 Mark Galdo
Mark Galdo

- Laboratory for Spinal Cord Development and Regeneration, Department of Neurological Surgery, University of California, San Francisco, CA, USA
Dehydroepiandrosterone (DHEA) is synthesized from cholesterol by activity of P450scc and P450c17, enzymes that we previously characterized in the developing nervous system. We describe the localization of P450c17 in the differentiated field of the ventral spinal cord in different motor neuron subtypes. We show that, during organogenesis, P450c17 activity is regulated along the antero/posterior axis of the spinal cord concomitantly with the gradient of neurogenesis. To examine whether DHEA may modulate this process, we measured proliferation and differentiation of ventral neural precursors in primary and explant cultures. Our results showed that DHEA-induced the expression of class II protein Nkx6.1, motor neuron precursor Olig-2, and definitive motor neuron marker Isl-1/2. DHEA also promoted proliferation of ventrally committed precursors in isolated spinal cord precursor cultures and in whole spinal cord explants. Both the proliferative and inductive effects of DHEA were dependent on sonic hedgehog signaling. The possibilities that the effects observed with DHEA were due to its metabolism into androgens or to activation of NMDA receptors were excluded. These results support the hypothesis that the tight regulation of DHEA biosynthesis may be a biologic clock restricting the period of ventral neuronal-precursor proliferation, thus controlling the number of pre-committed neurons in the developing neural tube.
Introduction
The topography of the central nervous system (CNS) results from an orchestrated control of cell proliferation and motility, maturation of neuronal and glial cells, axonal growth, and the establishment of proper synapses. Neural specification and patterning of the CNS are controlled by the combinatory expression of genes in the neural tube during development, establishing a code for neuronal identity in restricted specific regions (Rubenstein and Beachy, 1998). Sonic hedgehog (Shh) is one of the major morphogens controlling the expression of these homeodomain proteins and plays a primordial role in dictating cell fate in neural tube precursors through a graded signaling activity along the ventro-dorsal axis (Briscoe et al., 2000; Jessell, 2000; Lee and Pfaff, 2001).
Patterning activity is not the only function of Shh. Mitogenic activity of Shh has been observed in several tissues and organisms during their development (reviewed in Ingham and McMahon, 2001; Brito et al., 2002; Marti and Bovolenta, 2002). In addition, genetic alterations of Shh biogenesis or signal transduction lead to CNS hyperplasia (reviewed in Litingtung and Chiang, 2000; Ruiz i Altaba et al., 2002; Wetmore, 2003). In the neural tube, proliferative effects of Shh have been demonstrated in vitro and in vivo (Kalyani et al., 1998; Rowitch et al., 1999). However, environmental cues regulate the ability of precursor cells to proliferate in response to Shh (Rowitch et al., 1999; Barzi et al., 2010). We proposed that the neurosteroid dehydroepiandrosterone (DHEA) might be one such cue controlling the proliferative response of precursors to Shh.
DHEA is a secreted neurosteroid biosynthesized de novo in the developing CNS from the sequential action of two cytochrome P450 enzymes, P450scc and P450c17 (Compagnone and Mellon, 2000). To determine whether DHEA is produced in the regions where we have found P450c17 expressed and what its bioactivity may be, we focused our study on the developing spinal cord where P450c17 expression is first observed.
In this report, we establish a correlation between the expression and activity of P450c17 and show that P450c17 activity, and thus most likely DHEA biosynthesis, are regulated along the antero/posterior (A/P) axis of the developing spinal cord concomitantly with the gradient of neurogenesis. DHEA mimicked Shh signaling to promote the induction of motor neurons and sustained ventral progenitor proliferation in a Shh-dependent manner in vitro. The results of this work support the hypothesis that the tightly regulated biosynthesis of DHEA may be a biologic clock restricting the period of ventral neuronal-precursor proliferation and thus controlling the number of pre-committed neurons in the developing neural tube.
Materials and Methods
All experiments were reviewed and approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco, CA, USA.
Reagents and Chemicals
DHEA, BrdU, poly-D-lysine, progesterone, and streptavidin–FITC were purchased from Sigma/RBI (St. Louis, MO, USA); Collagenase H, insulin–transferrin–sodium selenium supplement, and the terminal deoxynucleotidyl transferase biotin d-UTP nick-end labeling (TUNEL) “In situ Cell Death Detection Kit,” Fluorescein from Roche Diagnostics (Indianapolis, IN, USA); and bFGF, Leibovitz’s L15 medium, high-glucose formula of DMEM, and HAM-F12 medium from Invitrogen/Gibco (Carlsbad, CA, USA). [3H]pregnenolone was obtained from NEN/Perkin-Elmer Life Sciences (Boston, MA, USA). The following antibodies were purchased from the Developmental Studies Hybridoma Bank (DSHB, University of Iowa, IA, USA): anti-Shh (5E1), Isl-1 (40.2D6), Lim1/2 (4F2), and Pax6. The anti-Shh (5E1), anti-Isl-1 (40.2D6), and anti-Lim1/2 (4F2) antibodies, developed by Dr. Thomas Jessell, the anti-Pax6 and the anti-Pax7 antibodies, developed by Dr. Atsushi Kawakami were fully characterized prior to their use in this paper, Hoechst, Alexa 546-coupled goat anti-mouse, Alexa 350-coupled goat anti-rabbit, neutravidin–Alexa 350, wheat-germ agglutinin, and the FluoroReporter Mini-biotin protein labeling kit were obtained from Molecular probes (Eugene, OR, USA). Goat anti-rabbit coupled to Phycoerythrin was bought from Chemicon (Temecula, CA, USA) and goat anti-mouse coupled to FITC from Jackson ImmunoResearch Laboratories Inc., (West Grove, PA, USA).
Analysis of Enzymatic Activity
Embryos were obtained from CD-1 mice (Charles River, Wilmington, MA, USA) at different ages from embryonic day (E) 10 to E16.5 (E0.5 = day of vaginal plug). Spinal cords were dissected in L15 medium containing 5% heat-inactivated horse serum. The spine was exposed after removal of the ventral organs. The notochord was peeled off and collected. Mesenchyme surrounding the spinal cord was carefully removed and collected, as were the dorsal root ganglia (DRG). The spinal cords were opened dorsally, flattened, and then cut into dorsal (roof plate + alar plate) and ventral (floor plate + motor columns) segments. Dorsal and ventral halves at all A/P levels were collected and analyzed separately. The genital ridges/gonads were collected as a positive control for P450c17 activity. Parts collected from three embryos were routinely pooled for the analyses and yielded reproducible and significant levels of DHEA biosynthesis.
The segments analyzed were homogenized in 0.25 M sucrose, 0.5 mM EDTA, and 10 mM Tris–Cl, pH 7.4, at 4°C and incubated with [3H]pregnenolone for 16 h at 37°C. Tissues were then extracted with isooctane:ethyl acetate (1:1, v:v) and dried under N2 before analysis with thin-layer chromatography (TLC), using a solvent mix of chloroform:ethyl acetate (1:3, v:v). Radiolabeled steroids were identified by using steroid standards and a purified preparation of P450c17-expressing yeast microsomes. Quantification of the percent conversion was obtained by using a Storm phospho-imager (Molecular Dynamics, Amersham Biosciences, Piscataway, NJ, USA) and Imagequant software. At least three independent experiments were performed for each segment and for each time point.
Spinal Cord Precursors: Primary Cultures
Dissections were performed in L15 medium. E10.5 CD-1 mouse embryos were transected below the otic vesicle and in the center of the hindlimb bud. The segment of the spinal cord used extended from the center of the hindlimb bud to three somites rostrally, and only the ventral and intermediate neural plates were retained. Dissected segments were mechanically separated from the mesenchyme and dissociated with collagenase H (0.045 U/ml). Isolated cells were cultured on poly-D-lysine-coated glass coverslips at 43,000 cells/cm2 in a serum-free medium (DMEM/F12) containing 50 ng/ml basic fibroblast growth factor (bFGF), 5 μg/ml insulin, 5 μg/ml transferrin, 3 × 10−8 M selenium, 2 × 10−8 M progesterone, and, when tested, DHEA at 10−10 M. When stated, cells were grown in the presence of the 3 μg/ml 5E1 anti-Shh antibody raised against the N-terminal fragment of Shh.
After various times in culture, ranging from 3 to 45 h, BrdU (10 μM) was incorporated for 1 h and cells were washed in PBS and fixed with glycine:ethanol (3:7, v:v) for 45 min at room temperature. DNA was denatured by using 4 M HCl for 10 min. Cells were washed and immunostained for BrdU (mouse anti-BrdU antibody used at 1:50), for NeuN (biotinylated mouse anti-NeuN antibody used at 1:200), for Isl-1 (rabbit anti-Isl-1/2 antibody, gift from Dr. Pfaff, used at 1:2,000), for Nkx6.1 (rabbit anti-Nkx6.1, gift from Dr. German, used at 1:4,000), and for Pax6 (DSHB, used at 1:20). Secondary antibodies were a goat anti-mouse-Alexa 546, a streptavidin–FITC, and a goat anti-rabbit-Alexa 350, which were used at 1:500, 1:200, and 1:500 respectively. TUNEL analysis was performed by using the “In situ Cell Death Detection Kit” as described previously (Hagedorn et al., 2000). Total cell number was determined on each slide after Hoechst or fluorescent-coupled wheat-germ agglutinin counter-staining.
At least five independent experiments were performed for each time point. Cells immunopositive for BrdU, NeuN, Isl-1, Nkx6.1, Pax6, Chx10, Eng-1, or O4 were counted in 10 randomly selected 1 mm2 fields/slide; three slides were counted per treatment and per experiment. A general factorial multiple analysis of variance was performed to determine the effects of the time in culture and of the presence of DHEA in the culture medium, as well as a possible interaction between these two factors.
Histochemistry
In situ hybridization and immunochemistry in mouse embryos were performed as described previously (Compagnone et al., 1995a).
