 Marià Alemany1,2*
Marià Alemany1,2*- 1 Faculty of Biology, Department of Nutrition and Food Science, University of Barcelona, Barcelona, Spain
- 2 CIBER Obesity and Nutrition, Institute of Health Carlos III, Madrid, Spain
The metabolic syndrome is basically a maturity-onset disease. Typically, its manifestations begin to flourish years after the initial dietary or environmental aggression began. Since most hormonal, metabolic, or defense responses are practically immediate, the procrastinated response do not seem justified. Only in childhood, the damages of the metabolic syndrome appear with minimal delay. Sex affects the incidence of the metabolic syndrome, but this is more an effect of timing than absolute gender differences, females holding better than males up to menopause, when the differences between sexes tend to disappear. The metabolic syndrome is related to an immune response, countered by a permanent increase in glucocorticoids, which keep the immune system at bay but also induce insulin resistance, alter the lipid metabolism, favor fat deposition, mobilize protein, and decrease androgen synthesis. Androgens limit the operation of glucocorticoids, which is also partly blocked by estrogens, since they decrease inflammation (which enhances glucocorticoid release). These facts suggest that the appearance of the metabolic syndrome symptoms depends on the strength (i.e., levels) of androgens and estrogens. The predominance of glucocorticoids and the full manifestation of the syndrome in men are favored by decreased androgen activity. Low androgens can be found in infancy, maturity, advanced age, or because of their inhibition by glucocorticoids (inflammation, stress, medical treatment). Estrogens decrease inflammation and reduce the glucocorticoid response. Low estrogen (infancy, menopause) again allow the predominance of glucocorticoids and the manifestation of the metabolic syndrome. It is postulated that the equilibrium between sex hormones and glucocorticoids may be a critical element in the timing of the manifestation of metabolic syndrome-related pathologies.
Introduction
Genetic (Loos and Bouchard, 2003), epigenetic (Gluckman and Hanson, 2008), environmental (Romao and Roth, 2008), and mainly dietary (Sclafani and Springer, 1976; Seidell, 1998; Mendoza et al., 2007) causes of the metabolic syndrome exert their effects principally through the modulation of the expression of proteins (enzymes, transporters, regulatory agents). These agents carry out their functions only within short periods of time because of their relatively fast turnover, since a short life is one of the basic premises of any regulatory agent. In the same way, wearing out, saturation, and exhaustion of metabolic pathways and control systems could not either extend too much far away in time from the actual active life of the agents implicated, i.e., the half-lives of proteins, or the life-span of the cells affected. In short, if the development of obesity (or other metabolic syndrome-related disease) depends on excessive energy or fat intake and the wearing out of the systems controlling fat deposition (Virtue and Vidal-Puig, 2009), then, it should be expected that the metabolic syndrome-induced derangements should appear shortly after the initiation of the inflammatory processes and regulatory systems’ alteration. However, the expected changes usually do not occur shortly after exposure to triggering agents. Often a number of years elapse before the symptoms of the metabolic syndrome appear and the cluster of related diseases fully develops (Gustafsson et al., 2011). In addition, the metabolic syndrome tends to appear more often and/or earlier in adult males than in females (Regitz-Zagrosek et al., 2007; Hong et al., 2009; Gustafsson et al., 2011), the differences in incidence decreasing sharply after menopause (Janssen et al., 2008). There are sex-related differences, but the regulatory mechanisms and metabolic pathways affected are the same. Then, why are there so marked sex-related differences in the incidence of the metabolic syndrome? And also, why does this delay in its full manifestation occurs?
There is also a growing incidence of infantile obesity (Bua et al., 2007; Patton et al., 2011), i.e., an accelerated presentation of metabolic syndrome diseases fairly shortly after exposure to environmental (i.e., diet, sedentary behavior) obesogenic conditions (Butte, 2009; Tamayo et al., 2010; Bleich et al., 2011; Guinhouya et al., 2011). Why the delay between triggering factors and disease manifestation is much shorter in children than in adults?
We cannot directly attribute the differences to genetic factors (usually shared), neither to the nature of pathogenic mechanisms: usually lack of exercise (Rey-López et al., 2008; Mitchell et al., 2009), social tensions and stress (Moschonis et al., 2010; Tamayo et al., 2010; Gundersen et al., 2011), and mainly, excess dietary energy and predominance of lipids in the diet (Kohen-Avramoglu et al., 2003; Zimmermann and Aeberli, 2008). The proinflammatory mechanisms are shared too (Valle et al., 2005; Kowalska et al., 2008; Rizvi, 2009; Maury and Brichard, 2010). In addition, the sex differences are practically inexistent in children (Dubose et al., 2006), but tend to become manifest already in adolescents (Duncan et al., 2004), a harbinger of the adult appearance of the metabolic syndrome.
The Protracted Development of the Metabolic Syndrome
The mechanisms explaining the metabolic alterations observed in the diseases of the metabolic syndrome cluster, when known, seldom refer to the delay in the process of presentation of the syndrome. This raises a number of key questions which are critical for our understanding of the (adult) metabolic syndrome, its roots, maintenance, development, and eventual treatment.
A. How can some people never develop obesity and others do it early under similar exposure to diet and environmental factors (Blundell et al., 2005)? This is apparently incongruous with our acceptance that genetic stock is not the key factor for most obesities (Loos and Bouchard, 2003). How can some people keep ingesting huge amounts of lipids (saturated, cholesterol-laden, etc.) with no apparent problems whatsoever, and other people develop obesity even ingesting supposedly very healthy and monastically scarce diets for life (Ravussin et al., 1988)? Can we find the differences in the genes?
B. We know (or consensually assume) the causes (diet, genetics, environment) of the metabolic syndrome, and we also know its results or consequences (obesity, diabetes, hypertension, etc.). Nevertheless, the effects do not immediately follow the exposure to the triggering agents. A fairly long period of time is usually needed for these factors to finally manifest their deleterious effects (Parker et al., 2003; Ramsay et al., 2008; Gustafsson et al., 2011). Then, how can our metabolic systems (which work reacting immediately to substrate or regulator concentrations, hormonal signals, gene expression, etc.) wait years to develop metabolic syndrome-related diseases. In addition, the effects of these uncovered illnesses cannot be easily reversed by exercise (Thomas et al., 2010), low energy diets, with even lower lipid (Straznicky et al., 2010) or the ultimate surgical removal of excess fat (Varela et al., 2008)? What is the basis for this double-sided inertia?
C. What is the limit line that links the original dietary aggression and the metabolic syndrome constellation of diseases? Why are there differences in the pattern of manifestation of metabolic syndrome diseases in different people? Is there a sequence of events or a shotgun massive development of parallel pathologies?
There is an enormous body of information on these issues, but there is also a huge void in place of serious integrative analysis of a disease that has been intuitively considered a unique pathognomic entity (Bruce and Byrne, 2009) even before serious studies on its pathogenicity began. The center of study is progressively descending into detail to the molecular level (Nicolson, 2007; Maury and Brichard, 2010), usually forfeiting the integration of these data within the context of a living being, and adding complexity and background noise to any contextual analysis of the situation (Miranda et al., 2005).
Metabolic and Regulative Changes Elicited by the Metabolic Syndrome
The mechanisms so far presented to explain the pathogenicity of the metabolic syndrome should induce significant changes in a very short period of time, since:
1. The rheologic alterations caused by red blood cells undeformability, a more than probable contributor to hypertension (Cicco and Pirrelli, 1999), can appear in a few months, since the mean half-life of an erythrocyte is in the range of 120 days. If the postulated modification of their membrane fluidity is set during the lifetime of the cell, then it should be apparent in a few months. Continuity would depend on new cells substituting the destroyed red cells and on them becoming again altered by glycosylation, oxidation, nitration, etc., losing membrane flexibility in the process. However, the conditions after a few months should remain unchanged for years to come, maintaining perhaps a hypertensive situation, but with no possible direct incidence whatsoever on further aggravating the situation.
2. The excess glucose and excess lipid ingested and not processed are finally accumulated into fat depots after all other outlets have been topped up (increased energy expenditure including thermogenesis, higher protein accumulation and turnover, higher levels of glycogen, etc.). Most of this lipid is found in white adipose tissue (WAT), and part in liver and muscle. However, WAT is the main site for storage of excess fat. Any maintained excess of energy would end being part of accumulated fat in WAT. However, this is not – again – the case. Usually, WAT does not grow indefinitely to monstrous proportions (Jo et al., 2009). In fact, many obese and overweight individuals maintain their (excessive) weight fairly well thanks to an operative (albeit misadjusted) ponderostat setting (Bradley, 1978). There is something that prevents WAT from growing indefinitely, a defense system based on the coordinated operation of immune system cells’ infiltration, which help produce cytokines that reorganize the body energy handling protocols (Cancello and Clément, 2006; Pang et al., 2008; Alemany, 2011b), but also prevent that WAT receives excess energy. In the end WAT informs the brain (probably through ponderostat signals) that the optimal level of fat stores is overcome (Bradley, 1978; van Itallie, 1990). In addition, WAT glycolyzes excess glucose (normally under hypoxic conditions) and produces lactate, resulting in local acidosis (Hagström et al., 1990; Muñoz et al., 2010); there is a marked vasoconstriction (largely cytokine or catecholamine-driven) than compounds the effects of acidosis to induce a marked hypoxia that exacerbates the secretion of cytokines and other signals (Wang et al., 2007, 2008), as well as increasing the glycolytic loop. As a consequence, WAT is defended against excess lipid accumulation. But all this sequence occurs in a few weeks, perhaps months, but not years! After WAT has grown to its limit and is fully invaded by cells of the immune system, its function as lipid storage depot and glycolytic outlet for excess plasma glucose remain unchanged, and could not be further increased (but can decrease) despite the maintenance of high levels of energy substrates. Thus, the pathogenic conditions observed in the WAT of a rat subjected to a high-lipid fattening diet should be the same in a few weeks than after a few months; nevertheless, the weight increase soon tappers off, often irrespective of dietary or surgical restrictive treatments (Sclafani and Springer, 1976; Greenwood et al., 1982).
3. The accumulation of fat in the liver due to deranged lipoprotein turnover, altered cholesterol metabolism, excess energy and overall inflammation (Wouters et al., 2008; Eguchi et al., 2011; Subramanian et al., 2011), occurs in parallel to the massive storage of fat in WAT, and has similarly insurmountable timelines blocking the accumulation of fat beyond the already surpassed limits of functionality. Possible deterioration of hepatocyte function encounters –again– the problem of cell cycle duration and cell component turnover. Most body cells are substituted (at different speeds, directly related to their activity and proclivity to damage) in relatively short periods, and within these cells, cell organelles, structures and molecules (largely proteins) are recycled at speeds ranging from minutes to months (but, again, not years or decades; Yuile et al., 1959; Millward et al., 1975). In consequence, damaged tissues are renewed, and maybe damaged again, but the level of damage could not be incremental (within certain limits) for so much dilated periods, especially when more often than not, exposure to triggering factors (such as diet) tends to decrease with time.
4. Changes in cell or tissue function may be the consequence of genetic, epigenetic, or developmental changes. It seems improbable that genetic design may be responsible for delayed modification of organ function out of line with the vital function plans. The epigenetic modifications, however, may play a more plausible role, in association with developmental changes [or, in our case, developmental regression to conditions found only during lactation (Alemany, 2011a)]. There is, simply, no information available on these issues. However, it is improbable that the changes would be established not to improve –or at least maintain– the basal original metabolic and regulatory conditions, but to deeply alter these in a way that plays havoc with energy homeostasis and severely endangers survival. This is not the way biological change works, and in fact tends to revert the direction (adaptability) of most inheritable epigenetic changes (Kaati et al., 2007; Lamm and Jablonka, 2008; Giraudo et al., 2010).
