

- Endocrinology Unit, Department of Clinical and Experimental Medicine, University Magna Graecia of Catanzaro, Catanzaro, Italy
A large body of evidences have shown that both the IGF-I receptor (IGF-IR) and the insulin receptor (IR) play a role in cancer development and progression. In particular, IR overactivation by IGF-II is common in cancer cells, especially in dedifferentiated/stem-like cells. In spite of these findings, until very recently, only IGF-IR but not IR has been considered a target in cancer therapy. Although several preclinical studies have showed a good anti-cancer activity of selective anti-IGF-IR drugs, the results of the clinical first trials have been disappointing. In fact, only a small subset of malignant tumors has shown an objective response to these therapies. Development of resistance to anti-IGF-IR drugs may include upregulation of IR isoform A (IR-A) in cancer cells and its overactivation by increased secretion of autocrine IGF-II. These findings have led to the concept that co-targeting IR together with IGF-IR may increase therapy efficacy and prevent adaptive resistance to selective anti-IGF-IR drugs. IR blockade should be especially considered in tumors with high IR-A:IGF-IR ratio and high levels of autocrine IGF-II. Conversely, insulin sensitizers, which ameliorate insulin resistance associated with metabolic disorders and cancer treatments, may have important implications for cancer prevention and management. Only few drugs co-targeting the IR and IGF-IR are currently available. Ideally, future IR targeting strategies should be able to selectively inhibit the tumor promoting effects of IR without impairing its metabolic effects.
Introduction
Several studies have convincingly shown that the insulin receptor (IR) pathway is more directly and intimately involved in cancer development and progression than previously thought. Overactivation of this pathway by both insulin and IGF-II is common in cancer cells, particularly in dedifferentiated/stem-like cells, and may represent an important factor of resistance to various anti-cancer drugs (Papa et al., 1990; Belfiore, 2007). Dysregulated expression of the IR isoform A (IR-A), which binds IGF-II with high affinity, has a crucial role in these mechanisms. Moreover, it has been shown that overactivation of the IGF-II/IR-A loop in cancer cells may represent a mechanism of adaptive resistance to anti-IGF-I receptor (anti-IGF-IR) drugs (Garofalo et al., 2011).
Oddly enough, type 2 diabetes mellitus (T2DM), a common disorder associated with insulin resistance (i.e., reduced glycometabolic effects of insulin), may also contribute to the unbalanced activation of the mitogenic effects of insulin through compensatory hyperinsulinemia (Jiang et al., 1999; Cusi et al., 2000). Accordingly, T2DM and other conditions associated with insulin resistance, such as obesity and metabolic syndrome, are now recognized as important risk factors for the development and progression of a variety of malignancies (Strickler et al., 2001; Kaaks and Lukanova, 2002; Vainio et al., 2002; Coughlin et al., 2004; Vigneri et al., 2006; Fair et al., 2007; Pisani, 2008).
Therefore, the IR pathway, traditionally representing the focus of antidiabetic therapies, has rapidly gained attention as a novel target in cancer. Learning how to exploit these new concepts for cancer prevention and therapy constitutes an important challenge. As far as therapy is concerned, we face the dilemma of how inhibiting the mitogenic IR pathway in cancer patients while minimizing or avoiding deterioration of the IR glucometabolic effects, which are of crucial importance for the normal functions of the entire organism.
The present review will focus on recent advances and new challenges in this rapidly evolving field.
IR and Cancer
IR Structure and Signaling
The insulin and the insulin-like growth factors I and II (IGF-I and IGF-II) signaling is mediated by hormone interaction with cognate tyrosine kinase receptors, IR and IGF-IR. Although these two receptors are highly homologous and are coupled to very similar intracellular substrate networks, in normal adult tissues insulin and IGFs stimulate specific functions, such as glucose metabolism for insulin and cell growth and proliferation for IGFs. However, in particular conditions, such as cancer, this signaling specificity is partially lost and both receptors may share similar biological functions. As the shared signaling pathway has an important role in cancer development and progression, both receptors have emerged as targets for cancer therapy.
Insulin receptor and IGF-IR are protein tyrosine kinases that belong to the IGF system and regulate many crucial aspects of cellular physiology (Ebina et al., 1985; Ullrich et al., 1985; Drakas et al., 2004). Both receptors are products of two distinct genes, which are believed to derive from a common ancestral gene through a duplication event. Reflecting this common heritage, IR and IGF-IR share a high degree of homology (Ullrich et al., 1985, 1986). Indeed, both receptors are expressed at the cellular surface in α2β2 configuration. The α subunit of each receptor contains the ligand binding sites, usually located at a cysteine-rich domain on the extracellular region (Andersen et al., 1995; Whittaker and Whittaker, 2005). In both receptors, the α subunit contains also a 16-aminoacid residue C-terminal sequence (CT peptide) that contributes to ligand binding. Contiguous to the IR CT peptide, a 12-aminoacid sequence (encoded by exon 11 of IR gene, which is alternatively spliced) is either excluded or included giving rise to isoform A (IR-A) or isoform B (IR-B), respectively (Moller et al., 1989; Mosthaf et al., 1990).
The β subunits of IR and IGF-IR include a large cytoplasmic region with tyrosine kinase activity. This region is the most conserved domain in IR and IGF-IR, showing approximately 85% similarity at the aminoacid level. The most structurally and functionally divergent region between the two receptors is the cytoplasmic carboxy-terminal domain within the intracellular β subunit. It shows three phospho-tyrosine residues in the IGF-IR, while two phospho-tyrosines are present in the corresponding region of the IR (Figure 1). This domain appears to mediate the transforming and differentiative response to IGF-IR but not its mitogenic or anti-apoptotic actions (Miura et al., 1995; O’Connor et al., 1997). Conversely, in IR, this region seems to mediate the mitogenic but not the metabolic functions (Faria et al., 1994).
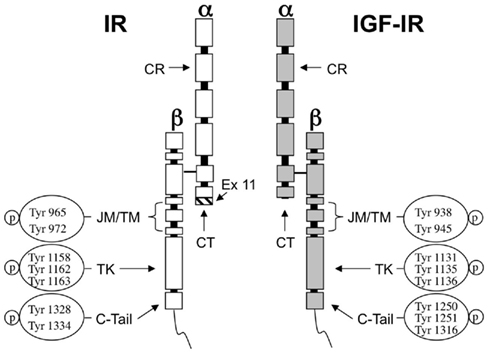
Figure 1. Structure of IR and IGF-IR and autophosphorylation sites. The ligand binding sites of both receptors are predominantly located at a cysteine-rich region (CR) in the extracellular α-subunit. The homology between IR and IGF-IR in this region ranges 45–65%. The CT peptide in the α-subunit contributes to the binding properties of both receptors. In IR the hatched fragment on the bottom of the CT region is encoded by exon 11 and is present in IR-B isoform but not in IR-A. The tyrosine kinase domain (TK) in the β-subunit is highly conserved showing 85% of similarity between both receptors. The most divergent region is the C-terminal domain (C-tail). JM, juxtamembrane domain; TM, trans-membrane domain.
As a consequence of the high level of homology of the two receptors, hybrid receptors (HRs) formed by an IR (IR-A or IR-B) αβ-hemireceptor and an IGF-IR αβ-hemireceptor (HR-A and HR-B, respectively) are also expressed in all tissues co-expressing IR and IGF-IR (Ullrich et al., 1986).
Insulin receptor, IGF-IR, and HRs bind the same ligands (insulin, IGF-I, and IGF-II), although with very different affinities (Table 1). Upon ligand binding, receptors become autophosphorylated on conserved tyrosine residues and activate similar intracellular signaling events. Tyrosine kinase domains within the β-subunit of both IR and IGF-IR catalyze the phosphorylation of specific substrates, such as the members of IR substrates family (IRS-1 to IRS-4), Gab-1, Cbl, and Shc. Following tyrosine phosphorylation, IRS proteins interact with GRB2 (growth factor receptor binding protein 2) and with the p85 regulatory subunit of phosphoinositide 3-kinase (PI3K). PI3K, in turn, regulates the activation of Akt via the phosphoinositide-dependent kinase 1 (PDK1). Akt activation is crucial in mediating metabolic effects by regulating metabolic enzymes, but also in mediating cell growth, proliferation, and survival (Hers et al., 2011).

Table 1. Relative ligand binding affinity of IR isoforms, IGF-IR, and HRs containing either IR-A (HR-A) or IR-B (HR-B) moieties.
Another pathway regulated by PI3K/Akt is the mammalian target of rapamycin (mTOR) pathway, which also has a central role in cell growth and metabolism (Hay and Sonenberg, 2004; Sarbassov et al., 2005). Downstream to mTOR, both p70S6K and 4EBP1 control mRNA translation and protein synthesis. The PI3K/Akt pathway is negatively regulated by the lipid phosphatase PTEN (phosphatase and tensin homolog), which dephosphorylates PI3K and inhibits cell growth and cell cycle progression through its interaction with cyclin D (Stambolic et al., 1998; Blanco-Aparicio et al., 2007). PTEN is the second most common tumor suppressor gene mutated in human cancer and the loss of its activity induces an increase in IR and IGF-IR signaling (Perks et al., 2007).
The second major signaling pathway downstream to IR and IGF-IR involves Ras, a GTP-binding protein, which cycles from the active (GTP-bound) to the inactive (GDP-bound) form. By binding to GRB2, IRS proteins couple GRB2 to IR or IGF-IR. Once associated with IRS proteins, GRB2 forms a complex with the Son of Sevenless (SOS) p21Ras. This event triggers the translocation of SOS to plasma membrane and activation of serine/threonine phosphorylation events that propagate insulin and IGFs signal through the Ras/Raf/MEK/ERK cascade. Activated ERK1/2 phosphorylate proteins in the cytosol (such as p90 ribosomal S6 kinases, RSKs; Roux and Blenis, 2004) and also translocate to the nucleus where they ultimately regulate important cellular processes including gene expression, cell motility, cell proliferation, cell survival, differentiation, and death (Ceresa and Pessin, 1998; Brunet et al., 1999; Roux and Blenis, 2004). A schematic representation of the main signaling pathways activated by both IR and IGF-IR is shown in Figure 2.
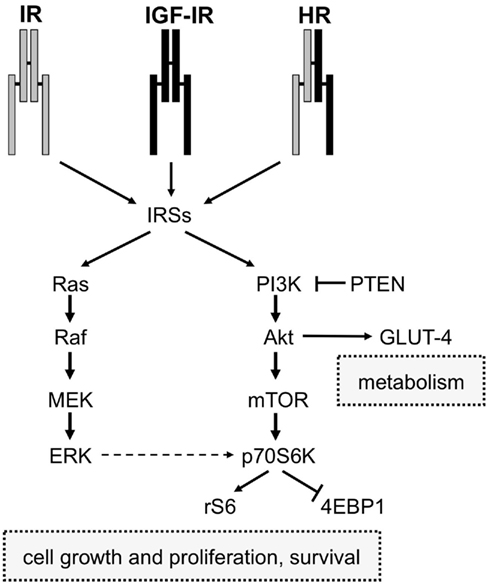
Figure 2. Schematic diagram of IR and IGF-IR signaling. IR, IGF-IR, and Hybrid receptors (HR) induce the phosphorylation of IRSs proteins after binding to their ligands (insulin and IGFs). Activated IRSs trigger the activation of two intracellular signaling networks: Ras/Raf/Mek/Erk and PI3K pathways. The first one is mainly involved in mediating the mitogenic effect of insulin and IGFs, while the PI3K pathway, via Akt, mediates both metabolic and cell growth responses. The Akt-mediated metabolic effects are induced by the activation of enzymes involved in gluconeogenesis, glucose uptake, protein synthesis, and lipogenesis, whereas the cell growth responses are mainly induced by the mTOR pathway.
The IR/IGF-IR Signaling Pathway and Its Involvement in Cancer
It is commonly accepted that insulin primarily mediates metabolic effects through IR and PI3K pathway activation, whereas IGFs mainly regulate growth processes through IGF-IR and mitogen-activated protein kinase (MAPK) signaling, which includes ERK1/2 (Nakae et al., 2001). Although this scheme may look simplistic, a complex and highly integrated network containing several points of regulation, signal divergence as well as overlap and crosstalk with other signaling cascades are involved in insulin and IGFs signaling. For example, the MAPK and PI3K pathways are interconnected and both converge on the mTOR/p70S6K axis, a crucial mediator of cell growth, survival and metabolism (Figure 2).
As previously mentioned, in physiological conditions and in adult life, IR and IGF-IR biological functions are distinct (Nakae et al., 2001). Whereas IR plays a key role in the regulation of a variety of metabolic functions, the IGF-IR has mainly a growth regulation function.
The molecular mechanisms involved in regulating signaling specificity between these two receptors are only partially understood and include: tissue-specific receptor expression, different ligand binding affinity and kinetics, different regulation in ligand secretion and bioavailability, differential expression and function of IR isoforms and HRs, differential abundance and sub-cellular distribution of signaling substrates.
However, in cancer IR/IGF-IR signaling is often deregulated with consequent loss of signaling specificity and overlap between IR and IGF-IR actions. The stimulation of two main intracellular cascades common to both receptors (i.e., the PI3K/mTOR and the MAPK cascades) may also become unbalanced with the consequent amplification of mitogenic signals. Various mechanisms may account for the disruption of the physiological specificity in IR and IGF-IR signaling. Cancer cells often overexpress both IGF-IR and IR, the latter being predominantly expressed as IR-A. IR-A contributes to the amplification of IGFs signaling by directly binding IGF-II and, to a lesser extent, IGF-I (Papa et al., 1990; Frasca et al., 1999; Vella et al., 2002; Samani et al., 2007; Belfiore and Frasca, 2008), and also by increasing the formation of HRs, which bind both IGF-I and IGF-II with high affinity (Table 1; Pandini et al., 1999). Moreover, cancer cells produce IGFs in an autocrine/paracrine manner (Samani et al., 2007). Lastly, a chronic increase in circulating insulin levels resulting from insulin resistance may affect cancer growth and progression through a prevalent activation of IR signaling along the mitogenic pathway and by increasing IGF-I bioavailability (Strickler et al., 2001; Kaaks and Lukanova, 2002; Vainio et al., 2002; Vigneri et al., 2009).
