


- 1 Orthopaedic Hospital Department of Orthopaedic Surgery, Orthopaedic Hospital Research Center, David Geffen School of Medicine, University of California Los Angeles, Los Angeles, CA, USA
- 2 Department of Obstetrics and Gynecology, David Geffen School of Medicine, University of California Los Angeles, Los Angeles, CA, USA
The roles of estrogens have been best studied in the breast, breast cancers, and in the female reproductive tract. However, estrogens have important functions in almost every tissue in the body. Recent clinical trials such as the Women’s Health Initiative have highlighted both the importance of estrogens and how little we know about the molecular mechanism of estrogens in these other tissues. In this review, we illustrate the diverse functions of estrogens in the bone, adipose tissue, skin, hair, brain, skeletal muscle and cardiovascular system, and how the loss of estrogens during aging affects these tissues. Early transcriptional targets of estrogen are reviewed in each tissue. We also describe the tissue-specific effects of selective estrogen receptor modulators (SERMs) used for the treatment of breast cancers and postmenopausal symptoms.
Introduction
Estrogens play a role in almost all cells and tissues in the body (Figure 1). Many of these roles have been observed in women who have a decrease in estrogens due to menopause or gonadectomy. Other effects have been observed due to differences in gender, such as autoimmune disease prevalence in either men or women. We are only starting to understand the molecular effects of estrogens in different tissues.
Figure 1. Tissues and organs affected by estrogen function. Estrogens play an important role in almost all cells and tissues in the body with gender-independent and gender-specific.
Estrogens signal through two nuclear receptors: estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ). ERα and ERβ are both expressed in most, if not all, tissues, though usually at lower levels than those found in reproductive tissues (Zallone, 2006). ERα and ERβ can bind to DNA at specific DNA motifs termed estrogen response elements (EREs). The “classical” ERE is a 13 base pair inverted palindromic sequence – GGTCANNNTGACC. In addition, ERα can indirectly activate or repress transcription by binding to other DNA binding proteins. Furthermore, estrogen can have non-genomic effects inducing the phosphorylation of components of various signaling pathways (e.g., the MAPK pathway) or calcium regulation (reviewed in reference Levin, 2005).
The Women’s Health Initiative (WHI) was a large-scale clinical trial designed to assess the risks and benefits of the use of estrogen or estrogen plus progestin as part of hormone replacement therapy (HRT) for postmenopausal women. This trial was stopped after 5 years of data collection due to adverse risks of taking hormone therapy. It was thought that estrogen would protect women against coronary heart disease, but the opposite was observed. Furthermore, estrogen plus progestin led to an increased number of invasive breast cancers. In contrast, women taking estrogen plus progestin had a decreased number of colorectal cancers and hip fractures. This trial demonstrated the tissue-specific effects of hormones in postmenopausal women and highlighted the need for a molecular understanding of estrogens and progestins in different tissues.
Selective estrogen receptor modulators (SERMs) are ER ligands that are antagonists in some tissues, but have either partial or full agonist activity in others. Tamoxifen, a first-generation SERM, has been clinically used for decades to treat ER-positive breast cancers. Raloxifene, a second-generation SERM is approved for the prevention of breast cancer and treatment of osteoporosis. Bazedoxifene and lasofoxifene are third generation SERMs that are FDA-approved for the treatment of postmenopausal osteoporosis. Each of these SERMs has a different tissue profile with both favorable and unfavorable effects (Table 1). Each SERM leads to a different conformational change in ER after ligand binding, causing a differential recruitment of coactivators, corepressors, and other transcriptional factors.
This review will focus on the effects of estrogens and SERMs in the bone, adipose tissue, skin, hair, brain, skeletal muscle and cardiovascular system, and how the loss of estrogens during aging affects these tissues. We will highlight the recent knowledge about aging effects and their consequences in these tissues and we will discuss the underlying biological mechanisms in both humans and experimental animal models. For this review we focus on direct transcriptional targets defined as genes regulated in 24 h or less upon E2 treatment because longer hormone treatment could result in the activation of indirect targets, e.g., induced by increased differentiation. The role of estrogen receptors (ERs) in cancers (such as breast cancer and endometrial cancer) is extremely important and has been thoroughly reviewed elsewhere (for example, see Shao and Brown, 2004; Barone et al., 2010; Zwart et al., 2011).
E2 in Bone and Aging
In 1940 Fuller Albright described postmenopausal osteoporosis and proposed that it was due to estrogen deficiency (Albright et al., 1940). During menopause, as estrogen levels decrease sharply, bone mineral density also decreases. Within the first 5 years after menopause women lose a large amount of trabecular bone, followed by a slower loss for the remaining years of life in both cortical and trabecular bone (Khosla et al., 2011). Treatment of women with HRT (either estrogen alone or estrogen plus progestin) has been shown to prevent this bone loss (Rossouw et al., 2002). In 2002 WHI showed that HRT prevents bone fractures (Rossouw et al., 2002). While we have long known about the beneficial effects of estrogen in bone, the molecular mechanism for the role of estrogen in bone cells is only beginning to be unraveled.
Postmenopausal loss of estrogens can be mimicked in ovariectomized (OVX) mice and ER knockout mice; each of these models has a decrease in cortical and cancellous bone mineral density. The bone phenotypes of ERα knockout (ERαKO), ERβ knockout (ERβKO), and double knockout (ERαβKO) have been described (Windahl et al., 2002). Female and male ERα null mice have a decrease in cortical bone mineral density, whereas ERβKO mice have normal cortical and cancellous bone mineral density. The ERαβKO mice have both decreased cortical and decreased cancellous bone mineral density, suggesting that ERα and ERβ can replace each other in cancellous bone, but they also have distinct roles. Furthermore, ERα has been specifically deleted in osteoclasts using a cathepsin K-cre (Nakamura et al., 2007) and a lysozyme M-cre (Martin-Millan et al., 2010). Both strains of mice with osteoclast specific deletions of ERα have trabecular bone loss, highlighting the importance of ERα in osteoclasts.
Overall, 17β-estradiol (E2) is protective in bone through its effects on the immune system, stromal cells, osteoblasts, and osteoclasts. Normally, when estrogens are present, osteoclasts undergo apoptosis, shifting the balance to an overall maintenance or building of bone. To this end, E2 induces the transcription of FasL specifically in osteoblasts, to induce osteoclast apoptosis in a paracrine manner (Krum et al., 2008a). Furthermore, loss of estrogens leads to an increase in the serum cytokines IL-1, IL-6, IL-7, and TNFα. These osteoclastogenic cytokines, produced in T-cells, stromal cells, and osteoblasts, lead to an overall decrease in bone mineral density and an increase in osteoclast number (Krum et al., 2010). Estrogens also decrease osteoclastogenesis by several additional mechanisms. Estrogens increase osteoprotegerin (OPG) from osteoblasts (Bord et al., 2003), the decoy receptor for receptor activator of NF-kappa-B ligand (RANKL), an essential cytokine for osteoclastogenesis, and decrease the activity of osteoclasts by decreasing cathepsin K and tartrate-resistant acid phosphatase (TRAP; Kremer et al., 1995).
Estrogens increase bone mineral density by affecting osteoblasts. Although the reports on the role of E2 in osteoblast proliferation and differentiation are confusing (reviewed in Krum, 2011), in vivo loss of E2 leads to an increase in osteoblast apoptosis (Kousteni et al., 2002). In the hormone replete state E2 induces the anti-apoptotic protein Bcl-2 to prevent osteoblast apoptosis (Pantschenko et al., 2005). Furthermore, E2 leads to activation of the Src/Shc/ERK pathway that blocks apoptosis (Kousteni et al., 2001). E2 also increases osteoblast differentiation by up-regulating bone morphogenic protein 2 (BMP-2; Zhou et al., 2003), which has been shown to stimulate bone formation in vitro and in vivo.
In the treatment of osteoporosis both HRT and SERMs are effective. The NSABP Study of Tamoxifen and Raloxifene Trial (STAR Trial) was conducted to directly compare tamoxifen with raloxifene in a population of women at increased risk for breast cancer. One of the endpoints was fracture reduction, and both tamoxifen and raloxifene treatments resulted in similar numbers of fractures. The genes regulated by tamoxifen and raloxifene in bone cells in vivo or in vitro are unknown, as is the reason for the agonist activity of SERMs in bone. Bazedoxifene and lasofoxifene are also approved for the treatment of postmenopausal osteoporosis. In summary, estrogens are protective in bone and several SERMs are already developed to treat osteoporosis. The knowledge of the molecular mechanism for the role of estrogen in bone may contribute to the development of an optimal treatment for women with a SERM to minimize the negative side effects of existing SERMs.
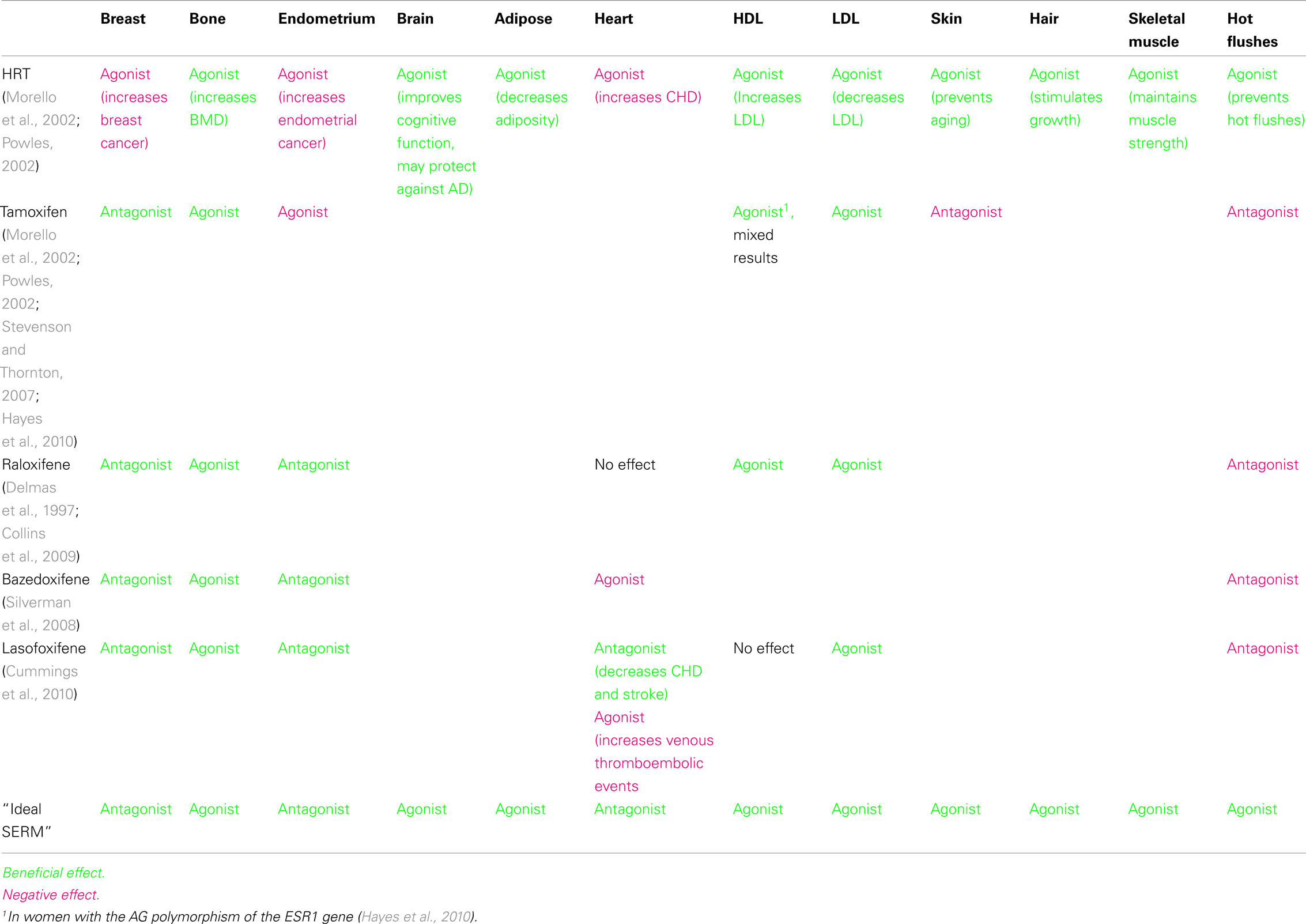
Table 1. Effects of selective estrogen receptor modulators in different tissues.