5. There may be an exhaustion of regulatory or control systems due to abusive extended exploitation of their capabilities at the limit of their possibilities, i.e., because of an excessive overload of nutrients. This theory has been used to explain the loss of control (and number) of pancreatic beta cells after massive secretion of insulin for years (Prentki and Nolan, 2006). This may be a possibility, but in most metabolic syndrome cases, there is a marked overproduction of insulin, which results in hyperinsulinemia, ineffective because of insulin resistance (Prager et al., 1987; Shanik et al., 2008). In some instances, beta cells die, and then type 1 diabetes develops as final stage of type 2 diabetes (Donath et al., 2005). The possibility of exhaustion or loss of function can be applied only to very special cells, such as neurons (and, perhaps, pancreatic beta cells; Prentki and Nolan, 2006; Ayarapeytanz et al., 2007), since most cells are subjected to turnover, and the body usually retains efficient mechanisms to compensate for deficits or excesses [e.g., muscle mass development/loss depending on use (van der Heijden et al., 2010), brown adipose tissue mass depending on cold exposure (Bukowiecki et al., 1982), or WAT masses, that are enlarged or shrunk depending on the availability of energy (Morkeberg et al., 1992)].
Schedules for Development of the Metabolic Syndrome in Adults
The dilated timing of development of most cases of the metabolic syndrome in adults fits fairly well with the patterns of change, over the long-term, of androgens and estrogens (as well as of the postulated estrone-derived ponderostat signal), and are far away from the rapid change associated with peptide hormones, catecholamines, and most metabolic regulative signals. Steroid hormones are also synthesized in the brain, and freely cross the blood–brain barrier (Pardridge and Mietus, 1979; Baulieu and Robel, 1990), their circulating half-lives are longer than those of peptide or protein hormones and have no difficulties in crossing through or interacting with the cell membranes (Rao, 1981; Oren et al., 2004); they may even have membrane-bound receptors (Nemere and Farach-Carson, 1998; Jain et al., 2005) or binding proteins (Fortunati et al., 1996).
A plausible way to explain the delay in the appearance in full regalia of the metabolic syndrome in adults is to consider the brain responsible for the timing and progressive alterations that characterize its appearance. This theory is based mainly in the possibility to change (or misadjust) the ponderostat setting by a number of (largely unknown) conditions, such as a psychiatric disorder (Tuthill et al., 2006), iatrogenic effects of some drugs [Schwartz et al., 2004; Virk et al., 2004; or environmental poisons (Irigaray et al., 2006)], hypercortisolism (Peeke and Chrousos, 1995; Björntorp and Rosmond, 2000), long time exposure to fattening food (nil for some people, very fast for others; Seidell, 1998; Blundell et al., 2005), coincidence of a combination of alleles from a long list of genes (Borecki et al., 1998; Swarbrick and Vaisse, 2003), in utero conditioning (Beall et al., 2004; DeSai et al., 2005), epigenetic inheritance (Whitaker et al., 2010), triggering of thrifty genes (Romao and Roth, 2008), etc. The ponderostat setting also changes along our lives, so that mature men tend to present wider girths than younger adults (Yoshinaga et al., 2002; Alley and Chang, 2010). These long-term cycles are essentially governed by the brain, which ultimately favors the secretion of androgens and estrogens (but also the stress-immunity hormones, glucocorticoids; Mauras et al., 1996; Demerath et al., 1999; Matchock et al., 2007).
The list of factors potentially modifying body fat set-point is longer: estrone (Ferrer-Lorente et al., 2005; and the ponderostat signal derived from estrone; Vilà et al., 2011) dehydroepiandrosterone (Tchernof and Labrie, 2004), progesterone, aldosterone. In addition, the possibilities of regulation include retinoids (Kriegsfeld and Silver, 2004), hydroxycalciferols (Wortsman et al., 2000), hormone-transporting globulins (Barat et al., 2005; Li et al., 2010), and especially, hormone receptors (Mauvais-Jarvis, 2011).
The Roles of Steroid Hormones
Androgens are closely related to insulin (or IGF-1; Sheffield-Moore, 2000) and growth hormone (Mauras, 2006) as anabolic protein-deposing agents (Griggs et al., 1989). Estrogens are antiinflammatory (Schaefer et al., 2005; Straub, 2007), antioxidant protective agents (Prokai et al., 2005; Reyes et al., 2006). And glucocorticoids are defense agents (and thus faster-acting; Orchinik et al., 1994) which prevent and correct damages (Hafezi-Moghadam et al., 2002), and control the immune system (Franchimont, 2004; Tait et al., 2008).
The roles for androgens and estrogens go much beyond their function as sexual and reproductive hormones. In the metabolic syndrome, androgens’ levels (Gooren, 2006; Gould et al., 2007; and to a lesser extent estrogens) decrease considerably (and corticosteroids increase; Duclos et al., 2005) in parallel to advancing age (Blouin et al., 2005). However, androgens decrease earlier and more markedly. This seems to be the main deviation from the pre-established normal (i.e., non-metabolic syndrome) pattern. The dependence of the pathologic manifestations of the metabolic syndrome on the secretion, availability, and function of steroid hormones can be speculatively established by:
A. Lower levels of dehydroepiandrosterone (Howard, 2007) give rise to lower levels of adrenal gland-derived androgens (Haring et al., 2009; Akishita et al., 2010); since the levels of estrogens are more or less maintained (at least until menopause in women, and even in old age in men), the drainage of androstenedione and testosterone must be even higher, causing hypoandrogenism. Glucocorticoid-induced leptin resistance (Ur et al., 1996; Zakrzewska et al., 1997) may diminish leptin effects on gonadotropin-releasing hormones (Barb et al., 2004), thus diminishing the secretion of sex hormones from testicle and ovary. In the testicle, this may result in decreased production of testosterone and, consequently, hypotestosteronemia; but in the ovary, extraovaric production of estrogen (WAT, breast; Klein et al., 1996) may partially compensate the decrease of ovaric synthesis, but at the expense of dwindling androgen levels. In addition, dehydroepiandrosterone plays a critical role in the intestinal partition of protein N (Wu, 1996) and is a powerful antiglucocorticoid (Kalimi et al., 1994). Low DHEA may alter the synthesis of intestinal carbamoyl-P (and thus the overall synthesis of urea; Marrero et al., 1991). DHEA levels are closely related with survival in adults (Barrett-Connor et al., 1986; Berr et al., 1996), a factor that gives weight to this essential purported role. In humans, dehydroepiandrosterone-sulfate is the main corticosteroid secreted to the bloodstream, and is thus the principal adrenal cortex-secreted hormone (Ebeling and Koivisto, 1994).
B. Hypoandrogenism decreases protein synthesis and turnover (Urban et al., 1995; Sheffield-Moore, 2000), thus lowering energy and amino acid needs. Low testosterone also affects insulin effectiveness, favoring insulin resistance in WAT (Corbould, 2007), but mainly decreasing overall insulin resistance associated to the metabolic syndrome (Kapoor et al., 2006; Traish et al., 2009). Hypoandrogenism results in smaller muscle mass, and consequently, in lower muscle energy needs, indirectly aggravating the problems of insulin resistance. In women, insulin or IGF-1 directly favors the synthesis of androgens in ovaries and adrenals (Nestler, 1997; Guercio et al., 2003), whilst in men, the main influence is exerted through stimulation of FSH production (Livingstone and Collison, 2002). Insulin resistance is associated to lower androgen levels (Grossmann et al., 2008; Yeap et al., 2009) in men, because of lower testicular production (Pitteloud et al., 2005), but not in post-menopausal women or those with polycystic ovary syndrome (Coviello et al., 2006; Al-Ozairi et al., 2008). The differences in androgen availability and handling may help explain some of the sex-related differences in the onset and development of the metabolic syndrome. In any case, the question is compounded by the inhibition of androgen synthesis induced by glucocorticoids (Dong et al., 2004), the increase in overall aromatase activity consequence of increased WAT mass, and the critical role of this tissue in the synthesis of estrogen (Cleland et al., 1985). Thus, obesity is directly related to decreasing androgens’ levels, aggravating the pathological situation by negative feedback.
C. Relative decrease in the availability of estrogen because of lower availability of SHBG (Heald et al., 2005; Weinberg et al., 2006), but also, in part, because of lower levels of DHEA precursor (Janson et al., 1982; Depergola et al., 1991; Turkmen et al., 2004; Howard, 2007). Low estrogen levels may reduce their protective effects on endothelial cells (Dai et al., 2004; Ling et al., 2006), nervous system cells (Pozzi et al., 2006; Suzuki et al., 2006), trauma-related inflammation (Yokoyama et al., 2003), and antiapoptotic effects (Hosoda et al., 2001; Liu et al., 2011).
D. High glucocorticoid activity negatively affects protein mass and its maintenance (Tomas et al., 1979; Löfberg et al., 2002), favoring amino acid oxidation (Block et al., 1987; England and Price, 1995). Glucocorticoids induce insulin resistance (Asensio et al., 2004; Ruzzin et al., 2005) and leptin resistance (Ur et al., 1996). They tend to counteract catecholamine effects (Mannelli et al., 1994; Lambert et al., 2010), allowing the deposition of fat in most cell types (Asensio et al., 2004).
E. Deranged maintenance of ponderostat settings caused by –or parallel to– altered secretion/detection of ponderostat signals, including sensitivity to peptides [leptin (Eikelis et al., 2007), adiponectin (Tomita et al., 2008)] and the postulated estrone-derived ponderostat signal (Vilà et al., 2011).
Globally, in the metabolic syndrome there is a decrease in all functions related to the androgen–estrogen system, with a parallel increase (by stress, counteraction, or previously established genetic design) of glucocorticoids. This is a rather sketchy explanation, which requires more data, since there is a considerable lack of information on the long-term changes of these hormones’ metabolism, the polycystic ovary syndrome being a significant exception (Kauffman et al., 2008; Guastella et al., 2010). The role of glucocorticoids in the genesis and maintenance of obesity has been widely studied (Livingstone et al., 2000; Achard et al., 2006), but seldom in relation to other types of steroid hormones.
Effects of Alterations in Steroid Hormone Equilibrium Balance
A very important environmental factor, that may play a significant role in the unbalancing of the steroid hormone equilibrium, is the massive use of steroids as drugs for the treatment of a large, diverse, and growing number of diseases, corticosteroids being a case in point (van der Laan and Meijer, 2008). Synthetic glucocorticoids are widely (albeit not always sagely) used in Medicine (Borchers et al., 2003). They elicit marked effects through classical glucocorticoid receptors (Rousseau et al., 1972), but they are not subjected to the normal (physiological) systems of control; i.e., dexamethasone may bind the glucocorticoid receptors and elicit a blockage response of corticotropin secretion (Pasquali et al., 2002), but it does not bind the corticosteroid-binding globulin (Westphal, 1969; Perogamvros et al., 2011), a key element in the control of glucocorticoids (Grasa et al., 2001; Perogamvros et al., 2011).
