci E, Chan JM, Tao Y, Hennekens CH, et al. Prospective study of colorectal cancer risk in men and plasma levels of insulin-like growth factor (IGF)-I and IGF-binding protein-3. J Natl Cancer Inst (1999) 91:620–5. doi:10.1093/jnci/91.23.2052
285. Yu H, Spitz MR, Mistry J, Gu J, Hong WK, Wu X. Plasma levels of insulin-like growth factor-I and lung cancer risk: a case-control analysis. J Natl Cancer Inst (1999) 91:151–6. doi:10.1093/jnci/91.2.151
286. Yu H, Rohan T. Role of the insulin-like growth factor family in cancer development and progression. J Natl Cancer Inst (2000) 92:1472–89. doi:10.1093/jnci/92.18.1472
287. Giovannucci E. Insulin, insulin-like growth factors and colon cancer: a review of the evidence. J Nutr (2001) 131:3109S–20S.
288. Renehan AG, Zwahlen M, Minder C, O’Dwyer ST, Shalet SM, Egger M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: systematic review and meta-regression analysis. Lancet (2004) 363:1346–53. doi:10.1016/S0140-6736(04)16044-3
289. Trayhurn P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol Rev (2013) 93:1–21. doi:10.1152/physrev.00017.2012
290. Gilbert CA, Slingerland JM. Cytokines, obesity, and cancer: new insights on mechanisms linking obesity to cancer risk and progression. Annu Rev Med (2013) 64:45–57. doi:10.1146/annurev-med-121211-091527
291. Yan W, Chang Y, Liang X, Cardinal JS, Huang H, Thorne SH, et al. High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology (2012) 55:1863–75. doi:10.1002/hep.25572
292. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer (2008) 8:618–31. doi:10.1038/nrc2444
293. Weigert A, Brune B. Nitric oxide, apoptosis and macrophage polarization during tumor progression. Nitric Oxide (2008) 19:95–102. doi:10.1016/j.niox.2008.04.021
294. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature (2008) 453:807–11. doi:10.1038/nature06905
295. Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood (2004) 104:2224–34. doi:10.1182/blood-2004-03-1109
296. Yamada S, Utsunomiya T, Morine Y, Imura S, Ikemoto T, Arakawa Y, et al. Expressions of hypoxia-inducible factor-1 and epithelial cell adhesion molecule are linked with aggressive local recurrence of hepatocellular carcinoma after radiofrequency ablation therapy. Ann Surg Oncol (2014). doi:10.1245/s10434-014-3575-z
297. Tsai YP, Wu KJ. Hypoxia-regulated target genes implicated in tumor metastasis. J Biomed Sci (2012) 19:102. doi:10.1186/1423-0127-19-102
298. Semenza GL. Molecular mechanisms mediating metastasis of hypoxic breast cancer cells. Trends Mol Med (2012) 18:534–43. doi:10.1016/j.molmed.2012.08.001
299. Zhao Y, Liu H, Riker AI, Fodstad O, Ledoux SP, Wilson GL, et al. Emerging metabolic targets in cancer therapy. Front Biosci (2011) 16:1844–60. doi:10.2741/3826
300. Kizaka-Kondoh S, Kuchimaru T, Kadonosono T. Pathophysiological response to hypoxia – from the molecular mechanisms of malady to drug discovery: hypoxia-inducible factor-1 (HIF-1)-active cells as a target for cancer therapy. J Pharmacol Sci (2011) 115:440–5. doi:10.1254/jphs.10R20FM
301. Nagasawa H. Pathophysiological response to hypoxia – from the molecular mechanisms of malady to drug discovery: drug discovery for targeting the tumor microenvironment. J Pharmacol Sci (2011) 115:446–52. doi:10.1254/jphs.10R25FM
302. Thomson CA, McCullough ML, Wertheim BC, Chlebowski RT, Martinez ME, Stefanick ML, et al. Nutrition and physical activity cancer prevention guidelines, cancer risk, and mortality in the women’s health initiative. Cancer Prev Res (2014) 7:42–53. doi:10.1158/1940-6207.CAPR-13-0258
303. Caponi PW, Lehnen AM, Pinto GH, Borges J, Markoski M, Machado UF, et al. Aerobic exercise training induces metabolic benefits in rats with metabolic syndrome independent of dietary changes. Clinics (2013) 68:1010–7. doi:10.6061/clinics/2013(07)20
304. Jakicic JM, Otto AD, Lang W, Semler L, Winters C, Polzien K, et al. The effect of physical activity on 18-month weight change in overweight adults. Obesity (2011) 19:100–9. doi:10.1038/oby.2010.122