E2 in Adipose Tissue and Aging
Adipose tissue distribution is dimorphic in humans (Cooke and Naaz, 2004). Compared to men, women have more subcutaneous fat, which develops in puberty and might be therefore preferentially promoted by estrogens (Cooke and Naaz, 2004). Interestingly, the increase of abdominal fat seems to be inhibited by estrogens in premenopausal women contrary to men who are prone to depot abdominal fat (Cooke and Naaz, 2004). Though, when E2 levels decline in postmenopausal women abdominal fat increases (Cooke and Naaz, 2004). The postmenopausal status is therefore typically associated with a gain in body weight, especially abdominal obesity, which increases the risk of developing a metabolic syndrome (reviewed in Carr, 2003; Teede et al., 2010).
In humans and rodents both ERα and ERβ are expressed in adipose tissue (Cooke and Naaz, 2004). 17β-estradiol (E2) can also signal through the yet not well-characterized membrane-associated ER in adipocytes (Dos Santos et al., 2002). Direct ERα and ERβ targets in adipocytes are still unknown, although potential targets are described here. A widely used in vivo model to investigate age-related changes in female body fat mass is ovariectomy (ovx) of female rodents, which become obese. Importantly, E2 treatment can reverse the post-ovariectomy-induced weight gain (Pedersen et al., 1992). ERαKO mice are also used to study the role of E2 in fat. During the whole life span ERαKO mice develop more white adipose tissue than wildtype animals, which is in contrast to normal body weight and adipose tissue of ERβKO mice (Ohlsson et al., 2000).
In humans leptin appears to be sexually dimorphic due to higher levels in women when compared to age-matched men (Rosenbaum et al., 1996; Saad et al., 1997). Leptin/obese knockout mice show excessive fat accumulation (Zhang et al., 1994) and ovariectomy (ovx) of these mice leads to a decrease of adipose tissue (Yoneda et al., 1998; Gui et al., 2004). Interestingly, this effect was reversible by E2 administration for 24 h (Machinal et al., 1999), but not after 12 h of E2 treatment (Gui et al., 2004).
The prevalence of the metabolic syndrome is increased in postmenopausal women relative to premenopausal women (Park et al., 2003). Studies analyzing adipose tissues from postmenopausal women demonstrated an increase of markers associated with insulin sensitivity (e.g., peroxisome proliferator-activated receptor γ; fatty acid transporter), a reduced expression of markers involved in fat turnover (e.g., acetyl CoA carboxylase a, long chain acyl CoA dehydrogenase, hormone sensitive lipase), and a reduction in aromatase (CYP19A1) mRNA levels (Misso et al., 2005a,b). Genetically manipulated mice with a loss in E2 signaling induced by Cyp19a1 or ERα deficiency are characterized by increased visceral adiposity, insulin resistance and impaired glucose tolerance, which among others are features of the metabolic syndrome (Carr, 2003).
Adiponectin (ADIPOQ) belongs to the group of adipose-derived hormones that are known as adipokines. ADIPOQ may play a role in the metabolic syndrome due to its inverse relationship to high fat amounts and increased insulin resistance (Carr, 2003). Studies on the adiponectin status in humans have shown either increased or equal levels in postmenopausal women (Nishizawa et al., 2002). When E2 is depleted in rodents, Adipoq is decreased in murine adipose tissue (Gui et al., 2004; Misso et al., 2005b). However, after E2 administration for 12 h no effect on Adipoq expression was observed (Gui et al., 2004). Interestingly, studies examining Adipoq expression in adipose tissue of ovx rodents came to different results because adiponectin was shown to be either down-regulated in CD1 mice or not changed in Sprague-Dawley rats (Gui et al., 2004; Fazliana et al., 2009). Therefore, additional studies must be done to determine if ADIPOQ is a true E2 target in humans and/or rodents.
Another adipokine, resistin (RETN), may be also involved in the development of the metabolic syndrome. Although the physiological relevance of resistin in obesity and insulin resistance in humans is controversially discussed (Kusminski et al., 2005), RETN is E2-dependently regulated in adipose tissue. Studies have shown that Retn is upregulated in ovx rodents (Gui et al., 2004; Fazliana et al., 2009) and down-regulated after 24 h E2 treatment (Huang et al., 2005). However, Retn is not affected after 12 h E2 administration, which implies a time-dependent action of E2 in the regulation of Retn or potentially that Retn is not a direct ER target gene (Gui et al., 2004). Studies about adipokines and their E2-dependent role in obesity or the metabolic syndrome remain still controversial due to results from a widely used in vitro model utilizing murine fibroblasts (3T3-L1), which express endogenous ERα and ERβ and can be differentiated into adipocytes (Student et al., 1980). Chen et al. (2006) showed that during this differentiation process E2 can lead to a dose- and time-dependent increase in Retn gene expression. However, Yi et al. (2008) found contrary results because treatment of 3T3-L1-derived mature adipocytes with E2 (10−5 to 10−9 M) for 24 h neither affected adipokine gene expression (e.g., resistin, adiponectin) nor the expression of ERα and ERβ. Furthermore, it was shown that E2 suppresses the expression of Adipoq only in early-differentiated 3T3-L1 fibroblasts but not in completely differentiated cells suggesting a time-dependent E2 activity (Murase et al., 2006).
Glucocorticoids might also play an important role in adipose tissue development and the onset of the metabolic syndrome. HSD11B1 (hydroxysteroid 11-beta-dehydrogenase 1) converts inactive cortisone into its active form cortisol, whereas the other isoform HSD11B2 catalyzes the back reaction. The enzyme HSD11B1 may play a role in adipose tissue development due to its increased mRNA levels and activity in most obese humans (White et al., 2008). The interplay between E2 and HSD11B1 was also studied by Engeli et al. evaluating the expression of different HSD11B isoforms in adipose tissue of aged women. Interestingly, the expression of HDS11B1 was increased, whereas HSD11B2 levels were decreased in centrally obese menopausal women compared to lean menopausal women. These data suggest that E2 might play a role in preventing obesity and the metabolic syndrome by controlling the expression of different protein isoforms (Engeli et al., 2004). In contrast to in vivo data, a 24 h E2 treatment of primary pre-adipocytes from aged women led to an increase in HSD11B1 expression (Dieudonne et al., 2006). Furthermore, studies using mouse models suggest an important role of HSD11B1 in metabolic syndrome. Over-expression of HSD11B1 under the aP2 promoter resulted in a phenotype very similar to the metabolic syndrome (Masuzaki et al., 2001), whereas mice deficient for HSD11B1 are protected from this disease (Kotelevtsev et al., 1997; Morton et al., 2001). The discrepancies about the E2-dependent down- or up-regulation of HSD11B1 in adipose tissue remain to be resolved and may contribute to clarify its role in the development of the metabolic syndrome.
To gain more insight into the mechanisms by which E2 is involved in the regulation of insulin sensitivity, glucose metabolism, body fat accumulation, and the metabolic syndrome, Macotela et al. (2009) examined the gene expression of glucose transporters and key lipogenic markers. They found a gender specific difference in the glucose and lipid metabolism of adipocytes, which are characterized by the increased expression of Glut4 [solute carrier family 2 (facilitated glucose transporter), member 4], Glut1, Acc1 (acetyl CoA carboxylase-1), and Fasn (fatty acid synthase) in female mice (Macotela et al., 2009). Interestingly, Glut4 and Acc1 are down-regulated in ovx mice and it remains to be elucidated whether the same gene regulation can be also found in postmenopausal women that have an increased prevalence to develop a metabolic syndrome (Park et al., 2003; Macotela et al., 2009).
As in other tissues, E2 regulates both genomic and non-genomic signaling in adipocytes. E2 administration in primary rat adipocytes for as little as 5–15 min leads to MAPK pathway and cAMP response element-binding protein (CREB) phosphorylation. Moreover, in vitro studies have shown that E2 can up-regulate the CREB-dependent expression of the AP-1 protein c-fos (FBJ osteosarcoma oncogene) in addition to increasing the AP-1 binding ability of c-fos in rat white adipocytes. These observations suggest a role for E2 in regulating non-genomic MAPK signaling in addition to increasing transcription of ER targets (such as uncoupling protein 2 – ucp2) in adipose (Dos Santos et al., 2002).
Taken together, E2 has been shown to play an important age-dependent role in the development of obesity and the metabolic syndrome. However, the direct target genes of estrogen and ERs remain largely unknown, as are the mechanisms of E2 action. In the future, a tissue-specific E2 or SERM might be an appropriate basis for the development of therapeutic regimens for obesity.
E2 in Skin and Hair Follicles and Aging
The skin and their accessory appendages (e.g., hair follicles, nails, and sweat glands) are derived from ectoderm during development and form the outermost barrier of the mammalian body. The skin plays an important barrier function against pathogens and other external factors, such as UV light. The effects of E2 are particularly obvious in the skin since a huge number of epidemiological studies have provided convincing evidence that E2 inhibits skin aging by preventing skin wrinkling and promoting: skin thickness, collagen content, and skin moisture (reviewed in Thornton, 2002; Verdier-Sevrain et al., 2006). In addition, E2 has been shown to play a supportive role in skin wound healing (reviewed in Emmerson and Hardman, 2011). The first study proving an effect of E2 on skin showed an improvement of acne and eczema upon E2 treatment; oral contraceptives are still used for the treatment of severe acne (Loeser, 1937; Arowojolu et al., 2009).
The effects of E2 in skin are facilitated by the ERs ERα and ERβ, which are both expressed in keratinocytes, fibroblasts, and melanocytes (Jee et al., 1994; Hughes et al., 1997; Haczynski et al., 2002). Moreover, the skin is not only a target for sexual hormones but also produces and releases E2 by enzymatic conversion of estrogen precursors (reviewed in Emmerson and Hardman, 2011).
During UV light-induced photoaging the expression of several matrix metalloproteinases (e.g., MMP1, MMP2, MMP3, and MMP9) are upregulated leading to collagen degradation, accompanied by the down-regulation of the expression of type I and III collagens (reviewed in Baumann, 2005). Consistently, ovx rats exposed to UVB radiation show an increase in deep wrinkles, decreased skin elasticity and advanced curling of dermal fibers compared to control animals (Tsukahara et al., 2001, 2004). Interestingly, the skin-protective effect of E2 seems to depend on active transforming growth factor-β1 (TGF-β1) signals, which implies a crosstalk between these two pathways in collagen homeostasis of the skin (Son et al., 2005).
Skin thickness has been demonstrated to positively correlate with E2 levels. Although many studies suggest that fibroblasts are the main target of E2 in the skin, E2 also stimulates the proliferation of keratinocytes. This effect is mediated by two different E2-dependent mechanisms: on the one hand E2 induces the transcriptional upregulation of the cell cycle regulators cyclin D1 (CCND1) and CCND2, which promotes proliferation. On the other, E2 inhibits keratinocyte apoptosis by the induction of Bcl-2 gene expression (reviewed in Verdier-Sevrain et al., 2006).
E2 plays another important role in skin wound healing. The inflammatory response to a wound is strongly regulated by growth factors and cytokines (Shaw and Martin, 2009). Decelerated wound healing is a critical indication in postmenopausal women (Ashcroft et al., 1997). Aged females show a notably decreased expression of TGF-β1 in skin fibroblasts, which play an important role in the wound healing process. In cultured fibroblasts obtained from postmenopausal women mRNA expression levels of both ERα and ERβ were increased after E2 treatment for 24 h (Surazynski et al., 2003). Furthermore, E2 increases the expression of insulin-like growth factor 1 receptor (IGF1R), which influences the collagen biosynthesis in these cells (Surazynski et al., 2003). These data highlight the important role of estrogen in improving wound healing in the elderly skin. In addition, not only E2 but also the phytoestrogen genistein can modulate wound healing. Treatment of ovx mice with genistein inhibits, like E2, the expression of pro-inflammatory cytokines, such as interleukin-6 (IL-6) and tumor necrosis factor alpha (TNFα; Emmerson et al., 2010). Consistently, Chung et al. (2000) found the cytokine target gene and transcription factor c-jun to be upregulated in elderly Caucasian skin compared to young skin samples. Macrophage migration inhibitory factor (MIF) also contributes to the inflammatory response in skin (Shimizu, 2005). In wounded ovx wildtype mice an increase in Mif gene expression could be detected (Ashcroft et al., 2003). Interestingly, in Mif-null mice E2 depletion had no delayed effect on wound healing (Ashcroft et al., 2003). These data suggest that MIF might be a mediator of excessive inflammation in the absence of E2. Therefore, the anti-inflammatory role of E2 via down-regulation of MIF should be also considered as an important effect in other disorders of postmenopausal women, such as osteoporosis or atherosclerosis.