Therefore, in cancer cells, the IR may stimulate more strongly the protumoral responses rather than the metabolic effects thus contributing to induce resistance to various anti-cancer therapies.
IR and IGF-IR: Two Different Team Players in Cancer?
As mentioned before, the main signaling pathways activated by IR and IGF-IR are very similar and interconnected. However, one may ask the question whether the IR role in cancer is unique or just overlaps with that of IGF-IR. A clear answer is not available yet, although several pieces of evidences suggest that the IR and IGF-IR are rather two different team players in cancer biology. Indeed, the two receptors may selectively phosphorylate different substrates or the shared substrates may be phosphorylated at different sites. Moreover, specific and different protein–protein interactions with both receptors may occur (Table 2; Kim and Accili, 2002). For example, carcinoembryonic antigen-related cell adhesion molecule 2 (CEACAM-2), a cell surface molecule that is involved in receptor trafficking, appears to be a preferential substrate of IR and not of IGF-IR (Najjar et al., 1997). Similarly, mitotic arrest deficient 2 (MAD2), a cell cycle regulator, interacts with IR but not with IGF-IR (O’Neill et al., 1997). In contrast, c-Crk proto-oncogene (an adapter protein belonging to the Ras pathway) as well as 14-3-3 (a scaffolding protein involved in apoptosis) and IIP-1 (a protein involved in cell motility) specifically bind IGF-IR but not IR (Craparo et al., 1997; Furlanetto et al., 1997; Ligensa et al., 2001). Yet, the adapter protein Growth factor receptor-bound protein 10 (Grb-10), although phosphorylated by both IR and IGF-IR, seems to interact differently with the two receptors (Laviola et al., 1997; He et al., 1998; Table 2).
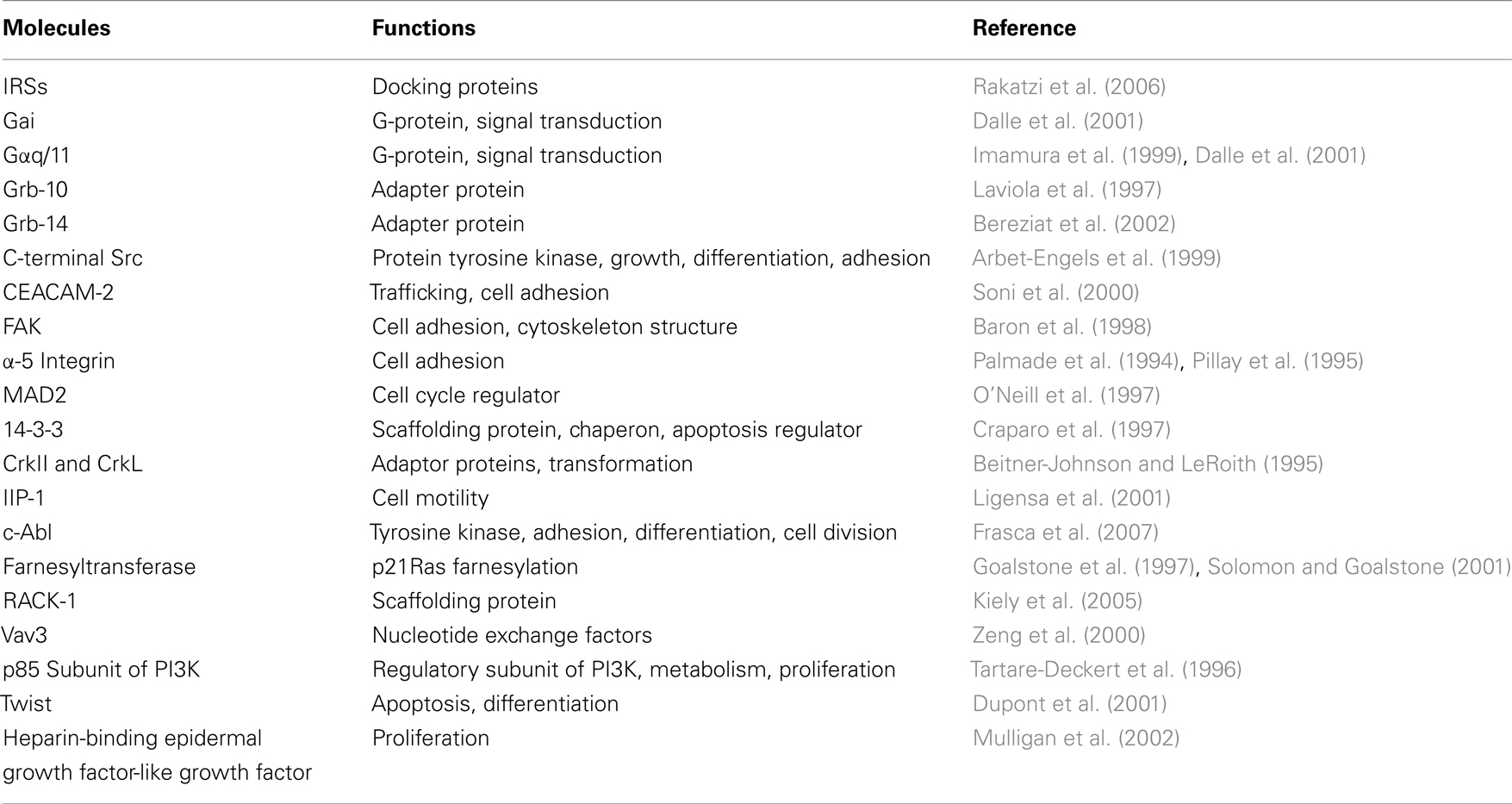
Table 2. Principal substrates found to be differentially involved by IR and IGF-IR activation and that may have a role in functional specificity of these two receptors.
Furthermore, gene expression studies suggest that IR and IGF-IR differentially regulate genes involved in the control of proliferation, apoptosis, differentiation, and cell adhesion. For example IGF-IR, but not IR, induces the expression of twist, a gene regulating the apoptotic process as well as the cell invasion and metastasis (Dupont et al., 2001). The two receptors may act differently toward α-5 integrin and focal adhesion kinase (Fak). In particular IGF-IR causes α-5 integrin upregulation and Fak phosphorylation (Palmade et al., 1994), while IR induces α-5 integrin downregulation and Fak dephosphorylation (Pillay et al., 1995; Table 2). Furthermore, it has been found that IGF-IR forms a ternary complex with α-5 integrin and E-cadherin (Canonici et al., 2008). This crosstalk between the IGF-IR and cell adhesion molecules could be of importance in sustaining alterations in cell adhesion and mobility that occur during the epithelial–mesenchymal transition (EMT) and cancer metastasis. A direct role in EMT has been described for IGF-IR and involves the activation of NF-kB, Snail, SLUG, and ZEB-1/2 transcription factors (Chua et al., 2007; Kim et al., 2007b; Sivakumar et al., 2009). The possible role of IR in EMT has not been validated yet. However, recent evidences highlight a novel role for IRS-1 and IRS-2 in the regulation of EMT process (Shi et al., 2009).
A unique feature of IR is the promotion of the phosphorylation and activation of farnesyltransferase, an enzyme responsible for p21Ras farnesylation. Farnesylation of Ras protein is essential for its translocation to the plasma membrane and subsequent activation by the various growth factors. Since the activation of the Ras pathway mediates cellular mitogenic responses, the effect of insulin in increasing the pool of farnesylated p21Ras amplifies the mitogenic effects of insulin itself and other growth factors, such as IGF-I. This mechanism could explain the exaggerated mitogenic response to growth factors in patients with high levels of circulating insulin (Goalstone et al., 1997; Solomon and Goalstone, 2001; Table 2).
To better understand the functional differences and overlaps between IR and IGF-IR, cell lines derived from mice lacking IR or IGF-IR have been used. In mouse fibroblasts deprived of endogenous IGF-IR (R-cells), IGF-IR but not IR showed a unique permissive effect on cell transformation (Sell et al., 1994; Valentinis et al., 1997). However, in NIH3T3 fibroblasts IR overexpression increases cell proliferation, chemotaxis, and induces a ligand-dependent transformed phenotype (Giorgino et al., 1991).
IR Isoform A: A Gateway to the Crosstalk between the IR and the IGF-IR Pathway
The generation of IR isoforms and their specific tissue distribution represents a link between the IR and the IGF-IR pathway. The IR-A is predominant in the fetal life while the IR-B predominates in most adult and differentiated tissues. Both IR isoforms may be overexpressed in cancer, but usually IR-A is the predominant subtype accounting for 60–100% of total IR. As already mentioned, the two isoforms show different ligand binding affinity. IR-A is a high-affinity receptor not only for insulin but also for IGF-II (Frasca et al., 1999) and IGF-I (Denley et al., 2007; Sacco et al., 2009), while IR-B may be considered a specific receptor for insulin (Table 1). Because of these differences in ligand affinity, kinetics of activation and trafficking, IR-B is preferentially associated with metabolic and differentiating signals. In contrast, IR-A mainly favors cell growth, proliferation, and survival. Given that IR-A binds IGF-II with similar affinity than IGF-IR (Frasca et al., 1999), in cells that express both receptors, IGF-II exerts its biological effects through both receptors according to their molar ratio. Thus, in cancer tissues showing a high IR-A:IGF-IR ratio, IGF-II mainly signals via the IR-A.
In contrast, IGF-I binds with high affinity to the IGF-IR and HRs but with much lower affinity to the IR-A (Frasca et al., 1999; Table 1). However, IGF-I may activate intracellular signaling and stimulate mitogenesis in IR-A overexpressing cells (Sacco et al., 2009). It is worth noting that HRs containing IR-A (HR-A) or IR-B (HR-A) moieties may differ for binding affinity (Table 1). The potential implications of HRs in cancer have been recently reviewed (Belfiore, 2007; Belfiore et al., 2009a).
Several evidences suggest that intracellular signals and biological effects mediated by IR-A in response to IGFs are partially different from those elicited by insulin. Studies conducted in vitro using mouse fibroblasts expressing only IR-A but not IGF-IR (R-/IR-A cells), have shown that IGF-II induces more potent mitogenic effects than insulin itself (Frasca et al., 1999). Yet, in human rhabdomyosarcoma cells expressing almost only IR-A and lacking functional IGF-IR, IGF-II is more potent than insulin in inducing migration (Sciacca et al., 2002). Global gene expression studies performed in R-/IR-A cells have confirmed that certain genes are differentially regulated by IGF-II and insulin (Pandini et al., 2003). Similarly, proteomics analysis revealed that several proximal effectors of the IR-A are selectively and differentially recruited by IGF-II or insulin (Morcavallo et al., 2011). The molecular mechanisms responsible for the potent mitogenic action exerted by IGF-II binding to IR-A are incompletely understood. In R-/IR-A cells IGF-II elicits unbalanced intracellular signaling as compared to insulin by favoring the activation of p70S6K and ERK rather than Akt activation (Sacco et al., 2009). This specific signaling pattern appears to involve a preferential activation of IRS-2 rather than IRS-1 and a reduction in the activation of negative feedbacks (Sacco et al., 2009). Furthermore, IR-A activation by IGF-II may differentially affect substrate recruitment to the receptor and receptor internalization and trafficking. The importance of isoform balance and IR-A/IGF-II loop in cancer has been also highlighted by studies conducted in stem/progenitors cells. These cells may undergo transformation and become cancer stem cells (CSC) through a dysregulation of the self-renewal program. CSC may drive tumor growth, progression and spread and be responsible for resistance to conventional anti-cancer therapies (Wicha et al., 2006). In human embryonic stem (ES) cells, it has been seen that self-renewal and pluripotency properties depend on IGF-II production by ES-cell-derived fibroblast-like cells that act as stem cell niche (Bendall et al., 2007). Consistent with these findings, we have recently found that IGF-II production by thyroid progenitor cells influence their self-renewal capacity. Furthermore, we have found that IR and IGF-IR were expressed at high levels in thyrospheres and markedly decreased in differentiated cells. IR-A was the predominant isoform in thyroid stem cells and its relative abundance was associated with characteristics of stemness and cancer, while a decrease in IR-A:IR-B ratio was associated with cell differentiation (Malaguarnera et al., 2011). Although the molecular mechanisms by which stemness and differentiation programs are influenced by IR isoforms, IGF-IR, and IGFs, are still unclear, it is undoubted that this emerging scenario will have important implication for cancer prevention and therapy.
The IR Pathway as a Candidate Target for Cancer Prevention and Therapy
Validation of IR as a Therapeutical Target in Preclinical Models: in vivo and in vitro Studies
A large body of evidences have demonstrated the importance of IR expression and chronic hyperinsulinemia in cell transformation and tumor development. Studies conducted in obese Zucker (fa/fa) rats, as well as in insulin-resistant patients (Jiang et al., 1999; Cusi et al., 2000), have shown that insulin resistance and hyperinsulinemia may cause abnormal stimulation of mitogenic intracellular signaling and increased cell proliferation and migration in spite of selective attenuation of the PI3K pathway and impaired glucose homeostasis. The first animal model linking insulin and cancer was a model of chemically induced colon tumor in rats, where the injection of insulin enhanced tumorigenesis whereas calorie restriction and low fat diet exerted a protective role (Steinbach et al., 1993; Tran et al., 1996; Corpet et al., 1997). In a different model of rat breast cancerogenesis, the induction of insulinopenic diabetes completely prevented mammary tumor formation or caused a rapid regression of established tumors (Heuson and Legros, 1972).