DHEA-producing neurons were identified by using an anti-P450c17 antibody (gift from Dr. Miller) used at 1:5,000. Motor neurons were immunodetected with a mouse anti-Isl-1 antibody at 1:50. When dual or triple labeling was performed, anti-Isl-1 antibody was biotinylated. Isl-1 immunoreactive neurons were then revealed with neutravidin–Alexa 350 used at 1:1,000. V2 interneurons were immunodetected by using an anti-Chx10 antibody (gift from Dr. Pfaff) at 1:50. The pool of ventrally committed precursors was immunodetected with an anti-Nkx6.1 antibody (gift from Dr. German) at 1:4,000, and the dorsally committed neurons were immunodetected with an anti-Pax6 antibody (DSHB) used at 1:20. Pools of motor neurons were identified by using anti-Isl-1/2 (40.2D6, DSHB) at 1:100, anti-Isl2 (gift from Dr. Pfaff) at 1:2,000, anti-Lim1/2 (4F2, DSHB) at 1:100, anti-Lim3 (gift from Dr. Pfaff) at 1:3,000, and anti-HB9 (gift from Dr. Pfaff) at 1:8,000. Secondary antibodies were an anti-rabbit-Phycoerythrin (Chemicon) and an anti-mouse-FITC (Jackson ImmunoResearch), both used at 1:200.
Three-Dimensional Spinal Cord Explant Cultures
Whole neural tube explants
The assay was performed as previously described (Colamarino and Tessier-Lavigne, 1995). Spinal cords were removed from E11.5 mouse embryos in L15 medium and, after elimination of the mesenchyme, the mid-lumbar, and sacral segments were collected. Explants were dissected out of the spinal cord, flattened, and then were cut to approximately 800 μm in length. They were placed in 0.04 ml collagen beads made fresh from rat-tail tendons, and cultures were carried out in the same medium as the spinal cord precursor cultures. When specified, 10−10 M of DHEA or 16-fluoro-5-androsten-17-one (fluasterone) and/or anti-Shh antibody (3 μg/ml) were added to the culture medium. After 21 h of culture, BrdU incorporation (10 μM) was carried out for a pulse of 3 h to permit diffusion of the reagent through the collagen gel. Explants were fixed with 4% paraformaldehyde as stated earlier. BrdU detection was performed as described earlier and quantified. Results were obtained from at least four independent experiments.
Intermediate Neural Plate Explants: Motor Neuron Induction Assay
Intermediate neural plates were dissected out of E10 mouse neural tubes following a procedure adapted from that described for the chick embryo (Roelink et al., 1995; Ericson et al., 1996; Incardona et al., 1998). Isolated intermediate neural plates were cut into 100 μm pieces and cultures in collagen gel matrices as described earlier. Explants were grown in DMEN-F12 1:1 medium containing 50 ng/ml FGF, 5 μg/ml insulin, 5 μg/ml transferrin, 3 × 10−8 M selenium, and 2 × 10−8 M progesterone for 12 h before receiving the N-terminal fragment of Shh (R&D System Inc., Minneapolis, MN, USA) at 2–20 nM ±DHEA or ±DHEA, and specific inhibitors of Shh signaling (5E1 antibody used 3 μg/ml or 1.2 μM cyclopamine (TRC Inc., Toronto, CA, USA). After 24 h in the experimental medium, explants were rinsed in PBS and fixed in 4% PFA for 24 h. Explants were processed for immunocytochemical determination of the expression of class II and class I homeodomain proteins (Nkx6.1 and Pax7, respectively), Olig-2, and Isl-1/2. Results were obtained from at least four independent experiments. A minimum of eight different explants per experimental condition were quantified. The quantification of Nkx6.1, Pax7, Olig-2, and Isl-1/2 immunopositive nuclei was performed using a computer assisted imaging software (Slidebook from Intelligent Imaging Innovation,) specialized in such morphometric analyses. This software allows the recognition of both in signal intensity and the shape (size) of the object stained allowing for a reduction of the bias when assessing the number of nuclei in field containing a high density of stained nuclei.
Statistical Analysis
Results are presented as means ± SEM. MANOVA and ANOVA algorithms were used to determine differences among groups using SPSS software (SPSS Inc., Chicago, IL, USA) and Bonferroni’s post hoc test (significance established at 95%). Asterisks indicate statistically significance levels, * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
Results
Expression of P450c17 in the Developing Motor Neurons
We previously identified the distribution of P450scc and P450c17, enzymes crucial for the production of DHEA, in the developing nervous system (Compagnone et al., 1995a,b). P450c17 expression is regionally and developmentally regulated in neurons of the CNS, including those in the position of motor neurons in the spinal cord (Compagnone et al., 1995a). We therefore asked if the expression pattern of P450c17 was temporally and regionally regulated within the different subtypes of neuronal populations in the developing neural tube. P450c17 was expressed early in development in the cervical neural tube (E9–E10) and later in the brachial/thoracic (E10–E11) and in the lumbar (E12.5) segments (data not shown). At E13.5, P450c17 was expressed at all levels of the developing spinal cord, with the weakest signal seen at the brachial level (Figure 1A). P450c17 was observed in both the ventral and dorsal regions of the spinal cord. In the dorsal region, P450c17 antibody labeled commissural neurons whose axons crossed the floor plate ventrally (not shown). In the ventral spinal cord, P450c17 was expressed in cell bodies and fibers in a ventro-medio-lateral position, suggesting that P450c17 could be localized either in precursor cells migrating out of the sub ventricular zone (SVZ) or in cells that have already acquired a differentiated phenotype but are still located close to the SVZ boundary. To determine the cellular localization of P450c17 in the ventral spinal cord, we performed dual immunostaining or dual immuno-hybrido-staining with Nkx2.2 and Nkx6.1, markers for ventrally committed precursors (Jessell, 2000; Lee and Pfaff, 2001). Although P450c17 and Nkx2.2 mRNA were observed in the same regions in the E10.5 spinal cord, they were not expressed in the same cells: Nkx2.2 mRNA was generally expressed in cells clustered in a more medial position than the cells expressing P450c17 (data not shown). P450c17 co-localized with Nkx6.1 only in cells migrating in the differentiated field in E10.5 mouse embryos (data not shown), but its expression domain was not restricted to Nkx6.1 immunopositive cells. P450c17 expression was also seen in cells in the differentiated field in the position of motor neurons. To identify the subpopulations of cells in which P450c17 was expressed, we used Isl-1 as a marker for motor neurons and Chx10 as a marker for V2 interneurons (Jessell, 2000; Lee and Pfaff, 2001). In E13.5 mouse embryos, we found P450c17 localized in a subset of Isl-1+ motor neurons but not in Chx10+ interneurons (data not shown). In an attempt to further characterize P450c17 expression in motor neuron pools, we used HB9 as a marker of pre-mitotic motor neurons, Isl-1 as a marker of post-mitotic motor neurons and Isl2, Lim1/2, and Lim3 as markers for the different columnar identities (Jessell, 2000). At E13.5, P450c17 was expressed in motor neuron immunopositives for Isl-1 or HB9 pre-mitotic neurons (Figures 1B,C). It co-localized with subsets of cells immunopositive for Isl2, Lim1/2, and Lim3 (Figures 1B,C). Our results showed that P450c17 did not segregate with a particular population of motor neurons, but rather that it was expressed by motor neurons in the ventro-medio-lateral region of the differentiated field (see Figure 1D). This finding suggests that DHEA may be secreted by this population of neurons and thus may act on neighboring cells.
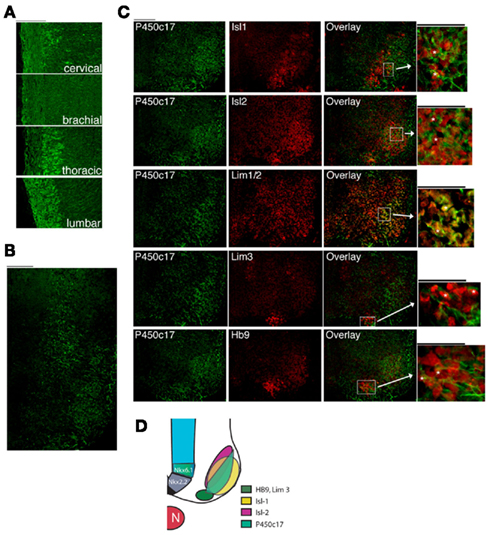
Figure 1. P450c17 expression in the developing spinal cord in the mouse. (A) Immunostaining of P450c17 in E13.5 sagittal sections of the spinal cord. (B) Co-expression of P450c17 (green) with Isl-1, Isl2, Lim1/2, Lim3, and HB9 (red) in an E13.5 transverse section of whole embryo (thoracic level of the spinal cord). For each marker, an overlay with P450c17 is shown, as well as a high magnification view. P450c17 being a microsomal protein, its staining is found both in cell bodies and in fibers, whereas Isl-1, Isl2, Lim1/2, Lim3, and HB9 are nuclear proteins. Asterisks indicate cells dually labeled. (A–C) Bars equal 100 μm. (D) Schematic representation of the expression domains of P450c17 in the different domains of motor neuron subtypes markers.
P450c17 Activity in the Developing Neural Tube
To establish a correlation between the protein expression and the enzymatic activity of P450c17, we performed TLC analysis of radiolabeled metabolites of [3H]pregnenolone, the substrate of P450c17 (Figure 2A). Conversion of pregnenolone into DHEA results from the sequential action of the two enzymatic activities of P450c17: 17α-hydroxylase and 17–20 lyase activities. Both activities are represented in Figure 2 as a percent conversion of pregnenolone into DHEA (Figures 2B,D) and into 17-OH pregnenolone (Figures 2C,D). As positive controls, we used gonadal ridges of similar developmental stages (averaging a basal percent conversion of pregnenolone into DHEA of 5%, not significantly modified during embryogenesis) and a preparation of yeast over-expressing P450c17 (Auchus et al., 1998) or adult male testis homogenates. To determine whether P450c17 activity was regulated in the D/V axis of the developing neural tube, we dissected ventral and dorsal regions of the brachial/thoracic spinal cord of mouse embryos at different embryonic ages and measured P450c17 activities (Figures 2B,C). Similarly, to determine if DHEA biosynthesis was regulated along the A/P axis, we dissected different regions of the developing spinal cord at different A/P levels (cervical, brachial, thoracic, and lumbar) in E10.5–E16.5 embryos (Figure 2D).
