 Daniel Edsgärd1
Daniel Edsgärd1 Marlene D. Dalgaard2
Marlene D. Dalgaard2 Nils Weinhold1
Nils Weinhold1 Agata Wesolowska-Andersen1 Ewa Rajpert-De Meyts2* Anne Marie Ottesen2 Anders Juul2 Niels E. Skakkebæk2 Thomas Skøt Jensen1
Agata Wesolowska-Andersen1 Ewa Rajpert-De Meyts2* Anne Marie Ottesen2 Anders Juul2 Niels E. Skakkebæk2 Thomas Skøt Jensen1 Ramneek Gupta1 Henrik Leffers2
Ramneek Gupta1 Henrik Leffers2 Søren Brunak1*
Søren Brunak1*- 1Department of Systems Biology, Technical University of Denmark, Lyngby, Denmark
- 2Department of Growth and Reproduction, Rigshospitalet, Copenhagen, Denmark
Testicular germ cell cancer (TGCC) is one of the most heritable forms of cancer. Previous genome-wide association studies have focused on single nucleotide polymorphisms, largely ignoring the influence of copy number variants (CNVs). Here we present a genome-wide study of CNV on a cohort of 212 cases and 437 controls from Denmark, which was genotyped at ∼1.8 million markers, half of which were non-polymorphic copy number markers. No association of common variants were found, whereas analysis of rare variants (present in less than 1% of the samples) initially indicated a single gene with significantly higher accumulation of rare CNVs in cases as compared to controls, at the gene PTPN1 (P = 3.8 × 10−2, 0.9% of cases and 0% of controls). However, the CNV could not be verified by qPCR in the affected samples. Further, the CNV calling of the array-data was validated by sequencing of the GSTM1 gene, which showed that the CNV frequency was in complete agreement between the two platforms. This study therefore disconfirms the hypothesis that there exists a single CNV locus with a major effect size that predisposes to TGCC. Genome-wide pathway association analysis indicated a weak association of rare CNVs related to cell migration (false-discovery rate = 0.021, 1.8% of cases and 1.1% of controls). Dysregulation during migration of primordial germ cells has previously been suspected to be a part of TGCC development and this set of multiple rare variants may thereby have a minor contribution to an increased susceptibility of TGCCs.
Introduction
Testicular germ cell cancer (TGCC) is the most common malignancy in young men aged 15−45 years. The incidence has increased over the last decades and is highest in the Nordic countries with 8–9 cases per 100,000, whereas the incidence in men of African and Asian ancestry is five-fold lower (Chia et al., 2010). Environmental exposure partly explains the increasing incidence and ancestral disparity (Skakkebaek et al., 2001), but there is also evidence of a substantial genetic contribution to TGCC susceptibility. The familial aggregation of TGCC is one of the highest among cancers. Brothers and sons of TGCC patients have an 8–10 times and 4–6 times higher risk to develop the disease, while the risk increases 75- and 35-fold for monozygotic and dizygotic twins, respectively (Swerdlow et al., 1997; Hemminki and Li, 2004). Despite the relatively high degree of heritability, genome-wide familial linkage analyses have not identified any loci predisposing for TGCC, and candidate studies have only found one rare deletion (2–3%) at the Y chromosome that confers a modest 2–3 fold increased risk (Nathanson et al., 2005). Recently, genome-wide association studies that search for common single nucleotide variants associated to TGCC have identified susceptibility loci at the genes KITLG, SPRY4, BAK1, DMRT1, TERT, and ATF7IP (Kanetsky et al., 2009; Rapley et al., 2009; Turnbull et al., 2010; Dalgaard et al., 2012). The strongest association was found at KITLG with a greater than 2.5-fold increased risk of disease. Consistent with the relatively high familial relative risk, this is the largest effect size found for any single loci among cancers. However, a considerable portion of the heritability remains to be explained.
Here we investigate constitutional DNA copy number variations (CNVs) as another source of genetic variability that may contribute to the development of TGCC. Recent studies have described associations of common CNVs with neuroblastomas (Diskin et al., 2009), systemic autoimmunity (Fanciulli et al., 2007), psoriasis (Hollox et al., 2007), and osteoporosis (Yang et al., 2008). Rare variants, typically originating from recent and de novo events, constitute a significant portion of genomic variation. The thousand genomes project indicates that there are about 20,000 CNVs with allele frequencies down to 1% (1000 Genomes Project Consortium, 2010; Mills et al., 2011). The contribution of such rare, or even rarer, variants, to complex disease susceptibility is to a large extent unknown, but they seem to play an important role in psychiatric disorders (International Schizophrenia Consortium, 2008; Pinto et al., 2010) and they have been indicated to influence childhood obesity (Glessner et al., 2010). Further, identification of de novo mutations is possible in studies of family-trios and recently three de novo CNVs were found in 3 out of 43 TGCC trios, a frequency higher than found in two other cancer types (Stadler et al., 2012), highlighting the paradigm of rare genetic events influencing susceptibility to TGCC. To date, case-control association studies of individual rare CNVs have insufficient power to identify disease-causing variants. To evaluate the impact of rare CNVs with respect to risk for TGCC, we therefore compared the genome-wide burden of rare CNVs and investigated whether any genes or pathways were targeted by multiple rare CNVs such that their aggregated frequency was higher in cases than in controls.
In summary, to assess the effect of CNVs on TGCC we genotyped a Danish case-control cohort (Dalgaard et al., 2012) and analyzed the resulting data with respect to the association of both common and rare germline CNVs to TGCC.
Results
To identify CNVs that confer a risk to TGCC, we analyzed common and rare variants in a genome-wide dataset of approximately 1.8 million markers in a Danish cohort constituting 212 TGCC cases and 437 controls. Application of stringent quality control criteria for reliable CNV identification (Figure 1) resulted in a final discovery set of 189 cases and 380 controls. Common variants were defined as CNVs present in more than 1% of the study population, and rare variants as CNVs present in no more than 1% of the studied subjects. Common variants were analyzed with respect to individual locus association, and rare variants with respect to overall genetic burden, gene association, and pathway association.