Recent studies carried out in transgenic type 2 diabetic mice (MKR) have confirmed and extended these findings. In MKR mice severe hyperinsulinemia affected breast cancer progression and tumor burden both of which were blocked by IR signaling inhibition with either an IR/IGF-IR tyrosine kinase inhibitor or by decreasing insulin circulating levels by the administration of a β3 adrenergic receptor agonist (Fierz et al., 2010; Novosyadlyy et al., 2010). In accordance with these findings, after implantation of Met-1 breast cancer cells into mice, the downregulation of IR signaling by short hairpin (sh)RNA prevented tumor formation (Novosyadlyy et al., 2009). Similarly, the downregulation of IR by shRNA inhibited anchorage independent growth of both LCC6 and T47D cells and tumor growth, angiogenesis and lymphangiogenesis in animals after LCC6 cells xenograft. LCC6-shIR cells formed fewer pulmonary metastases compared to LCC6 wild type cells, even in presence of functional IGF-IR. The concept that insulin may induce tumor development is reinforced from the finding that insulin AspB10 (X-10), an insulin analog never used in the clinical setting, is associated with increased occurrence of mammary tumors in rats and β-cell proliferation in transgenic mice (Drejer, 1992; Vincent et al., 1995). After those findings, other synthetic insulins, some of them used in T2DM therapy, have been evaluated for their mitogenic activity. In vitro studies conducted on this issue have shown that some of these insulin analogs (in particular glargine and detemir) are more potent than native insulin in inducing proliferative effects, probably because of a higher affinity for IGF-IR and preferential activation of mitogenic ERK pathway (Kurtzhals et al., 2000; Mayer et al., 2008). Clinical studies have so far focused the attention on cancer risk in diabetic patients using the long acting analog glargine. Three retrospective studies published in 2009 have suggested a possible increase in cancer risk in diabetics treated with glargine as compared to those treated with native insulin (Colhoun, 2009; Hemkens et al., 2009; Jonasson et al., 2009). A fourth study, instead, found an increased cancer risk for patients treated with all insulin preparations, with no difference with those treated with glargine (Currie et al., 2009). An independent analysis from all these studies have concluded that glargine does not carry any additional cancer risk as compared with other insulins (Pocock and Smeeth, 2009). Other recent studies have confirmed the lack of association between glargine use and cancer (Morden et al., 2011; Ruiter et al., 2011). Furthermore, glargine, when administered in vivo, is rapidly metabolized to M1 and M2 compounds with a mitogenic:metabolic ratio similar to that of native insulin (Sommerfeld et al., 2010). Overall these data indicate that glargine safety profile is similar to that of other insulins. Furthermore, in clinical setting, native insulin itself, at least in particular conditions, may be associated to increased risk of cancer. However, it is important to note that insulin analogs may interact differently with the two IR isoforms and/or with IGF-IR and/or IR/IGF-IR hybrids. Therefore, their metabolic:mitogenic ratio should be accurately investigated in order to assure a safe profile of the new insulin analogs. More details regarding this topic will be extensively discussed in a different chapter of this special issue.
Targeting the IR Pathway Dysregulation for Cancer Prevention
As mentioned before, the long-term exposure to hyperinsulinemia consequent to insulin resistance provides a link between diabetes, obesity, and cancer. Therefore, one may expect that insulin sensitizers, used as antidiabetic drugs, by lowering insulin levels, may correct the dysfunctional IR signaling associated with hyperinsulinemia thus contributing to prevent cancerogenesis. Biguanides and thiazolidinediones (TZDs) are the two classes of insulin sensitizers studied so far.
Metformin, the only biguanide used in the clinical setting, has shown encouraging antitumor effects and long-term safety profile. Although the mechanisms underlying the antineoplastic activity of metformin are not yet clearly defined, several studies have reported that this drug targets the complex I in mitochondrial electron transport chain, impairing ATP production. This event triggers the activation of 5′ adenosine monophosphate-activated protein kinase (AMPK), a component of a complex regulatory network (LKB1/AMPK/mTOR/Akt) involved in the control of cellular energy homeostasis and cell size. The activation of the LKB1/AMPK pathway causes downregulation of gluconeogenesis in the hepatocytes and consequent reduction in blood glucose and insulin levels. Furthermore, through the inhibition of the mTOR/AKT pathway, metformin negatively affects cell growth (Shaw et al., 2004; Zakikhani et al., 2006; Alimova et al., 2009). Thus, metformin has a dual negative impact on IR-driven cancer cells, not only through its insulin-lowering action but also through a direct inhibition of IR mitogenic signaling. These actions are shared with other AMPK activators, such as caloric restriction and physical exercise, both powerful physiological inhibitors of tumorigenesis.
Evidences from animal models have supported the potential of metformin to reduce tumor development and progression (Schneider et al., 2001; Hawley et al., 2003; Tomimoto et al., 2008; Hosono et al., 2010; Johnson and Bowker, 2010; Zhu et al., 2011). Most importantly, observational clinical studies have shown a substantial decrease in cancer risk in T2DM patients exposed to metformin as compared with those taking other treatments (Johnson et al., 2002; Bowker et al., 2006; Monami et al., 2009). In particular, a retrospective cohort study of T2DM patients exposed to metformin in monotherapy or in combination with other glucose-lowering therapies, including insulin, has shown that insulin users were more likely to develop solid tumors, especially colorectal (hazard ratio, HR = 1.69) or pancreatic cancers (HR = 4.63) than those taking metformin in monotherapy. Moreover, combination therapies containing metformin plus insulin were associated with a lower cancer risk (HR = 0.54; Currie et al., 2009). These data have been supported by a recent retrospective cohort study conducted in diabetic patients showing a lower cancer related mortality risk in the metformin group (HR = 0.56) as compared with insulin users (HR = 1.56; Bo et al., 2011). Further support for a metformin role in breast cancer prevention has been provided by a population-based case–control study in which metformin use was associated with a reduced cancer risk (HR = 0.77; Bosco et al., 2011).
In light of these promising observations, two pilot clinical trials are currently being conducted in non-diabetic women with the purpose to evaluate the possible impact of metformin in affecting breast cancer growth and/or progression (clinical trials: NCT00897884, NCT01101438).
TZDs, the second class of insulin sensitizers available for clinical use, belong to the group of PPARγ agonists and have raised much hope as antidiabetic drugs and potential anti-cancer agents (Tontonoz and Spiegelman, 2008). However, clinical results have been disappointing because of concern for long-term toxicity and for the controversial impact on cancerogenesis. TZDs may show both anti-cancer effects as well as tumor promoting responses depending on the cell context (Armengol et al., 2000; Butler et al., 2000; Mueller et al., 2000; Inoue et al., 2001; Heaney et al., 2002; Kawa et al., 2002; Shimada et al., 2002; Burstein et al., 2003; Li et al., 2003; Patel et al., 2005; Kim et al., 2007a). Like metformin, TZDs are able to ameliorate insulin resistance, although through different mechanisms, and decrease insulin and free IGF-I levels (Tontonoz and Spiegelman, 2008). TZDs also negatively impact on the IGF system signaling pathways, an intriguing characteristic suggesting potential antineoplastic activity (Aiello et al., 2006; Belfiore et al., 2009b). Indeed, in vitro studies have shown that TZDs are able to downregulate both MAPK and PI3K/mTOR/Akt pathways following IR/IGF-IR activation. These findings may suggest that the TZDs could be beneficial in cancers “addicted” to overactivated IGF system and in insulin resistant, hyperinsulinemic patients. However, this hypothesis has not been confirmed by the scanty data collected in the clinical setting. Recent studies have reported no significant variations of cancer risk in diabetic patients treated with TZDs (Monami et al., 2008; Ferrara et al., 2011), while other studies have even suggested an increased risk for bladder cancer in pioglitazone users (Lewis et al., 2011). Because of these latter data, in some countries the use of pioglitazone has been restricted. The second TZD, formerly extensively used in diabetic patients, rosiglitazone, has been removed from the market because of significant cardiac toxicity (Loke et al., 2011). To date, the full range of TZDs effects in IR-driven cancerogenesis is not well understood.
IR Involvement in Cancer Resistance: the Ultimate TK Receptor for Cancer Cells?
Although preclinical studies have provided strong evidences for the role of IR in cancer, until very recently the IR has not been a deliberate target of cancer therapies. Rather, the attention has been focused on the homologous IGF-IR. Indeed, several companies have developed drugs to block the IGF-IR in cancer and numerous trials have been designed (Gualberto and Pollak, 2009). Although a variety of in vitro and in vivo studies have showed a good anti-cancer activity of anti-IGF-IR drugs, especially in combination with chemotherapy (Pollak, 2008b), the results of the first clinical trials have been disappointing. In fact, only a small subset of malignant tumors, including approximately 10% Ewing sarcomas and a small percentage of NSCLC, have shown an objective response to these therapies (Hofmann and Garcia-Echeverria, 2005; Scotlandi, 2006; Gualberto and Pollak, 2009). These unsatisfactory results have contributed to turn our attention to the IR pathway as a factor in the development of resistance to selective anti-IGF-IR drugs and, therefore, as a possible new target. As documented for several malignancies, IGF-IR itself is involved in cancer resistance not only to traditional therapies (Dunn et al., 1997; Gooch et al., 1999; Casa et al., 2008) but also to old and new targeted therapies such as anti-estrogens and inhibitors of epidermal growth factor receptor/human epidermal growth factor-2 (EGFR/HER2) and of Akt/mTOR signaling (Carter et al., 1992; Lu et al., 2001; Nicholson et al., 2004; Wan et al., 2007; Guix et al., 2008; Tamburini et al., 2008). The model of breast cancer has been particularly studied and has suggested that the IR/IGF-IR pathway has unique importance. Breast cancer cells treated with anti-estrogen therapies may become estrogen-independent and exploit alternative pathways of growth and survival such as those mediated by EGFR and HER2. However, cells initially responsive to EGFR/HER2 inhibitors may, in turn, become resistant to these drugs and acquire addiction to IGF-IR signaling. However, anti-IGF-IR therapies may favor the emergence of IR-driven resistant cancer clones through several mechanisms that include: (a) increased cell sensitivity to insulin, as observed in cultured breast cancer cells treated with selective anti-IGF-IR small interfering RNAs (siRNAs; Zhang et al., 2007). These cells express enhanced IR homodimers possibly because IR hemidimers engaged in HRs formation are reduced; (b) increased IR-A and/or IGF-II gene expression by the cancer cells, as observed in Ewing’s sarcoma cells that have developed resistance to specific IGF-IR inhibitors, both antibodies and tyrosine kinase inhibitors (Garofalo et al., 2011); (c) development/worsening of hyperinsulinemia, caused by reduced IGF-I feedback at the pituitary level and consequent increase in plasma GH (Pollak, 2008b).
On the basis of these considerations it is likely that IR-based mechanisms may ultimately play a key role in cancer resistance to various treatments (Figure 3), an additional reason to consider IR as a suitable target in cancer.
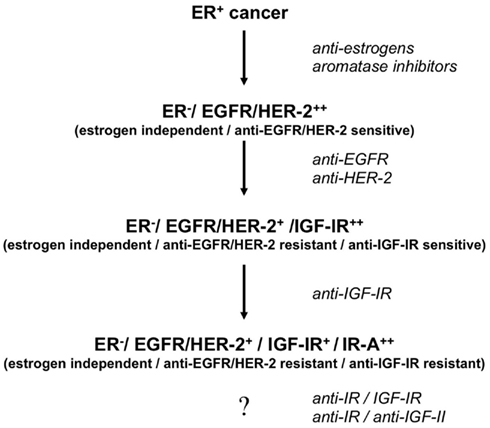
Figure 3. Proposed model of development of stepwise resistance to medical therapies and IR-A addiction in breast cancer. Following anti-estrogen therapy, cancers initially ER+ (Estrogen Receptor positive and estrogen-dependent/sensitive) may become ER− (Estrogen Receptor negative and estrogen-independent/resistant) and sensitive to anti-EGFR/HER2 blockade. Later on, tumor cells may acquire resistance to these agents and become sensitive to anti-IGF-IR therapies. Eventually, clones may develop resistance to selective IGF-IR blockade by upregulating both IR-A and IGF-II. Co-targeting IR and IGF-IR may prevent such evolution. In any case we need to develop efficacious strategies to target the IR-A/IGF-II loop in cancer.
Indications and Strategies for Targeting IR in Cancer Patients
Increasing IR Sensitivity, IR Blockade, or Both?
The specific indications for targeting the IR pathway in cancer treatment are still unclear but this issue is likely to be under intensive scrutiny in the near future. Given the relevance of insulin resistance not only in increasing cancer risk but also in worsen cancer prognosis (Duggan et al., 2011; Irwin et al., 2011), one may question whether insulin resistance should be always addressed in cancer patients. Insulin resistance in these patients may precede the diagnosis of cancer because of genetic factors, T2DM or obesity, or may be induced by various anti-cancer treatments, including anti-IGF-IR targeted therapies, anti-androgen therapy (Aggarwal et al., 2011), or by conditions such as chronic inflammation and malnutrition frequently associated with cancer (Van Cutsem and Arends, 2005; Gonda et al., 2009). In these cases the use of insulin sensitizers would have a strong rationale. Metformin has been recently found to improve pathologic complete response rates in diabetic patients with breast cancer receiving neoadjuvant chemotherapy (Jiralerspong et al., 2009) as well as progression-free survival for chemotherapy treated diabetic patients with advanced non-small cell lung cancer (Tan et al., 2011). Whether these effects are to be ascribed to the hyperinsulinemia lowering effect of metformin or to direct effects of metformin on cancer cells is still unclear. Moreover, metformin is able to control hyperglycemia associated with the use of anti-IGF-IR antibodies (Atzori et al., 2011). Again, whether metformin is sufficient to counteract the adverse effects of anti-IGF-IR drugs induced insulin resistance remains to be established. No such studies are available with other insulin sensitizers such as thiazolidinediones despite the promising preclinical anti-cancer activity of these compounds (Sertznig et al., 2007).
Taken together, available data strongly suggest that hyperinsulinemia should be diagnosed in all cancer patients and appropriately managed. Our arsenal of cancer-effective insulin sensitizers seems currently limited to metformin. However, there is hope that other insulin sensitizers, such as novel PPARγ agonists or Sirt-1 agonists (Herranz et al., 2010) could be soon developed for clinical use.
Insulin sensitizing therapy, however, while reducing the direct effect of circulating insulin on cancer cells is unlikely to normalize plasma insulin levels and cannot influence the effects of autocrine IGF-II binding on IR-A in cancer cells. Therefore, another question is when IR blockade should be considered and whether there is any place for the use of metformin or other insulin sensitizers together with IR blockade. There is no definite answer to these questions yet. However, it is reasonable to think that malignancies carrying an overactivated IGF-II/IR-A loop are best candidates for IR blocking therapies. As previously mentioned, not only IR-A is commonly overexpressed in cancer together with the IGF-IR, but it is also predominant in certain cancers (Belfiore et al., 2009a). Whenever both IGF-I and IGF-II are present in the tumor microenvironment, it seems reasonable to assume that IGF-IR is saturated by the high-affinity ligand IGF-I whereas IR-A is the principal receptor of IGF-II.
