


- Department of Experimental Medicine, Medical Physiopathology, Food Science and Endocrinology Section, Sapienza University of Rome, Rome, Italy
Androgen deprivation therapy (ADT) for prostate carcinoma (PCa) may cause cardiometabolic complications unless intermittent androgen blockade (IAB) is instituted. An 80-year-old caucasian man was diagnosed intermediate grade (Gleason 4 + 3) PCa and was treated with continuous ADT with triptorelin plus bicalutamide. After 6 months of treatment, he experienced an acute myocardial infarction and 1 month after hospitalization he came to our outpatient clinic for fatigue, weight gain, and hyperglycemia. Due to iatrogenic hypogonadism, we decided to proceed with IAB, but after 3 months ADT withdrawal his serum testosterone (T) was still 0.5 ng/mL. Due to very low concomitant PSA levels (0.1 ng/mL) he was then proposed intermittent T-gel supplementation (Tostrex®) which was initiated according to the following scheme: 6 months on and 3 months off. T-gel dose was titrated tri-weekly in order to achieve T plasma levels below 3.49 ng/mL. After 6 months on, his serum T raised to a mean value of about 2.0 ng/mL without increments in PSA. After overall 12 months on, his serum T peaked to a mean value of 3.0 ng/mL while a delay in PSA rise was seen after 24 months (0.6 ng/mL) but remained stable until the last observation carried forward (LOCF), at 45 months. No clinical and biochemical PCa progression were observed at LOCF. Reversion of iatrogenic metabolic syndrome started after 6 months of T supplementation without using any add-on treatment. This case provides support that once regression of PCa growth is attained, T supplementation may be administered in well differentiated PCa, especially if IAB is not successful in reverting iatrogenic hypogonadism and its associated cardiac and metabolic complications.
Case Presentation
An 80-years-old caucasian man (B.F.) with localized prostate carcinoma (PCa; Gleason score 4 + 3; stage pT1c) that was treated with androgen deprivation therapy (ADT) with triptorelin plus bicalutamide from 6 months was referred to our outpatient clinic for recent onset of fatigue, weight gain, and hyperglycemia. He was taking Suprefact® 1 injection every 3 months combined with oral Casodex® 50 mg daily. Before and after 6 months of ADT, his PSA dropped from 5.4 (free PSA = 0.81 ng/mL; ratio = 0.15%) to 0.1 while testosterone (T) dropped from 3.7 to 0.8 ng/mL, respectively; plasma LH level before treatment was normal (2.4 IU/L). As a result of this treatment, he experienced hospitalization for myocardial ischemia and underwent a revascularization procedure. Once discarded with specific therapy (low dose aspirin, angiotensin receptor blocker plus thiazides, and simvastatin 40 mg/day), he noted the appearance of progressive fatigue, loss of memory, dyspnea, tachycardia, weight gain (increase in waist circumference about two sizes), and diffuse bone pain. He was admitted at our outpatient clinic with the diagnosis of metabolic syndrome (Table 1) and iatrogenic hypogonadism and was prescribed a Whole Body Scintiscan that showed the absence of distant metastases from PCa. According to patient’s Framingham’s score, the 10 years estimated global cardiovascular risk was >30% so that intermittent androgen blockade (IAB) was recommended in order to minimize further increase in cardiovascular risk. However, 3 months after ADT withdrawal, the patient still complained same symptoms and his T plasma levels were still to 0.5 ng/mL, with an LH of 0.1 IU/L. Testicular size was reduced (6 mL bilaterally) and digital rectal examination revealed enlarged prostate with a palpable nodule (primary tumor) that had been already evidenced at ultrasound diagnosis of PCa without any change in size (2 cm diameter). Thus, add-on treatment with T supplementation was advised prior signing a detailed informed consent and the warning the patient on a possible progression of PCa after commencing treatment. Accordingly, same warning regarding the progression of atherosclerosis in severe hypogonadal patients and consequent increased mortality risk was acknowledged. We decided to use T-gel 2% (Tostrex®) because of the possibility to titrate the dosage every 2–3 weeks. The patient was started with a dose of 2 puff/day and total T and PSA were evaluated at 8:00 a.m. every 21 days, before the application of gel on same morning (mean dose administered during the observation period was 2.2 puff/day equaling 22 mg/day). After 6 months from commencing treatment, glico-metabolic parameters returned to normal (Table 1), as expected from the time course of T actions on target organs (Saad et al., 2011). We decided to maintain his plasma T level below 3.5 ng/mL because of consistency with biological effects on target organs as expected from delta increase from patients’ baseline levels after ADT. After 30 months, PSA peaked to 0.6 ng/mL then remaining stable throughout the entire period of observation (Figure 1) and prostate gland did not show further enlargements as suggested by control TC (data not shown). According to our previous data (Aversa et al., 2012), the patient was osteopenic at baseline and an increase in bone mineral density after 3 years was documented irrespective from vitamin D supplementation (Table 1), with reported improvement in bone pain commencing after 1 year. At present, the follow-up carried out at 45 months shows no further increments in PSA and no progression of PCa and the patient is still on intermittent T-gel supplementation.