Hair follicles represent a unique skin appendage since they periodically regenerate new hair during the hair cycle. E2 has been shown to inhibit hair growth in many mammalian species (Thornton, 2002). In contrast, in humans E2 may have a stimulatory effect on hair growth, most probably by shortening the resting phase and prolonging the hair growth phase of the hair cycle (Sinclair et al., 1999). Accordingly, treatments of females with the E2-antagonist tamoxifen or with aromatase-inhibitors result in hair thinning or recession (reviewed in Stevenson and Thornton, 2007). Recently, a study has shown that E2 treatment for 6 h significantly upregulates the expression of PRL (prolactin) and its receptor PRLR in the human hair follicle (Langan et al., 2010). These data provide new insights into the extra-pituitary expression patterns of PRL and PRLR and show that skin and hair follicles are both target and an extra-pituitary source of PRL, which vice versa also regulates hair growth.
In summary, a number of studies have established an essential function of E2 in skin biology. Therefore, E2 may offer an appropriate treatment option to prevent or delay age-associated alterations of the skin by affecting dermal fibroblasts, epidermal keratinocytes, or hair follicles.
E2 in the Brain and Aging
Several studies with estrogen replacement therapy have shown a significant effect of estrogens on brain biology (Craig and Murphy, 2007). These data raise the question if the loss of estrogens or the decline in ER expression contributes to this phenomenon.
In animal studies an age-related reduction of ERα and ERβ expression was observed in the female brain (Wilson et al., 2002; Thakur and Sharma, 2007). Interestingly, the decline in ERs was region specific when samples from cerebral cortex and hypothalamus were analyzed (Wilson et al., 2002; Thakur and Sharma, 2007). Studies with ERαKO, ERβKO, and double mutant ERαβKO mice revealed a receptor subtype-specific action of E2 depending on the brain area (Hill and Boon, 2009). In ovariectomized rats both receptors, ERα and ERβ, are expressed with either overlapping or region specific differences (Shughrue et al., 1997; Mohamed and Abdel-Rahman, 2000; Shughrue and Merchenthaler, 2000; Rose’Meyer et al., 2003). Both ERα and ERβ are regulated in E2-depleted mice depending on the brain region (Mohamed and Abdel-Rahman, 2000; Rose’Meyer et al., 2003).
The widely used model of ovariectomy to mimic depleted E2 levels has been used to investigate E2-dependent gene expression patterns in the brain of rodents. The expression of the cytoprotective heat shock protein (Hsp) and subtypes of the neuroprotective adenosine receptor (Adora) were decreased after ovariectomy suggesting the involvement of E2 in these neuroprotective effects (Rose’Meyer et al., 2003; Hou et al., 2010).
In the pituitary gland the role of E2 was analyzed by an in vitro model that uses GH3 cells derived from a rat pituitary tumor (Tashjian et al., 1968). In these cells a decrease in steroid receptor coactivator-1 (Src-1) expression was shown after E2 treatment for 24 h, whereas nuclear receptor corepressor (NCoR) mRNA expression levels were not altered in the presence of E2 (Misiti et al., 1998). In vivo studies with male rats receiving a single s.c. injection of E2 (5 μg/kg body weight) have revealed time-dependent changes in silencing mediators of retinoid and thyroid hormone receptors (Src-1 and Smrt) in the pituitary, but no effects in NCoR or p300 expression were found (Misiti et al., 1998). These data suggest an E2-specific modulation of transcriptional co-regulators in the pituitary. However, only the expression of transforming growth factor beta isoforms was analyzed in normal pituitary and in relation to age (Hentges et al., 2000). E2-regulated genes in the normal pituitary remain unknown.
During the last three decades it became clear that many severe brain-related diseases can be linked with E2 function. The development of Alzheimer’s disease (AD) might be estrogen-specific due to the higher prevalence for women than men (Henderson, 2006). Furthermore, the incidence for AD in women increases with age with a low risk at the age of 50 (Henderson, 2006). The hypothesis that E2 replacement therapy reduces the risk of AD was confirmed in several studies (Cutter et al., 2003; Brinton, 2005; Henderson, 2006). However, these data are in contrast to the findings of the Women’s Health Initiative Memory Study (WHIMS; reviewed in Candore et al., 2006; Henderson, 2006). In this study the risk of dementia was highly increased for women that received HRT (reviewed in Candore et al., 2006; Henderson, 2006). The hippocampus, which belongs to the limbic system, is an important brain structure for memory functions and might be an interesting region to study the role of E2 in normal function and AD (Eichenbaum et al., 1992). In postmenopausal women the expression of ERα was increased in hippocampal subareas (Ishunina et al., 2007). Additionally, Ishunina et al. (2007) analyzed samples from female patients with AD that showed a down-regulation of ERα and aromatase (CYP19A1) expression in the hippocampus. Furthermore, in the human brain ERα splice variants were found that changed with both age and AD progression (McEwen, 2001; Perlman et al., 2005; Ishunina et al., 2007; Ishunina and Swaab, 2008). These results may imply that elderly women have an increased risk for AD. The increasing amounts of data for AD and related gene expression patterns are publicly available and continuously updated in the AlzGene database (http://www.alzgene.org; Bertram et al., 2007).
Apolipoprotein E (ApoE) is a major risk factor in AD due to its high correlation with ApoE-4 allele in AD patients (Corder et al., 1993; Poirier, 1996). In cerebrospinal fluid of those patients a significant reduction of ApoE was observed (Blennow et al., 1994). The underlying mechanism for the connection between ApoE and AD is still unclear. One hypothesis suggests an impaired lipid transport via ApoE in the brain of AD patients resulting in a loss of plasticity and synaptic integrity (Poirier, 1996). Importantly, several studies have demonstrated an E2-dependent increase of ApoE expression in vitro and in vivo (Stone et al., 1997). Furthermore, the expression of glial fibrillary acidic protein (GFAP), which modifies astrocyte–neuron interactions, shows an estrus-dependent variability and has been studied in more detail (Olmos et al., 1989; Naftolin et al., 1996). After 24 h E2-treatment GFAP was increased in ApoE-synthesizing astrocytes (Stone et al., 1998). These data indicate that an E2-induced increase of ApoE and GFAP may contribute to an improvement in lipid transport and cell–cell-interactions in the brain and therefore suggests a neuroprotective action of E2.
Cholinergic neurons from basal forebrain, medial septum, and nucleus basalis magnocellularis innervate areas of the cortex and hippocampus and may be involved in learning and memory, while cognitive decline in AD is accompanied by reduced choline acetyl transferase (Chat) activity and loss of these neurons (Luine et al., 1986; Price, 1986). Neurotrophins, which include brain derived nerve factor (BDNF) and nerve growth factor (NGF) are accountable for the development and maintenance of cholinergic neurons and their signal transduction occurs via binding to tyrosine kinase receptor type 1 (TRKA). In ovx rats expression of Chat and TRKA in the basal forebrain and Bdnf and Ngf in the cortical and hippocampal brain are reduced (Singh et al., 1993, 1995; McMillan et al., 1996). Interestingly, the reduced expression of Chat and TRKA in the medial septum and nucleus basalis magnocellularis was only detectable 6 months after ovariectomy, but not 3 months post-ovariectomy (Gibbs, 1998).
Major signs for the manifestation of aging and age-associated disorders like Parkinson’s and AD are the loss of neurotransmitters and their receptors leading to the reduced responsiveness to neurotransmitters (reviewed in Enna et al., 1987). Degradation of the nigrostriatal dopaminergic pathway, a discrete brain region, leads to impaired sensorimotoric control and is therefore characteristic for Parkinson’s disease (PD; Gillies and McArthur, 2010). The development of PD is associated with age and gender due to its high prevalence over the age of 65 and the twofold higher risk in men, respectively (Gillies and McArthur, 2010). Therefore, it is not surprising that data from clinical studies show parkinsonian symptoms in postmenopausal women and a decline in risk and development of PD with E2 replacement therapy (reviewed in Gillies and McArthur, 2010). Concentrations of both striatal dopamine receptors, D1 and D2, decrease with age (Roth, 1990). These changes are associated with a reduced control in motor function and may be more related to the D2 receptor (O’Boyle and Waddington, 1984; Henry et al., 1986). Mesco et al. (1991) observed a 56% reduced expression of Drd2 (dopamine receptor D2) in old rats. Roth and Joseph (1994) suggested that administration of E2 to adult male or ovx female rats will lead to an increase of striatal dopamine receptors. These results imply that E2 or a SERM may be a suitable therapeutic for the treatment of PD considering the role of estrogen in the dopaminergic system (Roth and Joseph, 1994).
The process of normal aging coincides with a growth hormone (GH) deficiency (Lamberts et al., 1997). Adults with GH deficiency have characteristic symptoms and signs, like increased abdominal fat mass, depressed mood, or decrease in bone density, which are some features similar to symptoms of postmenopausal women (Lamberts et al., 1997; Carroll et al., 1998). The expression of Gh shows sexual dimorphism in pituitary and hypothalamic sections with higher mRNA levels in male rats and a decline during aging (Takahashi et al., 1990; Martinoli et al., 1991; Iruthayanathan et al., 2005). Treatment of pituitary cells with E2 for 24 h resulted in an increase of Gh (Iruthayanathan et al., 2005). Remarkably, in the hippocampus contrary results were observed regarding Gh expression with higher levels in females than males and an increase during age (Donahue et al., 2006). The stimulation of GH secretion occurs via GH releasing hormone (Ghrh), which is produced in the hypothalamus, and expression of Ghrh is reduced in pituitary with age in female rats (Iruthayanathan et al., 2005). Interestingly, somatostatin (Sst), which acts opposing to Ghrh, is also reduced with age in female rats (Martinoli et al., 1991). These data imply that age-related and therefore E2-dependent changes of Gh expression are region specific in the brain.
Data from the Colorado Thyroid Study showed a higher prevalence for hypothyroidism in postmenopausal women (Canaris et al., 2000). Thyrotropin-releasing hormone (TRH) is produced in hypothalamus and stimulates the release of thyroid-stimulating hormone in the pituitary. TRH is inactivated by TRH-degrading enzyme (THRDE). When E2 (5 μg/kg body weight) was s.c. injected into male rats, Trhde expression was decreased in the pituitary, whereas levels of Trhr mRNA were increased after 6 h followed by normalization of gene expression after 24 h (Schomburg and Bauer, 1997). In contrast, the expression of Trhde and Trhr was not altered in the hypothalamus after E2 administration (Schomburg and Bauer, 1997). These results suggest an organ-specific effect of E2 on gene expression and show that E2 may be used as an appropriate therapeutic in age-related hypothyroidism.
Studies from Watters and Dorsa suggest a signal-crosstalk between E2 and the cAMP signaling pathway in the brain. They could show that ovariectomy leads to a reduction of neurotensin/neuromedin (NT/N) expression in the mouse brain. Interestingly, this down-regulation is rapidly normalized 8 h after i.p. administration of 50 μg E2/kg body weight (Watters and Dorsa, 1998). The fact that the neurotensin promoter contains CREB recognition sites and that a single E2 dose given to ovx rats increases the immunoreactivity of phosphorylated CREB in neurons suggests a signal-crosstalk between the estrogen and cAMP pathways (Kislauskis et al., 1988; Zhou et al., 1996). Another possible signaling cascade interacting with E2 was described by Cabilla et al. (2006). Administration of 40 μg E2/kg body weight resulted in the regulation of NO-sensitive or soluble guanylyl cyclase subunit alpha 1 (sGCα1) and sGCβ1 in ovx rats, which are receptors of nitric oxide (NO) synthase implying that E2 may be involved in the NO/sGC/cyclic GMP pathway.
Taken together, these data highlight both a region specific and dose-dependent role of estrogen in the brain. While we know many genes regulated by E2 in the brain, it is still a challenge to create the optimal treatment for postmenopausal women developing brain-related diseases like AD or PD.