As we previously mentioned, it has become increasingly clear that, among other unknown factors, IR-A overexpression is likely to be a major factor in determining in vivo resistance to anti-IGF-IR therapies. Therefore, more recent studies have taken in serious consideration the concept that co-targeting IR together with IGF-IR, should be a more effective approach to prevent adaptive resistance to anti-IGF-IR therapies. The measurement of IR:IGF-IR ratio and HRs (Pandini et al., 2007), and IGF-II expression in tumor cells should be extremely helpful in identifying malignancies suitable to anti-IR/IGF-IR therapies. However, IR overexpression by itself may not be a guarantee for full IR functionality (Belfiore et al., 1996). Some studies have been proposed that phosphorylated IR/IGF-IR or IRS-1 are possible biomarkers of sensitivity to IR/IGF-IR blockade (Byron et al., 2006; Law et al., 2008). Finally, an IGF gene expression signature has been shown to correlate with response to a dual anti-IR/IGF-IR inhibitor, BMS-754807 (Litzenburger et al., 2011). Furthermore, using the IR/IGF-IR inhibitor OSI-906 (see also next paragraph) it has been found that cancer cell lines exhibiting an epithelial phenotype were more sensitive than tumor cells that have undergone EMT (Mulvihill et al., 2009).
Radiolabeled anti-IGF-IR antibodies have also been used to visualize in vivo IGF-IR expression using both Single-Photon Emission Computed Tomography and Positron Emission Tomography in preclinical models (Heskamp et al., 2011). In the future, similar techniques, possibly employing anti-IR antibodies, may help patient selection for IR/IGF-IR targeted therapy.
Finally, regarding the question whether metformin could be of any benefit if administered together with IR/IGF-IR inhibitors, there are simply no data yet, although there is a rationale for including metformin in the management of hyperglycemia and hyperinsulinemia that are frequent adverse effects of these drugs.
Currently Available Drugs Co-Targeting IR and IGF-IR
Only very few of currently available drugs, generally developed for inhibiting IGF-IR, actually share the ability of inhibiting also IR.
Monoclonal antibodies
Anti-IGF-IR antibodies may be selective for homodimeric IGF-IR, but often also block HRs to a various extent (Soos and Siddle, 1989). Double blockade of IGF-IR and HRs may results advantageous over selective IGF-IR blockade. In cancers with low IR:IGF-IR ratio this approach may significantly affect IR activity, as in these cancer cells most IR moieties are engaged in HRs and could, therefore, be blocked. Unfortunately, among the several anti-IGF-IR antibodies currently on trial, only dalotuzumab (formerly h7C10) has been characterized for its binding activity to HRs (Pandini et al., 2007). In fact, it has been observed that anti-IGF-IR antibodies binding to HRs may also cause some IR degradation (Sachdev et al., 2006). IR degradation was also seen by administering a combination of two anti-IGF-IR antibodies with diverse binding epitopes and ligand-blocking properties (competitive and allosteric), which also caused a more potent blockade of IGF-I and IGF-II compared with either antibody alone (Dong et al., 2010). However, this approach results insufficient in cancer with high IR:IGF-IR ratio, where most IRs occur as homodimers. In such cells, IR blockade was shown to inhibit cancer cell proliferation in response to exogenous and/or autocrine IGF-II (Sciacca et al., 1999; Vella et al., 2002) and to be more useful than IGF-IR blockade (Pandini et al., 2007).
A viable strategy for co-targeting IR-A and IGF-IR effects in cells with high IR:IGF-IR ratio could be to combine anti-IGF-IR antibodies showing high affinity to HRs with anti-IGF-II blocking antibodies. Antibodies recognizing both IGF-II and IGF-I have already been developed (Miyamoto et al., 2005; Feng et al., 2006) and have shown promising results on in vivo growth of IGF-I- or IGF-II-driven tumors (Gao et al., 2011). However, studies showing results of such combination therapy are not available.
Tyrosine kinase inhibitors
Small molecules with tyrosine kinase inhibitory activity for IGF-IR are also being actively investigated for their possible use in cancer patients. Owing to the high homology between the IGF-IR and the IR ATP-binding site of the kinase domain, these molecules, however, may also inhibit the IR to a variable degree (Buck and Mulvihill, 2011). Although this feature was initially unwanted because it contributes to deterioration of glucose metabolism, it was later recognized that co-targeting IGF-IR and IR may actually be advantageous in order to prevent IR-A driven adaptive resistance.
Some of these TK inhibitors may also affect various other TKs to some extent, with the result to be characterized by increased efficacy in some models, but also increased toxicity. Small molecule TK inhibitors can be given orally and also differ from anti-IGF-IR antibodies for having a faster kinetics that allow more flexibility when administered in combination with standard chemotherapy (Buck and Mulvihill, 2011).
Two of these molecules, Picropodophyllin (PPP) and Nordihydroguaiaretic acid (NDGA), described as IGF-IR inhibitors with antitumoral activity, actually do not appear to inhibit the IR. Their mode of action is rather complex and not well understood. Intriguingly, they do not cause hyperglycemia or insulin resistance.
PPP is a non-toxic cyclolignan, described as a selective IGF-IR inhibitor with an in vivo IC50 for IGF-IR inhibition of 40 nM (Girnita et al., 2004). The drug is a non-ATP-competitive IGF-IR inhibitor and targets amino acids outside of the activation loop, a region homologous between the two receptors. PPP induces also IGF-IR downregulation. This compound, therefore, is unable to co-target the IR. Accordingly, mice treated with PPP did not develop hyperglycemia, although showing inhibition of IGF-IR overexpressing tumors (Girnita et al., 2004). PPP is currently being tested as a single agent treatment in an open-label Phase I/II trial in patients with solid tumors that progress in spite of several lines of treatment. No changes in variables involved in glucose metabolism, such as blood glucose and insulin, have been reported in treated patients, confirming that the IR pathway is not inhibited (Economou et al., 2008).
NDGA, is a plant derived phenolic compound that has been shown to be an IGF-IR and HER2 inhibitor (Youngren et al., 2005) and to be able to revert breast cancer resistance to trastuzumab (Rowe et al., 2008). In addition, it also inhibits TGF-β effects at multiple levels (Li et al., 2009) and may function as a global transcription inhibitor (Rowe et al., 2008). NDGA does not inhibit the IR. Actually, it can even ameliorate insulin resistance by activating the AMPK (Lee et al., 2011).
While these two inhibitors share the interesting feature to inhibit the IGF-IR without inducing insulin resistance, they have the limitation to leave intact the IGF-II/IR-A loop, which is the principal mechanism of IGF system activation in certain malignancies. Further work is needed to better clarify their mode of action and their effects on glucose metabolism.
In contrast, two other inhibitors, BMS-754807 and OSI-906, are indeed able to co-target the IR and IGF-IR.
BMS-754807, a pyrrolotriazine designed to be an ATP-competitive antagonist of IGF-IR. It was found, however, to inhibit both IGF-1R and IR activity with very similar activity (IC50 was 1.8 and 1.7 nM, respectively; Carboni et al., 2009). The activity of downstream molecules, such as Akt and, to a lesser extent, ERK was also inhibited. However, this compound has also shown significant inhibiting activity toward other important TK receptors (Met, RON, TrkA, TrkB) and also toward Aurora A and B. In cell culture, however, inhibiting activity of BMS-754807 against IGF-IR was 9- to 29-folds higher than against Met and 25- to 64-folds higher than against TrkA. In proliferation assays of cell lines expressing high levels of IGF-IR, BMS-754807 resulted more efficacious than anti-IGF-IR antibody mAb391 and induced a distinct cell phenotype when compared to the anti-Aurora inhibitor VX-680 (Carboni et al., 2009). Cellular IC50, however, varied widely between 5 and 365 nM. In the animal model, BMS-754807, as a single agent, has shown antitumor activity in a variety of epithelial and mesenchymal cell xenografts. Major side effects were hyperglycemia and hyperinsulinemia.
BMS-754807 is currently being evaluated in Phase I/II clinical trials as a single agent (NCT00898716 and NCT00569036) and in combination therapies with cetuximab (NCT00908024), trastuzumab (NCT00788333), letrozole (NCT01225172), paclitaxel, and carboplatin (NCT00793897) in patients with various advanced or metastatic malignancies. Preliminary results of Phase I study appear encouraging showing a good rate of stable disease when BMS-754807 is used as a single agent (Evans et al., 2010). BMS-754807 was well tolerated and fatigue and hyperglycemia were the most frequent related adverse events. Hyperglycemia was manageable and did not lead to discontinuation of the therapy.
In an in vitro model, BMS-754807 was able to attenuate the hyperactivity of the IR pathway in murine fibroblasts that are null for the IGF-IR (Dinchuk et al., 2010). However, it is too early to have a clear picture on how the IR inhibiting activity of this compound relates to its in vivo antitumor activity.
While BMS-754807 has significant inhibitory effects on other kinases, OSI-906 is probably the most specific co-inhibitor of IR and IGF-IR. It was also developed in the context of drug discovery work aimed at identifying small molecule inhibitors against the IGF-IR. However, it was optimized against both IR and IGF-IR crystal structures and has shown the ability to inhibit ligand-stimulated IGF-IR phosphorylation with an IC50 of 19–35 nM. It also inhibits IGF-IR dependent ERK, AKT, and p70S6K activation with similar efficacy (Mulvihill et al., 2009). Interestingly, IC50 for IR was 39 nM, whereas IC50 for a variety of threonine/serine kinases was in the range of 1–10 μM. These unique characteristics qualify OSI-906 as a selective dual inhibitor for IGF-IR and IR. In in vitro experiments, 1 μM OSI-906 fully inhibited both IGF-IR and IR phosphorylation. However, when evaluated in a panel of cancer cell lines, EC50 for proliferation inhibition fell into a wider range (21–810 nM; Mulvihill et al., 2009). Tumor repression in preclinical models was correlated to the degree and duration of IGF-IR phosphorylation inhibition. The relationship between IR phosphorylation and tumor inhibition is less studied.
OSI-906 is now being evaluated in Phase I escalation studies as a single agent according to various administration schedules (intermittent or continuous; NCT00514306, NCT00514007). Overall, these studies have found encouraging disease control rates (Carden et al., 2010; Evans et al., 2010). Given the relatively short half-life of OSI-906 there is hope that intermittent administration may allow partial recovery from glucose metabolism deterioration while maintaining a good antitumor activity.
The drug is being evaluated as a single agent in patients with advanced hepatocellular carcinoma (Phase II study, NCT01101906), in patients with adrenocortical carcinoma (Phase III study, NCT00924989) and in patients with relapsed small cell lung cancer (Phase II study, NCT01387386). These malignancies are generally characterized by the overactivation of the IR/IGF-IR axis and, in particular, of the IGF-II/IR-A loop. Other ongoing Phase I/II studies aim to evaluate OSI-906 in combination with a variety of anti-cancer agents, including paclitaxel, irinotecan, erlotinib, hormone therapy.
Hyperglycemia has a major role as a dose-limiting toxicity factor (Macaulay et al., 2010). However, it is reversible after cessation of treatment and does not preclude clinical efficacy (Carden et al., 2010; Evans et al., 2010).
These studies will hopefully provide proof-of-concept evidences that dual inhibition of IGF-IR and IR may prevent resistance to selective anti-IGF-IR agents.
Monitoring the Efficacy and Adverse Effects of IR Blockade
A variety of pharmacodynamic markers, i.e., markers reflecting the actual ability of the therapy to hit the intended target, have been developed to monitor the efficacy of anti-IGF-IR drugs and are being also adopted for dual inhibitors of IR and IGF-IR (Gedrich et al., 2009). For instance, IR and IGF-IR autophosphorylation in peripheral blood mononuclear cells (PBMCs) has been used to monitor the efficacy of OSI-906. Similarly, monitoring of IGF-IR phosphorylation in PBMCs or in circulating tumor cells, has been described for anti-IGF-IR antibodies (de Bono et al., 2007; Tolcher et al., 2009). Fluorodeoxyglucose-positron emission tomography scans have been also used to assess the metabolic activity of tumor cells (Tolcher et al., 2009).
Hyperglycemia and elevations of circulating insulin represent the most frequent adverse effects of these therapies and have been also used as a measure of peripheral blockade of IR and IGF-IR. While insulin resistance is an obvious consequence of IR blockade by dual IR/IGF-IR inhibitors, it is also partially due to inhibition of IGF-I negative feedback at the level of pituitary with consequent plasma GH elevation (Pollak, 2008a). Plasma GH, therefore, may also provide a measure of IGF-IR inhibition at the pituitary level and correlates with insulin resistance, increased plasma IGF-I and IGF-BP3 (Gedrich et al., 2009).
Data on toxic effects of IR inhibition appear still very limited. In studies with OSI-906, most common adverse effects, other than hyperglycemia, were nausea, vomiting, lethargy, and fatigue (Carden et al., 2010; Evans et al., 2010). Although these effects were generally mild and did not prevent reaching a biologically efficacious dose, more data are needed to ensure that long-term treatment does not impair the function of organs sensitive to the non-metabolic effects of insulin, such as the central nervous system.
Future Developments
Anti-cancer therapies specifically targeting the IR or IR-specific substrates are lacking. In fact, the efforts of drug companies have been so far focused in increasing, and not diminishing, insulin secretion and action in order to cure diabetes. Therefore, the implementation of IR blocking therapies appears particularly worrisome given the crucial role of IR in glucose metabolism.
Future strategies may consider targeting selectively the IR-A isoform, in order to minimize the impact on glucose metabolism. However, blocking antibodies specifically recognizing the IR-A have not been described and their feasibility is uncertain. Given that IGF-II binding affinity for IR-A is much greater than for IR-B, it is conceivable to design an inverse agonist of IGF-II, able to interfere with the IGF-II/IR-A loop and possibly also with the IGF-II/IGF-IR loop. IGF-II may be also blocked by monoclonal antibodies, as mentioned above.
Another possibility is to devise IR isoform-specific insulin analogs and/or analogs with full metabolic activity but with blocking activity for the mitogenic branch of IR. The design of these analogs could be made possible by the recent advances in this technology and better understanding of IR structure (Jensen et al., 2007; Jiracek et al., 2010). Such analogs may be used in place of native insulin or of currently used insulin analogs for the therapy of diabetic patients in order to avoid the adverse effects of hyperinsulinemia on cancer cells.