305. Kraus WE, Houmard JA, Duscha BD, Knetzger KJ, Wharton MB, McCartney JS, et al. Effects of the amount and intensity of exercise on plasma lipoproteins. N Engl J Med (2002) 347:1483–92. doi:10.1056/NEJMoa020194
306. Halle M, Berg A, Konig D, Keul J, Baumstark MW. Differences in the concentration and composition of low-density lipoprotein subfraction particles between sedentary and trained hypercholesterolemic men. Metabolism (1997) 46:186–91. doi:10.1016/S0026-0495(97)90300-0
307. Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med (2002) 346:393–403. doi:10.1056/NEJMoa012512
308. Gibney J, Healy ML, Sonksen PH. The growth hormone/insulin-like growth factor-I axis in exercise and sport. Endocr Rev (2007) 28:603–24. doi:10.1210/er.2006-0052
309. Katzmarzyk PT, Janssen I, Ardern CI. Physical inactivity, excess adiposity and premature mortality. Obes Rev (2003) 4:257–90. doi:10.1046/j.1467-789X.2003.00120.x
310. Hu FB, Willett WC, Li T, Stampfer MJ, Colditz GA, Manson JE. Adiposity as compared with physical activity in predicting mortality among women. N Engl J Med (2004) 351:2694–703. doi:10.1056/NEJMoa042135
311. Bernstein L, Henderson BE, Hanisch R, Sullivan-Halley J, Ross RK. Physical exercise and reduced risk of breast cancer in young women. J Natl Cancer Inst (1994) 86:1403–8. doi:10.1093/jnci/86.18.1403
312. Chlebowski RT. Nutrition and physical activity influence on breast cancer incidence and outcome. Breast (2013) 22(Suppl 2):S30–7. doi:10.1016/j.breast.2013.07.006
313. Boyle T, Fritschi L, Platell C, Heyworth J. Lifestyle factors associated with survival after colorectal cancer diagnosis. Br J Cancer (2013) 109:814–22. doi:10.1038/bjc.2013.310
314. Song JH, Kim YS, Yang SY, Chung SJ, Park MJ, Lim SH, et al. Physical activity and other lifestyle factors in relation to the prevalence of colorectal adenoma: a colonoscopy-based study in asymptomatic Koreans. Cancer Causes Control (2013) 24:1717–26. doi:10.1007/s10552-013-0247-4
315. Rebillard A, Lefeuvre-Orfila L, Gueritat J, Cillard J. Prostate cancer and physical activity: adaptive response to oxidative stress. Free Radic Biol Med (2013) 60:115–24. doi:10.1016/j.freeradbiomed.2013.02.009
316. McMillan DC, Sattar N, McArdle CS. ABC of obesity. Obesity and cancer. BMJ (2006) 333:1109–11. doi:10.1136/bmj.39042.565035.BE1
317. Jing K, Wu T, Lim K. ω-3 polyunsaturated fatty acids and cancer. Anticancer Agents Med Chem (2013) 13:1162–77. doi:10.2174/18715206113139990319
318. Laviano A, Rianda S, Molfino A, Rossi Fanelli F. ω-3 fatty acids in cancer. Curr Opin Clin Nutr Metab Care (2013) 16:156–61. doi:10.1097/MCO.0b013e32835d2d99
319. Cockbain AJ, Volpato M, Race AD, Munarini A, Fazio C, Belluzzi A, et al. Anticolorectal cancer activity of the ω-3 polyunsaturated fatty acid eicosapentaenoic acid. Gut (2014). doi:10.1136/gutjnl-2013-306445
320. Gu Z, Suburu J, Chen H, Chen YQ. Mechanisms of ω-3 polyunsaturated fatty acids in prostate cancer prevention. Biomed Res Int (2013) 2013:824563. doi:10.1155/2013/824563
321. Chen Z, Odstrcil EA, Tu BP, McKnight SL. Restriction of DNA replication to the reductive phase of the metabolic cycle protects genome integrity. Science (2007) 316:1916–9. doi:10.1126/science.1140958
322. Silverman SJ, Petti AA, Slavov N, Parsons L, Briehof R, Thiberge SY, et al. Metabolic cycling in single yeast cells from unsynchronized steady-state populations limited on glucose or phosphate. Proc Natl Acad Sci U S A (2010) 107:6946–51. doi:10.1073/pnas.1002422107
323. Dang CV. Links between metabolism and cancer. Genes Dev (2012) 26:877–90. doi:10.1101/gad.189365.112
324. Friberg E, Wallin A, Wolk A. Sucrose, high-sugar foods, and risk of endometrial cancer – a population-based cohort study. Cancer Epidemiol Biomarkers Prev (2011) 20:1831–7. doi:10.1158/1055-9965.EPI-11-0402
325. Cust AE, Slimani N, Kaaks R, van Bakel M, Biessy C, Ferrari P, et al. Dietary carbohydrates, glycemic index, glycemic load, and endometrial cancer risk within the European prospective investigation into cancer and nutrition cohort. Am J Epidemiol (2007) 166:912–23. doi:10.1093/aje/kwm161