E2 in Skeletal Muscle and Aging
Several studies have demonstrated that lower circulating E2 levels, but not local E2 concentrations, are associated with loss in muscle strength and muscle mass (Pollanen et al., 2011 and reviewed in Lowe et al., 2010). This decrease in muscle can be prevented by HRT in peri- and post-menopausal woman (reviewed in Lowe et al., 2010). The beneficial role of E2 for muscle strength has been also verified in mouse models, where ovariectomy leads to 10–20% weaker muscles compared to normal or E2-treated mice (Lowe et al., 2010). Increased muscle strength, in combination with improved muscle composition (i.e., decreased fat, see above), improves overall performance, including a decrease in falls in the elderly (Sipila et al., 2001; Ronkainen et al., 2009). Long-term studies in postmenopausal women have suggested that HRT, alone or in combination with power training, affects gene transcription thereby regulating energy metabolism, contraction response, and mitochondrial function of the skeletal muscle (Pollanen et al., 2010; Ronkainen et al., 2010). It is also hypothesized that estrogens decrease oxidative damage in muscles (Lowe et al., 2010) and affect myosin function (Moran et al., 2007). Furthermore, data from animal studies suggest a gender and E2-specific influence on membrane stability and exercise-induced muscle damage (Enns and Tiidus, 2010).
Both estrogen receptors, ERα and ERβ, are expressed in skeletal muscles (Lemoine et al., 2003; Wiik et al., 2003, 2009a). E2 treatment of rat skeletal myotubes for 6 h enhances the mRNA expression of ERβ but not ERα (Wiik et al., 2009b). In addition, muscle contractions induced by electrostimulation were shown to increase ERβ expression suggesting that E2 in combination with exercising could influence skeletal muscle conditions (Wiik et al., 2009a). A second study showed that after E2 treatment ERα mRNA but not ERβ levels were increased (Dieli-Conwright et al., 2009). These conflicting studies in gene regulation of ERα and ERβ suggest the need for functional studies, such as chromatin immunoprecipitation (ChIP), to determine the roles for each receptor in skeletal muscle cells.
The skeletal muscle is an important site for insulin resistance and notably ERα−/− mice show adipocyte hyperplasia and hypertrophy, glucose intolerance, and insulin resistance (Heine et al., 2000). The latter is one symptom of the metabolic syndrome for which postmenopausal women have a higher risk compared to premenopausal women (see the above section on E2 in adipose tissue and aging; Park et al., 2003). It has been demonstrated that estrogen receptors play a subtype-specific role in muscle glucose metabolism. The expression of insulin-regulated glucose transporter GLUT4 is not affected by the loss of ERβ but is extremely low in ERα knockout mice (Barros et al., 2006). These data suggest a mechanism for the insulin resistance of ERα−/− mice and illustrate the essential function of ERα for glucose homeostasis in the skeletal muscle.
A study by Dieli-Conwright and colleagues investigated the influence of E2, tamoxifen (TAM) and raloxifene (RAL) on mRNA expression in human skeletal muscle cells. The expression of important co-regulators of estrogen receptors in skeletal muscle were first examined, such as the steroid receptor co-activator (SRC-1, NCOA1) and the silencing mediator for retinoid and thyroid hormone receptors (SMRT). A significant increase in SRC-1 and decrease in SMRT expression could be detected after 24 h treatment with E2. These changes in co-regulators probably have global effects in the response to E2, but was not investigated in this cell type. This report also described the role of E2 and SERMs on key muscle genes. The expression of MyoD, a stimulator for myoblast differentiation, was increased after E2 treatment but not after incubation with TAM or RAL. The expression of the insulin-regulated glucose transporter GLUT4 was increased with E2 and RAL, but decreased with TAM treatments (Dieli-Conwright et al., 2009). These studies highlight the E2-responsive nature of skeletal muscle and the tissue-specific and gene-specific differences in SERM action.
Further studies established the role of E2 in muscle development, during which differentiation and cell growth are reciprocally exclusive events in the myogenic lineage. It was shown that E2 promotes MyoD expression and differentiation in mouse skeletal muscle and dividing myoblasts. The release of the repressing AP-1 transcription factor from the MyoD promoter was identified as the molecular mechanism (Pedraza-Alva et al., 2009). Furthermore, hypertrophy of skeletal muscles is associated with an upregulation of Four and a Half LIM domain 1 (Fhl1; Loughna et al., 2000) and E2 down-regulates the expression of Fhl1 in rat myoblasts suggesting a growth-inhibitory role (Wang et al., 2010). Kahlert et al. (1997) showed that E2 could not stimulate myoblast growth. Taken together, these data suggest a role of E2 for the myogenic lineage by favoring differentiation during muscle cell development.
Apoptosis of muscle normally occurs during development to control the number of proliferating myoblasts. The protective role of E2 in murine skeletal muscle cells has been also linked to an anti-apoptotic effect (Vasconsuelo et al., 2008). This protective action is mediated by non-genomic pathways involving PI3K/Akt. Both ERα and ERβ were shown to be necessary for the activation of AKT. Interestingly, ERβ had a greater anti-apoptotic effect at the mitochondrial level (Vasconsuelo et al., 2008; Ronda et al., 2010).
In summary, E2 effects skeletal muscle differentiation and survival and we are just beginning to understand the gene regulation by E2 in this cell type. Overall, E2 has beneficial effects on skeletal muscle and this combined with its effects on bone, leads to an overall reduction in the risk of osteoporotic fractures. The clinical effects of SERMs in skeletal muscle is only beginning to be explored (Jacobsen et al., 2008).
E2 in the Cardiovascular System and Aging
Premenopausal women have a lower incidence of cardiovascular disease than men, whereas there is no difference after menopause, suggesting that estrogens may play a role. However the WHI showed that treatment of postmenopausal women with estrogen plus progestin did not prevent cardiovascular disease. Two additional trials – the Heart and Estrogen/Progestin Replacement Study (HERS; Hulley et al., 1998) and the estrogen replacement and atherosclerosis trial (ERA; Lakoski et al., 2005) also failed to show the protective effects of HRT in the cardiovascular system. The clinical implications of these studies are controversial and it has been suggested that perhaps lower doses of hormones and/or beginning HRT near the onset of menopause rather than years afterward may reduce the risks of HRT (Rozenbaum, 2006). Alternatively, estrogens may have protective effects earlier in life and may not be able to be used to treat cardiovascular disease. In contrast, data from a huge number of animal studies have demonstrated a cardio-protective effect of E2 (reviewed in Arnal et al., 2007; Deschamps et al., 2010).
Many of the effects of estrogens in the cardiovascular system are on the lipid profile. High serum lipid levels (especially LDL) contribute to atherosclerosis. Estrogens lead to decreased LDL and increased HDL. Tamoxifen and raloxifene also decrease LDL.
ERα and ERβ are both expressed in human and rat cardiac myocytes and fibroblasts. Studies have shown cardiac abnormalities in ERαKO and ERβKO mice, such as polarization defects, increased organ size and hypertension (Johnson et al., 1997; Zhu et al., 2002; Forster et al., 2004). Polymorphisms in ERα and ERβ are associated with increased risk of cardiovascular disease only in women, further demonstrating the role of estrogen signaling in the cardiovascular system. The rs2234693 single nucleotide polymorphism (SNP) is associated with significantly increased risk of myocardial infarction (MI) among postmenopausal women (Shearman et al., 2006). This SNP is not in a coding region of the ERα protein, and it is unknown how this SNP increases MI. Methylation of the ERα promoter, and a subsequent reduction of ERα protein, is also associated with cardiovascular disease (Post et al., 1999).
E2 has a direct role in cardiac myocytes. Spontaneously hypertensive rats that underwent ovariectomy showed increased myocardial apoptosis accompanied by the upregulation of Bcl-2-associated X protein (Bax), angiotensin converting enzyme (Ace), angiotensin II type 1 receptor (At1), and brain natriuretic peptide (Bnp), which demonstrates a protective effect of E2 on stressed myocardial cells (Fabris et al., 2011). Treatment of myocardial endothelial cells with E2 resulted in upregulation of Claudin 5 (Cldn5), which is an important integral membrane tight junction protein (Burek et al., 2010). These data show that E2, in addition to protection against apoptosis, might have a protective effect on the cytoskeletal integrity of cardiovascular cells.
Lipoprotein lipase (LPL) plays an important role in heart energy metabolism and fatty acid uptake. E2 increases LPL expression in the mouse heart (Liu et al., 2008). LPL is increased after 2 h of E2 treatment, suggesting that LPL is a direct ER target. A decrease in LPL can lead to decreased cardiac function, and thus after menopause cardiovascular disease is in part regulated by LPL.
A 1 h treatment with E2 was shown to increase transcription of adenine nucleotide translocator 1 (ANT1) in the heart of ovariectomized female rats, but not in male rats (Too et al., 1999). Alterations in ANT expression have been linked to cardiac disease. Furthermore, knockout mice deficient in ANT1 showed characteristics of heart disease (Graham et al., 1997). Two other proteins that show gender specific upregulation in the heart are connexin 43 and progesterone receptor; they have an increased induction in female cardiac myocytes over that observed in male cardiac myocytes (Grohe et al., 1997). The functional consequences of E2-mediated upregulation of these two proteins in the heart are not clear.
Estrogens regulate not only the heart, but also the vasculature. ERα and ERβ are both expressed in vascular endothelial cells and vascular smooth muscle cells. E2 regulates many genes in these cell types, such as adhesion molecules and genes involved in angiogenesis, vascular remodeling, and inflammation (reviewed in Klouche, 2006). Down-regulation of these E2-regulated genes is associated with atherosclerosis and other vascular diseases.
The protective effects in the vasculature are in part regulated by an E2-mediated increase in vasodilation of atherosclerotic arteries. E2 increases endothelial nitric oxide activity (eNOS), which is not mediated by ER transcription, but a rapid ERα-dependent non-genomic effect that occurs in as little as 5 min after E2 treatment (Chen et al., 1999). Interestingly, raloxifene, but not tamoxifen, also increases eNOS activity (Simoncini and Genazzani, 2000).
Estrogen receptor beta knockout mice have sustained systolic and diastolic hypertension (Zhu et al., 2002). Furthermore, vascular smooth muscle cells from these mice show abnormalities of ion channel function. These phenotypes suggest a protective role for ERβ in the cardiovascular system via the vasculature. Through microarray analysis Otsuki et al. (2003) indentified lipocalin-type prostaglandin D synthase (L-PGDS) as a direct target of ERβ in the heart. L-PGDS acts to catalyze the isomerization of prostaglandin H2, a common precursor of various prostanoids, to produce prostaglandin D2 (PGD2). PGD2 can act as a vasodilator and anticoagulant to protect the cardiovascular system. Fifteen other E2-regulated genes in the heart were identified by this study.
In summary, estrogen targets both the vasculature and the heart. While we know many genes regulated by E2 in the cardiovascular system, the optimal treatment of humans with HRT or SERMs still must be determined.
E2 and Hot Flushes and Aging
Hot flushes are a major unfavorable symptom during menopause that has been causally linked with different organs and tissues. Data from clinical trials indicate that 40–87% of postmenopausal women had experiences with vasomotor symptoms like hot flushes (Hagstad and Janson, 1986; Freedman, 2005). Although hot flushes occur concurrently with the loss of estrogens, estrogen levels do not correlate between women experiencing hot flushes and those not experiencing hot flushes (Hutton et al., 1978). However, HRT does alleviate hot flushes (Nelson, 2004). The molecular role of estrogens in the initiation of hot flushes and the genes regulated by E2 to prevent hot flushes are unknown.
Interestingly, several datasets suggest an association of obesity and the severity of hot flushes in postmenopausal women (Chiechi et al., 1997; Wilbur et al., 1998). In adipocytes of postmenopausal women the potential vasodilator adrenomedullin (ADM) was found to be upregulated (Gupta et al., 2007; Li et al., 2007). Furthermore, another vasodilator, calcitonin-gene-related peptide (CGRP) is also regulated in adipose tissue of postmenopausal women (Gupta et al., 2007). Unfortunately, no data about the body mass index of the fat sample donors was given. These data suggest a 17β-estradiol-dependent regulation of adipokines actions, either directly or indirectly, which are possibly involved in hot flushes of postmenopausal women.
In clinical trials norepinephrine in combination with serotonin was effective in the treatment of hot flushes in postmenopausal women (Hall et al., 2011). Furthermore, studies demonstrated peripheral heat loss and vasodilation accompanied with decline in body temperature after injection of norepinephrine into preoptic hypothalamus (Freedman, 2005). The facts that norepinephrine acts in part via a2-adrenergic receptor modulated by estrogens in the brain and the central activity of norepinephrine is regulated by gonadal steroids suggest a possible combination of agents in the treatment of vasomotor symptoms in postmenopausal women (Freedman, 2005).