Other possible approaches may target IR overexpression and/or abnormal IR gene splicing in cancer cells, although the mechanisms involved are incompletely characterized. Mutations of antioncogenes, such as p53 may have a role in IR overexpression (Webster et al., 1996). Other important factors may involve the common overexpression of HMGA1 proteins in cancer (Reeves et al., 2001). HMGA1 are non-histone nuclear proteins that function as architectural transcription factors and have been implicated in several aspects of malignant cells. Recently, an important role in the regulation of both IR and IGF-IR in normal and cancer cells has been recognized (Foti et al., 2003; Aiello et al., 2010). Targeting the transcriptional complex involving HMGA1 and driving aberrant IR transcription in cancer cells could also be an avenue worth to be explored.
RNA splicing factors involved in aberrant IR-A generation in cancer cells could also be a target. However, the regulation of such splicing factors in cancer is still poorly characterized (Paul et al., 2006; Belfiore et al., 2009a).
The IR-A substrates specifically activated by IGF-II have been sought in the attempt to identify novel molecular targets with an important role in malignancies with an overactivated IGF-II/IR-A (Morcavallo et al., 2011). Data obtained strongly support the concept that IGF-II, upon binding to IR-A, activates a unique signaling pattern that partially differs from that of insulin and have led to the identification of novel IR-A substrates specifically or predominantly activated by IGF-II and not by insulin. These studies suggest the possibility to block the mitogenic branch of IR signaling without affecting its metabolic function. Interestingly, among substrates selectively stimulated by IGF-II are discoidin domain receptors, involved in cell migration and tumor metastasis, and ephrin receptor B4, involved in bidirectional signaling upon cell–cell contact (Morcavallo et al., 2011). More work is needed to assess whether these substrates may prove to be suitable molecular targets in cancer therapy. Finally, IR-A trafficking appears significantly different when binding IGF-II, rather than insulin (our unpublished data). These differences may be exploited to selectively target IR-A activation and signaling after IGF-II.
Concluding Remarks
Most recent studies have supported the concept that co-targeting the IR together with IGF-IR in cancer would be a more useful therapeutic option than targeting the IGF-IR alone. Preclinical data and early clinical trials have provided evidences that IR/IGF-IR co-targeting may potentiate various chemotherapeutic regimens and prevent adaptive resistance to selective anti-IGF-IR drugs. Unfortunately, IR inhibition also blocks the glycometabolic cascade and generally results in glucose metabolism deterioration and T2DM worsening, effects also shared by selective IGF-IR inhibitors. T2DM is generally responsive to insulin sensitizers, which have the additional beneficial effect of lowering hyperinsulinemia, thus reducing the mitogenic effect of insulin.
However, only very few drugs acting as insulin sensitizers or IR inhibitors are currently available for clinical treatments and we expect that more options will be accessible in the near future. We are just starting to learn how and when using these two classes of drugs either alone or in combination in cancer patients. Ideally, we would need to target the mitogenic cascade of IR leaving relatively intact the metabolic cascade, a task that is obviously extremely challenging. A variety of different approaches are under scrutiny and hopefully will end up providing clinically viable treatments.
The use of biomarkers for appropriate patients selection and efficacy monitoring also awaits future developments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC) to Antonino Belfiore, and from PRIN-MIUR, grant 2008BKRFBH_005, to Antonino Belfiore.
References
Aggarwal, R. R., Ryan, C. J., and Chan, J. M. (2011). Insulin-like growth factor pathway: a link between androgen deprivation therapy (ADT), insulin resistance, and disease progression in patients with prostate cancer? Urol. Oncol.
Aiello, A., Pandini, G., Frasca, F., Conte, E., Murabito, A., Sacco, A., Genua, M., Vigneri, R., and Belfiore, A. (2006). Peroxisomal proliferator-activated receptor-gamma agonists induce partial reversion of epithelial-mesenchymal transition in anaplastic thyroid cancer cells. Endocrinology 147, 4463–4475.
Aiello, A., Pandini, G., Sarfstein, R., Werner, H., Manfioletti, G., Vigneri, R., and Belfiore, A. (2010). HMGA1 protein is a positive regulator of the insulin-like growth factor-I receptor gene. Eur. J. Cancer 46, 1919–1926.
Alimova, I. N., Liu, B., Fan, Z., Edgerton, S. M., Dillon, T., Lind, S. E., and Thor, A. D. (2009). Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 8, 909–915.
Andersen, A. S., Wiberg, F. C., and Kjeldsen, T. (1995). Localization of specific amino acids contributing to insulin specificity of the insulin receptor. Ann. N. Y. Acad. Sci. 766, 466–468.
Arbet-Engels, C., Tartare-Deckert, S., and Eckhart, W. (1999). C-terminal Src kinase associates with ligand-stimulated insulin-like growth factor-I receptor. J. Biol. Chem. 274, 5422–5428.
Armengol, G., Knuutila, S., Lluis, F., Capella, G., Miro, R., and Caballin, M. R. (2000). DNA copy number changes and evaluation of MYC, IGF1R, and FES amplification in xenografts of pancreatic adenocarcinoma. Cancer Genet. Cytogenet. 116, 133–141.
Atzori, F., Tabernero, J., Cervantes, A., Prudkin, L., Andreu, J., Rodriguez-Braun, E., Domingo, A., Guijarro, J., Gamez, C., Rodon, J., Di Cosimo, S., Brown, H., Clark, J., Hardwick, J., Beckman, R. A., Hanley, W., Hsu, K., Calvo, E., Rosello, S., Langdon, R. B., and Baselga, J. (2011). A phase I, pharmacokinetic and pharmacodynamic study of dalotuzumab (MK-0646), an Anti-IGF-1R monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 17, 6304–6312.
Baron, V., Calleja, V., Ferrari, P., Alengrin, F., and Van Obberghen, E. (1998). p125Fak focal adhesion kinase is a substrate for the insulin and insulin-like growth factor-I tyrosine kinase receptors. J. Biol. Chem. 273, 7162–7168.
Beitner-Johnson, D., and LeRoith, D. (1995). Insulin-like growth factor-I stimulates tyrosine phosphorylation of endogenous c-Crk. J. Biol. Chem. 270, 5187–5190.
Belfiore, A. (2007). The role of insulin receptor isoforms and hybrid insulin/IGF-I receptors in human cancer. Curr. Pharm. Des. 13, 671–686.
Belfiore, A., Costantino, A., Frasca, F., Pandini, G., Mineo, R., Vigneri, P., Maddux, B., Goldfine, I. D., and Vigneri, R. (1996). Overexpression of membrane glycoprotein PC-1 in MDA-MB231 breast cancer cells is associated with inhibition of insulin receptor tyrosine kinase activity. Mol. Endocrinol. 10, 1318–1326.
Belfiore, A., and Frasca, F. (2008). IGF and insulin receptor signaling in breast cancer. J. Mammary Gland Biol. Neoplasia 13, 381–406.
Belfiore, A., Frasca, F., Pandini, G., Sciacca, L., and Vigneri, R. (2009a). Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 30, 586–623.
Belfiore, A., Genua, M., and Malaguarnera, R. (2009b). PPAR-gamma agonists and their effects on IGF-I receptor signaling: implications for cancer. PPAR Res. 2009, 830501.
Bendall, S. C., Stewart, M. H., Menendez, P., George, D., Vijayaragavan, K., Werbowetski-Ogilvie, T., Ramos-Mejia, V., Rouleau, A., Yang, J., Bosse, M., Lajoie, G., and Bhatia, M. (2007). IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature 448, 1015–1021.
Benyoucef, S., Surinya, K. H., Hadaschik, D., and Siddle, K. (2007). Characterization of insulin/IGF hybrid receptors: contributions of the insulin receptor L2 and Fn1 domains and the alternatively spliced exon 11 sequence to ligand binding and receptor activation. Biochem. J. 403, 603–613.
Bereziat, V., Kasus-Jacobi, A., Perdereau, D., Cariou, B., Girard, J., and Burnol, A. F. (2002). Inhibition of insulin receptor catalytic activity by the molecular adapter Grb14. J. Biol. Chem. 277, 4845–4852.
Blanco-Aparicio, C., Renner, O., Leal, J. F., and Carnero, A. (2007). PTEN, more than the AKT pathway. Carcinogenesis 28, 1379–1386.
Bo, S., Ciccone, G., Rosato, R., Villois, P., Appendino, G., Ghigo, E., and Grassi, G. (2011). Cancer mortality reduction and metformin. A retrospective cohort study in type 2 diabetic patients. Diabetes Obes. Metab.
Bosco, J. L., Antonsen, S., Sorensen, H. T., Pedersen, L., and Lash, T. L. (2011). Metformin and incident breast cancer among diabetic women: a population-based case-control study in Denmark. Cancer Epidemiol. Biomarkers Prev. 20, 101–111.
Bowker, S. L., Majumdar, S. R., Veugelers, P., and Johnson, J. A. (2006). Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 29, 254–258.
Brunet, A., Roux, D., Lenormand, P., Dowd, S., Keyse, S., and Pouyssegur, J. (1999). Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 18, 664–674.
Buck, E., and Mulvihill, M. (2011). Small molecule inhibitors of the IGF-1R/IR axis for the treatment of cancer. Expert Opin. Investig. Drugs 20, 605–621.
Burstein, H. J., Demetri, G. D., Mueller, E., Sarraf, P., Spiegelman, B. M., and Winer, E. P. (2003). Use of the peroxisome proliferator-activated receptor (PPAR) gamma ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Res. Treat. 79, 391–397.
Butler, R., Mitchell, S. H., Tindall, D. J., and Young, C. Y. (2000). Nonapoptotic cell death associated with S-phase arrest of prostate cancer cells via the peroxisome proliferator-activated receptor gamma ligand, 15-deoxy-delta12,14-prostaglandin J2. Cell Growth Differ. 11, 49–61.
Byron, S. A., Horwitz, K. B., Richer, J. K., Lange, C. A., Zhang, X., and Yee, D. (2006). Insulin receptor substrates mediate distinct biological responses to insulin-like growth factor receptor activation in breast cancer cells. Br. J. Cancer 95, 1220–1228.
Canonici, A., Steelant, W., Rigot, V., Khomitch-Baud, A., Boutaghou-Cherid, H., Bruyneel, E., Van Roy, F., Garrouste, F., Pommier, G., and Andre, F. (2008). Insulin-like growth factor-I receptor, E-cadherin and alpha v integrin form a dynamic complex under the control of alpha-catenin. Int. J. Cancer 122, 572–582.
Carboni, J. M., Wittman, M., Yang, Z., Lee, F., Greer, A., Hurlburt, W., Hillerman, S., Cao, C., Cantor, G. H., Dell-John, J., Chen, C., Discenza, L., Menard, K., Li, A., Trainor, G., Vyas, D., Kramer, R., Attar, R. M., and Gottardis, M. M. (2009). BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther. 8, 3341–3349.
Carden, C. P., Kim, E. S., Jones, R. L., Alam, S. M., Johnson, F. M., Stephens, A. W., Poondru, S., Gedrich, R., Lippman, S. M., and Kaye, S. B. (2010). Phase I study of intermittent dosing of OSI-906, a dual tyrosine kinase inhibitor of insulin-like growth factor-1 receptor (IGF-1R) and insulin receptor (IR) in patients with advanced solid tumors. J. Clin. Oncol. 28, abstr. :2530.
Carter, P., Presta, L., Gorman, C. M., Ridgway, J. B., Henner, D., Wong, W. L., Rowland, A. M., Kotts, C., Carver, M. E., and Shepard, H. M. (1992). Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. U.S.A. 89, 4285–4289.
Casa, A. J., Dearth, R. K., Litzenburger, B. C., Lee, A. V., and Cui, X. (2008). The type I insulin-like growth factor receptor pathway: a key player in cancer therapeutic resistance. Front. Biosci. 13, 3273–3287.
Ceresa, B. P., and Pessin, J. E. (1998). Insulin regulation of the Ras activation/inactivation cycle. Mol. Cell. Biochem. 182, 23–29.
Chua, H. L., Bhat-Nakshatri, P., Clare, S. E., Morimiya, A., Badve, S., and Nakshatri, H. (2007). NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene 26, 711–724.
Colhoun, H. M. (2009). Use of insulin glargine and cancer incidence in Scotland: a study from the Scottish Diabetes Research Network Epidemiology Group. Diabetologia 52, 1755–1765.
Corpet, D. E., Jacquinet, C., Peiffer, G., and Tache, S. (1997). Insulin injections promote the growth of aberrant crypt foci in the colon of rats. Nutr. Cancer 27, 316–320.
Coughlin, S. S., Calle, E. E., Teras, L. R., Petrelli, J., and Thun, M. J. (2004). Diabetes mellitus as a predictor of cancer mortality in a large cohort of US adults. Am. J. Epidemiol. 159, 1160–1167.
Craparo, A., Freund, R., and Gustafson, T. A. (1997). 14-3-3 (Epsilon) interacts with the insulin-like growth factor I receptor and insulin receptor substrate I in a phosphoserine-dependent manner. J. Biol. Chem. 272, 11663–11669.
Currie, C. J., Poole, C. D., and Gale, E. A. (2009). The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 52, 1766–1777.
Cusi, K., Maezono, K., Osman, A., Pendergrass, M., Patti, M. E., Pratipanawatr, T., Defronzo, R. A., Kahn, C. R., and Mandarino, L. J. (2000). Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J. Clin. Invest. 105, 311–320.
Dalle, S., Ricketts, W., Imamura, T., Vollenweider, P., and Olefsky, J. M. (2001). Insulin and insulin-like growth factor I receptors utilize different G protein signaling components. J. Biol. Chem. 276, 15688–15695.
de Bono, J. S., Attard, G., Adjei, A., Pollak, M. N., Fong, P. C., Haluska, P., Roberts, L., Melvin, C., Repollet, M., Chianese, D., Connely, M., Terstappen, L. W., and Gualberto, A. (2007). Potential applications for circulating tumor cells expressing the insulin-like growth factor-I receptor. Clin. Cancer Res. 13, 3611–3616.
Denley, A., Bonython, E. R., Booker, G. W., Cosgrove, L. J., Forbes, B. E., Ward, C. W., and Wallace, J. C. (2004). Structural determinants for high-affinity binding of insulin-like growth factor II to insulin receptor (IR)-A, the exon 11 minus isoform of the IR. Mol. Endocrinol. 18, 2502–2512.