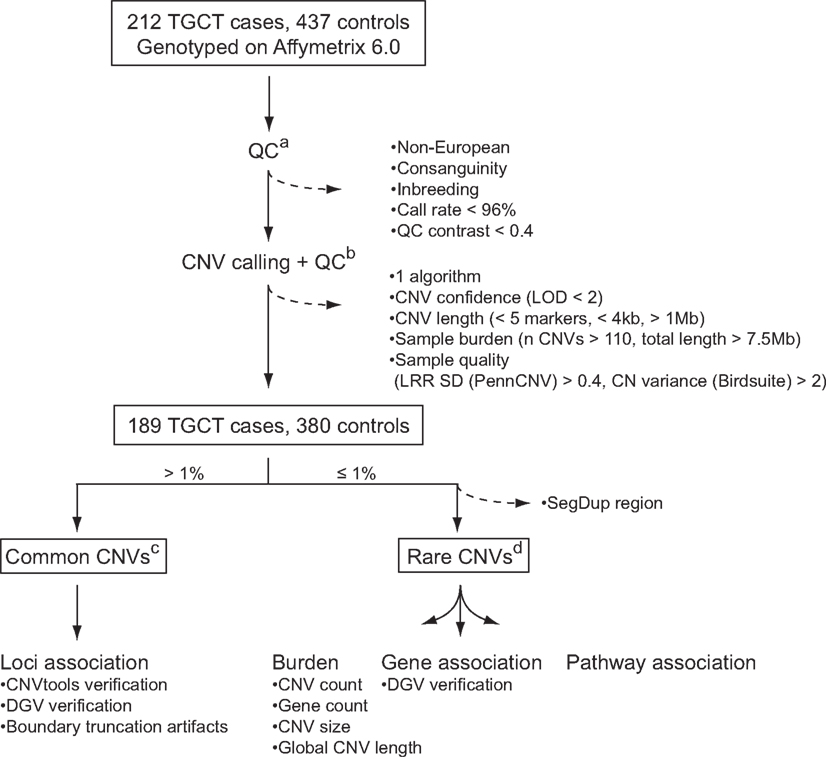
Figure 1. CNV quality control and analysis. Dashed arrows indicate CNVs and samples that were excluded from the analysis: (a) quality control of samples based on SNP calls; (b) quality control of CNVs and samples based on CNV calls; (c) association analysis of common CNVs; (d) association analysis of rare CNVs with respect to genomic burden, as well as genes and pathways with an excess of rare CNVs among cases. See “Materials and Methods” for further details. LOD, log odds; LRR SD, log R ratio standard deviation; DGV, database of genomic variants.
Locus Association Analysis
In order to identify common CNVs associated with TGCC, binary copy number state frequencies of the case and control cohorts were compared at all loci with CNV frequencies above 1%. We observed one genome-wide significant deletion at 1p13.3 covering the gene GSTM1 (P = 0.02, 37.2% cases, 19.5% controls), but downstream quality control by manual inspection of the copy number intensity histogram at this locus, and application of histogram-based association analysis (Barnes et al., 2008) suggested a false positive finding (nominal P = 0.26, 51.3% cases, 58.2% controls). A histogram of the copy number signal showed three distinct clusters with 51, 40, and 8% of the cases, respectively, and 58, 37, and 5% of the controls. The three clusters were assumed to correspond to copy numbers of 1, 2, and 3. Further, the deletion allele frequency at this locus has been estimated to be ∼40% in the International HapMap Phase 3 population study (Altshuler et al., 2010).
Given the varying deletion frequencies reported, and to assess the quality of the array-data CNV calling, we performed targeted sequencing of the GSTM1 (chr1p13) gene in 62 patients. The sequencing data showed three clusters with 52, 40, and 8% of the samples, and 55 out of the 62 patients were present in the same clusters as in the array-data. The CNV frequencies of the three clusters were in perfect agreement with those from the histogram analysis of the array-data, thereby corroborating that deletion of GSTM1 had no association to TGCC. The sequencing also revealed that the actual copy numbers were 0, 1, and 2 rather than 1, 2, and 3 since the majority of samples had no DNA present (0 sequence reads). The reason for that mis-assignment is that a copy number of 2 is generally assumed by CNV calling software to be the normal copy number state, having the major allele frequency, whereas in this case only 5–10% of the population carry two copies of the gene.
CNV Burden Association Analysis
Testing whether individuals with TGCC had a greater genomic burden of rare CNVs than controls, we observed a weak indication of increased burden with respect to the number of CNVs per sample, the number of affected genes per sample, and the average length of CNVs per sample (case/control ratio: 1.08, 1.10, and 1.11 respectively), and a significant difference with respect to the total length of all CNVs per sample (case/control ratio: 1.19, P = 0.03; Table 1).
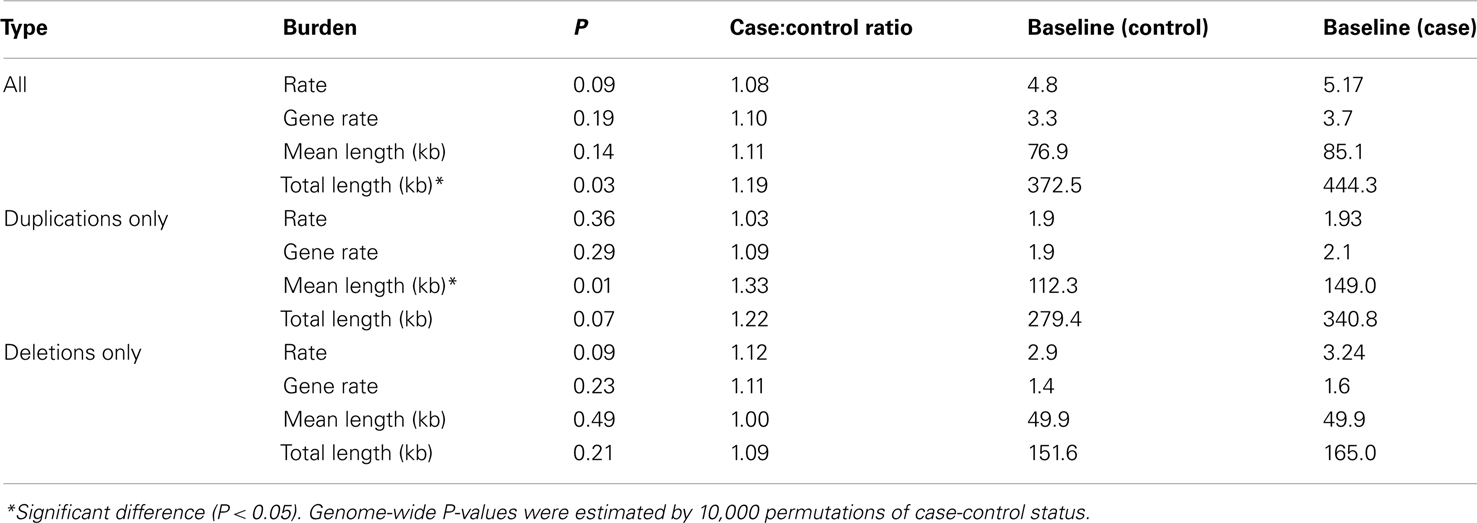
Table 1. Global burden of rare CNVs in cases versus controls.