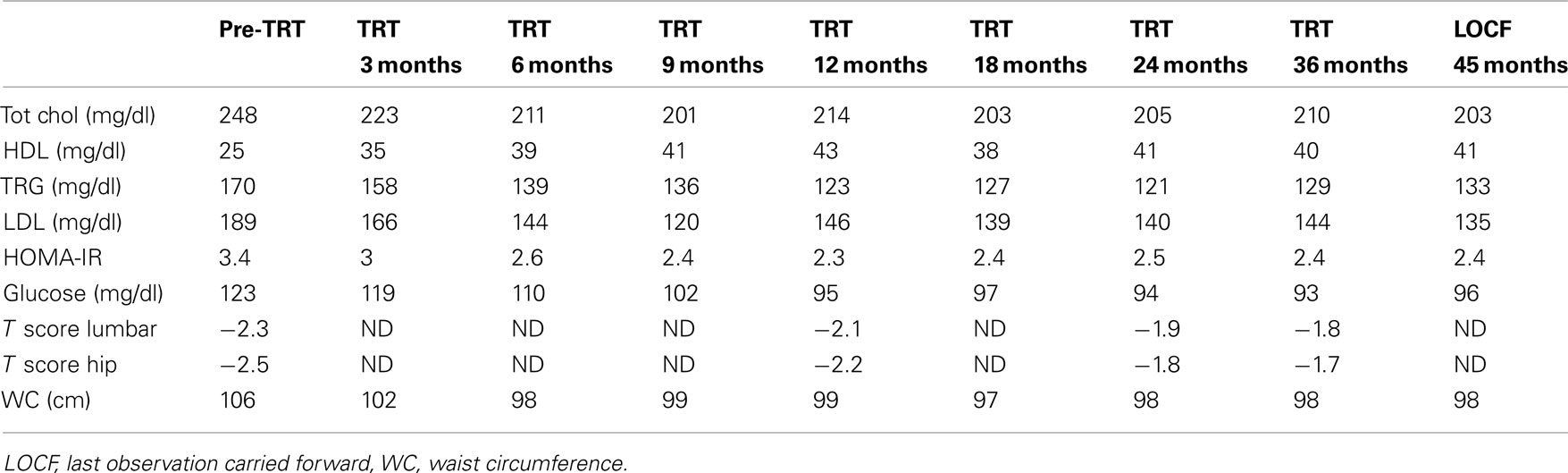
Table 1. Biochemical and DEXA measurements of the patient with PCa.
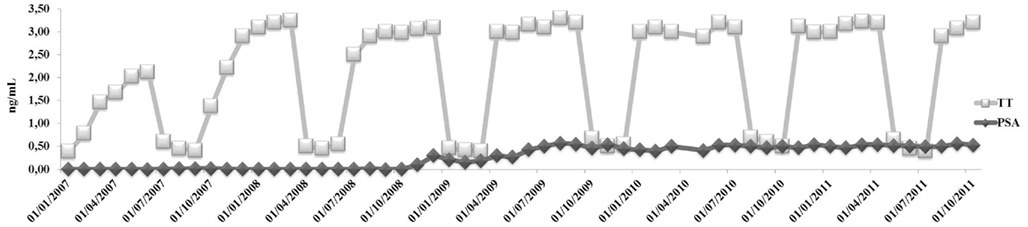
Figure 1. Testosterone and PSA course during 3 years follow-up.