Denley, A., Carroll, J. M., Brierley, G. V., Cosgrove, L., Wallace, J., Forbes, B., and Roberts, C. T. Jr. (2007). Differential activation of insulin receptor substrates 1 and 2 by insulin-like growth factor-activated insulin receptors. Mol. Cell. Biol. 27, 3569–3577.
Dinchuk, J. E., Cao, C., Huang, F., Reeves, K. A., Wang, J., Myers, F., Cantor, G. H., Zhou, X., Attar, R. M., Gottardis, M., and Carboni, J. M. (2010). Insulin receptor (IR) pathway hyperactivity in IGF-IR null cells and suppression of downstream growth signaling using the dual IGF-IR/IR inhibitor, BMS-754807. Endocrinology 151, 4123–4132.
Dong, J., Demarest, S. J., Sereno, A., Tamraz, S., Langley, E., Doern, A., Snipas, T., Perron, K., Joseph, I., Glaser, S. M., Ho, S. N., Reff, M. E., and Hariharan, K. (2010). Combination of two insulin-like growth factor-I receptor inhibitory antibodies targeting distinct epitopes leads to an enhanced antitumor response. Mol. Cancer Ther. 9, 2593–2604.
Drakas, R., Tu, X., and Baserga, R. (2004). Control of cell size through phosphorylation of upstream binding factor 1 by nuclear phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. U.S.A. 101, 9272–9276.
Drejer, K. (1992). The bioactivity of insulin analogues from in vitro receptor binding to in vivo glucose uptake. Diabetes Metab. Rev. 8, 259–285.
Duggan, C., Irwin, M. L., Xiao, L., Henderson, K. D., Smith, A. W., Baumgartner, R. N., Baumgartner, K. B., Bernstein, L., Ballard-Barbash, R., and Mctiernan, A. (2011). Associations of insulin resistance and adiponectin with mortality in women with breast cancer. J. Clin. Oncol. 29, 32–39.
Dunn, S. E., Hardman, R. A., Kari, F. W., and Barrett, J. C. (1997). Insulin-like growth factor 1 (IGF-1) alters drug sensitivity of HBL100 human breast cancer cells by inhibition of apoptosis induced by diverse anticancer drugs. Cancer Res. 57, 2687–2693.
Dupont, J., Fernandez, A. M., Glackin, C. A., Helman, L., and Leroith, D. (2001). Insulin-like growth factor 1 (IGF-1)-induced twist expression is involved in the anti-apoptotic effects of the IGF-1 receptor. J. Biol. Chem. 276, 26699–26707.
Ebina, Y., Edery, M., Ellis, L., Standring, D., Beaudoin, J., Roth, R. A., and Rutter, W. J. (1985). Expression of a functional human insulin receptor from a cloned cDNA in Chinese hamster ovary cells. Proc. Natl. Acad. Sci. U.S.A. 82, 8014–8018.
Economou, M. A., Andersson, S., Vasilcanu, D., All-Ericsson, C., Menu, E., Girnita, A., Girnita, L., Axelson, M., Seregard, S., and Larsson, O. (2008). Oral picropodophyllin (PPP) is well tolerated in vivo and inhibits IGF-1R expression and growth of uveal melanoma. Invest. Ophthalmol. Vis. Sci. 49, 2337–2342.
Evans, T., Lindsay, C. R., Chan, E., Tait, B., Michael, S. A., Day, S., Stephens, A. W., Franke, A., Poondru, S., and Puzanov, I. (2010). Phase I dose-escalation study of continuous oral dosing of OSI-906, a dual tyrosine kinase inhibitor of insulin-like growth factor-1 receptor (IGF-1R) and insulin receptor (IR), in patients with advanced solid tumors. J. Clin. Oncol. 28, abstr. :2531.
Fair, A. M., Dai, Q., Shu, X. O., Matthews, C. E., Yu, H., Jin, F., Gao, Y. T., and Zheng, W. (2007). Energy balance, insulin resistance biomarkers, and breast cancer risk. Cancer Detect. Prev. 31, 214–219.
Faria, T. N., Blakesley, V. A., Kato, H., Stannard, B., Leroith, D., and Roberts, C. T. Jr. (1994). Role of the carboxyl-terminal domains of the insulin and insulin-like growth factor I receptors in receptor function. J. Biol. Chem. 269, 13922–13928.
Feng, Y., Zhu, Z., Xiao, X., Choudhry, V., Barrett, J. C., and Dimitrov, D. S. (2006). Novel human monoclonal antibodies to insulin-like growth factor (IGF)-II that potently inhibit the IGF receptor type I signal transduction function. Mol. Cancer Ther. 5, 114–120.
Ferrara, A., Lewis, J. D., Quesenberry, C. P. Jr., Peng, T., Strom, B. L., Van Den Eeden, S. K., Ehrlich, S. F., and Habel, L. A. (2011). Cohort study of pioglitazone and cancer incidence in patients with diabetes. Diabetes Care 34, 923–929.
Fierz, Y., Novosyadlyy, R., Vijayakumar, A., Yakar, S., and Leroith, D. (2010). Insulin-sensitizing therapy attenuates type 2 diabetes-mediated mammary tumor progression. Diabetes 59, 686–693.
Foti, D., Iuliano, R., Chiefari, E., and Brunetti, A. (2003). A nucleoprotein complex containing Sp1, C/EBP beta, and HMGI-Y controls human insulin receptor gene transcription. Mol. Cell. Biol. 23, 2720–2732.
Frasca, F., Pandini, G., Malaguarnera, R., Mandarino, A., Messina, R. L., Sciacca, L., Belfiore, A., and Vigneri, R. (2007). Role of c-Abl in directing metabolic versus mitogenic effects in insulin receptor signaling. J. Biol. Chem. 282, 26077–26088.
Frasca, F., Pandini, G., Scalia, P., Sciacca, L., Mineo, R., Costantino, A., Goldfine, I. D., Belfiore, A., and Vigneri, R. (1999). Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 19, 3278–3288.
Furlanetto, R. W., Dey, B. R., Lopaczynski, W., and Nissley, S. P. (1997). 14-3-3 Proteins interact with the insulin-like growth factor receptor but not the insulin receptor. Biochem. J. 327(Pt 3), 765–771.
Gao, J., Chesebrough, J. W., Cartlidge, S. A., Ricketts, S. A., Incognito, L., Veldman-Jones, M., Blakey, D. C., Tabrizi, M., Jallal, B., Trail, P. A., Coats, S., Bosslet, K., and Chang, Y. S. (2011). Dual IGF-I/II-neutralizing antibody MEDI-573 potently inhibits IGF signaling and tumor growth. Cancer Res. 71, 1029–1040.
Garofalo, C., Manara, M. C., Nicoletti, G., Marino, M. T., Lollini, P. L., Astolfi, A., Pandini, G., Lopez-Guerrero, J. A., Schaefer, K. L., Belfiore, A., Picci, P., and Scotlandi, K. (2011). Efficacy of and resistance to anti-IGF-1R therapies in Ewing’s sarcoma is dependent on insulin receptor signaling. Oncogene 30, 2730–2740.
Gedrich, R., Poondru, S., Stephens, A., Logan, T., Hart, K., Buck, E., Carden, C., Lindsay, C., Kaye, S., Kim, E., Evans, J., Puzanov, I., Epstein, D., and Miglarese, M. (2009). Pharmacodynamic biomarkers for OSI-906, an insulin-like growth factor-1 receptor (IGF-1R) tyrosine kinase inhibitor, in cancer patients with advanced solid tumors. Mol. Cancer Ther. 8, abstr. :A46.
Giorgino, F., Belfiore, A., Milazzo, G., Costantino, A., Maddux, B., Whittaker, J., Goldfine, I. D., and Vigneri, R. (1991). Overexpression of insulin receptors in fibroblast and ovary cells induces a ligand-mediated transformed phenotype. Mol. Endocrinol. 5, 452–459.
Girnita, A., Girnita, L., Del Prete, F., Bartolazzi, A., Larsson, O., and Axelson, M. (2004). Cyclolignans as inhibitors of the insulin-like growth factor-1 receptor and malignant cell growth. Cancer Res. 64, 236–242.
Goalstone, M., Carel, K., Leitner, J. W., and Draznin, B. (1997). Insulin stimulates the phosphorylation and activity of farnesyltransferase via the Ras-mitogen-activated protein kinase pathway. Endocrinology 138, 5119–5124.
Gonda, T. A., Tu, S., and Wang, T. C. (2009). Chronic inflammation, the tumor microenvironment and carcinogenesis. Cell Cycle 8, 2005–2013.
Gooch, J. L., Van Den Berg, C. L., and Yee, D. (1999). Insulin-like growth factor (IGF)-I rescues breast cancer cells from chemotherapy-induced cell death – proliferative and anti-apoptotic effects. Breast Cancer Res. Treat. 56, 1–10.
Gualberto, A., and Pollak, M. (2009). Clinical development of inhibitors of the insulin-like growth factor receptor in oncology. Curr. Drug Targets 10, 923–936.
Guix, M., Faber, A. C., Wang, S. E., Olivares, M. G., Song, Y., Qu, S., Rinehart, C., Seidel, B., Yee, D., Arteaga, C. L., and Engelman, J. A. (2008). Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J. Clin. Invest. 118, 2609–2619.
Hawley, S. A., Boudeau, J., Reid, J. L., Mustard, K. J., Udd, L., Makela, T. P., Alessi, D. R., and Hardie, D. G. (2003). Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2, 28.
He, W., Rose, D. W., Olefsky, J. M., and Gustafson, T. A. (1998). Grb10 interacts differentially with the insulin receptor, insulin-like growth factor I receptor, and epidermal growth factor receptor via the Grb10 Src homology 2 (SH2) domain and a second novel domain located between the pleckstrin homology and SH2 domains. J. Biol. Chem. 273, 6860–6867.
Heaney, A. P., Fernando, M., Yong, W. H., and Melmed, S. (2002). Functional PPAR-gamma receptor is a novel therapeutic target for ACTH-secreting pituitary adenomas. Nat. Med. 8, 1281–1287.
Hemkens, L. G., Grouven, U., Bender, R., Gunster, C., Gutschmidt, S., Selke, G. W., and Sawicki, P. T. (2009). Risk of malignancies in patients with diabetes treated with human insulin or insulin analogues: a cohort study. Diabetologia 52, 1732–1744.
Herranz, D., Munoz-Martin, M., Canamero, M., Mulero, F., Martinez-Pastor, B., Fernandez-Capetillo, O., and Serrano, M. (2010). Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat. Commun. 1, 3.
Hers, I., Vincent, E. E., and Tavare, J. M. (2011). Akt signalling in health and disease. Cell. Signal. 23, 1515–1527.
Heskamp, S., Van Laarhoven, H. W., Molkenboer-Kuenen, J. D., Franssen, G. M., Versleijen-Jonkers, Y. M., Oyen, W. J., Van Der Graaf, W. T., and Boerman, O. C. (2011). ImmunoSPECT and immunoPET of IGF-1R expression with the radiolabeled antibody R1507 in a triple-negative breast cancer model. J. Nucl. Med. 51, 1565–1572.
Heuson, J. C., and Legros, N. (1972). Influence of insulin deprivation on growth of the 7,12-dimethylbenz(a)anthracene-induced mammary carcinoma in rats subjected to alloxan diabetes and food restriction. Cancer Res. 32, 226–232.
Hofmann, F., and Garcia-Echeverria, C. (2005). Blocking the insulin-like growth factor-I receptor as a strategy for targeting cancer. Drug Discov. Today 10, 1041–1047.
Hosono, K., Endo, H., Takahashi, H., Sugiyama, M., Uchiyama, T., Suzuki, K., Nozaki, Y., Yoneda, K., Fujita, K., Yoneda, M., Inamori, M., Tomatsu, A., Chihara, T., Shimpo, K., Nakagama, H., and Nakajima, A. (2010). Metformin suppresses azoxymethane-induced colorectal aberrant crypt foci by activating AMP-activated protein kinase. Mol. Carcinog. 49, 662–671.
Imamura, T., Vollenweider, P., Egawa, K., Clodi, M., Ishibashi, K., Nakashima, N., Ugi, S., Adams, J. W., Brown, J. H., and Olefsky, J. M. (1999). G alpha-q/11 protein plays a key role in insulin-induced glucose transport in 3T3-L1 adipocytes. Mol. Cell. Biol. 19, 6765–6774.
Inoue, K., Kawahito, Y., Tsubouchi, Y., Kohno, M., Yoshimura, R., Yoshikawa, T., and Sano, H. (2001). Expression of peroxisome proliferator-activated receptor gamma in renal cell carcinoma and growth inhibition by its agonists. Biochem. Biophys. Res. Commun. 287, 727–732.
Irwin, M. L., Duggan, C., Wang, C. Y., Smith, A. W., Mctiernan, A., Baumgartner, R. N., Baumgartner, K. B., Bernstein, L., and Ballard-Barbash, R. (2011). Fasting C-peptide levels and death resulting from all causes and breast cancer: the health, eating, activity, and lifestyle study. J. Clin. Oncol. 29, 47–53.
Jensen, M., Hansen, B., De Meyts, P., Schaffer, L., and Urso, B. (2007). Activation of the insulin receptor by insulin and a synthetic peptide leads to divergent metabolic and mitogenic signaling and responses. J. Biol. Chem. 282, 35179–35186.
Jiang, Z. Y., Lin, Y. W., Clemont, A., Feener, E. P., Hein, K. D., Igarashi, M., Yamauchi, T., White, M. F., and King, G. L. (1999). Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J. Clin. Invest. 104, 447–457.
Jiracek, J., Zakova, L., Antolikova, E., Watson, C. J., Turkenburg, J. P., Dodson, G. G., and Brzozowski, A. M. (2010). Implications for the active form of human insulin based on the structural convergence of highly active hormone analogues. Proc. Natl. Acad. Sci. U.S.A. 107, 1966–1970.
Jiralerspong, S., Palla, S. L., Giordano, S. H., Meric-Bernstam, F., Liedtke, C., Barnett, C. M., Hsu, L., Hung, M. C., Hortobagyi, G. N., and Gonzalez-Angulo, A. M. (2009). Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J. Clin. Oncol. 27, 3297–3302.