Because of the cardiovascular events associated with HRT, estrogens are no longer recommended for the treatment of hot flushes. No consistent results are obtained with phytoestrogens, such as soy in the treatment of hot flushes (Kronenberg and Fugh-Berman, 2002). Furthermore, SERMs such as raloxifene and tamoxifen do not alleviate hot flushes (see Table 1). Therefore, an “ideal” SERM needs to be developed to reduce the occurrence of hot flushes.
Conclusion
While estrogens and their physiological roles have been well-characterized in breast, breast cancer, and in the female reproductive tract, less is known about the direct targets of estrogens in other tissues, such as bone, adipose tissue, skin, hair, brain, skeletal muscle, and cardiovascular system. The WHI and other clinical trials and observational data have illustrated that estrogens affect many tissues throughout the body. Moreover, when we treat postmenopausal women with HRT or breast cancer patients with anti-estrogens there are many desirable and undesirable consequences. We must fully understand the molecular and cellular effects of estrogens in each of these tissues to best treat women.
We are only just beginning to understand how tissue specificity occurs on the molecular and transcriptional level. Of particular interest are findings on the functions of E2-interacting transcription factors. For instance, FoxA1 is a well-characterized “pioneer factor” for ERα in breast cancers, helping to recruit ERα to breast-specific enhancer regions (Carroll et al., 2005). However, FoxA1 is not expressed in all estrogen responsive tissues (for example it is not expressed in bone cells; Krum et al., 2008b). In addition, in osteoblasts, GATA4 is a pioneer factor for ERα and helps recruit ERα to osteoblast-specific enhancers (Miranda-Carboni et al., 2011). Yet, it is unknown how ERα is recruited to enhancers in other tissues but many other analogous examples will most likely exist and remain to be identified. Each tissue may have its own pioneer factor or there might be a small set of proteins that in different combinations determine where ERα is recruited in each cell type.
In parallel, tissue specificity is also determined in part by different coactivators expressed in each cell type. For example, the coactivator SRC-1 is necessary for agonist activity of tamoxifen in endometrial cells. In breast cancer cells SRC-1 is expressed at a lower level than in endometrial cells where tamoxifen is an ERα antagonist (Shang and Brown, 2002). Many more additional, unknown factors determine tissue specificity of the estrogen response. There are over 300 nuclear receptor coactivators and corepressors (O’Malley, 2008), suggesting an extremely complex mechanism of E2 tissue specificity and related effects during menopause and aging.
With the large number of diseases that can be potentially treated with estrogen agonists or antagonists, ideally, SERMs can be developed with a high level of target tissue specificity to best treat patients with minimal side effects (Table 1). But first we must understand the mechanism of estrogen action in each tissue.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a K12 BIRCWH grant from the NIH/ORWH (HD001400-08) and 1R56DK090231-01 to Susan A. Krum.
References
Albright, F., Bloomberg, E., and Smith, P. H. (1940). Postmenopausal osteoporosis. Trans. Assoc. Am. Physicians 55, 298–305.
Arnal, J. F., Scarabin, P. Y., Tremollieres, F., Laurell, H., and Gourdy, P. (2007). Estrogens in vascular biology and disease: where do we stand today? Curr. Opin. Lipidol. 18, 554–560.
Arowojolu, A. O., Gallo, M. F., Lopez, L. M., Grimes, D. A., and Garner, S. E. (2009). Combined oral contraceptive pills for treatment of acne. Cochrane Database Syst. Rev. 3, CD004425.
Ashcroft, G. S., Dodsworth, J., Van Boxtel, E., Tarnuzzer, R. W., Horan, M. A., Schultz, G. S., and Ferguson, M. W. (1997). Estrogen accelerates cutaneous wound healing associated with an increase in TGF-beta1 levels. Nat. Med. 3, 1209–1215.
Ashcroft, G. S., Mills, S. J., Lei, K., Gibbons, L., Jeong, M. J., Taniguchi, M., Burow, M., Horan, M. A., Wahl, S. M., and Nakayama, T. (2003). Estrogen modulates cutaneous wound healing by downregulating macrophage migration inhibitory factor. J. Clin. Invest. 111, 1309–1318.
Barone, I., Brusco, L., and Fuqua, S. A. (2010). Estrogen receptor mutations and changes in downstream gene expression and signaling. Clin. Cancer Res. 16, 2702–2708.
Barros, R. P., Machado, U. F., Warner, M., and Gustafsson, J. A. (2006). Muscle GLUT4 regulation by estrogen receptors ERbeta and ERalpha. Proc. Natl. Acad. Sci. U.S.A. 103, 1605–1608.
Baumann, L. (2005). A dermatologist’s opinion on hormone therapy and skin aging. Fertil. Steril. 84, 289–290; discussion 295.
Bertram, L., Mcqueen, M. B., Mullin, K., Blacker, D., and Tanzi, R. E. (2007). Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat. Genet. 39, 17–23.
Blennow, K., Hesse, C., and Fredman, P. (1994). Cerebrospinal fluid apolipoprotein E is reduced in Alzheimer’s disease. Neuroreport 5, 2534–2536.
Bord, S., Ireland, D. C., Beavan, S. R., and Compston, J. E. (2003). The effects of estrogen on osteoprotegerin, RANKL, and estrogen receptor expression in human osteoblasts. Bone 32, 136–141.
Brinton, R. D. (2005). Investigative models for determining hormone therapy-induced outcomes in brain: evidence in support of a healthy cell bias of estrogen action. Ann. N. Y. Acad. Sci. 1052, 57–74.
Burek, M., Arias-Loza, P. A., Roewer, N., and Forster, C. Y. (2010). Claudin-5 as a novel estrogen target in vascular endothelium. Arterioscler. Thromb. Vasc. Biol. 30, 298–304.
Cabilla, J. P., Diaz Mdel, C., Machiavelli, L. I., Poliandri, A. H., Quinteros, F. A., Lasaga, M., and Duvilanski, B. H. (2006). 17 beta-estradiol modifies nitric oxide-sensitive guanylyl cyclase expression and down-regulates its activity in rat anterior pituitary gland. Endocrinology 147, 4311–4318.
Canaris, G. J., Manowitz, N. R., Mayor, G., and Ridgway, E. C. (2000). The Colorado thyroid disease prevalence study. Arch. Intern. Med. 160, 526–534.
Candore, G., Balistreri, C. R., Grimaldi, M. P., Vasto, S., Listi, F., Chiappelli, M., Licastro, F., Lio, D., and Caruso, C. (2006). Age-related inflammatory diseases: role of genetics and gender in the pathophysiology of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1089, 472–486.
Carr, M. C. (2003). The emergence of the metabolic syndrome with menopause. J. Clin. Endocrinol. Metab. 88, 2404–2411.
Carroll, J. S., Liu, X. S., Brodsky, A. S., Li, W., Meyer, C. A., Szary, A. J., Eeckhoute, J., Shao, W., Hestermann, E. V., Geistlinger, T. R., Fox, E. A., Silver, P. A., and Brown, M. (2005). Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122, 33–43.
Carroll, P. V., Christ, E. R., Bengtsson, B. A., Carlsson, L., Christiansen, J. S., Clemmons, D., Hintz, R., Ho, K., Laron, Z., Sizonenko, P., Sonksen, P. H., Tanaka, T., and Thorne, M. (1998). Growth hormone deficiency in adulthood and the effects of growth hormone replacement: a review. Growth Hormone Research Society Scientific Committee. J. Clin. Endocrinol. Metab. 83, 382–395.
Chen, Y. H., Lee, M. J., Chang, H. H., Hung, P. F., and Kao, Y. H. (2006). 17 beta-estradiol stimulates resistin gene expression in 3T3-L1 adipocytes via the estrogen receptor, extracellularly regulated kinase, and CCAAT/enhancer binding protein-alpha pathways. Endocrinology 147, 4496–4504.
Chen, Z., Yuhanna, I. S., Galcheva-Gargova, Z., Karas, R. H., Mendelsohn, M. E., and Shaul, P. W. (1999). Estrogen receptor alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J. Clin. Invest. 103, 401–406.
Chiechi, L. M., Ferreri, R., Granieri, M., Bianco, G., Berardesca, C., and Loizzi, P. (1997). Climacteric syndrome and body-weight. Clin. Exp. Obstet. Gynecol. 24, 163–166.
Chung, J. H., Kang, S., Varani, J., Lin, J., Fisher, G. J., and Voorhees, J. J. (2000). Decreased extracellular-signal-regulated kinase and increased stress-activated MAP kinase activities in aged human skin in vivo. J. Invest. Dermatol. 115, 177–182.
Collins, P., Mosca, L., Geiger, M. J., Grady, D., Kornitzer, M., Amewou-Atisso, M. G., Effron, M. B., Dowsett, S. A., Barrett-Connor, E., and Wenger, N. K. (2009). Effects of the selective estrogen receptor modulator raloxifene on coronary outcomes in the raloxifene use for the heart trial: results of subgroup analyses by age and other factors. Circulation 119, 922–930.
Cooke, P. S., and Naaz, A. (2004). Role of estrogens in adipocyte development and function. Exp. Biol. Med. 229, 1127–1135.
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., Roses, A. D., Haines, J. L., and Pericak-Vance, M. A. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923.
Craig, M. C., and Murphy, D. G. (2007). Estrogen: effects on normal brain function and neuropsychiatric disorders. Climacteric 10(Suppl. 2), 97–104.
Cummings, S. R., Ensrud, K., Delmas, P. D., Lacroix, A. Z., Vukicevic, S., Reid, D. M., Goldstein, S., Sriram, U., Lee, A., Thompson, J., Armstrong, R. A., Thompson, D. D., Powles, T., Zanchetta, J., Kendler, D., Neven, P., and Eastell, R. (2010). Lasofoxifene in postmenopausal women with osteoporosis. N. Engl. J. Med. 362, 686–696.
Cutter, W. J., Craig, M., Norbury, R., Robertson, D. M., Whitehead, M., and Murphy, D. G. (2003). In vivo effects of estrogen on human brain. Ann. N. Y. Acad. Sci. 1007, 79–88.
Delmas, P. D., Bjarnason, N. H., Mitlak, B. H., Ravoux, A. C., Shah, A. S., Huster, W. J., Draper, M., and Christiansen, C. (1997). Effects of raloxifene on bone mineral density, serum cholesterol concentrations, and uterine endometrium in postmenopausal women. N. Engl. J. Med. 337, 1641–1647.
Deschamps, A. M., Murphy, E., and Sun, J. (2010). Estrogen receptor activation and cardioprotection in ischemia reperfusion injury. Trends Cardiovasc. Med. 20, 73–78.
Dieli-Conwright, C. M., Spektor, T. M., Rice, J. C., and Todd Schroeder, E. (2009). Oestradiol and SERM treatments influence oestrogen receptor coregulator gene expression in human skeletal muscle cells. Acta Physiol. (Oxf.) 197, 187–196.
Dieudonne, M. N., Sammari, A., Dos Santos, E., Leneveu, M. C., Giudicelli, Y., and Pecquery, R. (2006). Sex steroids and leptin regulate 11beta-hydroxysteroid dehydrogenase I and P450 aromatase expressions in human preadipocytes: sex specificities. J. Steroid Biochem. Mol. Biol. 99, 189–196.
Donahue, C. P., Kosik, K. S., and Shors, T. J. (2006). Growth hormone is produced within the hippocampus where it responds to age, sex, and stress. Proc. Natl. Acad. Sci. U.S.A. 103, 6031–6036.
Dos Santos, E. G., Dieudonne, M. N., Pecquery, R., Le Moal, V., Giudicelli, Y., and Lacasa, D. (2002). Rapid nongenomic E2 effects on p42/p44 MAPK, activator protein-1, and cAMP response element binding protein in rat white adipocytes. Endocrinology 143, 930–940.
Eichenbaum, H., Otto, T., and Cohen, N. J. (1992). The hippocampus – what does it do? Behav. Neural Biol. 57, 2–36.
Emmerson, E., Campbell, L., Ashcroft, G. S., and Hardman, M. J. (2010). The phytoestrogen genistein promotes wound healing by multiple independent mechanisms. Mol. Cell. Endocrinol. 321, 184–193.
Emmerson, E., and Hardman, M. J. (2011). The role of estrogen deficiency in skin ageing and wound healing. Biogerontology doi:10.1007/s10522-011-9322-y. [Epub ahead of print].