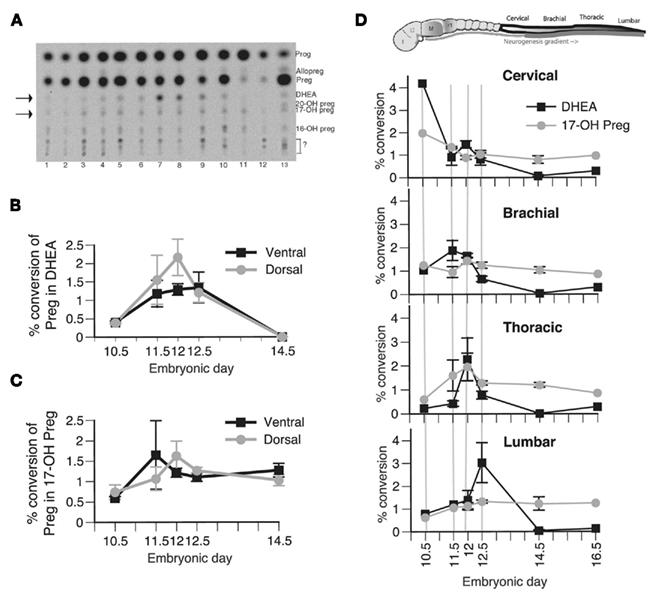
Figure 2. Biosynthesis of steroid products derived from P450c17 activity in vivo in the developing spinal cord. (A) Typical TLC plate from E12.5 embryos. 1: spinal cord (SpC) cervical ventral, 2: SpC cervical dorsal, 3: SpC brachial ventral, 4: SpC brachial dorsal, 5: SpC thoracic ventral, 6: SpC thoracic dorsal, 7: SpC lumbar ventral, 8: SpC lumbar dorsal, 9: genital ridge, 10: DRG, 11: notochord, 12: mesenchyme, 13: brain. Preg, pregnenolone; Prog, progesterone. Arrows point to DHEA and 17-OH pregnenolone. (B,C) Biosynthesis of DHEA (B) and 17-OH pregnenolone (C) was determined in the ventral (black line) and dorsal portion (gray line) of the brachial–thoracic segment of the spinal cord sampled at E10.5, E11.5, E12, E12.5, and E14.5. A general factorial analysis was performed to assess the significance of the position of the sample along the ventro-dorsal axis and the embryonic age on either the percent conversion of pregnenolone into DHEA or 17-OH pregnenolone. No difference was found. (D) Biosynthesis of DHEA (black line) and 17-OH pregnenolone (gray line) was determined in the cervical, brachial, thoracic, and lumbar segments of the spinal cord sampled at E10.5, E11.5, E12, E12.5, E14.5, and E16.5. The results show the average percent conversion of pregnenolone into DHEA and 17-OH pregnenolone of the ventral and dorsal measurements that were pooled for subsequent analysis, as no significant differences were observed between these two groups. A general factorial analysis showed a significant interaction between the segmental position along the A/P axis and the embryonic age only on DHEA biosynthesis (p < 0. 05) but not on 17-OH pregnenolone biosynthesis. One-way ANOVA showed that the peak of DHEA biosynthesis traveling along the A/P axis is significant for at least one of the two factors studied (either position along the A/P axis or embryonic age). Values represent the means ± SEM of 20 independent measures of samples from pools of three embryos.
Biosynthesis of DHEA was observed in both the ventral and the dorsal regions of the spinal cord (Figure 2B), consistent with P450c17 expression. No DHEA was produced in the mesenchyme or in the notochord, where P450c17 is not expressed (Figure 2A, lanes 11–12). No further conversion of DHEA into androgens was observed in the embryonic nervous system tissue.
In the brachial–thoracic region of the spinal cord, no significant differences were observed between dorsal and ventral biosynthesis of DHEA or 17-OH pregnenolone at the different embryonic ages analyzed (Figures 2B,C). However, DHEA biosynthesis was regulated along the A/P axis during the development of the spinal cord (Figure 2D). A two-way analysis of variance showed a significant interaction between the segmental position of the sample analyzed along the A/P axis and the embryonic age. We thus performed one-way analysis on the percent conversion of pregnenolone into DHEA for each of these factors and observed a significant peak of DHEA biosynthesis traveling along the A/P axis during embryogenesis (Figure 2D, black line). This peak was observed in the cervical region of the spinal cord at E10.5 (the earliest time point analyzed), in the brachial region at E11.5, in the thoracic part at E12, and in the lumbar portion at E12.5. The peak of DHEA biosynthesis was thus concomitant with the rostro-caudal gradient of neurogenesis described in the developing neural tube (Nornes and Carry, 1978). No significant changes were observed in the percent conversion of pregnenolone into 17-OH pregnenolone (Figure 2D, gray line). The average peak value of DHEA biosynthesis is about 2.85% (±1.02%), a value comparable to those obtained from the mouse adult testis, where DHEA serves as precursor for androgens (not shown). These low conversion levels were close to the sensitivity limit of the technique used but were readily quantifiable and reproducible, and DHEA production was confirmed by HPLC (not shown).
These results show that P450c17 expression was associated with P450c17 activity, and that DHEA was most likely produced in the ventral and dorsal spinal cord at a time consistent with its potential role in the modulation of neurogenesis or differentiation of neural precursors. DHEA is a lipophilic steroidal compound that can cross the plasma membrane. It may therefore act from a distance on cells in the SVZ. For the rest of this report we focused our investigations on the ventral regions of the neural tube in the aim to determine the role of DHEA.
Effect of DHEA Proliferation and Neuronal Differentiation in Isolated Spinal Cord Precursor Cultures
DHEA affects proliferation of ventrally committed precursors and differentiation of motor neurons in vitro
DHEA biosynthesis occurs in the developing spinal cord concomitantly with the gradient of neurogenesis and neuronal differentiation. DHEA may thus modulate proliferation or differentiation of cells present in the region where it is secreted. To test this possibility we cultured spinal cord precursors isolated from E10.5 ventral neural tube in a serum-free medium in the presence or in the absence of DHEA for 3–45 h. We studied the effect of DHEA at a concentration of 10−10 M, which has proved to be efficient as previously determined on embryonic neocortical cultures (Compagnone and Mellon, 1998). We first analyzed the effect of DHEA on proliferation, using BrdU incorporation performed as a 1 h pulse before fixation of the cells. DHEA significantly influenced the dynamics of the culture and increased both the number and the percent of cells proliferating over time when compared to control conditions (p < 0.0001, MANOVA). To obtain greater detail we compared the proliferation of cells cultured in the presence of DHEA to that of cells cultured in the absence of DHEA at different time points (every 3–5 h from 3 to 35 h and every 10 h thereafter) by using one-way ANOVA. The number of BrdU-incorporating cells increased in the presence of DHEA at nearly all time points examined (Figure 3A). The percentage of cells incorporating BrdU over the total cell number showed time periods during which proliferation was very significantly increased in the presence of DHEA (Figure 3B). Increased BrdU incorporation was particularly evident in DHEA-treated cultures after 7 h (with 38.4% ± 1.2 cells in S-phase), 18 h (with 48.3% ± 1.4 cells in S-phase), and 35 h (with 62.5% ± 7.5 cells in S-phase) as compared to a relatively stable 30% proliferation rate over time in the control conditions (28.6% ± 1.0, 33.5 ± 0.9, and 36.2 ± 2.7 cells in S-phase respectively). This pattern of BrdU incorporation was particularly intriguing and suggestive of active cycling in the DHEA-treated cultures. Consistent with increased proliferation in the DHEA-treated culture, the number of cells/cm2 increased over time to a higher extent in DHEA-treated cultures than in control cultures (Figure 3C). In the presence of DHEA, the growth rate abruptly dropped after 40–45 h in culture, which was indicative of cell death not seen in the control condition, where the growth curve had stabilized (Figure 3C). We then analyzed apoptosis in the culture by using the TUNEL assay. The apoptotic rate was indistinguishable between the two culture conditions from 3 to 40 h in culture. After 40–45 h in culture, a larger number of cells died from apoptosis in the cultures grown in the presence of DHEA. However, apoptosis did not fully account for the total number of lost cells at that time, so other cell death mechanisms may be involved. This result nonetheless demonstrated that DHEA does not sustain survival of the cells over time.