Johnson, J. A., and Bowker, S. L. (2010). Intensive glycaemic control and cancer risk in type 2 diabetes: a meta-analysis of major trials. Diabetologia 54, 25–31.
Johnson, J. A., Majumdar, S. R., Simpson, S. H., and Toth, E. L. (2002). Decreased mortality associated with the use of metformin compared with sulfonylurea monotherapy in type 2 diabetes. Diabetes Care 25, 2244–2248.
Jonasson, J. M., Ljung, R., Talback, M., Haglund, B., Gudbjornsdottir, S., and Steineck, G. (2009). Insulin glargine use and short-term incidence of malignancies-a population-based follow-up study in Sweden. Diabetologia 52, 1745–1754.
Kaaks, R., and Lukanova, A. (2002). Effects of weight control and physical activity in cancer prevention: role of endogenous hormone metabolism. Ann. N. Y. Acad. Sci. 963, 268–281.
Kawa, S., Nikaido, T., Unno, H., Usuda, N., Nakayama, K., and Kiyosawa, K. (2002). Growth inhibition and differentiation of pancreatic cancer cell lines by PPAR gamma ligand troglitazone. Pancreas 24, 1–7.
Kiely, P. A., Leahy, M., O’gorman, D., and O’connor, R. (2005). RACK1-mediated integration of adhesion and insulin-like growth factor I (IGF-I) signaling and cell migration are defective in cells expressing an IGF-I receptor mutated at tyrosines 1250 and 1251. J. Biol. Chem. 280, 7624–7633.
Kim, H. J., Hwang, J. Y., Choi, W. S., Lee, J. H., Chang, K. C., Nishinaka, T., Yabe-Nishimura, C., and Seo, H. G. (2007a). Expression of a peroxisome proliferator-activated receptor gamma 1 splice variant that was identified in human lung cancers suppresses cell death induced by cisplatin and oxidative stress. Clin. Cancer Res. 13, 2577–2583.
Kim, H. J., Litzenburger, B. C., Cui, X., Delgado, D. A., Grabiner, B. C., Lin, X., Lewis, M. T., Gottardis, M. M., Wong, T. W., Attar, R. M., Carboni, J. M., and Lee, A. V. (2007b). Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-kappaB and snail. Mol. Cell. Biol. 27, 3165–3175.
Kim, J. J., and Accili, D. (2002). Signalling through IGF-I and insulin receptors: where is the specificity? Growth Horm. IGF Res. 12, 84–90.
Kurtzhals, P., Schaffer, L., Sorensen, A., Kristensen, C., Jonassen, I., Schmid, C., and Trub, T. (2000). Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes 49, 999–1005.
Laviola, L., Giorgino, F., Chow, J. C., Baquero, J. A., Hansen, H., Ooi, J., Zhu, J., Riedel, H., and Smith, R. J. (1997). The adapter protein Grb10 associates preferentially with the insulin receptor as compared with the IGF-I receptor in mouse fibroblasts. J. Clin. Invest. 99, 830–837.
Law, J. H., Habibi, G., Hu, K., Masoudi, H., Wang, M. Y., Stratford, A. L., Park, E., Gee, J. M., Finlay, P., Jones, H. E., Nicholson, R. I., Carboni, J., Gottardis, M., Pollak, M., and Dunn, S. E. (2008). Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res. 68, 10238–10246.
Lee, M. S., Kim, D., Jo, K., and Hwang, J. K. (2011). Nordihydroguaiaretic acid protects against high-fat diet-induced fatty liver by activating AMP-activated protein kinase in obese mice. Biochem. Biophys. Res. Commun. 401, 92–97.
Lewis, J. D., Ferrara, A., Peng, T., Hedderson, M., Bilker, W. B., Quesenberry, C. P. Jr., Vaughn, D. J., Nessel, L., Selby, J., and Strom, B. L. (2011). Risk of bladder cancer among diabetic patients treated with pioglitazone: interim report of a longitudinal cohort study. Diabetes Care 34, 916–922.
Li, F., Pham, J. D., Anderson, M. O., and Youngren, J. F. (2009). Nordihydroguaiaretic acid inhibits transforming growth factor beta type 1 receptor activity and downstream signaling. Eur. J. Pharmacol. 616, 31–37.
Li, M. Y., Deng, H., Zhao, J. M., Dai, D., and Tan, X. Y. (2003). Peroxisome proliferator-activated receptor gamma ligands inhibit cell growth and induce apoptosis in human liver cancer BEL-7402 cells. World J. Gastroenterol. 9, 1683–1688.
Ligensa, T., Krauss, S., Demuth, D., Schumacher, R., Camonis, J., Jaques, G., and Weidner, K. M. (2001). A PDZ domain protein interacts with the C-terminal tail of the insulin-like growth factor-1 receptor but not with the insulin receptor. J. Biol. Chem. 276, 33419–33427.
Litzenburger, B. C., Creighton, C. J., Tsimelzon, A., Chan, B. T., Hilsenbeck, S. G., Wang, T., Carboni, J. M., Gottardis, M. M., Huang, F., Chang, J. C., Lewis, M. T., Rimawi, M. F., and Lee, A. V. (2011). High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy. Clin. Cancer Res. 17, 2314–2327.
Loke, Y. K., Kwok, C. S., and Singh, S. (2011). Comparative cardiovascular effects of thiazolidinediones: systematic review and meta-analysis of observational studies. BMJ 342, d1309.
Lu, Y., Zi, X., Zhao, Y., Mascarenhas, D., and Pollak, M. (2001). Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J. Natl. Cancer Inst. 93, 1852–1857.
Macaulay, V. M., Middleton, M. R., Eckhardt, S. G., Juergens, R. A., Stephens, A. W., Poondru, S., Mccarthy, S. P., and Gadgeel, S. M. (2010). Phase I study of OSI-906, dual tyrosine kinase inhibitor of insulin-like growth factor-1 receptor (IGF-1R) and insulin receptor (IR) in combination with erlotinib (E) in patients with advanced solid tumors. J. Clin. Oncol. 28, abstr. :3016.
Malaguarnera, R., Frasca, F., Garozzo, A., Giani, F., Pandini, G., Vella, V., Vigneri, R., and Belfiore, A. (2011). Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J. Clin. Endocrinol. Metab. 96, 766–774.
Mayer, D., Shukla, A., and Enzmann, H. (2008). Proliferative effects of insulin analogues on mammary epithelial cells. Arch. Physiol. Biochem. 114, 38–44.
Miura, M., Surmacz, E., Burgaud, J. L., and Baserga, R. (1995). Different effects on mitogenesis and transformation of a mutation at tyrosine 1251 of the insulin-like growth factor I receptor. J. Biol. Chem. 270, 22639–22644.
Miyamoto, S., Nakamura, M., Shitara, K., Nakamura, K., Ohki, Y., Ishii, G., Goya, M., Kodama, K., Sangai, T., Maeda, H., Shi-Chuang, Z., Chiba, T., and Ochiai, A. (2005). Blockade of paracrine supply of insulin-like growth factors using neutralizing antibodies suppresses the liver metastasis of human colorectal cancers. Clin. Cancer Res. 11, 3494–3502.
Moller, D. E., Yokota, A., Caro, J. F., and Flier, J. S. (1989). Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol. Endocrinol. 3, 1263–1269.
Monami, M., Lamanna, C., Balzi, D., Marchionni, N., and Mannucci, E. (2009). Sulphonylureas and cancer: a case-control study. Acta Diabetol. 46, 279–284.
Monami, M., Lamanna, C., Marchionni, N., and Mannucci, E. (2008). Rosiglitazone and risk of cancer: a meta-analysis of randomized clinical trials. Diabetes Care 31, 1455–1460.
Morcavallo, A., Gaspari, M., Pandini, G., Palummo, A., Cuda, G., Larsen, M. R., Vigneri, R., and Belfiore, A. (2011). Research resource: new and diverse substrates for the insulin receptor isoform a revealed by quantitative proteomics after stimulation with igf-ii or insulin. Mol. Endocrinol. 25, 1456–1468.
Morden, N. E., Liu, S. K., Smith, J., Mackenzie, T. A., Skinner, J., and Korc, M. (2011). Further Exploration of the relationship between insulin glargine and incident cancer: a retrospective cohort study of older Medicare patients. Diabetes Care 34, 1965–1971.
Mosthaf, L., Grako, K., Dull, T. J., Coussens, L., Ullrich, A., and Mcclain, D. A. (1990). Functionally distinct insulin receptors generated by tissue-specific alternative splicing. EMBO J. 9, 2409–2413.
Mueller, E., Smith, M., Sarraf, P., Kroll, T., Aiyer, A., Kaufman, D. S., Oh, W., Demetri, G., Figg, W. D., Zhou, X. P., Eng, C., Spiegelman, B. M., and Kantoff, P. W. (2000). Effects of ligand activation of peroxisome proliferator-activated receptor gamma in human prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 97, 10990–10995.
Mulligan, C., Rochford, J., Denyer, G., Stephens, R., Yeo, G., Freeman, T., Siddle, K., and O’rahilly, S. (2002). Microarray analysis of insulin and insulin-like growth factor-1 (IGF-1) receptor signaling reveals the selective up-regulation of the mitogen heparin-binding EGF-like growth factor by IGF-1. J. Biol. Chem. 277, 42480–42487.
Mulvihill, M. J., Cooke, A., Rosenfeld-Franklin, M., Buck, E., Foreman, K., Landfair, D., O’connor, M., Pirritt, C., Sun, Y., Yao, Y., Arnold, L. D., Gibson, N. W., and Ji, Q. S. (2009). Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med. Chem. 1, 1153–1171.
Najjar, S. M., Blakesley, V. A., Li Calzi, S., Kato, H., Leroith, D., and Choice, C. V. (1997). Differential phosphorylation of pp120 by insulin and insulin-like growth factor-1 receptors: role for the C-terminal domain of the beta-subunit. Biochemistry 36, 6827–6834.
Nakae, J., Kido, Y., and Accili, D. (2001). Distinct and overlapping functions of insulin and IGF-I receptors. Endocr. Rev. 22, 818–835.
Nicholson, R. I., Staka, C., Boyns, F., Hutcheson, I. R., and Gee, J. M. (2004). Growth factor-driven mechanisms associated with resistance to estrogen deprivation in breast cancer: new opportunities for therapy. Endocr. Relat. Cancer 11, 623–641.
Novosyadlyy, R., Lann, D. E., Vijayakumar, A., Rowzee, A., Lazzarino, D. A., Fierz, Y., Carboni, J. M., Gottardis, M. M., Pennisi, P. A., Molinolo, A. A., Kurshan, N., Mejia, W., Santopietro, S., Yakar, S., Wood, T. L., and Leroith, D. (2010). Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Res. 70, 741–751.
Novosyadlyy, R., Vijayakumar, A., Lann, D., Fierz, Y., Kurshan, N., and Leroith, D. (2009). Physical and functional interaction between polyoma virus middle T antigen and insulin and IGF-I receptors is required for oncogene activation and tumour initiation. Oncogene 28, 3477–3486.
O’Connor, R., Kauffmann-Zeh, A., Liu, Y., Lehar, S., Evan, G. I., Baserga, R., and Blattler, W. A. (1997). Identification of domains of the insulin-like growth factor I receptor that are required for protection from apoptosis. Mol. Cell. Biol. 17, 427–435.
O’Neill, T. J., Zhu, Y., and Gustafson, T. A. (1997). Interaction of MAD2 with the carboxyl terminus of the insulin receptor but not with the IGFIR. Evidence for release from the insulin receptor after activation. J. Biol. Chem. 272, 10035–10040.
Palmade, F., Sechoy-Chambon, O., Coquelet, C., and Bonne, C. (1994). Insulin-like growth factor-1 (IGF-1) specifically binds to bovine lens epithelial cells and increases the number of fibronectin receptor sites. Curr. Eye Res. 13, 531–537.
Pandini, G., Frasca, F., Mineo, R., Sciacca, L., Vigneri, R., and Belfiore, A. (2002). Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem. 277, 39684–39695.
Pandini, G., Medico, E., Conte, E., Sciacca, L., Vigneri, R., and Belfiore, A. (2003). Differential gene expression induced by insulin and insulin-like growth factor-II through the insulin receptor isoform A. J. Biol. Chem. 278, 42178–42189.
Pandini, G., Vigneri, R., Costantino, A., Frasca, F., Ippolito, A., Fujita-Yamaguchi, Y., Siddle, K., Goldfine, I. D., and Belfiore, A. (1999). Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: evidence for a second mechanism of IGF-I signaling. Clin. Cancer Res. 5, 1935–1944.
Pandini, G., Wurch, T., Akla, B., Corvaia, N., Belfiore, A., and Goetsch, L. (2007). Functional responses and in vivo anti-tumour activity of h7C10: a humanised monoclonal antibody with neutralising activity against the insulin-like growth factor-1 (IGF-1) receptor and insulin/IGF-1 hybrid receptors. Eur. J. Cancer 43, 1318–1327.
Papa, V., Pezzino, V., Costantino, A., Belfiore, A., Giuffrida, D., Frittitta, L., Vannelli, G. B., Brand, R., Goldfine, I. D., and Vigneri, R. (1990). Elevated insulin receptor content in human breast cancer. J. Clin. Invest. 86, 1503–1510.
Patel, K. M., Wright, K. L., Whittaker, P., Chakravarty, P., Watson, M. L., and Ward, S. G. (2005). Differential modulation of COX-2 expression in A549 airway epithelial cells by structurally distinct PPAR(gamma) agonists: evidence for disparate functional effects which are independent of NF-(kappa)B and PPAR(gamma). Cell. Signal. 17, 1098–1110.
Paul, S., Dansithong, W., Kim, D., Rossi, J., Webster, N. J., Comai, L., and Reddy, S. (2006). Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J. 25, 4271–4283.
Perks, C. M., Vernon, E. G., Rosendahl, A. H., Tonge, D., and Holly, J. M. (2007). IGF-II and IGFBP-2 differentially regulate PTEN in human breast cancer cells. Oncogene 26, 5966–5972.
Pillay, T. S., Sasaoka, T., and Olefsky, J. M. (1995). Insulin stimulates the tyrosine dephosphorylation of pp125 focal adhesion kinase. J. Biol. Chem. 270, 991–994.
Pisani, P. (2008). Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch. Physiol. Biochem. 114, 63–70.