There is also a growing challenge in environmental antiandrogens (Hotchkiss et al., 2008), and, especially, estrogenic compounds (Shappell, 2006; Foster, 2008), found almost anywhere as contaminants, that reinforce part of the actions of estrogens (but not along the whole spectrum of their functions; Knight and Eden, 1996; Ørgaard and Jensen, 2008), and are neither subject to their physiological mechanisms of control (Mueller et al., 2004; Wang et al., 2010). These effects are more marked in some tissues (gonads, mammary glands; Cassidy, 1999; Dang, 2009), and in the induction of estrogen-dependent cancer (Ososki and Kennelly, 2003), but we do not know much about their effects on brain and on endothelial inflammation processes. Environmental estrogens may diminish the secretion of gonadotropins (McGarvey et al., 2001), depriving the body of necessary estrogens acting on a wide spectrum of places, and substituting their finely tuned function by unspecific sexual hormone-like-related tissue stimulation. Food is an important source of unnecessary or unwanted estrogen (Remesar et al., 1999; Malekinejad et al., 2006; often with a wide spectrum of functions), promoting out of tune growth and fat deposition, as is the case of animal fats (Holmes et al., 2000), isoflavones (Suetsugi et al., 2003; Choi et al., 2008), and especially, dairy products (Malekinejad et al., 2006). Estrogen is present in the milk as a growth factor that helps the suckling to better use the milk nutrients (Remesar et al., 1999), but milk was not designed to feed adults… which thus receive repeated and potentially damaging doses of dietary estrogen (Qin et al., 2004) and other growth –including diabetogenic– factors (Martin et al., 1991; Wasmuth and Kolb, 2000).
A peculiarity of steroid hormones is their considerable ability to interconvert to different molecular species, forming families of compounds that show different global activity, but also marked differences in metabolic roles, such is the case of estrone and estradiol (Miller, 1969; Myking et al., 1986; Kirilovas et al., 2007). The simplified panorama described above must be completed with the influence of agents different from the typical estradiol for estrogen, testosterone for androgen and cortisol (corticosterone in rats) for glucocorticoids. Receptor dimerization and complexity [Sonoda et al., 2008; including their presence in the cell surface (Kim et al., 2008; Levin, 2009) and/or nucleus/cytoplasm (Welshons et al., 1984; Sebastian et al., 2004)], is compounded by the existence of converting enzymes, typically aromatase (Simpson, 2000; Miller, 2006), 17βOH- (Labrie et al., 1997; Adamski and Jakob, 2001) and 11βOH-steroid dehydrogenases (Agarwal, 2003; Isomura et al., 2006). Other hormones, such as progesterone, play a particular role under specific conditions (i.e., pregnancy; Spencer and Bazer, 2002; Gellersen et al., 2009) or act as supplemental signals for the three classical lines described, largely glucocorticoid (and antiglucocorticoid) effects (Pedersen et al., 2003; Zhang et al., 2007).
Childhood Manifestation of the Metabolic Syndrome
The existence, and growing incidence (Yoshinaga et al., 2004; Reilly, 2005), of infantile and juvenile obesity, compounded by other manifestations of the metabolic syndrome (Goran and Gower, 1998; Rosenbloom, 2003; Tauman and Gozal, 2006) apparently seems to be contradictory with the delayed appearance of the metabolic syndrome in full maturity. In some cases, obesity appears fairly early, even during infancy (Yoshinaga et al., 2004). This same phenomenon can be observed in rats, weaned on high-lipid cafeteria diet (Maxwell et al., 1988), which grow faster for a time and become obese at an early age (Bayol et al., 2005). The causes of childhood obesity are considered a combination of genetic (Wardle et al., 2008) and environmental factors: sedentary behavior (Tremblay and Willms, 2003), but mainly overnutrition, with abundance of lipid (Ailhaud and Guesnet, 1997; Macé et al., 2006). The role of diet, however, is far from being fully proven (Newby, 2007). The presence of growth factors in the diet, i.e., from milk (Remesar et al., 1999) or meats (Meyer, 2001) is a significant factor contributing to the early manifestation of obesity. Childhood obesity is parallel to serious disturbances of the endocrine system, including advanced menarche in girls (Freedman et al., 2003), hypogonadism or delayed puberty (Kaplowitz, 1998) and gynecomastia (Ersöz et al., 2002) in boys, and a generalized tendency to altered cortisol function (i.e., depression; Goodman and Whitaker, 2002; Dockray et al., 2009).
The rapid development of the metabolic syndrome in children exposed to obesogenic diets and other metabolic syndrome-inducing factors in fact is not too much different from the situation in mature adults: in both cases, androgens and estrogens have not yet achieved their adult levels of functionality (or are in frank decline in mature subjects). Both situations have in common the low levels/availability of these steroid hormones. In fact, in children there is a higher response to hormones inducing the accumulation of body fat, as is the case of estrone (Tang et al., 2001). As expected, the setting on of childhood obesity also alters the normal sexual development into adulthood (Esposito et al., 2008; Goldbeck-Wood, 2010). This condition affects boys more deeply than girls, especially when environmental or iatrogenic factors delay their gonadal development, affect their fertility and maintain low testosterone levels throughout (Rozati et al., 2002; Sikka and Wang, 2008).
The differences in the timing of metabolic syndrome manifestation in children and adults agree with low androgen and estrogen and high glucocorticoid levels in both sections of the life cycle. However, coincidence is not proof enough, and more studies on the interactive influence of sexual hormones in the development of the metabolic syndrome (or its arrest) are needed to fully sustain the hypothesis presented here. In the case of glucocorticoids, their direct implication in the development of the metabolic syndrome both in childhood and in maturity has been already proven (Björntorp and Rosmond, 2000; Wolf, 2002; Spencer and Tilbrook, 2011).
Open Questions
The hypothesis presented here does not cover all cases of manifestation or control of the metabolic syndrome, since there are a number of factors which may deeply change or modulate the presentation of the metabolic syndrome. Genetic factors may not only modulate the proclivity to favor the appearance of the metabolic syndrome (Goulart et al., 2009; Sookoian and Pirola, 2011), but may trigger responses inducing disease, such as obesity, through massive alteration of the mechanisms of control, as is the case of some genetic-based obesities such as the Prader–Willi syndrome (Cassidy, 1997). In animal models, such as the Zucker obese rats, containing the Leprfa/fa mutation (Phillips et al., 1996), we know the altered mechanism, the lack of responsiveness to leptin. Evidently, these cases could not be correlated with the hypothesis postulated here, in spite of altered glucocorticoid (Pesonen et al., 1992) and infertility observed in Zucker obese rats. In humans with the Prader–Willi syndrome there are alterations (with wide variations) in sexual maturation (Eiholzer et al., 2006), and hypothalamic steroid hormone regulation (Brandau et al., 2008). In both cases, despite their large differences in genetic cause and triggering agents, steroid hormones’ changes are not at the root of the problem, but nevertheless their function is deeply altered.
The ponderostat mechanism of control of body weight (Bradley, 1978) has been postulated as a homoeostatic system in which the brain regulated energy expenditure and energy intake in parallel to maintain the desired mass of body fat reserves (Bradley, 1978; Cabanac, 2001). This mechanism is maintained through signals from adipose tissue informing the brain of the actual fat mass. Leptin has been postulated as a possible ponderostat signal (Zhang et al., 1994); it is generally accepted that leptin participates in the control of energy balance (Gautron and Elmquist, 2011), but not in this role. Oleoyl-estrone has been also postulated as a ponderostat signal (Adán et al., 1999), and its structure and function is closely related to estrogen (Sanchis et al., 1996). Glucocorticoids inhibit its lipostatic effects (Grasa et al., 2007), and oleoyl-estrone modulates increases glucocorticoid synthesis (Romero et al., 2010) and decreases the synthesis of testosterone (Romero et al., 2009). However, recent studies have shown that oleoyl-estrone is not the active agent producing changes in ponderostat setting and metabolic changes, but a derivative of this compound, which structure has not yet been discovered but which is nonetheless related to estrogens too (Vilà et al., 2011).
The question of different intensities of manifestation of metabolic syndrome diseases (or their absence) in different individuals subjected to the same stimuli remains open. Genetic adaptive factors may play an important role, but so far they are not known, and neither the possible consequences this resistance plays on (or is played by) steroid hormones.
Conclusion
Steroid hormones are the only known medium- to long-term signals capable of eliciting procrastinated, often imperceptible, changes along time that shape our vital cycle. They are also closely controlled by the brain along pre-established patterns of change, and probably are also implied in the magnification of the problems caused by the metabolic syndrome. These effects in fact largely reflect the lifetime consequences of their own altered equilibrium.
The normal decrease in androgen and estrogen levels along with advancing age prevent these hormones to overcome the growing insult of environmental factors which elicit a immune system defense reaction countered by an increase in glucocorticoids which prevail over androgens (but also estrogens) to fully allow the metabolic syndrome and related pathologies to fully manifest. In consequence, I postulate that the timing of appearance of the metabolic syndrome-related pathologies is closely linked to parallel decreases in androgens and estrogens, and thus the syndrome fully flourished mainly under conditions of low sex hormone levels (infancy, advancing age in men, menopause in women; Figure 1).
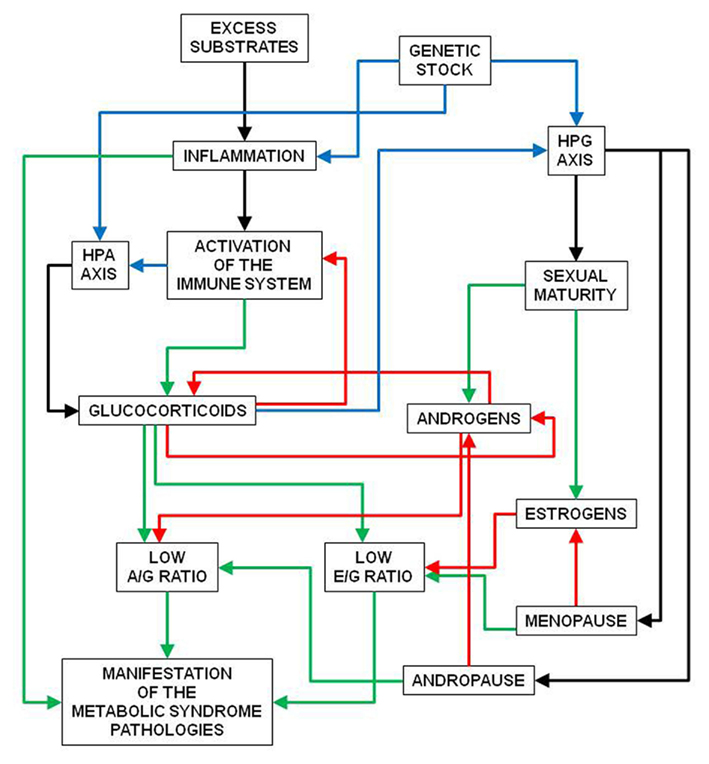
Figure 1. Interactions between glucocorticoids and sexual hormones in the regulation of the manifestation of the metabolic syndrome-related pathologies. Excess substrate availability, genetic predisposition, and other factors induce inflammation, which triggers the manifestation of the metabolic syndrome. This is favored by low androgen/glucocorticoid activity ratio (A/G ratio) and low estrogen/glucocorticoid activity ratio (E/G ratio). When sexual maturity is attained, secretion of both androgens and estrogens raise the ratios, both because of direct effects on glucocorticoid synthesis and decreased inflammation (estrogen). Inflammation and activation of the EPA axis tends to raise glucocorticoid activity (which negatively interacts with androgen and estrogen). Low levels of sexual hormones (andropause, menopause) decrease their capacity to limit the effects of glucocorticoids, decreasing both A/G and E/G ratios, and allowing the full manifestation of the metabolic syndrome pathologies. Blue lines: variable effects; green lines: increasing or enhancing effects; red lines: decreasing or inhibiting effects; black lines: direct relationships, consequences.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work supported by grant SAF2009-11739 of the Plan Nacional de Investigación en Biomedicina of the Government of Spain.