Gene Association Analysis
Next, we explored if there were any specific genes where rare CNVs were more common in cases than controls. This analysis did not require that CNVs found in different samples overlapped each other, rather, it was sufficient that they were located within the same genic region. Two genes were found to have genome-wide significance, PTPN1 (Pemp. = 0.038) and KCNB2 (Pemp. = 0.022), affecting 0.9 and 1.2% of cases, respectively, whereas no occurrence in controls was observed at these loci (Table 2). The CNV at PTPN1 involved five cases, all found to have a heterozygous deletion at the same intronic region (Figure 2). CNVs at KCNB2 were found at three different loci: four and one deletions at two different introns and one deletion and one duplication at the promoter (Figure 2). Several CNVs have previously been reported in healthy individuals at KCNB2, but not at PTPN1 (Database of Genomic Variants, v. 10), corroborating a true TGCC association at PTPN1, but weakening the possibility of an actual association to KCNB2.

Table 2. Genes with an association of rare CNVs.
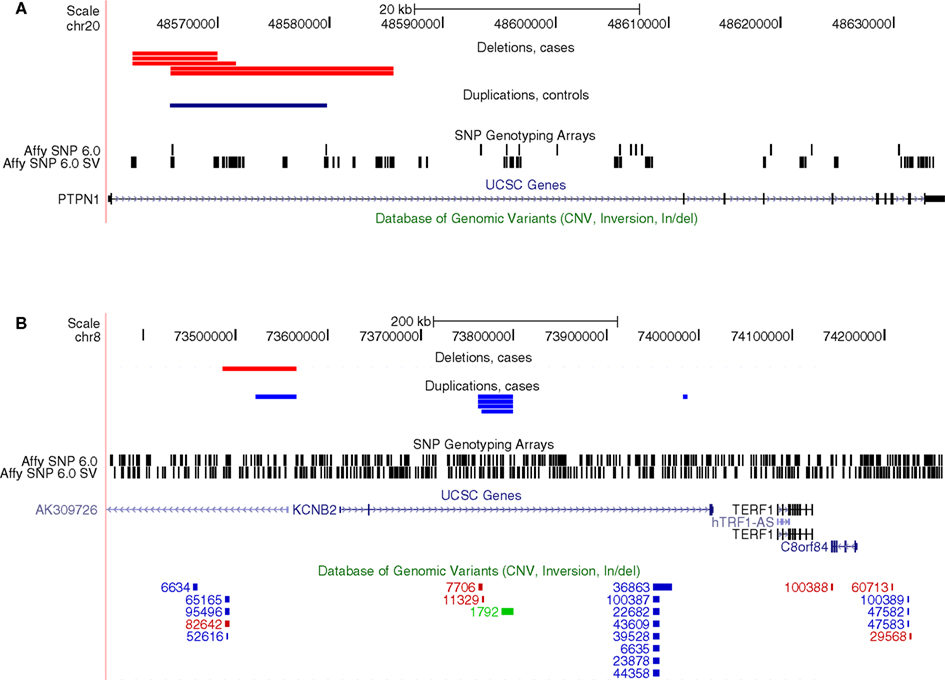
Figure 2. Genes with a significant excess of rare CNVs among cases as inferred from the genome-wide analysis of array-data. (A) Five cases with deletions and one control with a duplication at an intron of PTPN1, (B) Six cases with deletions and one case with a duplication at introns of KCNB2. The bottom track (Database of Genomic Variants) indicates that many CNVs have previously been observed at KCNB2 in healthy individuals, whereas none has been observed at PTPN1.
We attempted to verify the CNV at PTPN1 by performing qPCR on the affected samples, but all five samples with an indication of a heterozygous deletion in the array-data were observed to have two copies in the qPCR. Thus, the CNV calls from the array-data of PTPN1 are likely to be false positives and no association between PTPN1 and TGCC can be inferred at this stage.
Pathway Association Analysis
Proteins tend to act in concert and perturbations of different components in a set of proteins that are interacting in a network may result in a dysregulation with similar outcome (Lage et al., 2007; Navlakha and Kingsford, 2010; Pers et al., 2011). We therefore conducted association analysis on the level of pathways and protein–protein interaction networks. Association was assessed in the same fashion as for genes and loci described above, that is, by comparing case and control cohorts with respect to the total frequency of rare CNV events targeting a pathway. We compiled comprehensive collections of gene sets, where a set of genes either share function or operate in the same pathway, and performed one thousand random permutations (shuffling case and control status) to estimate a local false-discovery rate (FDR) of each gene set (Efron and Tibshirani, 2002). We observed a significant differential proportion of rare CNVs between cases and controls in 14 gene sets (FDR < 5%, Table 3). However, 11 of these associations were driven by the gene-specific association to PTPN1 described above. The three remaining gene sets, which did not include PTPN1, were: “regulation of cell migration” [go:0030334, 1.8% versus 1.1%, FDR = 0.021, OR = 3.47 (1.12−11.82)], “positive regulation of catalytic activity” [go:0043085, 1.4% versus 0.5%, FDR = 0.04, OR = 5.54 (1.31 − 32.78)], and “macromolecular complex disassembly” [go:0032984, 2.3% versus 1.9%, FDR = 0.047, OR = 2.47 (1.00−6.23)]. The most significant gene set among all sets tested was “regulation of cell migration.” A total of 16 individuals harbored CNVs that altogether overlapped 14 genes in this pathway, at 13 unique CNV loci (Table 4). Genes in this gene set that were affected in cases but not controls included: BCL2, CDH13, CORO1A, KDR, MUC2, MUC5AC, ONECUT2, and PTPRK.
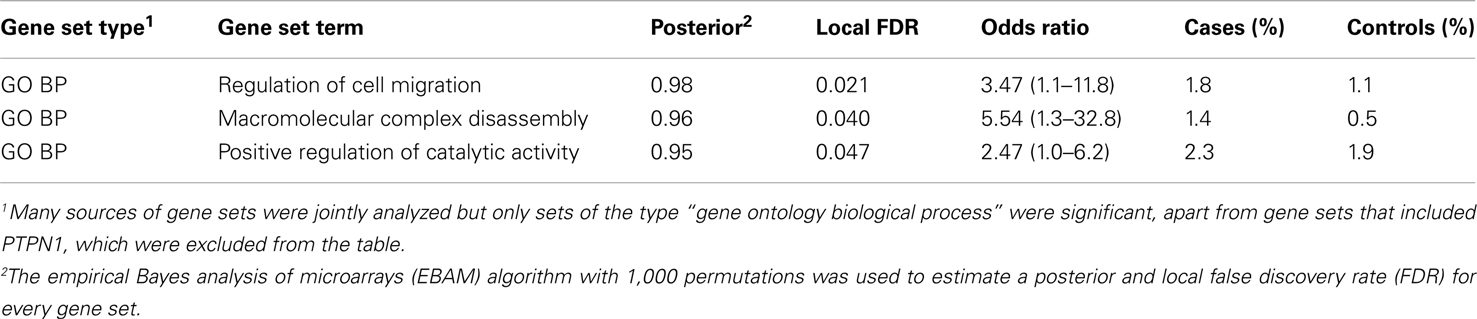
Table 3. Gene sets with an association of rare CNVs.