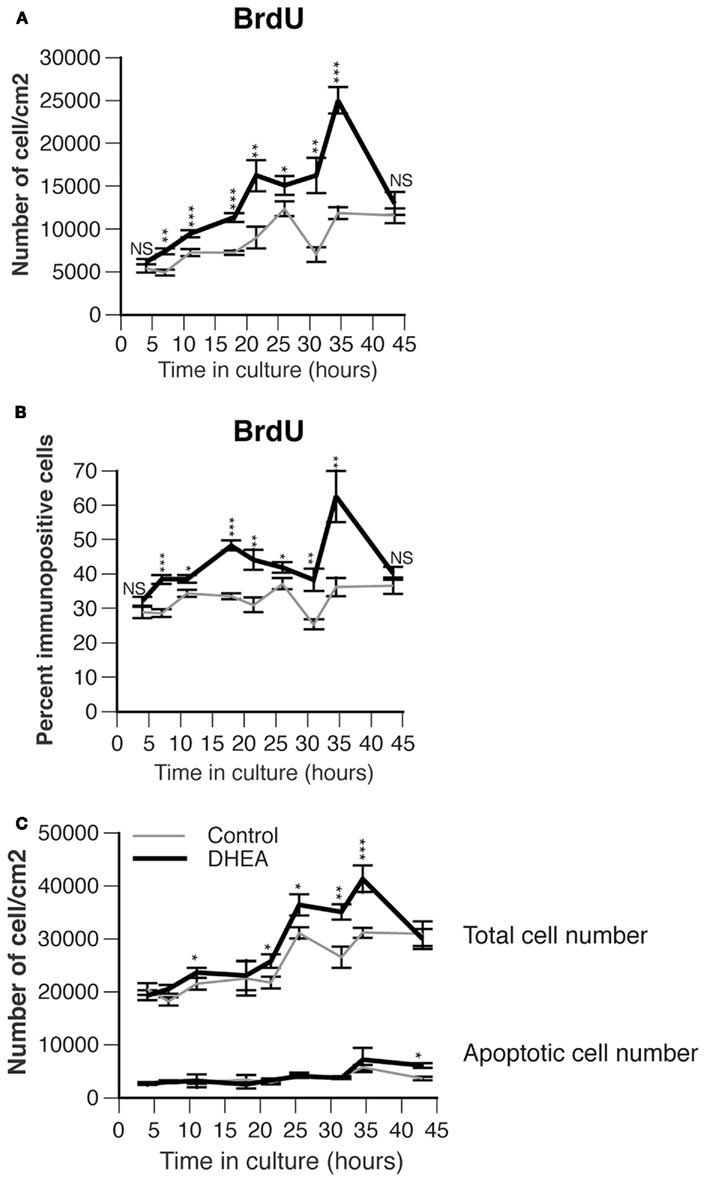
Figure 3. Effect of DHEA on proliferation of precursor cells isolated from E10.5 mouse neural tube lumbar region in primary culture. (A) Number of immunopositive cells for BrdU. Cells were incubated for different period of time in the absence (control, gray line) or presence of 10−10 M DHEA (black line) and immunostained for BrdU. Average values are expressed as a variable of time in vitro, error bars delineate the SEM. A multiple analysis of variance (MANOVA) was performed to assess the percent variance explained by the time in culture versus that explained by the presence of DHEA in the culture medium. Differences between groups for each of these factors were determined by using one-way ANOVA and Bonferroni’s post hoc test. Asterisks show significant differences between control and DHEA-treated cultures at each time point. (B) Percent of BrdU immunopositive cells per total cell number. Values represent the means ± SEM of five independent platings. Analyses similar to those in A were performed and asterisks show the significance level between control and DHEA-treated cultures at each time point. (C) Effect of DHEA on growth curve and number of apoptotic cells in primary culture. Cells were cultured in the presence (black line) or absence (control, gray line) of DHEA. The total cell number was counted after staining with Hoechst or wheat-germ agglutinin coupled to a fluorochrome and expressed as a number of cells/cm2. Apoptosis was assessed with TUNEL analysis and expressed as a number of apoptotic cells/cm2. Values represent the means ± SEM of five independent platings.
To determine if the proliferating cells were committing to a motor neuron fate in culture, we studied the expression of Nkx6.1 (a marker of ventrally committed precursors), Pax6 (a marker of dorsally committed precursors), and Isl-1/2 (a marker of definitive motor neurons). The presence of DHEA in the culture medium increased the percentage of Nkx6.1+ cells (Figures 4A,B), but not that of Pax6+ cells (Figure 4C), at the early time points, with highly significant differences between the DHEA-treated and control cultures at 7–10 h and 18–21 h). We were interested to find that the percentage of Nkx6.1+ cells was increased in DHEA-treated cells at the same time points when BrdU incorporation was observed, possibly indicating that Nkx6.1+ cells could incorporate BrdU. To test this hypothesis, we performed dual immunohistochemistry and found that the percentage of dually labeled cells for BrdU and Nkx6.1+ cells was significantly higher in DHEA-treated cultures after 7 h (35.3% ± 1.5) and 18 h (30.4% ± 1.4) in vitro compared to 21.4% ± 1.7 and 20.4% ± 0.8 in the control conditions respectively (Figure 4B), indicating that Nkx6.1+ populations represented the majority of cells incorporating BrdU at these time points. At later time points, the percentage of Nkx6.1+ cells incorporating BrdU was reduced in DHEA-treated culture and did not differ from control conditions, but they still accounted for about 20% of the cells incorporating BrdU. We were also interested that the percentage of Isl-1/2+ cells significantly increased in DHEA-treated culture compared to control cultures with a 8–9 h delay, as compared to the DHEA-mediated increase observed in Nkx6.1+ cells. Isl-1/2+ cells represented 30% of the total cell number after 18 h and 26 h in culture in the presence of DHEA, as compared to 13 and 15% respectively in control conditions (Figure 4D), suggesting that the Nkx6.1+ population may have been further committed into differentiating in motor neurons in the cultures grown in the presence of DHEA. These experiments indicate that DHEA promoted proliferation of ventrally committed precursors and sustained their differentiation into motor neurons in vitro.
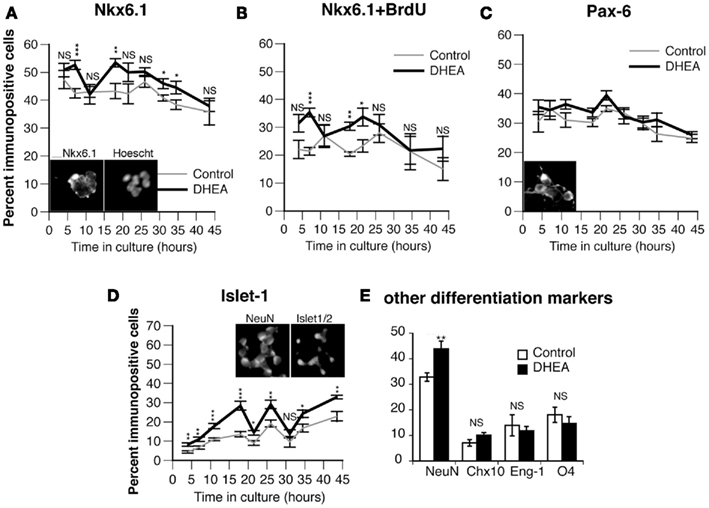
Figure 4. Effect of DHEA on the proliferation of committed precursors in cultures and on the generation of motor neurons in vitro. Cells isolated from E10.5 mouse neural tube lumbar region were cultured in the presence (black line) or in the absence (control, gray line) of DHEA for different periods of time and stained for BrdU and Nkx6.1. (A) Percent of Nkx6.1+ cells. (B) Percent of dividing Nkx6.1+ cells (dually labeled Nkx6.1 and incorporating BrdU). (C) Percent of cells expressing Pax6. (D) Percent of Isl-1+ cells. (E) Percent of other differentiated populations including NeuN+, Eng-1+, Chx10+, O4+ 30 h after seeding. Values represent the means ± SEM of five independent platings. Statistical analysis was performed as described in Figure 3. Inset pictures show a representative staining of Nkx6.1, Pax6, Isl-1/2, and NeuN. Scale above Nkx6.1 inset = 10 μm.
Our results show that BrdU incorporation increased in the presence of DHEA, even after a long period in culture (the last peak being observed between 30 and 40 h), but this increase did not correspond to an increase in the Nkx6.1+ population. Similarly, although the Isl-1/2+ population was still significantly increased in DHEA-treated cultures after 30–40 h in vitro, the percent of motor neurons produced in control conditions increased, reducing the difference between the two culture conditions. Moreover, the total number of cells decreased in the culture grown in the presence of DHEA, thus increasing the percentage of motor neurons that survived the culture conditions rather than being actively differentiated in vitro. These results indicate that cells produced during the last peak of proliferation are unlikely to be motor neurons. To identify the possible cell types produced in the cultures by the last peak of proliferation, we examined the expression of markers of definitive states of various ventral neurons, including Eng-1 (a marker of committed V1 interneurons) and Chx10 (a marker of committed V2 interneurons). We also studied the expression of NeuN (a marker of fully differentiated neurons) and that of 04 (a marker of immature oligodendrocytes) after 30 h in culture (Figure 4E). DHEA significantly increased the neuronal differentiation as assessed by an increased percentage of NeuN+ cells in the culture but did not significantly alter the differentiation of interneuron populations. Similarly, the population of immature oligodendrocytes was not significantly different in cultures grown in the presence or in the absence of DHEA. This result indicates that DHEA may affect not only the proliferation of motor neuron precursors and the differentiation of motor neurons, but also the proliferation of other neuronal populations that have yet to be identified.
The DHEA-mediated increase in neuronal differentiation requires the Shh pathway
To determine the mechanism by which DHEA increased Nkx6.1+ cell proliferation and motor neuron differentiation, we first examined the possibility of DHEA’s acting as an agonist of the NMDA receptor (Bergeron et al., 1996; Compagnone and Mellon, 1998). We found no inhibition of DHEA-mediated effects by the NMDA non-competitive agonist MK801 (not shown), which is consistent with the lack of expression of the NMDA receptor at this early stage (Maric et al., 2000). We then reasoned that the effects observed with DHEA recapitulated those observed with Shh (Yamada et al., 1993; Roelink et al., 1995). As Shh can be synthesized and secreted in our cultures containing induced floor plate cells, we investigated the Shh signaling pathway to see whether it was involved in DHEA-mediated effects by using the 5E1 antibody raised against Shh. 5E1 antibody blocks Shh signal transduction by interfering with the binding of Shh to Patched (Fuse et al., 1999; Pepinsky et al., 2000). We chose to study inhibition of motor neuron differentiation in the cultures at a relatively late stage (35 h after seeding) in order to obtain enough motor neurons in the control conditions to reliably observe inhibition and yet observe a significant difference between both experimental conditions. The addition of 5E1 antibody in the control conditions significantly blocked motor neuron differentiation but did not change the fraction of BrdU-incorporating cells (Figure 5A). When this antibody was added to cells grown in the presence of DHEA, we observed a total inhibition of DHEA-mediated effects, not only on motor neuron differentiation but also on BrdU incorporation (Figure 5A). These findings indicate that the proliferating and differentiating effects of DHEA are dependent on Shh signaling. As all of these events were possibly initiated earlier during culture by the DHEA-mediated increase in Nkx6.1+ cell proliferation, we studied dual expression of Nkx6.1 and BrdU incorporation in cultures grown in the presence of 5E1 antibody for a shorter period (21 h after seeding). Addition of 5E1 antibody had no effect on BrdU incorporation or on the Nkx6.1 precursor population in the control cultures (Figure 5B). In the presence of DHEA, however, addition of the 5E1 antibody reduced the number of BrdU+, Nkx6.1+, and dually labeled BrdU/Nkx6.1+ cells to the levels seen in the control cultures, indicating that: (i) the DHEA-mediated increase in Nkx6.1+ cell proliferation is dependent on Shh signaling pathway; and (ii) the proliferation of Nkx6.1+ cells precedes motor neuron differentiation in our culture system and reduction of this phenomenon leads to a significant reduction of the DHEA-mediated differentiation of motor neurons in vitro. The use of an unrelated antibody had no significant effects on any of these outcomes (Figure 5B), indicating specificity of the inhibition observed by 5E1.