Background
Testosterone is the fundamental steroid hormone that drives prostate growth but there is no proven evidence that T treatment may convert subclinical PCa to clinically detectable PCa (Rhoden and Averbeck, 2009; Jannini et al., 2011). After 3- to 6-months of treatment, ADT is known to cause metabolic imbalance, i.e., increase in fat mass, insulin resistance, hyperlipidemia, metabolic syndrome (Braga-Basaria et al., 2006), and/or full-blown diabetes mellitus, loss of bone mass, and fractures (Grossmann and Zajac, 2011), as well as increased cardiovascular risk, anemia, and loss of muscle (Punnen et al., 2011). All these metabolic changes put patients at an increased risk of cerebrovascular events, such as ischemic strokes and transient ischemic attacks, causing a significant increased risk of cardiovascular death (Levine et al., 2010). T is largely metabolized into its more potent effector, dihydrotestosterone (DHT), which promotes cellular proliferation of prostatic epithelium (Hsing, 2001). Results from the Prostate Cancer Prevention Trial indicated that inhibition of the conversion of T to DHT by finasteride reduced the occurrence of prostate cancer by approximately 25% during a 7-year follow-up; although the risk of high-grade tumors was higher in the treated group than in the untreated group (Canby-Hagino et al., 2007). The evidence that prostate cancer development has a hormonal component comes from a large range of sources, including the historical observation that most PCa respond to ADT until they invariably establish an androgen-independent growth mechanism, leading to a more aggressive, castrate-resistant phenotype (Lonergan and Tindall, 2011). The potential mechanisms involving the genesis and growth of androgen-independent PCa include super-expression or mutation of the androgen receptor (AR), which could determine resistance to the anti-androgenic therapy. However, most advanced PCa have no mutations or amplifications of the AR, suggesting a potential role of non-androgenic growth factors, such as epidermal growth factor (EGF), transforming growth factor alpha, insulin-like growth factor (IGF-1), and fibroblast growth factor. These factors, and their receptors like EGFR (HER-1) and HER-2/neu, may contribute to the proliferation and growth of prostate cancer through paracrine or autocrine mechanisms (Neto et al., 2010). Recently a collaborative analysis of 18 prospective studies on the associations between endogenous hormone concentrations and PCa risk indicates that the circulating concentrations of androgens or estradiol do not appear to be associated with the risk of prostate cancer. Nowadays, a history of PCa is an absolute contraindication to T supplementation therapy (Roddam et al., 2008; Heidenreich et al., 2011). In the last decade many authors reported their personal experience in treating men with confined PCa (Kaufman and Graydon, 2004; Agarwal and Oefelein, 2005) or after radical prostatectomy (Khera et al., 2009) or heterogeneous populations of prostate cancer survivors (Leibowitz et al., 2010) with different T preparations for different periods of time. Morgentaler and Traish recently proposed a saturation model that accounts for the seemingly contradictory results in human PCa studies; They suggested that the changes in serum T concentrations below the point of maximal androgen–androgen receptor (A–AR) binding elicit substantial changes in PCa growth, as seen with castration, or with T administration to previously castrated men. By contrast, once maximal A–AR binding is attained, the presence of additional androgen produces little further effect (Morgentaler and Traish, 2009). At present, there is limited experience and controversy about T use after successful treatment of PCa (Jannini et al., 2011). This is the first interventional report with T supplementation in a PCa patient who was submitted to prior ADT for localized PCa.