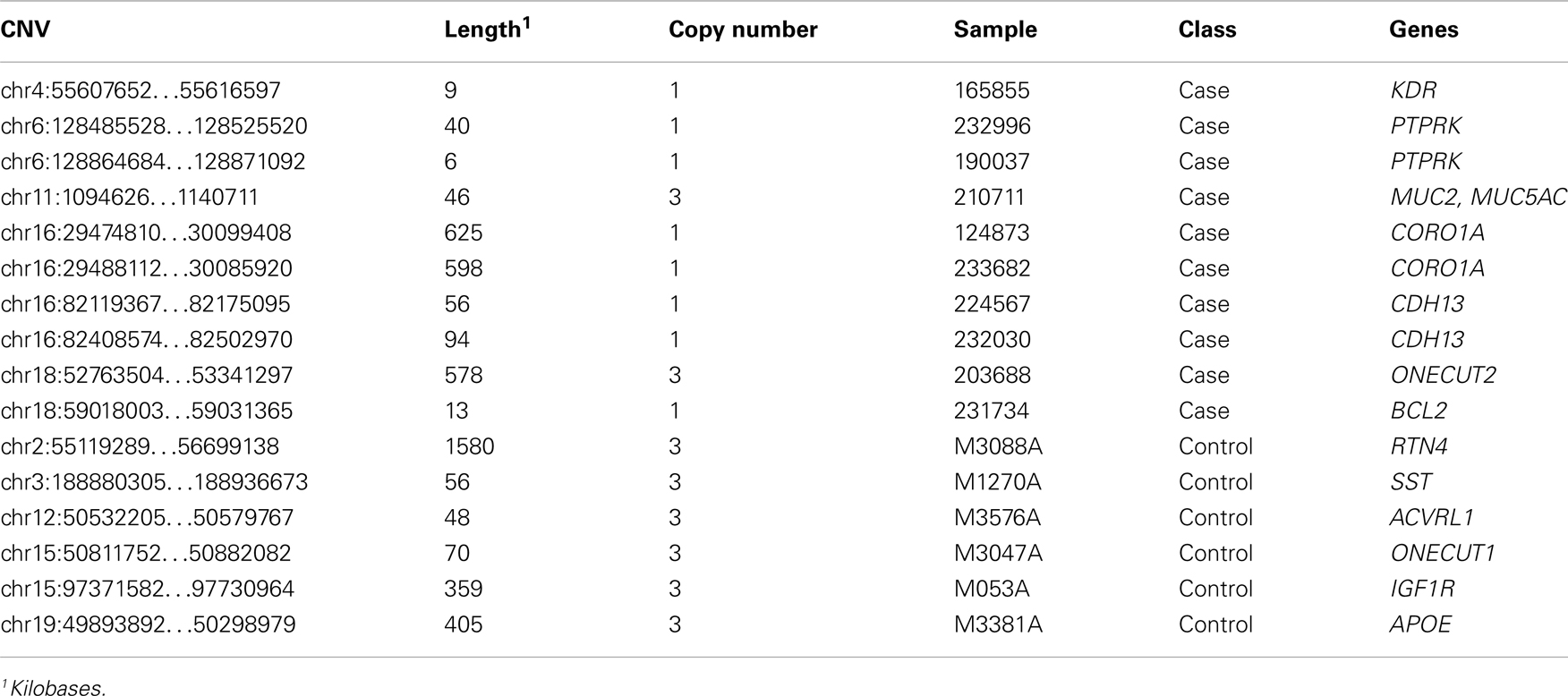
Table 4. CNVs targeting genes that are part of the gene set “Regulation of cell migration.”
Discussion
We assessed the effect of common and rare CNVs in a TGCC case-control cohort. No single locus was found to be associated to TGCC, but one potential gene network of interest with a weak significant association was identified, having an elevated frequency of rare CNVs among cases. The absence of any single locus CNV associated to TGCC is in line with the relatively few findings for other diseases, including a screening of ∼3,400 common CNVs in eight common diseases (Craddock et al., 2010). Furthermore, common CNVs are typically ancient variations, which are tightly correlated with single nucleotide polymorphisms (SNPs), and can therefore be detected by genome-wide association studies of common SNPs (Lander, 2011). However, one should not neglect the importance of common CNVs in gene-phenotype association studies, since there exists evidence that disease associated SNPs have a tendency to tag CNVs more often than random, and that such CNV-tagging SNPs are enriched for expression quantitative trait loci (eQTL; Gamazon et al., 2011). Further, rare CNVs are not tagged by common SNPs implicated by GWASs and they have been found to play a major role in neurodevelopmental disorders (Merikangas et al., 2009) and rare de novo CNVs were recently found in 3 out of 43 TGCC family-trios (Stadler et al., 2012). We have previously searched for CNVs associated to familial TGCTs, where the inheritance component is much higher than among sporadic cases, but failed to find a CNV that was significant across the small set of studied families. We only identified a handful of CNVs that segregated with TGCT, but these were either family-specific or relatively common variants, such as RLN1 (Edsgärd et al., 2011).
The rare association analysis presented in this study initially indicated two genes to be associated with TGCC, PTPN1, and KCNB2. KCNB2 was considered a false positive due to the amount of previously reported CNVs in independent control cohorts at this locus, being on par with that of the frequency in the case group of this study. PTPN1 appeared as a good candidate since it has previously been implicated with the progression of prostate cancer along with evidence that the androgen receptor is a transcriptional regulator of PTPN1 (Lessard et al., 2010, 2012). A loss of PTPN1 may thereby be associated with impaired responsiveness to androgens, which would be consistent with the fact that low androgen status during development is a risk factor for TGCT (Rajpert-De Meyts and Skakkebaek, 1993; Sonne et al., 2008). Further, CNVs at the 20q13 chromosomal region have been observed in several other cancers (Nishizaki et al., 1998; Schaid, 2004; Furukawa et al., 2006). Furthermore, all affected probands in this study presented a deletion, consistent with a tumor suppressing function of oncogenic kinases, and PTPN1 has been shown to be able to play both a pro- and anti-oncogenic role (Stuible et al., 2008). A considerable effort was made to ensure high quality of the CNV calls: by using two complementary CNV calling methods, by applying several strict QC criteria, and by manual inspection of the raw probe signals. However, despite the stringent QC the qPCR did not verify the heterozygous deletion of PTPN1 in any of the samples. This highlights the difficulty of assessing the reliability of CNV calls from genome-wide short-nucleotide microarrays.