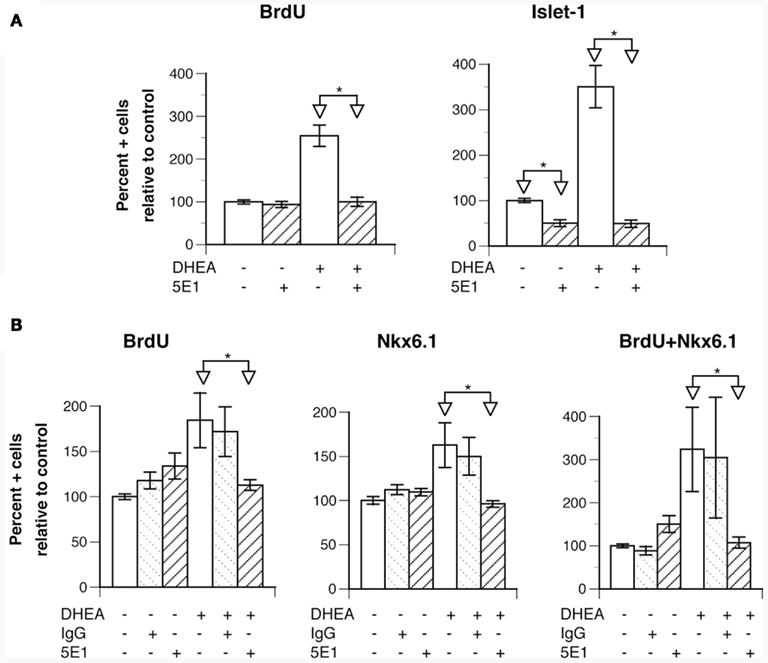
Figure 5. The effects of DHEA on proliferation and neuronal differentiation are dependent upon Shh signaling. Cells isolated from E10.5 mouse neural tube lumbar region were used. (A) Effect of the addition of the anti-Shh 5E1 blocking antibody (used at 3 μg/ml) in control and DHEA-treated cultures grown for 35 h on BrdU incorporation and Isl-1 expression. The DHEA-mediated increase in BrdU incorporation and Isl-1 expression is significant in comparison to the control conditions p ≤ 0.05). (B) Effect of addition of the 5E1 blocking antibody in control and DHEA-treated cultures grown for 21 h on BrdU incorporation and Nkx6.1 expression (The effect of DHEA on BrdU incorporation and Nkx6.1 expression is significant in comparison to the control conditions p ≤ 0.05). Average data from three independent platings are shown expressed as percent of control values, error bars are SEM. Statistical analysis was performed as described in Figure 3.
These results show a primary effect of DHEA on the proliferation of the Nkx6.1+ population that translates into an increased differentiation of motor neurons. This effect is clear during the first 30 h in culture, but then becomes more difficult to follow. In the late period of culture, increased proliferation does not occur in the Nkx6.1 population; nonetheless motor neuron differentiation is sustained, although to a lesser extent. This could be due to: (i) a loss of synchronization in the cell cycle of dividing precursors, making the proliferation and differentiation phases overlap; (ii) an increased survival of motor neurons in the presence of DHEA (even though the rate of apoptosis and the total cell number abruptly drop in the late cultures grown in the presence of DHEA); or (iii) a possible direct effect of DHEA on the induction of motor neurons superimposed on the proliferative effects of DHEA. Because the primary spinal cord precursor culture model does not allow us to discriminate between possible proliferative and inductive effects of DHEA in spinal cord ventral progenitor we have studied this question in more details using other models.
Effect of DHEA in Motor Neuron Specification Determined with Intermediate Neural Plate Explants
In vivo, induction of class I and class II protein expression in uncommitted precursors shortly precedes the proliferation of ventral progenitors. Precursors committed to become motor neurons express Olig-2 and exit the cell cycle (Novitch et al., 2001). The increase in the proportion of mature motor neurons (Isl-1+) in our primary cultures with DHEA may derive from the prior increase in the proliferation of the pool of ventral precursors, but alternatively it may possibly derive from a direct effect of DHEA on the induction of the expression of homeoprotein proteins specifying motor neuron development among these cells (Nkx6.1, Olig-2, and then Isl-1/2). To test this possibility, we isolated intermediate neural plate explants from E10 mouse embryos and cultured them first for 12 h in a serum-free medium to wash any residual induction factors, and then for an additional 24 h in fresh serum-free medium containing either no induction signal, or DHEA (10−10 M), or different concentrations (2, 7, and 20 nM) of Shh alone or with DHEA (10−10 M). We monitored the expression of class I homeoprotein Pax7 (Figures 6A,C), class II homeoprotein Nkx6.1, motor neuron progenitor state marker Olig-2, and definite post-mitotic motor neuron marker Isl-1/2. When grown alone, cells in explants expressed low levels of class II protein Nkx6.1 and high levels of class I protein Pax7. Very few cells or none expressed Olig-2 or Isl-1/2. In the presence of DHEA, expression of class I protein Pax7 was reduced whereas that of class II protein Nkx6.1 was increased to a level comparable to that of explants grown in the presence of 2 nM Shh. DHEA also induced expression of Olig-2 and Isl-1/2 (Figures 6A,C). Thus DHEA exerted a repressive action of class I protein expression and an activating action of class II protein expression in vitro, mimicking the action of Shh signaling. In this context, the number of class II protein expressing progenitors was increased to a higher extent than that of motor neuron progenitors and definitive motor neurons, suggesting that the extinction of class I and the increase in class II proteins induced by DHEA may have triggered the cascade of transcriptional repressors that directs motor neuron generation (Novitch et al., 2001, 2003; Shirasaki and Pfaff, 2002). The addition of DHEA-induced a potentiation of the effects of Shh alone when present at 2 nM in the cultures (Figure 6C). DHEA potentiated Shh effects at 7 nM on motor neuron precursor induction (p ≤ 0.0001) but not at 20 nM when Shh activity peaks and cannot be furthered by DHEA neither on induction of motor neuron precursors, definite motor neuron nor on the repression of class I proteins (Figure 6C). The effect of the combination of both signals was not synergic but rather additive and saturable, suggesting a mechanism involving a saturating membrane process. To study this issue more closely, and as we had previously observed that the effects mediated by DHEA were Shh-dependent in our primary culture model, we examined whether the DHEA-mediated induction of class II protein expression, the generation of motor neuron progenitors, and the generation of definitive motor neurons are dependent on the Shh signaling pathway by using the blocking antibody 5E1 and the Shh antagonist cyclopamine. In the presence of either 5E1 or cyclopamine, the induction of Nkx6.1 (not shown), Olig-2, and Isl-1/2 expression by DHEA was abolished (Figures 6B,C), suggesting that DHEA mediates its effects through a mechanism dependent on an intact Shh signaling pathway.
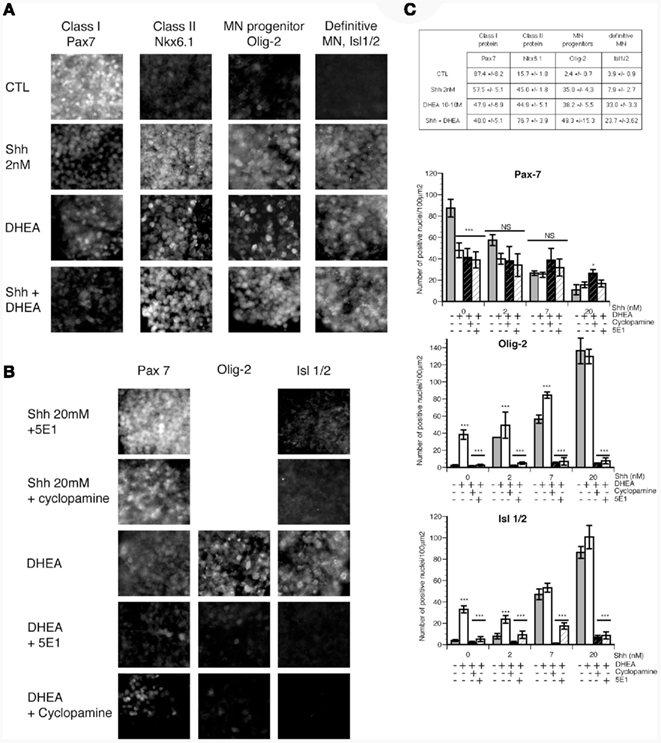
Figure 6. The effects of DHEA on induction of class I and II homeodomain protein, a motor neuron precursor marker, and a definitive motor neuron precursor in intermediate neural plate explants. Intermediate neural plates isolated from E10 thoracic mouse neural tubes were used. (A) Photomicrograph representative of explants cultured in control conditions (without inductive factors) in the presence of 2 nM of Shh, in the presence of 10−10 M DHEA, or in the presence of Shh 2 nM and 10−10 M DHEA. We studied expression of Nkx6.1, Pax7, Olig-2, and Isl-1/2. (B) Expression of Pax7, Olig-2, and Isl-1/2 in the presence of DHEA alone or with 5E1 (3 μg/ml), a blocking antibody against Shh, or cyclopamine, an antagonist of Shh signaling. (C) Quantification of (A) is shown in the table. Data are the number of immunopositive cells per 100 μm2 ± SEM. Quantification of (B) and of all the other conditions tested is shown in the different graphs representing the number of immunopositive cells per 100 μm2 ± SEM for Pax7, Olig-2, and Isl-1/2 respectively. A MANOVA analysis was performed to determine the influence of the dose of Shh used and that of the composition of the culture medium. Differences between the different medium compositions at every single dose of Shh were determined by ANOVA and Bonferroni post hoc analysis. * = p < 0.05, ** = p < 0.001, *** = p < 0.0001.
To explore whether this effect was also operative in the DHEA-mediated repression of class I protein expression, we studied the expression of Pax7 in the explants treated with DHEA in the presence of 5E1 or cyclopamine. Neither cyclopamine nor 5E1 blocked the repression of Pax7 expression in DHEA-treated explants (Figure 6B), indicating that DHEA mediates the repression of Pax7, and possibly that of other class I proteins, in a manner independent of Shh signaling. Similarly, the addition of DHEA to Shh-treated explants did not increase the repression of Pax7 above the level observed in explants treated with Shh alone.