Engeli, S., Bohnke, J., Feldpausch, M., Gorzelniak, K., Heintze, U., Janke, J., Luft, F. C., and Sharma, A. M. (2004). Regulation of 11beta-HSD genes in human adipose tissue: influence of central obesity and weight loss. Obes. Res. 12, 9–17.
Enna, S. J., Samirajski, T., and Beer, B. (1987). Brain Neurotransmitters and Receptors in Aging and Age-related Disorders. New York: Raven Press.
Enns, D. L., and Tiidus, P. M. (2010). The influence of estrogen on skeletal muscle: sex matters. Sports Med. 40, 41–58.
Fabris, B., Candido, R., Bortoletto, M., Toffoli, B., Bernardi, S., Stebel, M., Bardelli, M., Zentilin, L., Giacca, M., and Carretta, R. (2011). Stimulation of cardiac apoptosis in ovariectomized hypertensive rats: potential role of the renin-angiotensin system. J. Hypertens. 29, 273–281.
Fazliana, M., Wan Nazaimoon, W. M., Gu, H. F., and Ostenson, C. G. (2009). Labisia pumila extract regulates body weight and adipokines in ovariectomized rats. Maturitas 62, 91–97.
Forster, C., Kietz, S., Hultenby, K., Warner, M., and Gustafsson, J. A. (2004). Characterization of the ERbeta−/− mouse heart. Proc. Natl. Acad. Sci. U.S.A. 101, 14234–14239.
Freedman, R. R. (2005). Pathophysiology and treatment of menopausal hot flashes. Semin. Reprod. Med. 23, 117–125.
Gibbs, R. B. (1998). Impairment of basal forebrain cholinergic neurons associated with aging and long-term loss of ovarian function. Exp. Neurol. 151, 289–302.
Gillies, G. E., and McArthur, S. (2010). Independent influences of sex steroids of systemic and central origin in a rat model of Parkinson’s disease: a contribution to sex-specific neuroprotection by estrogens. Horm. Behav. 57, 23–34.
Graham, B. H., Waymire, K. G., Cottrell, B., Trounce, I. A., Macgregor, G. R., and Wallace, D. C. (1997). A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat. Genet. 16, 226–234.
Grohe, C., Kahlert, S., Lobbert, K., Stimpel, M., Karas, R. H., Vetter, H., and Neyses, L. (1997). Cardiac myocytes and fibroblasts contain functional estrogen receptors. FEBS Lett. 416, 107–112.
Gui, Y., Silha, J. V., and Murphy, L. J. (2004). Sexual dimorphism and regulation of resistin, adiponectin, and leptin expression in the mouse. Obes. Res. 12, 1481–1491.
Gupta, P., Harte, A. L., Da Silva, N. F., Khan, H., Barnett, A. H., Kumar, S., Sturdee, D. W., and Mcternan, P. G. (2007). Expression of calcitonin gene-related peptide, adrenomedullin, and receptor modifying proteins in human adipose tissue and alteration in their expression with menopause status. Menopause 14, 1031–1038.
Haczynski, J., Tarkowski, R., Jarzabek, K., Slomczynska, M., Wolczynski, S., Magoffin, D. A., Jakowicki, J. A., and Jakimiuk, A. J. (2002). Human cultured skin fibroblasts express estrogen receptor alpha and beta. Int. J. Mol. Med. 10, 149–153.
Hagstad, A., and Janson, P. O. (1986). The epidemiology of climacteric symptoms. Acta Obstet. Gynecol. Scand. Suppl. 134, 59–65.
Hall, E., Frey, B. N., and Soares, C. N. (2011). Non-hormonal treatment strategies for vasomotor symptoms: a critical review. Drugs 71, 287–304.
Hayes, D. F., Skaar, T. C., Rae, J. M., Henry, N. L., Nguyen, A. T., Stearns, V., Li, L., Philips, S., Desta, Z., and Flockhart, D. A. (2010). Estrogen receptor genotypes, menopausal status, and the effects of tamoxifen on lipid levels: revised and updated results. Clin. Pharmacol. Ther. 88, 626–629.
Heine, P. A., Taylor, J. A., Iwamoto, G. A., Lubahn, D. B., and Cooke, P. S. (2000). Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc. Natl. Acad. Sci. U.S.A. 97, 12729–12734.
Henderson, V. W. (2006). Estrogen-containing hormone therapy and Alzheimer’s disease risk: understanding discrepant inferences from observational and experimental research. Neuroscience 138, 1031–1039.
Henry, J. M., Filburn, C. R., Joseph, J. A., and Roth, G. S. (1986). Effect of aging on striatal dopamine receptor subtypes in Wistar rats. Neurobiol. Aging 7, 357–361.
Hentges, S., Pastorcic, M., De, A., Boyadjieva, N., and Sarkar, D. K. (2000). Opposing actions of two transforming growth factor-beta isoforms on pituitary lactotropic cell proliferation. Endocrinology 141, 1528–1535.
Hill, R. A., and Boon, W. C. (2009). Estrogens, brain, and behavior: lessons from knockout mouse models. Semin. Reprod. Med. 27, 218–228.
Hou, Y., Wei, H., Luo, Y., and Liu, G. (2010). Modulating expression of brain heat shock proteins by estrogen in ovariectomized mice model of aging. Exp. Gerontol. 45, 323–330.
Huang, S. W., Seow, K. M., Ho, L. T., Chien, Y., Chung, D. Y., Chang, C. L., Lai, Y. H., Hwang, J. L., and Juan, C. C. (2005). Resistin mRNA levels are downregulated by estrogen in vivo and in vitro. FEBS Lett. 579, 449–454.
Hughes, S. V., Robinson, E., Bland, R., Lewis, H. M., Stewart, P. M., and Hewison, M. (1997). 1,25-dihydroxyvitamin D3 regulates estrogen metabolism in cultured keratinocytes. Endocrinology 138, 3711–3718.
Hulley, S., Grady, D., Bush, T., Furberg, C., Herrington, D., Riggs, B., and Vittinghoff, E. (1998). Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/Progestin Replacement Study (HERS) Research Group. JAMA 280, 605–613.
Hutton, J. D., Jacobs, H. S., Murray, M. A., and James, V. H. (1978). Relation between plasma oestrone and oestradiol and climacteric symptoms. Lancet 1, 678–681.
Iruthayanathan, M., Zhou, Y. H., and Childs, G. V. (2005). Dehydroepiandrosterone restoration of growth hormone gene expression in aging female rats, in vivo and in vitro: evidence for actions via estrogen receptors. Endocrinology 146, 5176–5187.
Ishunina, T. A., Fischer, D. F., and Swaab, D. F. (2007). Estrogen receptor alpha and its splice variants in the hippocampus in aging and Alzheimer’s disease. Neurobiol. Aging 28, 1670–1681.
Ishunina, T. A., and Swaab, D. F. (2008). Estrogen receptor-alpha splice variants in the human brain. Gynecol. Endocrinol. 24, 93–98.
Jacobsen, D. E., Samson, M. M., Van Der Schouw, Y. T., Grobbee, D. E., and Verhaar, H. J. (2008). Efficacy of tibolone and raloxifene for the maintenance of skeletal muscle strength, bone mineral density, balance, body composition, cognitive function, mood/depression, anxiety and quality of life/well-being in late postmenopausal women >/= 70 years: study design of a randomized, double-blind, double-dummy, placebo-controlled, single-center trial. Trials 9, 32.
Jee, S. H., Lee, S. Y., Chiu, H. C., Chang, C. C., and Chen, T. J. (1994). Effects of estrogen and estrogen receptor in normal human melanocytes. Biochem. Biophys. Res. Commun. 199, 1407–1412.
Johnson, B. D., Zheng, W., Korach, K. S., Scheuer, T., Catterall, W. A., and Rubanyi, G. M. (1997). Increased expression of the cardiac L-type calcium channel in estrogen receptor-deficient mice. J. Gen. Physiol. 110, 135–140.
Kahlert, S., Grohe, C., Karas, R. H., Lobbert, K., Neyses, L., and Vetter, H. (1997). Effects of estrogen on skeletal myoblast growth. Biochem. Biophys. Res. Commun. 232, 373–378.
Khosla, S., Melton, L. J. III, and Riggs, B. L. (2011). The unitary model for estrogen deficiency, and the pathogenesis of osteoporosis: is a revision needed? J. Bone Miner. Res. 26, 441–451.
Kislauskis, E., Bullock, B., Mcneil, S., and Dobner, P. R. (1988). The rat gene encoding neurotensin and neuromedin N. Structure, tissue-specific expression, and evolution of exon sequences. J. Biol. Chem. 263, 4963–4968.
Kotelevtsev, Y., Holmes, M. C., Burchell, A., Houston, P. M., Schmoll, D., Jamieson, P., Best, R., Brown, R., Edwards, C. R., Seckl, J. R., and Mullins, J. J. (1997). 11Beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc. Natl. Acad. Sci. U.S.A. 94, 14924–14929.
Kousteni, S., Bellido, T., Plotkin, L. I., O’Brien, C. A., Bodenner, D. L., Han, L., Han, K., Digregorio, G. B., Katzenellenbogen, J. A., Katzenellenbogen, B. S., Roberson, P. K., Weinstein, R. S., Jilka, R. L., and Manolagas, S. C. (2001). Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell 104, 719–730.
Kousteni, S., Chen, J. R., Bellido, T., Han, L., Ali, A. A., O’Brien, C. A., Plotkin, L., Fu, Q., Mancino, A. T., Wen, Y., Vertino, A. M., Powers, C. C., Stewart, S. A., Ebert, R., Parfitt, A. M., Weinstein, R. S., Jilka, R. L., and Manolagas, S. C. (2002). Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science 298, 843–846.
Kremer, M., Judd, J., Rifkin, B., Auszmann, J., and Oursler, M. J. (1995). Estrogen modulation of osteoclast lysosomal enzyme secretion. J. Cell. Biochem. 57, 271–279.
Kronenberg, F., and Fugh-Berman, A. (2002). Complementary and alternative medicine for menopausal symptoms: a review of randomized, controlled trials. Ann. Intern. Med. 137, 805–813.
Krum, S. A. (2011). Direct transcriptional targets of sex steroid hormones in bone. J. Cell. Biochem. 112, 401–408.
Krum, S. A., Chang, J., Miranda-Carboni, G., and Wang, C. Y. (2010). Novel functions for NFkappaB: inhibition of bone formation. Nat. Rev. Rheumatol. 6, 607–611.
Krum, S. A., Miranda-Carboni, G. A., Hauschka, P. V., Carroll, J. S., Lane, T. F., Freedman, L. P., and Brown, M. (2008a). Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO J. 27, 535–545.
Krum, S. A., Miranda-Carboni, G. A., Lupien, M., Eeckhoute, J., Carroll, J. S., and Brown, M. (2008b). Unique ER{alpha} cistromes control cell type-specific gene regulation. Mol. Endocrinol. 22, 2393–2406.
Kusminski, C. M., Mcternan, P. G., and Kumar, S. (2005). Role of resistin in obesity, insulin resistance and Type II diabetes. Clin. Sci. 109, 243–256.
Lakoski, S. G., Brosnihan, B., and Herrington, D. M. (2005). Hormone therapy, C-reactive protein, and progression of atherosclerosis: data from the estrogen replacement on progression of coronary artery atherosclerosis (ERA) trial. Am. Heart J. 150, 907–911.
Lamberts, S. W., Van Den Beld, A. W., and Van Der Lely, A. J. (1997). The endocrinology of aging. Science 278, 419–424.
Langan, E. A., Ramot, Y., Hanning, A., Poeggeler, B., Biro, T., Gaspar, E., Funk, W., Griffiths, C. E., and Paus, R. (2010). Thyrotropin-releasing hormone and oestrogen differentially regulate prolactin and prolactin receptor expression in female human skin and hair follicles in vitro. Br. J. Dermatol. 162, 1127–1131.
Lemoine, S., Granier, P., Tiffoche, C., Rannou-Bekono, F., Thieulant, M. L., and Delamarche, P. (2003). Estrogen receptor alpha mRNA in human skeletal muscles. Med. Sci. Sports Exerc. 35, 439–443.
Levin, E. R. (2005). Integration of the extranuclear and nuclear actions of estrogen. Mol. Endocrinol. 19, 1951–1959.
Li, Y., Jiang, C., Wang, X., Zhang, Y., Shibahara, S., and Takahashi, K. (2007). Adrenomedullin is a novel adipokine: adrenomedullin in adipocytes and adipose tissues. Peptides 28, 1129–1143.