Our pathway analysis identified the gene set “regulation of cell migration” as having the highest difference in proportion of rare CNVs in cases compared to controls. There is a growing body of information that strongly suggests a crucial role of primordial germ cell (PGC) biology in TGCC oncogenesis. PGCs are embryonic cells which during mid-gestation migrate from the base of the yolk sac, along the hind-gut, to the genital ridge, one of the longest migrations of all mammalian cells. Four recent SNP GWAS TGCC studies associated KITLG and a number of other genes related to the KIT-KITLG pathway (Kanetsky et al., 2009; Rapley et al., 2009; Turnbull et al., 2010; Dalgaard et al., 2012), a regulatory network which is believed to be of crucial importance in the determination of the fate of PGCs (Rapley and Nathanson, 2010). For instance, mutations of the KIT receptor, or the KIT ligand, in the mouse, blocks PGC migration, resulting in infertility (Matzuk and Lamb, 2002). In addition, a disturbance of the migration of PGCs during early fetal development may cause extragonadal germ cell cancers along the midline of the body (Oosterhuis and Looijenga, 2005). One of the afflicted genes in the “regulation of cell migration” gene set was PTPRK, at which two samples had a deletion. Like PTPN1, PTPRK belongs to the family of protein tyrosine phosphatases, but it is a membrane-bound receptor. PTRPK has been implicated in TGFβ-signaling (Xu et al., 2010), and a recent GWAS study indicated the involvement of TGFβ superfamily signaling in testicular dysgenesis (Dalgaard et al., 2012). In mouse, PGCs divide rapidly under the influence of TGFβ signaling factors, and defects in PGC development is observed in knockout models of bone morphogenetic proteins (BMPs; Lawson et al., 1999; Ying et al., 2000). In total there were 14 genes which were part of the “regulation of cell migration” pathway and that harbored CNVs. It would be of high interest to further elucidate the role of these genes by studying how the CNVs affect the expression levels of the corresponding genes. What may complicate matters is if the critical period of time is the development of the fetus, which may force one to revert to the study of animal models.
In the analysis of common CNVs we made a further assessment of the GSTM1 CNV frequency by targeted sequencing. The main reason for this was to assess the quality of and verify the array-data CNV calls of a seemingly problematic locus. However, GSTM1 was also partly chosen due to that the risk of developing TGCC has been shown to increase by the exposure to certain environmental factors (Skakkebaek et al., 2001) and GSTM1 is known to be involved in the detoxification of xenobiotic compounds (Hengstler et al., 1998). Hypothetically, it is not unlikely that there exists a genetic variation that may affect the ability to metabolize environmental chemicals, which in turn affects the risk of TGCC. Apart from such gene-environment interactions it is probable that part of the unexplained heritability can be explained by gene–gene interactions, where a combination of several genetic variations cause a greater effect on the phenotype than the sum of their individual effects (epistasis). Further research is required to assess the effect of rare variants in varying genetic backgrounds.
In conclusion, this study corroborates the rejection of the hypothesis that a single CNV locus mediates a major risk for TGCC. It suggests that several rare CNVs may contribute to the oncogenesis of a subset of TGCC subjects, but the frequency after aggregation of CNVs on the implicated gene and pathways is still low, and these CNVs therefore only provide a minor contribution to the overall heritability. Larger cohorts are needed to further explore the impact of rare variants in TGCC.
Materials and Methods
Sample Collection
Two hundred and twelve men with TGCCs and 439 healthy young men with semen concentrations above 60 million sperm/ml were collected at the Department of Growth and Reproduction, Rigshospitalet, Denmark. All DNA samples were obtained from men of Danish ancestry, who provided informed consent for genetic analysis. The samples were coded during the entire analysis. The project has been approved by the Regional Medical Ethics Committee (Nr H-KF-265848) and the Danish Personal Data Protection Agency (Nr 2008-41-2158). This cohort has previously been analyzed with respect to single nucleotide variation (Dalgaard et al., 2012). Sixty-two of the TGCC patients were selected for sequencing of the GSTM1 gene.
Genotyping and SNP-Based Sample Quality Control
Germline DNA was extracted from peripheral blood using QuickGene DNA whole blood kits from FujiFilm Life Science according to the manufacturer’s manual. Samples were genotyped using the Affymetrix Genome-wide Human SNP Array 6.0. Here we present the analysis of CNVs, SNP association analysis and details of the cohort are described elsewhere (Dalgaard et al., 2012), but an initial sample quality control was performed using SNP genotypes called by the Birdseed algorithm [Affymetrix Power Tools (APT) v. 1.10.2]. We excluded samples based on a genotyping call rate below 96%; QC contrast below 0.4 as according to the Affymetrix GCOS software; non-European ancestry by inspection of a plot of the first two principal components of the cohort and the HapMap phase III samples (Altshuler et al., 2010); high degree of relatedness based on identity-by-descent where one individual was kept among related samples; and samples with an outlying inbreeding coefficient.
CNV Detection and Quality Control
For samples that passed the SNP quality control described above we ran two CNV calling algorithms, BirdSuite (v. 1.5.5) and PennCNV (v. 2010May01). PennCNV requires a signal intensity file and a SNP genotyping file and these were generated by quantile normalization of PM-only probes with median polish probe summarization (APT v. 1.12.0) and Birdseed (v. 2 in APT 1.12.0), respectively. We excluded CNVs which failed any of the four following criteria: (1) A CNV was not called by both algorithms. A histogram of the percentage of overlap indicates that the vast majority has >90% overlap, but we set the threshold to at least 10%. (2) A CNV log odds confidence score larger than two, as recommended by BirdSuite (Korn et al., 2008). (3) CNV size was less than five markers or four kilobases, in effect excluding the 25% short-length quantile of CNVs. (4) A CNV was longer than one megabase (three outliers based on histogram). Further, samples were excluded with respect to the three following criteria: (1) extreme sample burden in terms of more than 110 CNVs (four outliers); or (2) a total length of CNVs larger than 7.5 megabases (two outliers); (3) bad sample quality in terms of high variance of copy number signal (median copy number variance larger than two, as recommended by BirdSuite, or a Log R Ratio standard deviation (LRR SD) obtained from PennCNV larger than 0.4). LRR SD was set according to PennCNV guidelines when CNV calling are performed on Affymetrix samples. Finally, rare CNVs that had more than 50% overlap with segmental duplication regions (retrieved from UCSC hg18) were removed, since such regions have been shown to generate more false CNV calls (Pinto et al., 2010). A total of 189 TGCC cases and 380 controls remained after the completion of all quality control steps, harboring a total of 1008 and 1872 rare CNVs, respectively.