It appears, from these findings, that DHEA can induce expression of class II proteins and reduce the expression of class I proteins, mimicking Shh signaling. However, the DHEA-mediated induction of class I protein is dependent on the Shh pathway, whereas the DHEA-mediated repression of class I protein expression occurs independently from Shh. We have also shown that the DHEA-mediated induction of class II protein expression can trigger expression of motor neuron progenitor protein markers and expression of definitive motor neurons in vitro.
Effect of DHEA on Cell Proliferation Determined with Whole Spinal Cord Explants
To study the effects of DHEA in the context of the whole spinal cord, we used spinal cord explant cultures isolated from the lumbo-sacral region of E11 mouse embryos. At this A/P level and stage, DHEA is endogenously produced at very low levels (see Figure 2D). Thus the effects observed were due to the exogenous addition of DHEA in the culture medium for 24 h. We measured BrdU incorporation in explants grown in the presence or absence of DHEA (Figure 7). In the control condition, a low but consistent level of BrdU incorporation was observed (Figure 7A). In this in vitro model, in which the SVZ is exposed, BrdU incorporation was increased in the presence of DHEA in 80% of the explants studied (Figure 7A). Increased proliferation was mostly seen in the SVZ region surrounding the floor plate, and it gradually decreased from the ventral to the dorsal region of the flattened spinal cord (Figures 7B,C). In the dorsal spinal cord, no significant differences were observed between control and DHEA explants (Figure 7B,C). Only half of the explants grown in the presence of DHEA showed a low number of BrdU incorporated cells that extended in the dorsal domain. These observations strongly suggested that the proliferative response to DHEA depended on the gradient of Shh along the ventro-dorsal axis of the cord. To confirm this hypothesis, we used the 5E1 antibody and observed a complete abolition of DHEA-induced proliferation (Figures 7A–C). In all explants treated with the 5E1 antibody, the floor plate appeared to be uniformly labeled (Figure 7A). One possible explanation for this result is the detection of the monoclonal 5E1 antibody attached to the surface of floor plate cells. Because 5E1 and BrdU antibodies are both monoclonal antibodies, the secondary antibody used to detect BrdU in these explants will also bind to the 5E1 antibody when present. These results show that, in the presence of endogenous secretion of Shh by floor plate cells, DHEA-induced proliferation of neural precursors of the SVZ. To assess that this effect was direct, and not due to the possible metabolism of DHEA in other steroids, we used a non-androgenic fluorinated analog of DHEA, fluasterone (Schwartz and Pashko, 1995). As shown in Figure 7, the same Shh-dependent increase of BrdU incorporation in the ventral SVZ was seen with fluasterone as with DHEA. The effects observed are thus directly due to the presence of DHEA.
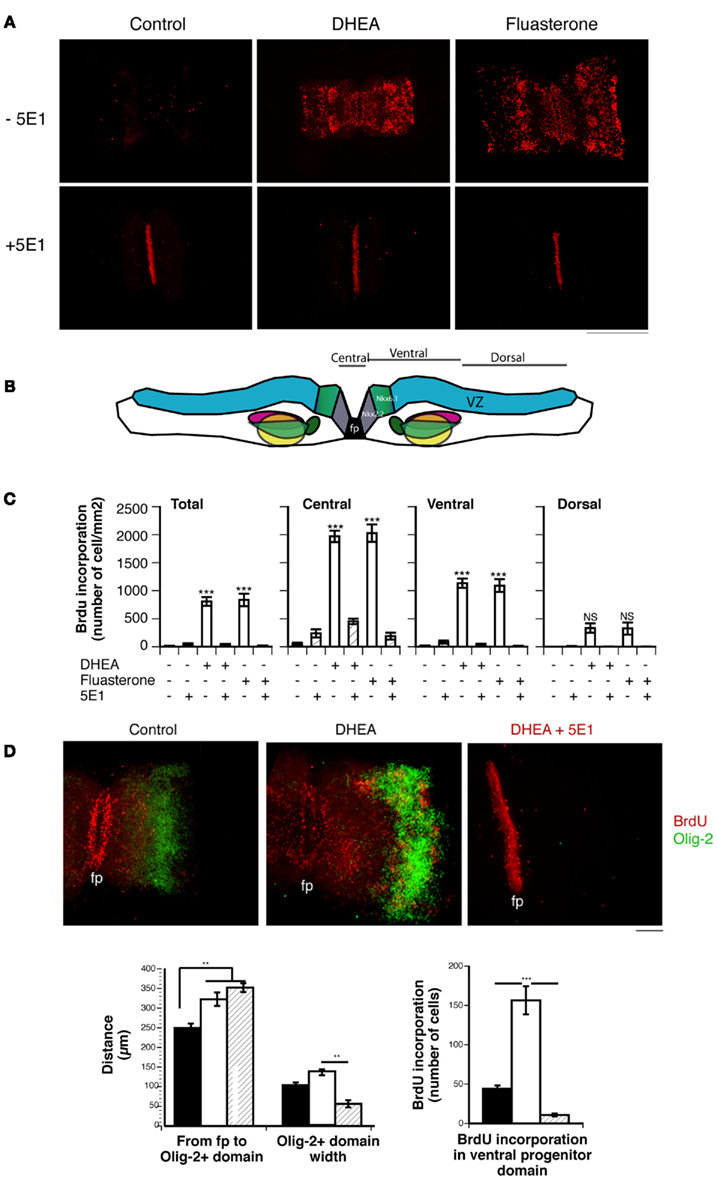
Figure 7. Effect of DHEA on cell proliferation in spinal cord explants. (A) BrdU immunostaining of E11.5 lumbar spinal cord explants cultured in the absence (Control) or presence of DHEA or its derivative fluasterone, with or without the 5E1 anti-Shh antibody. Bar = 0.5 mm. (B) Layout of the spinal cord explant in the collagen matrix. The different regions in which BrdU incorporation was quantified are shown. (C) Quantification of BrdU incorporation in the whole explant (Total), central part (∼100 μm around the floor plate), and ventral and dorsal domains. Results are expressed as a number of immunopositive BrdU cells/mm2 to correct for different sizes of explants. (D) Photomicrograph of whole spinal cord explants immunostained for BrdU (red) and Olig-2 (Green) showing the effect of DHEA on the expansion of the ventrally committed precursor domain and the dorsal shift of the Olig-2+ domain. These effects are antagonized in the presence of 5E1, which also abolished the Shh-mediated induction of Olig-2 expression. (fp = floor plate, bar = 100 μm). Quantification of the distance and width of the Olig-2+ motor neuron precursor domain is shown in each experimental condition. Quantification of the number of the BrdU-incorporating cells in the ventral committed precursor domain is shown. Values represent the means ± SEM of three independent platings. One-way ANOVA analysis was performed to assess significance of the treatment. Significant differences between groups were determined with Bonferroni post hoc analysis. * = p < 0.05, ** = p < 0.001, *** = p < 0.0001.
We then determined whether the ventro-dorsal patterning of the explants was modified under these conditions by studying Olig-2 expression. In the presence of DHEA, we observed an increased proliferation of the ventrally committed neural precursors, which resulted in a dorsal shift of the Olig-2+ motor neuron precursor domain (Figure 7D). Although some proliferative cells are observed in the olig-2+ domain, cells expressing Olig-2 do not incorporate BrdU, consistent with their post-mitotic state. DHEA also increased the level of Olig-2 expression and the size of the Olig-2 domain, indicating an increased induction of this protein expression in a larger number of motor neuron precursors (Figure 7D). These results show that physiological levels of DHEA allowed for a sustained proliferation of ventrally committed precursors in spinal cord explants. As a result of the expansion of the ventral progenitors, the domain of the motor neuron precursors shifts dorsally. Increased proliferation of ventrally committed precursors (possibly including Nkx6.1+ precursors) may also contribute to a larger pool of – motor neuron precursors observed in explants cultured in the presence of DHEA. DHEA-mediated BrdU incorporation and Olig-2 expression were abolished by adding 5E1 antibody to the culture medium, demonstrating the dependence of the effects observed on Shh signaling (Figure 7D).
Discussion
This study addresses how DHEA, a neurosteroid produced in the developing neural tube, may potentiate the biological response of ventral precursors to Shh. The results establish that DHEA is biosynthesized in the developing ventral neural tube at the time of neurogenesis and cell fate determination. In in vitro models, DHEA modulated the induction of expression of Nkx6.1, a class II homeodomain protein, as well as expression of the homeodomain proteins Olig-2, a specific motor neuron precursor marker, and Isl-1/2, a definitive motor neuron marker. Moreover, DHEA promoted proliferation of ventral neuronal progenitors committed into the derepression cascade leading to the generation of motor neurons. Both of these biological actions of DHEA are dependent on Shh. These results support the hypothesis that DHEA biosynthesis participates in the Shh-mediated induction of ventral neuronal populations and controls the proliferative response of neuronal progenitors to Shh in the mid-gestation ventral neural tube in vivo. We found, in addition, that DHEA represses expression of Pax7, a class I homeodomain protein, in intermediate neural plate explants in a manner independent from Shh. This observation remains to be further documented.
DHEA Biosynthesis in the Developing Neural Tube
In this study, P450c17, an enzyme crucial for the biosynthesis of DHEA, was expressed in the neural tube in a regulated spatial and temporal pattern. Expression of P450c17 in the developing neural tube was first seen at E10 in the differentiated field of the neural tube in the cervical region. It then extended to all A/P levels at E13 and became localized in subsets of motor neuron pools. These results establish that expression of P450c17 correlates with its activity in the developing neural tube. Moreover, this activity was regulated along the A/P axis of the neural tube. As P450c17 expression remains detectable several days after the biosynthesis of DHEA stops, it appears that the regulation of DHEA biosynthesis does not rely solely on transcriptional regulation of P450c17. Future studies must determine the mechanism regulating the temporal and regional activity of P450c17 among (i) the availability of specific cofactors, (ii) competition for its rate-limiting substrate, or (iii) further conversion of DHEA into androgens. The period of time during which DHEA is biosynthesized in the developing neural tube corresponds to the period during which neurogenesis occurs. Neural progenitors emerging from the SVZ remain highly proliferative while inductive signals – such as withdrawal of FGF, retinoic acid, and Shh secretion – dictate cell fate decisions through a transcriptional derepression strategy (Muhr et al., 1999). As the identity of repressor cascades in the ventral spinal cord have been largely defined for pathways of motor neuron and interneuron generation (Briscoe et al., 2001; Muhr et al., 2001), we focused on determining the biological action of DHEA in the ventral neural tube by using in vitro models recapitulating ventral neuron generation.