References
Achard, V., Boullu-Ciocca, S., Desbrière, R., and Grino, M. (2006). Perinatal programming of central obesity and the metabolic syndrome: role of glucocorticoids. Metab. Syndr. Relat. Disord. 4, 129–137.
Adamski, J., and Jakob, F. J. (2001). A guide to 17b-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 171, 1–4.
Adán, C., Cabot, C., Vilà, R., Grasa, M. M., Masanés, R. M., Esteve, M., Estruch, J., Fernández-López, J. A., Remesar, X., and Alemany, M. (1999). Oleoyl-estrone treatment affects the ponderostat setting differently in lean and obese Zucker rats. Int. J. Obes. 23, 366–373.
Agarwal, A. K. (2003). Cortisol metabolism and visceral obesity: role of 11 beta-hydroxysteroid dehydrogenase type I enzyme and reduced co-factor NADPH. Endocr. Res. 29, 411–418.
Ailhaud, G., and Guesnet, P. (1997). Fatty acid composition of fats is an early determinant of childhood obesity: a short review and an opinion. Obes. Res. 5, 21–26.
Akishita, M., Fukai, S., Hashimoto, M., Kameyama, Y., Nomura, K., Nakamura, T., Ogawa, S., Iijima, K., Eto, M., and Ouchi, Y. (2010). Association of low testosterone with metabolic syndrome and its components in middle-aged Japanese men. Hypertens. Res. 33, 587–591.
Alemany, M. (2011a). Different effects of hyperlipidic diets in lactation and adulthood: growth versus the development of obesity. Reprod. Biol. Endocrinol. 9, 101.
Alemany, M. (2011b). The defense of adipose tissue against excess substrate-induced hypertrophia: immune system cell infiltration and arrested metabolic activity. J. Clin. Endocrinol. Metab. 96, 66–68.
Alley, D. E., and Chang, V. W. (2010). Metabolic syndrome and weight gain in adulthood. J. Gerontol. A Biol. Sci. Med. Sci. 65, 111–117.
Al-Ozairi, E., Michael, E., and Quinton, R. (2008). Insulin resistance causing severe postmenopausal hyperandrogenism. Int. J. Gynaecol. Obstet. 100, 280–281.
Asensio, C., Muzzin, P., and Rohner-Jeanrenaud, F. (2004). Role of glucocorticoids in the physiopathology of excessive fat deposition and insulin resistance. Int. J. Obes. 28, S45–S52.
Ayarapeytanz, M. G., Yakovlev, A. A., Levshina, I. P., Vorontsova, O. N., Stepanichev, M. Y., Onufriev, M. V., Lazareva, N. A., and Gulyaeva, N. V. (2007). Studies of mechanisms involved in neuronal cell death induced by chronic stress in rats. Neurochem. J. 1, 86–92.
Barat, P., Duclos, M., Gatta, B., Roger, P., Mormede, P., and Moisan, M. P. (2005). Corticosteroid binding globulin gene polymorphism influences cortisol driven fat distribution in obese women. Obes. Res. 13, 1485–1490.
Barb, C. R., Barrett, J. B., and Kraeling, R. R. (2004). Role of leptin in modulating the hypothalamic-pituitary axis and luteinizing hormone secretion in the prepuberal gilt. Domest. Anim. Endocrinol. 26, 201–214.
Barrett-Connor, E., Khaw, K. T., and Ten, S. S. C. (1986). A prospective study of dehydroepiandrosterone sulfate, mortality, and cardiovascular disease. N. Engl. J. Med. 315, 1519–1524.
Baulieu, E. E., and Robel, P. (1990). Neurosteroids: a new brain function? J. Steroid Biochem. Mol. Biol. 37, 395–403.
Bayol, S. A., Simbi, B. H., and Stickland, N. C. (2005). A maternal cafeteria diet during gestation and lactation promotes adiposity and impairs skeletal muscle development and metabolism in rat offspring at weaning. J. Physiol. (Lond.) 567, 951–961.
Beall, M. H., El Haddad, M., Gayle, D., DeSai, M., and Ross, M. G. (2004). Adult obesity as a consequence of in utero programming. Clin. Obstet. Gynecol. 47, 957–966.
Berr, C., Lafont, S., Debuire, B., Dartigues, J. F., and Baulieu, E. E. (1996). Relationships of dehydroepiandrosterone sulfate in the elderly with functional, psychological, and mental status, and short-term mortality: a French community-based study. Proc. Natl. Acad. Sci. U.S.A. 93, 13410–13415.
Bleich, S. N., Ku, R., and Wang, Y. C. (2011). Relative contribution of energy intake and energy expenditure to childhood obesity: a review of the literature and directions for future research. Int. J. Obes. 35, 1–15.
Block, K. P., Richmond, W. B., Mehard, W. B., and Buse, M. G. (1987). Glucocorticoid-mediated activation of muscle branched-chain alpha-keto acid dehydrogenase in vivo. Am. J. Physiol. 252, E396–E407.
Blouin, K., Despres, J. P., Couillard, C., Tremblay, A., Prud’Homme, D., Bouchard, C., and Tchernof, A. (2005). Contribution of age and declining androgen levels to features of the metabolic syndrome in men. Metab. Clin. Exp. 54, 1034–1040.
Blundell, J. E., Stubbs, R. J., Golding, C., Croden, F., Alam, R., Whybrow, S., Le Noury, J., and Lawton, C. L. (2005). Resistance and susceptibility to weight gain: individual variability in response to a high-fat diet. Physiol. Behav. 86, 614–622.
Borchers, A. T., Keen, C. L., and Gershwin, M. E. (2003). Use and misuse of corticosteroids. Compr. Ther. 29, 157–165.
Borecki, I. B., Blangero, J., Rice, T., Pérusse, L., Bouchard, C., and Rao, D. C. (1998). Evidence for at least two major loci influencing human fatness. Am. J. Hum. Genet. 63, 831–838.
Bradley, P. (1978). The ponderostat and a physiological model of obesity. Am. J. Clin. Nutr. 31, 1976–1978.
Brandau, D. T., Theodoro, M., Garg, U., and Butler, M. G. (2008). Follicle stimulating and luteinizing hormones, estradiol and testosterone in Prader-Willi syndrome. Am. J. Med. Genet. A 146A, 665–669.
Bruce, K. D., and Byrne, C. D. (2009). The metabolic syndrome: common origins of a multifactorial disorder. Postgrad. Med. J. 85, 614–621.
Bua, J., Olsen, L. W., and Sørensen, T. I. A. (2007). Secular trends in childhood obesity in Denmark during 50 years in relation to economic growth. Obesity (Silver Spring). 15, 977–985.
Bukowiecki, L., Collet, A. J., Follea, N., Guay, G., and Jahjah, L. (1982). Brown adipose tissue hyperplasia: a fundamental mechanism of adaptation to cold and hyperphagia. Am. J. Physiol. 242, E353–E359.
Butte, N. F. (2009). Impact of infant feeding practices on childhood obesity. J. Nutr. 139, 412S–416S.
Cancello, R., and Clément, K. (2006). Is obesity an inflammatory illness? Role of low-grade inflammation and macrophage infiltration in human white adipose tissue. Br. J. Obstet. Gynaecol. 113, 1141–1147.
Cassidy, A. (1999). Potential tissue selectivity of dietary phytoestrogens and estrogens. Curr. Opin. Lipidol. 10, 47–52.
Choi, S. Y., Ha, T. Y., Ahn, J. Y., Kim, S. R., Kang, K. S., Hwang, I. K., and Kim, S. (2008). Estrogenic activities of isoflavones and flavones and their structure-activity relationships. Planta Med. 74, 25–32.
Cicco, G., and Pirrelli, A. (1999). Red blood cell (RBC) deformability RBC aggregability and tissue oxygenation in hypertension. Clin. Hemorheol. Microcirc. 21, 169–177.
Cleland, W. H., Simpson, E. R., and Mendelson, C. R. (1985). Effects of aging and obesity on aromatase activity of human adipose cells. J. Clin. Endocrinol. Metab. 60, 174–177.
Corbould, A. (2007). Chronic testosterone treatment induces selective insulin resistance in subcutaneous adipocytes of women. J. Endocrinol. 192, 585–594.
Coviello, A. D., Legro, R. S., and Dunaif, A. (2006). Adolescent girls with polycystic ovary syndrome have an increased risk of the metabolic syndrome associated with increasing androgen levels independent of obesity and insulin resistance. J. Clin. Endocrinol. Metab. 91, 492–497.
Dai, Z., Zhu, H. Q., Jiang, D. J., Jiang, J. L., Deng, H. W., and Li, Y. J. (2004). 17b-Estradiol preserves endothelial function by reduction of the endogenous nitric oxide synthase inhibitor level. Int. J. Cardiol. 96, 223–227.
Dang, Z. C. (2009). Dose-dependent effects of soy phyto-oestrogen genistein on adipocytes: mechanisms of action. Obes. Rev. 10, 342–349.
Demerath, E. W., Towne, B., Wisemandle, W., Blangero, J., Chumlea, W. C., and Siervogel, R. M. (1999). Serum leptin concentration, body composition, and gonadal hormones during puberty. Int. J. Obes. 23, 678–685.
Depergola, G., Giagulli, V. A., Garruti, G., Cospite, M. R., Giorgino, F., Cignarelli, M., and Giorgino, R. (1991). Low dehydroepiandrosterone circulating levels in premenopausal obese women with very high body mass index. Metab. Clin. Exp. 40, 187–190.
DeSai, M., Gayle, D., Babu, J., and Ross, M. G. (2005). Programmed obesity in intrauterine growth-restricted newborns: modulation by newborn nutrition. Am. J. Physiol. 288, R91–R96.
Dockray, S., Susman, E. J., and Dorn, L. D. (2009). Depression, cortisol reactivity, and obesity in childhood and adolescence. J. Adolesc. Health 45, 344–350.
Donath, M. Y., Ehses, J. A., Maedler, K., Schumann, D. M., Ellingsgaard, H., Eppler, E., and Reinecke, M. (2005). Mechanisms of B-cell death in type 2 diabetes. Diabetes 54, S108–S113.
Dong, Q., Salva, A., Sottas, C. M., Niu, E. M., Holmes, M., and Hardy, M. R. (2004). Rapid glucocorticoid mediation of suppressed testosterone biosynthesis in male mice subjected to immobilization stress. J. Androl. 25, 973–981.
Dubose, K. D., Stewart, E. E., Charbonneau, S. R., Mayo, M. S., and Donnelly, J. E. (2006). Prevalence of the metabolic syndrome in elementary school children. Acta Paediatr. 95, 1005–1011.
Duclos, M., Pereira, P. M., Barat, P., Gatta, B., and Roger, P. (2005). Increased cortisol bioavailability, abdominal obesity, and the metabolic syndrome in obese women. Obes. Res. 13, 1157–1166.
Duncan, G. E., Li, S. M., and Zhou, X. H. (2004). Prevalence and trends of a metabolic syndrome phenotype adolescents, 1999–2000. Diabetes Care 27, 2438–2443.
Ebeling, P., and Koivisto, V. A. (1994). Physiological importance of dehydroepiandrosterone. Lancet 343, 1479–1481.