326. Tasevska N, Jiao L, Cross AJ, Kipnis V, Subar AF, Hollenbeck A, et al. Sugars in diet and risk of cancer in the NIH-AARP diet and health study. Int J Cancer (2012) 130:159–69. doi:10.1002/ijc.25990
327. Longo VD, Fontana L. Calorie restriction and cancer prevention: metabolic and molecular mechanisms. Trends Pharmacol Sci (2010) 31:89–98. doi:10.1016/j.tips.2009.11.004
328. Brethauer SA, Aminian A, Romero-Talamas H, Batayyah E, Mackey J, Kennedy L, et al. Can diabetes be surgically cured? Long-term metabolic effects of bariatric surgery in obese patients with type 2 diabetes mellitus. Ann Surg (2013) 258:628–37. doi:10.1097/SLA.0b013e3182a5034b
329. Guo X, Liu X, Wang M, Wei F, Zhang Y. The effects of bariatric procedures versus medical therapy for obese patients with type 2 diabetes: meta-analysis of randomized controlled trials. Biomed Res Int (2013) 2013:410609. doi:10.1155/2013/410609
330. Nestvold TK, Nielsen EW, Lappegard KT. Bariatric surgery reduces risk factors for development of type 2 diabetes mellitus in morbidly obese, nondiabetic patients. Metab Syndr Relat Disord (2013) 11:441–6. doi:10.1089/met.2013.0085
331. Greenburg DL, Lettieri CJ, Eliasson AH. Effects of surgical weight loss on measures of obstructive sleep apnea: a meta-analysis. Am J Med (2009) 122:535–42. doi:10.1016/j.amjmed.2008.10.037
332. Laimer MW, Engl J, Tschoner A, Kaser S, Ritsch A, Tatarczyk T, et al. Effects of weight loss on lipid transfer proteins in morbidly obese women. Lipids (2009) 44:1125–30. doi:10.1007/s11745-009-3349-8
333. Moretto M, Kupski C, da Silva VD, Padoin AV, Mottin CC. Effect of bariatric surgery on liver fibrosis. Obes Surg (2012) 22:1044–9. doi:10.1007/s11695-011-0559-y
334. Athyros VG, Tziomalos K, Karagiannis A, Mikhailidis DP. Cardiovascular benefits of bariatric surgery in morbidly obese patients. Obes Rev (2011) 12:515–24. doi:10.1111/j.1467-789X.2010.00831.x
335. Ikramuddin S, Livingston EH. New insights on bariatric surgery outcomes. JAMA (2013) 310:2401–2. doi:10.1001/jama.2013.280927
336. Christou NV, Lieberman M, Sampalis F, Sampalis JS. Bariatric surgery reduces cancer risk in morbidly obese patients. Surg Obes Relat Dis (2008) 4:691–5. doi:10.1016/j.soard.2008.08.025
337. Adams TD, Stroup AM, Gress RE, Adams KF, Calle EE, Smith SC, et al. Cancer incidence and mortality after gastric bypass surgery. Obesity (2009) 17:796–802. doi:10.1038/oby.2008.610
338. Ashrafian H, Ahmed K, Rowland SP, Patel VM, Gooderham NJ, Holmes E, et al. Metabolic surgery and cancer: protective effects of bariatric procedures. Cancer (2011) 117:1788–99. doi:10.1002/cncr.25738
339. Sjöström L, Gummesson A, Sjöström CD, Narbro K, Peltonen M, Wedel H, et al. Effects of bariatric surgery on cancer incidence in obese patients in Sweden (Swedish Obese Subjects Study): a prospective, controlled intervention trial. Lancet Oncol (2009) 10:653–62. doi:10.1016/S1470-2045(09)70159-7
Keywords: adipose tissue, carcinogenesis, obesity, inflammation, adipokine, hypoxia, insulin resistance
Citation: Pérez-Hernández AI, Catalán V, Gómez-Ambrosi J, Rodríguez A and Frühbeck G (2014) Mechanisms linking excess adiposity and carcinogenesis promotion. Front. Endocrinol. 5:65. doi: 10.3389/fendo.2014.00065
Received: 04 March 2014; Paper pending published: 22 March 2014;
Accepted: 15 April 2014; Published online: 01 May 2014.
Edited by:
Nada Y. Kalaany, Harvard Medical School, USAReviewed by:
Marialuisa Appetecchia, Regina Elena National Cancer Institute IRCCS, ItalyBernhard Gregor Baumgartner, Paracelsus Medical School, Austria
Copyright: © 2014 Pérez-Hernández, Catalán, Gómez-Ambrosi, Rodríguez and Frühbeck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gema Frühbeck, Department of Endocrinology and Nutrition, Clínica Universidad de Navarra, Avda. Pío XII 36, Pamplona 31008, Spain e-mail: gfrühbeck@unav.es