CNV Association Analysis
Copy number variant association analysis was performed using plink (v. 1.07) and custom R (v. 2.12) scripts. Common and rare CNVs were defined as those with an allele frequency above or below 1%, respectively. The allele frequency of a CNV was determined using the locus within a CNV region with the maximum number of overlapping individual CNVs.
Locus Association
Common CNVs were evaluated by searching the whole genome for loci with a significantly higher degree of affected cases as compared to controls. Binary state frequencies were used, and a Fisher test was performed for deletion, amplification, and any type of aberration, respectively. Genome-wide significance was estimated by generating a null distribution based on one thousand case-control status permutations. For each permutation the minimal P-value was selected, thereby providing control of the family-wise error rate (FWER). Loci with significant associations were further verified by CNVtools (Barnes et al., 2008) which uses a complementary CNV calling strategy, since it employs a statistical model based on density-based clustering rather than a hidden Markov model. Furthermore, loci residing at the edges of a common CNV and associations from variation of boundary truncation were excluded.
Global Burden Analysis
The impact of rare CNVs was assessed by three approaches: genome-burden analysis, gene association, and pathway association analysis.
The global burden of rare CNVs in cases compared to controls were assessed with respect to (i) the number of CNVs per sample, (ii) the number of affected genes per sample, (iii) the average length of CNVs per sample, and (iv) the total length of CNVs per sample.
Gene Association
Gene association analysis was performed using the number of case and control samples harboring a rare CNV that overlapped the gene of interest. Genes were retrieved from UCSC (hg18). CNV frequencies were compared with Fisher’s test and multiple testing corrected P-values were obtained based on case-control permutation as described above for the locus association of common CNVs. Significant CNVs which were found to have a lower allele frequency in our case cohort than in the Database of Genomic Variants (DGV, v. 10), were considered false positives.
Pathway Association
Pathway association analysis was performed based on the number of case and control samples that had a rare CNV in any of the genes of a pathway. The R package “siggenes” was used to obtain P-values corrected for multiple testing across all tested gene sets. The package provides a FDR, based on randomized case-control sampling, as well as an adjustment of the variance of an individual pathway using information from the observed variances of all pathways (Efron and Tibshirani, 2002). Gene sets were retrieved from KEGG (Kyoto Encyclopedia of Genes and Genomes; Ogata et al., 1999), Reactome (D’Eustachio, 2011), BioCarta1, NCI-Nature curated pathways (Pathway Interaction Database; Schaefer et al., 2009), GO (Gene Ontology; Ashburner et al., 2000), COSMIC (Catalog of Somatic Mutations In Cancer; Forbes et al., 2011), Cyclebase (Gauthier et al., 2010), protein–protein interaction complexes (Lage et al., 2007), OMIM (Online Mendelian Inheritance in Man2), MGI (Mouse Genome Informatics3) and a set of candidate infertility genes from a recent review (Matzuk and Lamb, 2008). Terms annotating more than 700 or less than 5 genes were discarded, since they do not produce meaningful statistical results.
Verification of PTPN1 Copy Number
Copy number verification of PTPN1 was done using Quantitative PCR on the Mx3000P platform from Stratagene (Agilent Technologies, Santa Clara CA, USA). The protocol has been described previously (Ottesen et al., 2007). Primers for the CNV at PTPN1 were designed using Primer3 (Rozen and Skaletsky, 2000). Primer sequences for PTPN1 were: forward 5′-TTC AAC CCT AAC TAG GTA TGC A-3′ and reverse 5′-CTA AAA TGC TGG AGA CTT AGG T-3′ and primers for GAPDH: forward 5′-CTC CCC ACA CAC ATG CAC TTA-3′ and reverse 5′-TTG CCA AGT TGC CTG TCC TT-3′ (DNA Technology A/S, Aarhus, Denmark). GAPDH was used as endogenous control gene. Mixtures of forward and reverse primers were denatured for 3 min at 95°C and incubated on ice until use. DNA concentrations were 8–17 ng, 15 μL Brilliant SYBR Green QPCR Master Mix (Stratagene), 7.0 μL primer mixture of PTPN1 (final conc.: Fw and Rev 100 nm) or GAPDH (final conc.: Fw and Rev 100 nm), and a total volume of 30 μL. Conditions for amplification were as follows: one cycle at 95°C for 10 min and 40 cycles at 95°C for 1 min, 62°C for 1 min and 72°C for 1 min. The PTPN1:GAPDH ratio was calibrated to a normal male reference control of DNA, as previously described for other genes (Ottesen et al., 2007; Mau Kai et al., 2008). All specimens were analyzed in triplicate and the mean-ratio was used to infer integer copy number.
Targeted Sequencing of GSTM1
Design of targeted capture baits
Target capture was designed on the Agilent SureSelect capture system (Santa Clara CA, USA). Baits for capture were designed by tiling the genomic region harboring GSTM1 with an approximately 50% overlap between consecutive baits. The bait sequences were optimized to avoid regions with extreme GC content or highly variable regions, and to avoid possible cross-hybridization or self-folding of the baits, which could decrease the hybridization specificity. The principle of this method has been described previously (Wesolowska et al., 2011).
Library preparation, pooling, target enrichment, and sequencing
DNA shearing, library preparation, and pooling were performed using a modified protocol of the SureSelect Target Enrichment System (Agilent Technologies) to allow sample multiplexing as described elsewhere (Wesolowska et al., 2011). Three micrograms of genomic DNA was sheared by ultrasound (Covaris; Woburn, MA, USA) to yield fragments of an average size of ∼200 bp. The sheared fragments were ligated with custom made adaptors containing a unique four-base barcode, and subsequently purified and amplified by ligation-mediated PCR (LM-PCR). Amplified DNA was pooled in groups of 10 with equal amount of each, after which enrichment was performed by hybridization to custom SureSelect target capture baits (Agilent Technologies). The captured libraries were processed with Illumina Cluster Generation Station (Illumina, San Diego CA, USA) following the manufactures recommendations. We performed 100 nt single-end read sequencing on the Illumina HiSeq 2000 sequencing platform.