Eguchi, Y., Mizuta, T., Sumida, Y., Ishibashi, E., Kitajima, Y., Isoda, H., Horie, H., Tashiro, T., Iwamoto, E., Takahashi, H., Kuwashiro, T., Soejima, S., Kawaguchi, Y., Oda, Y., Emura, S., Iwakiri, R., Ozaki, I., Eguchi, T., Ono, N., Anzai, K., Fujimoto, K., and Koizumi, S. (2011). The pathological role of visceral fat accumulation in steatosis, inflammation, and progression of nonalcoholic fatty liver disease. J. Gastroenterol. 46, 70–78.
Eiholzer, U., l’Allemand, D., Rousson, V., Schumpf, M., Gasser, T., Girard, J., Grüters, A., and Simoni, M. (2006). Hypothalamic and gonadal components of hypogonadism in boys with Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 91, 892–898.
Eikelis, N., Wiesner, G., Lambert, G., and Esler, M. (2007). Brain leptin resistance in human obesity revisited. Regul. Pept. 139, 45–51.
England, B. K., and Price, S. R. (1995). Acidosis and glucocorticoids interact to provoke muscle protein and amino acid catabolism. Blood Purif. 13, 147–152.
Ersöz, H. Ö., Önde, M. E., Terekeci, H., Kurtoglu, S., and Tor, H. (2002). Causes of gynaecomastia in young adult males and factors associated with idiopathic gynaecomastia. Int. J. Androl. 25, 312–316.
Esposito, K., Giugliano, F., Ciotola, M., de Sio, M., d’Armiento, M., and Giugliano, D. (2008). Obesity and sexual dysfunction, male and female. Int. J. Impot. Res. 20, 358–365.
Ferrer-Lorente, R., García-Pelaez, B., Gómez-Ollés, S., Fernández-López, J. A., Remesar, X., and Alemany, M. (2005). Effects of oral estrone on rat energy balance. Steroids 70, 667–672.
Fortunati, N., Fissore, F., Comba, A., Becchis, M., Catalano, M. G., Fazzari, A., Berta, L., and Frairia, R. (1996). Sex steroid-binding protein and its membrane receptor in estrogen-dependent breast cancer: biological and pathophysiological impact. Horm. Res. 45, 202–206.
Foster, W. G. (2008). Environmental estrogens and endocrine disruption: importance of comparative endocrinology. Endocrinology 149, 4267–4268.
Franchimont, D. (2004). Overview of the actions of glucocorticoids on the immune response. A good model to characterize new pathways of immunosuppression for new treatment strategies. Ann. N. Y. Acad. Sci. 1024, 124–137.
Freedman, D. S., Khan, L. K., Serdula, M. K., Dietz, W. H., Srinivasan, S. R., and Berenson, G. S. (2003). The relation of menarcheal age to obesity in childhood and adulthood: the Bogalusa heart study. BMC Pediatr. 3, 3. doi:10.1186/1471-2431-3-3
Gautron, L., and Elmquist, J. K. (2011). Sixteen years and counting: an update on leptin in energy balance. J. Clin. Invest. 121, 2087–2093.
Gellersen, B., Fernandes, M. S., and Brosens, J. J. (2009). Non-genomic progesterone actions in female reproduction. Hum. Reprod. Update 15, 119–138.
Giraudo, S. Q., Della-Fera, M. A., Proctor, L., Wickwire, K., Ambati, S., and Baile, C. A. (2010). Maternal high fat feeding and gestational dietary restriction effects on offspring body weight, food intake and hypothalamic gene expression over three generations in mice. Pharmacol. Biochem. Behav. 97, 121–129.
Gluckman, P. D., and Hanson, M. A. (2008). Developmental and epigenetic pathways to obesity: an evolutionary-developmental perspective. Int. J. Obes. 32, S62–S71.
Goodman, E., and Whitaker, R. C. (2002). A prospective study of the role of depression in the development and persistence of adolescent obesity. Pediatrics 110, 497–504.
Gooren, L. (2006). Visceral obesity, the metabolic syndrome, androgens and estrogens. Aging Male 9, 75–79.
Goran, M. I., and Gower, B. A. (1998). Abdominal obesity and cardiovascular risk in children. Coron. Artery Dis. 9, 483–487.
Goulart, A. C., Rexrode, K. M., Cheng, S., Rose, L., Buring, J. E., Ridker, P. M., and Zee, R. Y. (2009). Association of genetic variants with the metabolic syndrome in 20,806 white women: the women’s health genome study. Am. Heart J. 158, 257–262.e1.
Gould, D. C., Kirby, R. S., and Amoroso, P. (2007). Hypoandrogen-metabolic syndrome: a potentially common and underdiagnosed condition in men. Int. J. Clin. Pract. 61, 341–344.
Grasa, M. M., Cabot, C., Fernández-López, J. A., Remesar, X., and Alemany, M. (2001). Modulation of corticosterone availability to white adipose tissue of lean and obese Zucker rats by corticosteroid-binding globulin. Horm. Metab. Res. 33, 407–411.
Grasa, M. M., Serrano, M., Fernández-López, J. A., and Alemany, M. (2007). Corticosterone inhibits the lipid-mobilizing effects of oleoyl-estrone in adrenalectomized rats. Endocrinology 148, 4056–4063.
Greenwood, M. R. C., Koopmans, H. S., Maggio, C. A., and Sclafani, A. (1982). Zucker fa/fa rats maintain their obese body composition 10 months after jejunoileal bypass surgery. Int. J. Obes. 6, 513–525.
Griggs, R. C., Kingston, W., Jozefowicz, R. F., Herr, B. E., Forbes, G., and Halliday, D. (1989). Effect of testosterone on muscle mass and muscle protein synthesis. J. Appl. Physiol. 66, 498–503.
Grossmann, M., Thomas, M. C., Panagiotopoulos, S., Sharpe, K., MacIsaac, R. J., Clarke, S., Zajac, J. D., and Jerums, G. (2008). Low testosterone levels are common and associated with insulin resistance in men with diabetes. J. Clin. Endocrinol. Metab. 93, 1834–1840.
Guastella, E., Longo, R. A., and Carmina, E. (2010). Clinical and endocrine characteristics of the main polycystic ovary syndrome phenotypes. Fertil. Steril. 94, 2197–2201.
Guercio, G., Rivarola, M. A., Chaler, E., Maceiras, M., and Belgorosky, A. (2003). Relationships between the growth hormone/insulin-like growth factor-1 axis, insulin sensitivity, and adrenal androgens in normal prepubertal and pubertal girls. J. Clin. Endocrinol. Metab. 88, 1389–1393.
Guinhouya, B. C., Samouda, H., Zitouni, D., Vilhelm, C., and Hubert, H. (2011). Evidence of the influence of physical activity on the metabolic syndrome and/or on insulin resistance in pediatric populations: a systematic review. Int. J. Pediatr. Obes. 6, 361–388.
Gundersen, C., Mahatmya, D., Garasky, S., and Lohman, B. (2011). Linking psychosocial stressors and childhood obesity. Obes. Rev. 12, e54–e63.
Gustafsson, P. E., Persson, M., and Hammarstrom, A. (2011). Life course origins of the metabolic syndrome in middle-aged women and men: the role of socioeconomic status and metabolic risk factors in adolescence and early adulthood. Ann. Epidemiol. 21, 103–110.
Hafezi-Moghadam, A., Simoncini, T., Yang, Z. Q., Limbourg, F. P., Plumier, J. C., Rebsamen, M. C., Hsieh, C. M., Chui, D. S., Thomas, K. L., Prorock, A. J., Laubach, V. E., Moskowitz, M. A., French, B. A., Ley, K., and Liao, J. K. (2002). Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat. Med. 8, 473–479.
Hagström, E., Arner, P., Ungerstedt, U., and Bolinder, J. (1990). Subcutaneous adipose tissue: a source of lactate production after glucose ingestion in humans. Am. J. Physiol. 258, E888–E893.
Haring, R., Völzke, H., Felix, S. B., Schipf, S., Dörr, M., Rosskopf, D., Nauck, M., Schöfl, C., and Wallaschofski, H. (2009). Prediction of metabolic syndrome by low serum testosterone levels in men results from the study of health in Pomerania. Diabetes 58, 2027–2031.
Heald, A. H., Anderson, S. G., Ivison, F., Riste, L., Laing, I., Cruickshank, J. K., and Gibson, J. M. (2005). Low sex hormone binding globulin is a potential marker for the metabolic syndrome in different ethnic groups. Exp. Clin. Endocrinol. Diabetes 113, 522–528.
Holmes, M. D., Spiegelman, D., Willett, W. C., Manson, J. A. E., Hunter, D. J., Barbieri, R. L., Colditz, G. A., and Hankinson, S. E. (2000). Dietary fat intake and endogenous sex steroid hormone levels in postmenopausal women. J. Clin. Oncol. 18, 3668–3676.
Hong, J., Stubbins, R. E., Smith, R. R., Harvey, A. E., and Núñez, N. P. (2009). Differential susceptibility to obesity between male, female and ovariectomized female mice. Nutr. J. 8, 11.
Hosoda, T., Nakajima, H., and Honjo, H. (2001). Estrogen protects neuronal cells from amyloid b-induced apoptotic cell death. Neuroreport 12, 1965–1970.
Hotchkiss, A. K., Ankley, G. T., Wilson, V. S., Hartig, P. C., Durhan, E. J., Jensen, K. M., Dalma, M., and Leon, E. G. Jr. (2008). Of mice and men (and mosquitofish): antiandrogens and androgens in the environment. Bioscience 58, 1037–1050.
Howard, J. M. (2007). Common factor of cancer and the metabolic syndrome may be low DHEA. Ann. Epidemiol. 17, 270.
Irigaray, P., Ogier, V., Jacquenet, S., Notet, V., Sibille, P., Méjean, L., Bihain, B. E., and Yen, F. T. (2006). Benzo[a]pyrene impairs b-adrenergic stimulation of adipose tissue lipolysis and causes weight gain in mice. A novel molecular mechanism of toxicity for a common food pollutant. FEBS J. 273, 1362–1372.
Isomura, Y., Mune, T., Morita, H., Suwa, T., Takada, N., Yamamoto, Y., and Takeda, J. (2006). Physiologic roles of 11b-hydroxysteroid dehydrogenase type 2 in kidney. Metab. Clin. Exp. 55, 1352–1357.
Jain, S., Li, Y. N., Kumar, A., and Sehgal, P. B. (2005). Transcriptional signaling from membrane raft-associated glucocorticoid receptor. Biochem. Biophys. Res. Commun. 336, 3–8.
Janson, P. O., Dennefors, B., and Hamberger, L. (1982). The ageing ovary. Acta Obstet. Gynecol. Scand. 61, 7–9.
Janssen, I., Powell, L. H., Crawford, S., Lasley, B., and Sutton-Tyrrell, K. (2008). Menopause and the metabolic syndrome. The study of women’s health across the nation. Arch. Intern. Med. 168, 1568–1575.
Jo, J., Gavrilova, O., Pack, S., Jou, W., Mullen, S., Sumner, E., Cushman, S. W., and Periwal, V. (2009). Hypertrophy and/or hyperplasia: dynamics of adipose tissue growth. PLoS Comp. Biol. 5, e1000324. doi:10.1371/journal.pcbi.1000324
Kaati, G., Bygren, L. O., Pembrey, M., and Sjöström, M. (2007). Transgenerational response to nutrition, early life circumstances and longevity. Eur. J. Hum. Genet. 15, 784–790.
Kalimi, M., Shafagoj, Y., Loria, R., Padgett, D., and Regelson, W. (1994). Anti-glucocorticoid effects of dehydroepiandrosterone (DHEA). Mol. Cell. Biochem. 131, 99–104.