Discussion
Although it is well documented that T administration in castrated men with advanced or metastatic PCa leads to progression of disease (Fowler and Whitmore Jr., 1981), this aspect has never been investigated in patients with localized PCa after ADT withdrawal. As far as we are aware, this is the first report of intermittent T supplementation in the long-term (45 months) in a patient with non-invasive PCa after ADT withdrawal and persistent severe secondary hypogonadism. Despite the long-standing prohibition against the use of T in men with PCa, preliminary data from three important publications suggests encouraging data. A phase-1 study reports no progression of disease due to T administration after 24 months of treatment in men with low risk castrate-resistant PCa (Szmulewitz et al., 2009). Another study suggests that even exogenous supraphysiological T levels are neutral on castrate-resistant PCa progression (Morris et al., 2009). Morgentaler (2009) reports on a case of PSA decline after 2 years of T supplementation in a man with untreated PCa. ADT-induced hypogonadism is an independent risk factor for the development of insulin resistance/metabolic syndrome, cardiovascular disease, and bone frailty (Foresta et al., 2011) with an increased male morbidity and mortality. Several clinical trials have examined whether longitudinal changes in cardiovascular risk factors may translate into a real increased risk of cardiovascular death. It is presently unclear whether there is a causal relationship between ADT and cardiovascular mortality. What is certain is that ADT may affect cardiovascular mortality in a subset of these study populations and it may be prudent to carefully consider the potential risks and benefits before commencing ADT, particularly in patients with coronary artery disease. Patients with cardiac disease who initiate ADT should receive particular attention to secondary preventive interventions (Levine et al., 2010). Keeping in mind these considerations, and due to the fact that benefits and risks deriving from ADT in our patient were not evaluated in advance, we decided to propose IAB in order to minimize further cardiometabolic complications during ADT. Unexpectedly, ADT withdrawal did not produce physiological increments in plasma T levels so that we decided to challenge the patient with intermittent T-gel supplementation considering encouraging results obtained by other clinical trials. We strictly monitored PSA levels and progression of disease over a long period of time (45 months) and were able to demonstrate for the first time that T supplementation after ADT did not determine serious adverse events but long-standing improvements from the metabolic, cardiovascular, and skeletal standpoints. We are aware that caution is necessary and conclusions regarding T supplementation in men undergoing ADT for localized PCa are far to be drawn, as well. However, due to increased surveillance in higher numbers of men with PCa, the request for correction of hypogonadism-related complications will be increasing. We hope that multicenter clinical trials investigating this aspect will be proposed in the near future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agarwal, P. K., and Oefelein, M. G. (2005). Testosterone replacement therapy after primary treatment for prostate cancer. J. Urol. 173, 533–536.
Aversa, A., Bruzziches, R., Francomano, D., Greco, E. A., Fornari, R., Di Luigi, L., Lenzi, A., and Migliaccio, S. (2012). Effects of long-acting testosterone undecanoate on bone mineral density in middle aged men with late-onset hypogonadism and metabolic syndrome: results from a 36 months controlled study. Aging Male (in press).
Braga-Basaria, M., Dobs, A. S., Muller, D. C., Carducci, M. A., John, M., Egan, J., and Basaria, S. (2006). Metabolic syndrome in men with prostate cancer undergoing long-term androgen-deprivation therapy. J. Clin. Oncol. 24, 3979–3983.
Canby-Hagino, E., Hernandez, J., Brand, T. C., and Thompson, I. (2007). Looking back at PCPT: looking forward to new paradigms in prostate cancer screening and prevention. Eur. Urol. 51, 27–33.
Foresta, C., Strapazzon, G., De Toni, L., Perilli, L., Di Mambro, A., Muciaccia, B., Sartori, L., and Selice, R. (2011). Bone mineral density and testicular failure: evidence for a role of vitamin D 25-hydroxylase in human testis. J. Clin. Endocrinol. Metab. 96, E646–E652.
Fowler, J. E., and Whitmore, W. F. Jr. (1981). The response of metastatic adenocarcinoma of the prostate to exogenous testosterone. J. Urol. 12, 372–375.
Grossmann, M., and Zajac, J. D. (2011). Management of side effects of androgen deprivation therapy. Endocrinol. Metab. Clin. North Am. 40, 655–671.
Heidenreich, A., Bellmunt, J., Bolla, M., Joniau, S., Mason, M., Matveev, V., Mottet, N., Schmid, H. P., van der Kwast, T., Wiegel, T., Zattoni, F., and European Association of Urology. (2011). EAU guidelines on prostate cancer. Part 1: screening, diagnosis, and treatment of clinically localised disease. Eur. Urol. 59, 61–71.
Jannini, E. A., Gravina, G. L., Morgentaler, A., Morales, A., Incrocci, L., and Hellstrom, W. J. (2011). Is testosterone a friend or a foe for the prostate? J. Sex. Med. 8, 946–955.
Kaufman, J. M., and Graydon, R. J. (2004). Androgen replacement after curative radical prostatectomy for prostate cancer in hypogonadal men. J. Urol. 172, 920–922.