Pocock, S. J., and Smeeth, L. (2009). Insulin glargine and malignancy: an unwarranted alarm. Lancet 374, 511–513.
Pollak, M. (2008a). Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 8, 915–928.
Pollak, M. (2008b). Targeting insulin and insulin-like growth factor signalling in oncology. Curr. Opin. Pharmacol. 8, 384–392.
Rakatzi, I., Stosik, M., Gromke, T., Siddle, K., and Eckel, J. (2006). Differential phosphorylation of IRS-1 and IRS-2 by insulin and IGF-I receptors. Arch. Physiol. Biochem. 112, 37–47.
Reeves, R., Edberg, D. D., and Li, Y. (2001). Architectural transcription factor HMGI(Y) promotes tumor progression and mesenchymal transition of human epithelial cells. Mol. Cell. Biol. 21, 575–594.
Roux, P. P., and Blenis, J. (2004). ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 68, 320–344.
Rowe, D. L., Ozbay, T., Bender, L. M., and Nahta, R. (2008). Nordihydroguaiaretic acid, a cytotoxic insulin-like growth factor-I receptor/HER2 inhibitor in trastuzumab-resistant breast cancer. Mol. Cancer Ther. 7, 1900–1908.
Ruiter, R., Visser, L. E., Van Herk-Sukel, M. P., Coebergh, J. W., Haak, H. R., Geelhoed-Duijvestijn, P. H., Straus, S. M., Herings, R. M., and Stricker, B. H. (2011). Risk of cancer in patients on insulin glargine and other insulin analogues in comparison with those on human insulin: results from a large population-based follow-up study. Diabetologia.
Sacco, A., Morcavallo, A., Pandini, G., Vigneri, R., and Belfiore, A. (2009). Differential signaling activation by insulin and insulin-like growth factors I and II upon binding to insulin receptor isoform A. Endocrinology 150, 3594–3602.
Sachdev, D., Singh, R., Fujita-Yamaguchi, Y., and Yee, D. (2006). Down-regulation of insulin receptor by antibodies against the type I insulin-like growth factor receptor: implications for anti-insulin-like growth factor therapy in breast cancer. Cancer Res. 66, 2391–2402.
Samani, A. A., Yakar, S., Leroith, D., and Brodt, P. (2007). The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr. Rev. 28, 20–47.
Sarbassov, D. D., Ali, S. M., and Sabatini, D. M. (2005). Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 17, 596–603.
Schneider, M. B., Matsuzaki, H., Haorah, J., Ulrich, A., Standop, J., Ding, X. Z., Adrian, T. E., and Pour, P. M. (2001). Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology 120, 1263–1270.
Sciacca, L., Costantino, A., Pandini, G., Mineo, R., Frasca, F., Scalia, P., Sbraccia, P., Goldfine, I. D., Vigneri, R., and Belfiore, A. (1999). Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene 18, 2471–2479.
Sciacca, L., Mineo, R., Pandini, G., Murabito, A., Vigneri, R., and Belfiore, A. (2002). In IGF-I receptor-deficient leiomyosarcoma cells autocrine IGF-II induces cell invasion and protection from apoptosis via the insulin receptor isoform A. Oncogene 21, 8240–8250.
Sell, C., Dumenil, G., Deveaud, C., Miura, M., Coppola, D., Deangelis, T., Rubin, R., Efstratiadis, A., and Baserga, R. (1994). Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol. Cell. Biol. 14, 3604–3612.
Sertznig, P., Seifert, M., Tilgen, W., and Reichrath, J. (2007). Present concepts and future outlook: function of peroxisome proliferator-activated receptors (PPARs) for pathogenesis, progression, and therapy of cancer. J. Cell. Physiol. 212, 1–12.
Shaw, R. J., Bardeesy, N., Manning, B. D., Lopez, L., Kosmatka, M., Depinho, R. A., and Cantley, L. C. (2004). The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 6, 91–99.
Shi, J., Wang, D. M., Wang, C. M., Hu, Y., Liu, A. H., Zhang, Y. L., Sun, B., and Song, J. G. (2009). Insulin receptor substrate-1 suppresses transforming growth factor-beta1-mediated epithelial-mesenchymal transition. Cancer Res. 69, 7180–7187.
Shimada, T., Kojima, K., Yoshiura, K., Hiraishi, H., and Terano, A. (2002). Characteristics of the peroxisome proliferator activated receptor gamma (PPARgamma) ligand induced apoptosis in colon cancer cells. Gut 50, 658–664.
Sivakumar, R., Koga, H., Selvendiran, K., Maeyama, M., Ueno, T., and Sata, M. (2009). Autocrine loop for IGF-I receptor signaling in SLUG-mediated epithelial-mesenchymal transition. Int. J. Oncol. 34, 329–338.
Slaaby, R., Schaffer, L., Lautrup-Larsen, I., Andersen, A. S., Shaw, A. C., Mathiasen, I. S., and Brandt, J. (2006). Hybrid receptors formed by insulin receptor (IR) and insulin-like growth factor I receptor (IGF-IR) have low insulin and high IGF-1 affinity irrespective of the IR splice variant. J. Biol. Chem. 281, 25869–25874.
Solomon, C. S., and Goalstone, M. L. (2001). Dominant negative farnesyltransferase alpha-subunit inhibits insulin mitogenic effects. Biochem. Biophys. Res. Commun. 285, 161–166.
Sommerfeld, M. R., Muller, G., Tschank, G., Seipke, G., Habermann, P., Kurrle, R., and Tennagels, N. (2010). In vitro metabolic and mitogenic signaling of insulin glargine and its metabolites. PLoS ONE 5, e9540.
Soni, P., Lakkis, M., Poy, M. N., Fernstrom, M. A., and Najjar, S. M. (2000). The differential effects of pp120 (Ceacam 1) on the mitogenic action of insulin and insulin-like growth factor 1 are regulated by the nonconserved tyrosine 1316 in the insulin receptor. Mol. Cell. Biol. 20, 3896–3905.
Soos, M. A., and Siddle, K. (1989). Immunological relationships between receptors for insulin and insulin-like growth factor I. Evidence for structural heterogeneity of insulin-like growth factor I receptors involving hybrids with insulin receptors. Biochem. J. 263, 553–563.
Stambolic, V., Suzuki, A., De La Pompa, J. L., Brothers, G. M., Mirtsos, C., Sasaki, T., Ruland, J., Penninger, J. M., Siderovski, D. P., and Mak, T. W. (1998). Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95, 29–39.
Steinbach, G., Kumar, S. P., Reddy, B. S., Lipkin, M., and Holt, P. R. (1993). Effects of caloric restriction and dietary fat on epithelial cell proliferation in rat colon. Cancer Res. 53, 2745–2749.
Strickler, H. D., Wylie-Rosett, J., Rohan, T., Hoover, D. R., Smoller, S., Burk, R. D., and Yu, H. (2001). The relation of type 2 diabetes and cancer. Diabetes Technol. Ther. 3, 263–274.
Tamburini, J., Chapuis, N., Bardet, V., Park, S., Sujobert, P., Willems, L., Ifrah, N., Dreyfus, F., Mayeux, P., Lacombe, C., and Bouscary, D. (2008). Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: rationale for therapeutic inhibition of both pathways. Blood 111, 379–382.
Tan, B. X., Yao, W. X., Ge, J., Peng, X. C., Du, X. B., Zhang, R., Yao, B., Xie, K., Li, L. H., Dong, H., Gao, F., Zhao, F., Hou, J. M., Su, J. M., and Liu, J. Y. (2011). Prognostic influence of metformin as first-line chemotherapy for advanced nonsmall cell lung cancer in patients with type 2 diabetes. Cancer 117, 5103–5111.
Tartare-Deckert, S., Murdaca, J., Sawka-Verhelle, D., Holt, K. H., Pessin, J. E., and Van Obberghen, E. (1996). Interaction of the molecular weight 85K regulatory subunit of the phosphatidylinositol 3-kinase with the insulin receptor and the insulin-like growth factor-1 (IGF-I) receptor: comparative study using the yeast two-hybrid system. Endocrinology 137, 1019–1024.
Tolcher, A. W., Sarantopoulos, J., Patnaik, A., Papadopoulos, K., Lin, C. C., Rodon, J., Murphy, B., Roth, B., Mccaffery, I., Gorski, K. S., Kaiser, B., Zhu, M., Deng, H., Friberg, G., and Puzanov, I. (2009). Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1. J. Clin. Oncol. 27, 5800–5807.
Tomimoto, A., Endo, H., Sugiyama, M., Fujisawa, T., Hosono, K., Takahashi, H., Nakajima, N., Nagashima, Y., Wada, K., Nakagama, H., and Nakajima, A. (2008). Metformin suppresses intestinal polyp growth in ApcMin/+ mice. Cancer Sci. 99, 2136–2141.
Tontonoz, P., and Spiegelman, B. M. (2008). Fat and beyond: the diverse biology of PPARgamma. Annu. Rev. Biochem. 77, 289–312.
Tran, T. T., Medline, A., and Bruce, W. R. (1996). Insulin promotion of colon tumors in rats. Cancer Epidemiol. Biomarkers Prev. 5, 1013–1015.
Ullrich, A., Bell, J. R., Chen, E. Y., Herrera, R., Petruzzelli, L. M., Dull, T. J., Gray, A., Coussens, L., Liao, Y. C., Tsubokawa, M., Mason, A., Seeburg, P. H., Grunfeld, C., Rosen, O. M., and Ramachandran, J. (1985). Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 313, 756–761.
Ullrich, A., Gray, A., Tam, A. W., Yang-Feng, T., Tsubokawa, M., Collins, C., Henzel, W., Le Bon, T., Kathuria, S., Chen, E., Jacobs, S., Francke, U., Ramachandran, J., and Fujita-Yamaguchi, Y. (1986). Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 5, 2503–2512.
Vainio, H., Kaaks, R., and Bianchini, F. (2002). Weight control and physical activity in cancer prevention: international evaluation of the evidence. Eur. J. Cancer Prev. 11(Suppl. 2), S94–S100.
Valentinis, B., Morrione, A., Taylor, S. J., and Baserga, R. (1997). Insulin-like growth factor I receptor signaling in transformation by src oncogenes. Mol. Cell. Biol. 17, 3744–3754.
Van Cutsem, E., and Arends, J. (2005). The causes and consequences of cancer-associated malnutrition. Eur. J. Oncol. Nurs. 9(Suppl. 2), S51–S63.
Vella, V., Pandini, G., Sciacca, L., Mineo, R., Vigneri, R., Pezzino, V., and Belfiore, A. (2002). A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J. Clin. Endocrinol. Metab. 87, 245–254.
Vigneri, P., Frasca, F., Sciacca, L., Frittitta, L., and Vigneri, R. (2006). Obesity and cancer. Nutr. Metab. Cardiovasc. Dis. 16, 1–7.
Vigneri, P., Frasca, F., Sciacca, L., Pandini, G., and Vigneri, R. (2009). Diabetes and cancer. Endocr. Relat. Cancer 16, 1103–1123.
Vincent, M. T., Carroll, R. J., Hammer, R. E., Chan, S. J., Guz, Y., Steiner, D. F., and Teitelman, G. (1995). A transgene coding for a human insulin analog has a mitogenic effect on murine embryonic beta cells. Proc. Natl. Acad. Sci. U.S.A. 92, 6239–6243.
Wan, X., Harkavy, B., Shen, N., Grohar, P., and Helman, L. J. (2007). Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 26, 1932–1940.
Webster, N. J., Resnik, J. L., Reichart, D. B., Strauss, B., Haas, M., and Seely, B. L. (1996). Repression of the insulin receptor promoter by the tumor suppressor gene product p53: a possible mechanism for receptor overexpression in breast cancer. Cancer Res. 56, 2781–2788.
Whittaker, J., and Whittaker, L. (2005). Characterization of the functional insulin binding epitopes of the full length insulin receptor. J. Biol. Chem. 280, 20932–20936.
Wicha, M. S., Liu, S., and Dontu, G. (2006). Cancer stem cells: an old idea – a paradigm shift. Cancer Res. 66, 1883–1890; discussion 1895–1886.
Yamaguchi, Y., Flier, J. S., Benecke, H., Ransil, B. J., and Moller, D. E. (1993). Ligand-binding properties of the two isoforms of the human insulin receptor. Endocrinology 132, 1132–1138.
Youngren, J. F., Gable, K., Penaranda, C., Maddux, B. A., Zavodovskaya, M., Lobo, M., Campbell, M., Kerner, J., and Goldfine, I. D. (2005). Nordihydroguaiaretic acid (NDGA) inhibits the IGF-1 and c-erbB2/HER2/neu receptors and suppresses growth in breast cancer cells. Breast Cancer Res. Treat. 94, 37–46.
Zakikhani, M., Dowling, R., Fantus, I. G., Sonenberg, N., and Pollak, M. (2006). Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 66, 10269–10273.
Zeng, L., Sachdev, P., Yan, L., Chan, J. L., Trenkle, T., Mcclelland, M., Welsh, J., and Wang, L. H. (2000). Vav3 mediates receptor protein tyrosine kinase signaling, regulates GTPase activity, modulates cell morphology, and induces cell transformation. Mol. Cell. Biol. 20, 9212–9224.
Zhang, H., Pelzer, A. M., Kiang, D. T., and Yee, D. (2007). Down-regulation of type I insulin-like growth factor receptor increases sensitivity of breast cancer cells to insulin. Cancer Res. 67, 391–397.
Keywords: insulin receptor, IGF-I receptor, cancer, insulin resistance, IGF-I, IGF-II, IR isoform A, target therapy
Citation: Malaguarnera R and Belfiore A (2011) The insulin receptor: a new target for cancer therapy. Front. Endocrin. 2:93. doi: 10.3389/fendo.2011.00093
Received: 19 August 2011; Paper pending published: 14 September 2011;
Accepted: 19 November 2011; Published online: 06 December 2011.
Edited by:
Annamaria Anita Livia Colao, University Federico II of Naples, ItalyCopyright: © 2011 Malaguarnera and Belfiore. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Antonino Belfiore, Endocrinology Unit, Department of Clinical and Experimental Medicine, University of Catanzaro, Campus Universitario, Viale Europa, località Germaneto, 88100 Catanzaro, Italy. e-mail:YmVsZmlvcmVAdW5pY3ouaXQ=