Data analysis
Sequence reads were trimmed and high quality reads were mapped to the human reference genome build 37 (GRCH37) using Burrow-Wheeler Alignment algorithm (Li and Durbin, 2010). Alignments with mapping score below 30 were discarded and PCR duplicates were removed with Picard MarkDuplicates4. Copy numbers were estimated by calculating a sequence depth ratio from the number of reads in the targeted genomic region, normalized by size of the region and total number of reads for the sample. A histogram of the sequence depth ratio from all individuals identified three distinct clusters corresponding to two copies and heterozygous and homozygous deletions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Betina F. Nielsen for skillful microarray work and clinicians for handling of patients and controls (especially Dr Niels Jørgensen and his team). We would like to thank the DTU Multi-Assay Core facility for automated library preparation infrastructure. The work was supported by the Villum Kann Rasmussen Foundation, the Danish childhood cancer research foundation, and the Danish Cancer Research Foundation.
Footnotes
References
1000 Genomes Project Consortium. (2010). A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073.
Altshuler, D., Gibbs, R., Peltonen, L., Dermitzakis, E., Schaffner, S., Yu, F., et al. (2010). Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58.
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29.
Barnes, C., Plagnol, V., Fitzgerald, T., Redon, R., Marchini, J., Clayton, D., et al. (2008). A robust statistical method for case-control association testing with copy number variation. Nat. Genet. 40, 1245–1252.
Chia, V., Quraishi, S., Devesa, S., Purdue, M., Cook, M., and McGlynn, K. (2010). International trends in the incidence of testicular cancer, 1973-2002. Cancer Epidemiol. Biomarkers Prev. 19, 1151–1159.
Craddock, N., Hurles, M., Cardin, N., Pearson, R., Plagnol, V., Robson, S., et al. (2010). Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 464, 713–720.
Dalgaard, M., Weinhold, N., Edsgärd, D., Silver, J., Pers, T., Nielsen, J., et al. (2012). A genome-wide association study of men with symptoms of testicular dysgenesis syndrome and its network biology interpretation. J. Med. Genet. 49, 58–65.
D’Eustachio, P. (2011). Reactome knowledgebase of human biological pathways and processes. Methods Mol. Biol. 694, 49–61.
Diskin, S., Hou, C., Glessner, J., Attiyeh, E., Laudenslager, M., Bosse, K., et al. (2009). Copy number variation at 1q21.1 associated with neuroblastoma. Nature 459, 987–991.
Edsgärd, D., Scheel, M., Hansen, N. T., Ralfkiaer, U., Jensen, T. S., Skakkebaek, N. E., et al. (2011). Heterozygous deletion at the RLN1 locus in a family with testicular germ cell cancer identified by integrating copy number variation data with phenome and interactome information. Int. J. Androl. 34, e175–e188.
Efron, B., and Tibshirani, R. (2002). Empirical bayes methods and false discovery rates for microarrays. Genet. Epidemiol. 23, 70–86.
Fanciulli, M., Norsworthy, P., Petretto, E., Dong, R., Harper, L., Kamesh, L., et al. (2007). FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat. Genet. 39, 721–723.
Forbes, S., Bindal, N., Bamford, S., Cole, C., Kok, C., Beare, D., et al. (2011). COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 39, D945–D950.
Furukawa, T., Sunamura, M., and Horii, A. (2006). Molecular mechanisms of pancreatic carcinogenesis. Cancer Sci. 97, 1–7.
Gamazon, E., Nicolae, D., and Cox, N. (2011). A study of CNVs as trait-associated polymorphisms and as expression quantitative trait loci. PLoS Genet. 7:e1001292. doi:10.1371/journal.pgen.1001292
Gauthier, N., Jensen, L., Wernersson, R., Brunak, S. R., and Jensen, T. (2010). Cyclebase.org: version 2.0, an updated comprehensive, multi-species repository of cell cycle experiments and derived analysis results. Nucleic Acids Res. 38, D699–D702.
Glessner, J., Bradfield, J., Wang, K., Takahashi, N., Zhang, H., Sleiman, P., et al. (2010). A genome-wide study reveals copy number variants exclusive to childhood obesity cases. Am. J. Hum. Genet. 87, 661–666.
Hemminki, K., and Li, X. (2004). Familial risk in testicular cancer as a clue to a heritable and environmental aetiology. Br. J. Cancer 90, 1765–1770.
Hengstler, J. G., Arand, M., Herrero, M. E., and Oesch, F. (1998). Polymorphisms of N-acetyltransferases, glutathione S-transferases, microsomal epoxide hydrolase and sulfotransferases: influence on cancer susceptibility. Recent Results Cancer Res. 154, 47–85.
Hollox, E., Huffmeier, U., Zeeuwen, P., Palla, R., Lascorz, J., Rodijk-Olthuis, D., et al. (2007). Psoriasis is associated with increased |[beta]|-defensin genomic copy number. Nat. Genet. 40, 23–25.
International Schizophrenia Consortium. (2008). Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455, 237–241.
Kanetsky, P., Mitra, N., Vardhanabhuti, S., Li, M., Vaughn, D., Letrero, R., et al. (2009). Common variation in KITLG and at 5q31.3 predisposes to testicular germ cell cancer. Nat. Genet. 41, 811–815.
Korn, J., Kuruvilla, F., McCarroll, S., Wysoker, A., Nemesh, J., Cawley, S., et al. (2008). Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 40, 1253–1260.
Lage, K., Karlberg, O., Storling, Z., Olason, P., Pedersen, A., Rigina, O., et al. (2007). A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 25, 309–316.
Lawson, K. A., Dunn, N. R., Roelen, B. A., Zeinstra, L. M., Davis, A. M., Wright, C. V., et al. (1999). Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev. 13, 424–436.
Lessard, L., Labbé, D., Deblois, G., Bégin, L., Hardy, S., Mes-Masson, A.-M., et al. (2012). PTP1B is an androgen receptor-regulated phosphatase that promotes the progression of prostate cancer. Cancer Res. 72, 1529–1537.
Lessard, L., Stuible, M., and Tremblay, M. (2010). The two faces of PTP1B in cancer. Biochim. Biophys. Acta 1804, 613–619.
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595.
Matzuk, M., and Lamb, D. (2002). Genetic dissection of mammalian fertility pathways. Nat. Med. 8, S33–S40.
Matzuk, M., and Lamb, D. (2008). The biology of infertility: research advances and clinical challenges. Nat. Med. 14, 1197–1213.
Mau Kai, C., Juul, A., McElreavey, K., Ottesen, A. M., Garn, I. D., Main, K. M., et al. (2008). Sons conceived by assisted reproduction techniques inherit deletions in the azoospermia factor (AZF) region of the Y chromosome and the DAZ gene copy number. Hum. Reprod. 23, 1669–1678.
Merikangas, A., Corvin, A., and Gallagher, L. (2009). Copy-number variants in neurodevelopmental disorders: promises and challenges. Trends Genet. 25, 536–544.