Khera, M., Grober, E. D., Najari, B., Colen, J. S., Mohamed, O., Lamb, D. J., and Lipshultz, L. I. (2009). Testosterone replacement therapy following radical prostatectomy. J. Sex. Med. 6, 1165–1170.
Leibowitz, R. L., Dorff, T. B., Tucker, S., Symanowski, J., and Vogelzang, N. J. (2010). Testosterone replacement in prostate cancer survivors with hypogonadal symptoms. BJU Int. 105, 1397–1401.
Levine, G. N., D’Amico, A. V., Berger, P., Clark, P. E., Eckel, R. H., Keating, N. L., Milani, R. V., Sagalowsky, A. I., Smith, M. R., Zakai, N., and American Heart Association Council on Clinical Cardiology and Council on Epidemiology and Prevention, the American Cancer Society, and the American Urological Association. (2010). Androgen-deprivation therapy in prostate cancer and cardiovascular risk: a science advisory from the American Heart Association, American Cancer Society, and American Urological Association: endorsed by the American Society for Radiation Oncology. Circulation 121, 833–840.
Lonergan, P. E., and Tindall, D. J. (2011). Androgen receptor signaling in prostate cancer development and progression. J. Carcinog. 10, 20.
Morgentaler, A. (2009). Two years of testosterone therapy associated with decline in prostate-specific antigen in a man with untreated prostate cancer. J. Sex. Med. 6, 574–577.
Morgentaler, A., and Traish, A. M. (2009). Shifting the paradigm of testosterone and prostate cancer: the saturation model and the limits of androgen-dependent growth. Eur. Urol. 55, 310–320.
Morris, M. J., Huang, D., Kelly, W. K., Slovin, S. F., Stephenson, R. D., Eicher, C., Delacruz, A., Curley, T., Schwartz, L. H., and Scher, H. I. (2009). Phase 1 trial of high-dose exogenous testosterone in patients with castration-resistant metastatic prostate cancer. Eur. Urol. 56, 237–244.
Neto, A. S., Tobias-Machado, M., Wroclawski, M. L., Fonseca, F. L., Pompeo, A. C., and Del Giglio, A. (2010). Molecular oncogenesis of prostate adenocarcinoma: role of the human epidermal growth factor receptor 2 (HER-2/neu). Tumori 96, 645–649.
Punnen, S., Cooperberg, M. R., Sadetsky, N., and Carroll, P. R. (2011). Androgen deprivation therapy and cardiovascular risk. J. Clin. Oncol. 29, 3510–3516.
Rhoden, E. L., and Averbeck, M. A. (2009). Testosterone therapy, and prostate carcinoma. Curr. Urol. Rep. 10, 453–459.
Roddam, A. W., Allen, N. E., Appleby, P., and Key, T. J. (2008). Endogenous sex hormones and prostate cancer: a collaborative analysis of 18 prospective studies. Endogenous Hormones and Prostate Cancer Collaborative Group. J. Natl. Cancer Inst. 100, 170–183.
Saad, F., Aversa, A., Isidori, A. M., Zafalon, L., Zitzmann, M., and Gooren, L. (2011). Onset of effects of testosterone treatment and time span until maximum effects are achieved. Eur. J. Endocrinol. 165, 675–685.
Keywords: testosterone gel, myocardial infarction, metabolic syndrome, localized prostate cancer
Citation: Aversa A, Francomano D and Lenzi A (2012) Cardiometabolic complications after androgen deprivation therapy in a man with prostate cancer: effects of 3 years intermittent testosterone supplementation. Front. Endocrin. 3:17. doi: 10.3389/fendo.2012.00017
Received: 07 November 2011; Paper pending published: 01 December 2011;
Accepted: 20 January 2012; Published online: 16 February 2012.
Edited by:
Claire Perks, University of Bristol, UKReviewed by:
Giovanni Vitale, Istituto Auxologico Italiano – Universita’ Degli Studi Di Milano, ItalyAndrea Garolla, Università di Padova, Italy
Copyright: © 2012 Aversa, Francomano and Lenzi. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Antonio Aversa, Department of Experimental Medicine, Section of Medical Physiopathology, Sapienza University of Rome, Viale Regina Elena 324, 00161 Rome, Italy. e-mail:YW50b25pby5hdmVyc2FAdW5pcm9tYTEuaXQ=