Kaplowitz, P. (1998). Delayed puberty in obese boys: comparison with constitutional delayed puberty and response to testosterone therapy. J. Pediatr. 133, 745–749.
Kapoor, D., Goodwin, E., Channer, K. S., and Jones, T. H. (2006). Testosterone replacement therapy improves insulin resistance, glycaemic control, visceral adiposity and hypercholesterolaemia in hypogonadal men with type 2 diabetes. Eur. J. Endocrinol. 154, 899–906.
Kauffman, R. P., Baker, T. E., Baker, V. M., DiMarino, P., and Castracane, V. D. (2008). Endocrine and metabolic differences among phenotypic expressions of polycystic ovary syndrome according to the 2003 Rotterdam consensus criteria. Am. J. Obstet. Gynecol. 198, 670.e1–670.e10.
Kim, K. H., Moriarty, K., and Bender, J. R. (2008). Vascular cell signaling by membrane estrogen receptors. Steroids 73, 864–869.
Kirilovas, D., Schedvins, K., Naessén, T., von Schoultz, B., and Carlström, K. (2007). Conversion of circulating estrone sulfate to 17b-estradiol by ovarian tumor tissue: a possible mechanism behind elevated circulating concentrations of 17b-estradiol in postmenopausal women with ovarian tumors. Gynecol. Endocrinol. 23, 25–28.
Klein, S., Coppack, S. W., Mohamed-Ali, V., and Landt, M. (1996). Adipose tissue leptin production and plasma leptin kinetics in humans. Diabetes 45, 984–987.
Knight, D. C., and Eden, J. A. (1996). A review of the clinical effects of phytoestrogens. Obstet. Gynecol. 87, 897–904.
Kohen-Avramoglu, R., Theriault, A., and Adeli, K. (2003). Emergence of the metabolic syndrome in childhood: an epidemiological overview and mechanistic link to dyslipidemia. Clin. Biochem. 36, 413–420.
Kowalska, I., Straczkowski, M., Nikolajuk, A., Adamska, A., Karczewska-Kupczewska, M., Otziomek, E., Kinalska, I., and Gorska, M. (2008). Insulin resistance, serum adiponectin, and proinflammatory markers in young subjects with the metabolic syndrome. Metab. Clin. Exp. 57, 1539–1544.
Kriegsfeld, L. J., and Silver, R. (2004). The skinny on body weight regulation: the role of retinoid signaling in photoperiod-mediated weight loss. Endocrinology 145, 11–12.
Labrie, F., Luu-The, V., Lin, S. X., Labrie, C., Simard, J., Breton, R., and Bélanger, A. (1997). The key role of 17b-hydroxysteroid dehydrogenases in sex steroid biology. Steroids 62, 148–158.
Lambert, G. W., Straznicky, N. E., Lambert, E. A., Dixon, J. B., and Schlaich, M. P. (2010). Sympathetic nervous activation in obesity and the metabolic syndrome-causes, consequences and therapeutic implications. Pharmacol. Ther. 126, 159–172.
Lamm, E., and Jablonka, E. (2008). The nurture of nature: hereditary plasticity in evolution. Philos. Psychol. 21, 305–319.
Levin, E. R. (2009). Membrane oestrogen receptor a signalling to cell functions. J. Physiol. 587, 5019–5023.
Li, C. Y., Giles, W. H., Ford, E. S., Liu, S. M., and Li, B. Y. (2010). Association of testosterone and sex hormone-binding globulin with metabolic syndrome and insulin resistance in men. Diabetes Care 33, 1618–1624.
Ling, S. H., Zhou, L. M., Li, H., Dai, A. Z., Liu, J. P., Komesaroff, P. A., and Sudhir, K. (2006). Effects of 17b-estradiol on growth and apoptosis in human vascular endothelial cells: influence of mechanical strain and tumor necrosis factor-a. Steroids 71, 799–808.
Liu, H., Pedram, A., and Kim, J. K. (2011). Oestrogen prevents cardiomyocyte apoptosis by suppressing p38 alpha-mediated activation of p53 and by down-regulating p53 inhibition on p38 beta. Cardiovasc. Res. 89, 119–128.
Livingstone, C., and Collison, M. (2002). Sex steroids and insulin resistance. Clin. Sci. 102, 151–166.
Livingstone, D. E. W., Jones, G. C., Smith, K., Jamieson, P. M., Andrew, R., Kenyon, C. J., and Walker, B. R. (2000). Understanding the role of glucocorticoids in obesity: tissue-specific alterations of corticosterone metabolism in obese Zucker rats. Endocrinology 141, 560–563.
Löfberg, E., Gutierrez, A., Wernerman, J., Anderstam, B., Mitch, W. E., Price, S. R., and Walker, B. R. (2002). Effects of high dose of glucocorticoids on free amino acids, ribosomes and protein turnover in human muscle. Eur. J. Clin. Invest. 32, 345–353.
Loos, R. J. F., and Bouchard, C. (2003). Obesity – is it a genetic disorder? J. Intern. Med. 254, 401–425.
Macé, K., Shahkhalili, Y., Aprikian, O., and Stan, S. (2006). Dietary fat and fat types as early determinants of childhood obesity: a reappraisal. Int. J. Obes. 30, S50–S57.
Malekinejad, H., Scherpenisse, P., and Bergwerff, A. A. (2006). Naturally occurring estrogens in processed milk and in raw milk (from gestated cows). J. Agric. Food Chem. 54, 9785–9791.
Mannelli, M., Lanzilotti, R., Pupilli, R., Ianni, L., Conti, A., and Serio, M. (1994). Adrenal medulla secretion in Cushing’s syndrome. J. Clin. Endocrinol. Metab. 78, 1331–1335.
Marrero, M., Prough, R. A., Putnam, R. S., and Milewich, L. (1991). Inhibition of carbamoyl phosphate synthetase-I by dietary dehydroepiandrosterone. J. Steroid Biochem. Mol. Biol. 38, 599–609.
Martin, J. M., Trink, B., Daneman, D., Dosch, H. M., and Robinson, B. (1991). Milk proteins in the etiology of insulin-dependent diabetes mellitus (IDDM). Ann. Med. 23, 447–452.
Matchock, R. L., Dorn, L. D., and Susman, E. J. (2007). Diurnal and seasonal cortisol, testosterone, and DHEA rhythms in boys and girls during puberty. Chronobiol. Int. 24, 969–990.
Mauras, N. (2006). Growth hormone and testosterone: effects on whole body metabolism and skeletal muscle in adolescence. Horm. Res. 66, 42–48.
Mauras, N., Rogol, A. D., Haymond, M. W., and Veldhuis, J. D. (1996). Sex steroids, growth hormone, insulin-like growth factor-1: neuroendocrine and metabolic regulation in puberty. Horm. Res. 45, 74–80.
Maury, E., and Brichard, S. M. (2010). Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol. Cell. Endocrinol. 314, 1–16.
Mauvais-Jarvis, F. (2011). Estrogen and androgen receptors: regulators of fuel homeostasis and emerging targets for diabetes and obesity. Trends Endocrinol. Metab. 22, 24–33.
Maxwell, G. M., Fourie, F., and Bates, D. J. (1988). The effect of restricted and unrestricted “cafeteria” diets upon the energy exchange and body-composition of weanling rats. Nutr. Rep. Int. 37, 629–637.
McGarvey, C., Cates, P. S., Brooks, N. A., Swanson, I. A., Milligan, S. R., Coen, C. W., and O’Byrne, K. T. (2001). Phytoestrogens and gonadotropin-releasing hormone pulse generator activity and pituitary luteinizing hormone release in the rat. Endocrinology 142, 1202–1208.
Mendoza, J. A., Drewnowski, A., and Christakis, D. A. (2007). Dietary energy density is associated with obesity and the metabolic syndrome in U.S. adults. Diabetes Care 30, 974–979.
Meyer, H. H. D. (2001). Biochemistry and physiology of anabolic hormones used for improvement of meat production. APMIS 109, 1–8.
Miller, B. G. (1969). The relative potencies of oestriol, oestradiol and oestrone on the uterus and vagina of the mouse. J. Endocrinol. 43, 563–570.
Miller, W. R. (2006). Aromatase and the breast: regulation and clinical aspects. Maturitas 54, 335–341.
Millward, D. J., Garlick, P. J., Stewart, R. J., Nnanyelugo, D. O., and Waterlow, J. C. (1975). Skeletal muscle growth and protein turnover. Biochem. J. 150, 253–243.
Miranda, P. J., DeFronzo, R. A., Califf, R. M., and Guyton, J. R. (2005). Metabolic syndrome: definition, pathophysiology, and mechanisms. Am. Heart J. 149, 33–45.
Mitchell, J. A., Mattocks, C., Ness, A. R., Leary, S. D., Pate, R. R., Dowda, M., Blair, S. N., and Riddoch, C. (2009). Sedentary behavior and obesity in a large cohort of children. Obesity (Silver Spring). 17, 1596–1602.
Morkeberg, J. C., Sheng, H. P., and Mersmann, H. J. (1992). Differences in energy intake affect body composition and adipose tissue accretion in young growing piglets. Nutr. Res. 12, 1503–1516.
Moschonis, G., Tanagra, S., Vandorou, A., Kyriakou, A. E., Dede, V., Siatitsa, P. E., Koumpitski, A., Androutsos, O., Grammatikaki, E., Kantilafti, M., Naoumi, A., Farmaki, A. E., Siopi, A., Papadopoulou, E. Z., Voutsadaki, E., Chlouveraki, F., Maragkopoulou, K., Argyri, E., Giannopoulou, A., and Manios, Y. (2010). Social, economic and demographic correlates of overweight and obesity in primary-school children: preliminary data from the Healthy Growth Study. Public Health Nutr. 13, 1693–1700.
Mueller, S. O., Simon, S., Chae, K., Metzler, M., and Korach, K. S. (2004). Phytoestrogens and their human metabolites show distinct agonistic and antagonistic properties on estrogen receptor a (ERa) and ERb in human cells. Toxicol. Sci. 80, 14–25.
Muñoz, S., Franckhauser, S., Elias, I., Ferré, T., Hidalgo, A., Monteys, A. M., Molas, M., Cerdán, S., Pujol, A., Ruberte, J., and Bosch, F. (2010). Chronically increased glucose uptake by adipose tissue leads to lactate production and improved insulin sensitivity rather than obesity in the mouse. Diabetologia 53, 2417–2430.
Myking, O., Aakvag, A., and Ohm, O. J. (1986). Splanchnic extraction of oestrone and oestradiol and production of oestrone sulphate in man. Acta Endocrinol. 112, 442–446.
Nemere, I., and Farach-Carson, M. C. (1998). Membrane receptors for steroid hormones: a case for specific cell surface binding sites for vitamin D metabolites and estrogens. Biochem. Biophys. Res. Commun. 248, 443–449.
Newby, P. K. (2007). Are dietary intakes and eating behaviors related to childhood obesity? A comprehensive review of the evidence. J. Law Med. Ethics 35, 35–60.
Nicolson, G. L. (2007). Metabolic syndrome and mitochondrial function: molecular replacement and antioxidant supplements to prevent membrane peroxidation and restore mitochondrial function. J. Cell. Biochem. 100, 1352–1369.
Orchinik, M., Moore, F. L., and Rose, J. D. (1994). Mechanistic and functional studies of rapid corticosteroid actions. Ann. N. Y. Acad. Sci. 746, 101–114.