Liu, D., Deschamps, A., Korach, K. S., and Murphy, E. (2008). Estrogen-enhanced gene expression of lipoprotein lipase in heart is antagonized by progesterone. Endocrinology 149, 711–716.
Loeser, A. A. (1937). The resorption and action of follicular hormone rubbed into the skin. J. Obstet. Gynaecol. Br. Emp. 44, 710–714.
Loughna, P. T., Mason, P., Bayol, S., and Brownson, C. (2000). The LIM-domain protein FHL1 (SLIM 1) exhibits functional regulation in skeletal muscle. Mol. Cell Biol. Res. Commun. 3, 136–140.
Lowe, D. A., Baltgalvis, K. A., and Greising, S. M. (2010). Mechanisms behind estrogen’s beneficial effect on muscle strength in females. Exerc. Sport Sci. Rev. 38, 61–67.
Luine, V. N., Renner, K. J., Heady, S., and Jones, K. J. (1986). Age and sex-dependent decreases in ChAT in basal forebrain nuclei. Neurobiol. Aging 7, 193–198.
Machinal, F., Dieudonne, M. N., Leneveu, M. C., Pecquery, R., and Giudicelli, Y. (1999). In vivo and in vitro ob gene expression and leptin secretion in rat adipocytes: evidence for a regional specific regulation by sex steroid hormones. Endocrinology 140, 1567–1574.
Macotela, Y., Boucher, J., Tran, T. T., and Kahn, C. R. (2009). Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes 58, 803–812.
Martin-Millan, M., Almeida, M., Ambrogini, E., Han, L., Zhao, H., Weinstein, R. S., Jilka, R. L., O’Brien, C. A., and Manolagas, S. C. (2010). The estrogen receptor-alpha in osteoclasts mediates the protective effects of estrogens on cancellous but not cortical bone. Mol. Endocrinol. 24, 323–334.
Martinoli, M. G., Ouellet, J., Rheaume, E., and Pelletier, G. (1991). Growth hormone and somatostatin gene expression in adult and aging rats as measured by quantitative in situ hybridization. Neuroendocrinology 54, 607–615.
Masuzaki, H., Paterson, J., Shinyama, H., Morton, N. M., Mullins, J. J., Seckl, J. R., and Flier, J. S. (2001). A transgenic model of visceral obesity and the metabolic syndrome. Science 294, 2166–2170.
McEwen, B. S. (2001). Invited review: estrogens effects on the brain: multiple sites and molecular mechanisms. J. Appl. Physiol. 91, 2785–2801.
McMillan, P. J., Singer, C. A., and Dorsa, D. M. (1996). The effects of ovariectomy and estrogen replacement on trkA and choline acetyltransferase mRNA expression in the basal forebrain of the adult female Sprague-Dawley rat. J. Neurosci. 16, 1860–1865.
Mesco, E. R., Joseph, J. A., Blake, M. J., and Roth, G. S. (1991). Loss of D2 receptors during aging is partially due to decreased levels of mRNA. Brain Res. 545, 355–357.
Miranda-Carboni, G. A., Guemes, M., Bailey, S., Anaya, E., Corselli, M., Peault, B., and Krum, S. A. (2011). gata4 regulates estrogen receptor-{alpha}-mediated osteoblast transcription. Mol. Endocrinol. 7, 1126–1136.
Misiti, S., Schomburg, L., Yen, P. M., and Chin, W. W. (1998). Expression and hormonal regulation of coactivator and corepressor genes. Endocrinology 139, 2493–2500.
Misso, M. L., Jang, C., Adams, J., Tran, J., Murata, Y., Bell, R., Boon, W. C., Simpson, E. R., and Davis, S. R. (2005a). Adipose aromatase gene expression is greater in older women and is unaffected by postmenopausal estrogen therapy. Menopause 12, 210–215.
Misso, M. L., Jang, C., Adams, J., Tran, J., Murata, Y., Bell, R., Boon, W. C., Simpson, E. R., and Davis, S. R. (2005b). Differential expression of factors involved in fat metabolism with age and the menopause transition. Maturitas 51, 299–306.
Mohamed, M. K., and Abdel-Rahman, A. A. (2000). Effect of long-term ovariectomy and estrogen replacement on the expression of estrogen receptor gene in female rats. Eur. J. Endocrinol. 142, 307–314.
Moran, A. L., Nelson, S. A., Landisch, R. M., Warren, G. L., and Lowe, D. A. (2007). Estradiol replacement reverses ovariectomy-induced muscle contractile and myosin dysfunction in mature female mice. J. Appl. Physiol. 102, 1387–1393.
Morello, K. C., Wurz, G. T., and Degregorio, M. W. (2002). SERMs: current status and future trends. Crit. Rev. Oncol. Hematol. 43, 63–76.
Morton, N. M., Holmes, M. C., Fievet, C., Staels, B., Tailleux, A., Mullins, J. J., and Seckl, J. R. (2001). Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice. J. Biol. Chem. 276, 41293–41300.
Murase, Y., Kobayashi, J., Nohara, A., Asano, A., Yamaaki, N., Suzuki, K., Sato, H., and Mabuchi, H. (2006). Raloxifene promotes adipocyte differentiation of 3T3-L1 cells. Eur. J. Pharmacol. 538, 1–4.
Naftolin, F., Mor, G., Horvath, T. L., Luquin, S., Fajer, A. B., Kohen, F., and Garcia-Segura, L. M. (1996). Synaptic remodeling in the arcuate nucleus during the estrous cycle is induced by estrogen and precedes the preovulatory gonadotropin surge. Endocrinology 137, 5576–5580.
Nakamura, T., Imai, Y., Matsumoto, T., Sato, S., Takeuchi, K., Igarashi, K., Harada, Y., Azuma, Y., Krust, A., Yamamoto, Y., Nishina, H., Takeda, S., Takayanagi, H., Metzger, D., Kanno, J., Takaoka, K., Martin, T. J., Chambon, P., and Kato, S. (2007). Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell 130, 811–823.
Nelson, H. D. (2004). Commonly used types of postmenopausal estrogen for treatment of hot flashes: scientific review. JAMA 291, 1610–1620.
Nishizawa, H., Shimomura, I., Kishida, K., Maeda, N., Kuriyama, H., Nagaretani, H., Matsuda, M., Kondo, H., Furuyama, N., Kihara, S., Nakamura, T., Tochino, Y., Funahashi, T., and Matsuzawa, Y. (2002). Androgens decrease plasma adiponectin, an insulin-sensitizing adipocyte-derived protein. Diabetes 51, 2734–2741.
O’Boyle, K. M., and Waddington, J. L. (1984). Loss of rat striatal dopamine receptors with ageing is selective for D-2 but not D-1 sites: association with increased non-specific binding of the D-1 ligand [3H]piflutixol. Eur. J. Pharmacol. 105, 171–174.
Ohlsson, C., Hellberg, N., Parini, P., Vidal, O., Bohlooly, Y. M., Rudling, M., Lindberg, M. K., Warner, M., Angelin, B., and Gustafsson, J. A. (2000). Obesity and disturbed lipoprotein profile in estrogen receptor-alpha-deficient male mice. Biochem. Biophys. Res. Commun. 278, 640–645.
Olmos, G., Naftolin, F., Perez, J., Tranque, P. A., and Garcia-Segura, L. M. (1989). Synaptic remodeling in the rat arcuate nucleus during the estrous cycle. Neuroscience 32, 663–667.
O’Malley, B. (2008). The year in basic science: nuclear receptors and coregulators. Mol. Endocrinol. 22, 2751–2758.
Otsuki, M., Gao, H., Dahlman-Wright, K., Ohlsson, C., Eguchi, N., Urade, Y., and Gustafsson, J. A. (2003). Specific regulation of lipocalin-type prostaglandin D synthase in mouse heart by estrogen receptor beta. Mol. Endocrinol. 17, 1844–1855.
Pantschenko, A. G., Zhang, W., Nahounou, M., Mccarthy, M. B., Stover, M. L., Lichtler, A. C., Clark, S. H., and Gronowicz, G. A. (2005). Effect of osteoblast-targeted expression of bcl-2 in bone: differential response in male and female mice. J. Bone Miner. Res. 20, 1414–1429.
Park, Y. W., Zhu, S., Palaniappan, L., Heshka, S., Carnethon, M. R., and Heymsfield, S. B. (2003). The metabolic syndrome: prevalence and associated risk factor findings in the US population from the Third National Health and Nutrition Examination Survey, 1988–1994. Arch. Intern. Med. 163, 427–436.
Pedersen, S. B., Borglum, J. D., Moller-Pedersen, T., and Richelsen, B. (1992). Effects of in vivo estrogen treatment on adipose tissue metabolism and nuclear estrogen receptor binding in isolated rat adipocytes. Mol. Cell. Endocrinol. 85, 13–19.
Pedraza-Alva, G., Zingg, J. M., Donda, A., and Perez-Martinez, L. (2009). Estrogen receptor regulates MyoD gene expression by preventing AP-1-mediated repression. Biochem. Biophys. Res. Commun. 389, 360–365.
Perlman, W. R., Matsumoto, M., Beltaifa, S., Hyde, T. M., Saunders, R. C., Webster, M. J., Rubinow, D. R., Kleinman, J. E., and Weickert, C. S. (2005). Expression of estrogen receptor alpha exon-deleted mRNA variants in the human and non-human primate frontal cortex. Neuroscience 134, 81–95.
Poirier, J. (1996). Apolipoprotein E in the brain and its role in Alzheimer’s disease. J. Psychiatry Neurosci. 21, 128–134.
Pollanen, E., Fey, V., Tormakangas, T., Ronkainen, P. H., Taaffe, D. R., Takala, T., Koskinen, S., Cheng, S., Puolakka, J., Kujala, U. M., Suominen, H., Sipila, S., and Kovanen, V. (2010). Power training and postmenopausal hormone therapy affect transcriptional control of specific co-regulated gene clusters in skeletal muscle. Age (Omaha) 32, 347–363.
Pollanen, E., Sipila, S., Alen, M., Ronkainen, P. H., Ankarberg-Lindgren, C., Puolakka, J., Suominen, H., Hamalainen, E., Turpeinen, U., Konttinen, Y. T., and Kovanen, V. (2011). Differential influence of peripheral and systemic sex steroids on skeletal muscle quality in pre- and post-menopausal women. Aging Cell 10, 650–660.
Post, W. S., Goldschmidt-Clermont, P. J., Wilhide, C. C., Heldman, A. W., Sussman, M. S., Ouyang, P., Milliken, E. E., and Issa, J. P. (1999). Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovasc. Res. 43, 985–991.
Powles, T. J. (2002). Anti-oestrogenic prevention of breast cancer – the make or breakpoint. Nat. Rev. Cancer 2, 787–794.
Ronda, A. C., Vasconsuelo, A., and Boland, R. (2010). Extracellular-regulated kinase and p38 mitogen-activated protein kinases are involved in the antiapoptotic action of 17beta-estradiol in skeletal muscle cells. J. Endocrinol. 206, 235–246.
Ronkainen, P. H., Kovanen, V., Alen, M., Pollanen, E., Palonen, E. M., Ankarberg-Lindgren, C., Hamalainen, E., Turpeinen, U., Kujala, U. M., Puolakka, J., Kaprio, J., and Sipila, S. (2009). Postmenopausal hormone replacement therapy modifies skeletal muscle composition and function: a study with monozygotic twin pairs. J. Appl. Physiol. 107, 25–33.
Ronkainen, P. H., Pollanen, E., Alen, M., Pitkanen, R., Puolakka, J., Kujala, U. M., Kaprio, J., Sipila, S., and Kovanen, V. (2010). Global gene expression profiles in skeletal muscle of monozygotic female twins discordant for hormone replacement therapy. Aging Cell 9, 1098–1110.
Rose’Meyer, R. B., Mellick, A. S., Garnham, B. G., Harrison, G. J., Massa, H. M., and Griffiths, L. R. (2003). The measurement of adenosine and estrogen receptor expression in rat brains following ovariectomy using quantitative PCR analysis. Brain Res. Brain Res. Protoc. 11, 9–18.
Rosenbaum, M., Nicolson, M., Hirsch, J., Heymsfield, S. B., Gallagher, D., Chu, F., and Leibel, R. L. (1996). Effects of gender, body composition, and menopause on plasma concentrations of leptin. J. Clin. Endocrinol. Metab. 81, 3424–3427.