Mills, R., Walter, K., Stewart, C., Handsaker, R., Chen, K., Alkan, C., et al. (2011). Mapping copy number variation by population-scale genome sequencing. Nature 470, 59–65.
Nathanson, K., Kanetsky, P., Hawes, R., Vaughn, D., Letrero, R., Tucker, K., et al. (2005). The Y deletion gr/gr and susceptibility to testicular germ cell tumor. Am. J. Hum. Genet. 77, 1034–1043.
Navlakha, S., and Kingsford, C. (2010). The power of protein interaction networks for associating genes with diseases. Bioinformatics 26, 1057–1063.
Nishizaki, T., Ozaki, S., Harada, K., Ito, H., Arai, H., Beppu, T., et al. (1998). Investigation of genetic alterations associated with the grade of astrocytic tumor by comparative genomic hybridization. Genes Chromosomes Cancer 21, 340–346.
Ogata, H., Goto, S., Sato, K., Fujibuchi, W., Bono, H., and Kanehisa, M. (1999). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29–34.
Oosterhuis, W., and Looijenga, L. (2005). Testicular germ-cell tumours in a broader perspective. Nat. Rev. Cancer 5, 210–222.
Ottesen, A. M., Garn, I. D., Aksglaede, L., Juul, A., and Rajpert-De Meyts, E. (2007). A simple screening method for detection of Klinefelter syndrome and other X-chromosome aneuploidies based on copy number of the androgen receptor gene. Mol. Hum. Reprod. 13, 745–750.
Pers, T., Hansen, N. T., Lage, K., Koefoed, P., Dworzynski, P., Miller, M. L., et al. (2011). Meta-analysis of heterogeneous data sources for genome-scale identification of risk genes in complex phenotypes. Genet. Epidemiol. 35, 318–332.
Pinto, D., Pagnamenta, A., Klei, L., Anney, R., Merico, D., Regan, R., et al. (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372.
Rajpert-De Meyts, E., and Skakkebaek, N. E. (1993). The possible role of sex hormones in the development of testicular cancer. Eur. Urol. 23, 54–59.
Rapley, E., and Nathanson, K. (2010). Predisposition alleles for testicular germ cell tumour. Curr. Opin. Genet. Dev. 20, 225–230.
Rapley, E., Turnbull, C., Al Olama, A. A., Dermitzakis, E., Linger, R., Huddart, R., et al. (2009). A genome-wide association study of testicular germ cell tumor. Nat. Genet. 41, 807–810.
Rozen, S., and Skaletsky, H. (2000). Primer3 on WWW for general users and for biologist programmers. Methods Mol. Biol. 132, 365–386.
Schaefer, C., Anthony, K., Krupa, S., Buchoff, J., Day, M., Hannay, T., et al. (2009). PID: the Pathway Interaction Database. Nucleic Acids Res. 37, D674–D679.
Schaid, D. (2004). The complex genetic epidemiology of prostate cancer. Hum. Mol. Genet. 13, R103–R121.
Skakkebaek, N. E., Rajpert-De Meyts, E., and Main, K. M. (2001). Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects. Hum. Reprod. 16, 972–978.
Sonne, S. B., Kristensen, D. M. B., Novotny, G., Olesen, I. A., Nielsen, J., Skakkebaek, N., et al. (2008). Testicular dysgenesis syndrome and the origin of carcinoma in situ testis. Int. J. Androl. 31, 275–287.
Stadler, Z., Esposito, D., Shah, S., Vijai, J., Yamrom, B., Levy, D., et al. (2012). Rare de novo germline copy-number variation in testicular cancer. Am. J. Hum. Genet. 91, 379–383.
Stuible, M., Doody, K., and Tremblay, M. (2008). PTP1B and TC-PTP: regulators of transformation and tumorigenesis. Cancer Metastasis Rev. 27, 215–230.
Swerdlow, A. J., De Stavola, B. L., Swanwick, M. A., and Maconochie, N. E. (1997). Risks of breast and testicular cancers in young adult twins in England and Wales: evidence on prenatal and genetic aetiology. Lancet 350, 1723–1728.
Turnbull, C., Rapley, E., Seal, S., Pernet, D., Renwick, A., Hughes, D., et al. (2010). Variants near DMRT1, TERT and ATF7IP are associated with testicular germ cell cancer. Nat. Genet. 42, 604–607.
Wesolowska, A., Dalgaard, M. D., Borst, L., Gautier, L., Bak, M., Weinhold, N., et al. (2011). Cost-effective multiplexing before capture allows screening of 25‚000 clinically relevant SNPs in childhood acute lymphoblastic leukemia. Leukemia 25, 1001–1006.
Xu, Y., Baker, D., Quan, T., Baldassare, J., Voorhees, J., and Fisher, G. (2010). Receptor type protein tyrosine phosphatase-kappa mediates cross-talk between transforming growth factor-beta and epidermal growth factor receptor signaling pathways in human keratinocytes. Mol. Biol. Cell 21, 29–35.
Yang, T.-L., Chen, X.-D., Guo, Y., Lei, S.-F., Wang, J.-T., Zhou, Q., et al. (2008). Genome-wide copy-number-variation study identified a susceptibility gene, UGT2B17, for osteoporosis. Am. J. Hum. Genet. 83, 663–674.
Keywords: copy number variation, rare variants, testicular germ cell cancers
Citation: Edsgärd D, Dalgaard MD, Weinhold N, Wesolowska-Andersen A, Rajpert-De Meyts E, Ottesen AM, Juul A, Skakkebæk NE, Skøt Jensen T, Gupta R, Leffers H and Brunak S (2013) Genome-wide assessment of the association of rare and common copy number variations to testicular germ cell cancer. Front. Endocrin. 4:2. doi: 10.3389/fendo.2013.00002
Received: 29 September 2012; Accepted: 07 January 2013;
Published online: 29 January 2013.
Edited by:
Alberto Ferlin, University of Padova, ItalyReviewed by:
Alessandra Mosca, University Hospital “Maggiore della Carità,” ItalyFrank Weber, University Hospital Essen, Germany
Copyright: © 2013 Edsgärd, Dalgaard, Weinhold, Wesolowska-Andersen, Rajpert-De Meyts, Ottesen, Juul, Skakkebæk, Skøt Jensen, Gupta, Leffers and Brunak. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Testis cancer related aspects: Ewa Rajpert-De Meyts, Department of Growth and Reproduction, Rigshospitalet, Blegdams vej 9, 2100 Copenhagen, Denmark. e-mail:ZXJtQHJoLmRr; Bioinformatic aspects: Søren Brunak, Department of Systems Biology, Technical University of Denmark, Kemitorvet 208, 2800 Lyngby, Denmark. e-mail:YnJ1bmFrQGNicy5kdHUuZGs=