Oren, I., Fleishman, S. J., Kessel, A., and Ben-Tal, N. (2004). Free diffusion of steroid hormones across biomembranes: a simplex search with implicit solvent model calculations. Biophys. J. 87, 768–779.
Ørgaard, A., and Jensen, L. (2008). The effects of soy isoflavones on obesity. Exp. Biol. Med. 233, 1066–1080.
Ososki, A. L., and Kennelly, E. J. (2003). Phytoestrogens: a review of the present state of research. Phytother. Res. 17, 845–869.
Pang, C., Gao, Z. G., Yin, J., Zhang, J., Jia, W. P., and Ye, J. P. (2008). Macrophage infiltration into adipose tissue may promote angiogenesis for adipose tissue remodeling in obesity. Am. J. Physiol. 295, E313–E322.
Pardridge, W. M., and Mietus, L. J. (1979). Transport of steroid hormones through the rat blood-brain barrier. Primary role of albumin-bound hormone. J. Clin. Invest. 64, 145–154.
Parker, L., Lamont, D. W., Unwin, N., Pearce, M. S., Bennett, S. M. A., Dickinson, H. O., White, M., Mathers, J. C., Alberti, K. G., and Craft, A. W. (2003). A lifecourse study of risk for hyperinsulinaemia, dyslipidaemia and obesity (the central metabolic syndrome) at age 49-51 years. Diabet. Med. 20, 406–415.
Pasquali, R., Ambrosi, B., Armanini, D., Cavagnini, F., Uberti, E. D., Del Rio, G., de Pergola, G., Maccario, M., Mantero, F., Marugo, M., Rotella, C. M., Vettor, R., and Study Group on Obesity of the Italian Society of Endocrinology. (2002). Cortisol and ACTH response to oral dexamethasone in obesity and effects of sex, body fat distribution, and dexamethasone concentrations: a dose-response study. J. Clin. Endocrinol. Metab. 87, 166–175.
Patton, G. C., Coffey, C., Carlin, J. B., Sawyer, S. M., Williams, J., Olsson, C. A., and Wake, M. (2011). Overweight and obesity between adolescence and young adulthood: a 10-year prospective cohort study. J. Adolesc. Health 48, 275–280.
Pedersen, S. B., Kristensen, K., and Richelsen, B. (2003). Anti-glucocorticoid effects of progesterone in vivo on rat adipose tissue metabolism. Steroids 68, 543–550.
Peeke, P. M., and Chrousos, G. P. (1995). Hypercortisolism and obesity. Ann. N. Y. Acad. Sci. 771, 665–676.
Perogamvros, I., Aarons, L., Miller, A. G., Trainer, P. J., and Ray, D. W. (2011). Corticosteroid-binding globulin regulates cortisol pharmacokinetics. Clin. Endocrinol. (Oxf.) 74, 30–36.
Pesonen, U., Koulu, M., Heikinheimo, O., and Huupponen, R. (1992). The glucocorticoid antagonist mifepristone reveals abnormal regulation of the adrenocortical system in obese Zucker rats. J. Endocrinol. 132, 425–431.
Phillips, M. S., Liu, Q. Y., Hammond, H. A., Dugan, V., Hey, P. J., Caskey, C. T., and Hess, J. F. (1996). Leptin receptor missense mutation in the fatty Zucker rat. Nat. Genet. 13, 18–19.
Pitteloud, N., Hardin, M., Dwyer, A. A., Valassi, E., Yialamas, M., Elahi, D., and Hayes, F. J. (2005). Increasing insulin resistance is associated with a decrease in Leydig cell testosterone secretion in men. J. Clin. Endocrinol. Metab. 90, 2636–2641.
Pozzi, S., Benedusi, V., Maggi, A., and Vegeto, E. (2006). Estrogen action in neuroprotection and brain inflammation. Ann. N. Y. Acad. Sci. 1089, 302–323.
Prager, R., Olefsky, J. M., and Wallace, P. (1987). Hyperinsulinemia does not compensate for peripheral insulin resistance in obesity. Diabetes 36, 327–334.
Prentki, M., and Nolan, C. J. (2006). Islet B cell failure in type 2 diabetes. J. Clin. Invest. 116, 1802–1812.
Prokai, L., Prokai-Tatrai, K., Perjési, P., and Simpkins, J. W. (2005). Mechanistic insights into the direct antioxidant effects of estrogens. Drug Dev. Res. 66, 118–125.
Qin, L. Q., Wang, P. Y., Kaneko, T., Hoshi, K., and Sato, A. (2004). Estrogen: one of the risk factors in milk for prostate cancer. Med. Hypotheses 62, 133–142.
Ramsay, S. E., Whincup, P. H., Morris, R., Lennon, L., and Wannamethee, S. G. (2008). Is socioeconomic position related to the prevalence of metabolic syndrome? Influence of social class across the life course in a population-based study of older men. Diabetes Care 31, 2380–2382.
Rao, G. S. (1981). Mode of entry of steroid and thyroid hormones into cells. Mol. Cell. Endocrinol. 21, 97–108.
Ravussin, E., Lillioja, S., Knowler, W. C., Christin, L., Freymond, D., Abbott, W. G. H., Boyce, V., Howard, B. V., and Bogardus, C. (1988). Reduced rate of energy expenditure as a risk factor for body weight gain. N. Engl. J. Med. 318, 467–472.
Regitz-Zagrosek, V., Lehmkuhl, E., and Mahmoozadelh, S. (2007). Gender aspects of the role of the metabolic syndrome as a risk factor for cardiovascular disease. Gend. Med. 4, S162–S177.
Reilly, J. J. (2005). Descriptive epidemiology and health consequences of childhood obesity. Best Pract. Res. Clin. Endocrinol. Metab. 19, 327–341.
Remesar, X., Tang, V., Ferrer, E., Torregrosa, C., Virgili, J., Masanés, R. M., Fernández-López, J. A., and Alemany, M. (1999). Estrone in food: a factor influencing the development of obesity? Eur. J. Nutr. 38, 247–253.
Reyes, M. R., Sifuentes-Alvarez, A., and Lazalde, B. (2006). Estrogens are potentially the only steroids with an antioxidant role in pregnancy: in vitro evidence. Acta Obstet. Gynecol. Scand. 85, 1090–1093.
Rey-López, J. P., Vicente-Rodríguez, G., Biosca, M., and Moreno, L. A. (2008). Sedentary behaviour and obesity development in children and adolescents. Nutr. Metab. Cardiovasc. Dis. 18, 242–251.
Rizvi, A. A. (2009). Cytokine biomarkers, endothelial inflammation, and atherosclerosis in the metabolic syndrome: emerging concepts. Am. J. Med. Sci. 338, 310–318.
Romao, I., and Roth, J. (2008). Genetic and environmental interactions in obesity and type 2 diabetes. J. Am. Diet. Assoc. 108, S24–S28.
Romero, M. M., Vilà, R., Fernández-López, J. A., Esteve, M., and Alemany, M. (2009). Influence of oleoyl-estrone treatment on circulating testosterone. Role of 17b-hydroxysteroid dehydrogenase isoenzymes. J. Physiol. Pharmacol. 60, 181–190.
Romero, M. M., Vilà, R., Fernández-López, J. A., Esteve, M., and Alemany, M. (2010). Oleoyl-estrone increases adrenal corticosteroid synthesis gene expression in overweight male rats. Steroids 75, 20–26.
Rosenbloom, A. L. (2003). Obesity, insulin resistance, beta-cell autoimmunity, and the changing clinical epidemiology of childhood diabetes. Diabetes Care 26, 2954–2956.
Rousseau, G. G., Baxter, J. D., and Tomkins, G. M. (1972). Glucocorticoid receptors: relations between steroid binding and biological effects. J. Mol. Biol. 67, 99–115.
Rozati, R., Reddy, P. P., Reddanna, P., and Mujtaba, R. (2002). Role of environmental estrogens in the deterioration of male factor fertility. Fertil. Steril. 78, 1187–1194.
Ruzzin, J., Wagman, A. S., and Jensen, J. (2005). Glucocorticoid-induced insulin resistance in skeletal muscles: defects in insulin signalling and the effects of a selective glycogen synthase kinase-3 inhibitor. Diabetologia 48, 2119–2130.
Sanchis, D., Balada, F., Grasa, M. M., Virgili, J., Peinado, J., Monserrat, C., Fernández-López, J. A., Remesar, X., and Alemany, M. (1996). Oleoyl-estrone induces the loss of body fat in rats. Int. J. Obes. 20, 588–594.
Schaefer, T. M., Wright, J. A., Pioli, P. A., and Wira, C. R. (2005). IL-1b-mediated proinflammatory responses are inhibited by estradiol via down-regulation of IL-1 receptor type I in uterine epithelial cells. J. Immunol. 175, 6509–6516.
Schwartz, T. L., Nihalani, N., Jindal, S., Virk, S., and Jones, N. (2004). Psychiatric medication-induced obesity: a review. Obes. Rev. 5, 115–121.
Sclafani, A., and Springer, D. (1976). Dietary obesity in adult rats: similarities to hypothalamic and human obesity syndromes. Physiol. Behav. 17, 461–471.
Sebastian, T., Sreeja, S., and Thampan, R. V. (2004). Import and export of nuclear proteins: focus on the nucleocytoplasmic movements of two different species of mammalian estrogen receptor. Mol. Cell. Biochem. 260, 91–102.
Seidell, J. C. (1998). Dietary fat and obesity: an epidemiologic perspective. Am. J. Clin. Nutr. 67, 546S–550S.
Shanik, M. H., Xy, Y. P., Skrha, J., Dankner, R., Zick, Y., and Roth, J. (2008). Insulin resistance and hyperinsulinemia. Is hyperinsulinemia the cart or the horse? Diabetes Care 31, S262–S268.
Shappell, N. W. (2006). Estrogenic activity in the environment: municipal wastewater effluent, river, ponds, and wetlands. J. Environ. Qual. 35, 122–132.
Sheffield-Moore, M. (2000). Androgens and the control of skeletal muscle protein synthesis. Ann. Med. 32, 181–186.
Sikka, S. C., and Wang, R. (2008). Endocrine disruptors and estrogenic effects on male reproductive axis. Asian J. Androl. 10, 134–145.
Sonoda, M. T., Martinez, L., Webb, P., Skaf, M. S., and Polikarpov, I. (2008). Ligand dissociation from estrogen receptor is mediated by receptor dimerization: evidence from molecular dynamics simulations. Mol. Endocrinol. 22, 1565–1578.
Sookoian, S., and Pirola, C. J. (2011). Metabolic syndrome: from the genetics to the pathophysiology. Curr. Hypertens. Rep. 13, 149–157.
Spencer, S. J., and Tilbrook, A. (2011). The glucocorticoid contribution to obesity. Stress 14, 233–246.
Spencer, T. E., and Bazer, F. W. (2002). Biology of progesterone action during pregnancy recognition and maintenance of pregnancy. Front. Biosci. 7, 1879–1898.
Straznicky, N. E., Lambert, E. A., Nestel, P. J., McGrane, M. T., Dawood, T., Schlaich, M. P., Masuo, K., Eikelis, N., de Courten, B., Mariani, J. A., Esler, M. D., Socratous, F., Chopra, R., Sari, C. I., Paul, E., and Lambert, G. W. (2010). Sympathetic neural adaptation to hypocaloric diet with or without exercise training in obese metabolic syndrome subjects. Diabetes 59, 71–79.