Rossouw, J. E., Anderson, G. L., Prentice, R. L., Lacroix, A. Z., Kooperberg, C., Stefanick, M. L., Jackson, R. D., Beresford, S. A., Howard, B. V., Johnson, K. C., Kotchen, J. M., and Ockene, J. (2002). Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 288, 321–333.
Roth, G. S., and Joseph, J. A. (1994). Cellular and molecular mechanisms of impaired dopaminergic function during aging. Ann. N. Y. Acad. Sci. 719, 129–135.
Rozenbaum, H. (2006). How to evaluate the risk-benefit ratio of the low-dose hormone replacement therapy? J. Steroid Biochem. Mol. Biol. 102, 256–260.
Saad, M. F., Damani, S., Gingerich, R. L., Riad-Gabriel, M. G., Khan, A., Boyadjian, R., Jinagouda, S. D., El-Tawil, K., Rude, R. K., and Kamdar, V. (1997). Sexual dimorphism in plasma leptin concentration. J. Clin. Endocrinol. Metab. 82, 579–584.
Schomburg, L., and Bauer, K. (1997). Regulation of the adenohypophyseal thyrotropin-releasing hormone-degrading ectoenzyme by estradiol. Endocrinology 138, 3587–3593.
Shang, Y., and Brown, M. (2002). Molecular determinants for the tissue specificity of SERMs. Science 295, 2465–2468.
Shao, W., and Brown, M. (2004). Advances in estrogen receptor biology: prospects for improvements in targeted breast cancer therapy. Breast Cancer Res. 6, 39–52.
Shearman, A. M., Cooper, J. A., Kotwinski, P. J., Miller, G. J., Humphries, S. E., Ardlie, K. G., Jordan, B., Irenze, K., Lunetta, K. L., Schuit, S. C., Uitterlinden, A. G., Pols, H. A., Demissie, S., Cupples, L. A., Mendelsohn, M. E., Levy, D., and Housman, D. E. (2006). Estrogen receptor alpha gene variation is associated with risk of myocardial infarction in more than seven thousand men from five cohorts. Circ. Res. 98, 590–592.
Shimizu, T. (2005). Role of macrophage migration inhibitory factor (MIF) in the skin. J. Dermatol. Sci. 37, 65–73.
Shughrue, P. J., Lane, M. V., and Merchenthaler, I. (1997). Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J. Comp. Neurol. 388, 507–525.
Shughrue, P. J., and Merchenthaler, I. (2000). Evidence for novel estrogen binding sites in the rat hippocampus. Neuroscience 99, 605–612.
Silverman, S. L., Christiansen, C., Genant, H. K., Vukicevic, S., Zanchetta, J. R., De Villiers, T. J., Constantine, G. D., and Chines, A. A. (2008). Efficacy of bazedoxifene in reducing new vertebral fracture risk in postmenopausal women with osteoporosis: results from a 3-year, randomized, placebo-, and active-controlled clinical trial. J. Bone Miner. Res. 23, 1923–1934.
Simoncini, T., and Genazzani, A. R. (2000). Raloxifene acutely stimulates nitric oxide release from human endothelial cells via an activation of endothelial nitric oxide synthase. J. Clin. Endocrinol. Metab. 85, 2966–2969.
Sinclair, R., Banfield, C., and Dawber, R. (1999). Handbook of Diseases of the Hair and the Scalp. New York: Blackwell, Oxford Press.
Singh, M., Meyer, E. M., Huang, F. S., Millard, W. J., and Simpkins, J. W. (1993). Ovariectomy reduces ChAT activity and NGF mRNA levels in the frontal cortex and hippocampus of the female Sprague-Dawley rat. Soc. Neurosci. Abstr. 19, 11.
Singh, M., Meyer, E. M., and Simpkins, J. W. (1995). The effect of ovariectomy and estradiol replacement on brain-derived neurotrophic factor messenger ribonucleic acid expression in cortical and hippocampal brain regions of female Sprague-Dawley rats. Endocrinology 136, 2320–2324.
Sipila, S., Taaffe, D. R., Cheng, S., Puolakka, J., Toivanen, J., and Suominen, H. (2001). Effects of hormone replacement therapy and high-impact physical exercise on skeletal muscle in post-menopausal women: a randomized placebo-controlled study. Clin. Sci. 101, 147–157.
Son, E. D., Lee, J. Y., Lee, S., Kim, M. S., Lee, B. G., Chang, I. S., and Chung, J. H. (2005). Topical application of 17beta-estradiol increases extracellular matrix protein synthesis by stimulating tgf-beta signaling in aged human skin in vivo. J. Invest. Dermatol. 124, 1149–1161.
Stevenson, S., and Thornton, J. (2007). Effect of estrogens on skin aging and the potential role of SERMs. Clin. Interv. Aging 2, 283–297.
Stone, D. J., Rozovsky, I., Morgan, T. E., Anderson, C. P., Hajian, H., and Finch, C. E. (1997). Astrocytes and microglia respond to estrogen with increased apoE mRNA in vivo and in vitro. Exp. Neurol. 143, 313–318.
Stone, D. J., Song, Y., Anderson, C. P., Krohn, K. K., Finch, C. E., and Rozovsky, I. (1998). Bidirectional transcription regulation of glial fibrillary acidic protein by estradiol in vivo and in vitro. Endocrinology 139, 3202–3209.
Student, A. K., Hsu, R. Y., and Lane, M. D. (1980). Induction of fatty acid synthetase synthesis in differentiating 3T3-L1 preadipocytes. J. Biol. Chem. 255, 4745–4750.
Surazynski, A., Jarzabek, K., Haczynski, J., Laudanski, P., Palka, J., and Wolczynski, S. (2003). Differential effects of estradiol and raloxifene on collagen biosynthesis in cultured human skin fibroblasts. Int. J. Mol. Med. 12, 803–809.
Takahashi, S., Kawashima, S., Seo, H., and Matsui, N. (1990). Age-related changes in growth hormone and prolactin messenger RNA levels in the rat. Endocrinol. Jpn. 37, 827–840.
Tashjian, A. H. Jr., Yasumura, Y., Levine, L., Sato, G. H., and Parker, M. L. (1968). Establishment of clonal strains of rat pituitary tumor cells that secrete growth hormone. Endocrinology 82, 342–352.
Teede, H. J., Lombard, C., and Deeks, A. A. (2010). Obesity, metabolic complications and the menopause: an opportunity for prevention. Climacteric 13, 203–209.
Thakur, M. K., and Sharma, P. K. (2007). Transcription of estrogen receptor alpha and beta in mouse cerebral cortex: effect of age, sex, 17beta-estradiol and testosterone. Neurochem. Int. 50, 314–321.
Too, C. K., Giles, A., and Wilkinson, M. (1999). Estrogen stimulates expression of adenine nucleotide translocator ANT1 messenger RNA in female rat hearts. Mol. Cell. Endocrinol. 150, 161–167.
Tsukahara, K., Moriwaki, S., Ohuchi, A., Fujimura, T., and Takema, Y. (2001). Ovariectomy accelerates photoaging of rat skin. Photochem. Photobiol. 73, 525–531.
Tsukahara, K., Nakagawa, H., Moriwaki, S., Kakuo, S., Ohuchi, A., Takema, Y., and Imokawa, G. (2004). Ovariectomy is sufficient to accelerate spontaneous skin ageing and to stimulate ultraviolet irradiation-induced photoageing of murine skin. Br. J. Dermatol. 151, 984–994.
Vasconsuelo, A., Milanesi, L., and Boland, R. (2008). 17Beta-estradiol abrogates apoptosis in murine skeletal muscle cells through estrogen receptors: role of the phosphatidylinositol 3-kinase/Akt pathway. J. Endocrinol. 196, 385–397.
Verdier-Sevrain, S., Bonte, F., and Gilchrest, B. (2006). Biology of estrogens in skin: implications for skin aging. Exp. Dermatol. 15, 83–94.
Wang, L. L., Qiu, G. R., Fu, W. N., Yuan, Z. W., and Sun, K. L. (2010). Transcriptional regulation of Fhl1 by estradiol in rat myoblastocytes. Steroids 75, 368–372.
Watters, J. J., and Dorsa, D. M. (1998). Transcriptional effects of estrogen on neuronal neurotensin gene expression involve cAMP/protein kinase A-dependent signaling mechanisms. J. Neurosci. 18, 6672–6680.
White, P. C., Rogoff, D., and Mcmillan, D. R. (2008). Physiological roles of 11 beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase. Curr. Opin. Pediatr. 20, 453–457.
Wiik, A., Ekman, M., Johansson, O., Jansson, E., and Esbjornsson, M. (2009a). Expression of both oestrogen receptor alpha and beta in human skeletal muscle tissue. Histochem. Cell Biol. 131, 181–189.
Wiik, A., Hellsten, Y., Berthelson, P., Lundholm, L., Fischer, H., and Jansson, E. (2009b). Activation of estrogen response elements is mediated both via estrogen and muscle contractions in rat skeletal muscle myotubes. Am. J. Physiol. Cell Physiol. 296, C215–220.
Wiik, A., Glenmark, B., Ekman, M., Esbjornsson-Liljedahl, M., Johansson, O., Bodin, K., Enmark, E., and Jansson, E. (2003). Oestrogen receptor beta is expressed in adult human skeletal muscle both at the mRNA and protein level. Acta Physiol. Scand. 179, 381–387.
Wilbur, J., Miller, A. M., Montgomery, A., and Chandler, P. (1998). Sociodemographic characteristics, biological factors, and symptom reporting in midlife women. Menopause 5, 43–51.
Wilson, M. E., Rosewell, K. L., Kashon, M. L., Shughrue, P. J., Merchenthaler, I., and Wise, P. M. (2002). Age differentially influences estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) gene expression in specific regions of the rat brain. Mech. Ageing Dev. 123, 593–601.
Windahl, S. H., Andersson, G., and Gustafsson, J. A. (2002). Elucidation of estrogen receptor function in bone with the use of mouse models. Trends Endocrinol. Metab. 13, 195–200.
Yi, K. W., Shin, J. H., Seo, H. S., Lee, J. K., Oh, M. J., Kim, T., Saw, H. S., Kim, S. H., and Hur, J. Y. (2008). Role of estrogen receptor-alpha and -beta in regulating leptin expression in 3T3-L1 adipocytes. Obesity 16, 2393–2399.
Yoneda, N., Saito, S., Kimura, M., Yamada, M., Iida, M., Murakami, T., Irahara, M., Shima, K., and Aono, T. (1998). The influence of ovariectomy on ob gene expression in rats. Horm. Metab. Res. 30, 263–265.
Zallone, A. (2006). Direct and indirect estrogen actions on osteoblasts and osteoclasts. Ann. N. Y. Acad. Sci. 1068, 173–179.
Zhang, Y., Proenca, R., Maffei, M., Barone, M., Leopold, L., and Friedman, J. M. (1994). Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425–432.
Zhou, S., Turgeman, G., Harris, S. E., Leitman, D. C., Komm, B. S., Bodine, P. V., and Gazit, D. (2003). Estrogens activate bone morphogenetic protein-2 gene transcription in mouse mesenchymal stem cells. Mol. Endocrinol. 17, 56–66.
Zhou, Y., Watters, J. J., and Dorsa, D. M. (1996). Estrogen rapidly induces the phosphorylation of the cAMP response element binding protein in rat brain. Endocrinology 137, 2163–2166.
Zhu, Y., Bian, Z., Lu, P., Karas, R. H., Bao, L., Cox, D., Hodgin, J., Shaul, P. W., Thoren, P., Smithies, O., Gustafsson, J. A., and Mendelsohn, M. E. (2002). Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science 295, 505–508.
Keywords: estrogen, SERM, tissue specificity, aging
Citation: Wend K, Wend P and Krum SA (2012) Tissue-specific effects of loss of estrogen during menopause and aging. Front. Endocrin. 3:19. doi: 10.3389/fendo.2012.00019
Received: 11 July 2011;
Accepted: 23 January 2012;
Published online: 08 February 2012.
Edited by:
Marc R. Blackman, National Institute of Health, USAReviewed by:
David George Monroe, Mayo Clinic College of Medicine, USASarianna Sipila, University of Jyväskylä, Finland
Copyright: © 2012 Wend, Wend and Krum. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Susan A. Krum, Orthopaedic Hospital Department of Orthopaedic Surgery, Orthopaedic Hospital Research Center, David Geffen School of Medicine, University of California Los Angeles, 615 Charles E. Young Dr. South, OHRC 410, Los Angeles, CA 90095, USA. e-mail:c2tydW1AbWVkbmV0LnVjbGEuZWR1