Induction and Proliferation: A Cooperative Mechanism to Control the MN Pool
Motor neurons and ventral interneurons are generated through a pathway that involves secretion of Shh from the notochord and floor plate (Patten and Placzek, 2000). Shh has diverse functions in the developing neural tube through a common signaling pathway (reviewed in Ingham and McMahon, 2001; Marti and Bovolenta, 2002). Shh signaling is critical for ventral neuronal differentiation. Dysfunction in the Shh signaling pathway in mice is associated with defects in ventral neural patterning (Chiang et al., 1996). In humans, it produces several pathologic conditions, such as holoprosencephaly, cyclopia, and impaired development of the neural tube and limbs, as well as various types of skin and CNS cancers, including medulloblastoma (reviewed in McMahon et al., 2003; Roessler and Muenke, 2003). In the neural tube, Shh has a primordial role in dictating cell fate in ventral precursors (Jessell, 2000; Briscoe et al., 2001; Lee and Pfaff, 2001). Shh is necessary and sufficient to specify ventral neuronal populations, as demonstrated through genetic ablation of its expression in the mouse (Chiang et al., 1996) and by using chick embryos electroporated with mutated forms of Shh responsible for human holoprosencephaly (Schell-Apacik et al., 2003). An elegant demonstration of dose-dependent gene activity in response to various concentrations of purified Shh has led to the interpretation that Shh controls ventralization of the neural tube through graded signals (Roelink et al., 1995; Ericson et al., 1997). A study of the mouse retina suggested Shh regulation in cell proliferation (Jensen and Wallace, 1997), and several other studies have clearly implicated Shh signaling in the developing mouse cerebellum, where the development of granule-cell precursors depends on the expression of Shh by Purkinje cells (Dahmane et al., 1997; Wallace, 1999; Weshcelard-Reya and Scott, 1999). Proliferative effects of Shh in the spinal cord have been shown both in vitro and in vivo (Kalyani et al., 1998; Rowitch et al., 1999). Rowitch et al. (1999) produced a transgenic mouse in which Shh was ectopically expressed in the dorsal neural tube and persisted beyond the time when normal neurogenesis occurred. In that model, the ectopic expression of Shh produced an increased proliferation of precursor cells only when induced in the embryo early, at a time when neurogenesis normally occurs. Ectopic expression of Shh at a later stage did not produce a proliferative response of precursors still present within its signaling range. It appears, then, that environmental cues participate in the modulation of the type of response of neural precursors to Shh signaling. Our experiments indicate that DHEA is capable of recapitulating Shh signaling by inducing Nkx6.1, Olig-2, and Isl-1/2 expression in E10 intermediate neural plate explants. While controlling cell fate decisions in neural precursors in a manner dependent on Shh, DHEA also controls proliferation of ventral neuronal-precursors both in isolated primary cultures and in E11 whole spinal cord explants. Evidence that the effects mediated by DHEA are abolished by antagonists of the Shh signaling pathway indicate that DHEA mediates its effects in a manner dependent on the Shh signaling pathway. Based on those findings, we propose that the time during which DHEA is biosynthesized in vivo may represent a temporal window of opportunity for ventral neuronal-precursor to proliferate in response to Shh, and its closing thus controls the number of pre-committed neurons. During that period, DHEA may also enhance the bioactivity of Shh in specifying the same ventral precursors that proliferate in response to DHEA and Shh into ventral neuronal subtypes, including those of motor neurons. These observations must be confirmed in an in vivo model.
An intriguing observation is the fact that DHEA repressed Pax7 expression in a manner independent of Shh signaling. This observation established in intermediate neural plate explants needs to be confirmed in models more closely related to the physiology of the developing dorsal neural tube. Nonetheless, if our observation was confirmed in models allowing the study of dorsal patterning under normal BMP signaling, DHEA being produced by commissural neurons, could participate in the control of Pax7 expression dorsally. In vivo, Pax7 expression is seen in the dorsal cord at the time when DHEA biosynthesis occurs, thus making the repressing effects of DHEA possible. But because in vivo inhibition of Shh biogenesis or signaling is enough to derepress expression of Pax7 in the ventral neural tube (Chiang et al., 1996; Incardona et al., 1998), it is unlikely that DHEA biosynthesis may be sufficient to repress Pax7 expression alone. This issue must be studied in greater details.
Mechanisms of Action
DHEA is a pleiotropic molecule that has several potential mechanisms of action (review in Compagnone and Mellon, 2000). In the developing embryo, it is mostly seen as acting either (i) as a weak NMDA agonist (Compagnone and Mellon, 1998) and GABAA antagonist (Majewska, 1995) or (ii) as a precursor of androgens, indirectly acting through the local biosynthesis of androstenedione, testosterone, or estradiol after aromatization. Evidence of a direct action of DHEA lies in our confirmatory results with fluasterone, which – as fluasterone cannot be metabolized into androgens or estrogens – eliminate the possibility that the effects observed could be due to the metabolism of DHEA into androgens or estrogens. The possibility that DHEA-mediated its effects by inducing NMDA receptor activation was also excluded, and our results are consistent with the fact that the units forming a functional NMDA receptor are not expressed at this early embryonic stage (Maric et al., 2000).
Our findings indicate that DHEA mediates motor neuron induction and ventral neuronal-precursor proliferation through a signaling pathway dependent on Shh. We can speculate that DHEA could interact with the Shh signaling pathway at several levels. Five possibilities need to be further analyzed: (1) DHEA may increase Shh expression in neural cells (2) DHEA could function as an agonist of the Shh pathway (3) DHEA may function in parallel and synergistically to Shh signaling (4) DHEA may function as an antagonist of BMP signaling (5) DHEA may modulate Shh range and activity gradient dependent on its lipid modification. Shh is dually lipid-modified, with palmitic acid at its N-terminus and cholesterol at its C-terminus (Mann and Beachy, 2004). Even though DHEA is present in vivo at concentrations in the femto- to pico-molar range – far below those needed for the interaction between cholesterol and Shh to occur (Porter et al., 1996), the last hypothesis is appealing because cholesterol modulation of Shh activity has been proposed to explain the differential effect of Shh in patterning the spinal cord versus telencephalon. Mice in which Shh lacks the cholesterol moiety show defect in ventral neuron specification and ganglionic eminence development in the telencephalon, while a full spectrum of the ventral cell type is observed in the spinal cord (Huang et al., 2007). DHEA is produced in an antero/posterior gradient in the spinal cord at the time of neural precursor proliferation. Therefore, it may substitute for cholesterol and interact with Shh in the spinal cord at the time of ventral precursor proliferation and specification. However, P450c17 expression is posterior to ventral neural precursors proliferation and specification by Shh signaling in the telencephalon (Compagnone and Mellon, 1998). These hypotheses need to be tested in other in vitro and in vivo models and are currently under investigation. In vivo models will require conditional knock-out of P450c17 enzyme since P450c17 knock-out mice have been developed and are embryonic lethal prior to gastrulation (Bair and Mellon, 2004).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
For the gift of reagents, we thank Dr. Walter Miller (University of California, San Francisco, CA, USA) for the anti-human P450c17 antibody, Dr. Mike German (University of California, San Francisco, CA, USA) for the anti-Nkx6.1 antibody, Dr. Samuel Pfaff (The Salk Institute, La Jolla, CA, USA) for the anti-Isl2, Lim3, HB9, and Chx10 antibodies, Dr. Thomas Jessell and Dr. Bennett Novitch (Columbia University, NY, USA) for Olig-2 antibody, Dr. Richard Auchus (UT Southwestern Medical Center, Dallas, TX, USA) for P450c17 enzymatic activity radiolabeled standard, derived from P450c17 over-expressing yeast, and Dr. Arthur Schwartz (Temple University, Philadelphia, PA, USA) for fluasterone. The anti-Shh (5E1), anti-Isl-1 (40.2D6), and anti-Lim1/2 (4F2) antibodies, developed by Dr. Thomas Jessell, the anti-Pax6 and the anti-Pax7 antibodies, developed by Dr. Atsushi Kawakami, were obtained from the Developmental Studies Hybridoma Bank (DSHB) developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA 52242, USA. We also thank Drs. Marc Tessier-Lavigne, John Rubenstein, and Oscar Marin, for fruitful discussions along the course of the redaction of this manuscript. This work was supported by a grant from the University of California, Research Evaluation and Allocation Committee, a grant from the Muscular Dystrophy Association and the generous gift from the Charitable Columbia Foundation to NAC. CF was supported by a fellowship from the Association for the Research on Cancer (ARC) and by grant NIH-NS41998 to NAC.
References
Auchus, R. J., Geller, D. H., Lee, T. C., and Miller, W. L. (1998). The regulation of human P450c17 activity: relationship to premature adrenarche, insulin resistance and the polycystic ovary syndrome. Trends Endocrinol. Metab. 9, 47–50.
Bair, S. R., and Mellon, S. H. (2004). Deletion of the mouse P450c17 gene causes early embryonic lethality. Mol. Cell. Biol. 24, 5383–5390.
Barzi, M., Berenguer, J., Menendez, A., Alvarez-Rodriguez, R., and Pons, S. (2010). Sonic hedgehog-mediated proloferation reguires the locaization of PKA to the cilium base. J. Cell. Sci. 123(Pt 1), 62–69.
Bergeron, R., de Montigny, C., and Debonnel, G. (1996). Potentiation of neuronal NMDA response induced by dehydroepiandrosterone and its suppression by progesterone: effects mediated via sigma receptors. J. Neurosci. 16, 1193–1202.
Briscoe, J., Chen, Y., Jessell, T. M., and Struhl, G. (2001). A hedgehog-insensitive form of patched provides evidence for direct long-range morphogen activity of sonic hedgehog in the neural tube. Mol. Cell 7, 1279–1291.