Subramanian, S., Goodspeed, L., Wang, S. R., Kim, J., Zeng, L. X., Ioannou, G. N., Haigh, W. G., Yeh, M. M., Kowdley, K. V., O’Brien, K. D., Pennathur, S., and Chait, A. (2011). Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor-deficient mice. J. Lipid Res. 52, 1626–1635.
Suetsugi, M., Su, L. L., Karlsberg, K., Yuan, Y. C., and Chen, S. (2003). Flavone and isoflavone phytoestrogens are agonists of estrogen-related receptors. Mol. Cancer Res. 1, 981–991.
Suzuki, S., Brown, C. M., and Wise, P. M. (2006). Mechanisms of neuroprotection by estrogen. Endocrine 29, 209–215.
Swarbrick, M. M., and Vaisse, C. (2003). Emerging trends in the search for genetic variants predisposing to human obesity. Curr. Opin. Clin. Nutr. Metab. Care 6, 369–375.
Tait, A. S., Butts, C. L., and Sternberg, E. M. (2008). The role of glucocorticoids and progestins in inflammatory, autoimmune, and infectious disease. J. Leukoc. Biol. 84, 924–931.
Tamayo, T., Herder, C., and Rathmann, W. (2010). Impact of early psychosocial factors (childhood socioeconomic factors and adversities) on future risk of type 2 diabetes, metabolic disturbances and obesity: a systematic review. BMC Public Health 10, 525. doi:10.1186/1471-2458-10-525
Tang, V., Torregrosa, C., Remesar, X., and Alemany, M. (2001). Dietary oleoyl-estrone affects the growth rate of young rats. Eur. J. Nutr. 40, 17–22.
Tauman, R., and Gozal, D. (2006). Obesity and obstructive sleep apnea in children. Paediatr. Respir. Rev. 7, 247–259.
Tchernof, A., and Labrie, F. (2004). Dehydroepiandrosterone, obesity and cardiovascular disease risk: a review of human studies. Eur. J. Endocrinol. 151, 1–14.
Thomas, T. R., Warner, S. O., Dellsperger, K. C., Hinton, P. S., Whaley-Connell, A. T., Rector, R. S., Liu, Y., Linden, M. A., Chockalingam, A., Thyfault, J. P., Huyette, D. R., Wang, Z., and Cox, R. H. (2010). Exercise and the metabolic syndrome with weight regain. J. Appl. Physiol. 109, 3–10.
Tomas, F. M., Munro, H. N., and Young, V. R. (1979). Effect of glucocorticoid administration on the rate of muscle protein breakdown in vivo in rats, as measured by urinary excretion of Nt-methylhistidine. Biochem. J. 178, 139–146.
Tomita, K., Oike, Y., Teratani, T., Taguchi, T., Noguchi, M., Suzuki, T., Mizutani, A., Yokoyama, H., Irie, R., Sumimoto, H., Takayanagi, A., Miyashita, K., Akao, M., Tabata, M., Tamiya, G., Ohkura, T., and Hibi, T. (2008). Hepatic AdipoR2 signaling plays a protective role against progression of nonalcoholic steatohepatitis in mice. Hepatology 48, 458–473.
Traish, A. M., Saad, F., and Guay, A. (2009). The dark side of testosterone deficiency: II. Type 2 diabetes and insulin resistance. J. Androl. 30, 23–32.
Tremblay, M. S., and Willms, J. D. (2003). Is the Canadian childhood obesity epidemic related to physical inactivity? Int. J. Obes. 27, 1100–1105.
Turkmen, S., Backstrom, T., and Idil, M. (2004). Reduction of dehydroepiandrosterone sulfate synthesis in women with polycystic ovary syndrome by human menopausal gonadotropin but not purified urinary follicle stimulating hormone: a comparative pilot study. Gynecol. Endocrinol. 19, 69–78.
Tuthill, A., Slawik, H., O’Rahilly, S., and Finer, N. (2006). Psychiatric co-morbidities in patients attending specialist obesity services in the UK. Q. J. Med. 99, 317–325.
Ur, E., Grossman, A., and Després, J. P. (1996). Obesity results as a consequence of glucocorticoid induced leptin resistance. Horm. Metab. Res. 28, 744–747.
Urban, R. J., Bodenburg, Y. H., Gilkison, C., Foxworth, J., Coggan, A. R., Wolfe, R. R., and Ferrando, A. (1995). Testosterone administration to elderly men increases skeletal muscle strength and protein synthesis. Am. J. Physiol. 269269, E820–E826.
Valle, M., Martos, R., Gascón, F., Cañete, R., Zafra, M. A., and Morales, R. (2005). Low-grade systemic inflammation, hypoadiponectinemia and a high concentration of leptin are present in very young obese children, and correlate with metabolic syndrome. Diabetes Metab. 31, 55–62.
van der Heijden, G. J., Wang, Z. Y. J., Chu, Z. L., Toffolo, G., Manesso, E., Sauer, P. J. J., and Sunehag, A. L. (2010). Strength exercise improves muscle mass and hepatic insulin sensitivity in obese youth. Med. Sci. Sports Exerc. 42, 1973–1980.
van der Laan, S., and Meijer, O. C. (2008). Pharmacology of glucocorticoids: beyond receptors. Eur. J. Pharmacol. 585, 483–491.
van Itallie, T. B. (1990). The glucostatic theory 1953–1988: roots and branches. Int. J. Obes. 14, 1–10.
Varela, J. E., Hinojosa, M. W., and Nguyen, N. T. (2008). Bariatric surgery outcomes in morbidly obese with the metabolic syndrome at US academic centers. Obes. Surg. 18, 1273–1277.
Vilà, R., Cabot, C., Villarreal, L., Monegal, A., Ayet, E., Romero, M. M., Grasa Mdel, M., Esteve, M., Fernández-López, J. A., Remesar, X., and Alemany, M. (2011). Oleoyl-estrone is a precursor of an estrone-derived ponderostat signal. J. Steroid Biochem. Mol. Biol. 124, 99–111.
Virk, S., Schwartz, T. L., Jindal, S., Nihalani, N., and Jones, N. (2004). Psychiatric medication induced obesity: an aetiologic review. Obes. Rev. 5, 167–170.
Virtue, S., and Vidal-Puig, A. (2009). Adipose tissue expandability, lipotoxicity and the metabolic syndrome – an allostatic perspective. Biochim. Biophys. Acta 1801, 338–349.
Wang, B., Wood, I. S., and Trayhurn, P. (2007). Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. Pflugers Arch. 455, 479–492.
Wang, B., Wood, I. S., and Trayhurn, P. (2008). Hypoxia induces leptin gene expression and secretion in human preadipocytes: differential effects of hypoxia on adipokine expression by preadipocytes. J. Endocrinol. 198, 127–134.
Wang, H., Li, J. A., Gao, Y., Xu, Y., Pan, Y., Tsuji, I., Sun, Z. J., and Li, X. M. (2010). Xeno-oestrogens and phyto-oestrogens are alternative ligands for the androgen receptor. Asian J. Androl. 12, 535–547.
Wardle, J., Carnell, S., Haworth, C. M. A., and Plomin, R. (2008). Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am. J. Clin. Nutr. 87, 398–404.
Wasmuth, H. E., and Kolb, H. (2000). Cow’s milk and immune-mediated diabetes. Proc. Nutr. Soc. 59, 573–579.
Weinberg, M. E., Manson, J. E., Buring, J. E., Cook, N. R., Seely, E. W., Ridker, P. M., and Rexrode, K. M. (2006). Low sex hormone-binding globulin is associated with the metabolic syndrome in postmenopausal women. Metab. Clin. Exp. 55, 1473–1480.
Welshons, W. V., Lieberman, M. E., and Gorski, J. (1984). Nuclear localization of unoccupied oestrogen receptors. Nature 307, 747–749.
Westphal, U. (1969). Assay and properties of corticosteroid-binding globulin and other steroid binding proteins. Meth. Enzymol. 15, 761–796.
Whitaker, K. L., Jarvis, M. J., Beeken, R. J., Boniface, D., and Wardle, J. (2010). Comparing maternal and paternal intergenerational transmission of obesity risk in a large population-based sample. Am. J. Clin. Nutr. 91, 1560–1567.
Wortsman, J., Matsuoka, L. Y., Chen, T. C., Lu, Z. R., and Holick, M. F. (2000). Decreased bioavailability of vitamin D in obesity. Am. J. Clin. Nutr. 72, 690–693.
Wouters, K., van Gorp, P. J., Bieghs, V., Gijbels, M. J., Duimel, H., Lutjohann, D., Kerksiek, A., van Kruchten, R., Maeda, N., Staels, B., van Bilsen, M., Shiri-Sverdlov, R., and Hofker, M. H. (2008). Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology 48, 474–486.
Wu, G. Y. (1996). An important role for pentose cycle in the synthesis of citrulline and proline from glutamine in porcine enterocytes. Arch. Biochem. Biophys. 336, 224–230.
Yeap, B. B., Chubb, S. A. P., Hyde, Z., Jamrozik, K., Hankey, G. J., Flicker, L., and Norman, P. E. (2009). Lower serum testosterone is independently associated with insulin resistance in non-diabetic older men: the health in men study. Eur. J. Endocrinol. 161, 591–598.
Yokoyama, Y., Kuebler, J. F., Matsutani, T., Schwacha, M. G., Bland, K. I., and Chaudry, I. H. (2003). Mechanism of the salutary effects of 17 b-estradiol following trauma-hemorrhage: direct downregulation of Kupffer cell proinflammatory cytokine production. Cytokine 21, 91–97.
Yoshinaga, M., Shimago, A., Koriyama, C., Nomura, Y., Miyata, K., Hashiguchi, J., and Arima, K. (2004). Rapid increase in the prevalence of obesity in elementary school children. Int. J. Obes. 28, 494–499.
Yoshinaga, M., Shimago, A., Noikura, Y., Kinou, S., Ohara, T., and Miyta, K. (2002). Bodyfat percentage in girls increased steadily with age and percentile rank of body mass index. Pediatr. Int. 44, 149–152.
Yuile, C. L., Lucas, E. V., Olson, J. P., and Shapiro, A. B. (1959). Plasma protein turnover and tissue exchange. Influence of dietary protein and protein depletion. J. Exp. Med. 109, 173–186.
Zakrzewska, K. E., Cusin, I., Sainsbury, A., Rohner-Jeanrenaud, F., and Jeanrenaud, B. (1997). Glucocorticoids as counterregulatory hormones of leptin – toward an understanding of leptin resistance. Diabetes 46, 717–719.
Zhang, S. M., Jonklaas, J., and Danielsen, M. (2007). The glucocorticoid agonist activities of mifepristone (RU486) and progesterone are dependent on glucocorticoid receptor levels but not on EC50 values. Steroids 72, 600–608.
Zhang, Y., Proenca, R., Maffei, M., Barone, M., Leopold, L., and Friedman, J. M. (1994). Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425–431.
Keywords: metabolic syndrome, obesity, androgens, estrogens, glucocorticoids
Citation: Alemany M (2012) Do the interactions between glucocorticoids and sex hormones regulate the development of the metabolic syndrome? Front. Endocrin. 3:27. doi: 10.3389/fendo.2012.00027
Received: 27 December 2011; Accepted: 06 February 2012;
Published online: 27 February 2012.
Edited by:
Chun Peng, York University, CanadaReviewed by:
Suraj Unniappan, York University, CanadaKaiping Yang, University of Western Ontario, Canada
Copyright: © 2012 Alemany. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Marià Alemany, Faculty of Biology, Department of Nutrition and Food Science, University of Barcelona, Av. Diagonal, 645, 08028 Barcelona, Spain. e-mail: malemany@ub.edu