Briscoe, J., Pierani, A., Jessell, T. M., and Ericson, J. (2000). A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell 101, 435–445.
Brito, J., Tannahill, D., and Keynes, R. (2002). A critical role for sonic hegehog signaling in the early expansion of the developing brain. Nat. Neurosci. 5, 103–110.
Chiang, C., Litingtung, Y., Lee, E., Young, K. E., Corden, J. L., Westphal, H., and Beachy, P. A. (1996). Cyclopia and defective axial patterning in mice lacking sonic hedgehog gene function. Nature 383, 407–413.
Colamarino, S. A., and Tessier-Lavigne, M. (1995). The axonal chemoattractant netrin-1 is also a chemorepellent for trochlear motor axons. Cell 81, 621–629.
Compagnone, N. A., Bulfone, A., Rubenstein, J. L., and Mellon, S. H. (1995a). Steroidogenic enzyme P450c17 is expressed in the embryonic central nervous system. Endocrinology 136, 5212–5223.
Compagnone, N. A., Bulfone, A., Rubenstein, J. L., and Mellon, S. H. (1995b). Expression of the steroidogenic enzyme P450scc in the central and peripheral nervous systems during rodent embryogenesis. Endocrinology 136, 2689–2696.
Compagnone, N. A., and Mellon, S. H. (1998). Dehydroepiandrosterone: a potential signalling molecule for neocortical organization during development. Proc. Natl. Acad. Sci. U.S.A. 95, 4678–4683.
Compagnone, N. A., and Mellon, S. H. (2000). Neurosteroids: biosynthesis and function of these novel neuromodulators. Front. Neuroendocrinol. 21, 1–60.
Dahmane, N., lee, J., robins heller, P., and ruiz-i-altaba, A. (1997). Activation of the transcirption factor Gli1 and the sonic hedgehog signaling pathway in skin tumors. Nature 389, 876–881.
Ericson, J., Briscoe, J., Rashbass, P., van Heyningen, V., and Jessell, T. M. (1997). Graded sonic hedgehog signaling and the specification of cell fate in the ventral neural tube. Cold Spring Harb. Symp. Quant. Biol. 62, 451–466.
Ericson, J., Morton, S., Kawakami, A., Roelink, H., and Jessell, T. M. (1996). Two critical periods of sonic hedgehog signaling required for the specification of motor neuron identity. Cell 87, 661–673.
Fuse, N., Maiti, T., Wang, B., Porter, J. A., Hall, T. M., Leahy, D. J., and Beachy, P. A. (1999). Sonic hedgehog protein signals not as a hydrolytic enzyme but as an apparent ligand for patched. Proc. Natl. Acad. Sci. U.S.A. 96, 10992–10999.
Hagedorn, L., Floris, J., Suter, U., and Sommer, L. (2000). Autonomic neurogenesis and apoptosis are alternative fates of progenitor cell communities induced by TGFbeta. Dev. Biol. 228, 57–72.
Huang, X., Litingtung, Y., and Chiang, C. (2007) , Region-specific requirement for cholesterol modification of sonic hedgehog in patterning the telencephalon and spinal cord. Development 134, 2095–2105.
Incardona, J. P., Gaffield, W., Kapur, R. P., and Roelink, H. (1998). The teratogenic vertrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 125, 3553–3562.
Ingham, P. W., and McMahon, A. P. (2001). Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15, 3059–3087.
Jensen, A. M., and Wallace, V. A. (1997). Expression of sonic hedgehog and its putative role as a precursor cell mitogen in the developing mouse retina. Development 124, 363–371.
Jessell, T. M. (2000). Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat. Rev. Genet. 1, 20–29.
Kalyani, A. J., Piper, D., Mujtaba, T., Lucero, M. T., and Rao, M. S. (1998). Spinal cord neuronal precursors generate multiple neuronal phenotypes in culture. J. Neurosci. 18, 7856–7868.
Lee, S. K., and Pfaff, S. L. (2001). transcriptional networks regulating neuronal identity in the developing spinal cord. Nat. Neurosci. 4, 1183–1191.
Litingtung, Y., and Chiang, C. (2000). Specification of ventral neuron types is mediated by an antagonistic interaction between Shh and Gli3. Nat. Neurosci. 3, 979–985.
Majewska, M. D. (1995). Neuronal actions of dehydroepiandrosterone. Possible roles in brain development, aging, memory, and affect. Ann. N. Y. Acad. Sci. 774, 111–120.
Mann, R. K., and Beachy, P. A. (2004). Novel lipid modifications of secreted protein signals. Annu. Rev. Biochem. 73, 891–923.
Maric, D., Liu, Q., Grant, G., Andreadis, J., Hu, Q., Chang, Y., Barker, J., Joseph, J., Stenger, D., and Ma, W. (2000). Functional ionotropic glutamate receptors emerge during terminal cell division and early neuronal differentiation of rat neuroepithelial cells. J. Neurosci. Res. 61, 652–662.
Marti, E., and Bovolenta, P. (2002). Sonic hedgehog in CNS development: one signal, multiple output. Trends Neurosci. 25, 89–96.
McMahon, A. P., Ingham, P. W., and Tabin, C. J. (2003). Developmental roles and clinical significance of hedgehog signaling. Curr. Top. Dev. Biol. 53, 1–114.
Muhr, J., Andersson, E., Persson, M., Jessell, T., and Ericson, J. (2001). Groucho-mediated transcriptional repression establishes progenitor cell pattern and neuronal fate in the ventral neural tube. Cell 104, 861–873.
Muhr, J., Graziano, E., Wilson, S., Jessell, T. M., and Edlund, T. (1999). Convergent inductive signals specify midbrain, hindbrain, and spinal cord identity in gastrula stage chick embryos. Neuron 23, 689–702.
Nornes, H. O., and Carry, M. (1978). Neurogenesis in spinal cord of mouse: an autoradiographic analysis. Brain Res. 159, 1–6.
Novitch, B., Wichterle, H., Jessell, T., and Sockanathan, S. (2003). A requirement for retinoic acid-mediated transcriptional activation in ventral neural patterning and motor neuron specification. Neuron 40, 81–95.
Novitch, B. G., Chen, A. I., and Jessell, T. M. (2001). Coordinate regulation of motor neuron subtype identity and pan-neuronal properties by the bHLH repressor Olig2. Neuron 31, 773–789.
Patten, I., and Placzek, M. (2000). The role of sonic hedgehog in neural tube patterning. Cell. Mol. Life Sci. 57, 1695–1708.
Pepinsky, R. B., Rayhorn, P., Day, E. S., Dergay, A., Williams, K. P., Galdes, A., Taylor, F. R., Boriack-Sjodin, P. A., and Garber, E. A. (2000). Mapping sonic hedgehog-receptor interactions by steric interference. J. Biol. Chem. 275, 10995–11001.
Porter, J. A., Young, K. E., and Beachy, P. A. (1996). Cholesterol modification of hedgehog signaling proteins in animal development. Science 274, 255–259. [Erratum in: Science 274, 1597].
Roelink, H., Porter, J. A., Chiang, C., Tanabe, Y., Chang, D. T., Beachy, P. A., and Jessell, T. M. (1995). Floor plate and motor neuron induction by different concentrations of the amino-terminal cleavage product of sonic hedgehog autoproteolysis. Cell 81, 445–455.
Roessler, E., and Muenke, M. (2003). How a hedgehog might see holoprosencephaly. Hum. Mol. Genet. 12, R15–R25.
Rowitch, D. H., St. Jacques, B., Lee, S. M. K., Flax, J. D., Snyder, E. Y., and McMahon, A. P. (1999). Sonic hedgehog regulates proliferation and inhibits differentiation of CNS precursor cells. J. Neurosci. 19, 8954–8965.
Rubenstein, J. L., and Beachy, P. A. (1998). Patterning of the embryonic forebrain. Curr. Opin. Neurobiol. 8, 18–26.
Ruiz i Altaba, A., Palma, V., and Dahmane, N. (2002). Hedgehog-Gli signalling and the growth of the brain. Nat. Rev. Neurosci. 31, 24–33.
Schell-Apacik, C., Rivero, M., Knepper, J. L., Roessler, E., Muenke, M., and Ming, J. E. (2003). Sonic hedgehog mutations causing human holoprosencephaly impair neural patterning activity. Hum. Genet. 113, 170–177.
Schwartz, A., and Pashko, L. (1995). Cancer prevention with dehydroepiandrosterone and non-androgenic structural analogs. J. Cell. Biochem. Suppl. 22, 210–217.
Shirasaki, R., and Pfaff, S. (2002). Transcriptional codes and the control of neuronal identity. Annu. Rev. Neurosci. 25, 251–281.
Wallace, V. A. (1999). Purkinje-cell-derived sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr. Biol. 9, 445–448.
Weshcelard-Reya, R. J., and Scott, M. P. (1999). Control of neuronal precursor proliferation in the cerebellum by sonic hedgehog. Neuron 22, 103–114.
Wetmore, C. (2003). Sonic hedgehog in normal and neoplastic proliferation: insight gained from human tumors and animal models. Curr. Opin. Genet. Dev. 13, 34–42.
Keywords: DHEA, neurosteroid, spinal cord, sonic hedgehog, proliferation, induction, neural precursors
Citation: Galdo M, Gregonis J, Fiore CS and Compagnone NA (2012) Dehydroepiandrosterone biosynthesis, role, and mechanism of action in the developing neural tube. Front. Endocrin. 3:16. doi: 10.3389/fendo.2012.00016
Received: 19 July 2011;
Paper pending published: 23 August 2011;
Accepted: 19 January 2012;
Published online: 07 February 2012.
Edited by:
Kazuyoshi Tsutsui, Waseda University, JapanReviewed by:
Synthia H. Mellon, University of California San Francisco, USAIshwar Parhar, Monash University, Malaysia
Copyright: © 2012 Galdo, Gregonis, Fiore and Compagnone. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Nathalie A. Compagnone, Innovative Concepts in Drug Development, Innovation Park Michel Caucik, BP2 100 rte des houilleres, 13590 Meyreuil, France. e-mail:bmNvbXBhZ25vbmVAaWNkZC1zYXMuY29t